

Abstract
An initial shift in our understanding of the basis of periodontal disease occurred early in the 2000s. The host response, rather than the bacterial burden, was the principal determinant of the disease. Microbial dysbiosis that occurs in periodontal disease results from a hyperinflammatory state in the host. A second shift in periodontal disease is taking place. This time in the realm of treatment strategies. Rather than targeting antimicrobials or inhibitors of individual inflammatory mediators, preclinical studies support using resolution pharmacology to convert the pro-inflammatory condition into a non-inflammatory one, thereby resolving both the local and systemic inflammation associated with periodontal disease. Here, I describe the bases for these shifts in paradigms.
Keywords: inflammation, lipoxin, microbial dysbiosis, periodontal disease, pharmacology, resolvin
1 |. FROM AN INFECTIOUS TO AN INFLAMMATORY DISEASE PARADIGM
Prior to the early 2000s, periodontal disease was considered an infectious disease. Exogenous pathogens, such as Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticola, were thought to cause the disease. Beginning in the late 1980s, studies of inflammatory markers suggested a role for the host response in disease pathogenesis (Figure 1). In a study of children and adults with periodontal disease, prostaglandin E2 (PGE2) in gingival crevicular fluid (GCF) correlated with disease severity. Additionally, children had higher levels of inflammation than did adults.1 A subsequent study showed that gingival crevicular fluid PGE2 concentrations were predictive of disease severity, which was measured as periodontal attachment loss.2 Indeed, patients with GCF-PGE2 concentrations >2 standard deviations above those of healthy individuals were 47 times more likely to have periodontal attachment loss.
FIGURE 1.
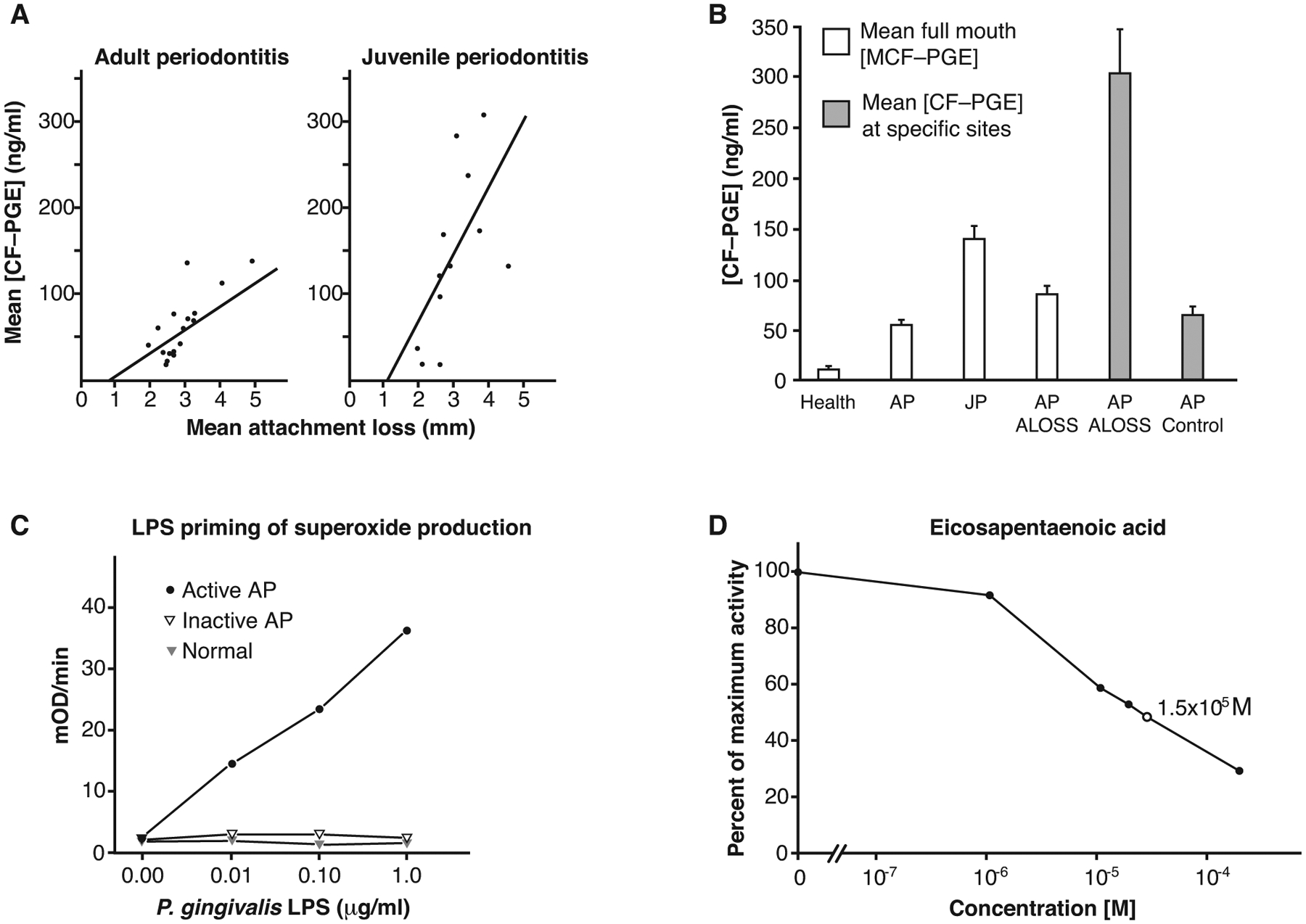
Inflammation is the determinant of periodontal disease. (A) Association between crevicular fluid PGE2 concentrations and disease severity in patients with periodontitis. The left side shows data from adults; the right side shows adolescents. CF-PGE, gingival crevicular fluid prostaglandin E2. Reproduced with permission from the American Academy of Periodontology.1 (B) In adult periodontitis, attachment loss is associated with high concentrations of crevicular fluid PGE2 (CF-PGE). ALOSS, attachment loss; AP, adult periodontitis; JP, juvenile periodontitis. Reproduced with permission from the American Academy of Periodontology.2 (C) Patients with active periodontitis. (D) Eicosapentaenoic acid inhibits PGE2 synthesis in human periodontal tissue homogenates. Reproduced from Springer Nature with permission19
