

Abstract
Lentiviral vectors are showing success in the clinic, but producing enough vector to meet the growing demand is a major challenge. Furthermore, next-generation gene therapy vectors encode multiple genes resulting in larger genome sizes, which is reported to reduce titers. A packaging limit has not been defined. The aim of this work was to assess the impact of genome size on the production of lentiviral vectors with an emphasis on producer cell mRNA levels, packaging efficiency, and infectivity measures. Consistent with work by others, vector titers reduced as genome size increased. While genomic infectivity accounted for much of this effect, genome sizes exceeding that of clinical HIV-1 isolates result in low titers due to a combination of both low genomic infectivity and decreased packaging efficiency. Manipulating the relative level of genomic RNA to gag-pol mRNA in the producer cells revealed a direct relationship between producer cell mRNA levels and packaging efficiency yet could not rescue packaging of oversized genomes, implying a de facto packaging defect. However, independent of genome size, an equimolar ratio between wild-type gag-pol mRNA and vector genomic RNA in producer cells was optimal for titer.
Keywords: HIV-1, lentivirus, gene therapy, vector, packaging, size, transduction, producer, titer
Graphical abstract
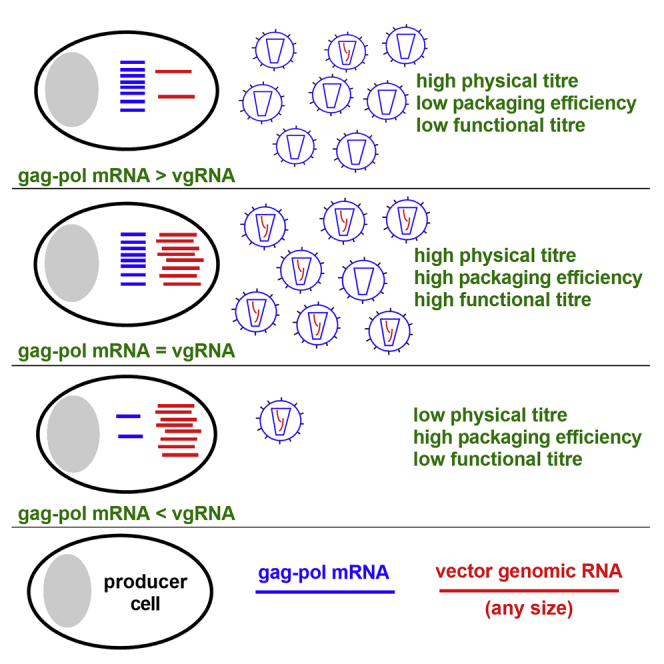
Lentiviral vector titers reduce as genome size increases. By studying producer cell mRNA, we find that, independent of genome size, an equimolar ratio of vector genomic RNA to gag-pol mRNA is optimal for packaging efficiency and titer but also reveals a de facto packaging defect for oversized vector genomes.
Introduction
Lentiviral vectors based on the human immunodeficiency virus type 1 (HIV-1) are widely used for gene therapy and are in the clinic.1 While gene therapy was initially limited to rare monogenic blood disorders that could be cured by replacement of a single defective gene, it is now also applied to polygenic diseases, diseases which demand more complex vector designs, as well as immunotherapies in which lentiviral vectors equip immune cells with a tumor-specific receptor. This greatly expands the number of patients who may benefit from gene therapies and therefore increases demand for vector supply, yet producing enough vector was already a bottleneck when patient numbers were small.2 Furthermore, immunomodulatory molecules are commonly added to vector designs in order to overcome the tumor-suppressive microenvironment,3 while safety switches to control transduced cells post-administration are also being explored.4, 5, 6, 7 However, the delivery of multiple transgenes increases the size of the vector genome, which reduces titer and further increases pressure on vector supply.
Previous work on HIV-1 vectors revealed a semi-logarithmic reduction in functional titer of approximately 3-fold per kb increase in genome size.8 No absolute packaging limit was defined, but reduced genome incorporation into virions reportedly accounted for the decrease in titer based on semiquantitative assays. These data contrast with work by others, which reported that both physical titer (the number of physical particles) and transduction efficiency (the proportion of target cells transduced at a given multiplicity of infection) were impacted by increasing vector genome size,9 while the accumulation of late reverse transcriptase (RT) products were also shown to be delayed when genome size increased.10 Similarly, Moloney murine leukemia virus was reported to lose titer as genome size increased due to a combination of packaging, entry, and reverse transcription defects,11 whereas replication-competent vectors based on the spleen necrosis virus were shown to have a replication defect at sizes greater than 9.4 kb and unable to replicate at sizes above 10 kb, reportedly due to a packaging defect.12 Foamy virus vectors lose functional titer with increasing genome size at half the rate of HIV-1-based vectors,13 supporting a role of reverse transcription to the overall impact of genome size on vector titer, since the foamy virus reverse transcriptase is reported to have higher processivity than that of HIV-1.14
The HIV-1 reference strain HXB2 has an RNA genome size of 9.2 kb that is selectively packaged as a dimer into assembling virions within infected cells. No HIV-1 isolates have been reported with an RNA genome size greater than 9.8 kb (https://www.hiv.lanl.gov/). Genetically engineered HIV-1 clones with oversized genomes have been constructed that explore the impact of genome size on viral replication. The HIV-1 replication was severely attenuated in Jurkat cells when RNA genome sizes were 9.9 and 10.1 kb. In contrast, a 9.8 kb genome was replication competent, albeit at a slower rate than the wild-type genome.15 These data are consistent with the observed genome size distribution from HIV-1 isolates. However, studies using fluorescently labeled HIV-1 capsid and viral RNA report 95% packaging efficiency into virus-like particles (VLPs) for genomes of both 9 kb and 17 kb,16 suggesting that any replication defect of genomes greater than 9.8 kb is not caused by a packaging limit. Comparatively, when Gag (or Gag-Pol) is expressed from a separate RNA to the genomic RNA, as would be the case for lentiviral vector, the observed packaging efficiency was lower and more variable. For example, one group reported 80% packaging efficiency of a 3 kb genome,16 whereas another reported just 35% packaging efficiency.17 This could be due to a fundamental difference between HIV-1 and lentiviral vector with respect to the relationship between gag-pol mRNA and viral genomic RNA (vgRNA). In HIV-1, vgRNA and gag-pol mRNA are the same molecule, such that the ratio between them is fixed at 1:1. In contrast, third-generation lentiviral vectors are produced using independently transcribed vgRNA and gag-pol mRNA,18 and therefore their ratio is not inherently fixed.
