

Abstract
Patients with coronavirus disease 2019 (COVID-19) who are critically ill develop vascular complications characterized by thrombosis of small, medium, and large vessels. Dysfunction of the vascular endothelium due to the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection has been implicated in the pathogenesis of the COVID-19 vasculopathy. Although initial reports suggested that endothelial injury was caused directly by the virus, recent studies indicate that endothelial cells do not express angiotensin-converting enzyme 2, the receptor that SARS-CoV-2 uses to gain entry into cells, or express it at low levels and are resistant to the infection. These new findings, together with the observation that COVID-19 triggers a cytokine storm capable of injuring the endothelium and disrupting its antithrombogenic properties, favor an indirect mechanism of endothelial injury mediated locally by an augmented inflammatory reaction to infected nonendothelial cells, such as the bronchial and alveolar epithelium, and systemically by the excessive immune response to infection. Herein we review the vascular pathology of COVID-19 and critically discuss the potential mechanisms of endothelial injury in this disease.
Following an initial outbreak of pneumonia in Wuhan, China, in December 2019,1 coronavirus disease 19 (COVID-19) has spread rapidly worldwide, infecting more than 186 million people (Johns Hopkins Coronavirus Resource Center, https://coronavirus.jhu.edu, last accessed July 12, 2021). Caused by a new type of coronavirus, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),2 the COVID-19 pandemic has put a major strain on the healthcare systems, causing a global health crisis of unparalleled proportions in modern times.3 Although most patients have recovered from the infection, many experienced a severe form of the disease that requires hospitalization and intensive care, and >3.2 million people have died. Individuals at greatest risk for the fatal complications of COVID-19 have been the elderly and those with underlying conditions, such as lung disease, hypertension, obesity, and diabetes.4
Clinical manifestations of COVID-19 in severely ill patients are adult respiratory distress syndrome and multiorgan system failure.4,5 The clinical course of the disease can be complicated by vascular events, including thrombosis of small, medium, and large blood vessels and thromboembolism.6,7 Although the primary target of SARS-CoV-2 is the respiratory and alveolar epithelium,8 the frequent occurrence of vascular complications in COVID-19 has led to the hypothesis that dysfunction of the endothelium, the inner lining of blood vessels, plays an important role in the progression of this disease into a debilitating and lethal condition.9 Two potential mechanisms have been hypothesized to explain how SARS-CoV-2 causes endothelial dysfunction and thrombosis. In the first scenario, SARS-CoV-2 directly infects the endothelium, disrupting its antithrombogenic and barrier properties. The second scenario invokes an indirect mechanism of endothelial injury mediated by the local and systemic inflammatory response to the viral infection.10,11 In this article, we briefly review the vascular pathology of COVID-19 and critically discuss the proposed mechanisms by which SARS-CoV-2 damages blood vessels, including recent studies that challenge the viral infection of endothelium hypothesis and strongly favor an indirect, inflammation-driven mechanism of endothelial injury.
Vascular Pathology of COVID-19
Our knowledge of lung pathology in COVID-19 is primarily based on autopsy studies. Histologically, COVID-19 lungs show features of diffuse alveolar damage, including injury/necrosis of the alveolar epithelium, intra-alveolar fibrin deposition and hemorrhage, alveolar edema, hyaline membranes, type 2 pneumocyte hyperplasia, interstitial inflammation, and organizing connective tissue in the alveolar septa.8,12,13 The microvasculature of COVID-19 lungs is abnormal and characterized by acute endothelial injury, thrombotic occlusion of alveolar capillaries, and neutrophilic capillaritis/endothelialitis.8,13,14 Injured alveolar capillaries become leaky and distorted, and undergo remodeling with formation of intraluminal endothelial pillars indicative of intussusceptive/splitting angiogenesis.12 COVID-19 lungs also exhibit pulmonary thromboembolism, pulmonary infarctions, and venous thrombosis.15, 16, 17 In one study, microvascular thrombosis was more prevalent in COVID-19 lungs compared with lungs of patients who died of influenza (H1N1).12 A meta-analysis of 27 diagnostic imaging studies found that the incidence of pulmonary emboli in patients admitted to the intensive care unit was considerably higher than the reported pulmonary incidence in intensive care unit patients with non–COVID-19 viral pneumonia.18 This study also found that pulmonary emboli were confined to peripheral arteries, whereas thrombosis of the leg deep veins was observed in less than half of these patients, which suggested in situ formation of thrombi in the pulmonary circulation.18 The higher incidence of vascular injury and thrombosis in COVID-19 lungs has been postulated to be the underlying cause of the ventilation-perfusion mismatch and impaired oxygen uptake that characterize the respiratory complications of COVID-19.11,19 Patients with COVID-19 have higher rates of strokes and myocardial infarctions compared with patients without COVID-19.7,20
Microvascular endothelial injury, thrombosis, and capillaritis have also been reported in the skin, heart, brain, liver, intestine, and kidneys of patients with COVID-19.14,21,22 Among the peripheral organs involved, the kidneys have been the most thoroughly studied because renal failure and proteinuria may complicate the clinical course of the disease. Kidney biopsies have revealed a spectrum of endothelial abnormalities, ranging from lifting of the glomerular endothelium from the basement membrane with the loss of endothelial fenestrations to endothelial cell swelling and thrombotic microangiopathy involving glomerular hilar arterioles and small arteries. These changes are often associated with a severe form of glomerular injury, known as collapsing glomerulopathy, and with acute tubular injury.23, 24, 25
A cutaneous condition called chilblains or COVID toes, which is associated with microvascular injury, has been diagnosed in children and young adults. Biopsies of these lesions have revealed endothelialitis with endothelial swelling and subendothelial infiltration of lymphocytes, lymphocytic vasculitis, and microthrombosis.21
Some severely ill patients with COVID-19 have been reported to have ischemic lesions of the upper and lower extremities (acroischemia), and in a few cases, patients have undergone below-the-knee amputations because of gangrene.26 Coagulation abnormalities in hospitalized and severely ill patients with COVID-19 are evidenced by elevated levels of circulating fibrinogen, fibrin degradation products, D-dimer, and von Willebrand factor (vWF).11,20,27
The occurrence of venous, arterial, and microvascular thrombosis in a single disease represents a rare finding that typifies COVID-19 vasculopathy and points to a distinct mechanism of disease.27 The many studies on the vascular complications of COVID-19 have reported that the endothelial lining of blood vessels is damaged by the SARS-CoV-2 infection, and endothelial injury has been implicated as an important contributor to the formation of thrombi in different vascular districts of patients with COVID-19.27 The following sections describe the direct and indirect mechanisms of SARS-CoV-2–mediated endothelial injury that have been advocated in the pathogenesis of the COVID-19 vasculopathy (Figure 1).
Figure 1.

Proposed causes of endothelial injury in coronavirus disease 2019 vasculopathy. A: Direct injury of the endothelium by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). B: Indirect injury of the endothelium by an excessive inflammatory reaction to the viral infection of alveolar and bronchiolar epithelial cells. The injured endothelium in both scenarios develops a proinflammatory phenotype and loses its antithrombogenic properties. The hypothesis that endothelial cells are directly injured by the virus has been challenged by recent studies reporting that endothelial cells do not express the angiotensin-converting enzyme 2 receptor or express it at low levels and are resistant to the SARS-CoV-2 infection. Image created with BioRender software (BioRender, San Francisco, CA).