Ultimately, a new paradigm in understanding the genesis of periodontal disease emerged. Although periodontal disease is associated with increased gingival pathogens, the biological basis for the disease is not infectious, but rather the result of uncontrolled, chronic inflammation. Earlier interpretations that a defective immune response allowed overgrowth of pathogens were replaced by data demonstrating that a hyperinflammatory response characterized the pathogenesis of disease (Figure 1).3,4 Offenbacher and colleagues focused on the role of PGE2 in tissue destruction, and Van Dyke and colleagues focused on the hyperresponsiveness of neutrophils in patients with periodontitis. Both groups proposed that the host response, rather than the bacterial burden, was the principal determinant of the disease. This represented a fundamental shift in understanding periodontal disease, a shift away from thinking of the disease from a purely infectious disease paradigm to an inflammatory disease paradigm.
2 |. INFLAMMATION, THE DRIVING FORCE OF BIOFILM DYSBIOSIS
Many of the studies of the microbiology of periodontal disease were cross-sectional population studies, which showed associations of bacteria with disease, but could not determine cause and effect. Some animal studies indicated that periodontal disease is driven by a “keystone” pathogen.5,6 However, a longitudinal study of people as they developed periodontal disease showed that the microbial dysbiosis occurred after disease developed.7 Animal studies showed that microbial dysbiosis was reversed simply by resolution or control of the inflammation, consistent with the model that inflammation was the driver of biofilm dysbiosis, and supporting the emerging data suggesting that inflammation is the underlying cause of the disease.8,9 Furthermore, in the presence of inflammation, the expression of virulence genes in all the gingival bacteria increases.10,11 Controlling the inflammation reverses this increase in virulence in the microbiome. Thus, the current model of periodontal disease is that inflammation creates the environment leading to microbial dysbiosis, and this induced dysbiosis is a sustaining inflammatory stimulus.8,9,12,13
There is evidence that inflammation is the driving force behind dysbiosis in several diseases in addition to periodontal disease. This connection between inflammation and dysbiosis has been reported for inflammatory bowel disease,14 cardiovascular disease,15,16 and diabetes.17,18
3 |. ANTI-INFLAMMATORY DRUGS, A SUBOPTIMAL TREATMENT
Based on the association of PGE2 with disease severity, initial pharmacological strategies were aimed at inhibiting prostaglandin production. Preliminary tests involved chemicals such as ibuprofen, indomethacin, α-tocopherol (a form of vitamin E), and the fatty acids docosahexaenoic acid and eicosapentaenoic acid (Figure 1).19 Some of these inhibited PGE2 with high potency in the low nanomolar or micromolar range. Subsequent studies showed that specific cyclooxygenase (COX) inhibitors effectively treated periodontal disease.20–22 However, such drugs cannot be used chronically; and, once the treatment is stopped, the disease returns.
