

Abstract
Ultrasensitive surface-enhanced Raman spectroscopy (SERS) still faces difficulties in the quantitative analysis because of its susceptibility to local optical field variations at plasmonic hotspots in metallo-dielectric nanostructures. Current SERS calibration approaches using Raman tags have inherent limitations due to spatial occupation competition with analyte molecules, spectral interference with analyte Raman peaks, and photodegradation. Here, we report that plasmon-enhanced electronic Raman scattering (ERS) signals can serve as an internal standard for spatial and temporal calibration of molecular Raman scattering (MRS) signals at the same hotspots, enabling rigorous quantitative SERS analysis. We observe a linear dependence between ERS and MRS signal intensities upon spatial and temporal variations of excitation optical fields, manifesting the |E|4 enhancements for both ERS and MRS processes at the same hotspots. Furthermore, we find that ERS calibration’s performance limit is caused by orientation variations of analyte molecules at hotspots.
Keywords: plasmonics, surface-enhanced Raman spectroscopy (SERS), quantitative SERS, SERS calibration, electronic Raman scattering
Graphical Abstract
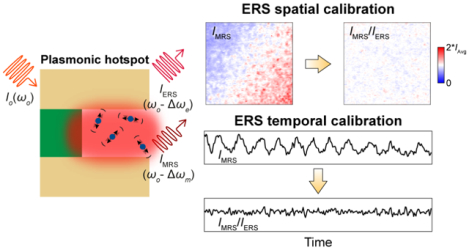
Surface-enhanced Raman spectroscopy (SERS) has emerged as an ultrasensitive molecular detection approach for biological and chemical analysis,1–5 and exploits plasmonic enhancement of both excitation and inelastic scattering processes for analyte molecules present in optical hotspots.6, 7 Due to the |E|4 plasmonic enhancement of molecular Raman scattering (MRS) processes,8 where |E| is the magnitude of the incident electric field, SERS signals are highly sensitive to the spatial and temporal variations of local optical fields at hotspots such as those originating from subtle changes of optical focusing conditions, nanoscale geometrical variations, and excitation laser power fluctuations. Therefore, despite enormous progress in high-performance SERS substrates,1, 9, 10 it is challenging to use SERS for accurate and reproducible quantitative analysis even with the same SERS substrates, which significantly limits its usage in real-world applications with less controlled experimental conditions.11, 12 Towards quantitative biological and chemical analysis, a significant effort in SERS research is to develop internal standards for SERS calibration by introducing exogenous Raman tags at plasmonic hotspots such as thiolate ligands,13–16 cucurbit[8]uril,17 4-mercaptobenzoic acid,15, 18 mercaptobenzimidazole,19 isotopes,20–22 and carbon-nanotubes.23 Since reference signals from Raman tags experience similar local field enhancements at the same hotspots, the calibrated ratiometric SERS signals for analyte molecules are significantly less sensitive to spatial variations of local optical fields as well as temporal fluctuations of laser excitation fields. However, SERS internal standards based on Raman tags face several inherent challenges for practical applications, including (i) spatial occupation competition between the analyte and exogenous reference molecules at hotspots; (ii) spectral interference of Raman bands; (iii) laser-induced degradation/desorption of Raman tags aggravated by plasmonic photochemical/photothermal effects; and (iv) the restricted shelf-life due to chronic chemical degradation of Raman tags. Notably, Raman tags can be embedded at hotspots inside core-molecule-shell nanoparticles to mitigate the issue of spatial occupation competition with analytes,9, 15, 16 but the other three challenging limitations still cannot be simply resolved in this way.
In this work, we report that electronic Raman scattering (ERS) background signals from metal nanostructures at plasmonic hotspots can serve as internal standards for spatial and temporal calibration in quantitative SERS analysis, which can eliminate the use of exogenous Raman tags and thus bypass their associated limitations. Plasmonic ERS signals originate from the surface-plasmon-enhanced inelastic light scattering of sp-band electrons in the metal (Figure 1A-left, Supporting Information) and are present in the broad SERS background with other possible emission sources, including photoluminescence signals by interband electronic transitions or intraband electronic transitions.24–32 Recent studies have shown that ERS signals dominate in the low-wavenumber range of the SERS background under continuous-wave laser excitation at near-infrared wavelengths where the interband transitions hardly occur due to the small photon energy, while intraband transitions are difficult to occur because of significant momentum mismatch in the sp-band dispersion.24, 27, 30 By using a long-pass filter to block the laser line (Rayleigh scattering), the filtered SERS background continuum exhibits an ERS pseudo-peak in the measured Raman spectra (Figure 1A-right).24, 27, 33 The plasmonic ERS intensity is proportional to the density of electron-hole pairs, ne−h, in metal nanostructures24, 27, 30 expressed as where Δωe is the Stokes-shifted frequency for the ERS process, ℏ is the Planck constant, kB is the Boltzmann constant, and T is the temperature. An ERS event involves the quantum transitions between a correlated pair of occupied and unoccupied electronic states. Therefore, the thermodynamics statistics of the ERS processes obey a Bose-Einstein distribution for a correlated electronic pair system with integer spin (s = 1), and the ERS intensity can exponentially increase when Δωe approaches zero. Compared to ERS signals showing a broad spectral feature because of the continuous electronic sp-bands of the metal, MRS signals in SERS carry many distinct narrow peaks in the measured spectra due to discrete inelastic energy shifts associated with discrete vibrational modes of a molecule (Figure 1B). While the previous studies suggest that the observed low-wavenumber pseudo-peaks in SERS measurements originate from the sideband amplified spontaneous emission in the excitation laser,34, 35 we experimentally rule out such possibility with control experiments using reference samples of Si substrates and flat Au films. Furthermore, this work introduces a rigorous theoretical analysis to interpret the origin of plasmonic ERS processes behind the observed low-wavenumber pseudo-peaks and the mechanism of how the ERS signals can act as the internal standard to correct spatial and temporal variations in SERS measurements. Notably, this work also provides the first study to investigate the molecule orientation variation effects on the SERS calibration performance using either ERS or MRS signals the internal standard.
Figure 1.
