

Summary
Transcription factor (TF) expression levels drive developmental programs, including cell fate and function, and their measurement by flow cytometry allows for robust downstream analysis. However, significant batch-to-batch variability between replicative experiments precludes direct comparison of absolute values across experimental conditions. Here, we present a flow cytometry protocol to measure the relative abundance of multiple TFs simultaneously in single cells, allowing for direct comparison across experimental conditions/time points. This protocol uses bone marrow cells but can be adapted for other cell types.
For complete details on the use and execution of this protocol, please refer to Manso et al. (2021) and Manso et al. (2019).
Subject areas: Cell Biology, Cell isolation, Single Cell, Flow Cytometry/Mass Cytometry, Immunology
Graphical abstract
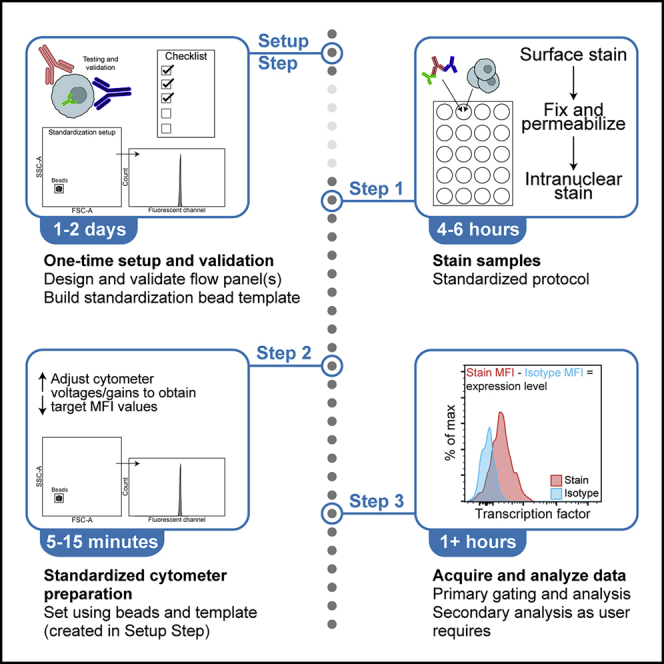
Highlights
-
•
Flow-cytometry-based protocol to simultaneously measure transcription factors (TFs)
-
•
Allows for direct comparison of relative TF levels across conditions/time points
-
•
Single-day experimental workflow can be used in a variety of downstream analyses
Transcription factor (TF) expression levels drive developmental programs, including cell fate and function, and their measurement by flow cytometry allows for robust downstream analysis. However, significant batch-to-batch variability between replicative experiments precludes direct comparison of absolute values across experimental conditions. Here, we present a flow cytometry protocol to measure the relative abundance of multiple TFs simultaneously in single cells, allowing for direct comparison across experimental conditions/time points. This protocol uses bone marrow cells but can be adapted for other cell types.
Before you begin
The protocol below describes the specific steps for using freshly isolated or cultured cells to determine cellular levels of transcription factors. Here, we describe the universal procedure the authors utilized to determine transcription factor levels in hematopoietic stem and progenitor cells (HSPCs) from human bone marrow as found in (Manso et al., 2021) and (Manso et al., 2019). However, this protocol can be adapted to any cell type and experimental model species (such as mouse) assuming the specific reagents required are available for that cell type and/or species.
CRITICAL: Before you begin, read the entire protocol as many individual steps have dependencies on preceding and subsequent steps. It is important to understand the entire protocol before beginning.
Note: This protocol assumes the user begins with a single-cell suspension of the cells of interest.
Reagent preparation and testing
Timing: 1–2 days (as needed)
-
1.
Prepare any buffers required (see materials and equipment, below).
-
2.
Ensure all reagents have been obtained in the required amounts.
-
3.
Appropriately titrate all flow cytometry antibodies to be used.
-
4.
Validate the staining of each cell surface and transcription factor antibody using cells that are known to be positive and negative for each targeted antigen.
-
5.
Determine the best intranuclear permeabilization kit for the specific transcription factor antibodies selected.