Other potential treatment strategies include using tumor necrosis factor α (TNFα) antagonists, such as etanercept or infliximab, or steroids. These treatments have serious side effects and are not ideal for long-term therapy. Statins, which have anti-inflammatory properties, also stop the progression of periodontal disease.23 Like the prostaglandin synthesis inhibitors, these are only effective during treatment and they do not reverse the effects of the disease. With their side effects, none of these are considered optimal therapeutics for periodontal disease.
4 |. NATURAL RESOLVERS OF INFLAMMATION
The body has a natural system for terminating inflammation. The acute phase of inflammation is initiated by microorganisms or injury activating proinflammatory mediators sequentially followed by neutrophil influx. Central to this early response are the eicosanoids; lipid mediators like prostaglandins and leukotrienes. After the acute phase, neutrophils begin to undergo apoptosis (programmed cell death) followed by an influx of mononuclear phagocytes and macrophages that phagocytize remaining bacteria and apoptotic neutrophils. As inflammation resolves, macrophages are cleared from the lesion through the lymphatics by a process called efferocytosis. Pathology results when acute inflammation does not resolve and becomes chronic. In the chronic lesion, neutrophils are not cleared, macrophages assume a proinflammatory phenotype and acquired immunity with the accumulation of various lymphocytes is activated. This chronic immune-inflammatory lesion is what characterizes periodontitis. The natural termination sequence of the acute phase of inflammation is initiated by a new set of eicosanoids collectively called specialized proresolving mediators (SPMs) comprising lipoxins, resolvins, protectins, and maresins.24 Whereas most prostaglandins and leukotrienes stimulate inflammatory responses; lipoxins, resolvins, protectins, and maresins terminate inflammatory responses and promote the resolution of inflammation.25 Like the pro-inflammatory molecules, the inflammation-resolving molecules are lipid mediators.24 The discovery of the molecular identity of these SPMs and their receptors launched the field of “resolution pharmacology.”26
Resolution pharmacology has key differences from inflammation inhibitor pharmacology. SPMs are naturally occurring molecules in the body that function as agonists for receptors. The receptors are only present when inflammation is present. Consequently, these are highly specific when used as drugs and promote a coordinated return to homeostasis through their impact on all cells expressing the receptor. This is in stark contrast to inhibitors or blockers that affect only one pathway or molecule. Inhibitors can never coordinate a return to homeostasis, and it is hypothesized that this is the reason they can slow disease, but do not resolve disease or promote regeneration. In fact, inhibitors of cyclooxygenases (ibuprofen, etc.) actually interfere with resolution of inflammation.24 Additionally, no contraindications to use have been identified, no adverse outcomes or side effects are known, and no tolerization has been observed. Exogenous application of SPMs promotes the clearance of bacteria; thus, there is no increased susceptibility to infection.27 Compared with the anti-inflammatory drugs, the SPMs have ideal properties for treatment of a chronic inflammatory condition, such as periodontitis.
Compared with individual inhibitors of inflammation, which block only one inflammatory event, SPMs promote anti-inflammatory events or block multiple pro-inflammatory events (Figure 2). For activated inflammatory cells, SPMs change the phenotype of the cells from pro-inflammatory to anti-inflammatory.28 For example, inflammasomes are disassembled and the production of interleukin 1β (IL-1β) is stopped, prostaglandin production is terminated, along with cessation of the expression of genes encoding inflammatory mediators, such as the cytokine TNFα and interleukin-12 (IL-12). Monocytes are activated in a non-inflammatory manner to promote resolution of the inflammation, and neutrophils and eosinophils are inhibited and driven into apoptosis. Furthermore, administration of one SPM stimulates the production of other SPMs. The net effect of SPMs is resolution of inflammation not just inhibition of inflammation.