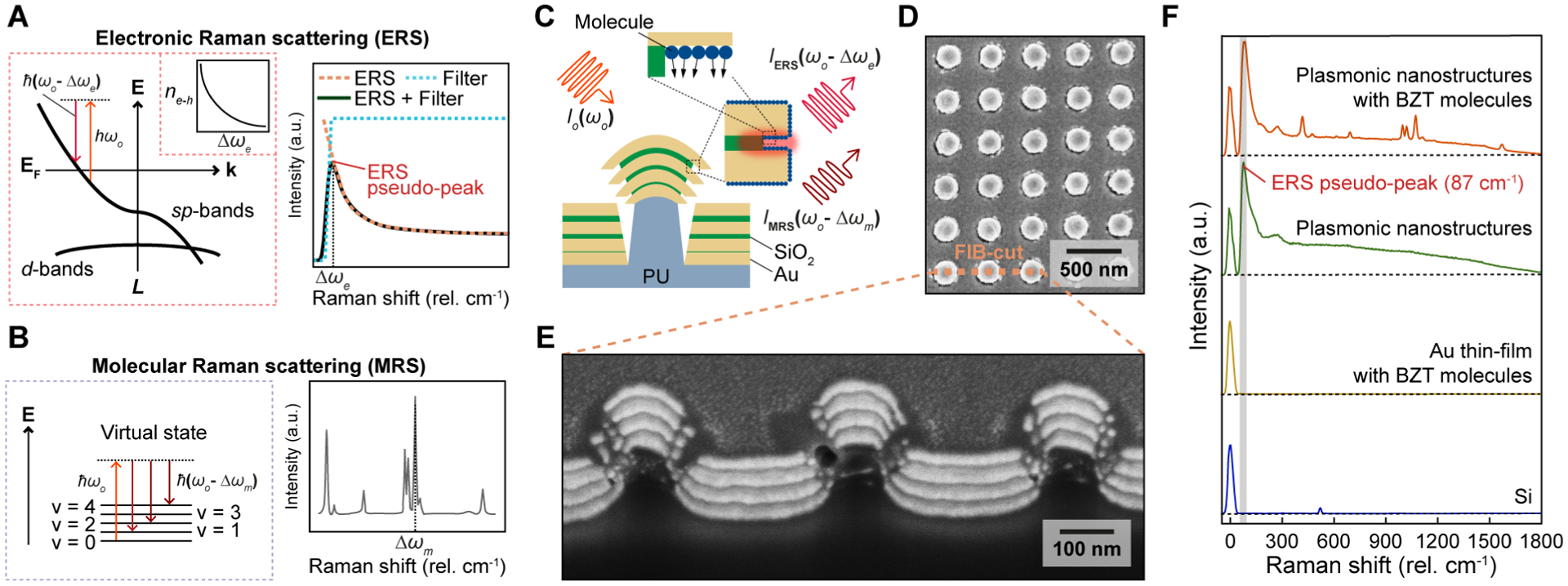
Plasmonic enhancement of ERS and MRS signals at hotspots. (A) Energy-diagram illustration of the ERS process (left) and the ERS pseudo-peak spectrum (right). Inset (left): the volume density spectrum of electron-hole pairs ne−h. (B) Energy-diagram illustration of the MRS process (left) and the measured Raman peaks of molecules (right). (C) Scheme of plasmon-enhanced ERS and MRS processes at hotspot regions from nanolaminate SERS substrates. PU, polyurethane; (D) Top and (E) cross-sectional view SEM images of the nanolaminate SERS substrates. FIB, focused-ion-beam; (F) The measured Raman spectra for four different samples.
Since the electromagnetic boundary condition relates the dominant perpendicular electric fields on the two sides of the metal-insulator interface at plasmonic hotspots (see details in Supporting Information and Figure S1), the ratio between MRS and ERS signals from the same plasmonic hotspots can be approximated as:
| (1) |
where εM and εI are the complex permittivity of metal and insulator, respectively, at the excitation laser frequency ωo, σERS and σMRS are the effective cross-sections for the ERS and MRS process, respectively, Δωm is the Stokes-shifted frequency for the MRS process, r (0< r <1) is the effective coefficient related to the orientation of transition dipole moment for a specific vibrational mode of analyte molecules,36 and N is concentration of analyte molecules. In eq. (1), except for r and N, all other terms can be grouped into a material-related parameter C, where . Therefore, by ERS calibration, the ratiometric value, IMRS/IERS, becomes less dependent on the spatial and temporal local field variations, and can better quantify the concentration of analyte molecules compared to the directly measured IMRS. Also, eq. (1) predicts that IMRS/IERS is still subject to orientation variations of the vibrational transition dipole moment for molecules at hotspots, setting the performance limit of the ERS calibration process for quantitative SERS analysis.
Here, we first test plasmonic ERS signals, IERS, as the internal standard for spatial calibration of MRS signals, IMRS, by using nanolaminate SERS substrates functionalized with a self-assembled monolayer of benzenethiol (BZT) molecules (Figure 1C). Nanolaminate SERS substrates consist of multilayered metal-insulator-metal (MIM) nanocavity arrays and MIM nanohole arrays separated by polymer nanopillar arrays (Figure 1D–E, and fabrication details in Supporting Information), and represents a generalized metallo-dielectric plasmonic system that supports multiple localized and delocalized plasmonic modes.37–39 To investigate plasmonic enhancement effects in ERS and MRS processes, we conducted Raman measurements for nanolaminate SERS substrates functionalized with BZT molecules and compared them with measurements on various reference samples, including bare nanolaminate SERS substrates, unpatterned Au thin-film (nominally 150 nm thick) with BZT molecules, and a bare silicon substrate (Figure 1F). Raman measurements performed under 785 nm laser excitation show several key results. First, BZT-functionalized nanolaminate SERS substrates exhibit distinct MRS signal of BZT molecules, while a BZT-functionalized flat Au film does not show MRS signals due to the lack of plasmonic modes needed for local field enhancement. Second, nanolaminate SERS substrates with and without BZT show a distinct ERS pseudo-peak at approximately 87 cm−1, whereas the silicon substrate and flat Au film do not show background ERS emission signals. While a few previous studies suggest that the low-wavenumber pseudo-peak in SERS background may originate from the inelastic backscattering signals from the amplified sideband spontaneous emission of the excitation laser source,34, 35 our measurements can rule out this possibility by showing the absence of such signals from the reference samples of the silicon and flat Au film. Third, the intensities of plasmonic ERS signals in the relatively high wavenumber range (>200 cm−1) can change slightly after introducing surface-modified molecules in plasmonic hotspots (Supporting Information and Figure S2). However, the low-wavenumber ERS-pseudo peaks show a similar intensity for the plasmonic nanostructured sample with and without surface-modified molecules. These observations suggest that when Δωe is small, ne−h(Δωe) in metal nanostructures can become much higher than the molecule density at hotspots to dominate over the effects due to the charge transfer process between the metal sp band and molecules. Therefore, the ERS calibration’s performance based on the low-wavenumber ERS-pseudo peak is generally not affected by the adsorbed molecules and related charge transfer process.
To verify that ERS-calibrated MRS signals are insensitive to variations of local field intensities from different hotspots, we analyzed the spatial-correlation of ERS and MRS signals from 10,000 pixels over a 100 μm × 100 μm area using BZT-functionalized nanolaminate SERS substrates. To generate additional spatial variations of local optical fields, we intentionally varied the excitation laser focusing condition by inducing geometric deformation of the sample (Figure 2A). Figure 2B shows the averaged SERS spectrum with one standard deviation (SD) expressed as gray shaded regions. Without deformation, the sample shows a high spatial uniformity with a low coefficient of variation (CV) value = 5.6 % (Figure S3), where CV is the ratio of the SD to the mean. With surface-immobilized BZT molecules on the Au surface, we can assume a uniform distribution of molecule concentration but with spatial variations of molecule orientations among different hotspots. For a facile comparison of relative spatial variations, two-dimensional (2D) map results were plotted with scale bar ranging from zero to two-times the average intensity values of each 2D map (Figure 2C–D). As shown in Figure 2C, the 2D maps of IERS, I422 and I1004 exhibit significant spatial variations but with spatially-correlated distribution patterns among them. In contrast, the ERS-calibrated MRS signals (I422/IERS and I1004/IERS) show much more uniform spatial distributions (Figure 2D), in agreement with the theoretical prediction from eq. (1) that ERS calibration can remove the spatial variations of local field intensities between different hotspots. Similar to ERS calibration, MRS-normalized signals (I1004/I422) also exhibit a more uniform 2D pattern by removing the effects of local field variations among hotspots. Notably, no matter by ERS calibration or MRS calibration, the 2D patterns of calibrated MRS signals still show residual random spatial variations due to the uncontrollable orientation variations of surface-assembled BZT molecules between different hotspots on the SERS substrates, as predicted in eq. (1). Therefore, while the ERS and MRS calibration approaches can significantly improve quantitative SERS analysis in 2D mapping by removing the spatial variations of excitation optical field intensities between different hotspots, they cannot calibrate the variations from the random orientations of analyte molecules.