Given the importance of lentiviral vectors to HIV research and gene therapy, improved understanding of how genome incorporation is impacted by different vector production systems is needed. Since viral particles both with and without a genome can enter a target cell, any difference in packaging efficiency could impact transduction efficiency and target cell biology. For example, genome-less virions could compete with genome-containing virions for entry receptors, contribute to the activation of immune sensing pathways, and deliver producer cell contents to the target cell. Furthermore, the production of genome-less virions that cannot become a transducing unit is inefficient use of producer cell resources. Maximizing functional output per cell will support efforts to generate a manufacturing process that can supply demand for gene therapy medicines at reduced costs.
The aim of this work was to assess the impact of genome size on the production of lentiviral vectors. The impact of genome size on producer cell mRNA content and packaging efficiency is described. A method to easily manipulate the ratio between vgRNA and gag-pol mRNA levels was established. This work has implications for the design and optimization of lentiviral vector production systems and contributes to understanding of how HIV-1 genome incorporation is regulated.
Results
Design of small, medium, and large lentiviral vector genomes
Three transfer constructs were designed to assess the relationship between lentiviral vector genome size and manufacturability (Figure 1). Each construct shares identical cis-acting sequences necessary for efficient packaging and transduction, in addition to a transgene cassette carrying the enhanced green fluorescence protein (EGFP) under the control of the human phosphoglycerate kinase promoter (PGK) and a woodchuck hepatitis virus post-transcriptional regulatory element (wPRE). The genome encoded by the small construct is 3,867 nucleotides (nt, from R-R) and is a standard third-generation self-inactivating lentiviral vector.18 To increase the genome size without disrupting cis-acting sequences or the transgene cassette, stuffer DNA was inserted within a multiple cloning site located between the cPPT and the PGK promoter. Native LacZ sequence, which has been used previously to increase lentiviral vector genome size,10 constituted stuffer DNA to create a medium genome (6,927 nt), while the large genome (10,093 nt) was generated by inserting a synthetic sequence generated by gene optimization of a 5′ portion of the human dystrophin open reading frame (ORF). Gene optimization includes the removal of predicted secondary structures and splice sites that could impact titer. The overall vgRNA GC content remained similar at 54%, 55%, and 55% for small, medium, and large vectors, respectively. The large construct was designed to exceed the largest reported HIV-1 isolate. While both stuffer DNA sequences are derived from coding sequences, they are not expected to be translated in producer cells, since they are not positioned to be the first 5′ open reading frame in transfer-vector-derived transcripts. In transduced cells, they are not expected to be transcribed, given their location upstream of the transgene cassette promoter.
Figure 1.

Design of small, medium, and large transfer vectors
A schematic of each genome size design shows key features in the vgRNA and its respective size from R-R. All constructs are identical except for the addition of stuffer DNA, as depicted. Ψ is the extended packaging signal including 5′ gag leader sequences. RRE is the Rev response element, and cPPT is the central polypurine tract including the central termination sequence. The 3′ LTR has a U3 deletion to render the vector self-inactivating.
Genome size, not plasmid size, affects lentiviral vector titer
Any increase to genome size causes an equal increase to the size of the transfer plasmid that encodes for the vector genome. Plasmid size has been reported to impact transfection efficiency, which could contribute to any observed effects from increasing genome size.19,20 Therefore the small, medium, and large transfer cassettes were transferred from plasmids to nanoplasmids,21 reducing backbone size from 3.8 to 0.5 kb. The packaging plasmid expression cassettes (encoding Gag-Pol, VSVg, and Rev) were also transferred to nanoplasmids to decrease the maximum construct size per transfection. The sizes of each construct are shown in Table 1. The reduction in backbone size offsets the incremental increases to genome size.
Table 1.
DNA construct sizes
| Plasmid size (kb) | Nanoplasmid size (kb) | |
|---|---|---|
| Small transfer | 8.5 | 5.1 |
| Medium transfer | 11.5 | 8.1 |
| Large transfer | 14.7 | 11.3 |
| Gag-Pol packaging | 9.4 | 6 |
| VSVg packaging | 7.2 | 3.8 |
| Rev packaging | 5.2 | 1.7 |
To evaluate these constructs, the transfer vectors in plasmids or nanoplasmids were co-transfected with third-generation packaging constructs in their respective format, and the resulting cell-culture supernatants were collected 2 days post-transfection. The functional, physical, and genomic titers were determined for each supernatant (Figure 2). For both formats, functional titer (in HEK293T transducing units per mL, TU/mL) decreased by between 3- and 4-fold per kb increase in genome size, which is consistent with previous reports.8 As intended, transgene expression levels were unaffected, since median fluorescence intensities were comparable in cells transduced by all lentiviral vector designs (Figure S1).
Figure 2.

Impact of DNA construct size on vector titer
(A–F) The functional (A and D), physical (B and E), and genomic (C and F) titers of small, medium, and large constructs when produced using standard plasmids (filled circles) or nanoplasmids (open squares) is shown according to the transfer plasmid size (A–C) and vector genome size (D–F). All data points represent the mean average from triplicate transfections, and error bars show the range. Data are representative of three independent experiments.
Functional titer is a combination of vector production efficiency and transduction efficiency, both of which may be impacted by altering the genome size. Both construct formats resulted in similar physical titers (Figures 2B and 2E) at all vector genome sizes. In contrast, the genomic titer (Figures 2C and 2F) decreased with increasing genome size. The decrease in genomic titer only accounted for some of the total decrease in functional titer, suggesting both vector production and transduction efficiency are impacted by genome size.
To assess whether reductions in titer were caused by the increasing size of the transfected DNA construct, vgRNA, or a combination of both, titers were plotted according to transfer construct size (Figures 2A–2C) and vector genome size (Figures 2D and 2E). The measures of titer were approximately superimposed when plotting against vector genome size, but not transfer construct size. Therefore, vector genome size is responsible for the impact to titer as opposed to transfer construct size. Consistent with this, physical titers were similar for all constructs in all formats, implying that transfection efficiencies were not impacted, since physical titer is dependent on Gag-Pol expression (independent of genomic RNA), and the transfected mass of packaging constructs were kept constant. Given the similar performance of plasmid and nanoplasmid formats, further work was conducted using constructs in the more typical plasmid format only.
Physical infectivity, genomic infectivity, and packaging efficiency
Normalizing functional titer to physical titer provides a measure of particle infectivity independent of variations in transfection efficiency, while normalizing functional to genomic titer provides a measure of genomic infectivity. In each case, a higher value indicates a higher transduction success rate per vector particle or per vector genome, respectively. Furthermore, the ratio between genomic and physical titer indicates the relative packaging efficiency (proportion of vector particles that contain vgRNA). These measures are shown in Figure 3.
Figure 3.

Impact of genome size on infectivity and packaging efficiency
(A–C) The physical infectivity (A), genomic infectivity (B), and packaging efficiency (C) were calculated from vector produced with transfer plasmids encoding small, medium, and large genomes. Each data point is shown from triplicate transfections, and the line passes through the mean average in each plot. Data are representative of three independent experiments.
