

Graphical abstract
Keywords: Riboseq, Bioinformatics, FAIR, Translation, Ribosome, Docker, Tool for ribosome analysis, Polyamines, OAZ1
Abstract
Ribosome profiling (RiboSeq) has emerged as a powerful technique for studying the genome-wide regulation of translation in various cells. Several steps in the biological protocol have been improved, but the bioinformatics part of RiboSeq suffers from a lack of standardization, preventing the straightforward and complete reproduction of published results. Too many published studies provide insufficient detail about the bioinformatics pipeline used. The broad range of questions that can be asked with RiboSeq makes it difficult to use a single bioinformatics tool. Indeed, many scripts have been published for addressing diverse questions. Here (https://github.com/equipeGST/RiboDoc), we propose a unique tool (for use with multiple operating systems, OS) to standardize the general steps that must be performed systematically in RiboSeq analysis, together with the statistical analysis and quality control of the sample. The data generated can then be exploited with more specific tools. We hope that this tool will help to standardize bioinformatics analyses pipelines in the field of translation.
1. Introduction
Translation is a key step in gene expression in which an mRNA is translated into protein by the ribosomes. When studying the regulation of a gene, it is crucial to determine its translational status. Indeed, several studies have revealed that the translatome and proteome are much better correlated than the transcriptome and proteome [1]. The regulation of translation plays a key role in controlling proteome homeostasis, with mRNA stored in various cytoplasmic structures while awaiting translation, and the restriction of translation until the mRNA is located in the right place in the cytoplasm [2]. The transcriptome (i.e. RNA level) is, thus, a poor proxy for evaluations of protein abundance.
The development of ribosome profiling (RiboSeq) techniques has paved the way for genome-wide studies of translational regulation [3]. Briefly, mRNA fragments protected from RNAse degradation (about 30 nucleotides) by the ribosomes are extracted and sequenced. These ribosome footprints (RPF) are then mapped onto a reference genome or transcriptome. The level of translation can be inferred from the density of footprints, providing information about the regulation of translation [4]. RiboSeq also provides large amounts of qualitative information (pausing sites, new reading frames, stop codon read through, ribosome residence time) through nucleotide resolution [5], [6], [7], but these analyses are highly dependent on data quality, which is addressed on a case-by-case basis.
Following the advent of RiboSeq, several bioinformatics tools were developed for analysis of the data generated [8]. Intuitively, one might imagine that RiboSeq and RNAseq data could be analyzed in the same way, but this is not the case due to certain specific features of RiboSeq data. Firstly, RPF are small (25–30 nucleotides), which complicates the mapping step, particularly for exon-intron boundaries. Secondly, RiboSeq can map ribosomes at the resolution of single nucleotides, which raises important new issues previously unfamiliar to non-specialists, such as the higher density of ribosomes in the coding sequence (CDS) than in UTRs, and periodicity. Unfortunately, this lack of knowledge has resulted in basic controls, such as checking that the data obtained do indeed correspond to active ribosome footprints, not being carried out. Among the various tools, we would like to highlight the RiboGalaxy platform [9] that is a very useful resource to analyse (or re-analyse) RiboSeq data. However, no matter how good the tools are, they still suffer from some flaws. They are either very computer-intensive or require data to be uploaded to remote servers (which can pose problems of industrial property), or do not meet the FAIR reproducibility criteria, which makes it difficult to reproduce the results obtained. The development of many “home-made scripts”, about which too little detail is provided for published results to be reproducible. In the rare cases in which these scripts are made publicly available, it is often difficult, if not impossible, to install them correctly on another computer, for various reasons. These problems have raised serious issues concerning the reproducibility of published data.
