

Abstract
Background
Members of the WRKY protein family, one of the largest transcription factor families in plants, are involved in plant growth and development, signal transduction, senescence, and stress resistance. However, little information is available about WRKY transcription factors in flax (Linum usitatissimum L.).
Results
In this study, comprehensive genome-wide characterization of the flax WRKY gene family was conducted that led to prediction of 102 LuWRKY genes. Based on bioinformatics-based predictions of structural and phylogenetic features of encoded LuWRKY proteins, 95 LuWRKYs were classified into three main groups (Group I, II, and III); Group II LuWRKYs were further assigned to five subgroups (IIa-e), while seven unique LuWRKYs (LuWRKYs 96–102) could not be assigned to any group. Most LuWRKY proteins within a given subgroup shared similar motif compositions, while a high degree of motif composition variability was apparent between subgroups. Using RNA-seq data, expression patterns of the 102 predicted LuWRKY genes were also investigated. Expression profiling data demonstrated that most genes associated with cellulose, hemicellulose, or lignin content were predominantly expressed in stems, roots, and less in leaves. However, most genes associated with stress responses were predominantly expressed in leaves and exhibited distinctly higher expression levels in developmental stages 1 and 8 than during other stages.
Conclusions
Ultimately, the present study provides a comprehensive analysis of predicted flax WRKY family genes to guide future investigations to reveal functions of LuWRKY proteins during plant growth, development, and stress responses.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-021-07697-w.
Keywords: Flax, Transcription factor, WRKY, Phylogenetic analysis, Expression patterns
Background
Flax (Linum usitatissimum L.) is an important industrial crop providing both stem fiber and linseed that are used to produce textiles fiber, edible oil, animal feed, and other industrial products [1]. As of 2011, flax was ranked as the third largest textile fiber crop and the fifth largest oil crop worldwide [2, 3]. Flax is a self-pollinating species with n = 15 chromosomes and a genome size of ~ 370 Mb [4, 5]. Bioinformatics analysis of an assembly of a flax whole-genome shotgun library predicted a total of 43,384 protein-coding genes [4]. Although genomic resources in flax are continuously accumulating to accelerate its varietal improvement program [6–11], the genetic basis for the flax fiber development and adaptation to environmental stress has not been fully explored. Therefore, a better understanding of the regulation mechanisms of flax development and stress resistance is critical to make progress and improvements in further flax breeding.
Transcription factors are clue elements in the regulation of signal transduction pathways in living organisms [12]. They often function as central regulators and molecular switches that activate or repress transcription of multiple target genes [13, 14]. The WRKY gene family, one of the largest families of transcription factors, has received increasing attention for its members’ roles in plant growth, regulation of defense responses, and stress responses [15–17]. WRKY proteins, which apparently exist exclusively in plants, share a WRKY domain (WD) that is comprised of about 60 amino acid residues [18]. Within the WRKY domain, two conserved sequences are present, a WRKYGQK sequence at the N-terminal end and a C2H2- or C2HC-type zinc-binding motif at the C-terminal end [19–21]. Zinc ions are required for WRKY binding to DNA target sequences, with impairment of binding observed in the presence of metal-chelating agents such as EDTA and 1,10-o-phenanthroline [22, 23]. The specific WRKYs-binding site within a gene promoter is referred to as the W-box. The W-box contains the consensus sequence (C/T)TGAC(T/C) that preferentially binds to all WRKY transcription factors (TFs) except for SPF1 [24]. WRKYs binding specificities for certain promoters may be influenced both by sequences flanking the W-box TGAC core motif and by distinct clustering patterns of functional W-boxes within promoters [24]. WRKY proteins are assigned to three groups (Group I, II, and III) based on number of WRKY domains and zinc finger motif structure [19]. Group I WRKYs contain two WRKY domains and two C–X4–5–C–X22–23–H–X–H (C2H2)-type zinc finger motifs. Group II WRKYs contain only one WRKY domain and a C2H2-type zinc finger motif and proteins of this group have been further subdivided into five subgroups based on phylogenetic relationships (IIa–e). Group III WRKYs contain one WRKY domain and a C–X7–C–X23–H–X–C (C2HC)-type zinc finger motif [19, 25].
Since the first WRKY gene, SPF1, was cloned from sweet potato, a large number of WRKY proteins have been identified in a variety of plant species [26–31]. WRKY proteins have been shown to play important roles in growth and development, signal transduction, senescence, and stress resistance [25]. For example, after the Panax ginseng gene PgWRKY6 was cloned and identified by Yang Y et al., it was shown to be upregulated during 2,4-dichlorophenoxyacetic acid (2,4-D)-induced embryogenic callus development; silencing of PgWRKY6 expression markedly reduced the embryogenic callus induction rate, highlighting the crucial role of this WRKY gene in P. ginseng hairy root somatic embryogenesis [32]. In Arabidopsis, biosynthesis of plant secondary cell walls (SCWs), which are composed mainly of cellulose, xylan, and lignin, has been shown to be regulated by a complex transcriptional network involving WRKYs activities [33, 34]. Specifically, AtWRKY12 was shown to function as a transcriptional repressor, while AtWRKY13 was shown to exert transactivation activity to induce stem lignin biosynthesis through direct NTS2 promoter binding [35]. Evidence for AtWRKY12 repression of SCW formation was obtained from experimental results showing enhanced SCW formation from pith cells in an Atwrky12 loss-of-function mutant, while in poplar, PtrWRKY19, a functional ortholog of AtWRKY12, also repressed SCW development from pith cells [36]. Additionally, over-expression of grape Group I VvWRKY2 in tobacco has been shown to alter expression of genes involved in the lignin biosynthetic pathway and cell wall formation [37].
In addition to their cell wall effects, WRKY proteins have been shown to control or modulate plant regulatory networks involving hormonal signaling mediators, including salicylic acid (SA), jasmonic acid (JA), gibberellic acid (GA), abscisic acid (ABA), and ethylene (ET) [38–41]. With regard to plant cell signaling, WRKY transcription factors (TFs), referred to as “jack-of-all-trades” factors, participate in both biotic and abiotic stress responses, with members of all WRKY subfamilies shown to be involved in responses to drought and salt stresses [18]. For example, AtWRKY18, AtWRKY40, and AtWRKY60 Group II subfamily IIa/IIb members negatively regulate transcription of receptor-like kinase CRK5 [41]. Meanwhile, Group I AtWRKY1 TF binds to promoters of MYB2, ABCG40, DREB1A, and ABI5 to regulate the drought response [42]. In addition, WRKYs can influence salt sensitivity, as Group I AtWRKY8 expression is significantly upregulated in plant roots under salt stress [43]. This observation aligns with results of a study showing that an AtWRKY8 knockout mutant exhibited greater salt sensitivity (manifesting as growth inhibition) after seed germination as compared to plants with a functional AtWRKY gene [44].
Other research has also suggested involvement of WRKYs in microbe-associated molecular pattern-triggered immunity, PAMP-triggered immunity, effector-triggered immunity, and system acquired resistance (SAR) [45]. For example, Group III WRKY PtrWRKY89, a regulator of a poplar SA-dependent defense-signaling pathway, has been implicated in plant pathogen resistance, as overexpression of its SA-inducible gene PtrWRKY89 led to enhanced expression of pathogen-related (PR) protein genes and improved transgenic poplar pathogen resistance [46]. Meanwhile in Arabidopsis, nearly all Group III WRKY members have been shown to respond to diverse biotic stresses, with AtWRKY28 and AtWRKY75 possibly acting via the JA/ET pathway to enhance plant resistance to oxalic acid and fungal infection [47].
The WRKY gene family has been suggested to play important and diverse roles in plant growth, development, and stresses tolerance [18]. However, no study to-date has been conducted to identify the WRKY genes in the flax genome. Therefore, a thorough investigation of the flax WRKY gene family might help to reveal critical molecular mechanisms of flax development and stresses tolerance. In the present study, a comprehensive genome-wide bioinformatics analysis was conducted to predict the flax WRKY gene family, yielding 102 LuWRKY members. Sequence features, conserved motifs, gene phylogeny, and expression patterns of LuWRKYs were also determined. Ultimately, the correlation and co-expression network analyses revealed comprehensive information describing the WRKY gene family in flax and provide guidance for future investigations to determine functions of LuWRKY genes during flax growth, development, and stress responses.
