

Abstract
Deposition of amyloid beta plaques in adult rat or human brain is associated with increased production of proinflammatory cytokines by associated glial cells that are responsible for degeneration of the diseased tissue. The expression of these cytokines is usually under check and is controlled at the post-transcriptional level via several microRNAs. Computational analysis of gene expression profiles of cortical regions of Alzheimer’s disease patients’ brain suggests ineffective target cytokine mRNA suppression by existing micro-ribonucleoproteins (miRNPs) in diseased brain. Exploring the mechanism of amyloid beta-induced cytokine expression, we have identified how the inactivation of the repressive miR-146 miRNPs causes increased production of cytokines in amyloid beta-exposed glial cells. In exploration of the cause of miRNP inactivation, we have noted amyloid beta oligomer-induced sequestration of the mTORC1 complex to early endosomes that results in decreased Ago2 phosphorylation, limited Ago2-miRNA uncoupling, and retarded Ago2-cytokine mRNA interaction in rat astrocytes. Interestingly, constitutive activation of mTORC1 by Rheb activator restricts proinflammatory cytokine production by reactivating miR-146 miRNPs in amyloid beta-exposed glial cells to rescue the disease phenotype in the in vivo rat model of Alzheimer’s disease.
Keywords: miRNAs, Astrocyte, Alzheimer’s disease, amyloid beta, cytokines, mTORC1 activation, neuroinflammation, cognition, Rheb, Endosome
Graphical abstract
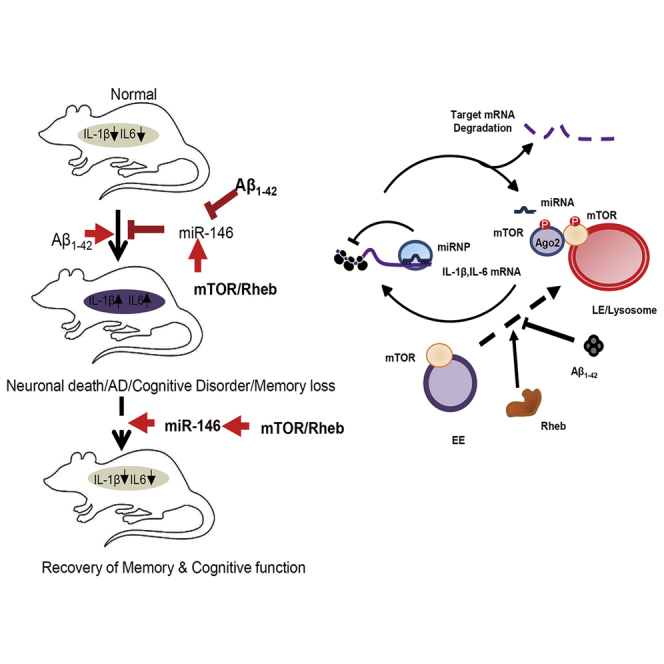
Inactivation of miRNPs is observed in amyloid beta-exposed glial cells in Alzheimer’s disease, which is linked with reduced mTORC1 activity. Activator protein Rheb restores mTORC1 activity to prevent the effect of Aβ1–42 oligomer in degenerating brain tissue and restores cognitive function by augmenting miR-146 activity.
Introduction
MicroRNAs (miRNAs) are 20- to 22-nt-long non-coding RNAs that can fine-tune the expression of their target mRNAs post-transcriptionally in metazoan cells by imperfect base pairing.1,2 As in other tissues, miRNAs are important gene regulators in the mammalian brain, where hundreds of miRNAs are known to control thousands of transcripts encoding important proteins that affect several physiological processes and events in the mammalian brain.3 Differentiation of embryonic stem cells to neuronal cells is also controlled by specific miRNAs such as miR-9,4 while miR-128 regulates the growth of the dendrites.5 There is also evidence of miRNA playing a significant role in neurodegenerative diseases (NDDs) such as in Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), or in amyotrophic lateral sclerosis (ALS).6 There have been reports that show the effect of conditional knockout of Dicer, an essential enzyme for miRNA biogenesis, on neuronal death.7 Additionally, differential miRNA expression profiles from postmortem brains of patients who died of AD have shown the possible importance of miRNA expression in the pathophysiology of this disease.8,9
Astrocytes are the most abundant cells in the mammalian brain. The increases in the number and complexity of this glial cell population during brain development support their possible role in regulating complex traits such as human cognition and behavior.10 Astrocytes provide support to the neuronal function by regulating ion homeostasis,11 or by controlling CNS blood flow,12 and they also play a crucial role in synaptic transmission by forming tripartite synapse.13,14 Apart from their supportive role, astrocytes are also immunomodulatory cells known to secrete various cytokines and chemokines, which play an important role in the pathophysiology of different diseases, including AD.15 In AD, increased production of inflammatory cytokines may lead to death of the neighboring neurons and is considered as the major path that amyloid beta (Aβ) oligomers activate in the glial cell population, leading to neuronal death.16 However, the exact mechanism of the excess cytokine production in glial cells by Aβ exposure is not clear. miRNAs are important regulators of the immune response, and they can control hyperresponsiveness of immune cells by reversibly regulating miRNA activity in activated macrophage cells.17, 18, 19 Thus, the expression and activity of miRNAs in astroglia, the major immune cells in brain context, are likely to have a role in controlling neuroinflammation during neurodegenerative diseases.
Argonaute (Ago) proteins play a crucial role in post-transcriptional gene silencing in eukaryotic cells. According to published reports, Ago proteins are responsible for the regulation of expression of most protein-coding genes primarily by associating with specific miRNAs having complementarities with those protein-coding genes.20,21 In mammals, there are four Ago proteins (Ago1–Ago4), and Ago2 is the major and most widely expressed Ago in different tissues, including the brain.22 Upon its association with miRNAs, it can cleave the target mRNAs, having perfect complementarities with bound miRNAs.23 Post-translational modification of Ago protein can also determine its function. Increased Ago stability and catalytic activity are associated with modifications such as hydroxylation and methylation of Ago.24,25 Among all of the modifications, phosphorylation at different positions may play a significant role in miRNA binding of Ago2 and its subcellular localization.26, 27, 28
Mammalian target of rapamycin (mTOR), a master regulator of cell growth, is comprised of mTORC1 and mTORC2, which are different forms of structural and functional complexes involving mTOR protein. Raptor and mLST8, along with mTOR, are the major subunits of mTORC1, which regulates cell growth, protein homeostasis and synthesis, and autophagy.29 mTORC2 constitutes mTOR, Rictor, mSin1, and mLST8. The major function of this complex includes lipogenetic regulation and that of the cytoskeletal structure.30 The mTOR pathway specializes in sensing multiple external stimuli such as growth factors and oxygen level, and it is known to respond by sensing amino acid and glucose concentrations as well. Important neuronal physiological phenomena such as synaptic plasticity, memory, and learning are often maintained by mTOR.31 Faulty mTORC1 signaling has been linked with AD previously, and TREM2-deficient microglial cells were shown to have a decrease in mTORC1 activity that contributed to high autophagic flux, leading to weakened microglial fitness.32 Studies have shown that in spite of known upstream and downstream regulatory factors, the major key to the mTOR-associated signaling lies in the compartmentalization of the mTORC1 to specific cellular sites.33,34 How disease conditions influence mTOR localization and whether mTOR localization plays a role in the pathophysiology of a disease are unresolved questions in the field.
In this study, we report how the exposure of an Aβ oligomer (Aβ1–42) in rat astrocytes decreases cellular mTORC1 activity by enhancing the sequestration of mTORC1 complexes to the early endosome. Aβ1–42 also restricts translocation of mTORC1 to the lysosome, which is found to be important for mTORC1 activation. mTOR inactivation reduces miRNA activity due to a decrease in Ago2 phosphorylation. Interestingly, reactivation of mTORC1 through the expression of the constitutive activator Rheb reverses both Aβ1–42-mediated pro-inflammatory cytokine production and restores behavioral abnormalities in animals along with a rescue of miRNA function, suggesting a pivotal role of miRNA activity modulation by mTORC1 in AD.
Results
Exposure to Aβ1–42 oligomer causes miRNA upregulation in human and adult rat brain
miRNAs are important regulators of most metazoan genes that are supposed to be upregulated and expressed profoundly upon inactivation or downregulation of cognate miRNAs. To obtain an insight into whether the miRNA and mRNA profiles get altered in the disease process, we analyzed the “regulator(s)-to-target(s)” (miRNA/mRNA) expression profiles in different brain regions of AD patients. The relationship between the differentially expressed miRNAs and their corresponding target genes were studied. A set of 30 miRNAs were found to be upregulated whereas 23 miRNAs were found to be downregulated in the prefrontal cortex of AD patients (Figure 1A; Table S1), and these miRNAs are likely to be related to AD etiology or progression. Clustering has been done for the analyzed miRNAs. Analysis of miRNA expression profiles in the prefrontal cortex of AD patients was performed by considering the samples provided in a GEO dataset (GEO: GSE48552).35 Based on clinical presentation and stage of the disease, the samples had been grouped into early stage and late-stage AD. Differentially expressed miRNA between these stages were determined by using the DESeq package in R. Normalized counts of the differentially expressed miRNAs identified when considering these samples have been depicted in a heatmap (Figure 1A).36 Furthermore, when considering mRNA expression profiling data, 2,092, 2,166, and 292 mRNAs were found to be upregulated while 1,253, 699, and 676 genes were found to be downregulated within the superior frontal gyrus, frontal cortex, and dorsolateral prefrontal cortex, respectively. Moreover, 271 genes (mRNAs) were found to be commonly upregulated among two or more of the different brain cortical regions considered herein for the analysis. The corresponding fraction of upregulated target mRNAs of each of the differentially expressed miRNAs was determined in different cortical regions (frontal cortex, superior frontal gyrus, and dorsolateral prefrontal cortex) of the human brain. Herein, we observed that in comparison to the downregulated miRNAs, nearly 79% of the upregulated miRNAs had a substantial fraction (0.6–0.8) of their target mRNAs also upregulated as in the frontal cortex (Figures 1C and 1D). Furthermore, the observation that in comparison to the downregulated miRNAs, a higher percentage of the upregulated miRNAs had a substantial fraction of their target mRNAs as upregulated is consistent within the other cortical regions considered in this study. Herein, in comparison to the downregulated miRNAs, 88% and 48% of the upregulated miRNAs had between 0.4 and 0.6 or between 0.2 and 0.4 fractions of their target mRNAs as upregulated within the superior frontal gyrus and dorsolateral prefrontal cortex, respectively (Figures 1C and 1D).
Figure 1.

Possible miRNA-to-mRNA expression relationships in Alzheimer’s disease (AD) and cancer
(A) Heatmap of differentially expressed miRNAs in prefrontal cortex of AD patients. The significance threshold for selection of differentially expressed miRNAs was considered as fold change of ±1.25 and a p value of ≤0.05. (B) Graphs showing expression levels of miR-146a targets in the prefrontal cortex of Alzheimer’s disease patients. The y axis represents fold change of individual mRNAs, and the x axis represents the –log10 p value. The table represents the number of miR-146a targets having at least 1.5-fold changes. (C) The probable relationship between differentially expressed miRNAs and mRNAs in AD considering the possible fraction of upregulated target mRNAs of each of the miRNAs as studied in different cortical regions (frontal cortex, superior frontal gyrus, and dorsolateral prefrontal cortex) is depicted here. (D) Percentages of upregulated or downregulated miRNAs that possibly have a fraction of their target genes as upregulated within the frontal cortex, superior frontal gyrus, and dorsolateral prefrontal cortex are shown here. (E) Comparison among the percentages of differentially expressed miRNAs that have different subsets or fractions of their target mRNAs as upregulated in degenerative tissues (AD) as opposed to highly proliferative tissues (cancer) was performed.