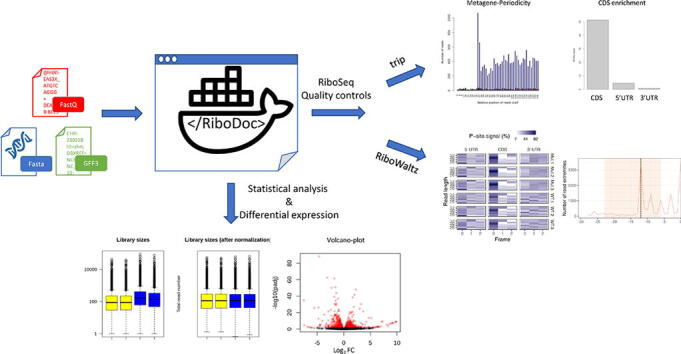
We present here a new open-source bioinformatics tool called RiboDoc. RiboDoc is designed to perform the first steps of RiboSeq data analysis, from the FastQ files to the differential expression analysis that must systematically be performed in all RiboSeq analyses. RiboDoc includes two different tools for qualitative analyses that can be selected by the user depending on the power of the computer: 1 – RiboWaltz [10] an R package for calculation of optimal P-site offsets, diagnostic analysis and visual inspection of ribosome profiling data. However, RiboWaltz requires a lot of computing power, and we wanted to make these preliminary analyses accessible in local to as many scientists as possible, including those without powerful computers. For this reason, we included a series of home-made scripts called TRiP, which provides controls (CDS enrichment and metagene-periodicity) for checking that the data correspond to active ribosome footprints, together with two statistical validations (principal component analysis (PCA) and Spearman’s correlation) to check that the replicates are suitable for the analysis (Fig. 1). RiboDoc complies with FAIR (Findable, Accessible, Interoperable, Reusable) principles [11] to ensure the highest degree of reproducibility, and is presented in a Docker container for easy and reproducible usage with Linux, Windows or MacOS operating systems. Once these initial steps have been performed, more specific tools can be used to address specific questions [8].
Fig. 1.

Overview of the full RiboDoc workflow. Every tool used for each step is specified near the corresponding arrow. The different elements are categorized and differentiated by color: yellow: operating systems compatible with RiboDoc; green: files to be provided by the user as input; purple: actions performed by the user; blue: steps performed in the analysis; red: main final output files available at the end of the analysis. The * indicates optional steps that can be performed if the user provides specific files. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2. Materials & methods
2.1. Technical considerations
RiboDoc can be deployed in the Linux, MacOS and Windows 10 operating systems. The minimal requirements are Dockercommunity edition v.19 or above (http://www.docker.com) warning for Docker Desktop on Windows: Ubuntu from Microsoft Store and WSL2 are required) and internet access (required for Docker image downloading only). The tool itself requires no additional installation and all the pipeline is managed by snakemake (v6.1.1). A step-by-step tutorial is available from the github repository (https://github.com/equipeGST/RiboDoc), and a summary is provided below.
2.2. Input reference files
For data alignment, a reference fasta file of the genome is required. This file may contain a reference transcriptome or a genome. For complex genomes, a transcriptome might be preferable as a lower complexity results in a faster analysis and less resources like RAM are needed. Another fasta file containing the sequences which the user doesn’t want to keep in the analysis (e.g. rRNA) is needed. We would advise the users to get those input files from the Ensembl website/database (http://www.ensembl.org) [12].
TRiP was designed to be used with specific transcriptomes for the qualitative analysis. This transcriptome file is created as follows:
-
1)
Select CDS, with 3′ and 5′ UTRs annotations from a GFF3 file of the reference genome
-
2)
Transform the resulting file into a BED file
-
3)
Convert the BED file into fasta format with “bedtools getfasta” [13]
-
4)
Concatenate the UTR and CDS sequence as transcripts
-
5)
Discard transcripts without a UTR
-
6)
For each gene, select the longest transcript by comparing the coordinates of the transcripts associated to each gene, so as to include only one transcript per gene
As a means of simplifying the use of RiboDoc, we provide, together with RiboDoc, a human transcriptome based on the Hg38 human genome and its associated GFF3 file, constructed as described above. This specific type of transcriptome file is only needed for qualitative analysis with the qualitative_analysis: “trip” option in the config.yaml file.
2.3. Folder preparation
Before running RiboDoc, users should prepare their data as follows.
After pulling the Docker image, the user should create a project folder, which must contain the duly completed config.yaml file available from the github repository (https://raw.githubusercontent.com/equipeGST/RiboDoc/main/config.yaml). The user should open this file and to save it in the project folder created above (for MacOS users, the file must be saved as a .txt file, and the txt extension must then be removed). Once this has been done, the user can complete the file with the necessary information. Two subfolders must also be created: i) “fastq” into which all the zipped fastQ files must be dropped. The file name format must be “sampleName.replicatNumber.fastq.gz” (e.g WT.1.fastq.gz) and ii) “database”, containing fasta (genome/transcriptome and undesired RNA sequences to align on) and a GFF annotation file for the genome of interest downloaded from http://www.ensembl.org. The command lines required to open RiboDoc from the Docker hub and to run RiboDoc are available as a README.txt file. The command line for running RiboDoc the --rm option which enables the removal of the container when it exists and uses a volume option. Indeed, it allows the creation of a storage space within the container that is separate from the rest of the container filesystem. With two arguments, this mounts the project directory on the host with the container, according to the specified path.