Results
Identification and analysis of LuWRKY genes
A total of 107 flax LuWRKY genes were predicted using PlantTFDB then their predicted protein sequences were subjected to Pfam and SMART analyses to confirm the presence of WRKY domains. All protein sequences were manually curated and those that did not contain a WRKY domain-like sequence (WRKY signature amino acid sequence with zinc finger motif) were discarded. Five sequences were excluded from further analysis due to their lack of a typical WRKY domain: Lus10001879, Lus10005131, Lus10005132, Lus10007326, and Lus10009969. Finally, 102 sequences were confirmed as flax WRKY genes (Table S1). Amino acid number, molecular weight, PI, chromosomal location, conserved motif, and domain pattern for each LuWRKY are listed in Table S1. Lengths of LuWRKY proteins ranged from 82 kD (Lus10022278) to 1199 kD (Lus10012030) amino acids and molecular weights fell between 9.29 kD (Lus10022278) and 132.77 kD (Lus10012030). Predicted PI values ranged from 4.61 to 10.76. Subcellular localization analysis showed that all LuWRKY proteins were localized to the nucleus. Although WRKY domains generally contained a highly conserved sequence (WRKYGQK) together with a zinc finger motif sequence at the N-terminus, numerous variants of the ‘WRKYGQK’ signature sequence were observed, including WRKYGHK, WRKYGKK, WKKYGQK, WRKYDQK, and WRKYHQK, which have altered DNA binding affinity. To facilitate understanding of LuWRKYs functions, already characterized orthologous genes in Arabidopsis are also shown in Table S1 based on PlantTFDB.
Phylogenetic analysis
To reveal evolutionary relationships of WRKY genes in flax and Arabidopsis, phylogenetic analyses of 101 LuWRKY and 67 AtWRKY protein sequences were conducted using the neighbor-joining method. Lus10011346 was excluded from the phylogenetic tree because it was too divergent from other sequences to achieve reliable alignment. Diversity was observed with greater prevalence outside rather than within the WD; therefore, full-length WRKY proteins were aligned to maximize the quality of alignments outside the WD and reduce dependency on manual adjustments. Ultimately, 95 LuWRKYs were identified that were assigned to three groups (Group I, II, and III) based on WRKY domain number and type of zinc finger motif (Fig. 1). Group I contained 22 protein sequences that all contained two WRKY domains. Group II and group III protein sequences contained one WRKY domain with various types of zinc finger motifs. The zinc finger motif sequence in Group II was C-X4–5-C-X22–23-H-X1-H (C2H2), while that found in Group III was C-X7-C-X23–27-H-T-C (C2HC). Of the 57 LuWRKYs assigned to Group II (based on the presence of one WRKY domain and a C2H2-type zinc finger motif), 4, 11, 19, 11 and 12 LuWRKYs were assigned to Group II subgroups IIa, IIb, IIc, IId and IIe, respectively. Meanwhile, 16 LuWRKYs, each with one WRKY domain and one C2HC-type zinc finger motif, were assigned to Group III. Surprisingly, seven LuWRKYs (Lus10012027, Lus10012029, Lus10012030, Lus10012678, Lus10016282, Lus10026409 and Lus10033000) were not assigned to any group, due to their unique structural features that precluded clear assignments into groups/subgroups. For example, Lus10026409 had only one WRKY domain but shared greater sequence homology with Group I members (with two WRKY domains), while Lus10012030 and Lus10016282 had more than two WRKY domains.
Fig. 1.

Phylogenetic tree of 101 flax WRKY proteins and 67 Arabidopsis WRKY proteins. The phylogenetic tree was constructed using MEGA 5.0 based on the neighbor-joining method, with bootstrap testing performed for 1000 replicates. The seven groups/subgroups are shown in different colors and unclassified proteins are indicated by red circles
Conserved motif identification
Conserved motifs of LuWRKY proteins were predicted using the MEME program. A total of eight distinct motifs were identified outside the WRKY domain. As shown in Fig. 2, Group I proteins contained two WRKY domains located at the N-terminus and C-terminus of the protein. Only the C-terminal WRKY domain was present in members of Groups II and III; the C-terminal WRKY domain possessed DNA binding functions. Most LuWRKY proteins within the same subgroup showed similar motif compositions, while high motif composition variability was observed between subgroups. For example, all LuWRKY proteins in Group I possessed motif 2, while all Group IId members contained motifs 6 and 7. Meanwhile, motif 3 and motif 1 were specific to Group I and Group III, respectively, while common motifs 5 and 8 were shared by Groups IIa and IIb and motif 4 was shared by most members of Groups I, IIb, and IIc.
Fig. 2.
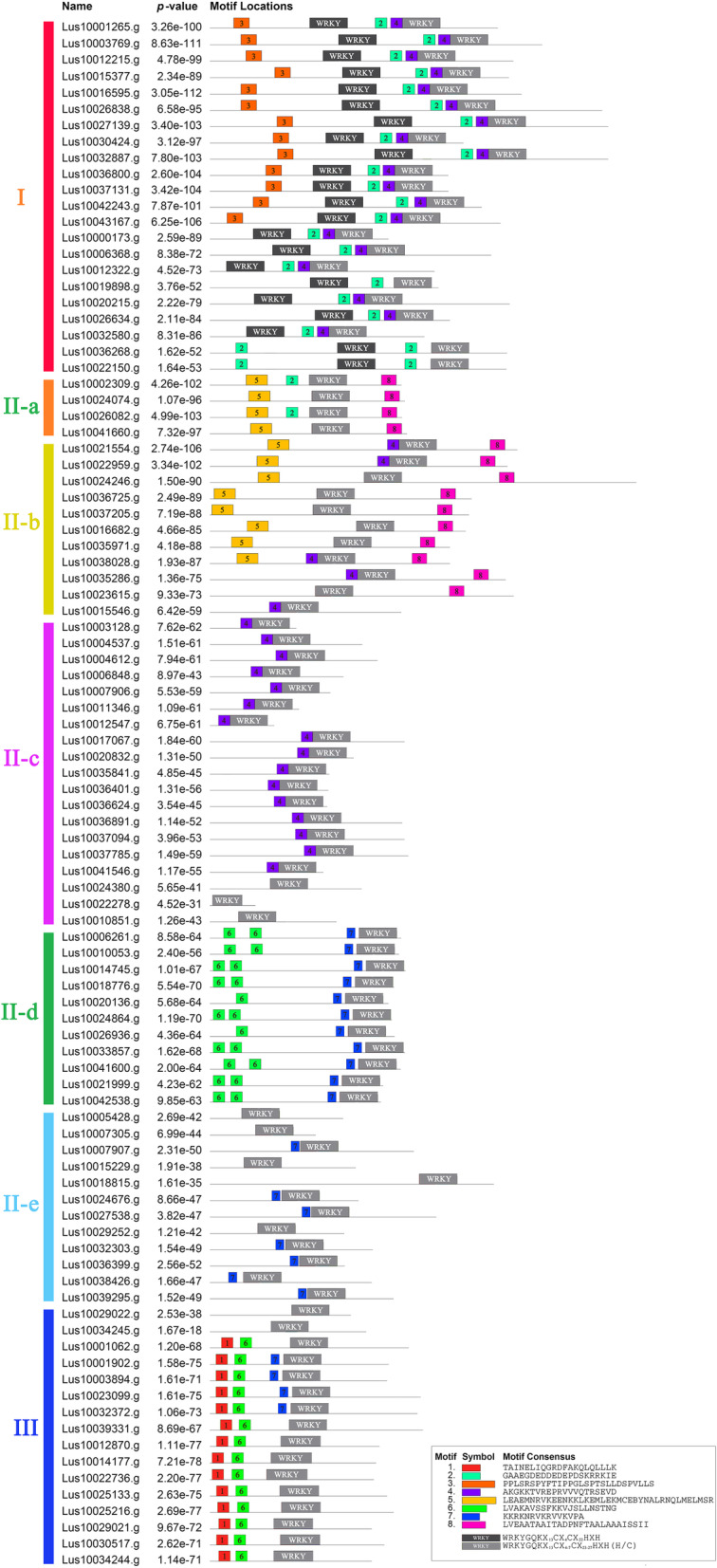
Distributions of conserved motifs in LuWRKY genes. Eight putative motifs are indicated in differently colored boxes. N-terminal and C-terminal WRKY domains are indicated in dark and light gray boxes respectively
Expression patterns of LuWRKY genes
The data that support the findings of this study have been deposited in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession number CNP0001606. Using RNA-seq data, expression patterns of 102 LuWRKYs were determined and FPKM values of genes encoding these LuWRKYs are shown in Table S2. Among the 102 LuWRKY genes, 14 showed very low levels of accumulated transcripts across all samples (FPKM < 1). These genes may be pseudogenes or they possibly may vary in spatial and temporal expression patterns. Heatmaps for LuWRKY genes showing FPKM values converted to log10 values were constructed using Heml software (Fig. 3).
