

Summary
MNase-seq (micrococcal nuclease sequencing) is used to map nucleosome positions in eukaryotic genomes to study the relationship between chromatin structure and DNA-dependent processes. Current protocols require at least two days to isolate nucleosome-protected DNA fragments. We have developed a streamlined protocol for S. cerevisiae and other fungi which takes only three hours. Modified protocols were developed for wild fungi and mammalian cells. This method for rapidly producing sequencing-ready nucleosome footprints from several organisms makes MNase-seq faster and easier, with less chemical waste.
Subject areas: Genomics, Sequencing, Model Organisms, Molecular Biology
Graphical abstract
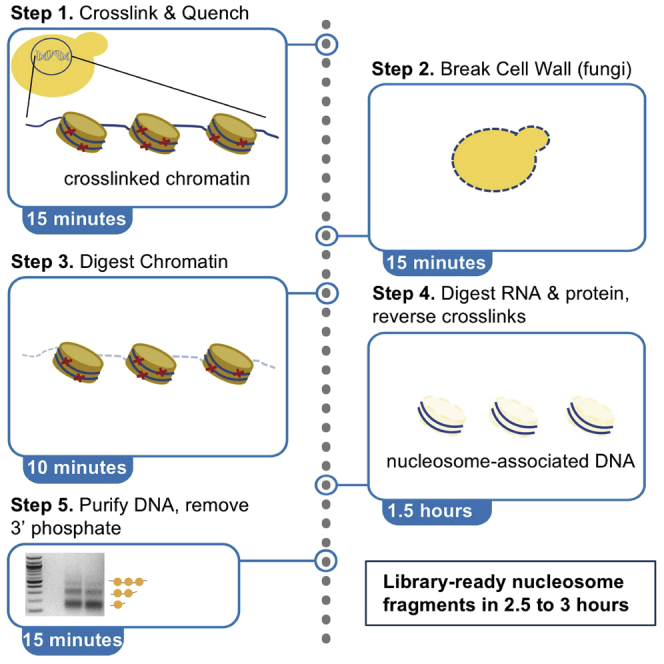
Highlights
-
•
A fast way to prepare micrococcal nuclease nucleosome footprints for MNase-seq
-
•
Eliminates use of phenol and chloroform and reduces the amount of cells required
-
•
Adaptable for a variety of organisms
MNase-seq is used to map nucleosome positions in eukaryotic genomes to study the relationship between chromatin structure and DNA-dependent processes. Current protocols require at least two days to isolate nucleosome-protected DNA fragments. We have developed a streamlined protocol for S. cerevisiae and other fungi which takes only three hours. Modified protocols were developed for wild fungi and mammalian cells. This method for rapidly producing sequencing-ready nucleosome footprints from several organisms makes MNase-seq faster and easier, with less chemical waste.
Before you begin
Timing: [0.5–2 h]
Here, we describe the protocol for use in budding yeast in liquid culture, which was optimized from a previously published protocol (Rodriguez et al, 2014). We have also adapted this streamlined protocol for use with S. cerevisiae growing on a plate, S. cerevisiae quiescent cells, S. pombe, N. crassa, wild mushrooms, C. elegans, D. rerio fin cells, and mammalian cells. The key changes to adapt the protocol to other cell stages or organisms are noted in the relevant step of the protocol.
-
1.If needed, prepare buffers (see materials and equipment for recipes).
-
a.The following stock solutions can be prepared ahead of time and stored for an extended period at 25°C: 2M sorbitol, 1M Tris pH 7.5, 2.5M glycine, STOP buffer.
-
b.Stock solutions of Proteinase K (20 mg/mL) and RNase A (10 mg/mL) can be prepared ahead of time and stored for an extended time at −20°C.
-
c.MNase (20 units/μL) and MNase digestion buffer can be aliquoted and stored long-term at −80°C. We have observed that MNase aliquots are stable for at least a year, but have not tested longer storage.
-
a.
-
2.
Ensure that the formaldehyde being used is less than 3 months old.
-
3.
2 days before the start of the experiment (day -2), streak the strain(s) of interest onto appropriate selection plates and incubate at 30°C. Sick strains, or strains grown in synthetic medium, may take longer to grow.
Note: If isolating quiescent (Q) cells, you will need an additional 3 days before starting the experiment, so cells need to be streaked out at least a week prior.
-
4.
The day before the experiment (day -1), start an overnight culture of yeast in YPD or other appropriate media; grow at 30°C with shaking for 18–20 h.
Note: If isolating Q cells, start an overnight culture on day -4 or earlier, as the culture must grow for at least 3 days to reach quiescence.
Note: If working from yeast on a plate, instead of an overnight culture, make a large patch (1cm × 3 cm) on a plate with appropriate selection if needed; sick strains may take up to 48 h to grow.
Preparing cell pellets
-
5.
On Day 0 (or Day 1, if proceeding directly to crosslinking) dilute an overnight culture of yeast to an OD600 = 0.2–0.3 in 25 mL of YPD, or other appropriate media. Grow at 30°C with shaking, to an OD600 = 0.8–1.
Note: For more slowly growing strains, a larger volume may be used and grown to a lower OD600; ensure at least one doubling takes place before pelleting. The goal is to have 20 OD∗mL of cells (i.e., 20ml of OD600 = 1, or 25ml of OD600 = 0.8).
-
6.
Pellet 20 OD∗mL of cells in a 50-mL conical tube at 3000×g for 5 min.
Note: See Table 2 for information on input of different cell types.
Table 2.