2.4. Fasta indexation
The conda (v4.9.2) (Anaconda Software Distribution. Computer software. Vers. 2-2.4.0. Anaconda, Nov. 2016. Web (https://anaconda.com)) environment is activated in Docker, and the input fasta files are then indexed: i) with Bowtie2 (v2.4.2) [14] only, for the non-coding RNA fasta data and ii) with both Bowtie2 and Hisat2 (v2.2.1) [15] for the reference genome.
2.5. FastQ file quality control with fastQC
The first step in our analysis is quality control for the raw data present in the fastQ files. This process is performed with FastQC (v0.11.9) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), which can spot multiple quality problems due to the sequencer or the starting library material. At the end of the quality control check, an HTML file appears in the subfolder RESULTS/fastqc in the project directory.
2.6. Adapter trimming and read length selection
CutAdapt (v3.4) [16] is used i) to trim reads, to remove the 3′ adapter sequence (error rate = 12.5%) and ii) to filter (maximum one N found in each read) and select reads on the basis of their length. Each read represents a RPF of about 28–30 bases in length [4]. The RiboDoc default parameters are slightly more flexible, retaining reads with lengths between 25 and 35 bases. Users can fix their own range, selecting fragments from 20 to 22 bases long for example, which correspond to another ribosome conformation during translation [13]. For data that have already been trimmed, we have added an option for selecting only sequences with a particular length of interest.
2.7. Unwanted RNA depletion
The trimmed reads are then mapped onto unwanted RNA sequences selected by the user with Bowtie2 (v2.4.2). rRNA sequences are recommended, but mitochondrial RNA and non-chromosomal sequences can also be used. All these sequencing can be found on http://www.ensembl.org. The alignment parameters of the tool are the default values. Only unaligned reads are retained for further analysis.
2.8. Double mapping
Hisat2 (v2.2.1) is then used to map reads onto the reference fasta file. This first tool provides a stringent alignment, taking introns into account (more efficient for complex genomes). Reads that are not aligned during this first step are mapped on the same fasta file with Bowtie2 (v2.4.2), with default parameters that are less strict and accept more complementarity errors (maximum of 1).
2.9. Generation of BAM files
The sequence alignment/map (SAM) files created by Hisat2 and Bowtie2 are merged and only the mapped reads are kept. Working with uniquely mapped reads only can be seen as a limitation, but in our opinion it is the safest procedure to be confident in our analysis. SAM files are then sorted and filtered with the selection of uniquely mapped reads only, and every read corresponding to the flag number 3844 is discarded (https://broadinstitute.github.io/picard/explain-flags.html) with the -F option. These files are then converted into binary alignment/map (BAM) with samtools (v1.12) [17].
2.10. Quality controls
Quality controls are required to ensure that the footprints obtained actually correspond to active ribosomes. Three quality controls are performed independently on each category of read, according to length (default parameters: 25–35 bases long). The first assesses the distribution of read lengths, the second assesses periodicity, and the third evaluates the density of reads in CDS vs. UTRs. Several technical issues may explain a failure of some data to pass these controls and in such cases, the data should be interpreted with considerable caution.
RiboDoc contains two pipelines available for this qualitative analysis. The first method is to operate with the integrated R package riboWaltz (v1.1.0) [10]. This package requires BAM files corresponding to reads aligned on a format specific reference fasta file and a gtf annotation file which are obtained with gffread (v0.12.1) [18] from the reference fasta and gff files provided by the user.
As riboWaltz requires a lot of resources and time, the user can choose to use TRiP which requires less computing power, while performing similar operations. TRiP requires less RAM but needs a specific gff format as input file. This second pipeline goes through these following steps:
2.11. Read length distribution
The SAM file is split on the basis of read length. One BAM file is created on the basis of read length, and is converted into a BED (browser extensible data) file with bedtools (v2.30.0) [13] for simplicity. File size is limited by merging reads mapping to exactly the same position. The final file contains the following six columns: chromosome, start coordinate on the chromosome, end coordinate on the chromosome, number of identical reads, comment, strand. A table is extracted from the BED files to summarize the distribution of reads between read lengths. An enrichment in reads of 27–28 bases is expected, although there have been several reports of RPF with different lengths [19], [20].
2.12. Metagene profile-periodicity
Periodicity should be calculated from a metagene profile. It provides the number of footprints relative to all annotated start and stop codons in a selected window. The window selected by default is −50/+100 nucleotides and −100/+50 nucleotides around the start and stop codons, respectively. These parameters can be modified by the user.