Fig. 3.

Hierarchical clustering of gene expression levels determined using RNA-seq at different fiber development stages (a) and in different tissues (b). FPKM values of LuWRKYs were transformed by log10. S1, seedling stage; S2, fir like stage; S3, early fast growing stage; S4, fast growing stage; S5, bud stage; S6, flowering stage; S7, green stage; S8, maturity stage. Upper, middle, and lower third zones of stem, root, and leaf at late fast growing stage are designated SU, SM, SD, R, and L, respectively
Next, expression profile data were divided into two parts, with one part related to different fiber development stages (Fig. 3a) and the other part related to relative expression level in different organs (Fig. 3b). As shown in Fig. 3a, 11 of the 102 genes (10.78%) were highly expressed (FPKM > 10) at all developmental stages in stems. In addition, many genes exhibited their highest expression levels at early or late stages of fiber development, including 22 genes (21.57%) at stage 1 and 57 (55.88%) at stage 8; 89 genes (87.25%) were expressed in all three organs (stem, root, and leaf) (Fig. 3b), while 29 genes showed predominant expression in only one tissue, including 3 (2.94%) in stem, 13 (12.75%) in root, and 13 (12.75%) in leaf. Meanwhile, 17 genes were differentially expressed in stem, with expression levels of 14 genes observed to proportionally increase with stem position (i.e., bottom > middle > top) and expression of three genes exhibiting the opposite pattern (i.e., top > middle > bottom).
Validation of RNA-seq data by quantitative RT-PCR (qRT-PCR)
To further verify the accuracy of flax digital gene expression (DGE) profiles, the expression levels of eight randomly selected genes were analyzed by qRT-PCR, including LuCesA8 (Lus10007296), LuCesA3 (Lus10007538), LuCesA4 (Lus10008225), LuWRKY83 (Lus10012870), LuNAC10 (Lus10013967), LuWRKY47 (Lus10020832), LuWRKY86 (Lus10023099) and LuMyb46 (Lus10039610). The results showed that expression levels of the eight genes determined by qRT-PCR agreed with the results of sequencing analysis and the RNA-seq data were reliable (Fig. 4).
Fig. 4.

Validation of RNA-seq data by qRT-PCR. The red line represents the value of FPKM in the DGE profile and the blue histogram represents the expression level of eight genes detected by qRT-PCR
Correlation analyses
After plant cellulose, hemicellulose, and lignin contents were determined at different developmental stages and in different tissues (Table S3), the correlations between the expression levels of LuWRKY genes and the contents of cellulose, hemicellulosic and lignin were analyzed (Fig. 5). Of the total 102 LuWRKY genes, expression levels of nine genes showed significantly positive correlations with cellulose content, while only LuWRKY49 (Lus10024380) was negatively correlated with cellulose content (p < 0.05). LuWRKY30 (Lus10022959) and LuWRKY71 (Lus10015229) were found to be positively and negatively correlated with hemicellulose content (p < 0.05), respectively. Meanwhile, expression levels of sixteen genes showed significant positive correlations with lignin content, and only LuWRKY10 (Lus10020215) negatively correlated with lignin content (p < 0.05). Importantly, these results suggested that correlation analysis was useful for identifying genes that potentially exerted key regulatory effects on cellulose, hemicellulose, and lignin synthesis in flax.
Fig. 5.

Correlation analyses between LuWRKY gene expression and cellulose, hemicellulose, and lignin contents. Pearson correlation coefficients were shown in the box. The level of significance was set to p < 0.05. * Correlation is significant at the 0.05 level (2-tailed). ** Correlation is significant at the 0.01 level (2-tailed)
Co-expression network analysis
A total of 42,886 genes detected in expression profiling data were subjected to weighted gene co-expression network analysis to reveal genes co-expressed with LuWRKYs (based on screening for proteins with scores above 0.5). After the co-expression network was constructed and visualized using Cytoscape (Fig. 6), seven LuWRKYs genes, including LuWRKY38 (Lus10003128), LuWRKY84 (Lus10014177), LuWRKY49 (Lus10024380), LuWRKY87 (Lus10025133), LuWRKY88 (Lus10025216), LuWRKY93 (Lus10034244), and LuWRKY37 (Lus10038028), were identified as hub genes with high co-expression correlations with 361 other genes. Table S4 lists co-expressed genes with correlation coefficients. Of 361 identified co-expressed genes, 228 were annotated using the GO database (Fig. 7). The GO term “binding” (GO: 0005488) best described the greatest number of genes (88), while the GO term “metabolic process” (GO: 0008152) best described 49 genes, and the GO term “cellular process” (GO:0009987) best described 38 genes.
Fig. 6.

Co-expression genes network of LuWRKY genes. The colors of the circle represent the different connectivities and ranges from green (genes with low connectivities) to red (genes with high connectivities)
Fig. 7.

Go enrichment analysis of 228 co-expression genes with LuWRKTs. The X-axis represents number of genes, and Y-axis represents different GO terms
Discussion
The WRKY TF family ranks as the seventh largest TF family in plants, after basic helix-loop-helix (bHLH), myeloblastosis-related (MYB), ethylene responsive factor (ERF), NAC (NAM, no apical meristem, ATAF1/2, and CUC2, cup-shaped cotyledon), basic leucine zipper (bZIP), and C2H2 TF families [45]. Although WRKY genes appear to exist in some diplomonads, social amoebae and other amoebozoa, and members of fungal class incertae sedis, WRKY genes are absent in other non-plant species [48]. The first cDNA encoding a WRKY protein, SPF1, was cloned from sweet potato (Ipomoea batatas) [26]. The WRKY family arose during evolution through tandem and segmental gene duplication. To date, 14,549 WRKY genes from 166 plant species have been deposited in PlantTFDB [47], including 72 WRKYs in Arabidopsis, 116 in cotton, 103 in rice, and 104 in poplar [45]. Genome information of flax (L. usitatissimum) revealed that whole-genome duplication (WGD) had occurred in the lineage of L. usitatissimum between 5 and 9 Mya [4] and subsequently gave rise to the 102 flax genes identified as WRKY genes in the present study.
Based on phylogenetic analyses and WRKY domain structures, 95 of the 102 LuWRKYs were assigned to seven groups (Groups I, IIa, IIb, IIc, IId, IIe, and III). Twenty-two WRKY proteins possessing two WRKY domains (at the N terminus and C terminus) were assigned to Group I, while those possessing a single C-terminal WRKY domain were assigned to Group II or III. Importantly, the two WRKY domains of Group I members have distinct functions; the C-terminal domain plays a major role in binding to the W-box, while the function of the N-terminal WRKY domain remained unclear and might influence promoter binding specificity and affinity [23, 49]. Notably, only the C-terminal WRKY domain is responsible for sequence-specific binding to DNA, as AtWRKY1 recognition of the W-box appears to mainly depend on the presence of the C-terminal WRKY domain, while the presence of the N-terminal WRKY domain only slightly influenced the protein–DNA interaction [23, 50]. Many variants of ‘WRKYGQK’ signature-sequence are present in LuWRKYs, including WRKYGHK, WRKYGKK, WKKYGQK, WRKYDQK, and WRKYHQK [51].WRKY domain show high affinity binding to a DNA sequence, termed the W-box sequence (C/T)TGAC(C/T), which is found in the promoter region of many genes [52].
In addition to the W box, a recent study indicates that the WRKY domain can also bind to SURE, a sugar responsive cis element, as a transcription activator [53]. In group-IId WRKYs, a plant zinc-cluster domain (PF10533) was present upstream of the WDs. The LuWRKY proteins in group III are typified mainly by having the less common CX7CX23–27HXC zinc binding motif [19]. It has been reported that substitutions of the WRKYGQK residues in the WRKY domain decreased the DNA-binding affinity, and any mutations of the conserved cysteine and histidine of the zinc-binding motif abolished the protein–DNA interaction [51].