Note: Specific commercially available kits must be used if this protocol is used to stain for FoxP3 or Ki-67. The user should refer to their selected kit’s manufacturer’s instructions to determine compatibility.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| While none are specified within, high-quality, flow cytometry validated monoclonal antibodies are required for the successful application of this protocol. See Manso et al. (2021) for specific antibodies used in the evaluation of human bone marrow hematopoietic stem and progenitor cells. | N/A | N/A |
| Biological samples | ||
| Human bone marrow | Mayo Clinic Department of Hematology specimen processing and Predolin Biobank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat#A7906 |
| Paraformaldehyde 10% solution, EM Grade | Electron Microscopy Sciences | Cat#15712-S |
| True-Nuclear™ Transcription Factor Buffer Set | BioLegend | Cat#424401 |
| 10× PBS, no calcium, no magnesium | Gibco | Cat#14200 |
| 0.5 M EDTA, pH 8.0 | Invitrogen | Cat#15575-020 |
| FcR Blocking Reagent, human | Miltenyi Biotec | Cat#130-059-901 |
| Software and algorithms | ||
| Flow cytometry data analysis software (most cytometer software used for sample acquisition can be used for rudimentary analysis, but a dedicated application, such as FlowJo version 10 (BD Biosciences) is highly recommended) | N/A | N/A |
| Downstream analysis software as required | N/A | N/A |
| Other | ||
| Rainbow Calibration Particles, 6th peak | Spherotech | Cat#RCP-30-5A-6 |
| Cell processing and counting materials as per user’s preference | N/A | N/A |
| 50 mL Conical tubes | N/A | N/A |
| 96-Well round bottom plates | N/A | N/A |
| 5 mL Flow cytometry tubes (or appropriate tube for placing sample on the flow cytometer) | N/A | N/A |
| Micropipettes and tips capable of 0.25 μL – 1000 μL volumes | N/A | N/A |
| Serological pipets | N/A | N/A |
| Centrifuge and rotor/buckets capable of using both 5 mL flow cytometry tubes (or appropriate tube for placing sample on the flow cytometer) and 96-well round bottom plates | N/A | N/A |
| Flow cytometer | N/A | N/A |
Materials and equipment
Flow cytometer setup
Beyond the normal setup of the flow cytometer to be used, additional one-time and daily setup is part of this standardized protocol and detailed in the Step-by-Step Method Details, below.
Note: This protocol is written in such a way that any flow cytometer with a sufficient number of available channels can be used. The requirements during the protocol are outlined, but note that the specific names used by each flow cytometer manufacturer may vary slightly.
Flow cytometer daily standardization bead alternatives
Many fluorescent beads are commercially available. When selecting a set of beads to use, make sure the beads generate a single peak (or a single peak can be cleanly isolated from a selection), are stable, and fluoresce in each channel of the cytometer. Further, the single fluorescent peak must be on scale for each flow cytometer channel.
Nuclear permeabilization reagent alternatives
Beyond the kit listed in the Key Resources Table (above), many other kits and variations have been used. When selecting a nuclear permeabilization kit/reagent, it is critical to ensure that it is designed for interrogation of nuclear, not cytoplasmic, antigens (commercially available kits will specify this in the product information). Further, commercially available kits will specifically list nuclear targets than cannot be detected with their product. However, caution should be used and proper testing implemented to ensure optimal staining (see reagent preparation and testing, above). This is particularly true of non-commercially-derived methods, just as placing cells in −20°C ethanol.
Buffers required
| 1× PBS - Store at 18°C–22°C for up to 3 months | Final concentration | Amount |
|---|---|---|
| 10× PBS | 1× | 50 mL |
| ddH2O | n/a | 450 mL |
| Total | n/a | 500 mL |
| Flow cytometry staining buffer - Store at 4°C for up to 3 months and keep on ice (4°C) when in use. | Final concentration | Amount |
|---|---|---|
| ddH2O | n/a | 890 mL |
| 10× PBS | 1× | 100 mL |
| 0.5 M EDTA, pH 8.0 | 0.005M | 10 mL |
| Bovine Serum Albumin (BSA) | 0.005 g/mL | 5 g |
| Total | n/a | 1 L |
Flow cytometry staining buffer alternatives
Many variations of flow cytometry staining buffers are used. To ensure standardization, the same buffer should be used in every experiment performed under this protocol.
| 1% paraformaldehyde (PFA) - Store at 4°C and keep on ice (4°C) when in use. 1% PFA expires 1 month after preparation. | Final concentration | Amount |
|---|---|---|
| 1× PBS | n/a | 45 mL |
| 10% paraformaldehyde | 1% | 5 mL |
| Total | n/a | 50 mL |
Note: If sterile staining/experiments are being conducted, all buffers listed above can be sterile filtered using a 0.22μm vacuum filter.
Step-by-step method details
Flow cytometry panel design
This protocol assumes the user has basic knowledge and ability to design flow cytometry panels. However, there are a few specific features of panel design that will be detailed here.
-
1.
Design flow cytometry panel as needed for both cell surface markers and intranuclear targets.
Note: When selecting antibody-conjugate combinations, the brightest fluorophore(s) should generally be reserved for transcription factors.
Note: When selecting transcription factor antibody-fluorophore combinations, it can be incredibly helpful to limit the selection of these to non-tandem dye combinations. This is due to the compensation requirement. When compensating, the user has two options: using antibody capture beads or cells. The authors opted for the use of cells for three reasons. First: the use of antibody capture beads can often lead to compensation control staining that is several logs brighter than the actual stain in the panel, potentially causing overcompensation. Additionally, the negative bead population is not representative of the negative cellular population. Second: most live/dead discrimination dyes cannot be used with antibody capture beads (as they are not antibodies), resulting in the use of both cells and beads during compensation. Third: compensating with cells allows for more accurate levels of positive and negative fluorescence to be accounted for and reveals basal cellular autofluorescence. When compensating, a single-stained compensation control is required for each antibody in the panel. However, transcription factor staining can be very dim and make for a poor compensation control. To circumvent this issue, a surrogate compensation control can be used for those fluorophores. This is done by using a different antibody that has the same fluorophore as the transcription factor antibody for the compensation control. To prevent potential overcompensation, using a surrogate compensation control that is 0.5–1 log brighter than the transcription factor antibody is optimal.