Comparison of parameters for preparation of mnase samples from different organisms
| Organism | Input | Spheroplasting (min) | Zymolyase (mg/mL) | MNase (μL of 20 U/μL) |
|---|---|---|---|---|
| S. cerevisiae liquid culture | 20 OD∗mL | 15 | 2 | 3 |
| S. cerevisiae agar plate | 30 OD∗mL | 30 | 2 | 1.5 |
| S. cerevisiae Q cells |
100 OD∗mL of dense fraction | 60 | 10 | 0.2 |
| S. pombe liquid culture | 20 OD∗mL | 15 | 2 | 3 |
| N. crassa | 50-mL culturea | 15 | 2 | 3 |
| Wild mushrooms | 50 mg of fruiting body | 15 | 2 | 3 |
| Mammalian cells | 106 cells | N/A | N/A | 3 |
| C. elegans | 3∗104 adult worms | N/A | N/A | 3 |
| D. rerio fin cells | Fins from 50 adult zebrafishb | N/A | N/A | 3 |
50-mL culture inoculated with 2.5∗107 cells harvested from freshly grown conidia, grown until at least 70% of conidia have germinated
treated with trypsin and liberase
-
7.
Carefully remove supernatant. Resuspend pellet in 1 mL of deionized water and transfer to a 1.5-mL microcentrifuge tube.
Optional: If time permits, you can proceed directly to the crosslinking protocol at this point instead of pelleting, flash freezing and storing the cells ahead of time; there is a Pause Point after crosslinking.
-
8.
Pellet at 20,000 × g for 1 minute in a microcentrifuge. Remove supernatant.
-
9.
Flash freeze the pellet in liquid nitrogen and store it at −80°C. Samples are stable for at least 2 or 3 months at −80°C; we have not tested longer storage times.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 37% Formaldehyde | Sigma-Aldrich | Cat# 252549 |
| Zymolyase (R) 100T | AMS Bio | Cat# 120493-1 |
| Nuclease, micrococcal | Worthington Biochemical | Cat# LS004798 |
| Exonuclease III | New England Biolabs | Cat# M0206S |
| RNase A | Fisher BioReagents | Cat# BP25391 |
| Proteinase K | Invitrogen | Cat# AM2544 |
| Quick CIP (calf intestinal phosphatase) | New England Biolabs | Cat# M0525L |
| Critical commercial assays | ||
| MinElute PCR Purification Kit | QIAGEN | Cat# 28006 |
| Ovation Ultralow System V2 | NuGEN | Cat# 0344NB-32 |
| Quant-IT PicoGreen dsDNA Assay Kit | Invitrogen | Cat# P7589 |
| Deposited data | ||
| Nucleosome positioning data (MNase-seq) from S. cerevisiae (exponentially growing, quiescent, solid patches), N. crassa, and S. pombe | This study | GEO: GSE141676 |
| Nucleosome positioning data (MNase-seq) from S. cerevisiae W303 | Donovan et al., 2019 | SRA: SRX5086833 |
| Nucleosome positioning data (MNase-seq) from isw2 S. cerevisiae | McKnight et al., 2016 | SRA: SRX1176421 |
| Nucleosome positioning data (MNase-seq) from S. pombe | Steglich et al., 2015 | SRA: SRX554384 |
| Nucleosome positioning data (MNase-seq) from S. pombe | DeGennaro et al., 2013 | SRA: SRX331943 |
| Nucleosome positioning data (MNase-seq) from N. crassa | Seymour et al., 2016 | SRA: SRX1596291 |
| Nucleosome positioning data (MNase-seq) from N. crassa | Klocko et al., 2019 | SRA: SRX2822417 |
| Experimental models: organisms/strains | ||
| S. cerevisiae W303-1A | Laboratory of Rodney Rothstein | ATCC: 208352 |
| Software and algorithms | ||
| Bowtie 2 | Langmead and Salzberg, 2012 | RRID:SCR_005476 |
| Integrated Genome Browser | Freese et al., 2016 | RRID:SCR_011792 |
Materials and equipment
2M Sorbitol
| Reagent | Final concentration | Add to 500 mL |
|---|---|---|
| Sorbitol | 2 M | 182.2 g |
| Deionized water | Bring to 500 mL |
After dissolving sorbitol in solution, sterile filter and store indefinitely at 25°C. Check periodically for contamination.
1M Tris pH 7.5
| Reagent | Final concentration | Add to 500 mL |
|---|---|---|
| Tris base | 1 M | 60.5 g |
| Deionized water | Bring to 500 mL |
After dissolving Tris base in ~300 mL of water, adjust the pH to 7.5 with concentrated HCl. Then bring the volume to 500 mL, and store indefinitely at 25°C.
2.5M Glycine
| Reagent | Final concentration | Add to 500 mL |
|---|---|---|
| Glycine | 2.5 M | 93.8 g |
| Deionized water | Bring to 500 mL |
After dissolving glycine in solution, sterile filter and store indefinitely at 25°C. Check periodically for contamination.
STOP buffer
| Reagent | Final concentration | Stock concentration | Add to 10 mL |
|---|---|---|---|
| EDTA pH 8.0 | 50 mM | 0.5 M | 1 mL |
| EGTA pH 8.0 | 50 mM | 0.5 M | 1 mL |
STOP Buffer can be stored indefinitely at 25°C.
MNase digestion buffer
| Reagent | Final concentration | Stock concentration | Add to 100 mL |
|---|---|---|---|
| Sorbitol | 1 M | 2 M | 50 mL |
| NaCl | 50 mM | 5 M | 1 mL |
| Tris pH 7.5 | 10 mM | 1 M | 1 mL |
| MgCl2 | 5 mM | 2 M | 250 μL |
| CaCl2 | 1 mM | 1 M | 100 μL |
| NP-40 (or Igepal) | 0.075% | 10% v/v | 740 μL |
| Spermidine | 0.5 mM | 250 mM | 200 μL |
| Beta-mercaptoethanol | 1 mM | 1.43 M (1:10 dilution) | 70 μL |
| Deionized water | Bring to 100 mL total |
MNase Digestion Buffer can be made ahead of time, aliquoted, and stored at −80°C indefinitely. We recommend 500 μL aliquots.