Endothelial Injury and Thrombosis in COVID-19: The Direct Viral Infection Hypothesis
The SARS-CoV-2 is an RNA virus composed of a nucleocapside, made of the viral genome and an N protein coat, and a membrane envelope. Club-shaped glycoprotein spikes located on its outer surface confer the virus a crownlike appearance.2 Angiotensin converting enzyme 2 (ACE2), a homologue of angiotensin-converting enzyme and regulator of the renin angiotensin system,28 has been identified as the main SARS-CoV-2 cell surface receptor.29 Binding of the SARS-CoV-2 spike (S) protein to the ACE2 receptor enables the virion to anchor to the cell membrane.29 Enzymatic cleavage of the S protein, leading to internalization of the nucleocapsid into the cell, is mediated by the proteases transmembrane serine protease 2 (TMPRSS2) and cathepsin B/L.29
ACE2 is also the receptor of SARS-CoV-1 (originally named SARS-CoV), which was responsible for the SARS epidemic in 2002 to 2003.30 Immunohistochemical studies, reported shortly after the SARS epidemic, localized the ACE2 protein to lung bronchial and alveolar epithelial cells, intestinal epithelial cells, renal tubular epithelial cells, and blood vessels/capillaries. The microvascular staining reaction was attributed to the endothelium, whereas arterial smooth muscle cells were also positive.31
Ultrastructural studies of lung tissue from COVID-19 autopsies document the presence of cytoplasmic particles, described as "viral particles"12,14 in alveolar capillary endothelial cells. A separate study reported the finding of “coronavirus-like particles” in glomerular endothelial cells but no evidence of such particles in the endothelium of other organs, including the lungs.13 "Coronavirus-like" or "viral particles" were also found in microvascular endothelial cells from postmortem brain tissue and skin biopsies.21,32
Immunohistochemical staining of postmortem lung tissue found viral proteins in alveolar capillary endothelial cells,12,16 but staining results were also reported as negative in these cells.13,17 Localization of SARS-CoV-2 proteins in glomerular endothelial cells from autopsy tissue by immunohistochemistry (IHC) was reported as positive33 or negative13,17,19 by different groups. However, multiple studies of kidney biopsies from patients with COVID-19 failed to confirm SARS-CoV-2 infection in the glomerular endothelium by electron microscopy, IHC, and in situ hybridization (ISH).23,25,34 In -one autopsy study, ISH revealed SARS-CoV-2 RNA in the microvasculature of endothelial cells and vessel walls of brainstem, leptomeninges, lung, heart, liver, kidney, and pancreas.35 A separate group identified SARS-CoV-2 RNA by ISH in the alveolar capillary endothelium but not in the endothelium of peripheral organs.16 In this study, SARS-CoV-2 proteins were detected in skin, brain, and liver microvessels by IHC but without corresponding viral RNA by ISH.16
Limitations to the Hypothesis that SARS-CoV-2 Infects Endothelial Cells
The many early reports on the pathology of COVID-19 have suggested that vascular injury and thrombosis may be caused by direct viral infection of the vascular endothelium. However, lack of reproducibility in different studies and failure to unequivocally confirm endothelial viral infection with different methods in individual studies have raised doubts about this hypothesis. Notably, although SARS-CoV-2 has been clearly identified by electron microscopy in the respiratory epithelium,8,17 many studies describing viral or coronavirus-like particles in endothelial cells have misinterpreted as coronavirus cell organelles, such as coated vesicles and multivesicular bodies. This pitfall has been highlighted by different groups in critical appraisals that have emphasized the need to use stringent criteria for the identification of the virus, which include detailed knowledge of coronavirus ultrastructure and morphogenesis and of subcellular organelles that can resemble the virus.36, 37, 38, 39, 40 Failure to rigorously adhere to these criteria and lack of expertise in virus morphology have resulted in confusion over differentiating coronavirus from normal structures within cells, leading to inaccurate interpretation of electron microscopy findings.
IHC and ISH can confirm the presence of viral components in specific cell types, and several studies have localized viral RNA and/or protein in putative endothelial cells as described in the previous section. However, the mere in situ demonstration of viral products by staining methods, in the absence of an obvious viral cytopathic effect and of definite ultrastructural evidence of replicating viral particles in the endothelium, is not considered sufficient evidence for productive viral infection in the stained cells.37 Furthermore, even in the presence of a specific IHC and/or ISH reaction, most studies have not used double immunostaining methods to accurately localize viral molecules of interest in the endothelial cells and exclude staining of pericytes, which are closely associated with the endothelium.
Emerging New Evidence that the SARS-CoV-2 Receptor ACE2 Is Not Expressed at Significant Levels in Endothelial Cells
Single-cell RNA sequencing (scRNA-seq), first developed in the middle 2000s, has enabled investigators to analyze the transcriptome of native individual cells from different organs. With this method, expression of genes of interest can be demonstrated in specific cell types following cluster analysis of gene expression profiles.41,42 By adopting this powerful new approach, several laboratories have now found that ACE2 is either not expressed in endothelial cells from mouse and human brain, heart, lungs, and skin43, 44, 45, 46 or is expressed at low levels.47,48 Epigenetic analysis of the chromatin landscape of human umbilical vein endothelial cells with genome-wide chromatin immunoprecipitation sequencing data for histone modifications and DNase I hypersensitivity from ENCODE (Encyclopedia of DNA Elements) has corroborated the scRNA-seq findings by revealing the absence of activation marks and the presence of repressive marks in the ACE2 locus.45 In one of our laboratories (G.L.), RNA-seq analysis found that freshly isolated mouse lung endothelial cells fail to express Ace2 and Tmprss2, the protease that participates in SARS-CoV-2 cell entry. Concordant with this observation, no accessible chromatin sites in the Ace2 and Tmprss2 gene loci were identified by Assay for Transposase-Accessible Chromatin sequencing analysis (G.L., unpublished observations). In addition, no ACE2 expression was identified in cultured human lung, brain, cardiac, and glomerular endothelial cells by quantitative RT-PCR and Western blot analysis.49 Moreover, double immunofluorescence confocal microscopy studies by He et al44 elegantly demonstrate strong staining for ACE2 in the pericytes of mouse heart and brain capillaries, with ACE2-negative endothelial cells. The expression of ACE2 in human and mouse brain and heart pericytes is confirmed by scRNA-seq analysis.44,45 In the mouse lungs, ACE2 is strongly expressed by alveolar type 2 cells and ciliated bronchial epithelial cells but not in endothelial cells by double immunofluorescence and scRNA-seq analysis. Most pericytes in the alveolar septa are ACE2 negative, although ACE2-positive pericytes have been identified in the bronchial walls and particularly trachea, where they are most abundant.44
Cultured Endothelial Cells Are Resistant to SARS-CoV-2 Infection
A critical aspect of the direct endothelial injury hypothesis is the capacity of the SARS-CoV-2 virus to infect the vascular endothelium. Until recently, however, no data were available in the vast COVID-19 literature regarding the endothelial cell susceptibility to SARS-CoV-2 viral infection. This gap has now been filled by recently published work by different groups in which isolated human endothelial cells were cultured in the presence of the SARS-CoV-2 virus. Using immunohistochemistry for the N or S protein and a viral progeny detection assay, Nascimento Conde et al50 found that SARS-CoV-2 is incapable of infecting primary human endothelial cells from lung, brain, and renal glomeruli as well as human umbilical vein endothelial cells. Ajmetaj-Shala et al49 demonstrated that blood outgrowth, lung, and aortic primary human endothelial cells were not susceptible to infection by SARS-CoV-2 or SARS-CoV-2 pseudovirus but could be infected with Ebola and vesicular stomatitis virus. McCracken et al45 were unable to achieve significant infection and replication levels of SARS-CoV-2 in primary human cardiac and pulmonary endothelial cells, which were instead permissive for the human coronavirus 229E virus that uses CD13 as its receptor. Schimmel et al47 found no morphologic alterations and no productive viral infection in human umbilical vein endothelial cells and human lung microvascular endothelial cells exposed to SARS-CoV-2. In this study, positive immunostaining for viral NP protein, suggestive of viral entry into the cell, was only obtained when endothelial cells were exposed to high viral titer. However, no infectious virions were detected in the endothelial cell supernatant, indicating failure of virus replication.47 A similar finding was obtained with cultures of human cardiac and pulmonary endothelial cells using supraphysiologic concentrations of the virus.45 These abortive infections obtained with high SARS-CoV-2 titers in endothelial cells that do not express the ACE2 receptor or express it weakly have been attributed to engagement by the virus of noncanonical receptors and lack of surface enzymes needed to cleave the S protein and mediate effective viral-host cell membrane fusion.43,45,47 Of note, endothelial cells became permissive to SARS-CoV-2 only after transduction with recombinant ACE2 receptor.50 Thus, in vitro studies with human endothelial cells isolated from different organs have consistently found that the vascular endothelium is resistant to SARS-CoV-2 infection. Although in vitro assays cannot fully reflect the complexity of the in vivo environment, the finding that endothelial cells resist infection by SARS-CoV-2 and the reproducibility of this finding in different laboratories argue against the hypothesis that endothelial injury in COVID-19 vasculopathy is caused directly by the virus.