The frequency of footprints at a specific coordinate is counted using a single footprint position (the 5′ end; transcript CDS start −50; transcript CDS start +100). A metagene profile graph presenting the frequency of each footprint at each position is obtained.
2.13. Read counts
Counts are performed on the BAM files with Htseq (v0.12.4) [21]. It uses the ‘intersection-strict’ mode on CDS regions to reduce biases provoked by potential noise in the UTR regions and piles of reads on start and stop codons caused by the association and dissociation times of the ribosomes at these specific positions. All count files are merged into a single count matrix. The header contains the sample names. If the UTR coverage option is turned on, then counts are also performed for the 3′- and 5′-UTRs. It is, therefore, possible to calculate a normalized count density (RPKM) for the CDS and UTRs from files containing CDS/3′-UTR/5′-UTR length.
2.14. Differential expression analysis
The differential expression analysis is performed with a custom-developed RMarkDown script using DESeq2 (v1.30.1), stringr (v1.4.0) and factoMineR (v2.4). The count matrix is first summarized, and the variations within and between biological conditions are then analyzed. The data are normalized and subjected to differential analysis.
2.15. Count matrix summary
The count matrix obtained as described above is loaded into R. It contains one column per sample and one row per feature. The total number of read counts in each sample is determined. Similar library sizes are advised to reduce the bias created by the normalization step although it is not mandatory.
2.16. Variation within and between biological conditions
Quantitative analysis is performed to highlight the variability between two (or more) sets of biological conditions. For this variability assessment, the replicates should be as close as possible. A pairwise scatter plot and a PCA are performed to check that this is the case. The scatter plot is associated with the coefficients of a Spearman correlation analysis, to determine the statistical relationship between two samples. The value must be as close to 1 as possible between replicates, and should be lower between different biological conditions. The PCA is also used to visualize variability. The first component should clearly separate samples from different biological conditions and group together replicates.
2.17. Data normalization
A DESeqDataSet (DDS) object is created from the raw counts for the different conditions.
The “counts” function of DESeq2 is used to normalize data through the DDS object. This step is necessary to eliminate technical biases and to make read counts comparable between samples even with different library sizes.
The DESeq2 normalization uses the median of ratios method. For each feature, a pseudo-reference sample is created (ref = sqrt(featureCount_sampleA*featureCount_sampleB)). The ratio of each sample to the reference is calculated (ratio = sample/ref). The normalization factor (or size factor) for each sample corresponds to the median value of all ratios for a given sample (normalization_factor_bySample = median(all_feature_ratio)). The raw counts for a sample are then divided by its size factor, to determine the normalized counts. The median of ratios method is based on the hypothesis that not all features are differentially expressed. As a result, the median is not influenced by outliers, which correspond to differentially expressed genes that do not distinguish between biological conditions.
Two graphs are plotted to check that normalization is performed correctly. One shows library size after normalization: all samples should have the same size. The second graph is a boxplot visualization of the difference in count distributions before and after normalization. The normalized counts should be almost identical between samples, whereas this is not the case for the raw data.
2.18. Differential analysis
The DESeq2 function is then run on the DDS object. The estimateSizeFactors subfunction first calculates the relative library depth of each sample. The estimateDispersions subfunction then estimates count dispersion for each feature. Finally, the nbinomWaldTest subfunction calculates the significance of coefficients in a negative binomial GLM, using the size and dispersion outputs.
The results are presented on seven graphs. The first represents the estimated dispersion against the mean of the normalized counts. The second graph shows the distribution of log2FC frequency, and the highest frequency on this graph is expected to be 0. Indeed, the vast majority of features should not be differentially expressed. The third and fourth figures represent the raw and adjusted p-value distributions, respectively. The mean normalized counts for each feature are then plotted against the log ratio of differential expression, to obtain an MA-plot highlighting differentially expressed features. The last two figures are a volcano plot and its magnification. They show each feature, according to its log2FC and its adjusted p-value. At the end of the differential expression analysis, tables showing which features are up- and downregulated are generated, together with an HTML report. The final graph displays all the figures and explanations described above.
2.19. Yeast strains and culture conditions
The two yeast strains used in this study are derivatives of the 74D-694 strain (MATa ade1-14 trp1-289 his3Δ200 leu2-3,112 ura3-52, [PIN+][psi−]). OAZif was constructed as previously described [22]. All strains were grown to mid-exponential growth phase in a synthetic medium, and growth was stopped by adding cycloheximide. RiboSeq experiments were performed as previously described [23]. The raw data (fasq files) and BAM files generated in this study have been deposited at GEO (under curation).