Compared to the small genome, the medium genome had approximately 10-fold lower physical infectivity, while the large genome was nearly 500-fold lower (Figure 3A). Physical infectivity can be reduced by both genomic infectivity (where a genome has lower probability of successfully completing transduction) and packaging efficiency (where a lower proportion of virions contain vgRNA). The relative contributions of these are shown in Figures 3B and 3C, respectively. The reduction in genomic infectivity was 8-fold between small and medium and over 25-fold between small and large, consistent with the proportional increase in genome size (0.8-fold increase between small and medium; 2.6-fold increase between small and large). In comparison, packaging efficiency was modestly affected between small and medium genomes, yet approximately 20-fold lower for the large genome. Hence, both vector production and transduction efficiency are negatively impacted by genome size, but the relative contribution of each is size dependent.
Manipulating the ratio of gag-pol mRNA and vgRNA in producer cells effectively alters packaging efficiency
To examine the relationship between genome size and packaging efficiency further, experiments were designed to manipulate packaging efficiency by overexpression of Gag-pol relative to vgRNA and vice versa. To facilitate this, all packaging components (Gag-pol, VSVg, and Rev) were assembled into modules on a single construct in a manner that maintained independent expression of each component (Figure 4A).22 This reduced the complexity of transient transfections from a 4-construct to a 2-construct system, enabling relative quantities of packaging components and transfer plasmid to be easily modified.
Figure 4.

Varying the packaging to transfer plasmid transfection ratio impacts packaging efficiency
(A) The single packaging construct used in co-transfections is illustrated. Striped ovals represent 2 copies of the chicken cSH4 element, while rectangle boxes are expression cassettes. The synGP, VSVg, and Rev expression cassettes are under the control of the TetR, which is expressed constitutively. Bacterial elements are not shown. (B and C) This construct was co-transfected at various ratios with transfer plasmids encoding small, medium, or large genomes, and the resulting supernatant was assayed for physical titer (B) and genomic titer (C). The impact of the construct ratios on packaging efficiency for each genome size is shown in (D). In plots (B)–(D), the data points show the mean average from triplicate transfections and the error bars show the range. Lines connect the mean averages for each genome size. Data are representative of two independent experiments.
Four packaging construct and transfer plasmid DNA mass ratios were selected that gave a range of relative packaging efficiencies for all genome sizes. The same total mass of DNA was used in all transfections. Hence, compared to small transfer plasmid, there were 1.4× and 1.7× fewer copies of medium and large transfer plasmid, respectively, at each ratio, while an equal quantity of the packaging construct was added for all sizes at each ratio. The ratios are presented as the percent of transfected DNA mass made up by the packaging construct. For example, 50% packaging construct means equal masses of packaging and transfer constructs were co-transfected. The tested ratios were 2%, 10%, 50%, and 90% packaging construct (and therefore 98%, 90%, 50%, and 10% transfer plasmid, respectively). As controls, 0% and 100% packaging construct ratios were also transfected. As expected, there was no detectable physical or genomic titer from the 0% packaging construct ratio in the supernatant, while there was normal physical titer but no detectable genomic titer in the 100% packaging construct control.
After transfection at the selected ratios, cell-culture supernatants were assayed for physical and genomic titers. The physical titer increased independently of genome size as a higher proportion of the packaging construct was transfected (Figure 4). Similarly, the genomic titer also increased over the first three ratios, then reduced at the final ratio. Although less transfer plasmid is transfected as the % packaging construct increases, the observed trend is consistent with a dependence on vector particle egress for the release of vgRNA into the cell-culture medium. The relative packaging efficiency (Figure 4D) was demonstrated to have decreased for all genome sizes as the proportion of packaging construct increased, but efficiency was impacted by genome size. The maximum packaging efficiency obtained with all genome sizes was achieved at the 2% and 10% packaging construct ratios. Medium-sized genomes were packaged with similar efficiency to small genomes at all ratios (<2-fold difference), while large genomes were around 10-fold less likely than small genomes to be packaged into virions at all ratios tested.
Low packaging efficiency would necessarily occur if the rate of vector particle egress exceeded the rate of vgRNA synthesis, since there would be insufficient genomes to supply all particles. Since Rev was expressed from the packaging construct, it may have become limiting when the % packaging construct transfected was low. The Rev protein is responsible for nuclear export of unspliced HIV-1 transcripts;23 hence, if Rev were limiting the observed splicing frequency at the major splice donor (MSD) is expected to increase. In contrast, our packaging construct expressed Gag-Pol from a Rev-independent synthetic Gag-Pol (synGP) sequence and would therefore be independent of Rev concentration.24 To quantify vgRNA transcripts that were spliced or not at the MSD, qPCR was performed on cDNA prepared from DNase-treated producer cell mRNA. Two primer sets that target overlapping regions within the packaging signal (Figure 5A) were employed: primer set qLVV is located 5′ of the MSD so quantifies total lentiviral vector transcripts (spliced and unspliced) initiated at the 5′ long terminal repeat (LTR), whereas qMSD specifically quantifies unspliced transcripts, since the primers span the MSD. Primers targeting the synGP transcript were also used in addition to the β-actin mRNA (ACTB), which served as an endogenous control to which all other targets were normalized. Amplification from no- controls was below the limit of detection for all samples.
Figure 5.

The impact of genome size on producer cell mRNA
(A) Schematic of the 5′ UTR of HIV-1 containing a major splice donor (MSD). Whether the MSD is used or not determines if the transcribed mRNA will be spliced or unspliced. The locations of primer/probe pairs that target the 5′ UTR upstream of the MSD (qLVV) or span the major splice donor (qMSD) and thus quantify total or unspliced mRNAs, respectively, are shown. The quantities of these transcripts, normalized to beta-actin mRNA, are shown in (B) and (C) at the various packaging construct ratios tested. The qLVV copies per qMSD are shown in (D) as a measure of MSD activity. A primer probe set targeting synGP was used to quantify gag-pol mRNA, and values normalized to beta-actin mRNA are shown in plot (E). In each plot (B–E), data points show the mean average from triplicate transfections and error bars show the range. Lines pass through the mean average for each genome size. Data are representative of two independent experiments.
Figure 5B shows that increasing the proportion of packaging construct (reducing transfer plasmid) inversely correlated with the abundance of total transfer vector transcripts (qLVV) in the transfected cells, as expected. In comparison, unspliced vgRNA transcripts (qMSD) peaked at 50% packaging construct, with fewer vgRNAs per cell at other ratios (Figure 5C). To understand this, the splicing frequency at the MSD was determined by dividing total transcripts (qLVV) by unspliced transcripts (qMSD), as shown in Figure 5D. In this measure, a value of one would indicate no splicing at the MSD, since unspliced transcripts are quantified by both assays. As higher proportions of packaging construct were transfected, splicing frequency reduced, consistent with the delivery of more Rev coding sequence. At the highest ratio tested (90% packaging construct), the total/unspliced value was approximately three, indicating that for every unspliced transcript there are two spliced transcripts. At the lowest ratio tested (2% packaging construct), there were 20–100 more spliced than unspliced transcripts. This was similar to the transfer-plasmid-only (0% packaging construct) controls (average 70 qLVV per qMSD). The spliced vgRNA for the large genome was approximately 2-fold more abundant than the small genome at all ratios tested. The quantity of synGP mRNA, which is Rev-independent, increased with increasing mass of packaging construct transfected and was similar for all genome sizes (Figure 5E), indicating that transfection efficiencies were consistent between transfer constructs. The packaging-construct-only transfections gave an average synGP per ACTB value of 2.9.