Endothelial Injury and Thrombosis in COVID-19: The Indirect Mechanism Hypothesis
The compelling new findings that vascular endothelial cells do not express the ACE2 receptor and are resistant to SARS-CoV-2 infection strongly implicate an indirect mechanism of injury in the pathogenesis of the COVID-19 vasculopathy. Mounting evidence suggests that in COVID-19 a self-amplifying cycle of excessive inflammation impairs the antithrombogenic properties of the vascular endothelium, in concert with the action of circulating prothrombotic factors, leads to formation of thrombi in capillaries, arteries, and veins.9,51 The therapeutic efficacy of corticosteroids further supports the hypothesis that disease progression, including its vascular complications, is fueled by an inflammatory reaction to the viral infection.52 The following sections describe how excessive inflammation triggered by SARS-CoV-2 infection of nonendothelial cells can lead to endothelial injury and thrombotic occlusion of blood vessels.
Mechanisms of Endothelial Injury and Thrombosis in the Pulmonary Vasculature
In the lungs, respiratory and alveolar epithelial cells infected by SARS-CoV-2 activate the type I/III interferon (IFN) response program.53,54 The IFN response to SARS-CoV-2 is, however, imbalanced and delayed, seemingly because of suppression of IFN signaling by SARS-CoV-2.53, 54, 55, 56 A reduced IFN response in patients with severe COVID-19 results in impaired IFN-β and low IFN-α production and activity, defective viral clearance from the infected cells, persistent blood viral load, and an NF-κB–driven exacerbated inflammatory response.56 Infected alveolar epithelial cells switch to an inflammatory phenotype and produce markedly elevated levels of cytokines and chemokines, including interleukin-6 (IL-6), IL-8, tumor necrosis factor (TNF)-α, and CXCL8.53 In addition, the SARS-CoV-2 S proteins can directly activate complement by preventing inhibition of the alternative pathway of complement convertase by its negative regulator factor H.57 Preliminary studies published online also suggest that the SARS-CoV-2 N protein can potentiate activation of the lectin pathway of complement by binding to mannose-binding lectin (MBL)–associated serine protease 2.58 The same study demonstrates strong staining for the complement components MBL, C3, C4, and the terminal attack complex C5b-9 in alveolar epithelial cells, leukocytes, and exudates in COVID-19 lungs.58 Complement activation becomes clinically manifest with elevated plasma levels of C5b-9 and other complement activation markers, which are higher in patients with COVID-19 compared with those of patients with influenza and non–COVID-19 respiratory failure.59, 60, 61 Leukocytes recruited to the site of infection by chemotactic factors such as C5a release inflammatory cytokines after activation of the NLR family pyrin domain containing 3 (NLRPR3) inflammasome and Toll-like receptors (TLRs) by SARS-CoV-2 products.62 Neutrophils accumulated in the alveolar capillaries form neutrophil extracellular traps (NETs), DNA- and enzyme-rich netlike structures that are toxic to the lung epithelium.63 Leukocytes also produce reactive oxygen species (ROS) and release proteolytic enzymes, which contribute to alveolar cell injury.64 Alveolar cells that are dying or dead because of viral cytopathic effect and inflammatory injury, including cytolytic effect by C5b-9, release damage-associated molecular patterns, which activate TLRs in adjacent cells and infiltrating leukocytes, leading to production of additional inflammatory cytokines and chemokines.65
The alveolar capillary endothelium exposed to a cytokine-rich milieu develops a proinflammatory phenotype, including up-regulated expression of cell adhesion molecules that promote leukocyte adhesion, transmigration, and influx into alveolar spaces.66 Endothelial cells also display features of complement activation evidenced by deposition in the alveolar capillaries of MBL, C4, C3, and C5b-9.67 Endothelial dysfunction and injury caused by inflammatory cytokines, ROS, NETs, and complement activation lead to disruption of the endothelial barrier function and vascular leakage.9,16
Endothelial injury is manifested by disruption of the endothelial glycocalyx, an antithrombogenic structure rich in glycosaminoglycans and proteoglycans.68,69 Degradation of the endothelial glycocalyx is evidenced by the presence of high circulating levels of chondroitin sulfate, syndecan 1, and hyaluronic acid in the plasma of severely ill patients with COVID-19.70 Breakdown of the endothelial glycocalyx leads to reduced production of nitric oxide (NO), a powerful vasodilator and inhibitor or platelet aggregation, and release of vWF, a potent platelet aggregating molecule, with resulting increase in vWF plasma levels.70 Concurrently elevated with glycocalyx breakdown products and vWF in COVID-19 plasma is P-selectin, an endothelial- and platelet-derived molecule that promotes platelet aggregation, platelet-endothelial interactions, and leukocyte recruitment.70 The plasma of patients with COVID-19 also contains increased levels of thrombomodulin, a glycocalyx-associated thrombin receptor that physiologically inhibits coagulation through the protein C-protein S pathway.71 Disruption of the glycocalyx can deplete the endothelium of additional anticoagulants, such as heparan sulfate proteoglycans, which bind antithrombin III, and CD39, an ecto-ADPase that inhibits platelet adhesion.72
Additional molecules whose loss in the injured endothelium may affect its antithrombogenic properties, are prostacyclin, tissue factor inhibitor, and tissue-type and urokinase-type plasminogen activators.9 Formation of thrombi can be further promoted by cytokine-induced production by endothelial cells of prothrombotic molecules, including thromboxane, a prostaglandin that stimulates platelet aggregation; plasminogen activator inhibitor 1, which antagonizes the endothelial fibrinolytic activity; and tissue factor, which initiates thrombin formation.9,73 In addition, formation of NETs within the alveolar capillaries has cytotoxic effects for the endothelium and promotes clotting by several mechanisms, including recruitment and activation of platelets.74 Activated platelets in turn regulate leukocyte activity, further contributing to leukocyte cytokine release.27 Platelet hyperactivation also results in microvesicle and granule release, platelet cytokine/chemokine secretion, platelet-leukocyte aggregates, and platelet apoptosis and/or aggregation, which contribute to inflammation and thrombosis.27 Thus, inflammation triggered by SARS-CoV-2–mediated injury of alveolar epithelial cells creates a permissive environment in the alveolar microvasculature for the clotting of blood, the aggregation of platelets, and the formation of thrombi.75
Mechanisms of Endothelial Injury and Thrombosis in the Vasculature of Peripheral Organs
Immune hyperreactivity or failure to resolve the inflammatory reaction because of immune dysregulation in the lungs leads to excessive production of cytokines/chemokines in severely ill patients, causing a cytokine storm characterized by elevated levels of circulating cytokines, systemic inflammatory symptoms, and multiorgan dysfunction.76, 77, 78 Inflammatory cytokines and chemokines that are elevated in the circulation of patients with COVID-19 include IL-1β, IL-6, TNF, IFN-γ, monocyte chemoattractant protein 1 (MCP1/CCL2), macrophage inflammatory protein 1α (MIP1α/CCL3), macrophage inflammatory protein 1β (MIP1β/CCL4), angiopoietin 2, and vascular endothelial growth factor.79 The cytokine storm is accompanied by aberrant activation of circulating monocytes, increase in neutrophils, and marked decrease in circulating T and B lymphocytes.27,78
Formation of thrombi in the peripheral organs of severely ill patients with COVID-19 with cytokine storm has been attributed to the concurrent action of multiple prothrombogenic factors, including endothelial injury, platelet hyperactivation, and an enhanced procoagulant state.75 Elevated levels of circulating cytokines, such as TNF, can impair the antithrombogenic properties of the endothelium by disrupting its glycocalyx.68 Cytokines such as TNF, IL-1β, and IFN- γ can also cause inflammatory cell death of the endothelium and activation of coagulation because of exposure of the subendothelial matrix.80 Circulating NETs and vWF, which are elevated in patients with COVID-19, acting in concert with tissue factor and other prothrombogenic molecules released locally by the injured endothelium that has lost its ability to maintain blood fluidity, can promote platelet aggregation and intravascular coagulation.70,75 Antiphospholipid antibodies, which are frequently detected in the plasma of hospitalized patients with COVID-19, contribute additional prothrombogenic stimuli by binding to and activating platelets and endothelial cells and triggering release of NETs by neutrophils.81
Pericytes, like alveolar epithelial cells, express the ACE2 receptor,44,45 can become infected and injured in patients with viremia, locally produce inflammatory cytokines, become the site of complement activation, and release tissue factor, triggering endothelial dysfunction and thrombosis.82 Although evidence of pericyte infection by SARS-CoV-2 is currently lacking, a recent study found that pericyte deficiency in mice leads to endothelial dysfunction and loss of antithrombogenic properties, resulting in increased endothelial expression and release of vWF, platelet aggregation, and fibrin formation.44 The same mechanism of indirect endothelial injury and resulting thrombosis in peripheral organs could be triggered by SARS-CoV-2 infection of other ACE2-positive nonendothelial cells, such as the kidney tubular epithelial cells and intestinal epithelial cells.31,83
Predisposing Functional Abnormalities in the Endothelium and Immune System of Patients at Risk for Severe COVID-19
Hospitalization and death attributable to COVID-19 are much more common among the elderly and those with underlying disorders such as diabetes, obesity, and hypertension.4 Impaired regulation of blood homeostasis by a dysfunctional endothelium in these patients may render blood vessels more susceptible to the thrombotic complications of COVID-19.