Figure 2.
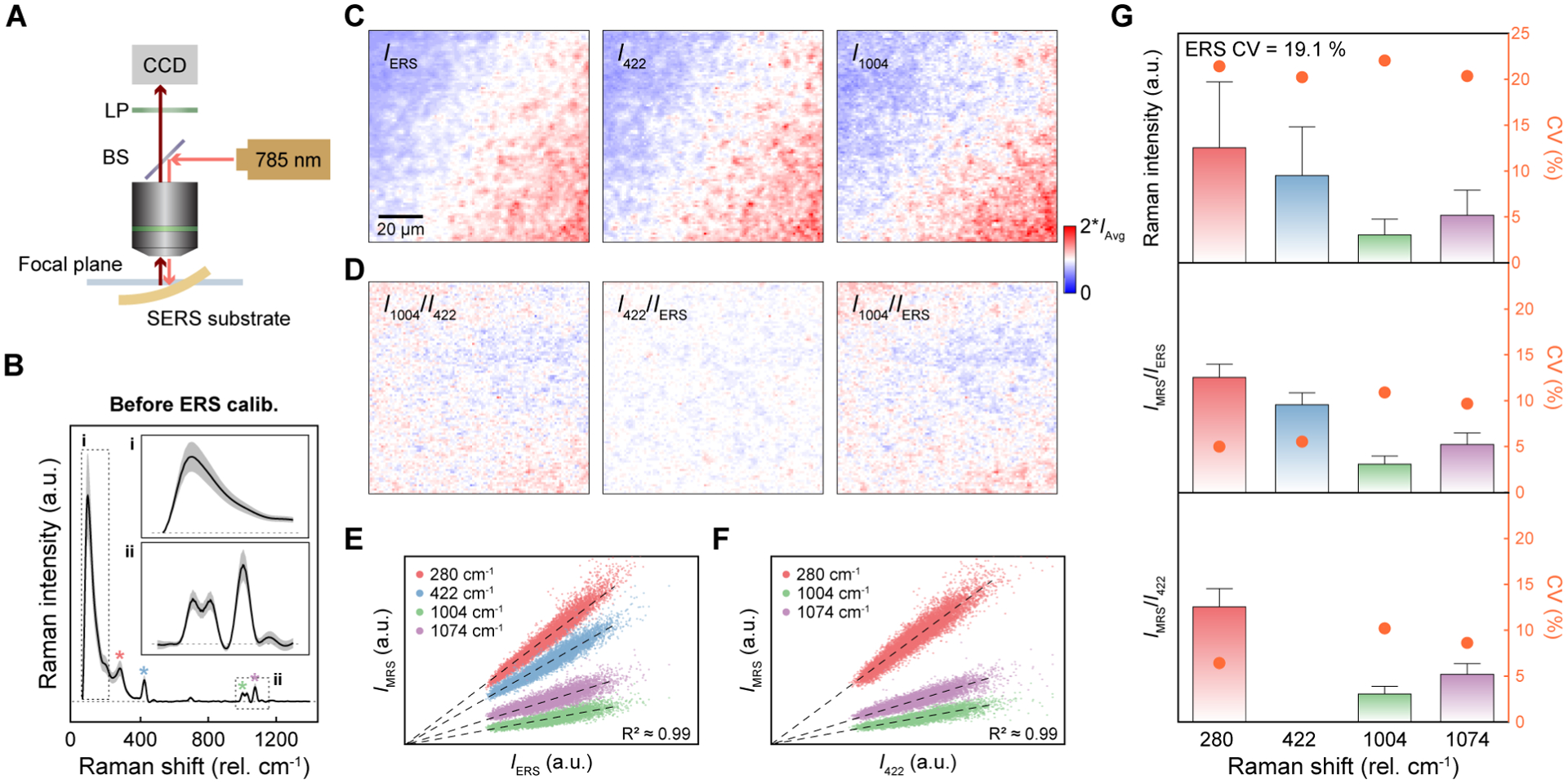
Spatially-correlated linear dependence between ERS and MRS signals. (A) Schematic illustration of the experimental setup for varying excitation laser focusing conditions by mechanical deformation of the SERS substrates. CCD, charge-coupled device; LP, long-pass filter; BS, beam splitter; (B) Average BZT SERS spectrum with SD (gray regions) from 10,000 pixels before ERS calibration. (C) 2D Raman images for IERS, I422 and I1004. (D) 2D Raman images of I1004/I422 by MRS calibration and I422/IERS and I1004/IERS by ERS calibration. (E–F) The scatter plots of IMRS as a function of (E) IERS and (F) I422. (G) Quantitative statistical analysis for before ERS calibration (top), after ERS calibration (middle), and MRS calibration by 422 cm−1 (bottom). The error bars for the intensity bars are 1.5 IQR from 10,000 pixels.
Figure 2E shows a statistical analysis of 10,000 pixels in 2D maps to examine the dependence of IMRS for MRS peaks at 280 cm−1, 422 cm−1, 1004 cm−1, and 1074 cm−1 on IERS, which can be compared with the dependence of IMRS for three MRS peaks at 280 cm−1, 1004 cm−1, and 1074 cm−1 on IMRS,422 of a specific MRS peak at 422 cm−1 in Figure 2F. For the quantitive assessment of the strength of the linear relationship, we calculated the coefficient of determination values (R2) between MRS and ERS signals for 10,000 pixels (Figure 2E). The observed high values of R2 (≈ 0.99) confirm the strong linear dependence between MRS and ERS signals at different locations with spatial variations of local optical field intensities, which means that both ERS and MRS signals follow |E|4 enhancements at the same hotspots.24, 27, 29–32, 40, 41 The different slopes for different MRS peaks are due to their different values of Raman scattering cross-sections, which reflects that different vibrational modes can have transition dipole moments of different amplitudes and orientations.42–44 In Figure 2F, the distribution plots display a linear relationship (R2 ≈ 0.99) between IMRS at 422 cm−1 and IMRS at 280 cm−1, 1004 cm−1, and 1074 cm−1, and also show a distribution spreading due to the orientation variations of surface-modified BZT molecules at hotspots.