Genome availability correlates with packaging efficiency for all genome sizes
If the gag-pol mRNA levels correlate with physical titer, gag-pol mRNA could serve as an indirect measure of virion release to which the relative abundance of vgRNA available in a cell could be compared. To assess this, the physical titer was plotted against the normalized synGP mRNA value for the small, medium, and large genomes at the four transfection ratios tested (Figure 6A). There was a strong correlation (R2 = 0.92) between synGP mRNA and physical titer across the observed range. Therefore, we define the intracellular ratio between vgRNA (quantified by qMSD) and gag-pol mRNA as “genome availability,” a relative measure of the amount of genomic RNA available compared to the number of virions being released. A high genome availability implies that genomes are in excess for the rate of virion production, while a low genome availability would necessarily cause low packaging efficiency, since virion release exceeds the number of vgRNAs available to be incorporated into the assembling particles.
Figure 6.

Genome availability correlates with packaging efficiency
(A) The relationship between physical titer released into the cell-culture medium and synGP mRNA levels in producer cells is shown on a scatterplot showing data points for all genome sizes and packaging construct ratios. The dotted line is the line of best fit from linear regression analysis (R2 = 0.92). (B) The impact of changing the packaging construct ratio to genome availability is shown for each construct size. (C) The correlation between genome availability and packaging efficiency is assessed for each genome size (data shown from all packaging construct ratios). In (A) and (C), all data points are shown, and lines are of best fit plotted according to linear regression analyses. In (B), data points represent mean average from triplicate transfections and error bars show the range; lines pass through the mean average for each genome size. Data are representative of two independent experiments.
Figure 6B shows that increasing the packaging construct ratio decreased genome availability at each increment. This reveals that despite the total qMSD per ACTB peaking at the 50% packaging construct ratio (Figure 5C), genome availability was still effectively manipulated using this method. Furthermore, the large construct consistently had 2- to 3-fold lower genome availability than the small construct at all ratios, although the majority of this is expected due to the 1.7-fold molar difference in transfer plasmid at each mass ratio resulting from different plasmid sizes (Table 1).
Supra-HIV-1-sized lentiviral vector genomes have a de facto packaging defect
To understand whether genome availability accounted for the differences in packaging efficiency (Figure 4C) between different transfection ratios and different vector genome sizes, the two measures were plotted against each other and subjected to linear regression analyses (Figure 6C). Genome availability positively correlated with packaging efficiency for all genome sizes and accounted for all differences between small and medium constructs. However, large genomes consistently packaged less efficiently than small/medium genomes, even when genome availability was similar. For equal packaging efficiency, the genome availability of a 10 kb genome must be approximately 50-fold higher than that of a small genome, according to the lines of best fit.
Genomic infectivity is not impacted by low packaging efficiency
Thus far, Gag-Pol was expressed from the Rev-independent synGP sequence. The HIV-1 controls genome availability at one genome per gag-pol mRNA, since they are the same transcript. We were interested in whether HIV-1 has evolved to favor quantity or quality (i.e., produce virus with maximum output or maximum packaging efficiency). Therefore, an alternative packaging construct was created where Gag-Pol was encoded by the wild-type sequence (WT-GP). As before, all other packaging components were encoded on the same construct but independently expressed (Figure 4A). This WT-GP containing packaging construct was used in co-transfections with the small transfer plasmid at varying ratios (5%, 10%, 20%, 50%, 80%, 90%, and 95% packaging construct by mass). The use of the small construct enabled functional titration of vector produced at sub-optimal transfection ratios. Enabling functional titer determination would additionally allow for any impact of low packaging efficiency on titer to be assessed against the hypothesis that a high proportion of genome-less virions could inhibit transduction by genome-containing virions.
Figure 7A shows the physical titer (RT activity/mL), recovered genomic titer (vgRNA/mL) and functional titer (TU/mL) of the transfected cell-culture supernatants. As expected, there were no detectable vgRNA copies or TUs from 100% or 0% packaging construct controls or from no-RT control reactions. Similarly, there was no detectable RT activity from the 0% packaging construct control. Consistent with data in Figure 4, physical titer increased with increasing mass of transfected packaging construct, while both genomic and functional titer peaked at the 50% packaging construct ratio. To understand whether an excess of genome-less virions impacted infectivity, the genomic and physical infectivities were compared (Figure 7B). As expected, the physical infectivity reduced as the % packaging construct increased. In contrast, the genomic titer was similar between 20% and 95% packaging construct ratios despite over 10-fold reduction in physical infectivity between these ratios. At the lowest packaging construct ratios (5% and 10%), both physical and genomic infectivity reduced 2-fold. It is possible that this is due to low concentrations of VSVg on the surface of transfected cells when small amounts of packaging construct are transfected.
Figure 7.

Titer and infectivity of lentiviral vector produced using WT-GP containing packaging construct and a small vector genome
(A) The physical, genomic, and functional titers are shown at the different packaging construct ratios tested. The genomic and physical infectivities are show in (B). (C) A scatterplot shows the relationship between genome availability and packaging efficiency for all samples at all packaging construct ratios. The vertical dotted line indicates the fixed genome availability used by HIV-1. In (A) and (B), data points are the mean average of triplicate transfections and error bars show the range. Lines connect the mean average at each packaging construct ratio. Data are representative of two independent experiments.
An equimolar vgRNA:gag-pol mRNA ratio is optimal for titer but not packaging efficiency
Producer cell mRNA was isolated from the transfected cells, DNase treated then quantified by qRT-PCR to determine the genome availability at each ratio and plotted together with the corresponding packaging efficiency (Figure 7C). As with synGP, there was a direct correlation between genome availability and packaging efficiency. Once packaging efficiency reached around 0.3 vgRNA per nU RT at a genome availability of approximately 5 qMSD/WT-GP mRNA copies, further increases to genome availability had no benefit, potentially because all virions contained genome at these ratios. Interestingly, equimolar qMSD/WT-GP amounts (genome availability of 1), as employed by HIV-1, corresponded to the 50% packaging construct ratio, which was also the ratio that achieved the highest functional titer. However, this was not the optimal ratio for packaging efficiency, which reached 3-fold higher at the 10% packaging construct ratio.