With aging, endothelial cells reduce their capacity to produce and release the vasodilating molecule NO,84,85 while increasing production of ROS.86 This results in defective regulation of vascular tone, increased oxidative stress, and enhanced platelet activation.87 Aging also promotes a proinflammatory endothelial phenotype characterized by greater nuclear translocation of NFκB and increased endothelial production of IL-6, TNF and MCP1.88 In addition, expression of PAI-1, which antagonizes endothelial fibrinolytic activity, is elevated in older individuals.89 Moreover, aging has been linked to thinning of the endothelial glycocalyx which can impair the capacity of endothelial cells to inhibit platelet adhesion and aggregation.70,90 These underlying endothelial abnormalities may further facilitate formation of thrombi in elderly patients infected by SARS-CoV-2.
In diabetes, endothelial cells manifest an impaired capacity to activate endothelial NO synthase (eNOS) in response to physiological stimuli leading to reduced production of NO.91 Bioavailability of NO is further compromised by increased oxidative stress due to production of ROS in response to elevated glucose levels.91,92 Patients with diabetes also have increased circulating levels of inflammatory cytokines, such as TNF, IL-6, and intercellular adhesion molecule 1, which indicates endothelial activation.93 Impaired regulation of blood homeostasis by the endothelium in diabetes is also attributable to disruption of the endothelial glycocalyx characterized by thinning and shedding into the circulation of its molecular components, including hyaluronic acid, syndecan 1, chondroitin sulfate, and heparan sulfate.94 Mediators of hyperglycemia-induced glycocalyx damage include ROS, advanced glycation end products, and glycocalyx-degrading enzymes, such as hyaluronidase and heparanase.94,95
Reduced NO bioavailability, increased inflammation, enhanced oxidative stress, and a prothrombotic phenotype are also features of endothelial dysfunction in obesity and hypertension.96,97 In patients with obesity, inflammation in the perivascular adipose tissue, particularly in visceral organs, has been implicated as the major source of locally released and circulating inflammatory cytokines responsible for impaired endothelial function.97
Among patients with predisposing conditions, there is great variability in the severity of COVID-19. Some recover from the infection, whereas others experience cytokine storm and multiorgan system failure.76 Recent studies have found that excessive systemic inflammation, which plays an important role in the COVID-19 vasculopathy, may be attributable to an ineffective IFN response to SARS-CoV-2 and misfiring of the immune system to the unabated viral infection. In one study, 3.5% of patients with life-threatening COVID-19 pneumonia were found to have inborn errors of TLR-3 and interferon regulatory transcription factor 7 (IRF7)–dependent type I IFN production and amplification due to loss-of-function mutations of loci that govern these genes.98 A separate study by the same group identified neutralizing autoantibodies against type I IFNs (IFN-α and IFN-ω) in 10.2% of patients with severe COVID-19 pneumonia.99 A defective IFN response, caused by loss-of-function variants of TLR7 and characterized by reduced expression of IFN-related genes, such as IRF7 and IFNB1, has also been reported in young and previously healthy male patients with severe COVID-19.100
An additional predisposing abnormality that can complicate the clinical course of COVID-19 has been identified in individuals of African ancestry who have developed renal failure, proteinuria, collapsing glomerulopathy, and thrombotic microangiopathy after SARS-CoV-2 infection. A high percentage of these patients carry the G1 and/or G2 high-risk apolipoprotein L1 (APOL1) alleles, which are evolutionarily enriched in this population because of their protective action against African trypanosomiasis.23, 24, 25 APOL1 is expressed by glomerular endothelial cells and podocytes and can be up-regulated by type I IFN.101 Although the mechanisms of collapsing glomerulopathy are poorly understood, APOL1 high-risk variants have been implicated in its pathogenesis for their ability to create pores in the cell membrane, which would result in injury of glomerular cells and collapse of the glomerular tuft.102 A similar mechanism of injury may be responsible for the thrombotic microangiopathy, which is often present in COVID-19–associated nephropathy.
Summary and Conclusion
Severe cases of COVID-19 are characterized by dysregulation of the immune system and an excessive inflammatory response to the viral infection. The clinical course of COVID-19 is often complicated by endothelial dysfunction, vasculopathy, and thrombosis of small, medium, and large blood vessels. Two mechanisms have been advocated for the vascular abnormalities of COVID-19: endothelial infection by SARS-CoV-2 and endothelial injury caused by excessive inflammation. Although initial reports, mostly based on autopsy material, favored the direct endothelial cell infection hypothesis, more recent molecular and microbiological studies by different laboratories have found that endothelial cells do not express significant levels of the ACE2 receptor, which SARS-CoV-2 uses to gain entry into cells, and are resistant to the infection. These new findings strongly suggest that COVID-19 vasculopathy is caused by an exaggerated immune response to viral infection of nonendothelial cells, which results in indirect injury of the vascular endothelium, loss of endothelial antithrombogenic properties, endothelial release of prothrombogenic factors, platelet hyperactivation, and coagulopathy, leading to thrombotic occlusion of blood vessels (Figure 2).
Figure 2.
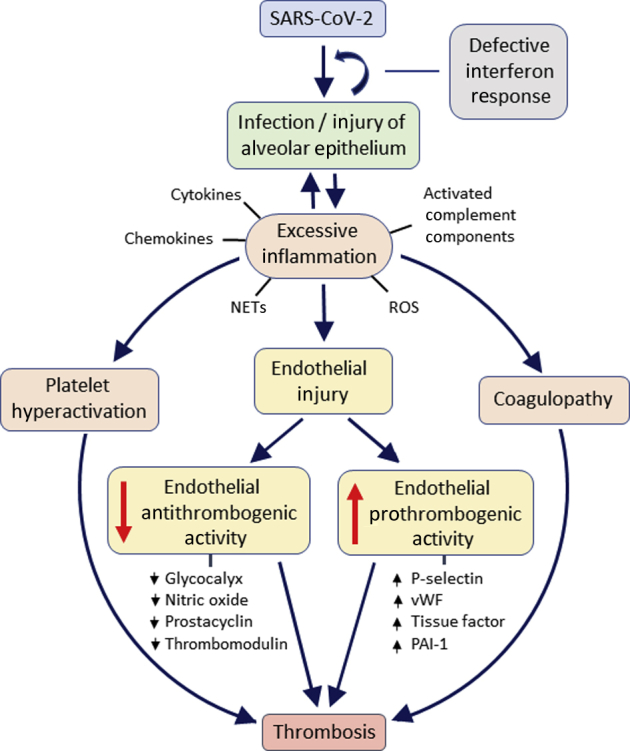
Putative mechanisms of endothelial injury and thrombosis in critically ill patients with coronavirus disease 2019 (COVID-19). The schematic summarizes key processes implicated in the pathogenesis of COVID-19 vasculopathy. Angiotensin-converting enzyme 2 (ACE2)–positive alveolar epithelial cells infected by severe acute respiratory syndrome coronavirus 2 trigger an inflammatory response, which, in the setting of defective antiviral interferon signaling, fails to suppress the infection and misfires, leading to overproduction of cytokines and chemokines, activation of complement, and recruitment of leukocytes, which release reactive oxygen species (ROS) and neutrophilic extracellular traps (NETs). Endothelial cells injured indirectly by the cytokines and other inflammatory products lose their antithrombogenic properties and release factors that promote coagulation and platelet aggregation. Hyperactivation of platelets and systemic coagulopathy induced by the inflammatory reaction to the infection contribute to the formation of thrombi. The same mechanisms of inflammation-mediated endothelial injury can create a permissive prothrombogenic environment in peripheral organs that contain ACE2-positive nonendothelial cells. PAI1, plasminogen activator inhibitor 1; vWF, von Willebrand factor.