In order to determine whether similar regulator(s)-to-target(s) relationship patterns could be prevalent in other diseases or cell types, we determined whether these deregulated mRNAs could be related to any other disease condition as well by performing a disease enrichment analysis that considered the set of commonly upregulated mRNAs. With the help of disease enrichment analysis, we found that these genes are also associated with cancer (Figures S1A and S1B). Therefore, subsequently we analyzed regulator(s)-to-target(s) (miRNA/mRNA) expression profiles in different cancer tissues. A substantial fraction of upregulated miRNAs had their target mRNAs as upregulated in oral squamous cell carcinoma, colorectal cancer, and breast cancer (Figures S1C–S1E). In this scenario, we observed that similar percentages of downregulated miRNAs and upregulated miRNAs have their target mRNA fractions as upregulated Figure S1 In particular, the upregulated miRNAs (65%) and downregulated miRNAs (67%) have similar fraction (between 0.4 and 0.6 or between 0.2 and 0.4) of their corresponding target mRNAs as upregulated in the cancer case studies considered herein (Figures 1E and S1F).
Importantly, we have considered multiple datasets to identify a common trend in the deregulated miRNA-mRNA interaction network by considering target mRNAs that are commonly differentially expressed in two or more of the different brain cortical regions. However, during the analysis we considered all of the target mRNAs that were significantly differentially expressed in each dataset to study the regulator(s)-to-target(s) (miRNA/mRNA) expression profile patterns in AD patients and compared the same. miR-146a-5p has consistently been found to exhibit this phenomenon across all of the conditions analyzed in the present study. Furthermore, miR-146a, which is known to regulate expression of a number of cytokines, was found to be upregulated.37 Expression of all of the probable miR-146a targets was also analyzed. Interestingly, most miR-146a targets were found to be upregulated in AD brain, indicating a possible loss of function of miR-146a on its target in the AD brain (Figure 1B; Table S2).
In the AD datasets considered herein for analysis, a substantial percentage of upregulated miRNAs have a large fraction of their corresponding target genes as upregulated. This is contrary to the general regulatory relationship between miRNAs and their target genes wherein the target genes of upregulated miRNAs are downregulated, as expected.2,38 However, this inverse relationship between the target genes and upregulated miRNAs has also been noted in other cell types other than those in the degenerative brain. In cancer cells, the trend of target gene upregulation in the backdrop of upregulated cognate miRNA expression has also been noted (Figure S1F). However, the upregulation of a target gene with an increase in cognate miRNAs has been more prominent in AD than in cancer (Figures 1D and 1E). Thus, we observed trends that in comparison to the downregulated miRNA, target mRNAs of upregulated miRNAs are mainly upregulated, and this phenomenon is more prominent in the AD condition than in cancer. Therefore, in the AD brain there is a possibility of the existence of a mechanism for gross inactivation of micro-ribonucleoproteins (miRNPs) that may make them ineffective to suppress their targets.
We wanted to explore the effect of the Aβ1–42 oligomer, the suspected causative link between amyloid plaque and the disease phenotype in AD patients, on cellular miRNA levels in rat brain. The preformed Aβ1–42 oligomer was stereotactically injected in the adult rat brain hippocampal region and animals were kept for 21 days, which is known to cause deposition of Aβ plaques in and around the injection area (Figures 2A and 2B).39 A deposition of Aβ plaques also resulted in glial fibrillary acidic protein (GFAP) activation, which is a marker for astrocyte proliferation as well as immune activation in rat brain (Figure 2B).40 Now there are several miRNAs present in rat brain that can regulate the expression profile of these cytokine mRNAs either directly or indirectly. For example, interleukin (IL)-6 is a direct target of let-7a, and miR-146a fine-tunes cytokine production by repressing the nuclear factor κB (NF-κB) pathway.41,42 Therefore, we wanted to check what are the expression patterns of those miRNAs that directly and indirectly regulate expression of pro-inflammatory cytokines in both rat brain as well as in primary rat astrocytes. mRNA levels of different proinflammatory cytokines, such as IL-1β and IL-6, were increased several fold when oligomeric Aβ1–42 was stereotactically infused in the hippocampus of adult rat brain, supporting an already reported Aβ1–42-induced immunoactivation reaction in affected brain (Figure 2C). Interestingly, as observed in human patient samples, expression profiles of several miRNAs, including the immunogenic miRNAs miR-21, miR-146a, and miR-155, also showed upregulation in adult rat brain injected with Aβ1–42 (Figure 2D). To identify the contribution of astrocytes in cytokine production in rat brain upon activation with the Aβ1–42 oligomer, we treated the primary rat astrocytes with Aβ1–42 oligomer ex vivo. The activation of the cells was scored by excess production of GFAP in rat primary astrocyte on Aβ1–42 oligomer exposure (Figures 2E and 2F). It was found that Aβ can activate astrocytes where mRNA levels of different cytokines were found to be increased several fold (Figures 2E and 2F). We noted a global increase in miRNA levels in Aβ1–42-treated astrocytes where 65%–70% of the miRNAs were found to be upregulated, including miR-146a, miR-155, miR-21, and let-7a. These miRNAs are direct and indirect regulators of IL-1β. IL-6 was also noted to have increased in treated astrocytes (Figure 2G). Interestingly, we found increases in several other miRNAs, such as miR-16, miR-29a, and miR-145, in both brain tissue as well as in primary glial cells upon amyloid exposure (Figures 2D and 2G). These miRNAs are not known to affect cytokine expression in mammalian cells, indicating the possibility that the phenomena we observed could have broader implications and affect several different types of miRNAs.
Figure 2.

Aβ1–42 induces an inflammatory response and increases miRNA levels in astrocytes
(A) Preformed Aβ1–42 oligomers in PBS were infused in the hippocampus at stereotaxic coordinates from bregma (AP, 3.6 mm; L, 2.1 mm; DV, 2.8 mm) in adult rat brain. PBS without Aβ1–42 was used for control injection. Animals were kept 21 days for plaque formation. (B) Cryosections of Aβ1–42-infused and PBS-infused control rat brain hippocampus were subjected to immunohistochemistry (IHC) analysis. Astrocytes were immunostained for GFAP (green), whereas Aβ was visualized in red and the nucleus was stained with Hoechst (blue). Relative intensities of the GFAP signals were measured by ImageJ software, and quantification is shown in the right panel. (C) qPCR-based quantification of total mRNA levels of IL-1β and IL-6 in PBS and Aβ1–42-infused adult rat hippocampus. The data were normalized against GAPDH mRNA levels. (D) Cellular miRNA levels in PBS- and Aβ1–42-infused adult rat brain hippocampus. TaqMan-based miRNA level quantification was done. Values were normalized with U6 snRNA level. Red represents the miRNAs that are known to regulate cytokine mRNAs, and blue represents miRNAs that are not known to regulate cytokine mRNAs. (E and F) Experimental strategy of isolation of rat primary astrocytes from 0- to 1-day-old rat pup brain followed by treatment of the primary cells with 1.5 μM Aβ1–42 oligomers ex vivo (E). Microscopic images showing effect of Aβ1–42 on primary cortical astrocytes stained with GFAP (green, F, upper panel). The intensity levels of the GFAP in both conditions were calculated by ImageJ software and plotted (F, bottom right panel). Graphs depict levels of cellular IL-1β and IL-6 mRNA levels in Aβ1–42-treated primary cortical astrocytes. The data were normalized against GAPDH mRNA level (F, lower left panel). (G) Cellular miRNA levels in Aβ1–42-treated primary cortical astrocytes. TaqMan-based miRNA level quantifications were done. Values were normalized with U6 snRNA. Red represents the miRNAs that are known to regulate cytokine mRNAs, and blue represents miRNAs that are not known to regulate cytokine mRNAs. For statistical significance, a minimum of three independent experiments were considered in each case unless otherwise mentioned, and error bars represent means ± SEM. p values were calculated with a Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.0001. ns, not significant.
mTOR-mediated Ago2 phosphorylation is required for miRNA-mediated repression of targets
To uncover the mechanism of non-functional miRNP accumulation in Aβ1–42-treated cells, we used C6 glioblastoma cells to treat with the Aβ1–42 oligomer before the biochemical analysis was done for the treated and control cells. To confirm the accumulation of nonfunctional miRNPs with the increase in total cellular miRNP levels observed in Aβ1–42 oligomer-treated cells, C6 glioma cells were transfected with Renilla luciferase (RL) reporter having the 3′ UTR of IRAK1 mRNA with two miR-146a binding sites (Figure 3A). A reduction in miR-146a activity was observed in Aβ1–42-treated cells compared to the DMSO-treated control cells (Figure 3B). The most probable cause of reduced miRNA activity could be the unbinding of miRNA with Ago2 proteins, which is known to happen in the mammalian macrophage during its activation phase and also in neuronal cells during the nerve growth factor-induced differentiation process.19,26 To check whether similar effects on Ago2 by Aβ1–42 may cause the downregulation of miRNA-Ago2 binding in Aβ1–42-treated astrocytes, we cultured primary astrocytes from rat pups and treated them with DMSO or Aβ1–42 for 24 h. Ago2 was immunoprecipitated (IP) followed by qPCR-based measurement of bound miRNAs. The RNA amount was normalized with the quantity of Ago2 protein recovered during the IP process. The quantification revealed, in Aβ1–42-treated cells, that there was an increased association of Ago2 with miR-146a and miR-155 compared to the control cells (Figure 3C). We also found an increase in both Ago2-associated miR-16 and miR-29a level upon Aβ1–42 treatment (Figure S2A). We performed similar experiments with primary neurons cultured ex vivo, where we could not detect a significant effect of Aβ1–42 oligomers on neuronal miRNA levels or Ago2-associated miRNA levels, suggesting an astrocyte-specific effect of Aβ1–42 on cellular miRNPs (Figures 3D and 3E). To check the possible role of any post-translational modification affecting miRNA activity in the Aβ treatment context, we measured phosphorylated Ago2 levels in Aβ1–42-treated cells. The reduced phosphorylated Ago2 level in Aβ1–42-treated cells was detected and compared to the control cells, and the results correlated with reduced interaction between Ago2 and mTOR after Aβ1–42 treatment (Figures 3F and 3G), indicating a possible role of mTOR in miRNA activity regulation via controlling Ago phosphorylation. This was suggestive from the earlier experiments in which an Aβ1–42-mediated decrease in Ago2 phosphorylation might hinder dissociation of miRNA from Ago2 protein but reduce miRNA activity. Therefore, we hypothesized that lack of Ago2 phosphorylation might perturb the repression of target mRNA in mammalian cells. Ago2 proteins have a MID domain that is found to be important for binding with the 5′ phosphate group of small RNAs.28 A phosphorylation at the Y529 position in the MID domain imparts a negative charge that dissociates miRNAs from Ago2 protein. The Ago2Y529F mutant, which cannot be phosphorylated at the Tyr529 position, remains bound to miRNA.19,28 C6 glioblastoma cells were transiently transfected with the Ago2-wild-type (WT) and Ago2-Y529F protein in the absence and presence of Aβ1–42 oligomers, and qPCR was performed to check the cellular mRNA levels of IL-1β and IL-6. We found a higher expression of both IL-1β and IL-6 mRNA levels in Ago2-Y529F-expressing cells even without Aβ1–42 exposure, which suggests the importance of Ago2 phosphorylation in cytokine mRNA expression (Figure 3H). However, the effect of Aβ1–42 on cytokine expression was absent in cells expressing Ago2-Y529E. This further suggests that cytokine expression is regulated primarily via Ago2 phosphorylation in glial cells. To confirm the involvement of mTORC1 in the miRNA activity regulation process possibly via Ago2 phosphorylation, cells were exposed to rapamycin, a known mTORC1 blocker.43 We found a significant decrease in miR-146a activity when treated with rapamycin (Figure 3I). We also found reduced amounts of Ago2 phosphorylation and an increase in Ago2-bound miR-146a levels upon rapamycin treatment (Figure 3J and 3K). These data signify the effects of mTORC1 inhibition in controlling miRNA activity and Ago2 phosphorylation that are also observed in astrocytes exposed to Aβ1–42. Experiments were also done in a heterogeneous context using HEK293 cells depleted for Raptor, an integral protein of the mTORC1 complex, and expressing exogenously a liver-specific miR-122 there.44 This was done to recheck the interlinking of mTOR inactivation with Ago2 phosphorylation and miRNA activity regulation even in non-astroglial cells. In HEK293 cells treated with siRaptor, we documented reduced miRNA activity and, as in Aβ1–42 treated cells, a reduction in Ago2 phosphorylation, confirming mTORC1 involvement in miRNA activity regulation in general in mammalian cells by targeting Ago2 phosphorylation (Figures S2B and S2C).