Discussion
In this study, the impact of lentiviral vector genome size on vector production was assessed by inserting heterologous stuffer DNA upstream of a PGK-GFP transgene cassette to generate vector genomes sized at approximately 4, 7, and 10 kb. The impact of these insertions on functional titer was found to be like that described by others,8,25 suggesting the specific sequences used to increase size were not detrimental to titer.
Potential mechanisms for how genome size could impact titer were considered. First, since increasing genome size would increase the size of the transfer plasmid encoding it, and plasmid size has been described to reduce transfection efficiency,19,20 it was hypothesized that the increase in transfer plasmid size reduces transfection efficiency, resulting in fewer vector particles being produced. Second, large genomes may be incorporated less frequently into vector particles than small genomes, resulting in low packaging efficiency (defined as the proportion of virions containing vgRNA). This could be caused by lower steady-state mRNA levels, for example because they are transcribed less efficiently or have reduced stability, resulting in insufficient vgRNA copies to supply all assembling virions. Alternatively, large genomes may be incorporated less efficiently due to physical constraints.12 Finally, large genomes could reduce the infectivity of each genome-containing virion by acting at one or more steps during transduction, such as uncoating, reverse transcription, nuclear import, and/or integration.11
We reduced the size of the plasmid backbone to investigate the relationship between total plasmid size and titer. However, no improvement to titer was observed using smaller plasmid backbones for any genome size (Figure 2), indicating that the reduction in titer is due to genome size, not plasmid size. Since we use bulk transfection methods, we could further rule out any impact of transfer plasmid size on transfection efficiency by showing that physical titer, dependent only on Gag-Pol expression, was similar for all sizes. Therefore, we exclude the first hypothesis (larger genomes indirectly cause low transfection efficiency) as a key contributor to the overall impact of genome size on titer.
Using sensitive and quantitative RT-PCR-based assays to determine both physical and genomic titer for all vector preparations, we described the relationship between genome size and particle infectivity (physical titer divided by functional titer), genomic infectivity (genomic titer divided by functional titer), and packaging efficiency (genomic titer divided by physical titer). As genome size increased, the particle infectivity reduced at a similar rate to functional titer (Figure 3). Genomic infectivity was negatively impacted by genome size but did not account for the entire loss in functional titer. While small and medium genomes were packaged with similar efficiency, approximately 10-fold fewer virions packaged the largest genome (Figure 4). Hence, the total loss in functional titer is due to impacts on both packaging and transduction efficiency. The relative contribution of these varies according to the size of the genome, since packaging efficiency of small genomes was only 2-fold higher than medium genomes but 10-fold higher than large genomes (Figure 4), while genomic infectivity reduced semi-logarithmically (Figure 3).
Given that genome size impacted packaging efficiency, we examined whether this was caused by an effect on steady-state genomic mRNA levels within the producer cell and/or a putative physical constraint that makes large genome incorporation inefficient. We demonstrated that the gag-pol encoding mRNA quantity correlated directly with physical titer (Figure 6A), and therefore it could be used as a surrogate for the rate of virion release. Hence, we used the ratio between genomic mRNA and gag-pol mRNA as a relative measure of genome availability—the number of genomes divided by the number of assembling virions. By this measure, low genome availability would mean the rate of virion release exceeds the number of genomes available to them and would result in low packaging efficiency. In contrast, high genome availability would ensure that there were enough genomes available to supply all assembling virions. If large genomes have similar genome availability to small genomes, but packaging efficiency is lower, this will indicate that genome incorporation is directly impacted by genome size.
To examine the contribution of genome availability to packaging efficiency, an approach to readily manipulate genome availability was developed. By combining all packaging components into a single construct, the packaging components could be titrated against transfer plasmid in two-plasmid co-transfection experiments. Demonstrating that this approach was effective, higher physical titers were obtained as increasing quantities of the packaging construct were transfected (Figure 5). Moreover, mRNA levels in the transfected producer cells directly correlated to the amount of plasmid transfected for both total transfer vector mRNA and gag-pol mRNA (Figure 5). Hence, as intended, genome availability was effectively manipulated by altering the ratio of packaging construct to transfer plasmid that was transfected into cells (Figure 6). Because all packaging components were present on a single construct, the concentration of Rev is expected to have been low at low packaging construct ratios. Consistent with this, unspliced vector transcripts made up a higher proportion of total vector transcripts as more Rev-encoding packaging construct was transfected (Figure 5D). Nevertheless, genome availability still decreased at each incremental packaging construct ratio increase to give over 50-fold range in genome availability for all genome sizes.
The large genome was never packaged as efficiently as small or medium genomes. Reduced genome availability only played a minor role in this defect, since, even at equivalent genome availabilities, small and medium genomes were packaged with similar efficiency, while the large genome was packaged 5- to 10-fold less efficiently than small or medium genomes (Figure 6C). This finding is consistent with reports that HIV-1 genomes engineered to be larger than 9.8 kb have a severe replication defect in vitro.15
In addition to describing the direct relationship between genome availability and packaging efficiency, our data on genomic titer revealed the compromise between a maximum output in genome-containing virions, which peaked at the 50% packaging construct ratio (Figure 4B), and a maximum packaging efficiency, which was achieved at 2% and 10% packaging construct ratios (Figure 4C). As HIV-1 has a fixed genome availability of 1, we were interested in whether HIV-1 has evolved to favor quantity or quality (i.e., produce virus with maximum output or maximum packaging efficiency). Therefore, we used an alternative packaging construct containing WT-GP rather than synGP, such that Gag-Pol translation efficiency, and therefore virion assembly and release, would be expected to be like that of HIV-1-infected cells. This packaging construct was titrated against the small transfer plasmid to enable functional titer to be detectable even at suboptimal ratios. The equimolar genome availability used by HIV-1 was achieved at a packaging construct ratio of 50%, which corresponded to the ratio at which the highest genomic and functional titer was obtained, but it was not the ratio with the highest packaging efficiency. This suggests that HIV-1 has evolved to balance Gag-Pol expression (and therefore rate of assembly and release) to achieve maximum infectious titer rather than maximum packaging efficiency. Such a strategy would indicate that the production of genome-less virions is not detrimental to the success of genome-containing HIV-1 virions in vivo. To test for any impact of low packaging efficiency on genomic infectivity of our VSVg pseudotyped lentiviral vector, functional titer was determined for vector produced by all transfection ratios. The genomic infectivity on HEK293T cells was unchanged by increasing the proportion of genome-less virions by 10-fold. However, we cannot rule out an impact on infectivity to other cell types, such as primary cells, where entry receptor competition or the triggering of innate immune sensors may adversely affect transduction.