To quantitatively evaluate ERS calibration, we show the intensity histograms of raw (Figure 2G-top), ERS-calibrated (Figure 2G-middle), and MRS-normalized (Figure 2G-bottom, using 422 cm−1) data with CV values for different vibrational modes. After ERS calibration, the CV values for vibrational modes at 280 cm−1, 422 cm−1, 1004 cm−1 and 1074 cm−1 are reduced from 21.4 %, 20.2 %, 22 %, and 20.4 % to 5 %, 5.5 %, 10.8 %, and 9.7 %, respectively. CV is a quantified value representing the uncertainty of measured data, and the reported uncertainty of MRS signals for BZT data in Figure 2 was calculated using one SD obtained from 10,000 individual measurements unless otherwise noted. Accordingly, the 1.5 interquartile ranges (IQR), presented as error bars in Figure 2G, are also significantly reduced by ERS calibration. IQR was calculated by subtracting the first quartile from the third quartile to statistically determine the thresholds for outliers. We can observe such improvements in the spectra with reduced SD by a factor of ≈ 3 and a histogram plot with a better fitting to a normal distribution curve (Figure S4). As expected, the ratiometric values of IMRS/I422 by calibration with the MRS peak at 422 cm−1 also show significant reductions of CV values to 6.4 %, 10.2 %, and 8.6 % for 280 cm−1, 1004 cm−1, and 1074 cm−1, respectively. However, the MRS calibration method, because of the introduction of reference molecules, has intrinsic limitations in spatial occupation competition at hotspots, spectral interference of Raman bands and involves tedious work to mix reference molecules with known concentrations in solution samples accurately.13–23 To support ERS calibration’s general utility for different SERS substrates, we have performed the same experiment for a bacterial cellulose SERS substrate coated with aggregation-based gold nanoparticles (Figure S5). ERS calibration shows a significant improvement of CV value from 63.6 % to 18.1 %, and the higher CV value for after ERS calibration compared to periodic nanolaminate SERS substrate is due to the randomly available sites of gold surface for thiol-based BZT molecules.
To demonstrate ERS calibration’s applicability for quantitative SERS analysis, we performed label-free concentration-dependent SERS measurements of Rhodamine 6G (R6G) analytes in phosphate-buffered-saline (1× PBS) solutions. 2D Raman mapping measurements with 20 pixels × 20 pixels were conducted over a 100 μm × 100 μm area for the SERS substrates immersed in the R6G solutions. In Figure 3A, without ERS calibration, the SERS spectra and corresponding 2D images for four representative concentrations of 6 μmol/L, 10 μmol/L, 40 μmol/L and 80 μmol/L reveal large SDs (gray region) and spatial variations with CV values of 30.8 %, 28 %, 18.7 % and 22.7 %, respectively. The reported MRS signal uncertainty for R6G data in Figure 3 was calculated by using one SD obtained from 400 individual measurements unless otherwise noted. Furthermore, as the R6G concentration increases from 10 μmol/L to 40 μmol/L, the measured average SERS intensities decrease instead of increasing, revealing that the variations of local field intensities at different locations of the large (≈ 16 cm2) sample will bias the quantitative SERS analysis of analyte concentrations in different measurements. After ERS calibration, the CV values of MRS peaks decrease to 7.3 %, 6.5 %, 5.4 %, and 5.5 %, respectively (Figure 3B). Furthermore, the ERS-calibrated SERS signals show gradually increased R6G peak intensities with increasing R6G concentrations. By ERS calibration, 2D Raman images also clearly show a gradual increase of Raman intensity with significantly reduced spatial variation.
Figure 3.
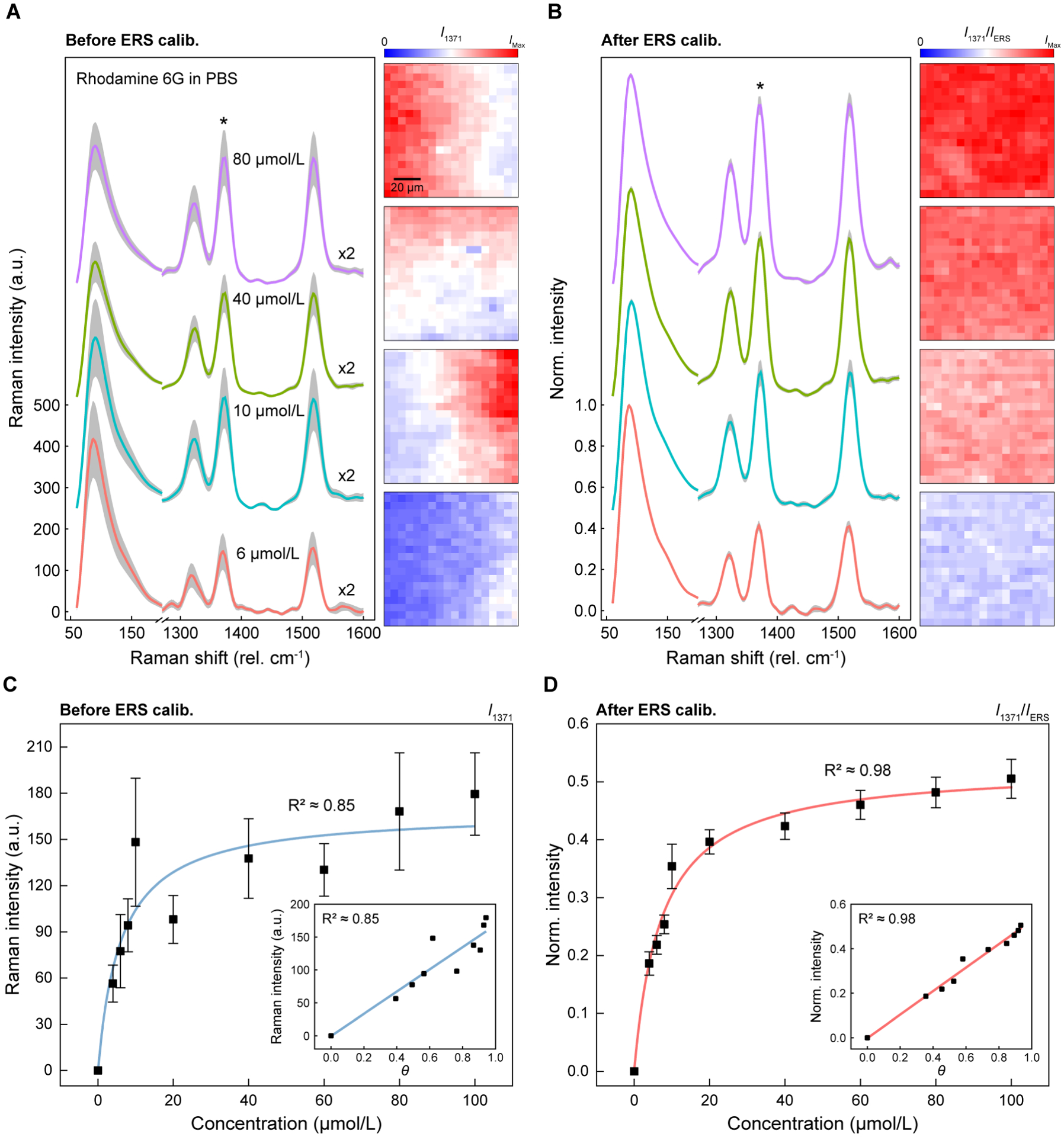
ERS spatial calibration for quantitative SERS analysis of R6G molecules with different concentrations. (A–B) Spatially averaged Raman spectra with SDs (gray regions) from 400 pixels for R6G molecules with different concentrations in PBS solution (A) before and (B) after ERS calibration. Intensities in the MRS region between 1300 cm−1 and 1600 cm−1 are multiplied by two, and the spectra are offset in the y-axis for clarity. (C–D) Working curves of R6G molecules with different concentrations from 4 μmol/L to 100 μmol/L using R6G peak at 1371 cm−1 (C) before and (D) after ERS calibration. The error bars show one standard deviation from 400 pixels. The inset shows the calculated surface coverage (θ) (C) before and (D) after ERS calibration.