Figure 3.

mTOR-mediated Ago2 phosphorylation is required for miRNA activity
(A) Schematic design of different Renilla luciferase (RL) reporters used for scoring miRNA activity. The 3′ UTR of IRAK1 mRNA with two miR-146a binding sites was cloned on the 3′ UTR of the Renilla mRNA. (B) Effect of Aβ1–42 treatment on cellular miR-146a activity and level. Specific activity of miR-146a was calculated by normalizing the fold repression of target reporter mRNA against the corresponding miRNA expression level. An increased cellular miR-146a level in C6 glioblastoma cells upon Aβ1–42 treatment is shown. The data were obtained by qRT-PCR-based quantification and were normalized against the expression of U6 snRNA. (C) Increase in Ago2-miRNA association in rat primary astrocytes upon Aβ1–42 treatment. Endogenous Ago2 from control- and 1.5 μM Aβ1–42-treated primary rat astrocytes was immunoprecipitated and the levels of Ago2-associated miR-146a and miR-155 were estimated by qRT-PCR. Values were normalized against the amount of Ago2 obtained in each immunoprecipitation reaction. (D and E) No change in miRNP levels in rat cortical neurons exposed to 1.5 μM Aβ1–42. Primary neuronal cells were treated with Aβ1–42 oligomers for 24 h, and the amounts of endogenous miRNAs and miRNAs associated with immunoprecipitated Ago2 were estimated and plotted after normalization against U6 snRNA and Ago2 levels, respectively. (F and G) Reduced mTOR-Ago2 interaction and Ago2 phosphorylation in Aβ1–42 oligomer-treated C6 glioblastoma cells. Western blot data show the amount of Ago2 phosphorylation (F) and the amount of mTOR pulled down with Ago2 (G). C6 glioblastoma cells were transiently transfected with FH-Ago2 and then treated with 2.5 μM Aβ1–42 for 24 h. Immunoseparation of FH-Ago2 was done with FLAG beads. The amount of Ago2 pulled down was detected by anti-HA antibody, and levels of phosphorylated Ago2 (Tyr phosphorylation) were detected by anti-phosphorylated (p-)Tyr antibody 4G10. (H) Effect of expression of Y529F mutant of Ago2 on cellular IL-1β and IL-6 mRNA levels on C6 cells. The qPCR data were normalized against GAPDH mRNA level. (I) Graph representing miR-146a-specific activity upon rapamycin (100 nM) treatment in C6 glioblastoma cells. The fold repression of miR-146a was normalized with the miR-146a expression level. (J) Western blot data showing level of Ago2 phosphorylation and Ago2-mTOR interaction upon rapamycin (100 nM) treatment. (K) qPCR data showing Ago2-associated miR-146a and miR-16 levels upon rapamycin (100 nM) treatment. qPCR data were normalized with the amount of Ago2 pulled down from the immunoprecipitation reaction. For statistical significance, a minimum of three independent experiments were considered in each case unless otherwise mentioned, and error bars represent means ± SEM. p values were calculated by utilizing a Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.0001.
Sequestration of mTORC1 to early endosome causes miRNA inactivation and increased miRNP formation in glial cells
We found a decrease in phosphorylated p70-S6 kinase along with active phosphorylated mTOR levels in Aβ1–42-treated astrocytes (Figures 4A and 4B). The expression of mTOR is tightly regulated in mammalian cells where different nutrients and growth factor-like glucose, amino acids, and insulin can activate mTORC1 depending on the metabolic state of the cell.45 Interestingly, it is not only the presence of upstream signals, but mTORC1 also shows localization-based activation. It was reported earlier that translocation of mTOR to the lysosomal membrane is important for mTORC1 activation, where it can be activated by the GTPase Rheb.34 Thus, to check for the possibility that Aβ1–42-mediated mTORC1 deactivation may happen because of the reduced mTORC1 localization to the lysosomal membrane, we microscopically looked at the localization of mTOR in control and treated cell. Confocal images were taken from both control and Aβ1–42-treated C6 glioblastoma cells, where lysosomes were stained with LysoTracker (red) and mTOR (green) was visualized by indirect immunofluorescence (Figure 4C). There was a decrease in colocalization between mTOR and lysosomes indicating a faulty translocation of mTOR in amyloid-treated cells (Figure 4D). Since mTOR was not translocating at the lysosomal surface, it was important to find out the altered localization of mTOR upon Aβ1–42 exposure. We found a significant increase in colocalization between mTOR and early endosomes (Figures 4E and 4F). Cells were transiently transfected with yellow fluorescent protein (YFP)-Endo, which marks early endosomes, and then activated with oligomeric Aβ1–42 for 24h. Higher colocalization between early endosomes and mTOR suggests that Aβ1–42 restricts mTOR to the early endosomes, which retards its translocation to the lysosomal surfaces to get activated. To determine whether mTOR sequestration in early endosome can affect miRNA activity or miRNP formation, we expressed a constitutively active GTPase-deficient Rab5 mutant, Rab5Q79C (Rab5-CA), that is known to cause mTOR sequestration in early endosomes (Figures 4G and 4H).46 With overexpression of the Rab5-CA mutant, we observed a decrease in specific activity of let-7a (Figure 4I) and an increase in both cellular and Ago2-associated miR-146a (Figures 4J and 4K). These results connect the mTOR compartmentalization defects in Aβ1–42-treated cells to functional inactivation of increased miRNPs observed.
Figure 4.

Sequestration to endosome causes mTORC1 inactivation linked with increased miRNA-Ago2 interaction in glial cells
(A and B) Lowering of p-mTOR in Aβ1–42 oligomer-treated C6 glioblastoma cells. Western blot data showing the levels of cellular p-mTOR levels along with downstream substrate of mTOR p-S6K levels in C6 glioblastoma cells (A) and in primary astrocytes (B) treated with Aβ1–42. (C and D) Decreased lysosome targeting of mTOR after Aβ1–42 oligomer treatment. Confocal images depicting localization of mTOR to lysosome in control and treated cells are shown in (C). Endogenous mTOR was visualized indirectly with immunofluorescence (green), and lysosomes are labeled with LysoTracker (red) in C6 glioblastoma cells. A Pearson’s coefficient of colocalization was used to measure the amount of mTOR translocating to lysosomes in control and Aβ1–42-treated C6 glioblastoma cells (D). (E and F) Increased endosome-mTOR localization after Aβ1–42 oligomer treatment. Confocal images show localization of endosomes and mTOR. Endosomes were tagged with YFP-Endo (green), and endogenous mTOR (red) was detected by indirect immunofluorescence. Colocalization between endosomes and mTOR was visualized as yellow (E). A Pearson’s coefficient of colocalization of early endosomes and mTOR in Aβ1–42 oligomer-treated C6 glioblastoma cells is shown in (F). (G and H) Effect of Rab5-CA on mTOR localization in C6 glioblastoma cells. Colocalization of mTOR in C6 cells expressing YFP-Endo upon Rab5-CA expression was done. The cells with YFP-Endo (green) and mTOR (red) were visualized in control and Rab5-CA-expressing cells (G). A Pearson’s coefficient of colocalization of mTOR and early endosome is shown in (H). (I) Alteration of miRNA activity and levels in mammalian cells defective for endosome maturation due to expression of the constitutively active form of Rab5 protein. A drop in cellular activity of let-7a miRNA in C6 cells upon expression of constitutively activated Rab5 mutant Rab5-CA is shown. The RL reporter with three let-7a miRNA binding sites was used to score the effect of Rab5-CA expression on miRNA repressive activity. (J and K) Levels of cellular and Ago2-associated miR-146a upon Rab5-CA expression in C6 glioblastoma cells. The cellular miR-146 level was normalized against the U6 snRNA level (J). The amount of Ago2 immunoprecipitated from control and Rab5-CA-expressing cells were used for normalization of the amount of miRNAs associated with Ago2 (K). For statistical significance, a minimum of three independent experiments were considered in each case unless otherwise mentioned, and error bars represent means ± SEM. p values were calculated by utilizing a Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.0001.Scale bar represents 10μm in panels C, G, E.
mTORC1 activation rescues cytokine repression by targeting the miRNP reactivation pathway
Based on the results described so far, mTOR translocation to lysosome could be important for Ago2 phosphorylation leading to sustained miRNP recycling and activity in astrocytes. To confirm this mode of action of mTOR in glial cells, C6 cells were transfected with plasmids encoding Rheb-Myc before the Aβ1–42 oligomer treatment. Rheb is a constitutive activator of mTOR that can activate mTOR independently of growth factor or amino acid stimulation.47,48 Results from Rheb-expressing cells suggest rescue of mTOR localization upon Aβ1–42 treatment in Rheb-Myc-expressing cells (Figure 5A). We observed about a 1.6-fold reduction in mTOR sequestration in endosomes, suggesting that there was mobilization of mTOR from endosomal compartments (Figures 5A and 5B). To investigate a possible role of mTOR reactivation in miRNA activity, we checked the status of Ago2-positive bodies in Aβ1–42-treated cells expressing Rheb-Myc (Figure 5C). Interestingly, in Rheb-expressing cells, we found that fewer Ago2-positive bodies colocalized with processing bodies or PBs compared to that in Aβ1–42-treated cells (Figure 5D). These data clearly suggest a role of mTOR signaling in mobilizing the Ago2 from an otherwise inactive compartment into an active compartment where it could repress the target cytokines. Activation of mTOR upon Rheb-Myc expression was confirmed by measuring the levels of phosphorylated mTOR and S6 kinase in untreated and Aβ1–42-treated cells before transfection with control or Rheb-Myc expression plasmids (Figure 5E). To get further evidence whether this process restores miRNA activity per se, we checked the different cytokine levels. Quantitative estimation revealed that there has been a 2.7-fold decrease in IL-1β mRNA level and about a 1.5-fold decrease in IL-6 mRNA level compared to the Aβ1–42-treated cells upon Rheb-Myc expression (Figure 5F). Again, Rheb-Myc-expressing cells showed a decrease in cellular miR-146a and miR-155 levels compared to the only Aβ1–42-treated cells (Figure 5G). Rheb-mediated mTOR reactivation also increases Ago2 phosphorylation in Aβ1–42-treated cells, reducing the Ago2-associated miR-146a and miR-155 levels (Figures 5H and 5I). We also observed a similar effect in cellular and Ago2-associated miR-16 and miR-29a levels when cells were expressing Rheb-Myc (Figures S3A–S3C). Since miR-16 and miR-29a do not directly regulate expression of any proinflammatory cytokines, there is a possibility that the Aβ1–42-mediated mTOR inactivation mechanism may affect the function of quite a few miRNAs.
Figure 5.