Spheroplast buffer
| Reagent | Final concentration | Stock concentration | Add to 10 mL |
|---|---|---|---|
| Sorbitol | 1 M | 2 M | 5 mL |
| Tris pH 7.5 | 50 mM | 1 M | |
| Beta-mercaptoethanol | 5 mM | 1.43 M (1:10 dilution) | 35 μL |
| Zymolyase | 2 mg/mL | N/A | 20 mg |
| Deionized water | Bring to 10 mL total |
Spheroplast buffer must be made fresh before use; 1 mL of buffer is needed per sample. Be sure to precisely measure the desired amount of zymolyase for accuracy – it may be easier to weigh the zymolyase first and adjust the total volume of buffer if necessary to get a final concentration of 2 mg/mL of zymolyase.
Step-by-step method details
Prepare cross-linked cells
In this step, formaldehyde is used to capture interactions between DNA and nucleosomes. The crosslinking reaction is then quenched by glycine. For yeast, cells are generally crosslinked with formaldehyde to maintain nucleosome positions throughout the subsequent steps, though it has been debated whether these crosslinks can efficiently trap nucleosome positions without introducing artifacts (Henikoff et al., 2011; Cole et al., 2012). It may be preferable to crosslink cells on the same day as growth, though we have achieved good results with the method below. Due to natural variation in cells, we recommend performing the experiment on three biological replicates.
MNase-seq of mammalian cells does not require crosslinking. We performed experiments on the cell line PLB-895 (Tucker et al., 1987) both with and without crosslinking and there was no noticeable difference in visualization of nucleosome footprints (Figure 6), though we recommend doing a side-by-side comparison when working with a new cell type for the first time, if possible.
-
1.
Thaw MNase digestion buffer on ice; you will need 100 μL per sample plus another 10 μL per 10 samples to dilute the ExoIII.
-
2.
Resuspend cell pellet in 1 mL of deionized water.
Note: Crosslinking is optional for mammalian cells; if skipping this step, proceed directly to step 8
-
3.Add 27 μL of 37% formaldehyde to a final concentration of 1%. Rotate end-over-end for 15 min at 25°C.
-
a.While the cells are cross-linking, prepare spheroplast buffer; you will need 1 mL per sample with 2 mg zymolyase per mL.
-
a.
-
4.
To quench the crosslinking reaction, add 50 μL of 2.5M glycine for a final concentration of 125 mM.
-
5.
Pellet at 20,000 × g for 1 minute in a microcentrifuge and remove supernatant carefully.
Pause point: At this point, cells can be flash frozen in liquid nitrogen and stored at −80°C. Samples are stable for at least 2 or 3 months at −80°C; we have not tested longer storage times.
Figure 6.

The rapid MNase protocol can be performed on human cells with or without crosslinking
(A) Cartoon schematic showing rapid MNase protocol and associated nucleosome footprints for 1 million human cells from the diploid myeloid leukemia cell line PLB-895. The protocol is identical to the yeast liquid culture protocol except that there is no zymolyase treatment.
(B) Cartoon schematic showing rapid MNase protocol for PLB-895 cells and associated nucleosome footprints when the crosslinking and crosslinking reversal steps are omitted and other steps are shortened. The crosslink-free protocol can provide nucleosome footprints that are ready for library construction in less than an hour. Left lane: 1.5 million cells input; right lane: 1 million cells input.
Make spheroplasts and digest chromatin
In this step, the cell walls of fungi are broken and nuclei are permeabilized to allow micrococcal nuclease (MNase) to access and digest extranucleosomal DNA. Permeabilization is required for fungi but not for mammalian cells, C. elegans, or D. rerio; for non-fungal samples proceed directly to step 8. [Troubleshooting Problem 1:]
After permeabilization, accessible regions of genomic DNA are digested by MNase, and the ends of the resulting nucleosome fragments are trimmed by Exonuclease III to reduce variation in size, which enables consistent, high-resolution identification of the nucleosome dyad at the center of the core particle. [Troubleshooting Problem 2:]
Cellular RNA is removed, formaldehyde crosslinks are reversed, and protein is digested prior to DNA purification. Finally, the residual 3’ phosphates (from MNase cleavage activity) are removed prior to genomic library construction.
-
6.Resuspend the pellet of crosslinked cells (fresh or previously frozen) in 1 mL spheroplast buffer plus 2 mg zymolyase to permeabilize the cell walls. Rotate at 25°C for 15 min.
-
a.While the sample incubates, dilute ExoIII 1:10 (from 100 U/μL stock) in MNase digestion buffer; you will need 3 μL of diluted ExoIII per sample. Keep on ice.
-
b.Thaw MNase (3 μL per sample), RNase (3 μL per sample), and Proteinase K (10 μL per sample) on ice.
-
a.
Note: For yeast taken directly from a plate, increase spheroplasting incubation time to 30 min. For purified Q cells, spheroplast for 60 min and increase the amount of zymolyase to 10 mg.
-
7.
Pulse-pellet (15 seconds at 20,000 × g); carefully remove supernatant.
-
8.
Add 100 μL digestion buffer + 3 μL MNase (20 U/μL; 60U total) + 3 μL ExoIII (10 U/μL). Incubate at 37°C for 10 min to digest chromatin.
Note: For yeast from a plate, use 1.5μl MNase. For Q cells, use 0.2μl MNase and incubate for 2–10 min (see troubleshooting 3 for more information on optimizing this protocol for Q cells).
CRITICAL: Exact timing of digestion across samples is essential for consistency. To achieve an identical start time for digest, we recommend pipetting the MNase into the open lid of the microcentrifuge tube, and then briefly spinning all the tubes down so that the enzyme reaches each sample at precisely the same time.
-
9.
Stop digestion with 12.5 μL STOP buffer.