Although considerable advances have been made since the initial outbreak of COVID-19 in China, more studies are needed to better characterize the causes of the excessive inflammatory response to the infection, identify patients more at risk for the dysfunctional immune response and its vascular complications, and establish key molecular targets for effective medical treatment. To that end, in vitro models with cultured human cells or tissue organoids and in vivo animal models have been developed to investigate the mechanisms of SARS-CoV-2 infection and test new therapies.103, 104, 105 Clinical trials are evaluating the safety and efficacy of drugs directed against cytokines and their receptors, cytokine signaling pathways, complement components, and other immune targets implicated in the hyperinflammatory response.106 As more knowledge is gained on the dysregulated immune response to the SARS-CoV-2 infection and its deleterious effects on the vasculature of the lungs and peripheral organs, clinical outcomes are expected to improve with the introduction of new therapies.
Acknowledgments
We thank Dr. Charles E. Alpers and Dr. Kelly D. Smith, Department of Laboratory Medicine and Pathology, University of Washington, Seattle, WA, and Dr. Santo V. Nicosia, Morsani College of Medicine, University of South Florida, Tampa, FL, for their careful review of the manuscript and helpful suggestions.
Footnotes
Supported by NIHNational Heart, Lung, and Blood Institute grant HL142596 (G.L.), NIHNational Center for Advancing Translational Sciences grant 3UH3TR002158-04S1 (S.A.), American Lung Association Dalsemer Award (N.C.), and Boehringer Ingelheim Discovery Award in Idiopathic Pulmonary Fibrosis/Interstitial Lung Disease (N.C.).
Disclosures: None declared.
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W., China Novel Coronavirus Investigating and Research Team A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollard C.A., Morran M.P., Nestor-Kalinoski A.L. The COVID-19 pandemic: a global health crisis. Physiol Genomics. 2020;52:549–557. doi: 10.1152/physiolgenomics.00089.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ejaz H., Alsrhani A., Zafar A., Javed H., Junaid K., Abdalla A.E., Abosalif K.O.A., Ahmed Z., Younas S. COVID-19 and comorbidities: deleterious impact on infected patients. J Infect Public Health. 2020;13:1833–1839. doi: 10.1016/j.jiph.2020.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X., Guan L., Wei Y., Li H., Wu X., Xu J., Tu S., Zhang Y., Chen H., Cao B. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dolhnikoff M., Duarte-Neto A.N., de Almeida Monteiro R.A., da Silva L.F.F., de Oliveira E.P., Saldiva P.H.N., Mauad T., Negri E.M. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J Thromb Haemost. 2020;18:1517–1519. doi: 10.1111/jth.14844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klok F.A., Kruip M., van der Meer N.J.M., Arbous M.S., Gommers D., Kant K.M., Kaptein F.H.J., van Paassen J., Stals M.A.M., Huisman M.V., Endeman H. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb Res. 2020;191:148–150. doi: 10.1016/j.thromres.2020.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martines R.B., Ritter J.M., Matkovic E., Gary J., Bollweg B.C., Bullock H., Goldsmith C.S., Silva-Flannery L., Seixas J.N., Reagan-Steiner S., Uyeki T., Denison A., Bhatnagar J., Shieh W.J., Zaki S.R., COVID-19 Pathology Working Group Pathology and pathogenesis of SARS-CoV-2 associated with fatal coronavirus disease, United States. Emerg Infect Dis. 2020;26:2005–2015. doi: 10.3201/eid2609.202095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Libby P., Luscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020;41:3038–3044. doi: 10.1093/eurheartj/ehaa623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basta G. Direct or indirect endothelial damage? an unresolved question. EBioMedicine. 2021;64:103215. doi: 10.1016/j.ebiom.2021.103215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaur S., Tripathi D.M., Yadav A. The enigma of endothelium in COVID-19. Front Physiol. 2020;11:989. doi: 10.3389/fphys.2020.00989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ackermann M., Verleden S.E., Kuehnel M., Haverich A., Welte T., Laenger F., Vanstapel A., Werlein C., Stark H., Tzankov A., Li W.W., Li V.W., Mentzer S.J., Jonigk D. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N Engl J Med. 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradley B.T., Maioli H., Johnston R., Chaudhry I., Fink S.L., Xu H., Najafian B., Deutsch G., Lacy J.M., Williams T., Yarid N., Marshall D.A. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: a case series. Lancet. 2020;396:320–332. doi: 10.1016/S0140-6736(20)31305-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varga Z., Flammer A.J., Steiger P., Haberecker M., Andermatt R., Zinkernagel A.S., Mehra M.R., Schuepbach R.A., Ruschitzka F., Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elsoukkary S.S., Mostyka M., Dillard A., Berman D.R., Ma L.X., Chadburn A., Yantiss R.K., Jessurun J., Seshan S.V., Borczuk A.C., Salvatore S.P. Autopsy findings in 32 patients with COVID-19: a single-institution experience. Pathobiology. 2021;88:56–68. doi: 10.1159/000511325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magro C.M., Mulvey J., Kubiak J., Mikhail S., Suster D., Crowson A.N., Laurence J., Nuovo G. Severe COVID-19: a multifaceted viral vasculopathy syndrome. Ann Diagn Pathol. 2021;50:151645. doi: 10.1016/j.anndiagpath.2020.151645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaefer I.M., Padera R.F., Solomon I.H., Kanjilal S., Hammer M.M., Hornick J.L., Sholl L.M. In situ detection of SARS-CoV-2 in lungs and airways of patients with COVID-19. Mod Pathol. 2020;33:2104–2114. doi: 10.1038/s41379-020-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suh Y.J., Hong H., Ohana M., Bompard F., Revel M.P., Valle C., Gervaise A., Poissy J., Susen S., Hekimian G., Artifoni M., Periard D., Contou D., Delaloye J., Sanchez B., Fang C., Garzillo G., Robbie H., Yoon S.H. Pulmonary embolism and deep vein thrombosis in COVID-19: a systematic review and meta-analysis. Radiology. 2021;298:E70–E80. doi: 10.1148/radiol.2020203557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gattinoni L., Chiumello D., Caironi P., Busana M., Romitti F., Brazzi L., Camporota L. COVID-19 pneumonia: different respiratory treatments for different phenotypes? Intensive Care Med. 2020;46:1099–1102. doi: 10.1007/s00134-020-06033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wise J. Covid-19 and thrombosis: what do we know about the risks and treatment? BMJ. 2020;369:m2058. doi: 10.1136/bmj.m2058. [DOI] [PubMed] [Google Scholar]
- 21.Garrido Ruiz M.C., Santos-Briz A., Santos-Briz A., Sanchez A., Alonso-Riano M., Burgos J., Medina-Miguelanez M., Puebla L., Roman-Curto C., Roncero-Riesco M., Garcia R., Ortiz P.L., Rodriguez-Peralto J.L. Spectrum of clinicopathologic findings in COVID-19-induced skin lesions: demonstration of direct viral infection of the endothelial cells. Am J Surg Pathol. 2021;45:293–303. doi: 10.1097/PAS.0000000000001634. [DOI] [PubMed] [Google Scholar]