Reactivation of mTOR by Rheb-Myc mobilizes mTOR from endosomes and reduces cytokine production by targeting the miRNA pathway
(A and B) Altered mTOR localization upon expression of Rheb-Myc in Aβ1–42 oligomer-treated C6 glioblastoma cells. Confocal images showing colocalization of endosomes and mTOR. The C6 glioblastoma cells expressing YFP-Endo and thus having endosomes tagged with YFP-Endo (green). Endogenous mTOR was stained by indirect immunofluorescence (red). Yellow is used to show the extent of colocalization between endosomes and mTOR (A). A plot is shown depicting the amount of colocalization between mTOR and endosomes in control and Aβ1–42 oligomer-treated C6 glioblastoma cells transfected rather with control vector or with Rheb-Myc expression plasmid. The amount of colocalization was measured with a Pearson’s coefficient of colocalization (B). (C and D) Ago2 localization to PBs gets affected in cells expressing Rheb-Myc. Confocal microscopic images of Ago2 bodies stained with GFP-Ago2 (green) co-stained for endogenous Rck/p54 (red) are shown. Colocalized Ago2 and Rck/p54 bodies were considered as PBs (yellow) (C). Plots depicting number of PBs per cell in C6 glioblastoma cells that were untreated or treated with Aβ1–42. In both conditions, the effect of expression of Rheb-Myc was scored (D). (E) Effect of Rheb-Myc expression on mTOR activation in control and Aβ1–42 oligomer-treated C6 glioblastoma cells. Western blot data show the levels of cellular p-mTOR and p-S6K levels in untreated and cells expressing either control or Rheb-Myc expression vectors. (F and G) Expression of Rheb-Myc restores cytokine and miRNA expression to control untreated levels in Aβ1–42 oligomers exposed to C6 glioblastoma cells. Quantitative data show the effect of Rheb-Myc expression on cellular IL-1β and IL-6 mRNA levels in both control- and Aβ1–42-treated C6 glioblastoma cells. The cytokine mRNA levels were normalized against the GAPDH mRNA level (F). The effect of Rheb-Myc expression on cellular miR-146a and miR-155 levels in both control- and Aβ1–42-treated C6 glioblastoma cells is shown (G). The data were normalized against U6 snRNA level. (H–J) Reversal of Ago2-miRNA and Ago2-cytokine mRNA interaction upon Rheb-Myc expression. Ago2-associated miR-146a and miR-155 levels and IL-1β and IL-6 mRNA levels were measured in C6 glioblastoma cells pre-treated with Aβ1–42 oligomers (I and J). The RNA level was normalized by the amount of Ago2 pulled down in the immunoprecipitation reaction (H). (K) Effect of Rheb-Myc expression on IL-1β and IL-6 mRNA levels when the cells were pre-treated either with control or by miR-146a- and miR-155-specific antagomiRs. In all of the sets, C6 glioblastoma cells were activated with 2.5 μM Aβ1–42 after 48 h of Rheb-Myc and anti-miR transfection. The data were normalized against GAPDH mRNA level. For statistical significance, a minimum of three independent experiments were considered in each case unless otherwise mentioned, and error bars represent means ± SEM. p values were calculated with a Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.0001. Scale bar represents 10μm in panel A and C.
Rheb-Myc expression has also increased the fraction of IL-1β and IL-6 mRNA bound to Ago2 in Aβ1–42-treated cells, confirming the availability of active miRNPs for repression of target messages (Figure 5J). miR-146 and miR-155 are the two major miRNAs reported as primary controllers of cytokine expression, and by trying the inactivation of both of these miRNAs with inhibitory antisense oligomers, we can expect to find the reduced response of Rheb-Myc expression in Aβ1–42-treated cells. We found a drop in the IL-1β level when Aβ1–42-exposed cells expressed Rheb-Myc (Figure 5F). Now, we wanted to check whether Rheb-Myc-mediated rescue in the cytokine mRNA repression depends on the repressive activity of the miRNAs. In the experiment shown in Figure 5K, all of the C6 glioblastoma cells were treated with Aβ1–42. Cells expressing control vector, control anti-miR-122, and exposed to 2.5 μM Aβ1–42 were taken as the control set. Cellular cytokine mRNA levels of the control set were taken as 1, and the rest of the experimental samples were plotted against it. Supporting the notion that the response of Rheb-Myc expression on cytokine expression is through reactivation of miR-155 and miR-146 in Aβ1–42-exposed cells, we have noted no significant reduction in cytokine production upon Rheb-Myc expression when the same cells were transfected with anti–miR-146 and anti-miR-155 oligomers in combination (Figure 5K). This is expected as, due to the lack of functional miR-146 and miR-155, the cytokines become non-responsive to mTOR activation, and thus mTOR must have an affect primarily through these miRNAs on expression of the cytokines in glial cells. These results suggest that activation of mTOR signaling causes miRNP activity restoration in Aβ1–42-treated glial cells.
Rescue of cognitive function in Aβ1–42 oligomer-infused adult rat brain upon Rheb-Myc expression
Inactivation of the mTORC1 pathway has been reported in AD brain.32,49 In the in vitro cell culture, we have documented restoration of mTORC1 activation to rescue cytokine repression by miRNPs. We checked whether overexpression of Rheb-Myc in an Aβ1–42 oligomer-induced AD model of the disease can result in reduction of pro-inflammatory cytokine expression to cause improvement in cognitive function of treated animal against the control group of animals. It has been shown before that Aβ1–42 oligomer infusion in the brain resulted in neuronal death and deposition of Aβ plaque in the vicinity of the infusion site with defective cognitive functions.39 To check the effect of Rheb-Myc on Aβ1–42-induced neuroinflammation and cognitive functions, we injected Aβ1–42 oligomers along with the Rheb-Myc expression cassette or control vector bilaterally in the hippocampal area of the brain of adult rats and kept them for 15 days. Following this infusion, several memory function tests were performed to assess cognitive functions in the experimental group of animals (Figure 6A).
Figure 6.

Rescue of cognitive function in Aβ1–42-infused adult rat brain upon Rheb-Myc expression
(A) Experimental flowchart shows a bilateral infusion of Aβ1–42 oligomers and Rheb-Myc plasmid that was done in adult rat hippocampus. After 15 days of incubation, different behavioral assays were carried out followed by sacrificing the animals. (B–D) Rescue of wait latency in Aβ1–42-infused adult rat brain upon Rheb-Myc expression. Passive avoidance assessment of memory performance was done, and the graph shows the acquisition process of experimental subjects. The rats that were able to enter the dark chamber within 300 s were selected as eligible for testing. The bar diagram reveals the wait latency in different experimental sets of rats. Rats having Rheb-Myc plasmid infused along with Aβ1–42 showed higher wait latency on the probing day, as compared to the animals having only Aβ1–42 infusion (D) (n = 7). (E–H) Novel object recognition test to show that the short-term memory recognition capability has been restored by Rheb-Myc expression in Aβ1–42-injected animals. Aβ1–42 and Rheb-Myc expression plasmid co-infused animals showed improvement in novel object identification capacity as compared to the Aβ1–42-infused rodents (F). The Rheb-Myc-Aβ1–42 co-infused animals also showed an increased preferential index and discrimination index as compared to the Aβ1–42-infused group of animals, indicating improvement in learning and memory function (G and H) (n = 7). (I and J) The elevated plus maze analysis showed improvement in cognitive ability in Rheb-Myc-expressing animals. The retention transfer latency (RTL) was measured in all control and Aβ1–42-infused groups, and the difference of initial transfer latency (ITL) and RTL was plotted along with the ITL and RTL separately (n = 7). For statistical significance, a minimum of three independent experiments were considered in each case unless otherwise mentioned. Animal data are represented as means ± SEM and analyzed by one-way ANOVA followed by Bonferroni’s test. p value <0.05 corresponds to ∗. In EPM data in panel J, # stands for significance between all the RTL data between all the animal groups, ∗ denotes significance between (ITL-RTL) data between all the animal groups.
We performed the passive avoidance test, which is used to understand learning and memory defects of rodents. This test showed that intra-hippocampal Aβ1–42 infusion significantly reduced wait latency compared to that in the control group of animals. Wait latency represents the time each animal spends in the light chamber before entering into the dark chamber where they experience a shock upon entering (experimental details are described in Materials and methods) during the 300-s period following 5-min habituation (Figure 6B). The probe test was performed 24 h after the training test. Compared to the control group, injection of Aβ1–42 markedly reduced the retention of fear memory, as they showed lower wait latency on the probe test as they moved to a dark chamber within a few seconds. However, rats having Rheb-Myc plasmid infused in the hippocampus along with Aβ1–42 oligomer showed higher wait latency on the probe stage as compared to the animals having only Aβ1–42 infused (Figures 6C and 6D). After behavioral studies, the animals were sacrificed. Brain tissues were fixed, and immunohistochemistry was done to check the expression of both Rheb-Myc and ZsGreen (ZsGreen-encoding plasmid was co-injected with Rheb-Myc encoding plasmid) at the infusion site. We found colocalization between both ZsGreen and Rheb-Myc at the infection site, confirming their expression (Figures S4A and S4B). This result indicates that Rheb-Myc could reverse the declining cognitive function in Aβ1–42-infused rats.
Next, we performed the novel object recognition test and observed that Aβ1–42-infused animals had no preference toward the novel object as compared to control group. In contrast, this short-term memory recognition capability in Aβ1–42 co-infused animals was significantly improved upon Rheb-Myc expression. The Rheb-Myc-Aβ-infused animals also showed an increased preferential index and discrimination index as compared to those in the Aβ1–42-infused group of animals (Figures 6E–6H). Additionally, the elevated plus maze (EPM) analysis showed improvement in cognitive ability in the above-mentioned group of animals. In Figure 6I, latency was the measure of the time taken by the animal to move from the open to the closed arm in the EPM. In the Aβ-infused group, retention transfer latency (RTL) was higher than initial transfer latency (ITL), which was significantly reduced in other groups. The difference between ITL and RTL (ITL − RTL) was used to assess memory and learning in the EPM test. Notably, a negative difference was obtained in the Aβ1–42-infused animals (Figure 6I). However, Aβ1–42-infused rats injected with Rheb-Myc plasmid showed a positive difference between ITL and RTL compared to the Aβ-infused group (Figures 6I and 6J).
The immunohistochemistry of Aβ1–42-infused adult rat brains showed astrogliosis, depicting an increase in GFAP in astrocytes, but in Rheb-Myc plasmid and Aβ co-infused rat brain, enhanced GFAP was found to decrease as compared to the Aβ1–42 oligomer-infused rat brain. However, no decrease in Aβ accumulation was found in Rheb-expressed, Aβ1–42-infused brains, suggesting that the effect of Rheb-Myc was on glial cell response and cytokine production rather than an effect on Aβ deposition (Figures 7A and 7B). These results indicate that ectopic expression of Rheb protein in Aβ1–42-infused rat brain causes improvement in cognitive behavior and spatial memory. While checking the differential expression status of cytokine mRNA levels in the infused rat brains, an almost 2-fold decrease in proinflammatory IL-6 and IL-1β was observed in the Rheb-Myc-Aβ1–42 co-infused rats as compared to the Aβ1–42-only infused rats (Figures 7C and 7D). Rheb-Myc-expressed animals showed a decrease in both the cellular and Ago2-associated miR-146a levels compared to the only Aβ1–42-injected animal tissue (Figures 7E and 7F). Therefore, by activating mTOR and miRNP, recycling the cytokine expression could be controlled to rescue the memory loss and the related phenotype in the AD model by expressing Rheb (Figure 7G).
Figure 7.