In addition to a potential impact of low packaging efficiency on infectivity, lentiviral vectors have been shown to package cellular RNA.26,27 When genomic RNA is unavailable, producer cell mRNA is non-selectively packaged instead.28 Genome-less virions pseudotyped with VSVg would still be fusion competent and able to transiently deliver their contents (including cellular mRNA and proteins) into the target cells. Since lentiviral vector producer cells typically express oncogenes such as E1A and LTA,29 the delivery of such proteins or their mRNA to primary cells destined for clinical use is a potential safety concern. However, the risk of any specific mRNA being delivered in harmful quantities by non-selective packaging into genome-less virions is likely to be very low, and any expression would be transient due to mRNA turnover. Nevertheless, lentiviral vector production systems should be optimized to achieve high titer, packaging efficiency, and infectivity for any given genome size in order to minimize the transfer of producer cell contents into target cells.
The data obtained in this work on the impact of genome size on titer is consistent with the general trends reported previously.8,9,25 However, using fully quantitative methods to evaluate the impact of size on packaging efficiency and genome availability, we have shown that there is a packaging defect when the genome size exceeds that of HIV-1. This defect was not rescued to sub-HIV-1 genome size levels by increasing genome availability. In contrast, others have shown that fluorescently labeled HIV-1 VLPs can package engineered HIV-1 genomes up to at least 17 kb as efficiently as 8 kb HIV-1 genomes.16 While packaging in these experiments benefitted from fixed genome availability (gag-pol mRNA and vgRNA were the same molecule), our data show that vectors larger than wild-type HIV-1 suffer a packaging defect even when genome availability is accounted for. Our approach benefitted from specifically detecting unspliced vgRNA, but we cannot rule out stuffer-specific effects in either system, or that sequences within the HIV-1 genome that are absent from our lentiviral vector design may enhance the packaging of large genomes. A combination of fluorescent labeling and molecular approaches applied to the same samples may enable full understanding of this discrepancy.
The data presented in this manuscript provide a target genome availability value, which stable producer cell lines and transfection ratios should target for all genomic RNAs, independent of genome size, in order to achieve high packaging efficiency and functional titer. When optimal genome availability is achieved by the producer cell, the benefit of efforts applied to further enhancing genome packaging are limited to 2-fold for genome sizes smaller than that of HIV-1. Using our repertoire of assays, we were able to distinguish between the contribution of packaging efficiency and genomic infectivity and report that genomic infectivity is the primary driver for the overall trend of increased genome size resulting in low titer. Hence, attention would be better applied to reducing total genome size, for example by minimizing the HIV-1 sequences and regulatory sequences. Although exceeding HIV-1 genome size causes a packaging defect in addition to low genomic infectivity, staying within HIV-1 genome size is not sufficient to prevent severe titer losses using the current lentiviral vector systems. Future efforts to reduce the burden of increased genome size should focus on enhancing genomic infectivity, potentially by engineering reverse transcriptase for improved processivity.10 Until this is realized, the cost-effectiveness of manufacturing multiple small vectors in favor of a large multi-cistronic design should be carefully considered where a co-transduction approach to delivering multiple genes is appropriate. Alternatively, this biological constraint of HIV-1 may be avoided by selecting an alternative integrating vector system, such as foamy viral vectors, which have been shown to tolerate large inserts with reduced impact on titer.13
This work contributes to the lentiviral vector field by introducing a measure of genome availability and demonstrating that this correlates closely with packaging efficiency. A genome availability of 1 (where gag-pol mRNA and vgRNA are present in equimolar amounts) is optimal for titer independent of genome size, but we demonstrate that genomes larger than that of HIV-1 package poorly even when genome availability is accounted for. We also rule out a role for increasing plasmid size as a contributor to the inverse relationship between genome size and titer. Additionally, we provide a method to readily manipulate and quantify relative packaging efficiency, which enabled us to show that low packaging efficiency does not impact infectivity of genome-containing vector particles on HEK293T cells. Vector production for any genome size can be optimized by altering transfer and packaging construct expression ratios to achieve maximal genomic titer. However, vector production only accounted for some of the loss to functional titer from increased genome size, the extent of which varied with size. Genomic infectivity was the main contributor to losses in titer for genome sizes below that of HIV-1, and further efforts to minimize the impact of size should focus on maintaining infectivity in target cells as genome size increases.
Materials and methods
DNA constructs
The small transfer plasmid, pG3-ns0, encodes a third-generation self-inactivating lentiviral vector genome using sequences described by others18,30 and contains the transgene cassette PGK-GFP-WPRE. The construct was synthesized by GeneArt (Thermo Fisher Scientific, Germany). The LacZ open reading frame was amplified by the PCR using NEB Q5 2× hotstart master mix (NEB, UK) and inserted between AclI and ClaI restriction sites located between the cPPT/CTS and PGK promoter in pG3-ns0 by Gibson assembly (NEBuilder HiFi master mix, NEB) to create pG3-ns0L (medium transfer plasmid). The 6,228 bp of the 5′ DMD ORF were gene optimized and synthesized by GeneArt, then used to replace LacZ in pG3-ns0L by restriction digestion using AclI and ClaI to create pG3-ns0D (large transfer plasmid). The pG3 plasmids were transformed into E. coli NEB10beta-competent cells (NEB, UK) and transfection-grade DNA prepared using the QIAGEN Endofree giga kit (QIAGEN, UK). To generate the equivalent constructs in nanoplasmid format, the genomic RNA expression cassettes from pG3-ns0, pG3-ns0L, and pG3-ns0D were amplified in 2 fragments each by the PCR, transferred to the NTC9 nanoplasmid backbone21 by Gibson assembly,31 and transformed into NTC-NP-CC4-competent E. coli (Nature Technology, Lincoln, NE, USA). Research-grade nanoplasmid preparations were purchased from Nature Technology. The identities of all constructs were verified by Sanger sequencing.
The design and construction of BAC1-pax (WT-GP containing packaging construct) has been described previously (patent WO 2017/089307). Briefly, the Tet repressor is encoded by the constitutive cytomegalovirus enhancer/promoter, while WT-GP, Rev, and VSVg coding sequences are under the control of the cytomegalovirus/TO promoter. BAC2-pax (synGP containing packaging construct) was constructed by gene synthesis (SGI-DNA, La Jolla, CA, USA) and is similar to BAC1-pax, except that it uses synGP rather than WT-GP.
Lentiviral vector production
HEK293T-adherent cells, used for all transfections with synGP, were cultured in growth medium (DMEM supplemented with 10% fetal bovine serum [FBS]). For transfection, cells were seeded into 12-well dishes at 4 × 105 cells per well in 1 mL growth medium on day 0 and incubated in a static cell culture incubator at 37°C with 5% CO2 and 60% humidity. On day 1, cells were transfected by adding 1 mg DNA complexed with 1.3 mg of PEIpro (Polyplus-transfection) in fresh growth medium (420 μL final volume). The next day, medium was replaced with 350 μL of growth medium supplemented with 5 mM sodium butyrate. For transfections using BAC2-pax, doxycycline was also added to 2 μg/mL at this stage. Vector-containing supernatant was collected on day 3 and clarified by centrifugation at 1,000 × g for 3 min. Supernatant was stored at −80°C for titration assays. Where producer cells were collected, cells were washed with PBS and then dissociated in 300 μL of TrypLE (Thermo Fisher Scientific). Collected cells were centrifuged at 1,000 × g for 3 min and the supernatant aspirated. The cell pellets were stored at −80°C.