The WRKY genes are believed to have originated approximately 1.5 to 2 billion years ago in eukaryotes prior to the divergence of plant phyla; this phylogeny clearly aligns with results of a recent report outlining the evolution of WRKY genes in flowering plants [48]. Based on our phylogenetic motif analyses here, we propose four major WRKY transcription factor lineages in flax: Groups I + IIc, Groups IIa + IIb, Groups IId + IIe, and Group III. These lineages align with a previous hypothesis asserting that a proto-WRKY ancestral gene with a single WRKY domain underwent domain duplication to produce Group I WRKY genes; thus, an ancestral Group I WRKY would have given rise to all WRKY genes. Subsequent loss of the N-terminal WRKY domain led to Group IIc genes. In the present study, along with clear division of most LuWRKY proteins, some exceptions were also present. For example, the WRKY domain structures of LuWRKY96 (Lus10012027), LuWRKY97 (Lus10012029), LusLuWRKY98 (Lus10012030), LuWRKY99 (Lus10012678) and LuWRKY102 (Lus10033000) were intermediate type between group I and group IIc. The LuWRKY101 (Lus10026409) proteins belong to group-IIc based on WD structure, but it was clustered with the group-I members in phylogeny. The results were consistent with the previous reports and indicated that the group-IIc was more evolutionarily close to group-I than other groups [54]. The presence of this PR intron in Group IId, IIe and III WRKY genes supports the hypothesis that these groups evolved from the group I C-terminal domain [48, 55]. In addition, Group III genes appear to share a common motif 7 with the Group IId + IIe, which indicated that the three groups were adjacent in evolutionary relationship. The recent work on the evolution of the WRKY gene family, proposed Group IIe genes predate Group IId genes [24] and Group III genes evolved earlier than groups IIa and IIb [56]. Group IIa genes are the group with the smallest number of members in flax. However, the lack of clustering of members of flax Group IIa and Arabidopsis Group IIa implies that diversification occurred after the divergence of monocots and dicots. In addition, all of the main groups of WRKY genes that are present in flowering plants are present in Selaginella moellendorffii except for Group IIa genes which were therefore the last to evolve and might appear to have arisen from Group IIb genes [48].
Numerous studies have shown that WRKY genes play crucial roles in diverse physiological and developmental processes [19, 41, 57]. Specifically, AtWRKY2 and AtWRKY34 are redundantly involved in pollen formation, pollen tube elongation, seed germination, and early growth after germination [58]. Lus10027139 and Lus10032887, homologues of AtWRKY2, are predominately expressed in plant stems and show a high level of expression during fiber development in flax. This implies that these homologues might be potential regulators of fiber formation in addition to their role in regulating pollen formation and seed germination. Almost all plant cells possess primary cell walls; however, some specialized cells, such as fiber cells, form thickened secondary cell walls. The deposition of SCWs provides mechanical strength, enhanced water-conducting capabilities [59, 60], and a defense structure to prevent pathogen entry into cells. In flax, bast fibers are produced that have very thick SCWs that contain high amounts of cellulose (> 70%) with low lignin content (2–7%). Indeed, the synthesis of SCWs in numerous plant species involves activity of WRKYs. For instance, SCW biosynthesis in potatoes is regulated via a complex transcriptional network [34], with StWRKY1 exerting direct control over secondary cell wall thickening through its action on the promoters of hydroxycinnamic acid amide (HCAA) biosynthetic genes, encoding 4-coumarate-CoA ligase (4-CL) and tyramine hydroxycinnamoyl transferase (THT). In grape plants, WRKY2 plays a role in regulating lignification, while tobacco plants over-expressing VvWRKY2 exhibit altered expression of genes involved in lignin biosynthesis and cell wall formation [37]. In Arabidopsis, AtWRKY13 has been reported to bind the AtNST2 promoter and regulate AtNST2 gene expression during SCW synthesis associated with sclerenchyma cell development [35, 61]. In the present study, correlation analysis indicated that expression levels of 29 LuWRKYs could be significantly correlated with cellulose, hemicellulose, or lignin content. These genes were mainly categorized into Groups I + IIc or Groups IId + IIe, with expression profiling data showing that most of them were expressed predominantly in stem. Both Lus10020832 and Lus10012678, homologues of AtWRKY13, showed significantly positive correlations with cellulose content, implying their putative roles in SCW formation. Lus10024380, a homologue of AtWRKY49, exhibited significantly negative correlations with cellulose content, which may function as a negative regulator and repress SCW biosynthesis in flax. Moreover, Lus10006368, Lus10016595, Lus10037094, Lus10033857, and Lus10042538, homologues of AtWRKY1, 20, 57, 21, and 74, respectively, displayed similar expression patterns and up-regulated expression during flax fiber development. Their expression levels were usually lowest in the top part of the stem, gradually increased in the middle, and were highest at the bottom part of the stem. The results strongly suggest that these genes likely play key roles during fiber development [62].
In addition to SCW synthesis, WRKY genes have been also shown to play important roles in responses to various abiotic stresses, including drought, salt, heat, and osmotic stresses [45]. For example, AtWRKY15 modulates plant growth and mediates salt/osmotic stress responses in Arabidopsis [63]. CRK5, a receptor-like protein kinase, is involved in abscisic acid (ABA) signaling and drought tolerance. AtWRKY18/40/60 negatively regulates the transcription of CRK5 in Arabidopsis thaliana. Meanwhile, AtWRKY25/26/33 genes have demonstrated to participate in heat-induced signal transduction [64]. Corresponding to these characterized Arabidopsis WRKYs, transcripts of the flax orthologues of AtWRKY15 (Lus10006261, Lus10041600), AtWRKY33 (Lus10001265, Lus10012215, Lus10042243, Lus10026409), and AtWRKY40 (Lus10002309, Lus10024074, Lus10026082) showed significant induction under saline-alkaline stress [65]. In addition, AtWRKY46/54/70 genes belong to Group III and encode important signaling components that regulate BR-regulated growth and osmotic stress [66]. Among the orthologous genes of AtWRKY46/54/70 in flax, Lus10012870, Lus10025133, Lus10025216, and Lus10030517, identified in response to BR [67], Lus10012870 and Lus10030517 were involved in flax osmotic resistance [68]. Expression profile data indicate that the genes, including Lus10001265, Lus10002309, Lus10012215, Lus10012870, Lus10024074, Lus10026082, Lus10026409, and Lus10043167, exhibited very similar expression patterns in the current work. They showed distinctly higher expression levels at stages 1 and 8 than the other stages and were predominantly expressed in leaves. The majority of these genes are classified into Groups I, IIa, and III. However, these genes have yet to be functionally characterized in flax successfully. We speculate that the genes might be promising candidate regulators involved in stress tolerance in flax.
WRKYs have also been shown to function as a hub to integrate signaling of multiple plant defensive phytohormones (JA, SA, ABA, GA, ET) during regulation of disease resistance and biotic stress responses [69]. Expression of AtWRKY7, a negative regulator of plant defense signaling during infection with bacterial pathogen Pseudomonas syringae, is induced by SA and P. syringae [70], while AtWRKY57 expression, which also negatively regulates plant defense signaling to infection, increases susceptibility of plants to Botrytis cinerea [71]. Conversely, AtWRKY4 enhances plant resistance to both necrotrophic and biotrophic pathogens, while upregulated OsWRKY71 levels induced by SA, methyl jasmonate (MeJA), or pathogen infection leads to enhanced resistance to Xanthomonas oryzae, as observed for an OsWRKY71 overexpression mutant [72]. With regard to plant antifungal defenses, AtWRKY28 and AtWRKY75 may enhance plant resistance to fungal infection through the JA/ET pathway [73], while LuWRKY36, a homolog of AtWRKY33, appears to promote secoisolariciresinol biosynthesis in response to Fusarium oxysporum elicitors [74].
Lignin is both developmentally deposited and pathogen-induced in the secondary thickened cell wall [75]. As a defensive chemical barrier, lignin plays important roles in preventing pathogen invasion. In fact, defense-induced lignification is a conserved basal defense mechanism employed by a wide range of plant species. In cotton (Gossypium hirsutum), quantitative analysis of resistance to wilt fungus Verticillium dahliae revealed an association between increased lignification in stems upon infection and resistance against wilt [76]. Meanwhile, transgenic tobacco plants constitutively overexpressing lignification-enhancing phenylalanine ammonia lyase (PAL) genes exhibited greater resistance to pathogens Cercospora nicotianae and Phytophthora parasitica cv. Nicotianae [77, 78], while RNAi-mediated suppression of expression of the gene encoding cinnamyl alcohol dehydrogenase (CAD) (normally expressed during normal vascular cell wall lignification) increased flax susceptibility to vascular fungus Fusarium oxysporum [79]. Fundamentally, little is known regarding the distinction between vascular cell wall lignification and defense-induced lignification in plants and their precise regulatory mechanisms. Nevertheless, in this study, the expressions of Lus10026634, Lus10004537, Lus10004612, Lus10036891, Lus10037094, Lus10041546, and Lus10010053 belonging to Group I + IIC were correlated with lignin biosynthesis, while their homologues, such as AtWRKY4, 7, 57, 71, and 75, in Arabidopsis are known to play important roles in plant response to various biotic stresses. It remains unclear if these LuWRKYs also participate in the regulation of defense-induced lignification in flax. Therefore, further studies are needed to determine the function of the LuWRKYs in immune regulation.