FIGURE 2.
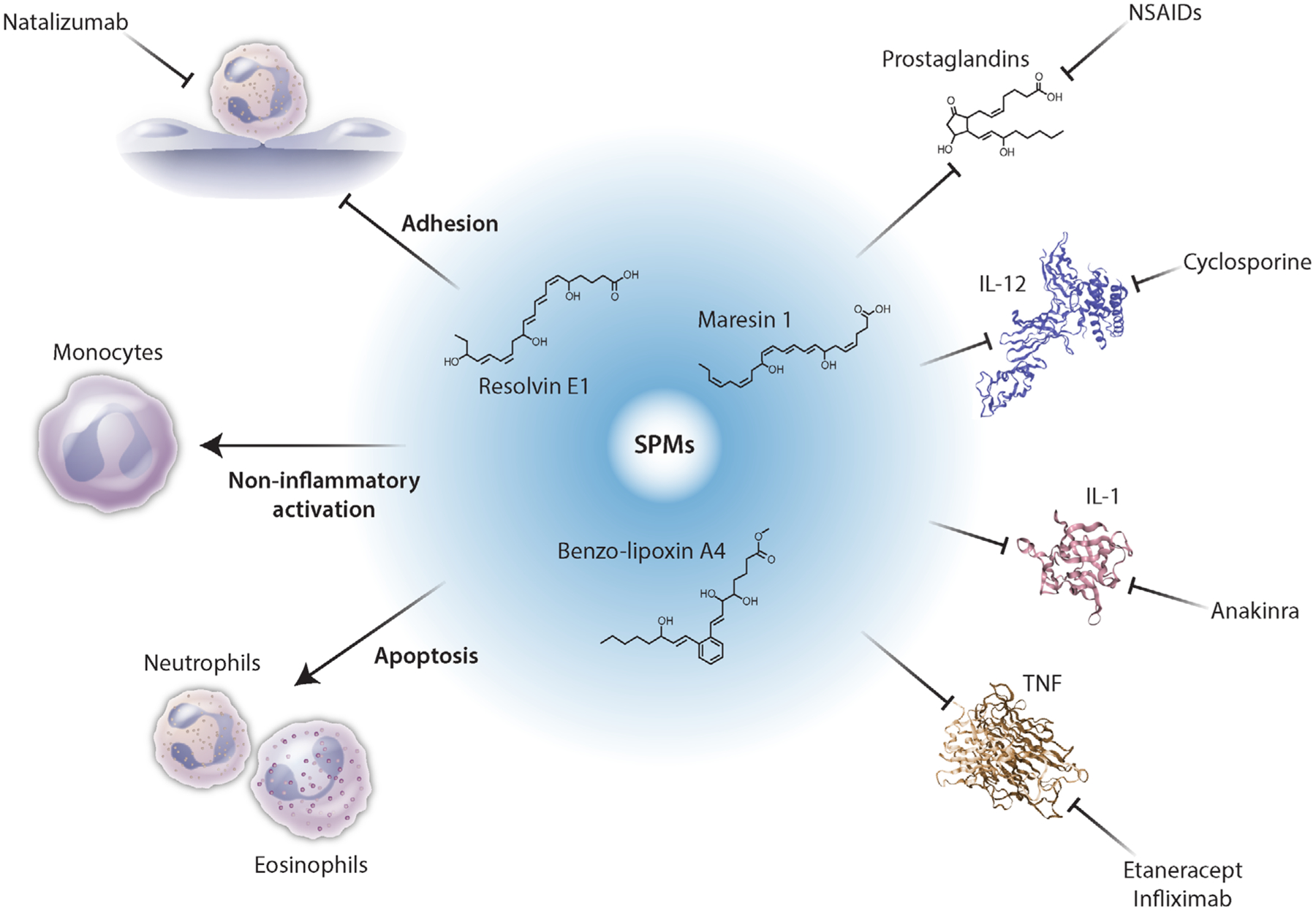
Specialized pro-resolving mediators (SPMs) promote resolution of inflammation. Whereas individual inhibitors of inflammation block one aspect of inflammation, SPMs, such as BLXA4, RvE1, and maresin 1, change the phenotypes of the immune cells thereby blocking production of inflammatory mediators to promote resolution of inflammation, not just inhibition of inflammation. [Credit: Heather McDonald, BioSerendipity, LLC, Elkridge, MD]
5 |. PRE-CLINICAL EVIDENCE OF THE BENEFITS OF RESOLVING INFLAMMATION
The SPMs are lipid mediators synthesized from arachidonic acid, eicosapentaenoic acid, and docosahexaenoic acid (Figure 3).25 The effects of resolvins, lipoxins, and maresins have been tested in preclinical studies. These include resolvin E1 (RvE1), benzo-lipoxin A4 (BLXA4), and maresin 1.