Suspension-adapted HEK293T cells were used for transfections with BAC1-pax (WT-GP containing packaging construct) and cultured in BalanCD HEK293 medium (Irvine Scientific) supplemented with 2× GlutaMAX and 0.1% Pluronic F-68 (Thermo Fisher Scientific), then incubated at 37°C, 5% CO2, and 70% humidity in a shaking incubator set at 140 rpm. For transfection, cells were seeded at 2e6 cells in 1 mL per well of a 24-well dish. Within 2 h, cells were transfected with 1 mg DNA complexed with 1.3 mg of PEIpro. The next day cells were supplemented with 5 mM sodium butyrate and 2 μg/mL doxycycline. Two days post-transfection, cells and media were clarified by centrifugation at 1,000 × g for 3 min. Media and cell pellets were separated and stored at −80°C.
Determination of functional, physical, and genomic titers
Functional titer was determined by mixing vector with 2 × 104 HEK293Tsa cells in a total volume of 200 μL per well of a 96-well plate and incubated at 37°C, 5% CO2, for 3–4 days. The percent of cells expressing GFP, determined by flow cytometry using an Accuri C6 (Becton Dickinson) using the gating strategy shown in Figure S1, was multiplied by the starting cell number and normalized to 1 mL of vector supernatant. Units are HEK293Tsa transducing units per mL (TU/mL).
The physical titer was determined using a SYBR-Green product-enhanced reverse transcriptase (SG-PERT) assay according to a published protocol,32 but with the following reagents: PowerUp Sybr Green Master Mix (Thermo Fisher Scientific) and recombinant HIV-1 RT supplied by Abcam.
Genomic titer was determined by extracting RNA from 50 μL of cell-culture medium using a Purelink pro 96 viral RNA/DNA purification kit (Thermo Fisher) according to the manufacturer’s protocol, including the optional on-column DNase step using PureLink DNase set (Thermo Fisher Scientific) and eluting in 100 μL of water. Superscript IV VILO master mix was used to prepare cDNA (Thermo Fisher Scientific). Absolute quantification of the recovered viral genomes per mL was performed by digital droplet PCR (ddPCR) using published primer/probe sequences spanning the MSD33 in conjunction with Bio-Rad QX200 automated droplet generator and droplet reader using ddPCR supermix for probes (Bio-Rad). The absolute copy number was normalized to mL of starting material in the final ddPCR volume. Although recovery efficiency is not known, it was found to be consistent for samples processed in parallel (not shown). All samples within an experiment were processed in parallel.
Producer cell RNA quantification
The PureLink pro 96 total RNA purification kit (Thermo Fisher) was used to isolate RNA from cell pellets according to the manufacturer’s protocol. Contaminating DNA was removed by treatment with Turbo DNA-free kit. Superscript IV VILO master mix was used for cDNA synthesis and determination of absolute copy number of various transcripts by ddPCR (WT-GP) or a combination of ddPCR and qPCR (others). Where combined, ddPCR was used to determine the absolute quantity of a reference sample, and other samples were determined by qPCR with TaqMan Fast Advanced master mix (Thermo Fisher Scientific) using the ΔCt method. Absolute quantities were normalized to the beta-actin housekeeping gene. All negative and no-RT controls had undetectable target copies. Primer/probe sequences for synGP were synGP_F 5′-AAGACTGAGCTGCAGGCC-3′; synGP_R 5′-GCATACTGAGAGTCTGTCACGA-3′ and synGP_P 5′-FAM-CCTCGCTTTGCAGGACTCGGGC-MGB-3′. For WT-GP, sequences were WT-GP_F 5′-CCAGCTGTGATAAATGTCAG-3′; WT-GP_R 5′-AATGTGTACAATCTAGCTGC-3′, and WT-GP_P 5′-FAM-AGGGGAAGCCATGCATGGACAAGTA-MGB-3′. Primer/probe sequences for ACTB,34 MSD,33 and 5LVV35 have been previously described.
Statistical analyses
Graphs were plotted and linear regression analyses performed using Graphpad Prism 7.04.
Acknowledgments
We are grateful to Steven Howe and Martijn Brugman for critically reviewing the manuscript.
Author Contributions
N.P.S. designed and conducted the experiments. N.P.S. authored the manuscript. C.A.V. reviewed the manuscript and supervised the study.
Declaration of interests
The authors are employees and shareholders of GlaxoSmithKline.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.04.007.