Conclusion
In this study, we conducted a genome-wide search for flax WRKY gene family members that led to identification of a total of 102 WRKY genes. Subsequent bioinformatics-based analyses of WRKY proteins revealed LuWRKYs amino acid numbers, molecular weights, predicted isoelectric point (PI) values, chromosomal locations, domain patterns, and conserved motifs. LuWRKYs were phylogenetically classified into three groups (Groups I, II, III), with Group II further divisible into five subgroups, for a total of seven LuWRKYs subgroups. Using RNA-seq data, expression patterns of LuWRKYs were determined at different developmental stages in diverse tissues. Notably, expression of 10, 2, and 17 genes were found to be significantly correlated with the cellulose, hemicellulose, and lignin contents, respectively. Moreover, many LuWRKYs were also shown to play important roles in responses to various biotic and abiotic stresses. The results of this study present comprehensive information describing the WRKY gene family in flax and provide useful clues to guide future investigations to determine functions of LuWRKY genes during flax growth, development, and stress responses.
Methods
Plant materials
The fiber flax variety ‘Diana’ was used in this study. Plants were grown in the experimental field of the Industrial Crops Institute of Heilongjiang Academy of Agricultural Sciences (Harbin, P.R. China) under natural conditions. They were planted in a 4-m2 (2,0 m × 2,0 m) plot with 2000 plants per m2 with a raw spacing of 20 cm. The soil of the experimental plot was chernozem (pH of 6.8). Hand weeding was used and there are no irrigation or fertilization treatments.
For analysis of differential gene expression profiles and determination of fiber chemical composition, plant samples were collected at different stages. The middle third of the stem was collected at eight different stages of flax fiber development: seedling stage (4th pair of true leaves unfolded), fir-like stage (stem, 10% of final length), early fast growing stage (stem, 30% of final length), fast growing stage (stem, 50% of final length), bud stage (visible flower buds), flowering stage (50% of flower open), green stage (seeds green and undeveloped) and maturity stage (plants are developed for harvesting of fiber type). In addition, the upper (9–15 cm from the shoot apex), middle (33–39 cm from the shoot apex), and lower (57–63 cm from the shoot apex) sections of the stems, roots (main root and fine root), and leaves (middle section) were also collected during the late fast growing stage (stem, 80% of final length, length of plant 72 cm).
For DGE analysis, thirteen samples were collected, and three biological replicates were produced for each sample. The samples used for chemical composition determination were prepared in triplicate, and 5 individual plants were pooled as one replicate. After samples were collected, they were immediately frozen in liquid nitrogen then stored at − 80 °C.
RNA extraction
Each plant tissue sample (kept frozen in liquid nitrogen) was ground into a fine powder using mortar and pestle. Plant total RNA was extracted using the cetyl trimethylammonium bromide (CTAB) method. For each sample, 4 μg of total RNA was digested in a 25-μl total volume with DNase I (Promega, Madison, WI, USA) to remove genomic DNA contamination. RNA quality was checked via 1.0% agarose gel electrophoresis followed by RNA visualization and quantification using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
DGE library preparation and sequencing
Sequencing data was filtered using SOAPnuke (v1.5.2) [41] by (1) removing reads containing sequencing adapter; (2) removing reads whose low-quality base rate (base quality less than or equal to 5) was > 20%; (3) removing reads whose unknown base (‘N’ base) frequency was > 5%. After filtering, clean reads were stored in FASTQ format then were mapped to the reference genome using HISAT2 (v2.0.4) [80]. Bowtie2 (v2.2.5) [81] was applied to align clean reads to the reference coding gene set then expression levels of genes were calculated using RSEM (v1.2.12) [82].
RNA-sequencing (RNA-seq) data analysis
High-throughput sequencing analysis software (HTSeq-v0.5.3) was used to enumerate the number of fragments mapped to each gene. Based on gene lengths and fragment counts mapped per gene, fragments per kilobase per million mapped fragments (FPKM) values were calculated for each gene in conjunction with sequencing depth and gene length ranges for fragment counts. Ultimately, FPKM values were used to estimate gene expression levels [83].
Identification of LuWRKY genes in flax
Protein sequences and DNA-binding domains of WRKY proteins were obtained from the Plant Transcription Factor Database (PlantTFDB) at (http://planttfdb.cbi.pku.edu.cn/). All candidate genes were further examined by confirming they contained WRKY core sequences using PFAM (http://pfam.xfam.org) and SMART (http://smart.embl-heidelberg.de/) online tools. Basic information about these genes, including amino acid numbers, molecular weights, predicted isoelectric points (PIs), conserved motifs, and domain patterns, were acquired through PlantTFDB. The chromosomal location was obtained through the Phytozome12 website (https://phytozome.jgi.doe.gov/). Subcellular localization was predicted using Cell-PLoc 2.0 website tools (http://www.csbio.sjtu.edu.cn/bioinf/Cell-PLoc-2/).
Analysis of phylogenetic relationship and conserved motifs
Full-length amino acid sequences of WRKYs derived from Arabidopsis were obtained using online phytozome12. Multiple alignments of 101 LuWRKYs and 67 AtWRKYs protein sequences were performed via ClustalW (version 1.83) using default parameters. A phylogenetic tree was constructed using the neighbor-joining method of MEGA 5.0 with 1000 bootstrap replicates. Conserved motifs of LuWRKYs were identified via the MEME program (version 5.1.1, http://meme-suite.org/tools/meme) using the following parameters: any number of repetitions, maximum of 10 motifs, and an optimum motif width of 6 to 60 amino acid residues.
Expression pattern analysis of LuWRKY genes
RNA-seq data expressed as FPKM was downloaded to study expression patterns of LuWRKY genes. To render the data suitable for cluster displays, absolute FPKM values were divided by the mean of all values then ratios were transformed into log10 values. HemI 1.0 software was used to generate the heatmap then heatmap analysis was performed using OmicShare tools, a free online platform for data analysis (http://www.omicshare.com/tools).
Quantitative RT-PCR analysis
cDNA synthesis was performed using 1 μg of DNase I-treated RNA using the PrimeScript RT Reagent Kit (TaKaRa, Japan) according to the manufacturer’s protocol. qRT-PCR was performed to determine transcript levels with quantification performed using an Opticon machine (Biorad, Hercules, CA, USA) after amplification using a real-time PCR Mix Kit with SYBR Green fluorescent dye (TOKOBO). To normalize variance among samples, the stably expressed GAPDH, EF1A and ETIF5A genes were used as internal controls [84]. The middle third of the stem at seedling stage was used as the sample normalizer. The relative expression levels were calculated from the threshold cycle according to the 2-ΔΔCt method [85] and the experiments were carried out in triplicate to ensure reproducibility of each sample. Gene-specific primers were designed using Primer 5.0 software and primer sequences are shown in Table S5.
Correlation analyses
Cellulose, hemicellulose, and lignin contents were detected in plant samples using the contents detection kits (QIYI, Shanghai) via UV spectrophotometry. The procedures were performed following the instructions of the kits. Cellulose content was measured by the anthrone method, and hemicellulose content was detected using the 3,5-Dinitrosalicylic Acid (DNS) method. The acetylbromide method was employed to determine the lignin content.
Co-expression network analysis
Normalization and processing of expression profile data were performed using the R software package. The normalized dataset was modularized using a weighted gene co-expression network analysis (WGCNA) algorithm. Genes co-expressed with LuWRKYs were screened based on threshold value > 0.5 then filtered genes were used to construct the correlation network. The network was visualized using Cytoscape version 3.6.1 (www.cytoscape.org).
Supplementary Information
Additional file 1: Table S1. The character of the WRKY proteins identified in flax.
Additional file 2: Table S2. The FPKM values of the WRKY genes in flax.
Additional file 3: Table S3. The contents of cellulose, hemicellulose, and lignin at different developmental stages and in different tissues.
Additional file 4: Table S4. The co-expressed genes with the LuWRKY genes.
Additional file 5 Table S5. Primers used for qRT-PCR analysis.