Note: We have eliminated SDS from the STOP buffer and switched the order of the Proteinase K and RNAse A steps in order to streamline the protocol. Traditionally, SDS was included in the STOP buffer and then Proteinase K was added (Rodriguez et al, 2014); both of these components are used to break down protein in the sample, but they also inactivate RNAse, necessitating an additional purification step before proceeding to the RNAse step. In our version, EDTA and EGTA are sufficient to halt crosslinking, and no extra purification is needed.
-
10.
Add 3 μL RNase A (10 mg/mL stock). Incubate at 42°C for 30 min.
-
11.
Add 12.5 μL 10% SDS + 10 μL Proteinase K (20 mg/mL stock) to digest protein and reverse crosslinks. Incubate at 65°C for 45 min.
Note: For uncrosslinked mammalian cells, the incubation time is reduced to 15 min
-
12.
Add 700 μL Buffer PB (from Qiagen kit). Purify using a single Qiagen MinElute PCR column. Follow protocol instructions, with the exception of the elution step. Elute with 12 μL 1× CutSmart buffer (NEB; diluted from 10× stock)
Note: For wild mushrooms, after adding Buffer PB centrifuge the sample for 2 min at 20,000 ×g to further clear remaining cellular debris. Apply the supernatant to the MinElute column.
-
13.
Add 1 μL Quick CIP. Incubate for 10 min at 37°C.
-
14.
Add 7 μL Orange Loading Dye (NEB) and run the entire sample on a 2% agarose gel.
Note: Using a 7cm × 10cm or 15cm × 10cm TAE-agarose gel in TAE, it takes approximately 40 min at 100V to get proper separation of bands
-
15.
Cut out the mononucleosome band, gel extract with a Qiagen MinElute column. Follow protocol instructions, with the exception of the elution step. Elute with 12 μL EB.
-
16.
Quantify with PicoGreen or other preferred method; store at −80°C. DNA yield typically ranges from 10-50 ng total. This sample is now ready for preparing libraries; we use 5 ng of input with the Ovation Ultralow Library System V2 (from NuGEN), though we have successfully sequenced libraries prepared from as little as 0.5 ng input. There are a variety of options for preparation of libraries, and this protocol can likely feed into whatever pipeline you are currently using. We have not tested how long samples are stable at −80°C, but we usually prepare library samples within a month.
Expected outcomes
The first step toward validating the nucleosome footprints obtained using this protocol is assessing the quality of the digests on agarose gel (Step 14). Figure 1 shows an example of well-digested DNA, where bands are evident for the tri-, di-, and mono-nucleosomal fragments. Some variation is acceptable, but over- or under-digested DNA may yield poor quality data (see troubleshooting, Problem 2).
Figure 1.

Sample gel showing properly digested nucleosome footprints
Agarose gel showing nucleosome footprints recovered from liquid culture of S. cerevisiae using the rapid MNase protocol. (A) and (B) are replicates from the same WT yeast strain.
After preparing libraries from the S. cerevisiae samples obtained using our new rapid protocol and a previously published protocol (Rodriguez et al., 2014), we sequenced them and analyzed the data using our standard protocol (McKnight and Tsukiyama 2015; Donovan et al., 2019). We compared our data to previously published datasets (Donovan et al., 2019; McKnight et al., 2016) for wild type and isw2 yeast (Figure 2). We chose Isw2-deficient yeast because Isw2 is required for nucleosome shifts at specific target loci, which allows us to determine if our protocol can recapitulate Isw2-specific nucleosome positions at transcription start sites. For all samples, nucleosome organization at transcription start sites (TSSs) displayed the stereotypical structure, with a nucleosome-depleted region flanked by packed nucleosome arrays (Figure 2A). Comparison of nucleosome positions at Isw2 targets showed that nucleosomes were detected at strain-specific but not protocol-specific locations (Figure 2B). Both the rapid protocol and the standard protocol recovered strain-specific nucleosome positioning at Ume6 binding sites, a known Isw2-recruitment protein (Goldmark et al., 2000) across the genome (Figures 2C and 2D).
Figure 2.

Rapid MNase can accurately map nucleosome positions in S. cerevisiae cells
(A) Nucleosome dyad signal at 4655 yeast transcription start sites (TSSs) are plotted for WT and isw2 nucleosomes harvested using a standard or rapid protocol.
(B) Example Genome Browser image showing the standard method and rapid method can map Isw2-directed nucleosome positions similar to published data sets at the RAD51 locus. Dashed lines indicate Isw2-positioned nucleosomes.
(C) Nucleosome dyad signal at 202 intergenic Ume6 target sites showing rapid and standard MNase methods can accurately identify global changes in nucleosome structure at an Isw2 recruitment motif.
(D) Genome Browser image showing all methods correctly identify Isw2-positioned nucleosomes at the MEI4-ACA1 locus, a Ume6 target site. Dashed lines indicate Ume6- and Isw2- positioned nucleosomes.
We also applied the rapid MNase protocol to purified quiescent cells (Allen et al., 2006), or Q cells. As previously observed, quiescent yeast required longer spheroplasting with an increased amount of zymolyase (McKnight et al., 2015) to compensate for a highly fortified cell wall (Li et al., 2015). We also tested different amounts of time for the MNase digest (Figure 3A), as we found our Q cell protocol required more adjustments to get acceptable footprints, which were not entirely consistent across samples of Q cells prepared on different days. We hypothesize that this is because isolation of Q cells involves several steps and therefore produces more heterogeneous cell samples. For this reason we recommend testing different MNase digest times and/or amounts of MNase when processing Q cells to obtain properly-digested nucleosomes. We found that we could also reproducibly recover nucleosome footprints from patches of yeast grown on agar plates, or “colony MNase” (Figure 3B) and the captured nucleosome positions accurately reflect nucleosome positioning across the yeast genome (Figures 3C and 3D). Similar to liquid culture, the rapid colony MNase protocol can also detect Isw2-specific nucleosome events genome-wide (Figures 3E and 3F). It is important to note that colony MNase is not appropriate for all types of experiments, and researchers should consider whether stationary or log-phase cells are more appropriate for their needs.