Rescue of cytokine level in Aβ1–42-infused adult rat brain upon Rheb-Myc expression
(A and B) Histology sections of Aβ1–42-infused and control rat brains. Cryosections that were subjected to IHC are shown. Confocal images of the sections of Aβ1–42-infused adult rat brains show astrogliosis depicted by an increase in GFAP-positive astrocytes. This was reversed in Rheb-Myc-Aβ-infused rat brain, where enhanced GFAP was found to decrease as compared to the Aβ1–42-infused rat brain (A). The graph depicting the intensity of GFAP in rat hippocampus is shown in (B). (C and D) Expression of IL-1β (C) and IL-6 mRNA (D) levels in rat hippocampal tissue samples upon Rheb-Myc expression. The data were normalized against GAPDH mRNA level. (E) Cellular miR-146a levels in the infused rat brains. The values were obtained by qRT-PCR and normalized against the U6 snRNA level. (F) Levels of Ago2-associated miR-146a are restored by Rheb-Myc in Aβ1–42-infused rat hippocampus tissue. Immunoprecipitation was done for endogenous Ago2, and associated miRNA was measured with qRT-PCR. The value was normalized against Ago2 band intensity as measured from the western blot. (G) The model depicts how Aβ exposure causes mTOR entrapment with endosomes restricting miRNP target RNA interaction. This causes inactivation of miRNPs to elevate proinflammatory cytokine production in glial cells. Interestingly, ectopic activation of mTOR by Rheb restores miRNP function in diseased brain, rescuing the cognitive function in adult rat brain. The mechanism of action of Aβ1–42 on miRNA via mTOR is shown in the right panel. For statistical significance, a minimum of three independent experiments were considered in each case unless otherwise mentioned, and error bars represent means ± SEM. p values were calculated with a Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.0001.
Discussion
Several miRNAs are expressed in brain and thought to regulate thousands of transcripts within the brain.3 In the present analysis, we could identify deregulated miRNA and mRNA within the cortical regions of AD patients, which may be associated with AD development or progression. Interestingly, in our analysis, a substantial percentage of upregulated miRNAs had a higher fraction of their corresponding target mRNAs as upregulated. This trend was consistently observed in late-stage AD cases considered from multiple studies. These observations suggest that contrary to the general regulatory relationship between miRNAs and their target mRNAs wherein miRNAs mainly downregulate mRNAs, other regulator-to-target relationships might also exist in tissues under disease conditions. In particular, miR-146a-5p is one such miRNA that exhibited this phenomenon wherein increased production of miR-146a-5p resulted in the upregulation of multiple target mRNAs.
Therefore, any change in the activity of the miRNA pathway could play a significant role during development or during any pathological condition. Also, the molecular mechanism behind Aβ-mediated neuronal death is still elusive, and an interplay between cellular structure, molecular biological processes, and the resulting immune response of the disease could contribute to the reason why AD is so difficult to treat. Another important aspect was to look at the involvement of glial cells in the buildup and disease progression, as glial cells easily outnumber neurons in the brain.50 Thus, in the early stage of the disease, glial cells might be involved in rectifying the problems of the neurons due to Aβ1–42 oligomer deposition, but they eventually contribute to the prognosis of the disease.
We found that mTOR sequesters into early endosome with exposure to Aβ1–42 in glial cells. mTORC1 is a very important protein complex that can regulate neuronal differentiation and brain activity. Depletion of an essential mTORC1 component, Raptor, is known to reduce the brain size considerably.51 mTORC1 signaling can also play a role in phosphorylation of Ago2 protein, which eventually can affect the miRNP function. Rheb, which is a GTPase and an activator of mTORC1, can reverse this process and results in a decrease in Ago2 miRNP content. mTORC1 activity-driven pathways may regulate the miRNA maturation process as well. The tuberous sclerosis complex (TSC) is an autosomal-dominant disorder and is caused by mutations in TSC1 or TSC2 that are connected with mTOR hyperactivity. TSC knockout cells, which have increased mTORC1 activity, have a reduced mature miRNA level.52 Accordingly, mTOR activation by Rheb can bring down the maturation process and also recycle existing miRNPs, and both of these pathways may contribute to the miRNA activity rescue process. Rheb knockout animals are embryonic lethal.53,54 Also, conditional Rheb knockout adult mice have a limited lifespan, indicating that Rheb is an essential protein factor that could play a major role in brain development and function. In the present study, by injecting Rheb-expressing plasmid into animal brain, we could improve motor function as well as memory of the AD animals. Similarly, researchers have shown that Rheb expression in brain of HD mouse model resulted in improved motor function.55 Also in PD, Rheb expression in dopaminergic neurons promoted axonal regrowth.56 Therefore, Rheb can affect brain function at multiple points. For example, mTOR-mediated translation, a Rheb-dependent process, is important at synapse for maintaining synaptic plasticity and at the cell body for maintaining memory.57,58 Also, Rheb expression can increase the acetyl choline level in the brain that can improve brain cognitive functions.59 Although all of these factors may play a role, our data suggest that miRNA activity regained by Rheb expression may be the most important factor in the recovery of behavioral, cognitive, and memory function in AD.
We have stereotactically injected Aβ1–42 oligomers in adult rat brain to produce amyloid plaques in the brain. This model has several advantages, as we can monitor the early cellular events during plaque formation and the effects it has on the miRNA machinery during the early phase of the disease. Also, oligomers can be taken up by glial cells in both the in vivo and in vitro cell culture system, which can have additional physiological effects on the cellular structure and machinery. This allows the limited and spatiotemporal-specific exposure of brain cells to Aβ1–42 and thus is advantageous over the transgenic mouse models that have a mutated Aβ gene (APP) or related protease gene, which may have some compensatory effect on both structure and physiology in the adult brain. Additionally, the transgene is expressed at a very early stage and in all of the cells of the brain. Importantly, the number of AD cases reported with genetic mutations in APP gene is very limited compared to the higher number of cases noted with sporadic Aβ expression.60 We have also injected Rheb expression vector with Aβ1–42 oligomer in brain hippocampus. This is to ensure only that a portion of the brain that is exposed to Aβ1–42 oligomer only takes up the Rheb-expressing plasmid and not the entire brain, which could complicate the analysis. This method can be useful in introducing specific miRNA or antisense RNA in a specific part of the brain with the help of co-injected Aβ1–42 oligomer.
Although we have only observed the effect that Aβ1–42 oligomer has on miRNA machinery on astroglial cells, it would be interesting to see what effect it may have on the miRNAs in other glial cells. Glial cells outnumber neurons at a 10:1 ratio, and among them almost 40% of cells are astrocytes, making them one of the most abundant cells of the brain. Also, Aβ1–42 oligomer-injected brain tissues showed considerable astrocyte activation along Aβ deposition. Primary astrocytes show elevated cytokine mRNA levels when exposed to Aβ1–42 oligomer. We did find some changes in the neuronal miRNA expression level, but a drop in miRNA activity resulting in cytokine overproduction was found to be specific for astroglial cells and not for neurons. In LPS-activated macrophages, miRNAs play a significant role in reducing pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α, IL-1β, or IL-6 so that anti-inflammatory IL-10 production can be initiated.19 This is a very important step in the inflammation process where miRNA fine-tunes the balance between pro-inflammatory and anti-inflammatory cytokine levels to maintain homeostasis of the process. The astrocytes exposed to Aβ1–42 oligomer show a loss in miRNA activity whereas loss of Ago2 phosphorylation leads to building up inactive miRNPs in astrocytes that restrict their encounter with the target mRNA. This leads to uncontrolled production of pro-inflammatory cytokines that eventually create neuroinflammation, leading to the loss of neurons.
If the mechanism is happening through Ago2 phosphorylation, then this mechanism should affect other miRNAs as well. We found that the patterns of data of miR-16 and miR-29a are very similar to what we also got for miR-146a and miR-155. Since miR-16 and miR-29a do not directly regulate expression of pro-inflammatory cytokines, we think that there is a possibility that this mechanism affects the functions of other miRNAs as well. In this context, we note that in primary astrocytes and rat brain (also in AD patient brain data) we have documented increased expression of many miRNAs that are not even astrocyte specific or not a known regulator of cytokines. This also signifies that the common pathway of miRNA activity modulation by mTORC1 has been targeted by Aβ1–42 oligomers. However, there are miRNAs that may be regulated at the transcriptional level in a significant way, while there could be a subset of miRNAs that also have regulated export and turnover in Aβ1–42-treated cells, and thus these factors may contribute significantly to the overall cellular levels of those miRNAs to show a reduction in the overall expression level.
The identification of important miRNAs in the gene regulation process in AD brain was initially a challenge, and thus we studied tissue-specific miRNA expression patterns in different parts of the brain tissue, with the help of TissueAtlas.61 Interestingly, in comparison to basal levels, miR-146a expression in the cortical part is preferentially enriched in AD brain. Subsequently, we wanted to focus on miRNAs specifically associated with the effect of Aβ oligomers in neural cell subsets. Correspondingly, with the help of a previous study, we determined that miRNA expression patterns can vary substantially across cell types, such as neurons and astrocytes. In particular, miR-146a and miR-21 are highly enriched in astrocytes in comparison to neurons in primary cultures from the rat cortex.62 Furthermore, based on our previous computational analysis and the miRNA distribution patterns in different human brain regions we selected miRNAs for our further analyses.
How does astrocyte activation affect neuronal health? We observed that rat primary astrocytes cultured in the in vitro condition secrete extra-cellular vesicles 70–100 nm in size known as the exosomes. These exosomes carry miRNAs and different RNA-binding proteins from astrocytes to recipient neuronal cells (D.D, unpublished data). Functional assays carried out in recipient cells showed that these exported miRNAs are stable and functional in neuronal cells, where they can regulate the gene expression of recipient cells. Exposure to Aβ1–42 increases miR-146a secretion through exosomes along with other RNA-binding proteins such as Ago2 and GW182 (D.D, unpublished data). We think that these exosomes could play an important role in both normal physiology and in pathological conditions such as AD, where astrocytes can regulate gene expression of neighboring neuronal cells and through these also fine-tune complex physiological processes such as learning and memory formation.
Several reports suggest the role of astroglia in improving cognition and memory by demonstrating that the reduction in their activity leads to memory impairment.63 Interestingly, human astrocyte grafting in immunodeficient mice improved memory in these cognition-deficient animals.64 Studies have also featured the necessity of astrocyte activation in maintaining synaptic plasticity and causing N-methyl-d-aspartate (NMDA)-mediated long-term potentiation and memory development in the hippocampus.65
Is astroglia the primary contributor in the high cytokine profile observed in AD? In the AD condition, Aβ is known to cause disruption in gliotransmission by elevating the calcium signaling levels in astrocytes.66 Besides during AD, interrupted glutamate uptake capacity and astrocytic calcium signaling can be disrupted by Aβ as well.67,68 Also, hippocampal astrocytes exposed to Aβ helped in improving the frequency of NMDA receptor-mediated slow inward currents, along with calcium elevations.69 Precisely, due to this wide array of functional properties, we thought of astrocytes as an interesting target for studying brain pathologies such as AD, where miRNA dysregulation in astrocytes may play a significant role in both neuroinflammation and cognition of the animal.
Postmortem neuropathological studies have shown that the number of reactive astrocytes in the vicinity of amyloid plaques increases as the disease advances.70 We also found astrogliosis at the amyloid deposition site in adult rat brain hippocampus (Figure 2B). Interestingly, microglial cells are also found in amyloid deposition sites along with astrocytes, and in APP/PS1: GFAP/vimentin double knockout mice where astrocyte activation is attenuated had increased numbers of microglial cells found around plaques.70 Also, microglia secrete inflammatory cytokines, which can convert the neuroprotective resting astrocytes into neurotoxic cells. Conversely, astrocytes secrete C3 complement fraction via NF-κB in oligomeric Aβ treatment, and C3 can in turn activate microglia through its C3R receptor.71 Studies also reported that astrocytes appear to influence the degree of microglial reactivity in mouse models of brain β-amyloidosis.72
Our findings suggest that restoring mTOR activity in the glial cells can rescue the disease phenotype and also cognition and memory in adult animals. Previous studies in the field of neurodegenerative diseases suggest maintenance of homeostasis of mTOR activity could be beneficial for disease management. For example, in the case of autism, inactivation of mTOR is found to be beneficial, as in autism patient hyperactivation of mTOR is common. Again, in ALS, where mTOR activity is reduced, restoring the activity is found to be advantageous in disease prognosis.73,74 In a nutshell, our study emphasizes the complex circuit between metabolic pathways, with gene expression regulation having an effect on disease pathology. An intervention with gene therapy or with small molecules to sustain normal miRNA metabolic pathways in brain will have a therapeutic value in a complex disease such as AD.