Example: mouse anti-human PU.1 IgG1, κ Alexa Fluor 647® transcription factor antibody could have a surrogate compensation control in the form of mouse anti-human CD3 IgG1, κ Alexa Fluor 647® antibody (titered to ensure the fluorescence expression is 0.5–1 logs brighter than the transcription factor antibody).
-
2.Design isotype control panel variants for transcription factors.
-
a.For each full staining panel, an additional panel with isotype controls in place of the transcription factor antibodies must be used.
-
a.
Note: This standardization protocol makes use of isotype controls to accurately measure levels of transcription factors by flow cytometry. The isotype controls allow for normalization of the output data by controlling for non-specific binding and inherent background staining common among transcription factor antibodies.
First, the antibody isotype of the isotype control, and its host species, must be identical to the primary antibody.
Example: Mouse anti-human PU.1 IgG1, κ primary antibody needs to be paired with a mouse IgG1, κ isotype control antibody.
Second, the antibody-fluorophore conjugation of the isotype control must be identical to that of the primary antibody.
Example: Mouse anti-human PU.1 IgG1, κ Alexa Fluor 647® needs to be paired with a mouse IgG1, κ Alexa Fluor 647® isotype control. Similar, but not identical pairings destroy the integrity of this standardized protocol (i.e., an APC isotype control paired with an Alexa Fluor 647® antibody or an Alexa Fluor 488® isotype control paired with a FITC primary antibody is forbidden).
Third, the isotype control must be used at the same protein concentration as the primary antibody.
Example: Mouse anti-human PU.1 IgG1, κ Alexa Fluor 647® is used at 1 μg per stain. The mouse IgG1, κ Alexa Fluor 647® isotype control must be used at the same concentration. It is worth noting that often, but not always, isotype control antibodies are provided at a higher concentration than many primary antibodies, requiring a different dilution to achieve the same protein concentration.
Panel testing and flow cytometer standardization setup
This setup step prepares the cytometer for standardized use with the optimized flow cytometry panel(s) designed by the user.
Note: Portions of this setup step are simplified from previously published protocols (Perfetto et al., 2012) and (Perfetto et al., 2006).
-
3.Using primary cells of the same type that will be experimentally evaluated, stain the cells using the standardized protocol outlined in the sections below.
-
a.If possible, test stains across multiple individuals/sources to account for minor staining variability within patients/individual sample sources.
-
b.Include appropriate isotype controls as separate stains.
-
a.
-
4.
Collect the data on the flow cytometer and immediately analyze.
-
5.
Assess if the panel allows for clean separation of cellular population(s), resolution of transcription factor(s), isotype controls that are negative, and appropriate compensation values.
-
6.Upon validation of the test panel(s), the cytometer can now be further setup for standardized analysis (performed on the same day immediately following step 5):
-
a.Prepare a worksheet template on the flow cytometer for the fluorescent beads (Figure 1).
-
b.Set the voltages/gains of each channel to that used in step 4, above.
-
c.Prepare the beads by adding 2–3 drops to 1 mL 1× PBS or flow cytometer sheath fluid.
-
d.Load the beads on the machine and begin acquiring events with a flow rate of approximately 150–250 events per second.
-
e.In the cytometer worksheet, create a graph of forward scatter area (FSC-A) vs. side scatter area (SSC-A) and draw a gate around the beads (Gate 1).
-
i.Note that only the FSC and SSC voltages can be changed at this point to ensure visualization of the beads.
-
i.
-
f.Create a histogram plot (as child graphs from Gate 1) for the fluorescent channel/detector on the cytometer that is closest to SSC.
-
i.Check the cytometer’s configuration to determine the channel closest to SSC. For most machines, SSC is collected on the 488 nm (blue) laser, indicating that the channel that detects the lowest wavelength on the 488 laser should be selected at this step. This channel is typically designated as FITC, GFP, 488B, etc.
-
i.
-
g.On the histogram of the channel closest to SSC, draw a gate around the main bead peak only (Gate 2), avoiding any smaller side peaks (indicative of bead doublets/aggregates)
-
i.This step ensures evaluation of single bead events.
-
i.
-
h.Create a series of histogram plots (as child graphs from Gate 2) for each remaining fluorescent channel on the cytometer (from all lasers).
-
i.Create a statistics view within the worksheet that evaluates the median fluorescent intensity (MFI) statistic of each channel on the flow cytometer based on Gate 2 (the single beads gate).
-
j.Record 10,000 events.
-
k.Within the worksheet, or on a master document, record the MFI for each channel following the bead acquisition.
-
i.These MFI values are the target values to be obtained each day prior to flow cytometer acquisition for this protocol.
-
ii.Note that the target MFI values can be rounded to the nearest multiplier of 50 (i.e., an MFI of 15,256 could be rounded to 15,250).
-
i.
-
l.Export/save the bead template and channel/detector MFI values.
-
a.
Figure 1.