Figure 3.

Rapid MNase can recover nucleosome footprints from isolated quiescent cells and yeast patches
(A) Representative gel showing nucleosome footprints recovered from purified quiescent cells using the rapid MNase protocol.
(B) Representative gel (right) showing nucleosome footprints recovered from a fresh patch of yeast collected from YPD-Agar (left).
(C) Nucleosome dyad signal at transcription start sites (TSSs) comparing standard and patch- recovered “colony” MNase footprints.
(D) Example Genome Browser image showing “colony” MNase footprints can accurately identify Isw2-directed nucleosome positions at the ESC8 locus compared to the standard MNase protocol and published data sets.
(E) Nucleosome dyad signal at 202 intergenic Ume6 target sites showing “colony” MNase can accurately identify global changes at an Isw2 recruitment motif.
(F) Genome Browser image showing colony MNase can similarly identify Isw2-positioned nucleosomes at the YIG1-CSM4 locus, an example Ume6 target site. Dashed lines indicate Ume6- and Isw2-positioned nucleosomes.
Without any protocol modifications, we were able to recover a well-defined and appropriately digested nucleosome ladder from wild type S. pombe cells (Figure 4A). We compared our sequencing data set with previously published MNase-seq data sets from S. pombe (DeGennaro et al., 2013; Steglich et al., 2015). Genomic nucleosome dyad positions from samples prepared by the rapid MNase protocol were the same as seen previously (Figure 4B). In addition, global nucleosome positioning at S. pombe TSSs was nearly identical across data sets (Figure 4C). We also performed the rapid MNase protocol on crosslinked, germinated N. crassa (Figure 4D). Importantly, we verified that the genomic nucleosome positions we obtained were similar to previously published N. crassa data sets (Seymour et al., 2016; Klocko et al., 2019) (Figure 4E).
Figure 4.

Rapid MNase can accurately map nucleosome positions in S. pombe and N. crassa
(A) Agarose gel showing example nucleosome footprints recovered from S. pombe using the rapid MNase protocol.
(B) Genome Browser image comparing nucleosome dyad positions on S. pombe chrII recovered for the rapid MNase protocol (top) and previously- published data sets.
(C) Alignment of nucleosome dyads at 11,350 transcription start sites (TSSs) for rapid MNase-recovered nucleosome footprints and previously-published data sets.
(D) Agarose gel showing example nucleosome footprints recovered from N. crassa using the rapid MNase protocol.
(E) Genome Browser image comparing nucleosome dyad positions at the N. crassa NCU3995-NCU3994 locus for the rapid MNase protocol (top) and previously-published data sets.
Table 1 summarizes some of the key differences between standard MNase protocols for various model fungi and our rapid protocol. In each instance, significantly less starting material is required for the rapid protocol, and in most cases the standard protocol uses phenol/chloroform extraction followed by ethanol precipitation for purification of the DNA; the use of a MinElute column shortens the time for purification considerably. The rapid protocol eliminates phenol and chloroform and also usually uses less formaldehyde, significantly decreasing the amount of hazardous reagents used overall and also reducing hazardous waste. Additionally, standard protocols for N. crassa require isolation of nuclei or a chromatin fraction, along with the use of protease inhibitors. The rapid protocol uses conidia without further preparation, greatly reducing the time and effort involved in preparing the starting material, and protease inhibitors are not required. Notably, one standard method for preparing MNase samples from N. crassa (Klocko et al, 2019) does not use formaldehyde, but we have observed that crosslinking improves the quality of the sequencing data and provides more distinct peaks for nucleosome positions (Figure 4E, top row versus bottom row). No standard method exists for MNase of S. cerevisiae or S. pombe from a patch of cells on a plate, so we could not make a comparison to our rapid “colony MNase”.
Table 1.
Comparison of sample input, reagents, and time required for standard versus rapid protocols for fungi
| Organism | Input | Formaldehyde per sample | DNA purification method | Days for protocola |
|---|---|---|---|---|
|
S. cerevisiae Standardb |
200 mL liquid culture | 5.5 mL | Phenol/chloroform extraction & ethanol precipitation | 3 |
|
S. cerevisiae Rapid |
20 OD∗mL pellet (from 25 mL culture) | 27 μL | MinElute column | 1.5 |
|
S. pombe Standardc |
100 mL liquid culture | 2.7 mL | Phenol/chloroform extraction & ethanol precipitation | 3 |
|
S. pombe Rapid |
20 OD∗mL pellet (from 25 mL culture) | 27 μL | MinElute column | 3 |
|
N. crassa Standard Ad |
Isolated nuclei from 500 mL conidia | None | MinElute column | 2 |
|
N. crassa Standard Be |
Isolated chromatin fraction from 50 mL conidia | 275 μL | Phenol/chloroform extraction & ethanol precipitation | 3 |
|
N. crassa Rapid |
50 mL conidia | 1.4 mL | MinElute column | 1 |
Number of days required to isolate mononucleosomal fragments from starting material (input)
We wished to design a rapid protocol that was standardized across species, so we performed experiments on a variety of locally foraged wild fungi and were able to obtain well-spaced nucleosome footprints (Figure 5). It is important to note that the size of the mononucleosome band is usually between 100 and 200 bp, but this product might run slightly larger in different organisms, as in lane 2 of Figure 5A. Because we did not sequence this sample, we cannot be absolutely certain that this band reflects mononucleosomal fragments, but we are reasonable sure. We also tested C. elegans, D. rerio fin cells, and the cell line PLB-895 (Tucker et al., 1987); while we do not have gel images for the C. elegans or D. rerio samples, we saw a prominent mononucleosome band for both of these organisms after a single test using the same protocol as for mammalian cells (with crosslinking), and we are confident that this protocol can be easily adapted for sequencing-quality nucleosome footprints. For PLB-895 cells, we performed the protocol with crosslinking in order to better compare it to our other samples (Figure 6A), but we also prepared samples without crosslinking as this is the more standard practice for MNase of mammalian cells (Figure 6B); there is no obvious difference visually. A 1-day MNase protocol already exists for mammalian cells (Ramani et al., 2019), but ours is significantly shorter and provides a promising standardized alternative to previously published protocols across organisms. We did not sequence these nucleosome fragments in the interest of time and cost, along with the lack of annotated genomes for wild fungi, but we encourage users to test the rapid protocol in their model organism of choice.