Supplemental information
References
- 1.Milone M.C., O’Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32:1529–1541. doi: 10.1038/s41375-018-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 3.Mardiana S., Solomon B.J., Darcy P.K., Beavis P.A. Supercharging adoptive T cell therapy to overcome solid tumor-induced immunosuppression. Sci. Transl. Med. 2019;11:eaaw2293. doi: 10.1126/scitranslmed.aaw2293. [DOI] [PubMed] [Google Scholar]
- 4.Wong N.M.L., Wong W.W. Engineering a Dual Small Molecule Gated ZAP70 Switch in T Cells. ACS Synth. Biol. 2018;7:969–977. doi: 10.1021/acssynbio.7b00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cartellieri M., Feldmann A., Koristka S., Arndt C., Loff S., Ehninger A., von Bonin M., Bejestani E.P., Ehninger G., Bachmann M.P. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016;6:e458. doi: 10.1038/bcj.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Stasi A., Tey S.-K., Dotti G., Fujita Y., Kennedy-Nasser A., Martinez C., Straathof K., Liu E., Durett A.G., Grilley B. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey S.R., Maus M.V. Gene editing for immune cell therapies. Nat. Biotechnol. 2019;37:1425–1434. doi: 10.1038/s41587-019-0137-8. [DOI] [PubMed] [Google Scholar]
- 8.Kumar M., Keller B., Makalou N., Sutton R.E. Systematic determination of the packaging limit of lentiviral vectors. Hum. Gene Ther. 2001;12:1893–1905. doi: 10.1089/104303401753153947. [DOI] [PubMed] [Google Scholar]
- 9.Canté-Barrett K., Mendes R.D., Smits W.K., van Helsdingen-van Wijk Y.M., Pieters R., Meijerink J.P. Lentiviral gene transfer into human and murine hematopoietic stem cells: size matters. BMC Res. Notes. 2016;9:312. doi: 10.1186/s13104-016-2118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cosnefroy O., Murray P.J., Bishop K.N. HIV-1 capsid uncoating initiates after the first strand transfer of reverse transcription. Retrovirology. 2016;13:58. doi: 10.1186/s12977-016-0292-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shin N.H., Hartigan-O’Connor D., Pfeiffer J.K., Telesnitsky A. Replication of lengthened Moloney murine leukemia virus genomes is impaired at multiple stages. J. Virol. 2000;74:2694–2702. doi: 10.1128/jvi.74.6.2694-2702.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gélinas C., Temin H.M. Nondefective spleen necrosis virus-derived vectors define the upper size limit for packaging reticuloendotheliosis viruses. Proc. Natl. Acad. Sci. USA. 1986;83:9211–9215. doi: 10.1073/pnas.83.23.9211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sweeney N.P., Meng J., Patterson H., Morgan J.E., McClure M. Delivery of large transgene cassettes by foamy virus vector. Sci. Rep. 2017;7:8085. doi: 10.1038/s41598-017-08312-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rinke C.S., Boyer P.L., Sullivan M.D., Hughes S.H., Linial M.L. Mutation of the catalytic domain of the foamy virus reverse transcriptase leads to loss of processivity and infectivity. J. Virol. 2002;76:7560–7570. doi: 10.1128/JVI.76.15.7560-7570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terwilliger E.F., Godin B., Sodroski J.G., Haseltine W.A. Construction and use of a replication-competent human immunodeficiency virus (HIV-1) that expresses the chloramphenicol acetyltransferase enzyme. Proc. Natl. Acad. Sci. USA. 1989;86:3857–3861. doi: 10.1073/pnas.86.10.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nikolaitchik O.A., Dilley K.A., Fu W., Gorelick R.J., Tai S.H., Soheilian F., Ptak R.G., Nagashima K., Pathak V.K., Hu W.S. Dimeric RNA recognition regulates HIV-1 genome packaging. PLoS Pathog. 2013;9:e1003249. doi: 10.1371/journal.ppat.1003249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jouvenet N., Simon S.M., Bieniasz P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA. 2009;106:19114–19119. doi: 10.1073/pnas.0907364106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dull T., Zufferey R., Kelly M., Mandel R.J., Nguyen M., Trono D., Naldini L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kreiss P., Cameron B., Rangara R., Mailhe P., Aguerre-Charriol O., Airiau M., Scherman D., Crouzet J., Pitard B. Plasmid DNA size does not affect the physicochemical properties of lipoplexes but modulates gene transfer efficiency. Nucleic Acids Res. 1999;27:3792–3798. doi: 10.1093/nar/27.19.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hornstein B.D., Roman D., Arévalo-Soliz L.M., Engevik M.A., Zechiedrich L. Effects of Circular DNA Length on Transfection Efficiency by Electroporation into HeLa Cells. PLoS ONE. 2016;11:e0167537. doi: 10.1371/journal.pone.0167537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams J.A. Vector Design for Improved DNA Vaccine Efficacy, Safety and Production. Vaccines (Basel) 2013;1:225–249. doi: 10.3390/vaccines1030225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y.H., Pallant C., Sampson C.J., Boiti A., Johnson S., Brazauskas P., Hardwicke P., Marongiu M., Marinova V.M., Carmo M. Rapid Lentiviral Vector Producer Cell Line Generation Using a Single DNA Construct. Mol. Ther. Methods Clin. Dev. 2020;19:47–57. doi: 10.1016/j.omtm.2020.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pollard V.W., Malim M.H. The HIV-1 Rev protein. Annu. Rev. Microbiol. 1998;52:491–532. doi: 10.1146/annurev.micro.52.1.491. [DOI] [PubMed] [Google Scholar]
- 24.Kotsopoulou E., Kim V.N., Kingsman A.J., Kingsman S.M., Mitrophanous K.A. A Rev-independent human immunodeficiency virus type 1 (HIV-1)-based vector that exploits a codon-optimized HIV-1 gag-pol gene. J. Virol. 2000;74:4839–4852. doi: 10.1128/jvi.74.10.4839-4852.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Counsell J.R., Asgarian Z., Meng J., Ferrer V., Vink C.A., Howe S.J., Waddington S.N., Thrasher A.J., Muntoni F., Morgan J.E., Danos O. Lentiviral vectors can be used for full-length dystrophin gene therapy. Sci. Rep. 2017;7:79. doi: 10.1038/s41598-017-00152-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eckwahl M.J., Arnion H., Kharytonchyk S., Zang T., Bieniasz P.D., Telesnitsky A., Wolin S.L. Analysis of the human immunodeficiency virus-1 RNA packageome. RNA. 2016;22:1228–1238. doi: 10.1261/rna.057299.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Telesnitsky A., Wolin S.L. The Host RNAs in Retroviral Particles. Viruses. 2016;8:235. doi: 10.3390/v8080235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rulli S.J., Jr., Hibbert C.S., Mirro J., Pederson T., Biswal S., Rein A. Selective and nonselective packaging of cellular RNAs in retrovirus particles. J. Virol. 2007;81:6623–6631. doi: 10.1128/JVI.02833-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pear W.S., Nolan G.P., Scott M.L., Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sirven A., Pflumio F., Zennou V., Titeux M., Vainchenker W., Coulombel L., Dubart-Kupperschmitt A., Charneau P. The human immunodeficiency virus type-1 central DNA flap is a crucial determinant for lentiviral vector nuclear import and gene transduction of human hematopoietic stem cells. Blood. 2000;96:4103–4110. [PubMed] [Google Scholar]
- 31.Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., 3rd, Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 32.Vermeire J., Naessens E., Vanderstraeten H., Landi A., Iannucci V., Van Nuffel A., Taghon T., Pizzato M., Verhasselt B. Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS ONE. 2012;7:e50859. doi: 10.1371/journal.pone.0050859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christodoulou I., Patsali P., Stephanou C., Antoniou M., Kleanthous M., Lederer C.W. Measurement of lentiviral vector titre and copy number by cross-species duplex quantitative PCR. Gene Ther. 2016;23:113–118. doi: 10.1038/gt.2015.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mori R., Wang Q., Danenberg K.D., Pinski J.K., Danenberg P.V. Both beta-actin and GAPDH are useful reference genes for normalization of quantitative RT-PCR in human FFPE tissue samples of prostate cancer. Prostate. 2008;68:1555–1560. doi: 10.1002/pros.20815. [DOI] [PubMed] [Google Scholar]
- 35.Mortellaro A., Hernandez R.J., Guerrini M.M., Carlucci F., Tabucchi A., Ponzoni M., Sanvito F., Doglioni C., Di Serio C., Biasco L. Ex vivo gene therapy with lentiviral vectors rescues adenosine deaminase (ADA)-deficient mice and corrects their immune and metabolic defects. Blood. 2006;108:2979–2988. doi: 10.1182/blood-2006-05-023507. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.