To evaluate quantitative SERS analysis by ERS calibration in a broad range of concentrations, we plotted the working curve from 4 μmol/L to 100 μmol/L using the Raman peak at 1371 cm−1 before (Figure 3C) and after (Figure 3D) ERS calibration. After ERS calibration, the working curve becomes smoother and can fit better to the Langmuir adsorption function with the R2 value improved from 0.85 to 0.98, and the equilibrium constant KT = (1.38 × 105 L/mol ± 0.171 × 105 L/mol) from fitting also shows a significantly reduced CV value from 38.6 % (1.62 × 105 L/mol ± 0.627 × 105 L/mol) to 12.3 %. The reported equilibrium constant uncertainty corresponds to one SD derived from Langmuir adsorption fitting. By ERS calibration, the ratiometric SERS signals show reduced SDs in the 2D Raman measurements for all different R6G concentrations. We calculated the surface coverage (θ) at different concentrations using KT (insets of Figure 3C–D). ERS calibration can lead to better linearity between R6G surface coverage and the ratiometric SERS signals, with the R2 value improved from 0.85 to 0.98. Furthermore, the working curve for the smaller range of R6G concentrations between 20 μmol/L and 100 μmol/L shows a linear relationship (Figure S6), and the linearity of the working curve is improved by ERS calibration, with the R2 value increased from 0.89 to 0.99. Besides, we demonstrate that ERS calibration for quantitative SERS analysis can work for different Raman peaks with similar performance (Figure S7), revealing the unique advantage of no spectral interference between the ERS internal standard at low wavenumbers (<100 cm−1) and different Raman peaks of analytes.
To examine the feasibility of using the ERS internal standard for temporal SERS calibration, we investigated the temporal-correlation between ERS and MRS signals in response to static and dynamic laser excitation fluctuations (Figure 4). Figure 4A shows a schematic illustration of hotspots’ microscopic environment containing analyte molecules in solutions under laser excitation of high or low power. With stronger laser excitation, the hotspots can generate higher intensities of both ERS and MRS signals as they follow the same |E|4 enhancement. Notably, the thermal activation can cause translational and rotational movements of individual molecules at hotspots in the solution, causing dynamical and stochastical perturbation of the SERS signals at a constant analyte concentration. The strength of thermal activation processes depends on the excitation laser power due to the local heating by plasmonic photothermal effects at hotspots.45, 46 In many real-world SERS applications targeted at wearable biochemical sensing47–50 and invivo biomedical monitoring,4, 51–53 the temporal fluctuations of local optical field intensities at hotspots can occur due to perturbed excitation conditions during experiments, such as optical path changes from body movement and power fluctuations of the laser source.
Figure 4.
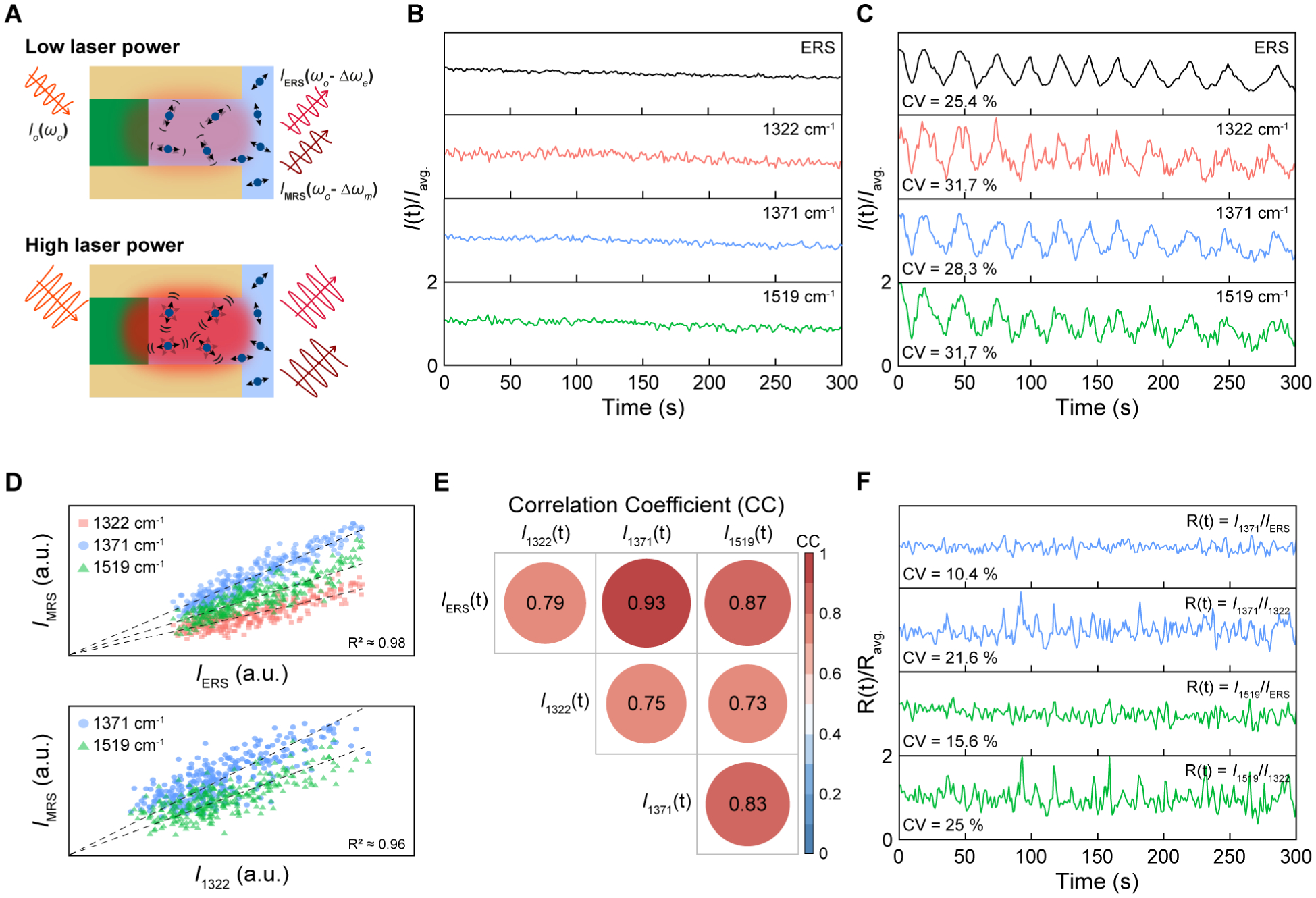
ERS temporal calibration for time-resolved SERS measurements. (A) Schematic illustration of the microscopic environment of the hotspots under laser excitation of low and high powers. (B–C) Time-trajectories of ERS signals at 87 cm−1 and three MRS signals at 1322 cm−1, 1371 cm−1, and 1519 cm−1 (B) without and (C) with dynamic modulations of laser excitation powers. (D) The scatter plots of IMRS as a function of IERS (top) and I1322 (bottom). (E) A matrix of calculated correlation coefficients (CC) among ERS and MRS signals. (F) Time-trajectories of 1371 cm−1 and 1519 cm−1 modes by ERS calibration at 87 cm−1 and the MRS calibration at 1322 cm−1.