Flow cytometer standardization bead worksheet template
The beads are first gated on FSC-A vs. SSC-A (Gate 1). Next, a histogram of the beads for the channel closest to SSC is created. Gating tightly on the main bead peak in the histogram eliminates bead aggregates (Gate 2). Additional histograms of each remaining fluorescent parameter are then created (easily sorted by laser). A statistics view is then created evaluating the MFI for each fluorescent channel based off Gate 2. As the beads are acquired, the cytometer voltages/gains are adjusted until the MFI from Gate 2 matches the Target MFI (as determined during the initial setup). Beads are then recorded and the voltage/gain values applied to that day’s flow cytometry experiment.
Daily flow cytometer setup prior to collecting samples
This daily step prepares the flow cytometer for standardized event collection.
-
7.Each day, before running any samples using this protocol:
-
a.Load the bead template and a new tube of flow cytometer setup beads on the machine.
-
b.Acquire, but don’t record, the beads at approximately 150–250 events per second.
-
c.Adjust the voltage/gain for the channel closest to SSC until the target MFI is reached when Gate 2 is properly placed.
-
d.Adjust the voltages/gains for each remaining channel on the cytometer until the target MFI values (as previously determined) for each channel are reached.Note: The voltages/gains flow cytometers use are arbitrary values that can vary day-to-day. The power of this protocol is to set each channel/detector to a specific fluorescence sensitivity value that can be exactly recapitulated each day.
-
e.Record 10,000 events.
-
f.The exact voltage/gain values determined during this step are to be used for that day’s flow cytometry experiments ran under this protocol (and cannot be carried forward to any other day’s experiments).
-
a.
Note: As mentioned above and throughout, any changes to any part of this protocol will affect the target MFI values and data integrity.
Example 1
A new lot of an antibody is to be used. Titrate the new antibody lot as normal. In the same staining procedure, include an additional sample that uses the current antibody lot at its current titer. Upon analysis, compare the newly titrated antibody against the previous lot and select the titer that matches the fluorescence expression level of the current antibody lot. Experiments can now continue.
Example 2
A new lot of the flow cytometer setup beads is to be used. Prepare a tube of the current and new flow cytometer beads. Use the current lot to adjust all voltages to obtain the current MFI target values for each channel. Then, load the new bead lot and record 10,000 events. Update the target MFI values as needed with those obtained from the new bead lot.
Note: Continually titrating new antibody lots and updating bead lot targets is a common feature of this protocol. To alleviate some of this work, consider obtaining appropriate volumes of reagents that are from the same lot for the duration of the study.
Staining cells for transcription factor expression level and comparative determination across multiple time points via standardized flow cytometry
The following steps detail the exact staining procedure for transcription factor staining.
Note: Variations to staining protocols are common and may need to be modified for each user’s exact application. However, following testing and determination of the staining protocol to use, no deviations from that protocol are allowed to maintain the integrity of the standardization. The protocol presented below reflects that which was used in (Manso et al., 2021) and (Manso et al., 2019) for freshly isolated, primary human bone marrow cells.
Note: Keep all reagents and cells on ice (4°C) unless otherwise indicated.
Note: This step assumes the user begins with a single-cell suspension of the cells of interest. There are a variety of methods used to generate single-cell suspensions and considerations including tissue source and cellular population(s) of interest dictate the method used.
-
8.Determine the number of cells required for each staining panel.
-
a.This includes both the full stain panel and the paired isotype control panel.
-
b.The author’s utilized 5x106 cells per individual stain (10x106 total for stain and isotype control panels) as they were targeting rare cell populations (see (Manso et al., 2021)).
-
c.Note that this number can be adjusted to the user’s requirements, but should remain constant throughout the entire use of this protocol to maintain standardization.
-
a.
-
9.
Count viable cells and remove an appropriate volume to obtain the live cell numbers required.
-
10.
Place cells in a 50 mL conical tube and add 5–10 mL flow cytometry staining buffer.
-
11.
Spin cells at 700 xg (RCF) for 5 min at 4°C.
-
12.
Decant supernatant.
-
13.
Re-suspend the cell pellet(s) in 300 μL/108 cells of flow cytometry staining buffer.
-
14.Add 100 μL/108 cells of Human Fc block, mix well, and place on ice (4°C) for 20 min.
-
a.Note, different blocking reagents can be used and may require different volumes and ratios. However, as with any modification, once determined, those values cannot change to maintain standardization.
-
a.
-
15.
Add 20 mL flow cytometry staining buffer to the sample(s).
-
16.
Spin cells at 700 xg (RCF) for 5 min at 4°C.
-
17.
Decant supernatant.
-
18.
Re-suspend samples at 25x106 cells/mL in flow cytometry staining buffer.
-
19.Plate 200 μL cells (5x106) into the appropriate wells of a labeled round-bottom 96-well plate.
-
a.For example, if one panel is being stained for, one well needs to be plated for the staining panel and a second well for the paired isotype control panel (per sample).
-
a.
-
20.Using remaining/extra cells, plate approximately 1–5x106cells/well for compensation controls.
-
a.A single-stained compensation control is required for each antibody-fluorophore combination and dye in the panel. This includes:
-
i.Unstained cells.