Acknowledgements
Not applicable.
Abbreviations
- ABA
Abscisic acid
- bHLH
Basic helix-loop-helix
- bZIP
Basic leucine zipper
- CAD
Cinnamyl alcohol dehydrogenase
- CTAB
Cetyl trimethylammonium bromide
- DGE
Digital gene expression
- DNS
3,5-Dinitrosalicylic Acid
- ERF
Ethylene responsive factor
- ET
Ethylene
- FRDL4
Ferric reductase defective-like 4
- GA
Gibberellin
- HCAA
Hydroxycinnamic acid amide
- JA
Jasmonic acid
- MeJA
Methyl jasmonate
- Myb
Myeloblastosis
- NAC
NAM–ATAF1,2–CUC2
- PR
Pathogen-related
- qRT-PCR
Quantitative RT-PCR
- SA
Salicylic acid
- SAR
System acquired resistance
- SCWs
Secondary cell walls
- THT
Tyramine hydroxycinnamoyl transferase
- WGCNA
Weighted gene co-expression network analysis
- WGD
Whole-genome duplication
Authors’ contributions
GW and XY designed the study. HY, WG and LZ carried out the data analyses, and drafted the manuscript. YY1, SC, LT, LC, QK, XS and JW implemented experimental work. XX were involved in directing the experiments and proofreading the manuscript. YY2, WH, YW and YL finally revised the manuscript. All authors approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31701480, 31471546), Heilongjiang Academy of Agricultural Sciences Foundation (2019KYJL016, 2020FJZX032), Bast fiber research system of Heilongjiang province (YYM19STX-22) and National bast fiber research system of China (CARS-19-S6). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Xie D, Dai Z, Yang Z, Tang Q, Sun J, Yang X, Song X, Lu Y, Zhao D, Zhang L, Su J. Genomic variations and association study of agronomic traits in flax. BMC Genomics. 2018;19(1):512. doi: 10.1186/s12864-018-4899-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.You FM, Jia G, Xiao J, Duguid SD, Rashid KY, Booker HM, Cloutier S. Genetic variability of 27 traits in a Core collection of flax (Linum usitatissimum L) Front Plant Sci. 2017;8:1636. doi: 10.3389/fpls.2017.01636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He L, Xiao J, Rashid KY, Yao Z, Li P, Jia G, Wang X, Cloutier S, You FM. Genome-wide association studies for Pasmo resistance in flax (Linum usitatissimum L) Front Plant Sci. 2019;9:1982. doi: 10.3389/fpls.2018.01982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z, Hobson N, Galindo L, Zhu S, Shi D, McDill J, Yang L, Hawkins S, Neutelings G, Datla R, Lambert G, Galbraith DW, Grassa CJ, Geraldes A, Cronk QC, Cullis C, Dash PK, Kumar PA, Cloutier S, Sharpe AG, Deyholos MK. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012;72(3):461–473. doi: 10.1111/j.1365-313X.2012.05093.x. [DOI] [PubMed] [Google Scholar]
- 5.Ragupathy R, Rathinavelu R, Cloutier S. Physical mapping and BAC-end sequence analysis provide initial insights into the flax (Linum usitatissimum L) genome. BMC Genomics. 2011;12(1):217. doi: 10.1186/1471-2164-12-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dash PK, Cao Y, Jailani AK, Gupta P, Venglat P, Xiang D, Rai R, Sharma R, Thirunavukkarasu N, Abdin MZ, Yadava DK, Singh NK, Singh J, Selvaraj G, Deyholos M, Kumar PA, Datla R. Genome-wide analysis of drought induced gene expression changes in flax (Linum usitatissimum) GM Crops Food. 2014;5(2):106–119. doi: 10.4161/gmcr.29742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta P, Dash PK. Molecular details of secretory phospholipase A2 from flax (Linum usitatissimum L.) provide insight into its structure and function. Sci Rep. 2017;7(1):11080. doi: 10.1038/s41598-017-10969-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dash PK, Rai R, Mahato AK, Gaikwad K, Singh NK. Transcriptome landscape at different developmental stages of a drought tolerant cultivar of flax (Linum usitatissimum) Front Chem. 2017;5:82. doi: 10.3389/fchem.2017.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorshkova T, Chernova T, Mokshina N, Gorshkov V, Kozlova L, Gorshkov O. Transcriptome analysis of intrusively growing flax fibers isolated by laser microdissection. Sci Rep. 2018;8(1):14570. doi: 10.1038/s41598-018-32869-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saha D, Mukherjee P, Dutta S, Meena K, Sarkar SK, Mandal AB, Dasgupta T, Mitra J. Genomic insights into HSFs as candidate genes for high-temperature stress adaptation and gene editing with minimal off-target effects in flax. Sci Rep. 2019;9(1):5581. doi: 10.1038/s41598-019-41936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Roy J, Blervacq AS, Créach A, Huss B, Hawkins S, Neutelings G. Spatial regulation of monolignol biosynthesis and laccase genes control developmental and stress-related lignin in flax. BMC Plant Biol. 2017;17(1):124. doi: 10.1186/s12870-017-1072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arce AL, Cabello JV, Chan RL. Patents on plant transcription factors. Recent Pat Biotechnol. 2008;2(3):209–217. doi: 10.2174/187220808786241024. [DOI] [PubMed] [Google Scholar]
- 13.Liu L, White MJ, MacRae TH. Transcription factors and their genes in higher plants functional domains, evolution and regulation. Eur J Biochem. 1999;262(2):247–257. doi: 10.1046/j.1432-1327.1999.00349.x. [DOI] [PubMed] [Google Scholar]
- 14.Warren AJ. Eukaryotic transcription factors. Curr Opin Struct Biol. 2002;12(1):107–114. doi: 10.1016/S0959-440X(02)00296-8. [DOI] [PubMed] [Google Scholar]
- 15.Rushton PJ, Somssich IE, Ringler P, Shen QJ. WRKY transcription factors. Trends Plant Sci. 2010;15(5):247–258. doi: 10.1016/j.tplants.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Liu LP, Zhang ZQ, Dong JL, Wang T. Overexpression of MtWRKY76 increases both salt and drought tolerance in Medicago truncatula. Environ Exp Bot. 2016;123:50–58. doi: 10.1016/j.envexpbot.2015.10.007. [DOI] [Google Scholar]
- 17.Zhu H, Zhou Y, Zhai H, He S, Zhao N, Liu QA. Novel Sweetpotato WRKY transcription factor IbWRKY2 positively regulates drought and salt tolerance in transgenic Arabidopsis. Biomolecules. 2020;10(4):506. doi: 10.3390/biom10040506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bakshi M, Oelmüller R. WRKY transcription factors: Jack of many trades in plants. Plant Signal Behav. 2014;9(2):e27700. doi: 10.4161/psb.27700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eulgem T, Rushton PJ, Robatzek S, Somssich IE. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000;5(5):199–206. doi: 10.1016/S1360-1385(00)01600-9. [DOI] [PubMed] [Google Scholar]
- 20.Eulgem T, Somssich IE. Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol. 2007;10(4):366–371. doi: 10.1016/j.pbi.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 21.Xu YH, Sun PW, Tang XL, Gao ZH, Zhang Z, Wei JH. Genome-wide analysis of WRKY transcription factors in Aquilaria sinensis (Lour) Gilg. Sci Rep. 2020;10(1):3018. doi: 10.1038/s41598-020-59597-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rushton PJ, Macdonald H, Huttly AK, Lazarus CM, Hooley R. Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of alpha-Amy2 genes. Plant Mol Biol. 1995;29(4):691–702. doi: 10.1007/BF00041160. [DOI] [PubMed] [Google Scholar]
- 23.de Pater S, Greco V, Pham K, Memelink J, Kijne J. Characterization of a zinc-dependent transcriptional activator from Arabidopsis. Nucleic Acids Res. 1996;24(23):4624–4631. doi: 10.1093/nar/24.23.4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang S, Gao Y, Liu J, Peng X, Niu X, Fei Z, Cao S, Liu Y. Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Mol Gen Genomics. 2012;287(6):495–513. doi: 10.1007/s00438-012-0696-6. [DOI] [PubMed] [Google Scholar]
- 25.Wu B, Li MY, Xu ZS, Wang F, Xiong AS. Genome-wide analysis of WRKY transcription factors and their response to abiotic stress in celery (Apium graveolens L) Biotechnol Biotechnol Equip. 2018;32(2):293–302. doi: 10.1080/13102818.2017.1413954. [DOI] [Google Scholar]