We first examined time-traces of solution-based 100 μmol/L R6G molecules by acquiring signals for 300 s with 1 s integration time without extrinsic laser modulations (Figure 4B). To have a side-by-side comparison of temporal fluctuations, we plotted time-trajectories of ERS and MRS signals with a scale from zero to two-times the average intensity value for modes at 1322 cm−1, 1371 cm−1, and 1519 cm−1 (Figure 4B). Compared to the MRS signals, the ERS signals show a smaller temporal fluctuation level since the ERS process is not susceptible to temporal variations of molecule concentrations and orientations at hotspots from their random movements and rotations in the solution. To temporally modulate the laser excitation conditions, we changed laser excitation powers by adjusting the aperture size in the laser beam path, which resulted in twelve cycles of gradual increase and decrease in laser power between 100 μW and 200 μW over 300 s (Figure 4C). Due to the temporal power fluctuations, we can observe high CV values of 25.4 %, 31.7 %, 28.3 %, and 31.7 % for the ERS peak at 87 cm−1 and the three MRS modes at 1322 cm−1, 1371 cm−1, and 1519 cm−1, respectively. The reported signal uncertainty for time-dependent measurements using R6G in Figure 4 was calculated by using one SD obtained from 300 individual measurements unless otherwise noted. Time-averaged spectra with dynamic power fluctuations show ≈ 3.5 times larger SD compared to the static excitation condition, representing difficulties for quantitative SERS analysis under dynamically fluctuating excitation conditions (Figure S8). We can also observe that ERS signals show a smaller CV value than all MRS signals with less noise, in agreement with the static excitation condition (Figure 4B). To investigate the temporal relation between ERS and MRS signals under dynamic laser excitation fluctuations, we performed statistical analysis to assess the time-dependent relationship between IMRS(t) for different MRS modes and IERS(t) for the ERS pseudo-peak at 87 cm−1 (Figure 4D-top) in comparison with the time-dependent relationship between IMRS(t) for different MRS modes and one specific IMRS,1322(t) at 1322 cm−1 (Figure 4D-bottom). We observe statistically linear relationships in both scatter plots of IMRS(t) vs. IERS(t) (R2 ≈ 0.98) and IMRS(t) vs. IMRS,1322(t) (R2 ≈ 0.96). However, compared to IMRS(t) vs. IERS(t), the scatter plot of IMRS(t) vs. IMRS,1322(t) exhibits a much wider spreading of scatter distributions because the temporal calibration standard of IMRS,1322(t), unlike IERS(t), is additionally subjected to dynamic molecular orientation variations due to thermally-activated random rotational movements of molecules in the solution. As expected, under static laser excitation, the scatter plots for time-dependent measurements of IMRS(t) vs. IERS(t) (R2 ≈ 0.99) and IMRS(t) vs. IMRS,1322(t) (R2 ≈ 0.99) also show statistically linear relationships with a wider spreading for IMRS(t) vs. IMRS,1322(t) (Figure S9A). To further quantitatively compare temporal-correlations of IMRS(t) vs. IERS(t) and IMRS(t) vs. IMRS,1322(t), we calculated the correlation coefficient (CC) (Pearson) values between them in the time-resolved measurements with dynamic power fluctuations (Figure 4E), which can provide a numerical measure of the statistical relationship between two time-dependent variables in the range from −1 to 1. Compared to CC values of 0.79, 0.93, and 0.87 between the ERS signal at 87 cm−1 and one MRS signal among the three different MRS modes (CCE-M), the CC values between any of two MRS signals among the three different MRS modes (CCM-M) show slightly lower values of 0.73, 0.75, and 0.83. CC values were calculated with a confidence level of 95 %. We observe a similar trend for the results under the static excitation condition (Figure S9B). In Figure 4F, ERS calibration can eliminate the incident light-induced signal fluctuations with significantly reduced CV values of 10.4 % and 15.6 % for 1371 cm−1 and 1519 cm−1, respectively. As shown in Figure 4B–4E, MRS calibration using the MRS peak at 1322 cm−1 can also correct the dynamic excitation power fluctuations, and the CV values of MRS signals at 1371 cm−1 and 1519 cm−1 can be reduced from 28.3 % and 31.7 % to 21.6 % and 25 %, respectively. Compared to ERS calibration, MRS calibration exhibits a lower performance since the MRS-based calibration standards carry the additional dynamic variations due to random rotation movements of molecules in the solution (Supporting Information). Furthermore, upon abrupt changes to the laser power (Figure S10), we demonstrate that ERS calibration can significantly reduce CV value from 36.7 % to 6.5 %, highly desirable for quantitative analysis in time-resolved SERS measurements.
In summary, we report a new SERS calibration method based on the ERS signals at plasmonic hotspots. With theoretical analysis, 2D Raman mapping of surface-immobilized molecules shows a spatially-correlated linear dependence between ERS and MRS signals, revealing that they experience the same local field enhancements at the hotspots. We experimentally demonstrate ERS calibration’s utility for improved quantitative SERS analysis using R6G analyte solutions, verifying that ERS-calibrated SERS signals can serve better as analytical values to reflect actual molecule concentration at hotspots with significantly reduced spatial and temporal variations, beneficial to diverse SERS applications. In agreement with theoretical predictions, experimental measurements show that the ERS calibration process cannot remove the effects from spatial or temporal orientation variations of analyte molecules at hotspots, setting the performance limit of ERS calibration. Compared to existing Raman tag-based SERS calibration methods, the new ERS calibration strategy can provide unique advantages to overcome fundamental limitations of internal molecular standards, such as no spectral interference from additional Raman bands, no spatial occupation competition with analyte molecules, and excellent photochemical/photothermal stability. Unlike label-based Raman tags with limited selection, the ERS signals can serve as a more general internal standard to directly calibrate label-free SERS signals for multiplexed monitoring of different analyte molecules in complicated biological and chemical systems, which can offer significant benefits to many analytical and translational applications.
Supplementary Material
ACKNOWLEDGMENT
The authors gratefully acknowledge funding support from the Air Force Office of Scientific Research (AFOSR) Young Investigator Award FA9550-18-1-0328 and National Institute of Standards and Technology (NIST) grants 70NANB18H201 and 70NANB19H163. A. A. and W. Z. acknowledge support under the Cooperative Research Agreement between the University of Maryland and the National Institute of Standards and Technology Physical Measurement Laboratory, Award 70NANB14H209, through the University of Maryland.
Footnotes
Supporting Information. The following files are available free of charge.
Theoretical derivation of ERS calibration; Fabrication of nanolaminate SERS substrates; Experimental setup; Chemical interface damping for ERS signals; Theoretical comparison between ERS and MRS calibrations for solution-based molecules; 2D Raman image using BZT without sample deformation; BZT SERS spectrum after ERS calibration; Histogram comparison of BZT signals before and after ERS calibration; Working curve from 20 μmol/L to 100 μmol/L of R6G solution before and after ERS calibration; Working curve of R6G using 619 cm−1 before and after ERS calibration; Time-averaged SERS spectra with SDs under static and dynamic excitation condition; Scatter plots of IMRS as a function of IERS and other IMRS without extrinsic laser modulation; CC values without extrinsic laser modulation; ERS calibration for abrupt changes of laser power;
The authors declare no competing financial interest.