3. Results & discussion
As we only implemented the published RiboWaltz package into RiboDoc we did not use this option here to demonstrate how RiboDoc works. We selected the trip option for the following analysis applied to two datasets: a previously published dataset for human cells [24] and an unpublished dataset for yeast from our own group. The human dataset was selected because quality controls had been rigorously performed, making it possible for us to compare RiboDoc with data analyzed with different scripts. We also wished to use RiboDoc with a different genome, and for this purpose we chose to use our own data for the yeast Saccharomyces cerevisiae. These two datasets are very different in size, making it possible to assess the efficacy of RiboDoc with different sample sizes (Table 1). RiboDoc was implemented with Docker. Docker technology is a state-of-the-art platform for deploying software applications without the need to worry about dependency issues. It can be used to deploy an application in a sandbox mode, called a container, on the host system. This is a crucial point for reproducibility. Indeed, all the tools present in RiboDoc have a fixed version number, to prevent discrepancies that could arise later, due to the updating of these tools.
Table 1.
Quantity of reads in each sample.
| Sample Name | Raw reads | Filtered reads | Multimapping | Unique map | Reference |
|---|---|---|---|---|---|
| OAZwt-1 | 135,961,717 | 99,746,557 | 11,274,105 | 32,284,163 | This study |
| OAZwt-2 | 78,562,493 | 59,335,275 | 4,432,144 | 11,246,378 | This study |
| OAZif-1 | 76,478,033 | 69,519,100 | 2,729,784 | 3,185,359 | This study |
| OAZif-2 | 79,111,449 | 70,933,589 | 4,094,929 | 8,414,254 | This study |
| WT F9-1 | 13,355,848 | 1,919,861 | 28,660 | 245,071 | [24] |
| WT F9-2 | 13,451,867 | 4,659,296 | 62,462 | 542,919 | [24] |
| WT F9-3 | 12,092,292 | 3,979,976 | 42,333 | 362497 | [24] |
| CO F9-1 | 14,600,572 | 3,043,591 | 56,786 | 461,568 | [24] |
| CO F9-2 | 12,292,279 | 2,046,318 | 33,728 | 275,252 | [24] |
| CO F9-3 | 14,541,142 | 2,946,107 | 47,431 | 394,833 | [24] |
We tested RiboDoc by determining whether replacing the +1 programmed frameshifting (PRF) site of the yeast OAZ1 gene with an in-frame version affected gene expression. We have shown that this +1 PRF is under the control of the yeast prion [PSI+] [23]. During this study, we also showed that replacing the naturally frameshifted OAZ1 gene with an in-frame allele led to a decrease in polyamine levels [23]. However, we did not, at the time, address the issue of the consequences of overexpressing Oaz1p for global gene expression. We used RiboDoc to address this question here.
Table 1 describes the reads that passed the various selection steps. It also provides us with an idea of the total number of reads passing the filter (reads containing adapters, reads that were too long or too short). Users should pay attention to this table, which may also indicate whether enough reads passed the filter for analysis. For example, we can see in Table 1 that for HEK293T cells (CO F9 and WT F9), only 245,071–542,929 reads were uniquely mapped onto the human transcriptome, whereas, according to our data, more than three million reads mapped uniquely onto the yeast genome. The number of reads obtained from HEK293T cells is sufficient for quantitative analysis (differential expression analysis), but we consider this number to be too low for some aspects of a qualitative analysis, such as the calculation of ribosome residence time (RRT). Indeed, RiboSeq data are notoriously heterogeneous and, as such, require much greater coverage than standard transcriptomic analyses. Table 2 provides an example of the time needed to perform the analyses depending on the computer resources available.
Table 2.
Computing Time.