Materials and methods
Expression profile analysis of miRNAs and their corresponding target mRNAs in AD and cancer
Expression profiles of miRNAs and mRNAs were utilized to study the possible regulatory relationships between miRNAs and their corresponding target mRNAs in multiple cortical regions of AD patients as opposed to normal deceased individuals. With the help of R packages (R Core Team, 2017),75 differentially expressed (fold change of ±1.25 and p value of ≤0.05) miRNAs within the pre-frontal cortex and mRNA within different regions of the frontal cortex were determined in the brains of AD patients. The differentially expressed miRNAs and mRNAs identified in this manner considering patient samples are likely to be associated with the development or progression of AD (GEO: GSE48552, GSE5281, GSE53697, and GSE15222).35,76, 77, 78, 79, 80 Utilizing miRNA-target regulatory information from TarBase and miRTarBase databases, regulator(s)-to-target(s)” (miRNA/mRNA) maps for the differentially expressed miRNAs and mRNAs were determined.81,82 Subsequently, we have compared the fractions of target mRNAs of each differentially expressed miRNA that are significantly upregulated or downregulated. Additionally, to ascertain whether such regulator(s)-to-target(s) patterns could be occurring in other disease conditions, we performed a disease enrichment analysis considering only the genes that were commonly upregulated among the AD datasets considered.83
In order to study regulator(s)-to-target(s) patterns in cancer, we have analyzed miRNA and mRNA expression profiling data from different cancer tissue samples. With the help of R packages, differentially expressed (fold change of ±1.25 and p value of ≤0.05) miRNAs and mRNAs were determined in breast cancer, colorectal cancer, and oral squamous cell carcinoma by considering relevant GEO datasets (GEO: GSE40056/GSE40057, GSE35982, and GSE70664/GSE70665).75,84, 85, 86 Subsequently, the fractions of significantly upregulated or downregulated target mRNAs of each of the differentially expressed miRNAs under each condition were compared.
siRNA and plasmid constructs
Plasmid information about pRL-con, pRL-3XBulge-let7a, pRL-3XBulge-miR-122, and firefly (FF) plasmids were previously explained in Pillai et al. and were a kind gift from Witold Filipowicz. miR-122 was expressed from pmiR-122 plasmid, which was previously described.87 FLAG-hemagglutinin (HA)-Ago2 and FLAG-HA-Ago2-Y529F were obtained as a kind gift from Gunter Meister. The plasmids Rab5-CA and Rheb-Myc were previously used and were a kind gift from J.M. Backer.88 All small interfering RNAs (siRNAs) were purchased from Dharmacon (ON-TARGETplus siRNA).
Cell culture and reagents
HEK293 and C6 glioblastoma cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 2 mM l-glutamine and 10% heat-inactivated fetal bovine serum (FBS).
Primary astrocytes were cultured from 0-1 day-old Sprague-Dawley rat pups. The whole brain was dissected out and meninges were removed carefully. The neocortex parts were isolated and cut into pieces. The small cortical tissue pieces were minced and subjected to trypsinization for 30 min. Trypsinized brain tissue was triturated in DMEM (Gibco) medium supplemented with 10% FBS (Gibco) and passed through nylon mesh to avoid clumps. This single-cell suspension was then added onto a poly-d-lysine (PDL)-coated plate (Sigma-Aldrich, St. Louis, MO, USA) and incubated for 2–3 min for preferential sticking of neurons. Next, the unattached cells were collected and harvested by centrifugation. Cells were resuspended in a fresh medium and seeded at a density of 1.2 million/35-mm plate or 0.4 million/well of a 24-well plate. Cells were maintained for 13 days in vitro (DIV) with a medium change given every alternate day.
Plasmid and siRNAs were transfected with Lipofectamine 2000 (Invitrogen) and RNAiMAX (Invitrogen), respectively, following the manufacturer’s protocol. All of the siRNA and plasmid co-transfection was done using Lipofectamine 2000. For activation of primary rat astrocytes or neurons 1.5 μM Aβ1–42 was used, whereas 2.5 μM Aβ1–42 was used for activating C6 glioblastoma cells. 100 nM rapamycin (Sigma) was used for blocking mTORC1.
Preparation of Aβ
High-pressure liquid chromatography (HPLC)-purified Aβ1–42 was purchased from American Peptide (Sunnyvale, CA, USA), and oligomeric Aβ1–42 was prepared as described previously.39 Briefly, lyophilized Aβ1–42 was reconstituted in 100% 1,1,1,3,3,3 hexafluoro-2-propanol (HFIP) to 1 mM. HFIP was removed by evaporation in a SpeedVac (Eppendorf, Hamburg, Germany), then resuspended in 5 mM anhydrous DMSO. This stock was then stored at 80°C. The stock was diluted with PBS to a final concentration of 400 mM, and SDS was added to a final concentration of 0.2%. The resulting solution was incubated at 37°C for 18–24 h. The preparation was diluted again with PBS to a final concentration of 100 mM and incubated at 37°C for 18–24 h before use.
Brain stereotaxic surgery
Adult male Sprague Dawley rats weighing 270g-330g were kept in a room controlled around 25°C and humidity 65%, providing food and water to ad-libitum. A 12h light-dark cycle was provided in the animal house of CSIR-Indian Institute of Chemical Biology, Kolkata. All the studies were performed with guidelines provided by the Committee for the Purpose of Control and Supervision of Experiments on Animals (Animal Welfare Divisions, Ministry of Environments and Forests, Govt. of India) along with the approval from the Institutional Animal Ethics Committee (IAEC). Male Sprague-Dawley rats (280–320 g) were anesthetized by injecting 50 mg/kg thiopentone (thiosol sodium, Neon Laboratories, Mumbai, India) and placing them on a stereotaxic frame (Stoelting, USA). Injection was done by using a 27G Hamilton syringe. A volume of 5 μL of 100 μM Aβ in PBS and 3 μg (4 μL) of plasmid was infused at a flow rate of 0.5 μL/min in the hippocampus at stereotaxic coordinates from bregma (AP, 3.6 mm; L, 2.1 mm; DV, 2.8 mm) according to the rat brain atlas. An equal volume of PBS was injected in control animals. Proper postoperative care was taken to maintain proper health conditions of the animal. Behavioral tests were performed 14 days after stereotaxic surgery of the animal. Animals were sacrificed on 20th days after behavioral analysis. The brains were dissected out, following cardiac perfusion, and fixed in 4% paraformaldehyde (PFA) for 24 h. They were then incubated in a 30% sucrose solution for another 24 h before proceeding for cryosectioning. Cryosections of the brain were done in a cryotome (Thermo Shandon, Pittsburgh, PA, USA).
Immunohistochemistry of brain slices
20-μm coronal cryosections of the brain from Aβ-infused or PBS-infused rats were blocked with 4% BSA in PBS containing 0.43% Triton X-100 for 1 h 20 min at room temperature. Brain slices were incubated in primary antibody in a blocking solution overnight at 4°C. Sections were washed three times with PBST and incubated with a fluorescence-tagged secondary antibody for 2 h at room temperature. Following three washes with PBS and Hoechst staining for the nucleus, the sections were mounted with Dibutylphthalate Polysterene Xylene (DPX) and observed in a confocal microscope.
RNA isolation and qPCR
Total RNA was isolated by using TRIzol or TRIzol LS reagent (Invitrogen) according to the manufacturer’s protocol. miRNA assays by real-time PCR were performed using specific TaqMan primers (Invitrogen). U6 small nuclear RNA (snRNA) was used as an endogenous control. Real-time analyses by two-step RT-PCR were performed for quantification of miRNA levels on a Bio-Rad CFX96 real-time system using an Applied Biosystems TaqMan chemistry-based miRNA assay system. One third of the reverse transcription mix was subjected to PCR amplification with TaqMan universal PCR master mix, no AmpErase (Applied Biosystems) and the respective TaqMan reagents for target miRNAs. Samples were analyzed in triplicates. The comparative Ct method, which included normalization by the U6 snRNA, was used for relative quantification. For quantification of mRNA levels, 200 ng of total cellular RNA was subjected to cDNA preparation followed by qPCR by the SYBR Green method. Each sample was analyzed in triplicates using the comparative Ct method. mRNA levels were normalized with GAPDH as the loading control.
IP assay
For IP of Ago2, cells were either transfected with FLAG-HA-tagged Ago2 or endogenous Ago2 and were pulled down from primary cells and tissue samples. For IP reactions, cells were lysed in lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM KCl, 5 mM MgCl2, 1 mM DTT, 10 mM sodium orthovanadate, 15 mM NaF), 0.5% Triton X-100, 0.5% sodium deoxycholate, and 1× EDTA-free protease inhibitor cocktail (Roche) for 30 min at 4°C, followed by three sonication pulses of 10 s each. The lysates, clarified by centrifugation, were incubated with HA antibody (at a concentration of 1:100) pre-bound protein G agarose beads (Invitrogen) or with pre-blocked anti-FLAG M2 beads and rotated overnight at 4°C. Subsequently, the beads were washed three times with IP buffer (20 mM Tris-HCl [pH 7.5], 150 mM KCl, 5 mM MgCl2, 1 mM DTT, 10 mM sodium orthovanadate,15 mM NaF). Washed beads were divided into two equal parts, and each part was analyzed for bound proteins and RNAs by western blot and qPCR, respectively.
Luciferase assay
miRNA repression was observed by performing a dual-luciferase assay. 10 ng of RL-con and RL-3xB-let-7a, RL-3xB-122, or RL-IRAK1 encoding plasmids was co-transfected with 100 ng of firefly encoding plasmid per well of a 24-well plate to study endogenous let-7a or exogenous miR-122 repression. Cells were lysed with 1× passive lysis buffer (PLB, Promega) before being subjected to a dual-luciferase assay (Promega, Madison, WI, USA) following the supplier’s protocol on a VICTOR X3 plate reader with injector (PerkinElmer, Waltham, MA, USA). RL expression levels for control and reporter were normalized with the firefly expression level for each reaction. All samples were done in triplicates.
Immunoblotting
SDS-polyacrylamide gel electrophoresis was performed with the samples, which included cell lysates, membrane fractions, and IP proteins, followed by transfer of the same to polyvinylidene fluoride (PVDF) nylon membranes. Specific required antibodies were used to probe the blot for at least 16 h at 4°C. This antibody-associated overnight incubation was followed by three washes with TBST (Tris buffered saline with Tween 20), after which at room temperature the membranes were incubated for 1 h with horseradish peroxidase-conjugated secondary antibodies (1:8,000 dilutions). Images of developed western blots were taken using an UVP BioImager 600 system equipped with VisionWorks Life Science software (UVP) v6.80.
Immunofluorescence and confocal imaging
For immunofluorescence studies, primary and C6 glioblastoma cells were grown on 18-mm coverslips coated with PDL and gelatin, respectively. Cells were transfected on the coverslips as discussed previously and fixed for 30 min in the dark with 4% PFA solution after 48 h of transfection. Cells were blocked and permeabilized with 3% BSA containing 0.1% Triton X-100 for 30 min, followed by overnight incubation with specific primary antibodies at 4°C. After the incubation, cells were washed three times with PBS and probed with respective secondary antibodies (Life Technologies) attached with specific fluorophores for 1 h at room temperature. For detection of lysosomes, 100 nM of LysoTracker Red DND-99 was added in the growing cells for 1 h before fixation. Images were captured using a Zeiss LSM800 confocal microscope and analyzed with Imaris7 software. All of the interactions between organelles were measured by calculating Pearson’s and Mander’s coefficient of colocalization using the coloc plug-in of Imaris7 software.