- 22.Maccio U., Zinkernagel A.S., Shambat S.M., Zeng X., Cathomas G., Ruschitzka F., Schuepbach R.A., Moch H., Varga Z. SARS-CoV-2 leads to a small vessel endotheliitis in the heart. EBioMedicine. 2021;63:103182. doi: 10.1016/j.ebiom.2020.103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akilesh S., Nast C.C., Yamashita M., Henriksen K., Charu V., Troxell M.L., Kambham N., Bracamonte E., Houghton D., Ahmed N.I., Chong C.C., Thajudeen B., Rehman S., Khoury F., Zuckerman J.E., Gitomer J., Raguram P.C., Mujeeb S., Schwarze U., Shannon M.B., De Castro I., Alpers C.E., Najafian B., Nicosia R.F., Andeen N.K., Smith K.D. Multicenter clinicopathologic correlation of kidney biopsies performed in COVID-19 patients presenting with acute kidney injury or proteinuria. Am J Kidney Dis. 2021;77:82–93.e1. doi: 10.1053/j.ajkd.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su H., Yang M., Wan C., Yi L.X., Tang F., Zhu H.Y., Yi F., Yang H.C., Fogo A.B., Nie X., Zhang C. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020;98:219–227. doi: 10.1016/j.kint.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu H., Larsen C.P., Hernandez-Arroyo C.F., Mohamed M.M.B., Caza T., Sharshir M., Chughtai A., Xie L., Gimenez J.M., Sandow T.A., Lusco M.A., Yang H., Acheampong E., Rosales I.A., Colvin R.B., Fogo A.B., Velez J.C.Q. AKI and collapsing glomerulopathy associated with COVID-19 and APOL 1 high-risk genotype. J Am Soc Nephrol. 2020;31:1688–1695. doi: 10.1681/ASN.2020050558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suarez-Valle A., Fernandez-Nieto D., Diaz-Guimaraens B., Dominguez-Santas M., Carretero I., Perez-Garcia B. Acro-ischaemia in hospitalized COVID-19 patients. J Eur Acad Dermatol Venereol. 2020;34:e455–e457. doi: 10.1111/jdv.16592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu S.X., Tyagi T., Jain K., Gu V.W., Lee S.H., Hwa J.M., Kwan J.M., Krause D.S., Lee A.I., Halene S., Martin K.A., Chun H.J., Hwa J. Thrombocytopathy and endotheliopathy: crucial contributors to COVID-19 thromboinflammation. Nat Rev Cardiol. 2021;18:194–209. doi: 10.1038/s41569-020-00469-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burrell L.M., Johnston C.I., Tikellis C., Cooper M.E. ACE2, a new regulator of the renin-angiotensin system. Trends Endocrinol Metab. 2004;15:166–169. doi: 10.1016/j.tem.2004.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., Muller M.A., Drosten C., Pohlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamming I., Timens W., Bulthuis M.L., Lely A.T., Navis G., van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paniz-Mondolfi A., Bryce C., Grimes Z., Gordon R.E., Reidy J., Lednicky J., Sordillo E.M., Fowkes M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) J Med Virol. 2020;92:699–702. doi: 10.1002/jmv.25915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puelles V.G., Lutgehetmann M., Lindenmeyer M.T., Sperhake J.P., Wong M.N., Allweiss L., Chilla S., Heinemann A., Wanner N., Liu S., Braun F., Lu S., Pfefferle S., Schroder A.S., Edler C., Gross O., Glatzel M., Wichmann D., Wiech T., Kluge S., Pueschel K., Aepfelbacher M., Huber T.B. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med. 2020;383:590–592. doi: 10.1056/NEJMc2011400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peleg Y., Kudose S., D'Agati V., Siddall E., Ahmad S., Nickolas T., Kisselev S., Gharavi A., Canetta P. Acute kidney injury due to collapsing glomerulopathy following COVID-19 infection. Kidney Int Rep. 2020;5:940–945. doi: 10.1016/j.ekir.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhatnagar J., Gary J., Reagan-Steiner S., Estetter L.B., Tong S., Tao Y., Denison A.M., Lee E., DeLeon-Carnes M., Li Y., Uehara A., Paden C.R., Leitgeb B., Uyeki T.M., Martines R.B., Ritter J.M., Paddock C.D., Shieh W.J., Zaki S.R. Evidence of severe acute respiratory syndrome coronavirus 2 replication and tropism in the lungs, airways, and vascular endothelium of patients with fatal coronavirus disease 2019: an autopsy case series. J Infect Dis. 2021;223:752–764. doi: 10.1093/infdis/jiab039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akilesh S., Nicosia R.F., Alpers C.E., Tretiakova M., Hsiang T.Y., Gale M., Jr., Smith K.D. Characterizing viral infection by electron microscopy: lessons from the coronavirus disease 2019 pandemic. Am J Pathol. 2021;191:222–227. doi: 10.1016/j.ajpath.2020.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bullock H.A., Goldsmith C.S., Miller S.E. Best practices for correctly identifying coronavirus by transmission electron microscopy. Kidney Int. 2021;99:824–827. doi: 10.1016/j.kint.2021.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dittmayer C., Meinhardt J., Radbruch H., Radke J., Heppner B.I., Heppner F.L., Stenzel W., Holland G., Laue M. Why misinterpretation of electron micrographs in SARS-CoV-2-infected tissue goes viral. Lancet. 2020;396:e64–e65. doi: 10.1016/S0140-6736(20)32079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hopfer H., Herzig M.C., Gosert R., Menter T., Hench J., Tzankov A., Hirsch H.H., Miller S.E. Hunting coronavirus by transmission electron microscopy - a guide to SARS-CoV-2-associated ultrastructural pathology in COVID-19 tissues. Histopathology. 2021;78:358–370. doi: 10.1111/his.14264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller S.E., Goldsmith C.S. Caution in identifying coronaviruses by electron microscopy. J Am Soc Nephrol. 2020;31:2223–2224. doi: 10.1681/ASN.2020050755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bainbridge M.N., Warren R.L., Hirst M., Romanuik T., Zeng T., Go A., Delaney A., Griffith M., Hickenbotham M., Magrini V., Mardis E.R., Sadar M.D., Siddiqui A.S., Marra M.A., Jones S.J. Analysis of the prostate cancer cell line LNCaP transcriptome using a sequencing-by-synthesis approach. BMC Genomics. 2006;7:246. doi: 10.1186/1471-2164-7-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolodziejczyk A.A., Kim J.K., Svensson V., Marioni J.C., Teichmann S.A. The technology and biology of single-cell RNA sequencing. Mol Cell. 2015;58:610–620. doi: 10.1016/j.molcel.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 43.Ganier C., Du-Harpur X., Harun N., Wan B., Arthurs C., Luscombe N., Watt F., Lynch M. CD147 (BSG) but not ACE2 expression is detectable in vascular endothelial cells within single cell RNA sequencing datasets derived from multiple tissues in healthy individuals. bioRxiv. 2020 doi: 10.1101/2020.05.29.123513. [DOI] [Google Scholar]
- 44.He L., Mäe M.A., Muhl L., Sun Y., Pietilä R., Nahar K., Liébanas E.V., Fagerlund M.J., Oldner A., Liu J., Genové G., Zhang L., Xie Y., Leptidis S., Mocci G., Stritt S., Osman A., Anisimov A., Hemanthakumar K.A., Räsänen M., Mirabeau O., Hansson E., Björkegren J., Vanlandewijck M., Blomgren K., Mäkinen T., Peng X.-R., Arnold T.D., Alitalo K., Eriksson L.I., Lendahl U., Betsholtz C. Pericyte-specific vascular expression of SARS-CoV-2 receptor ACE2 – implications for microvascular inflammation and hypercoagulopathy in COVID-19. bioRxiv. 2020 doi: 10.1101/2020.05.11.088500. [DOI] [Google Scholar]
- 45.McCracken I.R., Saginc G., He L., Huseynov A., Daniels A., Fletcher S., Peghaire C., Kalna V., Andaloussi-Mae M., Muhl L., Craig N.M., Griffiths S.J., Haas J.G., Tait-Burkard C., Lendahl U., Birdsey G.M., Betsholtz C., Noseda M., Baker A.H., Randi A.M. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation. 2021;143:865–868. doi: 10.1161/CIRCULATIONAHA.120.052824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh M., Bansal V., Feschotte C. A single-cell RNA expression map of human coronavirus entry factors. Cell Rep. 2020;32:108175. doi: 10.1016/j.celrep.2020.108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schimmel L., Chew K.Y., Stocks C., Yordanov T., Essebier T., Kulasinghe A., Monkman J., dos Santos Miggiolaro A.F.R., de Noronha L., Lagendijk A.K., Schroder K., Labzin L., Gordon E.J., Short K.R. Endothelial cells elicit a pro-inflammatory response to SARS-CoV-2 without productive viral infection. bioRxiv. 2021 doi: 10.1101/2021.02.14.431177. [DOI] [Google Scholar]
- 48.Zhao Y., Zhao Z., Wang Y., Zhou Y., Ma Y., Zuo W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. Am J Respir Crit Care Med. 2020;202:756–759. doi: 10.1164/rccm.202001-0179LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmetaj-Shala B., Peacock T.P., Baillon L., Swann O.C., Gashaw H., Barclay W.S., Mitchell J.A. Resistance of endothelial cells to SARS-CoV-2 infection in vitro. bioRxiv. 2020 doi: 10.1101/2020.11.08.372581. [DOI] [Google Scholar]
- 50.Nascimento Conde J., Schutt W.R., Gorbunova E.E., Mackow E.R. Recombinant ACE2 expression is required for SARS-CoV-2 to infect primary human endothelial cells and induce inflammatory and procoagulative responses. mBio. 2020;11:e03185-20. doi: 10.1128/mBio.03185-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dupont A., Rauch A., Staessens S., Moussa M., Rosa M., Corseaux D., Jeanpierre E., Goutay J., Caplan M., Varlet P., Lefevre G., Lassalle F., Bauters A., Faure K., Lambert M., Duhamel A., Labreuche J., Garrigue D., De Meyer S.F., Staels B., Vincent F., Rousse N., Kipnis E., Lenting P., Poissy J., Susen S., Lille Covid Research Network (LICORNE) Vascular endothelial damage in the pathogenesis of organ injury in severe COVID-19. Arterioscler Thromb Vasc Biol. 2021;41:1760–1773. doi: 10.1161/ATVBAHA.120.315595. [DOI] [PubMed] [Google Scholar]