-
ii.Fixable viability dye (FVD).
-
iii.Each cell surface antibody.
-
iv.Each transcription factor antibody.
-
i.
-
a.
Note: See comments in Flow cytometry panel design (above) for notes about compensation controls for transcription factor antibodies.
-
21.
Spin plate at 700 xg (RCF) for 3 min at 4°C.
-
22.
Flick plate to remove supernatant.
Note: To appropriately flick the plate, quickly invert the plate and flick once with the wrist using one fluid motion. While still inverted, blot the plate on a stack of paper towels to remove excess liquid.
-
23.Prepare fixable viability dye (FVD) live/dead discrimination dilution:
-
a.Each well requires 100 μL FVD working solution.
-
b.Make enough to have 2 wells extra.
-
c.In an appropriately sized tube, add 1× PBS following this formula:
-
i.[Number of sample wells + 2] × 100 μL = total volume 1× PBS.
-
i.
-
d.Dilute stock FVD to the manufacturer’s recommended dilution (refer to the manufacturer’s technical data sheet).
-
i.Example: the FVD used in (Manso et al., 2021) and (Manso et al., 2019) required a 1:1000 dilution, adding 1 μL stock FVD for every 1 mL 1× PBS.
-
i.
-
a.
Note: Fixable viability dyes work by staining free amine groups found in proteins. Therefore, buffers that contain protein (such as flow cytometry staining buffer) may quench or diminish dye staining. A protein-free buffer, such as 1× PBS, should be used at this step.
Note: Many live/dead alternatives exist and may have additional and/or different staining requirements. As with everything else, once a live/dead dye has been selected, it and its application cannot be changed to maintain the standardization integrity.
-
24.
Re-suspend each sample well in 100 μL FVD solution, mixing the cells well.
-
25.
Re-suspend the FVD compensation well in 100 μL FVD solution.
-
26.
Re-suspend all other compensation wells in 100 μL 1× PBS.
-
27.
Incubate protected from light on ice (4°C) for 20 min.
-
28.Prepare cell surface staining panel(s):
-
a.In a tube of an appropriate size, add flow cytometry staining buffer and the correct volumes of each cell surface staining antibody such that each well will get 100 μL staining cocktail with the appropriate final titer of antibodies. Mix well and keep protected from light at 4°C until use.
-
a.
Note: Being as accurate as possible for volumes used when preparing the staining panel(s) allows the user to achieve the desired concentration of each antibody. For example:
4 samples × 100 μL = 400 μL total staining cocktail required
Antibody A: 2.5 total μL
Antibody B: 4.75 total μL
Antibody C: 8.25 total μL
Volume flow cytometry staining buffer to add: 384.5 μL
Note: Each sample and staining panel requires its own, transcription factor(s)-paired isotype control(s) panel.
-
29.
Add 100 μL flow cytometry staining buffer to each well.
-
30.
Spin plate at 700 xg (RCF) for 3 min at 4oC.
-
31.
Flick and blot plate to remove supernatant.
-
32.
Add 100 μL cell-surface staining cocktail to each well as appropriate and mix well.
-
33.
Re-suspend all compensation wells in 100 μL flow cytometry staining buffer and mix well.
-
34.To each appropriate compensation control, add the single antibody for that control and mix well.
-
a.If using a surrogate compensation control (in place of a transcription factor antibody) that binds a cell surface antigen, stain it at this step. If using the actual intranuclear transcription factor antibody, no additional steps are required beyond re-suspension in flow cytometry staining buffer. That compensation control(s) will be stained in step 62.
-
a.
Note: See comments in Flow cytometry panel design (above) for notes about compensation controls for transcription factor antibodies.
-
35.
Incubate protected from light on ice (4°C) for 30 min.
-
36.
Add 100 μL flow cytometry staining buffer to each well.
-
37.
Spin plate at 700 xg (RCF) for 3 min at 4°C.
-
38.
Flick and blot plate to remove supernatant.
-
39.
Add 200 μL flow cytometry staining buffer to each well.
-
40.
Spin plate at 700 xg (RCF) for 3 min at 4°C.
-
41.
Flick and blot plate to remove supernatant.
-
42.Prepare BioLegend True-NuclearTM Transcription Factor Buffer fixation/permeabilization solution:
-
a.Always prepare this buffer fresh, no more than 1 h prior to use.
-
b.Prepare buffer following this formula: [total number of wells (samples and compensation controls) +2] × 500 μL = total volume required.
-
c.The 4× fixation/permeabilization solution needs to be diluted 1:4 in the fixation/permeabilization diluent.
-
d.Example: for 10 mL total volume, add 2.5 mL 4× fixation/permeabilization to 7.5 mL fixation/permeabilization diluent to obtain a working 1× solution.
-
a.
Note: The BioLegend True-NuclearTM Transcription Factor Buffer fixation/permeabilization kit was used in our studies (Manso et al., 2021) as it provided the cleanest staining with the best resolution. As mentioned above, the user must determine the optimal kit(s) for their studies and reagents.
Note: If a different cell number other than 5x106 total cells are being used in each stain, this volume may need to be adjusted as determined during the user’s initial testing of reagents.