Figure 5.

Nucleosome footprints can be rapidly recovered from wild mushroom samples
(A) Images of locally-foraged wild mushrooms that were subjected to the rapid MNase protocol (top). Sample 5-6 consists of a distinct surface fungal specimen (5) growing on a host specimen (6). Recovered nucleosome footprints for corresponding mushrooms are shown (bottom). Speculative identities for these samples are (1) Panaeolus foenisceii, (2) unknown, (3) Craterellus tubaeformis, (4) Cantharellus formosus, (5) Hypomyces lactifluorum, (6) Russula brevines, (7) Lycoperdon perlatum.
(B) Optimized rapid MNase for specimen 4 (chanterelle) was achieved using 50 mg of tissue leading to well-digested nucleosome footprints (top). The optimized protocol was performed on 50 mg of a previously-untested specimen leading to well-digested nucleosome footprints (bottom). Speculative identity of these samples are Cantharellus formosus (top) and Agricus xanthodermus (bottom).
Quantification and statistical analysis
Libraries were sequenced at the University of Oregon’s Genomics and Cell Characterization Core Facility on an Illumina NextSeq500 on the 37 cycle, paired-end, High Output setting, yielding approximately 10–20 million paired reads per sample. MNase sequencing data were analyzed as described previously (McKnight and Tsukiyama 2015). Paired-end reads were aligned to the S. cerevisiae sacCer3 (Yates et al, 2019), S. pombe (Wood et al., 2002), or N. crassa (Galagan et al., 2003) reference genome with Bowtie 2 (Langmead and Salzberg 2012), and filtered computationally for unique fragments between 100 and 200 bp. Dyad positions were calculated as the midpoint of alignment coordinates, then dyad coverage was normalized across the genome for an average read/bp of 1.0. Nucleosome alignment to the Ume6 binding site, WNGGCGGCWW, was performed by taking average dyad signal at each position relative to all intergenic instances of a motif center. Intergenic instances of the Ume6 motif were found using the Saccharomyces Genome Database Pattern Matching tool (https://yeastgenome.org/nph-patmatch). Transcription start sites were obtained from published datasets for S. cerevisiae (Nagalakshmi et al., 2008) and S. pombe (Thodberg et al., 2019). Previously-published S. cerevisiae data (SRX5086833, (Donovan et al., 2019); SRX1176421, (McKnight et al., 2016)), S. pombe data (SRX554384 (Steglich et al., 2015); SRX331943, (DeGennaro et al 2013)), and N. crassa data (SRX1596291, (Seymour et al., 2016); SRX2822417, (Klocko et al., 2019)) were downloaded from the Sequence Read Archive (SRA) and analyzed using our computational pipeline to identify nucleosome dyad positions. Data were visualized using Integrated Genome Browser (Freese et al., 2016). Sequencing data from this work can be accessed at the GEO database under accession code GSE141676.
Limitations
When testing our protocol on new organisms, we adjusted the following parameters to obtain acceptable nucleosome footprints: input amount (number of cells), spheroplasting time (for fungi), amount of zymolyase, and amount of MNase. Table 2 contains a comparison of these parameters across the different sample types that we tested successfully, which can be used as a guide when trying new samples. More detailed protocols for preparation of input samples from other organisms is available upon request.
Troubleshooting
Problem 1
Genomic DNA is not being digested, as evidenced by a very high molecular weight band on the agarose gel and little or no signal for nucleosome footprints, as in Figure 7.
Figure 7.

Sample gel showing intact genomic DNA and partial nucleosome footprints from non-permeabilized cells
Agarose gel showing example nucleosome footprints recovered from patches of S. cerevisiae using unoptimized “rapid colony MNase” protocol (2 mg of zymolyase for 15 mi) using an input of (A) 30 OD pellet of yeast, or (B) 20 OD pellet of yeast.
Potential solution
This most likely is the result of cells not being permeabilized. We suggest extending the incubation time for the spheroplasting step (Step 6) or increasing the amount of zymolyase per mL of buffer in 1-mg increments. Another option is to perform this step at 37°C rather than at 25°C. After spheroplasting, the solution should be much less opaque than it was prior to this step, so it is possible to troubleshoot this step without completing the entire protocol. Furthermore, there are likely organisms that may require additional steps to help permeabilize cells, particularly if the cells possess zymolyase-resistant cell walls. Previous work has demonstrated success using cryogrinding as the cell-breaking step (Gonzalez and Scazzocchio 1997; Givens et al., 2011).
Problem 2
The DNA is over- or under-digested, as evidenced by too much or too little signal for the mononucleosome band. Examples of under-digested samples are shown in lanes B, C, and E of Figure 8. Examples of over-digested samples are shown in lanes D and F. Lane A shows a sample where fewer than 20 OD of cells were pelleted initially.
Figure 8.

Sample gel showing under- and over-digested nucleosome footprints
Agarose gel showing example nucleosome footprints recovered from S. cerevisiae liquid culture using the rapid MNase protocol
(A) Only 15 OD of cells were used as input.
(B) Under-digested sample.
(C) Under-digested sample.
(D) Over-digested sample.
(E) Under-digested sample.
(F) Over-digested sample.