REFERENCES
- 1.Langer J; Jimenez de Aberasturi D; Aizpurua J; Alvarez-Puebla RA; Auguie B; Baumberg JJ; Bazan GC; Bell SEJ; Boisen A; Brolo AG; Choo J; Cialla-May D; Deckert V; Fabris L; Faulds K; Garcia de Abajo FJ; Goodacre R; Graham D; Haes AJ; Haynes CL; Huck C; Itoh T; Kall M; Kneipp J; Kotov NA; Kuang H; Le Ru EC; Lee HK; Li JF; Ling XY; Maier SA; Mayerhofer T; Moskovits M; Murakoshi K; Nam JM; Nie S; Ozaki Y; Pastoriza-Santos I; Perez-Juste J; Popp J; Pucci A; Reich S; Ren B; Schatz GC; Shegai T; Schlucker S; Tay LL; Thomas KG; Tian ZQ; Van Duyne RP; Vo-Dinh T; Wang Y; Willets KA; Xu C; Xu H; Xu Y; Yamamoto YS; Zhao B; Liz-Marzan LM, Present and Future of Surface-Enhanced Raman Scattering. ACS Nano 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kneipp J; Kneipp H; Kneipp K, SERS--a single-molecule and nanoscale tool for bioanalytics. Chem Soc Rev 2008, 37 (5), 1052–60. [DOI] [PubMed] [Google Scholar]
- 3.Laing S; Gracie K; Faulds K, Multiplex in vitro detection using SERS. Chem Soc Rev 2016, 45 (7), 1901–1918. [DOI] [PubMed] [Google Scholar]
- 4.Laing S; Jamieson LE; Faulds K; Graham D, Surface-enhanced Raman spectroscopy for in vivo biosensing. Nature Reviews Chemistry 2017, 1 (8). [Google Scholar]
- 5.Zong C; Xu M; Xu LJ; Wei T; Ma X; Zheng XS; Hu R; Ren B, Surface-Enhanced Raman Spectroscopy for Bioanalysis: Reliability and Challenges. Chem Rev 2018, 118 (10), 4946–4980. [DOI] [PubMed] [Google Scholar]
- 6.Moskovits M, Surface-enhanced Raman spectroscopy: a brief retrospective. Journal of Raman Spectroscopy 2005, 36 (6–7), 485–496. [Google Scholar]
- 7.Le Ru EC; Etchegoin PG, Rigorous justification of the [E](4) enhancement factor in Surface Enhanced Raman Spectroscopy. Chem Phys Lett 2006, 423 (1–3), 63–66. [Google Scholar]
- 8.Jiang J; Bosnick K; Maillard M; Brus L, Single molecule Raman spectroscopy at the junctions of large Ag nanocrystals. J Phys Chem B 2003, 107 (37), 9964–9972. [Google Scholar]
- 9.Lim DK; Jeon KS; Hwang JH; Kim H; Kwon S; Suh YD; Nam JM, Highly uniform and reproducible surface-enhanced Raman scattering from DNA-tailorable nanoparticles with 1-nm interior gap. Nat Nanotechnol 2011, 6 (7), 452–60. [DOI] [PubMed] [Google Scholar]
- 10.Ding S-Y; Yi J; Li J-F; Ren B; Wu D-Y; Panneerselvam R; Tian Z-Q, Nanostructure-based plasmon-enhanced Raman spectroscopy for surface analysis of materials. Nature Reviews Materials 2016, 1 (6). [Google Scholar]
- 11.Fang Y; Seong NH; Dlott DD, Measurement of the distribution of site enhancements in surface-enhanced Raman scattering. Science 2008, 321 (5887), 388–392. [DOI] [PubMed] [Google Scholar]
- 12.Lindquist NC; de Albuquerque CDL; Sobral-Filho RG; Paci I; Brolo AG, High-speed imaging of surface-enhanced Raman scattering fluctuations from individual nanoparticles. Nat Nanotechnol 2019, 14 (10), 981–987. [DOI] [PubMed] [Google Scholar]
- 13.Loren A; Engelbrektsson J; Eliasson C; Josefson M; Abrahamsson J; Johansson M; Abrahamsson K, Internal standard in surface-enhanced Raman spectroscopy. Analytical Chemistry 2004, 76 (24), 7391–7395. [DOI] [PubMed] [Google Scholar]
- 14.Chen HY; Lin MH; Wang CY; Chang YM; Gwo S, Large-Scale Hot Spot Engineering for Quantitative SERS at the Single-Molecule Scale. Journal of the American Chemical Society 2015, 137 (42), 13698–13705. [DOI] [PubMed] [Google Scholar]
- 15.Shen W; Lin X; Jiang C; Li C; Lin H; Huang J; Wang S; Liu G; Yan X; Zhong Q; Ren B, Reliable Quantitative SERS Analysis Facilitated by Core-Shell Nanoparticles with Embedded Internal Standards. Angew Chem Int Ed Engl 2015, 54 (25), 7308–12. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Y; Ding R; Joshi P; Zhang P, Quantitative surface-enhanced Raman measurements with embedded internal reference. Anal Chim Acta 2015, 874, 49–53. [DOI] [PubMed] [Google Scholar]
- 17.Kasera S; Biedermann F; Baumberg JJ; Scherman OA; Mahajan S, Quantitative SERS using the sequestration of small molecules inside precise plasmonic nanoconstructs. Nano Lett 2012, 12 (11), 5924–8. [DOI] [PubMed] [Google Scholar]
- 18.Fales AM; Vo-Dinh T, Silver embedded nanostars for SERS with internal reference (SENSIR). Journal of Materials Chemistry C 2015, 3 (28), 7319–7324. [Google Scholar]
- 19.Ansar SM; Li X; Zou S; Zhang D, Quantitative Comparison of Raman Activities, SERS Activities, and SERS Enhancement Factors of Organothiols: Implication to Chemical Enhancement. J Phys Chem Lett 2012, 3 (5), 560–5. [DOI] [PubMed] [Google Scholar]
- 20.Zhang DM; Xie Y; Deb SK; Davison VJ; Ben-Amotz D, Isotope edited internal standard method for quantitative surface-enhanced Raman spectroscopy. Analytical Chemistry 2005, 77 (11), 3563–3569. [DOI] [PubMed] [Google Scholar]
- 21.Deb SK; Davis B; Knudsen GM; Gudihal R; Ben-Amotz D; Davisson VJ, Detection and relative quantification of proteins by surface enhanced Raman using isotopic labels. Journal of the American Chemical Society 2008, 130 (30), 9624–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleinman SL; Ringe E; Valley N; Wustholz KL; Phillips E; Scheidt KA; Schatz GC; Van Duyne RP, Single-molecule surface-enhanced Raman spectroscopy of crystal violet isotopologues: theory and experiment. J Am Chem Soc 2011, 133 (11), 4115–22. [DOI] [PubMed] [Google Scholar]
- 23.Jie Z; Zenghe Y; Xiaolei Z; Yong Z, Quantitative SERS by electromagnetic enhancement normalization with carbon nanotube as an internal standard. Opt Express 2018, 26 (18), 23534–23539. [DOI] [PubMed] [Google Scholar]
- 24.Carles R; Bayle M; Benzo P; Benassayag G; Bonafos C; Cacciato G; Privitera V, Plasmon-resonant Raman spectroscopy in metallic nanoparticles: Surface-enhanced scattering by electronic excitations. Physical Review B 2015, 92 (17). [Google Scholar]
- 25.Haug T; Klemm P; Bange S; Lupton JM, Hot-Electron Intraband Luminescence from Single Hot Spots in Noble-Metal Nanoparticle Films. Phys Rev Lett 2015, 115 (6), 067403. [DOI] [PubMed] [Google Scholar]
- 26.Huang D; Byers CP; Wang LY; Hoggard AL; Hoenee B; Dominguez-Medina S; Chen SS; Chang WS; Landes CF; Link S, Photoluminescence of a Plasmonic Molecule. Acs Nano 2015, 9 (7), 7072–7079. [DOI] [PubMed] [Google Scholar]