-
43.
Mix the fixation/permeabilization working solution well and keep at 18°C–22°C until use.
-
44.
Label 5 mL FACS tubes for each sample and compensation control well.
-
45.
Add 300 μL of the 1× fixation/permeabilization solution to each tube.
-
46.
Add 200 μL of the 1× fixation/permeabilization working solution to each well of the plate and mix well.
-
47.Transfer each well to its appropriately labeled 5 mL FACS tube
-
a.The total volume is now 500 μL
-
a.
Note: Internal testing has found this volume and this size of sample tube to be optimal for limiting background staining while optimizing signal intensity.
-
48.
Gently mix using a pipette.
-
49.
Incubate protected from light at 18°C–22°C for 1 h.
Note: Everything from this point forward will be at 18°C–22°C.
Note: Do not vortex tubes to mix as cells can become more fragile following this protocol.
-
50.Prepare BioLegend True-NuclearTM Transcription Factor Buffer 1× permeabilization/wash buffer:
-
a.Always prepare this buffer fresh, no more than 1 h prior to the first use.
-
b.Prepare buffer following this formula: [total number of wells (samples and compensation controls) +2] × 500 μL × 5 = total volume required.
-
c.The 10× permeabilization/wash buffer needs to be diluted 1:10 in dH2O.
-
d.Example: for 50 mL total volume, add 5 mL 10× permeabilization/wash buffer to 45 mL dH2O to obtain a working 1× solution.
-
a.
-
51.
Mix the working permeabilization/wash buffer well and keep at 18°C–22°C.
-
52.
Spin the sample tubes at 700 ×g (RCF) for 5 min at 18°C–22°C.
-
53.
Decant tubes by inverting (do not shake or flick) and blot the mouth of the tube on a paper towel to remove residual liquid.
-
54.
Add 500 μL 1× permeabilization/wash buffer to each tube.
-
55.
Spin the sample tubes at 700 xg (RCF) for 5 min at 18°C–22°C.
-
56.
Decant tubes by inverting (do not shake or flick) and blot the mouth of the tube on a paper towel to remove residual liquid.
-
57.
Add 500 μL 1× permeabilization/wash buffer to each tube.
-
58.
Spin the sample tubes at 700 xg (RCF) for 5 min at 18°C–22°C.
-
59.Decant tubes by inverting (do not shake or flick) and blot the mouth of the tube on a paper towel to remove residual liquid.
-
a.A total of 3 washes post-fixation/permeabilization should take place.
-
a.
-
60.Prepare intranuclear staining cocktails in the 1× permeabilization/wash buffer:
-
a.In a tube of an appropriate size, add 1× permeabilization/wash buffer and the correct volumes of each transcription factor staining antibody or isotype control such that each tube will get 250 μL staining cocktail. Mix well and keep protected from light at 18°C–22°C until use.
-
a.
-
61.
Following the third wash, re-suspend the cell pellets in 250 μL of the appropriate staining or isotype control cocktail and mix well by pipetting.
-
62.Re-suspend the compensation controls in 250 μL 1× permeabilization/wash buffer and add any transcription factor single-stain compensation controls to the appropriate tube(s).
-
a.Depending on the use of surrogate compensation controls for transcription factor antibodies, this compensation control staining step may have already been completed.
-
a.
-
63.
Incubate protected from light at 18°C–22°C for 30 min.
-
64.
Add 250 μL 1× permeabilization/wash buffer to each tube.
-
65.
Spin the sample tubes at 700 xg (RCF) for 5 min at 18°C–22°C.
-
66.
Decant tubes by inverting (do not shake or flick) and blot the mouth of the tube on a paper towel to remove residual liquid.
-
67.
Add 500 μL 1× permeabilization/wash buffer to each tube.
-
68.
Spin the sample tubes at 700 xg (RCF) for 5 min at 18°C–22°C.
-
69.
Decant tubes by inverting (do not shake or flick) and blot the mouth of the tube on a paper towel to remove residual liquid.
-
70.
Re-suspend cell pellets in 400 μL 1% PFA.
-
71.
Place all tubes on ice (or in the refrigerator, 4°C) and protect from light until flow cytometry analysis.
Note: For optimal results, samples should be analyzed on the flow cytometer as quickly as possible, with the acquisition taking place no longer than 18–24 h following the completion of staining.
Expected outcomes
Following successful completion of this protocol, samples will be ready for flow cytometry acquisition in a standardized format that, following data analysis, allows for direct comparison of results across multiple experiments and collection days/times. Furthermore, we have successfully utilized this protocol to simultaneously evaluate three distinct transcription factors in a single staining cocktail. The latter is particularly important when a testing sample may be limiting, such as human bone marrow. This protocol provides for simultaneous interrogation of multiple TFs across immunophenotypically defined cellular subsets with single primary and isotype control stains.
Quantification and statistical analysis
Analysis of this data will allow the user to determine many features of their cells, including the percent positivity for each transcription factor in each cell population(s) of interest. Additionally, the expression values of each transcription factor can be directly compared. The user may also apply this standardized data to a number of downstream applications as desired.