| Operating System | CPU | RAM (GB) | Time (h)/Size of the fastq file |
|---|---|---|---|
| Linux | 18 | 32 | 15 h/13 GB |
| MacOS | 3 | 16 | 5.5 h/770 MB |
3.1. RiboSeq quality controls
When RiboDoc has completed the analysis, the first important control is confirming that the data actually come from active ribosomes. RPF have been reported to be about 28 nucleotides long for eukaryotic ribosome, although smaller fragments may correspond to an alternative ribosome conformation [20]. The read-length distribution (Fig. 2A) provides a visual representation of the numbers of each RPF. For the chosen example, we can clearly see that the majority of reads are 28 nucleotides long. However, this is not sufficient to be sure that we are really looking at active ribosomes. Indeed, such fragments could arise from the binding of mRNA to non-translating ribosomes or to ribosomes’ proteins. It is therefore necessary to check for an accumulation of signals in the CDS and to ensure that this signal displays a clear periodicity of 3, corresponding to the movement of the ribosome along the mRNA in discrete steps of three nucleotides (codon). These two parameters are specific to active ribosomes, and these checks therefore unambiguously confirm that the data correspond to active ribosomes. RiboDoc obtains the necessary parameters by performing a metagene analysis in which all the annotated CDS are pooled and the number of RPF 5′ ends at each nucleotide position in a window that can be defined by the user (by default, −50/+100 nucleotides around the start position and −100/+50 nucleotides around the stop position) is determined. Ansample is presented in Fig. 3A and B, showing clear periodicity for 28 nucleotide-long RPF, starting 12 nucleotides before the start codon and ending 15 nucleotides before the stop codon (the offset corresponds to the distance between the 5′ extremity of the RPF and the position of the P and A sites for the start and stop codons, respectively). This offset may vary with RPF size, if the 5′ ends are heterogeneous. A comparison of Fig. 3A and B clearly shows that, in yeast, more ribosomes are present downstream from the natural stop codon, in the 3′UTR of the genes, as previously described [25]. Periodicity provides a clear demonstration that the data correspond to genuinely active ribosomes, but this demonstration is highly dependent on the quality of the digestion step before RPF extraction from the ribosomes. Any under- or overdigestion will inevitably lead to poor periodicity without necessarily resulting in any indication that the data are also of poor quality. It is also important to highlight another potential issue with this representation. Indeed, if the genome contains a large number of overlapping CDS (as in bacteria), these overlapping CDS must be removed to prevent poor periodicity. For this reason, it is also possible to check the overrepresentation of footprints within the CDS (Fig. 2B).
Fig. 2.

Read length distribution. A) The number of reads, according to length (between 25 and 35 nucleotides) for the two yeast samples, OAZwt and OAZif, is represented. The length of the main population is indicated. B) Read enrichment in CDS vs UTR for HEK293T samples.
Fig. 3.

Metagene periodicity. Each graph shows the coverage of the CDS and UTR regions of the metagene for a specific read length (here 28 nucleotides), determined by representing all the reads starting at a specific relative position. Each of the three possible reading frames is represented in a different color (blue, green and red). The coverage is shown according to the window chosen by the user. In this example: from 50 nucleotides before the start codon to 100 nucleotides after the start codon, and then from 100 nucleotides before the stop codon to 50 nucleotides after the stop codon. To help the reader, we manually added the red arrows to indicate the position of the START and STOP codons and the offset observed that is due to only the first nucleotide on the 5′ side of the reads being counted. A) Periodicity for the OAZwt yeast strain. B) Periodicity for the HEK293T cell line. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. Statistical validation
Once the initial QC steps have been performed, the reproducibility of the replicates must be verified. RiboSeq data are much more heterogeneous than classical RNAseq data. It is, therefore, essential to ensure a high level of reproducibility between duplicates or triplicates. Based on our own experience, most of the variability observed with human cell lines is due to cell culture parameters, although the nature of the cells also plays an important role. Two statistical analyses are included within RiboDoc. The first is a pairwise sample-by-sample comparison in which Spearman’s correlation coefficients are displayed. The second is a PCA analysis. For the example used, we can see in Fig. 4 that the Spearman coefficient for correlation between duplicates is around 0.93 and 0.95 for HEK293T data and between 0.97 and 0.99 for yeast data. These results indicate that the duplicates are of very high quality, and the differential expression analysis can, therefore, be performed. It is important to highlight that, in situations in which there are few differences between the samples tested, PCA may incorrectly cluster samples together.
Fig. 4.

Statistical analysis. Scatter plots for the HEK293T cell and yeast data, with Spearman’s correlation coefficients for the relationship between each pair of samples. Each scatter plot shows the log10 read counts for every gene in a sample relative to those for another sample.
If one sample is of insufficient quality (low Spearman’s correlation coefficient, or a shift in the PCA graph), the user should remove it from the analysis. This will require removal of the final_report.html file too, together with the fasq file corresponding to the sample removed. RiboDoc can then be rerun, but only performing the steps for which recalculation is required.