- 52.Group R.C., Horby P., Lim W.S., Emberson J.R., Mafham M., Bell J.L., Linsell L., Staplin N., Brightling C., Ustianowski A., Elmahi E., Prudon B., Green C., Felton T., Chadwick D., Rege K., Fegan C., Chappell L.C., Faust S.N., Jaki T., Jeffery K., Montgomery A., Rowan K., Juszczak E., Baillie J.K., Haynes R., Landray M.J. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. 2021;384:693–704. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang J., Hume A.J., Abo K.M., Werder R.B., Villacorta-Martin C., Alysandratos K.D., Beermann M.L., Simone-Roach C., Lindstrom-Vautrin J., Olejnik J., Suder E.L., Bullitt E., Hinds A., Sharma A., Bosmann M., Wang R., Hawkins F., Burks E.J., Saeed M., Wilson A.A., Muhlberger E., Kotton D.N. SARS-CoV-2 infection of pluripotent stem cell-derived human lung alveolar type 2 cells elicits a rapid epithelial-intrinsic inflammatory response. Cell Stem Cell. 2020;27:962–973.e7. doi: 10.1016/j.stem.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamers M.M., van der Vaart J., Knoops K., Riesebosch S., Breugem T.I., Mykytyn A.Z., Beumer J., Schipper D., Bezstarosti K., Koopman C.D., Groen N., Ravelli R.B.G., Duimel H.Q., Demmers J.A.A., Verjans G., Koopmans M.P.G., Muraro M.J., Peters P.J., Clevers H., Haagmans B.L. An organoid-derived bronchioalveolar model for SARS-CoV-2 infection of human alveolar type II-like cells. EMBO J. 2021;40:e105912. doi: 10.15252/embj.2020105912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Moller R., Jordan T.X., Oishi K., Panis M., Sachs D., Wang T.T., Schwartz R.E., Lim J.K., Albrecht R.A., tenOever B.R. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–10345.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Pere H., Charbit B., Bondet V., Chenevier-Gobeaux C., Breillat P., Carlier N., Gauzit R., Morbieu C., Pene F., Marin N., Roche N., Szwebel T.A., Merkling S.H., Treluyer J.M., Veyer D., Mouthon L., Blanc C., Tharaux P.L., Rozenberg F., Fischer A., Duffy D., Rieux-Laucat F., Kerneis S., Terrier B. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu J., Yuan X., Chen H., Chaturvedi S., Braunstein E.M., Brodsky R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood. 2020;136:2080–2089. doi: 10.1182/blood.2020008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao T., Hu M., Zhang X., Li H., Zhu L., Liu H. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv. 2020 doi: 10.1101/2020.03.29.20041962. [DOI] [Google Scholar]
- 59.Holter J.C., Pischke S.E., de Boer E., Lind A., Jenum S., Holten A.R., Tonby K., Barratt-Due A., Sokolova M., Schjalm C., Chaban V., Kolderup A., Tran T., Tollefsrud Gjolberg T., Skeie L.G., Hesstvedt L., Ormasen V., Fevang B., Austad C., Muller K.E., Fladeby C., Holberg-Petersen M., Halvorsen B., Muller F., Aukrust P., Dudman S., Ueland T., Andersen J.T., Lund-Johansen F., Heggelund L., Dyrhol-Riise A.M., Mollnes T.E. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc Natl Acad Sci U S A. 2020;117:25018–25025. doi: 10.1073/pnas.2010540117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma L., Sahu S.K., Cano M., Kuppuswamy V., Bajwa J., McPhatter J.N., Pine A., Meizlish M., Goshua G., Chang C.-H., Zhang H., Price C., Bahel P., Rinder H., Lei T., Day A., Reynolds D., Wu X., Schriefer R., Rauseo A.M., Goss C.W., O'Halloran J.A., Presti R.M., Kim A.H., Gelman A.E., Cruz C.D., Lee A.I., Mudd P., Chun H.J., Atkinson J.P., Kulkarni H.S. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. bioRxiv. 2021 doi: 10.1101/2021.02.22.432177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Noris M., Benigni A., Remuzzi G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int. 2020;98:314–322. doi: 10.1016/j.kint.2020.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodrigues T.S., de Sa K.S.G., Ishimoto A.Y., Becerra A., Oliveira S., Almeida L. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. 2021;218:e20201707. doi: 10.1084/jem.20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Veras F.P., Pontelli M.C., Silva C.M., Toller-Kawahisa J.E., de Lima M., Nascimento D.C. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. 2020;217:e20201129. doi: 10.1084/jem.20201129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laforge M., Elbim C., Frere C., Hemadi M., Massaad C., Nuss P., Benoliel J.J., Becker C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat Rev Immunol. 2020;20:515–516. doi: 10.1038/s41577-020-0407-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cicco S., Cicco G., Racanelli V., Vacca A. Neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs): two potential targets for COVID-19 treatment. Mediators Inflamm. 2020;2020:7527953. doi: 10.1155/2020/7527953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tong M., Jiang Y., Xia D., Xiong Y., Zheng Q., Chen F., Zou L., Xiao W., Zhu Y. Elevated expression of serum endothelial cell adhesion molecules in COVID-19 patients. J Infect Dis. 2020;222:894–898. doi: 10.1093/infdis/jiaa349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Magro C., Mulvey J.J., Berlin D., Nuovo G., Salvatore S., Harp J., Baxter-Stoltzfus A., Laurence J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13. doi: 10.1016/j.trsl.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramnath R., Foster R.R., Qiu Y., Cope G., Butler M.J., Salmon A.H., Mathieson P.W., Coward R.J., Welsh G.I., Satchell S.C. Matrix metalloproteinase 9-mediated shedding of syndecan 4 in response to tumor necrosis factor alpha: a contributor to endothelial cell glycocalyx dysfunction. FASEB J. 2014;28:4686–4699. doi: 10.1096/fj.14-252221. [DOI] [PubMed] [Google Scholar]
- 69.Yilmaz O., Afsar B., Ortiz A., Kanbay M. The role of endothelial glycocalyx in health and disease. Clin Kidney J. 2019;12:611–619. doi: 10.1093/ckj/sfz042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fraser D.D., Patterson E.K., Slessarev M., Gill S.E., Martin C., Daley M., Miller M.R., Patel M.A., Dos Santos C.C., Bosma K.J., O'Gorman D.B., Cepinskas G. Endothelial injury and glycocalyx degradation in critically ill coronavirus disease 2019 patients: implications for microvascular platelet aggregation. Crit Care Explor. 2020;2:e0194. doi: 10.1097/CCE.0000000000000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goshua G., Pine A.B., Meizlish M.L., Chang C.H., Zhang H., Bahel P., Baluha A., Bar N., Bona R.D., Burns A.J., Dela Cruz C.S., Dumont A., Halene S., Hwa J., Koff J., Menninger H., Neparidze N., Price C., Siner J.M., Tormey C., Rinder H.M., Chun H.J., Lee A.I. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020;7:e575–e582. doi: 10.1016/S2352-3026(20)30216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bochenek M.L., Schafer K. Role of endothelial cells in acute and chronic thrombosis. Hamostaseologie. 2019;39:128–139. doi: 10.1055/s-0038-1675614. [DOI] [PubMed] [Google Scholar]