- 27.Hugall JT; Baumberg JJ, Demonstrating photoluminescence from Au is electronic inelastic light scattering of a plasmonic metal: the origin of SERS backgrounds. Nano Lett 2015, 15 (4), 2600–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin K-Q; Yi J; Hu S; Sun J-J; Zheng J-T; Wang X; Ren B, Intraband Hot-Electron Photoluminescence from Single Silver Nanorods. ACS Photonics 2016, 3 (7), 1248–1255. [Google Scholar]
- 29.Lin KQ; Yi J; Zhong JH; Hu S; Liu BJ; Liu JY; Zong C; Lei ZC; Wang X; Aizpurua J; Esteban R; Ren B, Plasmonic photoluminescence for recovering native chemical information from surface-enhanced Raman scattering. Nat Commun 2017, 8, 14891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mertens J; Kleemann ME; Chikkaraddy R; Narang P; Baumberg JJ, How Light Is Emitted by Plasmonic Metals. Nano Lett 2017, 17 (4), 2568–2574. [DOI] [PubMed] [Google Scholar]
- 31.Roloff L; Klemm P; Gronwald I; Huber R; Lupton JM; Bange S, Light Emission from Gold Nanoparticles under Ultrafast Near-Infrared Excitation: Thermal Radiation, Inelastic Light Scattering, or Multiphoton Luminescence? Nano Lett 2017, 17 (12), 7914–7919. [DOI] [PubMed] [Google Scholar]
- 32.Cai YY; Sung E; Zhang R; Tauzin LJ; Liu JG; Ostovar B; Zhang Y; Chang WS; Nordlander P; Link S, Anti-Stokes Emission from Hot Carriers in Gold Nanorods. Nano Lett 2019, 19 (2), 1067–1073. [DOI] [PubMed] [Google Scholar]
- 33.Huang J; Wang W; Murphy CJ; Cahill DG, Resonant secondary light emission from plasmonic Au nanostructures at high electron temperatures created by pulsed-laser excitation. Proc Natl Acad Sci U S A 2014, 111 (3), 906–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei HR; McCarthy A; Song J; Zhou W; Vikesland PJ, Quantitative SERS by hot spot normalization - surface enhanced Rayleigh band intensity as an alternative evaluation parameter for SERS substrate performance. Faraday Discuss 2017, 205, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei HR; Leng WN; Song J; Willner MR; Marr LC; Zhou W; Vikesland PJ, Improved Quantitative SERS Enabled by Surface Plasmon Enhanced Elastic Light Scattering. Analytical Chemistry 2018, 90 (5), 3227–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Ru EC; Blackie E; Meyer M; Etchegoin PG, Surface enhanced Raman scattering enhancement factors: a comprehensive study. J Phys Chem C 2007, 111 (37), 13794–13803. [Google Scholar]
- 37.Nam W; Ren X; Tali SAS; Ghassemi P; Kim I; Agah M; Zhou W, Refractive-Index-Insensitive Nanolaminated SERS Substrates for Label-Free Raman Profiling and Classification of Living Cancer Cells. Nano Lett 2019, 19 (10), 7273–7281. [DOI] [PubMed] [Google Scholar]
- 38.Song J; Nam W; Zhou W, Scalable High‐Performance Nanolaminated SERS Substrates Based on Multistack Vertically Oriented Plasmonic Nanogaps. Advanced Materials Technologies 2019, 4 (5). [Google Scholar]
- 39.Ren X; Nam W; Ghassemi P; Strobl JS; Kim I; Zhou W; Agah M, Scalable nanolaminated SERS multiwell cell culture assay. Microsyst Nanoeng 2020, 6 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Itoh T; Biju V; Ishikawa M; Kikkawa Y; Hashimoto K; Ikehata A; Ozaki Y, Surface-enhanced resonance Raman scattering and background light emission coupled with plasmon of single Ag nanoaggregates. J Chem Phys 2006, 124 (13), 134708. [DOI] [PubMed] [Google Scholar]
- 41.Farcau C; Astilean S, Evidence of a surface plasmon-mediated mechanism in the generation of the SERS background. Chem Commun (Camb) 2011, 47 (13), 3861–3. [DOI] [PubMed] [Google Scholar]
- 42.Moskovits M, Surface selection rules. The Journal of Chemical Physics 1982, 77 (9), 4408–4416. [Google Scholar]
- 43.Le Ru EC; Meyer M; Blackie E; Etchegoin PG, Advanced aspects of electromagnetic SERS enhancement factors at a hot spot. Journal of Raman Spectroscopy 2008, 39 (9), 1127–1134. [Google Scholar]
- 44.Le Ru EC; Meyer SA; Artur C; Etchegoin PG; Grand J; Lang P; Maurel F, Experimental demonstration of surface selection rules for SERS on flat metallic surfaces. Chem Commun 2011, 47 (13), 3903–3905. [DOI] [PubMed] [Google Scholar]
- 45.Alvarez-Puebla RA, Effects of the Excitation Wavelength on the SERS Spectrum. J Phys Chem Lett 2012, 3 (7), 857–66. [DOI] [PubMed] [Google Scholar]
- 46.Baffou G; Cichos F; Quidant R, Applications and challenges of thermoplasmonics. Nat Mater 2020, 19 (9), 946–958. [DOI] [PubMed] [Google Scholar]
- 47.Xu W; Ling X; Xiao J; Dresselhaus MS; Kong J; Xu H; Liu Z; Zhang J, Surface enhanced Raman spectroscopy on a flat graphene surface. Proc Natl Acad Sci U S A 2012, 109 (24), 9281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jeong JW; Arnob MM; Baek KM; Lee SY; Shih WC; Jung YS, 3D Cross-Point Plasmonic Nanoarchitectures Containing Dense and Regular Hot Spots for Surface-Enhanced Raman Spectroscopy Analysis. Adv Mater 2016, 28 (39), 8695–8704. [DOI] [PubMed] [Google Scholar]
- 49.Liu X; Wang J; Tang L; Xie L; Ying Y, Flexible Plasmonic Metasurfaces with User-Designed Patterns for Molecular Sensing and Cryptography. Advanced Functional Materials 2016, 26 (30), 5515–5523. [Google Scholar]
- 50.Xu K; Zhou R; Takei K; Hong M, Toward Flexible Surface-Enhanced Raman Scattering (SERS) Sensors for Point-of-Care Diagnostics. Adv Sci (Weinh) 2019, 6 (16), 1900925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qian X; Peng XH; Ansari DO; Yin-Goen Q; Chen GZ; Shin DM; Yang L; Young AN; Wang MD; Nie S, In vivo tumor targeting and spectroscopic detection with surface-enhanced Raman nanoparticle tags. Nat Biotechnol 2008, 26 (1), 83–90. [DOI] [PubMed] [Google Scholar]
- 52.Zavaleta CL; Smith BR; Walton I; Doering W; Davis G; Shojaei B; Natan MJ; Gambhir SS, Multiplexed imaging of surface enhanced Raman scattering nanotags in living mice using noninvasive Raman spectroscopy. P Natl Acad Sci USA 2009, 106 (32), 13511–13516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Henry AI; Sharma B; Cardinal MF; Kurouski D; Van Duyne RP, Surface-Enhanced Raman Spectroscopy Biosensing: In Vivo Diagnostics and Multimodal Imaging. Anal Chem 2016, 88 (13), 6638–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.