Potential solution
MNase activity can vary across lots and vendors, so it is critical to calibrate the MNase concentration to give the desired extent of digestion. We recommend performing a test of 3 samples with either 2 μL, 3 μL, or 4 μL of MNase (20 U/μL stock) in Step 6 for S. cerevisiae; different amounts of MNase should be chosen for other types of samples based on Table 2. When running these tests, it is not necessary to treat samples with CIP, so Step 13 of the protocol can be skipped. An ideal digest will result in signal for mono-, di-, and tri-nucleosome fragments, with the most signal for the mono-nucleosome band, as in Figure 1.
Sometimes it is necessary to adjust the amount of MNase used in the protocol based on your specific enzyme activity, particularly when working with an organism that we have not tested. It is worth noting that often it is still possible to excise the mononucleosome band and obtain high-quality data of over-digested samples, but it is preferable to work with appropriately digested footprints, and the extent of digestion should be consistent across samples that are being compared in a particular experiment.
If the ratio of mono- to di-nucleosome bands appears to be correct, the issue may actually be that too few cells were used, as in lane A of Figure 8. Within an organism, the number of cells used could vary depending on the growth conditions or media composition, as evidenced by the adjustments needed for colony MNase (Table 2). It therefore possible that specific conditions or mutant strains may require subtle changes to digestion or input amount, particularly if it is challenging to accurately quantify the initial number of cells.
Problem 3
Quiescent cells produce improperly-digested footprints. Similar to Problem 2, if DNA appears under- or over-digested for quiescent cells, there are additional steps we took to optimize these samples.
Potential solution
Assuming that the MNase activity has been calibrated as in Problem 2, there are additional considerations when processing quiescent cells. We found that quiescent cells isolated on different days were not consistently digested, so we recommend preparing multiple MNase samples as standard practice, and testing different incubation times for MNase digest in Step 8, as shown in Figure 3A. For the sample shown in Figure 3A, the mononucleosome band from the 5-minute sample was excised, but for other Q cells the 5-minute time point was over- or under-digested. As indicated in the protocol, using multiple different quiescent cells, we observed a range of 2–10 min to yield appropriate footprints, but due to the variability we have seen, it is conceivable that the optimum range might be shifted for quiescent samples prepared in another lab, as the technique seems sensitive to small changes. In our hands, once we optimized the amount of MNase, the same amount of MNase worked for a variety of Q cell samples.
Problem 4
Unexpectedly low DNA yield, as evidenced by faint bands or no signal on the agarose gel.
Potential solution
Increase the input amount when preparing the sample. Assuming calculations for input were correct, we recommend doubling or even tripling the amount of sample as a test. Yeast cells in log phase versus quiescence, for example, give slightly different OD readings, and sometimes sick strains, certain mutants, or yeast grown in different medium vary in size, so the OD reading and pellet size do not accurately reflect the number of cells and therefore amount of DNA in the sample. Furthermore, the amount of input required can vary drastically for different organisms, so for a sample type that we have not tested, optimization of the amount of input will be necessary.
Problem 5
Instead of nucleosome footprints, only a large smear is visible on the agarose gel.
Potential solution
This is likely due to RNA and/or residual protein in the sample. We most often see this with improperly digested RNA and suggest first increasing the incubation time with RNAse A in Step 10. If that is not effective, increase the incubation time with Proteinase K in Step 11. Another possibility is that the RNAse A and/or Proteinase K stocks have lost activity, and these may need to be re-made.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Laura McKnight (lmcknigh@uoregon.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The datasets generated during this study are available at GEO under accession code GSE141676.
Acknowledgments
The authors thank the Genomics Core (GC3F) at the University of Oregon for high-throughput sequencing services. The authors also thank Gabe Zentner (Indiana University), Christine Cucinotta (Fred Hutchinson Cancer Research Center), and Andrew Klocko (University of Colorado, Colorado Springs) for helpful discussions related to this work. Mushroom identification was assisted by Andrew Kern (University of Oregon), Nicole Liachko (University of Washington), Richard Ebright (Rutgers University), and Anthoni Goodman (University of Alabama, Birmingham). This work was supported by a National Institutes of Health training grant T32 GM007759 (to O.G.B.B., K.N.O., G.L.W., and D.A.D.), by a National Institutes of Health training grant T32 GM007413 (to V.N.T.), by NIGMS GM093061 (to E.U.S.), and by NIGMS R01 GM129242 (to J.N.M.).
Author contributions
Conceptualization, J.G.C. and J.N.M.; methodology, L.E.M., J.G.C., T.B.B., V.N.T., and J.N.M.; investigation, L.E.M., T.B.B., O.G.B.B., K.N.O., V.N.T., E.T.W., G.L.W., and J.N.M.; resources, G.L.W., E.T.W., D.A.D., S.D.H., and E.U.S.; writing – original draft, L.E.M. and J.N.M.; writing – reviewing and editing, L.E.M., J.G.C., T.B.B., O.G.B.B., K.N.O., V.N.T., E.T.W., G.L.W., D.A.D., S.D.H., E.U.S., and J.N.M.; visualization, J.N.M.; supervision, S.D.H., E.U.S., and J.N.M.; project administration, L.E.M. and J.N.M.; funding acquisition, J.N.M.
Declaration of interests
The authors declare no competing interests.