- 73.Yau J.W., Teoh H., Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc Disord. 2015;15:130. doi: 10.1186/s12872-015-0124-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Skendros P., Mitsios A., Chrysanthopoulou A., Mastellos D.C., Metallidis S., Rafailidis P., Ntinopoulou M., Sertaridou E., Tsironidou V., Tsigalou C., Tektonidou M., Konstantinidis T., Papagoras C., Mitroulis I., Germanidis G., Lambris J.D., Ritis K. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest. 2020;130:6151–6157. doi: 10.1172/JCI141374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jayarangaiah A., Kariyanna P.T., Chen X., Jayarangaiah A., Kumar A. COVID-19-associated coagulopathy: an exacerbated immunothrombosis response. Clin Appl Thromb Hemost. 2020;26 doi: 10.1177/1076029620943293. 1076029620943293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fajgenbaum D.C., June C.H. Cytokine storm. N Engl J Med. 2020;383:2255–2273. doi: 10.1056/NEJMra2026131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J., HLH Across Speciality Collaboration UK COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang J., Jiang M., Chen X., Montaner L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol. 2020;108:17–41. doi: 10.1002/JLB.3COVR0520-272R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pum A., Ennemoser M., Adage T., Kungl A.J. Cytokines and chemokines in SARS-CoV-2 infections-therapeutic strategies targeting cytokine storm. Biomolecules. 2021;11:91. doi: 10.3390/biom11010091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karki R., Sharma B.R., Tuladhar S., Williams E.P., Zalduondo L., Samir P., Zheng M., Sundaram B., Banoth B., Malireddi R.K.S., Schreiner P., Neale G., Vogel P., Webby R., Jonsson C.B., Kanneganti T.D. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184:149–168.e17. doi: 10.1016/j.cell.2020.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zuo Y., Estes S.K., Ali R.A., Gandhi A.A., Yalavarthi S., Shi H., Sule G., Gockman K., Madison J.A., Zuo M., Yadav V., Wang J., Woodard W., Lezak S.P., Lugogo N.L., Smith S.A., Morrissey J.H., Kanthi Y., Knight J.S. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci Transl Med. 2020;12:eabd3876. doi: 10.1126/scitranslmed.abd3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hoffman M. Some things i thought i knew about tissue factor that turn out to be wrong. Thromb Res. 2008;122 Suppl 1:S73–S77. doi: 10.1016/S0049-3848(08)70024-0. [DOI] [PubMed] [Google Scholar]
- 83.To K.F., Tong J.H., Chan P.K., Au F.W., Chim S.S., Chan K.C., Cheung J.L., Liu E.Y., Tse G.M., Lo A.W., Lo Y.M., Ng H.K. Tissue and cellular tropism of the coronavirus associated with severe acute respiratory syndrome: an in-situ hybridization study of fatal cases. J Pathol. 2004;202:157–163. doi: 10.1002/path.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Caporarello N., Meridew J.A., Aravamudhan A., Jones D.L., Austin S.A., Pham T.X., Haak A.J., Moo Choi K., Tan Q., Haresi A., Huang S.K., Katusic Z.S., Tschumperlin D.J., Ligresti G. Vascular dysfunction in aged mice contributes to persistent lung fibrosis. Aging Cell. 2020;19:e13196. doi: 10.1111/acel.13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Taddei S., Virdis A., Ghiadoni L., Salvetti G., Bernini G., Magagna A., Salvetti A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension. 2001;38:274–279. doi: 10.1161/01.hyp.38.2.274. [DOI] [PubMed] [Google Scholar]
- 86.Donato A.J., Eskurza I., Silver A.E., Levy A.S., Pierce G.L., Gates P.E., Seals D.R. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ Res. 2007;100:1659–1666. doi: 10.1161/01.RES.0000269183.13937.e8. [DOI] [PubMed] [Google Scholar]
- 87.Seals D.R., Jablonski K.L., Donato A.J. Aging and vascular endothelial function in humans. Clin Sci (Lond) 2011;120:357–375. doi: 10.1042/CS20100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Donato A.J., Black A.D., Jablonski K.L., Gano L.B., Seals D.R. Aging is associated with greater nuclear NF kappa B, reduced I kappa B alpha, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell. 2008;7:805–812. doi: 10.1111/j.1474-9726.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yamamoto K., Takeshita K., Kojima T., Takamatsu J., Saito H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res. 2005;66:276–285. doi: 10.1016/j.cardiores.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 90.Machin D.R., Bloom S.I., Campbell R.A., Phuong T.T.T., Gates P.E., Lesniewski L.A., Rondina M.T., Donato A.J. Advanced age results in a diminished endothelial glycocalyx. Am J Physiol Heart Circ Physiol. 2018;315:H531–H539. doi: 10.1152/ajpheart.00104.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tabit C.E., Chung W.B., Hamburg N.M., Vita J.A. Endothelial dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev Endocr Metab Disord. 2010;11:61–74. doi: 10.1007/s11154-010-9134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tesfamariam B., Cohen R.A. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am J Physiol. 1992;263:H321–H326. doi: 10.1152/ajpheart.1992.263.2.H321. [DOI] [PubMed] [Google Scholar]
- 93.Goldberg R.B. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J Clin Endocrinol Metab. 2009;94:3171–3182. doi: 10.1210/jc.2008-2534. [DOI] [PubMed] [Google Scholar]
- 94.Nieuwdorp M., van Haeften T.W., Gouverneur M.C., Mooij H.L., van Lieshout M.H., Levi M., Meijers J.C., Holleman F., Hoekstra J.B., Vink H., Kastelein J.J., Stroes E.S. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. 2006;55:480–486. doi: 10.2337/diabetes.55.02.06.db05-1103. [DOI] [PubMed] [Google Scholar]
- 95.Dogne S., Flamion B., Caron N. Endothelial glycocalyx as a shield against diabetic vascular complications: involvement of hyaluronan and hyaluronidases. Arterioscler Thromb Vasc Biol. 2018;38:1427–1439. doi: 10.1161/ATVBAHA.118.310839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dharmashankar K., Widlansky M.E. Vascular endothelial function and hypertension: insights and directions. Curr Hypertens Rep. 2010;12:448–455. doi: 10.1007/s11906-010-0150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Iantorno M., Campia U., Di Daniele N., Nistico S., Forleo G.B., Cardillo C., Tesauro M. Obesity, inflammation and endothelial dysfunction. J Biol Regul Homeost Agents. 2014;28:169–176. [PubMed] [Google Scholar]
- 98.Zhang Q., Bastard P., Liu Z., Le Pen J., Moncada-Velez M., Chen J. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:eabd4570. doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bastard P., Rosen L.B., Zhang Q., Michailidis E., Hoffmann H.H., Zhang Y. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585. doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.van der Made C.I., Simons A., Schuurs-Hoeijmakers J., van den Heuvel G., Mantere T., Kersten S., van Deuren R.C., Steehouwer M., van Reijmersdal S.V., Jaeger M., Hofste T., Astuti G., Corominas Galbany J., van der Schoot V., van der Hoeven H., Hagmolen Of Ten Have W., Klijn E., van den Meer C., Fiddelaers J., de Mast Q., Bleeker-Rovers C.P., Joosten L.A.B., Yntema H.G., Gilissen C., Nelen M., van der Meer J.W.M., Brunner H.G., Netea M.G., van de Veerdonk F.L., Hoischen A. Presence of genetic variants among young men with severe COVID-19. JAMA. 2020;324:1–11. doi: 10.1001/jama.2020.13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nichols B., Jog P., Lee J.H., Blackler D., Wilmot M., D'Agati V., Markowitz G., Kopp J.B., Alper S.L., Pollak M.R., Friedman D.J. Innate immunity pathways regulate the nephropathy gene apolipoprotein L1. Kidney Int. 2015;87:332–342. doi: 10.1038/ki.2014.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Friedman D.J. COVID-19 and APOL1: understanding disease mechanisms through clinical observation. J Am Soc Nephrol. 2021;32:1–2. doi: 10.1681/ASN.2020111629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Leist S.R., Schafer A., Martinez D.R. Cell and animal models of SARS-CoV-2 pathogenesis and immunity. Dis Model Mech. 2020;13:dmm046581. doi: 10.1242/dmm.046581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Monteil V., Kwon H., Prado P., Hagelkruys A., Wimmer R.A., Stahl M., Leopoldi A., Garreta E., Hurtado Del Pozo C., Prosper F., Romero J.P., Wirnsberger G., Zhang H., Slutsky A.S., Conder R., Montserrat N., Mirazimi A., Penninger J.M. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181:905–913.e7. doi: 10.1016/j.cell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wahl A., Gralinski L.E., Johnson C.E., Yao W., Kovarova M., Dinnon K.H., 3rd, Liu H., Madden V.J., Krzystek H.M., De C., White K.K., Gully K., Schafer A., Zaman T., Leist S.R., Grant P.O., Bluemling G.R., Kolykhalov A.A., Natchus M.G., Askin F.B., Painter G., Browne E.P., Jones C.D., Pickles R.J., Baric R.S., Garcia J.V. SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature. 2021;591:451–457. doi: 10.1038/s41586-021-03312-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Campbell C.M., Guha A., Haque T., Neilan T.G., Addison D. Repurposing immunomodulatory therapies against coronavirus disease 2019 (COVID-19) in the era of cardiac vigilance: a systematic review. J Clin Med. 2020;9:2935. doi: 10.3390/jcm9092935. [DOI] [PMC free article] [PubMed] [Google Scholar]