FIGURE 3.
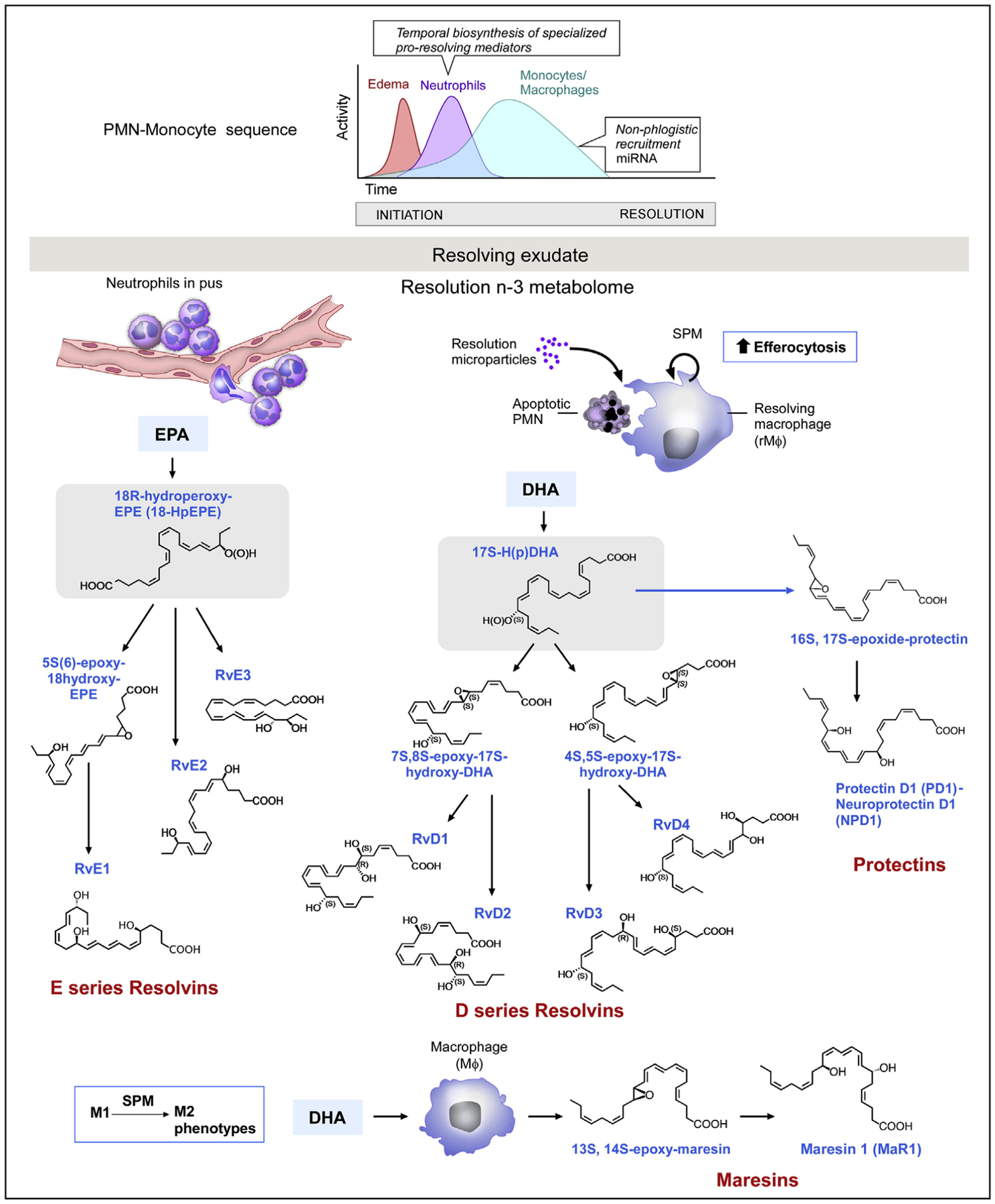
Resolution of inflammation begins with the emergence of specialized pro-resolving mediators (SPMs) after the acute phase. The upper graph represents the phases of the inflammatory response. Synthesis of lipid mediators of the resolution phase of inflammation. The diagram shows the production of E-series resolvins from eicosapentaenoic acid (EPA), and protectins, maresins, and D-series resolvins from docosahexaenoic acid (DHA). Not shown is the production of lipoxins from arachidonic acid. Reprinted with permission from Immunity25
Initial studies were performed with New Zealand white rabbits. Unlike mouse or dog models of periodontal disease, tying a ligature around a molar or pre-molar in a rabbit does not cause periodontal disease. This is because this rabbit model does not seem to have a pathogenic oral microbiome, and the ligature does not create a pathogenic dysbiosis. Thus, in the rabbit model, experimental periodontal disease is a two-step process: First ligature the tooth, then add a pathogen, such as Porphyromonas gingivalis (P. gingivalis) or another Gram-negative strain of bacteria to create dysbiosis. After 6 weeks of application of the pathogen, the rabbits develop periodontal disease.
Using this rabbit model, RvE1 was administered as an oral topical monotherapy every other day for 6 weeks after establishment of a significant chronic periodontitis. Local RvE1 therapy not only blocked progression of the disease, but promoted resolution of the disease and tissue regeneration.8 The pocket depths were reduced from 4 mm before treatment started to <1 mm after 6 weeks of treatment. Furthermore, the gingival tissue re-attached, and bone loss was reversed, which was confirmed to be “true” tissue regeneration with new cementum and periodontal ligament formation. Disease progression can be halted with an inhibitor of inflammation, such as ibuprofen, but there is no tissue regeneration or reversal of the disease state. These beneficial actions of an SPM are not limited to RvE1 but are also observed with BLXA4, D series resolvins, and maresins.