- 26.Ishiguro S, Nakamura K. Characterization of a cDNA encoding a novel DNA-binding protein SPF1 that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and beta-amylase from sweet potato. Mol Gen Genet. 1994;244(6):563–571. doi: 10.1007/BF00282746. [DOI] [PubMed] [Google Scholar]
- 27.Song H, Wang P, Nan Z, Wang X. The WRKY transcription factor genes in Lotus japonicas. Int J Genomics. 2014;2014:420128. doi: 10.1155/2014/420128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Y, Wu N, Song W, Yin G, Qin Y, Yan Y, Hu Y. Soybean (Glycine max) expansin gene superfamily origins: segmental and tandem duplication events followed by divergent selection among subfamilies. BMC Plant Biol. 2014;14(1):93. doi: 10.1186/1471-2229-14-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang C, Wang D, Yang C, Kong N, Shi Z, Zhao P, Nan Y, Nie T, Wang R, Ma H. Chen Q genome-wide identification of the potato WRKY transcription factor family. PLoS One. 2017;12(7):e0181573. doi: 10.1371/journal.pone.0181573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie T, Chen C, Li C, Liu J, Liu C, He Y. Genome-wide investigation of WRKY gene family in pineapple: evolution and expression profiles during development and stress. BMC Genomics. 2018;19(1):490. doi: 10.1186/s12864-018-4880-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song H, Sun W, Yang G, Sun J. WRKY transcription factors in legumes. BMC Plant Biol. 2018;18(1):243. doi: 10.1186/s12870-018-1467-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Y, Wang N, Zhao S. Functional characterization of a WRKY family gene involved in somatic embryogenesis in Panax ginseng. Protoplasma. 2020;257(2):449–458. doi: 10.1007/s00709-019-01455-2. [DOI] [PubMed] [Google Scholar]
- 33.Zhong R, Ye ZH. Regulation of cell wall biosynthesis. Curr Opin Plant Biol. 2007;10(6):564–572. doi: 10.1016/j.pbi.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 34.Wang HZ, Dixon RA. On-off switches for secondary cell wall biosynthesis. Mol Plant. 2012;5(2):297–303. doi: 10.1093/mp/ssr098. [DOI] [PubMed] [Google Scholar]
- 35.Li W, Tian Z, Yu D. WRKY13 acts in stem development in Arabidopsis thaliana. Plant Sci. 2015;236:205–213. doi: 10.1016/j.plantsci.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 36.Yang L, Zhao X, Yang F, Fan D, Jiang Y, Luo K. PtrWRKY19 a novel WRKY transcription factor contributes to the regulation of pith secondary wall formation in Populus trichocarpa. Sci Rep. 2016;6(1):18643. doi: 10.1038/srep18643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guillaumie S, Mzid R, Méchin V, Léon C, Hichri I, Destrac-Irvine A, Trossat-Magnin C, Delrot S, Lauvergeat V. The grapevine transcription factor WRKY2 influences the lignin pathway and xylem development in tobacco. Plant Mol Biol. 2010;72(1–2):215–234. doi: 10.1007/s11103-009-9563-1. [DOI] [PubMed] [Google Scholar]
- 38.Zhang ZL, Xie Z, Zou X, Casaretto J, Ho TH, Shen QJ. A rice WRKY gene encodes a transcriptional repressor of the gibberellin signaling pathway in aleurone cells. Plant Physiol. 2004;134(4):1500–1513. doi: 10.1104/pp.103.034967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Antoni R, Rodriguez L, Gonzalez-Guzman M, Pizzio GA, Rodriguez PL. News on ABA transport protein degradation and ABFs/WRKYs in ABA signaling. Curr Opin Plant Biol. 2011;14(5):547–553. doi: 10.1016/j.pbi.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 40.Rushton DL, Tripathi P, Rabara RC, Lin J, Ringler P, Boken AK, Langum TJ, Smidt L, Boomsma DD, Emme NJ, Chen X, Finer JJ, Shen QJ, Rushton PJ. WRKY transcription factors: key components in abscisic acid signaling. Plant Biotechnol J. 2012;10(1):2–11. doi: 10.1111/j.1467-7652.2011.00634.x. [DOI] [PubMed] [Google Scholar]
- 41.Lu K, Liang S, Wu Z, Bi C, Yu YT, Wang XF, Zhang DP. Overexpression of an Arabidopsis cysteine-rich receptor-like protein kinase CRK5 enhances abscisic acid sensitivity and confers drought tolerance. J Exp Bot. 2016;67(17):5009–5027. doi: 10.1093/jxb/erw266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiao Z, Li CL, Zhang W. WRKY1 regulates stomatal movement in drought-stressed Arabidopsis thaliana. Plant Mol Biol. 2016;91(1–2):53–65. doi: 10.1007/s11103-016-0441-3. [DOI] [PubMed] [Google Scholar]
- 43.Ma Q, Xia Z, Cai Z, Li L, Cheng Y, Liu J, Nian H. GmWRKY16 enhances drought and salt tolerance through an ABA-mediated pathway in Arabidopsis thaliana. Front Plant Sci. 2019;9:1979. doi: 10.3389/fpls.2018.01979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu Y, Chen L, Wang H, Zhang L, Wang F, Yu D. Arabidopsis transcription factor WRKY8 functions antagonistically with its interacting partner VQ9 to modulate salinity stress tolerance. Plant J. 2013;74(5):730–745. doi: 10.1111/tpj.12159. [DOI] [PubMed] [Google Scholar]
- 45.Chen F, Hu Y, Vannozzi A, Wu K, Cai H, Qin Y, Mullis A, Lin Z, Zhang LS. The WRKY transcription factor family in model plants and crops. Crit Rev Plant Sci. 2017;36(5):311–335. doi: 10.1080/07352689.2018.1441103. [DOI] [Google Scholar]
- 46.Jiang Y, Duan Y, Yin J, Ye S, Zhu J, Zhang F, Lu W, Fan D, Luo K. Genome-wide identification and characterization of the Populus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J Exp Bot. 2014;65(22):6629–6644. doi: 10.1093/jxb/eru381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, Gao G. PlantTFDB 40: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017;45(D1):D1040–D1045. doi: 10.1093/nar/gkw982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rinerson CI, Rabara RC, Tripathi P, Shen QJ, Rushton PJ. The evolution of WRKY transcription factors. BMC Plant Biol. 2015;15(1):66. doi: 10.1186/s12870-015-0456-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eulgem T, Rushton PJ, Schmelzer E, Hahlbrock K, Somssich IE. Early nuclear events in plant defence signalling: rapid gene activation by WRKY transcription factors. EMBO J. 1999;18(17):4689–4699. doi: 10.1093/emboj/18.17.4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamasaki K, Kigawa T, Inoue M, Tateno M, Yamasaki T, Yabuki T, Aoki M, Seki E, Matsuda T, Tomo Y, Hayami N, Terada T, Shirouzu M, Tanaka A, Seki M, Shinozaki K, Yokoyama S. Solution structure of an Arabidopsis WRKY DNA binding domain. Plant Cell. 2005;17(3):944–956. doi: 10.1105/tpc.104.026435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maeo K, Hayashi S, Kojima-Suzuki H, Morikami A, Nakamura K. Role of conserved residues of the WRKY domain in the DNA-binding of tobacco WRKY family proteins. Biosci Biotechnol Biochem. 2001;65(11):2428–2436. doi: 10.1271/bbb.65.2428. [DOI] [PubMed] [Google Scholar]
- 52.Yu D, Chen L, Zhang L. Transcription factor WRKY superfamily: origin, structure and function. Plant Divers. 2006;1(1):69–77. [Google Scholar]
- 53.Sun C, Palmqvist S, Olsson H, Borén M, Ahlandsberg S, Jansson CA. Novel WRKY transcription factor SUSIBA2 participates in sugar signaling in barley by binding to the sugar-responsive elements of the iso1 promoter. Plant Cell. 2003;15(9):2076–2092. doi: 10.1105/tpc.014597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar K, Srivastava V, Purayannur S, Kaladhar VC, Cheruvu PJ, Verma PK. WRKY domain-encoding genes of a crop legume chickpea (Cicer arietinum): comparative analysis with Medicago truncatula WRKY family and characterization of group-III gene(s) DNA Res. 2016;23(3):225–239. doi: 10.1093/dnares/dsw010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Wang L. The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol. 2005;5(1):1. doi: 10.1186/1471-2148-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, Nishiyama T, Perroud PF, Lindquist EA, Kamisugi Y, Tanahashi T, Sakakibara K, Fujita T, Oishi K, Shin-I T, Kuroki Y, Toyoda A, Suzuki Y, Hashimoto S, Yamaguchi K, Boore JL. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science. 2008;319(5859):64–69. doi: 10.1126/science.1150646. [DOI] [PubMed] [Google Scholar]