To determine the expression values of each transcription factor:
-
1.
Export and save flow cytometry data from the flow cytometer.
-
2.In a flow cytometry analysis program, perform all necessary gating steps to gate on the cell type(s) of interest.
-
a.This includes, but is not limited to the following standardized flow cytometry analysis gates: time, singlets, and live cells.
-
a.
Note: The authors’ studies utilized FlowJo version 10.5.3 (BD Biosciences) for all gating and extraction of data.
Note: At this point, it is good practice to check the compensation of each parameter against every other parameter to ensure the staining panel(s) are performing optimally.
-
3.
Within the gated cell population(s) of interest, evaluate the expression pattern(s) of the transcription factor(s) included for that staining combination (Figure 2).
-
4.If the expression pattern of the transcription factor is unimodal:
-
a.On the gated cell population(s) of interest, obtain the MFI values of each transcription factor and associated isotype control.
-
b.Subtract the MFI of the isotype control from the MFI of the fully stained sample.
-
c.The resulting value is the relative expression level of that transcription factor.
-
i.Example: Mouse anti-human PU.1 IgG1, κ Alexa Fluor 647® MFI = 1578.36. Mouse IgG1, κ Alexa Fluor 647® isotype control MFI = 657.48. Level of PU.1 expression = 920.88 (1578.36 - 657.48).
-
i.
-
a.
-
5.If the expression pattern is bimodal:
-
a.Within the gated cell population(s) of interest, determine the positive population (for that transcription factor) using the expression pattern of the transcription factor and that of the isotype control.Note: MFI values can only be applied to unimodal distributions. Any population(s) expressing bi- or higher orders of modality require different analysis methods, such as isolating a unimodal positive population/peak.
-
b.Gate on the positive population such that the isotype control would be less than 0.1% positive of the parent population.
-
c.Obtain the MFI values for the positive transcription factor population only.
-
i.This value is the relative expression level of that transcription factor.
-
i.
-
a.
Note: If the isotype control MFI value is equal to or greater than the MFI of the stained sample, by definition the interrogated transcription factor is not expressed, or expressed below the lower limit of detection. See Troubleshooting (below) if detection was expected.
Figure 2.

Example transcription factor analysis
Following collection and analysis of the standardized flow cytometry data, the population(s) of interest can be interrogated for transcription factor expression levels. For transcription factors with a unimodal expression pattern (top), the MFI of the transcription factors in the specified cell populations from the stained and isotype samples are extracted from the analysis software. The MFI of the isotype control is subtracted from the MFI of the experimental sample to obtain the relative expression of that specific transcription factor (graphically illustrated as an overlaid histogram). For transcription factors with bimodal expression patterns (bottom), the positive population is first gated on using the isotype control to help set the lower gate bounds. The MFI of the specific transcription factor in the positive population gate is then extracted as the relative expression level of that transcription factor. A portion of this figure is reprinted with permission from Manso et al., 2021.
Additional analysis applications
The generation of standardized flow cytometry data where MFI expression levels are directly comparable can be used in a number of additional/supplementary methods per the user’s needs. One key application is being able to use this data in dimensionality reduction (i.e., t-distributed stochastic neighbor embedding (tSNE) or Uniform Manifold Approximation and Projection (UMAP)) analysis tools. As an example, the authors utilized standardized flow cytometry data as an input for UMAP analysis of human bone marrow samples (ex vivo and in vitro) as can be seen in (Manso et al., 2021).
Limitations
Many notes and points of critical consideration throughout this protocol highlight the various limitations of this procedure. Beyond the technical limitations and considerations, other limitations must be noted. First, this protocol allows for the standardized analysis and simultaneous comparison of relative levels of multiple transcription factors but is unable to determine the exact protein or copy amount of each transcription factor in a given cell. Second, comparisons made using this protocol can only be made between identical transcription factors. That is, the relative level of transcription factor A can only be directly compared to the relative level of transcription factor A in other cell populations and/or samples. In no situation can transcription factor A be directly compared to transcription factor B. However, ratios of expression of transcription factors A and B could be compared as that is an indirect, not a direct, comparison.
Troubleshooting
Problem 1
Isotype control staining is equal to or greater than the full stained sample, and a positive transcription factor signal is expected (i.e., positive control).
Potential solution
Failure to follow each step of this protocol can lead to suboptimal staining resolution. Further, expired or degraded antibodies or other reagents can negatively affect staining quality. Tracking expiration dates and running appropriate positive and negative controls will help identify any source of suboptimal staining.
Additionally, the fixation/permeabilization kit/method used may not be optimal for that antibody, fluorophore, cell type, and/or antigen target. Alternative kits/methods should be tested.
See the before you begin and reagent preparation and testing sections.
Problem 2
Compensation controls result in wrong/incorrect compensation values for my panel.