3.3. Identification of differentially expressed genes
Once all the previous steps have been performed successfully, RiboDoc uses DESeq2 [26] to normalize and compare the data. As presented in Fig. 5, two graphs are generated: i) The MA-plot represents the log2 fold change according to the number of normalized counts for each gene. Genes with a statistically significant change are indicated in red (the threshold for statistical significance can be adjusted by the user). ii) The volcano plot giving the log2 fold change according to the adjusted p-value. All genes above the significance threshold are indicated in red (Fig. 5). In parallel, RiboDoc also generates tables (.txt) summarizing all the information from the differential analysis that will serve as the primary results for the RiboSeq analysis. In Fig. 5, only one gene is differentially expressed between OAZwt and OAZ-if (volcano plot padj < 0.01), whereas five genes are differentially expressed according to the MA-plot, generated with a different significance threshold (padj < 0.1). These results indicate that replacing the naturally frameshifted OAZ1 gene by an in-frame OAZ1 gene has no major impact on gene expression in yeast, despite its clear impact on polyamine levels, as demonstrated in our previous study [22]. The only gene strongly downregulated in the presence of OAZ-if was the PGM2(YMR105c) gene, encoding the major phosphoglucomutase isoform involved in glycolysis, the pentose phosphate shunt, and the metabolism of glycogen, trehalose, and galactose [27]. In the absence of a transcriptome, we cannot draw any firm conclusions as to whether the observed variation occurs at the level of translation or at the RNA level. The reason for the significantly lower level of expression of this gene in the presence of a low level of polyamine (OAZif strain) is unclear. However, interestingly, a null PGM2 mutant displays has been shown to be more sensitive to polyamines, identifying a link between Pgm2p and intracellular polyamine concentration [28].
Fig. 5.

MA-plots and volcano plots. MA-plots display the difference in expression between two conditions (log2FC) as a function of the number of read counts for each gene. Volcano plots represent the difference in expression as a function of the −log10 adjusted p-value obtained by DESeq2 for the differential analysis for each gene. Red dots indicate genes that are differentially expressed, whereas black dots show genes that are not differentially regulated. The user can define the threshold for statistical significance. For example, for yeast data, the significance threshold for the MA-plots and volcano plots is set at 0.01. For HEK293T cells, the same threshold (0.01) was used for both graphs. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We observe more variations between the two conditions tested in HEK293T cells expressing two different versions of the F9 gene (one with optimized codons). We found that 23 genes were downregulated, and 78 genes were upregulated in CO F9 cells relative to WT cells. It is surprising that codon optimization of the F9 gene induces so many changes in the expressome of the cells. However, expression of the optimized version of the F9 gene probably requires a more efficient diversion of the cellular machinery than expression of the unoptimized version, potentially accounting for this finding. These findings may be important for biotechnological or synthetic biology approaches, in which the diverted cell resources are rarely taken into account.
4. Conclusions
We describe here a new tool (RiboDoc) that efficiently performs the initial steps of RiboSeq analysis, regardless of the operating system used (Linux, Windows, MacOS). RiboDoc is provided as a Docker package including all the necessary bioinformatics tools for simplified installation and reproducible analyses. RiboDoc meets the need for standardization in the analysis of RiboSeq data, including two tools depending on the computer power available for the analysis. We checked that RiboDoc worked correctly, by using the “trip” option to reanalyze high-quality data previously published by others [19] and to analyse new data generated by our own laboratory. The results obtained demonstrate that RiboDoc works correctly, simplifying and standardizing the initial steps of the analysis. RiboDoc also provides controls for checking the quality of the data, which can be used to determine whether the data are of sufficiently high quality for a qualitative analysis requiring single-nucleotide resolution.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The English of this manuscript was corrected by Alex Edelman & Associates. The authors wish to thank Claire Toffano-Nioche and Thomas Denecker for providing information about FAIR, Snakemake and Docker. We also wish to thank Fabrice Leclerc for his advice on the conda environment. We thank the high-throughput sequencing facility of I2BC for providing sequencing and bioinformatics expertise. We also acknowledge support from the ANR Rescue_ribosome program (17-CE12-0024-01).