References
- Allen C., Buttner S., Aragon A.D., Thomas J.A., Meirelles O., Jaetao J.E., Benn D., Ruby S.W., Veenhuis M., Madeo F. Isolation of quiescent and nonquiescent cells from yeast stationary-phase cultures. J. Cell Biol. 2006;174:89–100. doi: 10.1083/jcb.200604072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cam H.P., Whitehall S. Micrococcal nuclease digestion of schizosaccharomyces pombe chromatin. Cold Spring Harb. Protoc. 2016;2016:996–1000. doi: 10.1101/pdb.prot091538. [DOI] [PubMed] [Google Scholar]
- Cole H.A., Howard B.H., Clark D.J. Genome-wide mapping of nucleosomes in yeast using paired-end sequencing. Methods Enzymol. 2012;513:145–168. doi: 10.1016/B978-0-12-391938-0.00006-9. [DOI] [PubMed] [Google Scholar]
- DeGennaro C.M., Alver B.H., Marguerat S., Stepanova E., Davis C.P., Bähler J., Park P.J., Winston F. Spt6 regulates intragenic and antisense transcription, nucleosome positioning, and histone modifications genome-wide in fission yeast. Mol. Cell Biol. 2013;33:4779–4792. doi: 10.1128/MCB.01068-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan D.A., Crandall J.G., Banks O.G.B., Jensvold Z.D., Truong V., Dinwiddie D., McKnight L.E., McKnight J.N. Engineered chromatin remodeling proteins for precise nucleosome positioning. Cell Rep. 2019;29:2520–2535.e4. doi: 10.1016/j.celrep.2019.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freese N.H., Norris D.C., Loraine A.E. Integrated genome browser: visual analytics platform for genomics. Bioinformatics. 2016;32:2089–2095. doi: 10.1093/bioinformatics/btw069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan J.E., Calvo S.E., Borkovich K.A., Selker E.U., Read N.D., Jaffe D., FitzHugh W., Ma L.J., Smirnov S., Purcell S. The genome sequence of the filamentous fungus Neurospora crassa. Nature. 2003;422:859–868. doi: 10.1038/nature01554. [DOI] [PubMed] [Google Scholar]
- Givens R.M., Mesner L.D., Hamlin J.L., Buck M.J., Huberman J.A. Integrity of chromatin and replicating DNA in nuclei released from fission yeast by semi-automated grinding in liquid nitrogen. BMC Res. Notes. 2011;4:499. doi: 10.1186/1756-0500-4-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmark J.P., Fazzio T.G., Estep P.W., Church G.M., Tsukiyama T. The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell. 2000;103:423–433. doi: 10.1016/s0092-8674(00)00134-3. [DOI] [PubMed] [Google Scholar]
- Gonzalez R., Scazzocchio C. A rapid method for chromatin structure analysis in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res. 1997;25:3955–3956. doi: 10.1093/nar/25.19.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff J.G., Belsky J.A., Krassovsky K., MacAlpine D.M., Henikoff S. Epigenome characterization at single base-pair resolution. Proc. Natl. Acad. Sci. U S A. 2011;108:18318–18323. doi: 10.1073/pnas.1110731108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klocko A.D., Uesaka M., Ormsby T., Rountree M.R., Wiles E.T., Adhvaryu K.K., Honda S., Selker E.U. Nucleosome positioning by an evolutionarily conserved chromatin remodeler prevents aberrant DNA methylation in neurospora. Genetics. 2019;211:563–578. doi: 10.1534/genetics.118.301711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Miles S., Breeden L.L. A genetic screen for saccharomyces cerevisiae mutants that fail to enter quiescence. G3 (Bethesda) 2015;5:1783–1795. doi: 10.1534/g3.115.019091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight J.N., Boerma J.W., Breeden L.L., Tsukiyama T. Global promoter targeting of a conserved lysine deacetylase for transcriptional shutoff during quiescence entry. Mol. Cell. 2015;59:732–743. doi: 10.1016/j.molcel.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight J.N., Tsukiyama T. The conserved HDAC Rpd3 drives transcriptional quiescence in S. cerevisiae. Genom. Data. 2015;6:245–248. doi: 10.1016/j.gdata.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight J.N., Tsukiyama T., Bowman G.D. Sequence- targeted nucleosome sliding in vivo by a hybrid Chd1 chromatin remodeler. Genome Res. 2016;26:693–704. doi: 10.1101/gr.199919.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani V., Qiu R., Shendure J. High sensitivity profiling of chromatin structure by MNase-SSP. Cell Rep. 2019;26:2465–2476.e4. doi: 10.1016/j.celrep.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J., McKnight J.N., Tsukiyama T. Genome-wide analysis of nucleosome positions, occupancy, and accessibility in yeast: nucleosome mapping, high-resolution histone ChIP, and NCAM. Curr. Protoc. Mol. Biol. 2014;108:21.28.1–21.28.16. doi: 10.1002/0471142727.mb2128s108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour M., Ji L., Santos A.M., Kamei M., Sasaki T., Basenko E.Y., Schmitz R.J., Zhang X., Lewis Z.A. Histone H1 limits DNA methylation in neurospora crassa. G3 (Bethesda) 2016;6:1879–1889. doi: 10.1534/g3.116.028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steglich B., Stralfors A., Khorosjutina O., Persson J., Smialowska A., Javerzat J.P., Ekwall K. The Fun30 chromatin remodeler Fft3 controls nuclear organization and chromatin structure of insulators and subtelomeres in fission yeast. PLoS Genet. 2015;11:e1005101. doi: 10.1371/journal.pgen.1005101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thodberg M., Thieffry A., Bornholdt J., Boyd M., Holmberg C., Azad A., Workman C.T., Chen Y., Ekwall K., Nielsen O. Comprehensive profiling of the fission yeast transcription start site activity during stress and media response. Nucleic Acids Res. 2019;47:1671–1691. doi: 10.1093/nar/gky1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker K.A., Lilly M.B., Heck L., Jr., Rado T.A. Characterization of a new human diploid myeloid leukemia cell line (PLB-985) with granulocytic and monocytic differentiating capacity. Blood. 1987;70:372–378. [PubMed] [Google Scholar]
- Wood V., Gwilliam R., Rajandream M.A., Lyne M., Lyne R., Stewart A., Sgouros J., Peat N., Hayles J., Baker S. The genome sequence of Schizosaccharomyces pombe. Nature. 2002;415:871–880. doi: 10.1038/nature724. [DOI] [PubMed] [Google Scholar]
- Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R. Ensembl 2020. Nucleic Acids Res. 2019;48:D682–D688. doi: 10.1093/nar/gkz966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during this study are available at GEO under accession code GSE141676.