Potential solution
When preparing compensation controls, they need to be treated exactly the same as the samples to ensure equal staining variability. Further, using the exact same antibodies for the compensation controls as those in the staining panel ensure correct staining. Additionally, compensation controls need to be as bright or brighter than those found in the actual panel. Therefore, when staining compensation controls, doubling the titer volume for the compensation sample can result in improved compensation. Additionally, if the compensation error involves a non-tandem dye, a surrogate compensation control could be used. If that fails to work, or the compensation error is with the use of a tandem dye, antibody capture beads could be considered as an alternative. As mentioned above, to prevent potential overcompensation, ensure the comp controls are 0.5–1 log brighter than the fluorescence expression obtained in the experimental panel(s). Lastly, additional testing may be required to ensure good panel design, proper titration, etc.
See before you begin and reagent preparation and testing sections and step-by-step method details (steps 1–5, 34, and 62).
Problem 3
A commercially available TF antibody is not staining a known positive cell population or is not generating the predicted staining pattern.
Potential solution
First, an assumption that an antibody that works in non-flow cytometry applications (western blot, ChIP, etc.) cannot be made that the antibody will work for flow cytometry. All TF antibodies must be validated for flow cytometry applications with negative and positive cell lines or primary cells with known expression status. Second, if the commercially available antibody is designated as appropriate for flow cytometry by the manufacturer, and staining is still not observed, the user is encouraged to contact the technical staff of the company from which the antibody was purchased to obtain the internal validation staining protocol, cell type(s) used, and flow cytometry data.
See before you begin and reagent preparation and testing sections and step-by-step method details (steps 1–5).
Problem 4
The isotype control MFI is less than zero.
Potential solution
First, ensure that all compensation values are correct as incorrect compensation (often overcompensation) can result in MFI values falling below zero. Second, if the isotype control antibody is very clean (i.e., no background staining), compensation values (even small ones) can cause the MFI to fall below zero. To correct for this, perform a single stain with the isotype control only and with the panel compensation applied. If the MFI of the isotype control with no compensation is at or above zero, yet falls below zero upon application of compensation values, then compensation is slightly modulating the values around zero. Raise the voltage for that channel until the isotype control MFI remains above zero (note that this will require different bead settings and verification that the positive staining is still on scale). Third, if acquisition has already been completed and negative isotype control values are observed, the user may consider setting those isotype control values to zero as raw flow cytometry data can never be negative (even if application of compensation adjusts the values around zero to be slightly negative).
See before you begin and reagent preparation and testing sections, step-by-step method details (steps 1–5), and quantification and statistical analysis (step 4).
Problem 5
I am often unable to perform flow cytometry acquisition within 18–24 h following staining.
Potential solution
In the time post-staining, there is an increase in debris and loss of certain antibody signals. Further, certain cell types/morphological compositions may be susceptible to lysis in extended periods of refrigerated cell suspension (even post-fixation). However, the amount of degradation and compromised sample will largely depend on cell type, antibody quality, and fluorophores used. The user would need to test if their specific application is stable for longer periods of time between staining and acquisition.
See step-by-step method details (step 71).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kay Medina (medina.kay@mayo.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate or analyze datasets. Further, it did not produce any original code.
Acknowledgments
We thank the Mayo Clinic Department of Hematology specimen processing and the Predolin Biobanking team for providing the samples used as examples in this protocol. This study was supported by Mayo Clinic NIH Grant Relief funding to K.L.M. and NIH T32 funding (NIH T32 AI07425-23) awarded to B.A.M.
Author contributions
B.A.M. and K.L.M. wrote the manuscript. B.A.M. performed and analyzed all experiments used as examples within.
Declaration of interests
B.A.M. is currently affiliated with the University of California Santa Cruz, Santa Cruz, CA, USA. This protocol is submitted in support of work done while affiliated with the Mayo Clinic, Rochester, MN, USA.
References
- Manso B.A., Krull J.E., Gwin K.A., Lothert P.K., Welch B.M., Novak A.J., Parikh S.A., Kay N.E., Medina K.L. Chronic lymphocytic leukemia B-cell-derived TNFalpha impairs bone marrow myelopoiesis. iScience. 2021;24:101994. doi: 10.1016/j.isci.2020.101994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manso B.A., Zhang H., Mikkelson M.G., Gwin K.A., Secreto C.R., Ding W., Parikh S.A., Kay N.E., Medina K.L. Bone marrow hematopoietic dysfunction in untreated chronic lymphocytic leukemia patients. Leukemia. 2019;33:638–652. doi: 10.1038/s41375-018-0280-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfetto S.P., Ambrozak D., Nguyen R., Chattopadhyay P., Roederer M. Quality assurance for polychromatic flow cytometry. Nat. Protoc. 2006;1:1522–1530. doi: 10.1038/nprot.2006.250. [DOI] [PubMed] [Google Scholar]
- Perfetto S.P., Ambrozak D., Nguyen R., Chattopadhyay P.K., Roederer M. Quality assurance for polychromatic flow cytometry using a suite of calibration beads. Nat. Protoc. 2012;7:2067–2079. doi: 10.1038/nprot.2012.126. [DOI] [PubMed] [Google Scholar]
- Roederer M. Compensation in flow cytometry. Curr. Protoc. Cytom. 2002;1:Unit 1 14. doi: 10.1002/0471142956.cy0114s22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate or analyze datasets. Further, it did not produce any original code.