Passive avoidance test
First, the rats were subjected to passive avoidance (shuttle box) training. Rodents have a natural tendency to move toward dark. The passive avoidance device had two identical compartments comprising 25 × 25 × 20 cm, which were divided by a guillotine door. One of the chambers was lighted and the other was dark. Electric shocks were conveyed to the grid floor by an isolated stimulator. At the beginning of the experiment rats were habituated in the device for 5 min in the lighted chamber facing away from the entrance to the dark side. On acquisition day the guillotine door was raised, and the latency to enter the dark chamber was recorded. When the animals did not enter the dark compartment within 5 min, they were eliminated from the experiment. Once they entered the dark chamber, they received a foot shock of 0.8 mA, which forced them to return to the light chamber. On the next day, defined as the probe day, the same rats were kept in the light chamber and time taken by them to enter the dark chamber was noted as the latency time. When the rats did not enter the dark chamber by 300 s, the successful acquisition of passive avoidance response was said to occur. The experimental work and data recording was performed by SHUTAVOID software by Panlab.
Novel object recognition (NOR)
The NOR task evaluates the rodents’ ability to recognize a novel object in the environment. The task procedure consists of three phases: habituation, familiarization, and test phase. In the habituation phase, each animal is allowed to freely explore the open-field arena in the absence of any objects. After the familiarization phase, a single animal is placed in the open-field arena containing two identical sample objects (A & A),for 5 min. After 24 h, the animal was returned to the open-field arena with two objects, one of which was identical to the sample and the other one was novel (A & B). The test was performed for 5 min. During both the familiarization and the test phase, objects were located in the opposite and symmetrical corners of the arena, and location of the novel versus familiar object was counterbalanced. The wait latency of the animals observed in each of the objects on probe day was calculated and designated as TN and TF for the novel and familiar objects, respectively. The discrimination index (DI) and preference index (PI) were derived according to the following formulae: DI = (TN − TF)/(TN + TF) and PI = [TN/(TN + TF)] × 100.
Elevated Plus Maze (EPM) test
The EPM was used for the assessment of memory processes in the animals on the 15th day following the method of that used in rats.89 The apparatus was made of a wooden plus-shaped maze elevated 60 cm from the floor having two open arms measuring 50 × 10 cm, crossed at right angles by two arms of the same dimensions enclosed by 50-cm-high walls. A 1-cm-high plywood edge surrounding the open arms was added to prevent the animals from falling off the maze. A rat was placed on the open arm facing away from the center and the transfer latency (TL; time in which the rat moves from the open arm to closed arms) was recorded on the day 1 and again on the following day. The time taken by the animal on day 1 to move from open arm to the closed arm was recorded as ITL, and that on day 2 was recorded as RTL. A duration of 5 min was used for each subject to record the data.
Statistical analysis
GraphPad Prism 5.00 (GraphPad, San Diego, CA, USA) was used to analyze the experiments performed in triplicate unless mentioned otherwise. The p values were obtained with the help of a non-parametric Student’s t test. Animal data are represented as means ± SEM and analyzed by one-way ANOVA followed by a Bonferroni’s test.
Data availability
Information related to antibodies, primers, and miRNA assays are available as Tables S3–S5.
Acknowledgments
We acknowledge Witold Filipowicz, Gunter Meister, and J.M. Backer for different plasmid constructs. S.N.B. is supported by The Swarnajayanti Fellowship (DST/SJF/LSA-03/2014-15) from the Department of Science and Technology, Government of India, while D.D., S.G., and I.M. received support from CSIR, India. This work was supported by funds from a High Risk High Reward Project Grant (HRR/2016/000093) from the Department of Science and Technology, Government of India, and by CEFIPRA fund 6003-J.
Author contributions
S.N.B. conceptualized the project, designed research, and analyzed data; D.D., I.M., S.G., and R.K.P. performed research and conducted animal experiments. S.N.B., D.D., and I.M., along with S.C.B. and S.C., analyzed data; and S.N.B., D.D., S.C.B., S.C., and I.M. wrote the paper.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.04.008.
Supplemental information
References
- 1.Bartel D.P. Metazoan microRNAs. Cell. 2018;173:20–51. doi: 10.1016/j.cell.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Filipowicz W., Bhattacharyya S.N., Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 3.Petri G., Expert P., Turkheimer F., Carhart-Harris R., Nutt D., Hellyer P.J., Vaccarino F. Homological scaffolds of brain functional networks. J. R. Soc. Interface. 2014;11:20140873. doi: 10.1098/rsif.2014.0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y., Wang F., Lee J.A., Gao F.B. MicroRNA-9a ensures the precise specification of sensory organ precursors in Drosophila. Genes Dev. 2006;20:2793–2805. doi: 10.1101/gad.1466306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruno I.G., Karam R., Huang L., Bhardwaj A., Lou C.H., Shum E.Y., Song H.W., Corbett M.A., Gifford W.D., Gecz J. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol. Cell. 2011;42:500–510. doi: 10.1016/j.molcel.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maciotta S., Meregalli M., Torrente Y. The involvement of microRNAs in neurodegenerative diseases. Front. Cell. Neurosci. 2013;7:265. doi: 10.3389/fncel.2013.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawase-Koga Y., Otaegi G., Sun T. Different timings of dicer deletion affect neurogenesis and gliogenesis in the developing mouse central nervous system. Dev. Dyn. 2009;238:2800–2812. doi: 10.1002/dvdy.22109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lukiw W.J., Zhao Y., Cui J.G. An NF-κB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 2008;283:31315–31322. doi: 10.1074/jbc.M805371200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sethi P., Lukiw W.J. Micro-RNA abundance and stability in human brain: Specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 2009;459:100–104. doi: 10.1016/j.neulet.2009.04.052. [DOI] [PubMed] [Google Scholar]
- 10.Molofsky A.V., Krencik R., Ullian E.M., Tsai H.H., Deneen B., Richardson W.D., Barres B.A., Rowitch D.H. Astrocytes and disease: A neurodevelopmental perspective. Genes Dev. 2012;26:891–907. doi: 10.1101/gad.188326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazzanti M., Sul J.Y., Haydon P.G. Glutamate on demand: Astrocytes as a ready source. Neuroscientist. 2001;7:396–405. doi: 10.1177/107385840100700509. [DOI] [PubMed] [Google Scholar]
- 12.Koehler R.C., Roman R.J., Harder D.R. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009;32:160–169. doi: 10.1016/j.tins.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Araque A., Parpura V., Sanzgiri R.P., Haydon P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- 14.Perea G., Navarrete M., Araque A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–431. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Cordiglieri C., Odoardi F., Zhang B., Nebel M., Kawakami N., Klinkert W.E., Lodygin D., Lühder F., Breunig E., Schild D. Nicotinic acid adenine dinucleotide phosphate-mediated calcium signalling in effector T cells regulates autoimmunity of the central nervous system. Brain. 2010;133:1930–1943. doi: 10.1093/brain/awq135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinney J.W., Bemiller S.M., Murtishaw A.S., Leisgang A.M., Salazar A.M., Lamb B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. (N. Y.) 2018;4:575–590. doi: 10.1016/j.trci.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Neill L.A., Sheedy F.J., McCoy C.E. MicroRNAs: The fine-tuners of Toll-like receptor signalling. Nat. Rev. Immunol. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- 18.Goswami A., Mukherjee K., Mazumder A., Ganguly S., Mukherjee I., Chakrabarti S., Roy S., Sundar S., Chattopadhyay K., Bhattacharyya S.N. MicroRNA exporter HuR clears the internalized pathogens by promoting pro-inflammatory response in infected macrophages. EMBO Mol. Med. 2020;12:e11011. doi: 10.15252/emmm.201911011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazumder A., Bose M., Chakraborty A., Chakrabarti S., Bhattacharyya S.N. A transient reversal of miRNA-mediated repression controls macrophage activation. EMBO Rep. 2013;14:1008–1016. doi: 10.1038/embor.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fabian M.R., Sonenberg N., Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 21.Friedman R.C., Farh K.K., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dueck A., Ziegler C., Eichner A., Berezikov E., Meister G. MicroRNAs associated with the different human Argonaute proteins. Nucleic Acids Res. 2012;40:9850–9862. doi: 10.1093/nar/gks705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu C.G., Calin G.A., Meloon B., Gamliel N., Sevignani C., Ferracin M., Dumitru C.D., Shimizu M., Zupo S., Dono M. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc. Natl. Acad. Sci. USA. 2004;101:9740–9744. doi: 10.1073/pnas.0403293101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qi H.H., Ongusaha P.P., Myllyharju J., Cheng D., Pakkanen O., Shi Y., Lee S.W., Peng J., Shi Y. Prolyl 4-hydroxylation regulates Argonaute 2 stability. Nature. 2008;455:421–424. doi: 10.1038/nature07186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirino Y., Mourelatos Z. Mouse Piwi-interacting RNAs are 2′-O-methylated at their 3′ termini. Nat. Struct. Mol. Biol. 2007;14:347–348. doi: 10.1038/nsmb1218. [DOI] [PubMed] [Google Scholar]
- 26.Patranabis S., Bhattacharyya S.N. Phosphorylation of Ago2 and subsequent inactivation of let-7a RNP-specific microRNAs control differentiation of mammalian sympathetic neurons. Mol. Cell. Biol. 2016;36:1260–1271. doi: 10.1128/MCB.00054-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng D., Ezzeddine N., Chen C.Y., Zhu W., He X., Shyu A.B. Deadenylation is prerequisite for P-body formation and mRNA decay in mammalian cells. J. Cell Biol. 2008;182:89–101. doi: 10.1083/jcb.200801196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rüdel S., Meister G. Phosphorylation of Argonaute proteins: Regulating gene regulators. Biochem. J. 2008;413:e7–e9. doi: 10.1042/BJ20081244. [DOI] [PubMed] [Google Scholar]
- 29.Ma X.M., Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 30.Cybulski N., Hall M.N. TOR complex 2: A signaling pathway of its own. Trends Biochem. Sci. 2009;34:620–627. doi: 10.1016/j.tibs.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 31.Casadio A., Martin K.C., Giustetto M., Zhu H., Chen M., Bartsch D., Bailey C.H., Kandel E.R. A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell. 1999;99:221–237. doi: 10.1016/s0092-8674(00)81653-0. [DOI] [PubMed] [Google Scholar]
- 32.lland T.K., Song W.M., Huang S.C., Ulrich J.D., Sergushichev A., Beatty W.L., Loboda A.A., Zhou Y., Cairns N.J., Kambal A. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell. 2017;170:649–663.e13. doi: 10.1016/j.cell.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saito K., Araki Y., Kontani K., Nishina H., Katada T. Novel role of the small GTPase Rheb: Its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J. Biochem. 2005;137:423–430. doi: 10.1093/jb/mvi046. [DOI] [PubMed] [Google Scholar]
- 34.Sancak Y., Peterson T.R., Shaul Y.D., Lindquist R.A., Thoreen C.C., Bar-Peled L., Sabatini D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lau P., Bossers K., Janky R., Salta E., Frigerio C.S., Barbash S., Rothman R., Sierksma A.S., Thathiah A., Greenberg D. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol. Med. 2013;5:1613–1634. doi: 10.1002/emmm.201201974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tahamtan A., Teymoori-Rad M., Nakstad B., Salimi V. Anti-inflammatory microRNAs and their potential for inflammatory diseases treatment. Front. Immunol. 2018;9:1377. doi: 10.3389/fimmu.2018.01377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasudevan S. Posttranscriptional upregulation by microRNAs. Wiley Interdiscip. Rev. RNA. 2012;3:311–330. doi: 10.1002/wrna.121. [DOI] [PubMed] [Google Scholar]