- 57.Chen L, Song Y, Li S, Zhang L, Zou C, Yu D. The role of WRKY transcription factors in plant abiotic stresses. Biochim Biophys Acta. 2012;1819(2):120–128. doi: 10.1016/j.bbagrm.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 58.Jiang W, Yu D. Arabidopsis WRKY2 transcription factor mediates seed germination and postgermination arrest of development by abscisic acid. BMC Plant Biol. 2009;9(1):96. doi: 10.1186/1471-2229-9-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Rybel B, Mähönen AP, Helariutta Y, Weijers D. Plant vascular development: from early specification to differentiation. Nat Rev Mol Cell Biol. 2016;17(1):30–40. doi: 10.1038/nrm.2015.6. [DOI] [PubMed] [Google Scholar]
- 60.Huang C, Zhang R, Gui J, Zhong Y, Li L. The receptor-like kinase AtVRLK1 regulates secondary Cell Wall thickening. Plant Physiol. 2018;177(2):671–683. doi: 10.1104/pp.17.01279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang H, Avci U, Nakashima J, Hahn MG, Chen F, Dixon RA. Mutation of WRKY transcription factors initiates pith secondary wall formation and increases stem biomass in dicotyledonous plants. Proc Natl Acad Sci U S A. 2010;107(51):22338–22343. doi: 10.1073/pnas.1016436107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim WC, Ko JH, Kim JY, Kim J, Bae HJ, Han KH. MYB46 directly regulates the gene expression of secondary wall-associated cellulose synthases in Arabidopsis. Plant J. 2013;73(1):26–36. doi: 10.1111/j.1365-313x.2012.05124.x. [DOI] [PubMed] [Google Scholar]
- 63.Vanderauwera S, Vandenbroucke K, Inzé A, van de Cotte B, Mühlenbock P, De Rycke R, Naouar N, Van Gaever T, Van Montagu MC, Van Breusegem F. AtWRKY15 perturbation abolishes the mitochondrial stress response that steers osmotic stress tolerance in Arabidopsis. Proc Natl Acad Sci. 2012;109(49):20113–20118. doi: 10.1073/pnas.1217516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ohama N, Sato H, Shinozaki K, Yamaguchi-Shinozaki K. Transcriptional regulatory network of plant heat stress response. Trends Plant Sci. 2017;22(1):53–65. doi: 10.1016/j.tplants.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 65.Yu Y, Huang W, Chen HWG, Yuan H, Song X, Kang Q, Zhao D, Jiang W, Liu Y, Wu J, Cheng L, Yao Y, Guan F. Identification of differentially expressed genes in flax (Linum usitatissimum L) under saline-alkaline stress by digital gene expression. Gene. 2014;549(1):113–122. doi: 10.1016/j.gene.2014.07.053. [DOI] [PubMed] [Google Scholar]
- 66.Chen J, Nolan TM, Ye H, Zhang M, Tong H, Xin P, Chu J, Chu C, Li Z, Yin Y. Arabidopsis WRKY46 WRKY54 and WRKY70 transcription factors are involved in Brassinosteroid-regulated plant growth and drought responses. Plant Cell. 2017;29(6):1425–1439. doi: 10.1105/tpc.17.00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yuan H, Ning K, Guo W, Tao L, Yu Y, Zhao L, Wu Y, Zhang L, Wu J, Guan F, Yang X, Wu G. Assessment of agronomic parameters and gene expression profiling of flax (Linum usitatissimum L) upon treatment with brassinosteroid and its biosynthetic inhibitor. Ind Crop Prod. 2019;128:270–281. doi: 10.1016/j.indcrop.2018.11.011. [DOI] [Google Scholar]
- 68.Wu J, Zhao Q, Sun D, Zhao Q, Wu G, Zhang L, Yuan H, Yu Y, Zhang SH, Yang Y, Li ZH, Jiang T. Transcriptome analysis of flax ( Linum usitatissimum L) undergoing osmotic stress. Ind Crop Prod. 2018;116:215–223. doi: 10.1016/j.indcrop.2018.02.035. [DOI] [Google Scholar]
- 69.Zhao P, Yao X, Cai C, Li R, Du J, Sun Y, Wang M, Zou Z, Wang Q, Kliebenstein DJ, Liu SS, Fang RX, Ye J. Viruses mobilize plant immunity to deter nonvector insect herbivores. Sci Adv. 2019;5(8):eaav9801. doi: 10.1126/sciadv.aav9801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim KC, Fan B, Chen Z. Pathogen-induced Arabidopsis WRKY7 is a transcriptional repressor and enhances plant susceptibility to Pseudomonas syringae. Plant Physiol. 2006;142(3):1180–1192. doi: 10.1104/pp.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jiang Y, Yu D. The WRKY57 transcription factor affects the expression of Jasmonate ZIM-domain genes transcriptionally to compromise Botrytis cinerea resistance. Plant Physiol. 2016;171(4):2771–2782. doi: 10.1104/pp.16.00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu X, Bai X, Wang X, Chu C. OsWRKY71 a rice transcription factor is involved in rice defense response. J Plant Physiol. 2007;164(8):969–979. doi: 10.1016/j.jplph.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 73.Chen X, Liu J, Lin G, Wang A, Wang Z, Lu G. Overexpression of AtWRKY28 and AtWRKY75 in Arabidopsis enhances resistance to oxalic acid and Sclerotinia sclerotiorum. Plant Cell Rep. 2013;32(10):1589–1599. doi: 10.1007/s00299-013-1469-3. [DOI] [PubMed] [Google Scholar]
- 74.Markulin L, Corbin C, Renouard S, Drouet S, Durpoix C, Mathieu C, Lopez T, Auguin D, Hano C, Lainé É. Characterization of LuWRKY36 a flax transcription factor promoting secoisolariciresinol biosynthesis in response to Fusarium oxysporum elicitors in Linum usitatissimum L hairy roots. Planta. 2019;250(1):347–366. doi: 10.1007/s00425-019-03172-9. [DOI] [PubMed] [Google Scholar]
- 75.Boerjan W, Ralph J, Baucher M. Lignin biosynthesis. Annu Rev Plant Biol. 2003;54(1):519–546. doi: 10.1146/annurev.arplant.54.031902.134938. [DOI] [PubMed] [Google Scholar]
- 76.Xu L, Zhu L, Tu L, Liu L, Yuan D, Jin L, Long L, Zhang X. Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA-Seq-dependent transcriptional analysis and histochemistry. J Exp Bot. 2011;62(15):5607–5621. doi: 10.1093/jxb/err245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Way HM. Constitutive expression of a phenylalanine ammonia-lyase gene from Stylosanthes humilis in transgenic tobacco leads to enhanced disease resistance but impaired plant growth. Physiol Mol Plant Pathol. 2002;60(6):275–282. doi: 10.1016/S0885-5765(02)90407-8. [DOI] [Google Scholar]
- 78.Way HM, Birch RG, Manners JM. A comparison of individual and combined l-phenylalanine ammonia lyase and cationic peroxidase transgenes for engineering resistance in tobacco to necrotrophic pathogens. Plant Biotechnol Rep. 2011;5(4):301–308. doi: 10.1007/s11816-011-0183-2. [DOI] [Google Scholar]
- 79.Wróbel-Kwiatkowska M, Starzycki M, Zebrowski J, Oszmiański J, Szopa J. Lignin deficiency in transgenic flax resulted in plants with improved mechanical properties. J Biotechnol. 2007;128(4):919–934. doi: 10.1016/j.jbiotec.2006.12.030. [DOI] [PubMed] [Google Scholar]
- 80.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 84.Huis R, Hawkins S, Neutelings G. Selection of reference genes for quantitative gene expression normalization in flax (Linum usitatissimum L.) BMC Plant Biol. 2010;10(1):71. doi: 10.1186/1471-2229-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. The character of the WRKY proteins identified in flax.
Additional file 2: Table S2. The FPKM values of the WRKY genes in flax.
Additional file 3: Table S3. The contents of cellulose, hemicellulose, and lignin at different developmental stages and in different tissues.
Additional file 4: Table S4. The co-expressed genes with the LuWRKY genes.
Additional file 5 Table S5. Primers used for qRT-PCR analysis.
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files.