References
- 1.Blevins W.R., Tavella T., Moro S.G., Blasco-Moreno B., Closa-Mosquera A., Díez J. Extensive post-transcriptional buffering of gene expression in the response to severe oxidative stress in baker's yeast. Sci Rep. 2019;9:11005. doi: 10.1038/s41598-019-47424-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dalla Costa I., Buchanan C.N., Zdradzinski M.D., Sahoo P.K., Smith T.P., Thames E. The functional organization of axonal mRNA transport and translation. Nat Rev Neurosci. 2021;22:77–91. doi: 10.1038/s41583-020-00407-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ingolia N.T. Ribosome footprint profiling of translation throughout the genome. Cell. 2016;165:22–33. doi: 10.1016/j.cell.2016.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivanov I.P., Shin B.S., Loughran G., Tzani I., Young-Baird S.K., Cao C. Polyamine control of translation elongation regulates start site selection on antizyme inhibitor mRNA via ribosome queuing. Mol Cell. 2018;70:254–264.e6. doi: 10.1016/j.molcel.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka M., Sotta N., Yamazumi Y., Yamashita Y., Miwa K., Murota K. The minimum open reading frame, AUG-stop, induces boron-dependent ribosome stalling and mRNA degradation. Plant Cell. 2016;28:2830–2849. doi: 10.1105/tpc.16.00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thiaville P., Legendre R., Rojas-Benitez D., Baudin-Baillieu A., Hatin I., Chalancon G. Global translational impacts of the loss of the tRNA modification t(6)A in yeast. Microb Cell. 2016;3:29–45. doi: 10.15698/mic2016.01.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiniry S.J., Michel A.M., Baranov P.V. Computational methods for ribosome profiling data analysis. Wiley Interdiscip Rev RNA. 2020;11 doi: 10.1002/wrna.1577. [DOI] [PubMed] [Google Scholar]
- 9.Michel A.M., Mullan J.P.A., Velayudhan V., O'Connor P.B.F., Donohue C.A., Baranov P.V. RiboGalaxy: a browser based platform for the alignment, analysis and visualization of ribosome profiling data. RNA Biol. 2016;13:316–319. doi: 10.1080/15476286.2016.1141862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lauria F., Tebaldi T., Bernabò P., Groen E.J.N., Gillingwater T.H., Viero G. riboWaltz: optimization of ribosome P-site positioning in ribosome profiling data. PLoS Comput Biol. 2018;14:e1006169. doi: 10.1371/journal.pcbi.1006169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkinson M.D., Dumontier M., Aalbersberg I.J., Appleton G., Axton M., Baak A. The FAIR guiding principles for scientific data management and stewardship. Sci Data. 2016;3:160018. doi: 10.1038/sdata.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J. Ensembl 2020. Nucleic Acids Res. 2020;48:D682–D688. doi: 10.1093/nar/gkz966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. [Google Scholar]
- 17.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pertea G., Pertea M. GFF utilities: GffRead and GffCompare. F1000Res. 2020;9 doi: 10.12688/f1000research.23297.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu C.-C., Zinshteyn B., Wehner K.A., Green R. High-resolution ribosome profiling defines discrete ribosome elongation states and translational regulation during cellular stress. Mol Cell. 2019;73:959–970.e5. doi: 10.1016/j.molcel.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lareau L.F., Hite D.H., Hogan G.J., Brown P.O. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife. 2014;3 doi: 10.7554/eLife.01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anders S., Pyl P.T., Huber W. HTSeq – a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Namy O., Galopier A., Martini C., Matsufuji S., Fabret C., Rousset J.-P. Epigenetic control of polyamines by the prion [PSI+] Nat Cell Biol. 2008;10:1069–1075. doi: 10.1038/ncb1766. [DOI] [PubMed] [Google Scholar]
- 23.Baudin-Baillieu A., Legendre R., Kuchly C., Hatin I., Demais S., Mestdagh C. Genome-wide translational changes induced by the prion [PSI+] Cell Rep. 2014;8:439–448. doi: 10.1016/j.celrep.2014.06.036. [DOI] [PubMed] [Google Scholar]
- 24.Alexaki A., Kames J., Hettiarachchi G.K., Athey J.C., Katneni U.K., Hunt R.C. Ribosome profiling of HEK293T cells overexpressing codon optimized coagulation factor IX. F1000Res. 2020;9:174. doi: 10.12688/f1000research.22400.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guydosh N.R., Green R. Dom34 rescues ribosomes in 3′ untranslated regions. Cell. 2014;156:950–962. doi: 10.1016/j.cell.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boles E., Liebetrau W., Hofmann M., Zimmermann F.K. A family of hexosephosphate mutases in Saccharomyces cerevisiae. Eur J Biochem. 1994;220:83–96. doi: 10.1111/j.1432-1033.1994.tb18601.x. [DOI] [PubMed] [Google Scholar]
- 28.Mulet J.M., Alejandro S., Romero C., Serrano R. The trehalose pathway and intracellular glucose phosphates as modulators of potassium transport and general cation homeostasis in yeast. Yeast. 2004;21:569–582. doi: 10.1002/yea.1126. [DOI] [PubMed] [Google Scholar]