- 39.Sanphui P., Biswas S.C. FoxO3a is activated and executes neuron death via Bim in response to β-amyloid. Cell Death Dis. 2013;4:e625. doi: 10.1038/cddis.2013.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frost G.R., Li Y.M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017;7:170228. doi: 10.1098/rsob.170228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iliopoulos D., Hirsch H.A., Struhl K. An epigenetic switch involving NF-κB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boldin M.P., Taganov K.D., Rao D.S., Yang L., Zhao J.L., Kalwani M., Garcia-Flores Y., Luong M., Devrekanli A., Xu J. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011;208:1189–1201. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsang C.K., Qi H., Liu L.F., Zheng X.F. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov. Today. 2007;12:112–124. doi: 10.1016/j.drudis.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 44.Sabatini D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA. 2017;114:11818–11825. doi: 10.1073/pnas.1716173114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hay N., Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 46.Flinn R.J., Backer J.M. mTORC1 signals from late endosomes: Taking a TOR of the endocytic system. Cell Cycle. 2010;9:1869–1870. doi: 10.4161/cc.9.10.11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Long X., Lin Y., Ortiz-Vega S., Yonezawa K., Avruch J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 48.Long X., Ortiz-Vega S., Lin Y., Avruch J. Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J. Biol. Chem. 2005;280:23433–23436. doi: 10.1074/jbc.C500169200. [DOI] [PubMed] [Google Scholar]
- 49.Lafay-Chebassier C., Paccalin M., Page G., Barc-Pain S., Perault-Pochat M.C., Gil R., Pradier L., Hugon J. mTOR/p70S6k signalling alteration by Aβ exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem. 2005;94:215–225. doi: 10.1111/j.1471-4159.2005.03187.x. [DOI] [PubMed] [Google Scholar]
- 50.von Bartheld C.S., Bahney J., Herculano-Houzel S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016;524:3865–3895. doi: 10.1002/cne.24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cloëtta D., Thomanetz V., Baranek C., Lustenberger R.M., Lin S., Oliveri F., Atanasoski S., Rüegg M.A. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J. Neurosci. 2013;33:7799–7810. doi: 10.1523/JNEUROSCI.3294-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ye P., Liu Y., Chen C., Tang F., Wu Q., Wang X., Liu C.G., Liu X., Liu R., Liu Y., Zheng P. An mTORC1-Mdm2-Drosha axis for miRNA biogenesis in response to glucose- and amino acid-deprivation. Mol. Cell. 2015;57:708–720. doi: 10.1016/j.molcel.2014.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goorden S.M., Hoogeveen-Westerveld M., Cheng C., van Woerden G.M., Mozaffari M., Post L., Duckers H.J., Nellist M., Elgersma Y. Rheb is essential for murine development. Mol. Cell. Biol. 2011;31:1672–1678. doi: 10.1128/MCB.00985-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goorden S.M., Abs E., Bruinsma C.F., Riemslagh F.W., van Woerden G.M., Elgersma Y. Intact neuronal function in Rheb1 mutant mice: Implications for TORC1-based treatments. Hum. Mol. Genet. 2015;24:3390–3398. doi: 10.1093/hmg/ddv087. [DOI] [PubMed] [Google Scholar]
- 55.Lee J.H., Tecedor L., Chen Y.H., Monteys A.M., Sowada M.J., Thompson L.M., Davidson B.L., Ruegg M.A. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron. 2015;85:303–315. doi: 10.1016/j.neuron.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim S.R., Kareva T., Yarygina O., Kholodilov N., Burke R.E. AAV transduction of dopamine neurons with constitutively active Rheb protects from neurodegeneration and mediates axon regrowth. Mol. Ther. 2012;20:275–286. doi: 10.1038/mt.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swiech L., Perycz M., Malik A., Jaworski J. Role of mTOR in physiology and pathology of the nervous system. Biochim. Biophys. Acta. 2008;1784:116–132. doi: 10.1016/j.bbapap.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 58.Gkogkas C., Sonenberg N., Costa-Mattioli M. Translational control mechanisms in long-lasting synaptic plasticity and memory. J. Biol. Chem. 2010;285:31913–31917. doi: 10.1074/jbc.R110.154476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jeon M.T., Nam J.H., Shin W.H., Leem E., Jeong K.H., Jung U.J., Bae Y.S., Jin Y.H., Kholodilov N., Burke R.E. In vivo AAV1 transduction with hRheb(S16H) protects hippocampal neurons by BDNF production. Mol. Ther. 2015;23:445–455. doi: 10.1038/mt.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barykin E.P., Mitkevich V.A., Kozin S.A., Makarov A.A. Amyloid β modification: A key to the sporadic Alzheimer’s disease? Front. Genet. 2017;8:58. doi: 10.3389/fgene.2017.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ludwig N., Leidinger P., Becker K., Backes C., Fehlmann T., Pallasch C., Rheinheimer S., Meder B., Stähler C., Meese E., Keller A. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016;44:3865–3877. doi: 10.1093/nar/gkw116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jovičić A., Roshan R., Moisoi N., Pradervand S., Moser R., Pillai B., Luthi-Carter R. Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J. Neurosci. 2013;33:5127–5137. doi: 10.1523/JNEUROSCI.0600-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao V., Suzuki A., Magistretti P.J., Lengacher S., Pollonini G., Steinman M.Q., Alberini C.M. Astrocytic β2-adrenergic receptors mediate hippocampal long-term memory consolidation. Proc. Natl. Acad. Sci. USA. 2016;113:8526–8531. doi: 10.1073/pnas.1605063113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han X., Chen M., Wang F., Windrem M., Wang S., Shanz S., Xu Q., Oberheim N.A., Bekar L., Betstadt S. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell. 2013;12:342–353. doi: 10.1016/j.stem.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adamsky A., Kol A., Kreisel T., Doron A., Ozeri-Engelhard N., Melcer T., Refaeli R., Horn H., Regev L., Groysman M. Astrocytic activation generates de novo neuronal potentiation and memory enhancement. Cell. 2018;174:59–71.e14. doi: 10.1016/j.cell.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 66.Lee L., Kosuri P., Arancio O. Picomolar amyloid-β peptides enhance spontaneous astrocyte calcium transients. J. Alzheimers Dis. 2014;38:49–62. doi: 10.3233/JAD-130740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vincent A.J., Gasperini R., Foa L., Small D.H. Astrocytes in Alzheimer’s disease: Emerging roles in calcium dysregulation and synaptic plasticity. J. Alzheimers Dis. 2010;22:699–714. doi: 10.3233/JAD-2010-101089. [DOI] [PubMed] [Google Scholar]
- 68.Matos M., Augusto E., Machado N.J., dos Santos-Rodrigues A., Cunha R.A., Agostinho P. Astrocytic adenosine A2A receptors control the amyloid-β peptide-induced decrease of glutamate uptake. J. Alzheimers Dis. 2012;31:555–567. doi: 10.3233/JAD-2012-120469. [DOI] [PubMed] [Google Scholar]
- 69.Pirttimaki T.M., Codadu N.K., Awni A., Pratik P., Nagel D.A., Hill E.J., Dineley K.T., Parri H.R. α7 Nicotinic receptor-mediated astrocytic gliotransmitter release: Aβ effects in a preclinical Alzheimer’s mouse model. PLoS ONE. 2013;8:e81828. doi: 10.1371/journal.pone.0081828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vehmas A.K., Kawas C.H., Stewart W.F., Troncoso J.C. Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol. Aging. 2003;24:321–331. doi: 10.1016/s0197-4580(02)00090-8. [DOI] [PubMed] [Google Scholar]
- 71.Lian H., Litvinchuk A., Chiang A.C., Aithmitti N., Jankowsky J.L., Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J. Neurosci. 2016;36:577–589. doi: 10.1523/JNEUROSCI.2117-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rodriguez G.A., Tai L.M., LaDu M.J., Rebeck G.W. Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J. Neuroinflammation. 2014;11:111. doi: 10.1186/1742-2094-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsai P.T., Hull C., Chu Y., Greene-Colozzi E., Sadowski A.R., Leech J.M., Steinberg J., Crawley J.N., Regehr W.G., Sahin M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488:647–651. doi: 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saxena S., Roselli F., Singh K., Leptien K., Julien J.P., Gros-Louis F., Caroni P. Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron. 2013;80:80–96. doi: 10.1016/j.neuron.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 75.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liang W.S., Reiman E.M., Valla J., Dunckley T., Beach T.G., Grover A., Niedzielko T.L., Schneider L.E., Mastroeni D., Caselli R. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA. 2008;105:4441–4446. doi: 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liang W.S., Dunckley T., Beach T.G., Grover A., Mastroeni D., Walker D.G., Caselli R.J., Kukull W.A., McKeel D., Morris J.C. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol. Genomics. 2007;28:311–322. doi: 10.1152/physiolgenomics.00208.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Readhead B., Haure-Mirande J.-V., Funk C.C., Richards M.A., Shannon P., Haroutunian V., Sano M., Liang W.S., Beckmann N.D., Price N.D. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron. 2018;99:64–82.e7. doi: 10.1016/j.neuron.2018.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scheckel C., Drapeau E., Frias M.A., Park C.Y., Fak J., Zucker-Scharff I., Kou Y., Haroutunian V., Ma’ayan A., Buxbaum J.D., Darnell R.B. Regulatory consequences of neuronal ELAV-like protein binding to coding and non-coding RNAs in human brain. Elife. 2016;5:e10421. doi: 10.7554/eLife.10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Webster J.A., Gibbs J.R., Clarke J., Ray M., Zhang W., Holmans P., Rohrer K., Zhao A., Marlowe L., Kaleem M., NACC-Neuropathology Group Genetic control of human brain transcript expression in Alzheimer disease. Am. J. Hum. Genet. 2009;84:445–458. doi: 10.1016/j.ajhg.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vlachos I.S., Paraskevopoulou M.D., Karagkouni D., Georgakilas G., Vergoulis T., Kanellos I., Anastasopoulos I.L., Maniou S., Karathanou K., Kalfakakou D. DIANA-TarBase v7.0: Indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015;43:D153–D159. doi: 10.1093/nar/gku1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chou C.H., Chang N.W., Shrestha S., Hsu S.D., Lin Y.L., Lee W.H., Yang C.D., Hong H.C., Wei T.Y., Tu S.J. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;44(D1):D239–D247. doi: 10.1093/nar/gkv1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu Y., Ye R.D. Microglial Aβ receptors in Alzheimer’s disease. Cell. Mol. Neurobiol. 2015;35:71–83. doi: 10.1007/s10571-014-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Luo D., Wilson J.M., Harvel N., Liu J., Pei L., Huang S., Hawthorn L., Shi H. A systematic evaluation of miRNA:mRNA interactions involved in the migration and invasion of breast cancer cells. J. Transl. Med. 2013;11:57. doi: 10.1186/1479-5876-11-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fu J., Tang W., Du P., Wang G., Chen W., Li J., Zhu Y., Gao J., Cui L. Identifying microRNA-mRNA regulatory network in colorectal cancer by a combination of expression profile and bioinformatics analysis. BMC Syst. Biol. 2012;6:68. doi: 10.1186/1752-0509-6-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shi W., Yang J., Li S., Shan X., Liu X., Hua H., Zhao C., Feng Z., Cai Z., Zhang L., Zhou D. Potential involvement of miR-375 in the premalignant progression of oral squamous cell carcinoma mediated via transcription factor KLF5. Oncotarget. 2015;6:40172–40185. doi: 10.18632/oncotarget.5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chang J., Nicolas E., Marks D., Sander C., Lerro A., Buendia M.A., Xu C., Mason W.S., Moloshok T., Bort R. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106–113. doi: 10.4161/rna.1.2.1066. [DOI] [PubMed] [Google Scholar]
- 88.Flinn R.J., Yan Y., Goswami S., Parker P.J., Backer J.M. The late endosome is essential for mTORC1 signaling. Mol. Biol. Cell. 2010;21:833–841. doi: 10.1091/mbc.E09-09-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Itoh J., Nabeshima T., Kameyama T. Utility of an elevated plus-maze for the evaluation of memory in mice: Effects of nootropics, scopolamine and electroconvulsive shock. Psychopharmacology (Berl.) 1990;101:27–33. doi: 10.1007/BF02253713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Information related to antibodies, primers, and miRNA assays are available as Tables S3–S5.