Additionally, the SPM treatment normalized the oral microbial populations. By 12 weeks of treatment, the pathogen P. gingivalis, which was used to induce periodontal disease, was eliminated. Other pathogenic bacteria that became detectable only in the inflamed state were also eliminated. The elimination of P. gingivalis occurred rapidly within 1 week of treatment. The mechanism for microbiome modification appears to be mediated by the bacterial microenvironment. As the inflammation is resolved, the food source for the P. gingivalis (essential amino acids from collagen breakdown) becomes unavailable. These organisms are assaccharolytic and require the collagen peptides from tissue destruction as their primary source of nutrients. These same types of experiments have now been reported in mice, rats, and pigs where dysbiosis and periodontitis was created with ligature alone. Reversal of dysbiosis and regeneration of lost periodontium was observed in all. Interestingly, dysbiosis in rodents does not involve P. gingivalis or any other bacterium recognized as a periodontal pathogen in humans.
Thus, the pre-clinical studies support the application of SPMs as a topical oral treatment for periodontal disease. Not only were the SPMs effective in halting the disease, but they also reversed the damage and induced regeneration, benefits not observed with inhibitors of inflammation.
6 |. THE FUTURE OF PERIODONTAL DISEASE THERAPY
The first paradigm shift in periodontal disease was the change in thinking of it as an infectious disease or microbial dysbiosis to thinking of it as a disease of inflammation with secondary microbial dysbiosis. The second paradigm shift could be moving away from using inhibitors of inflammation to using mediators of inflammation resolution as treatment (Figure 4). This second shift will likely be as profound as the change in understanding the etiology of the disease has been. With the potential of safe, effective, easy-to-deliver treatments that not only resolve inflammation in the mouth, but also have the potential to reset systemic inflammatory responses, lipoxin analogs could revolutionize treatment of many diseases that have chronic inflammation as a core component.
FIGURE 4.
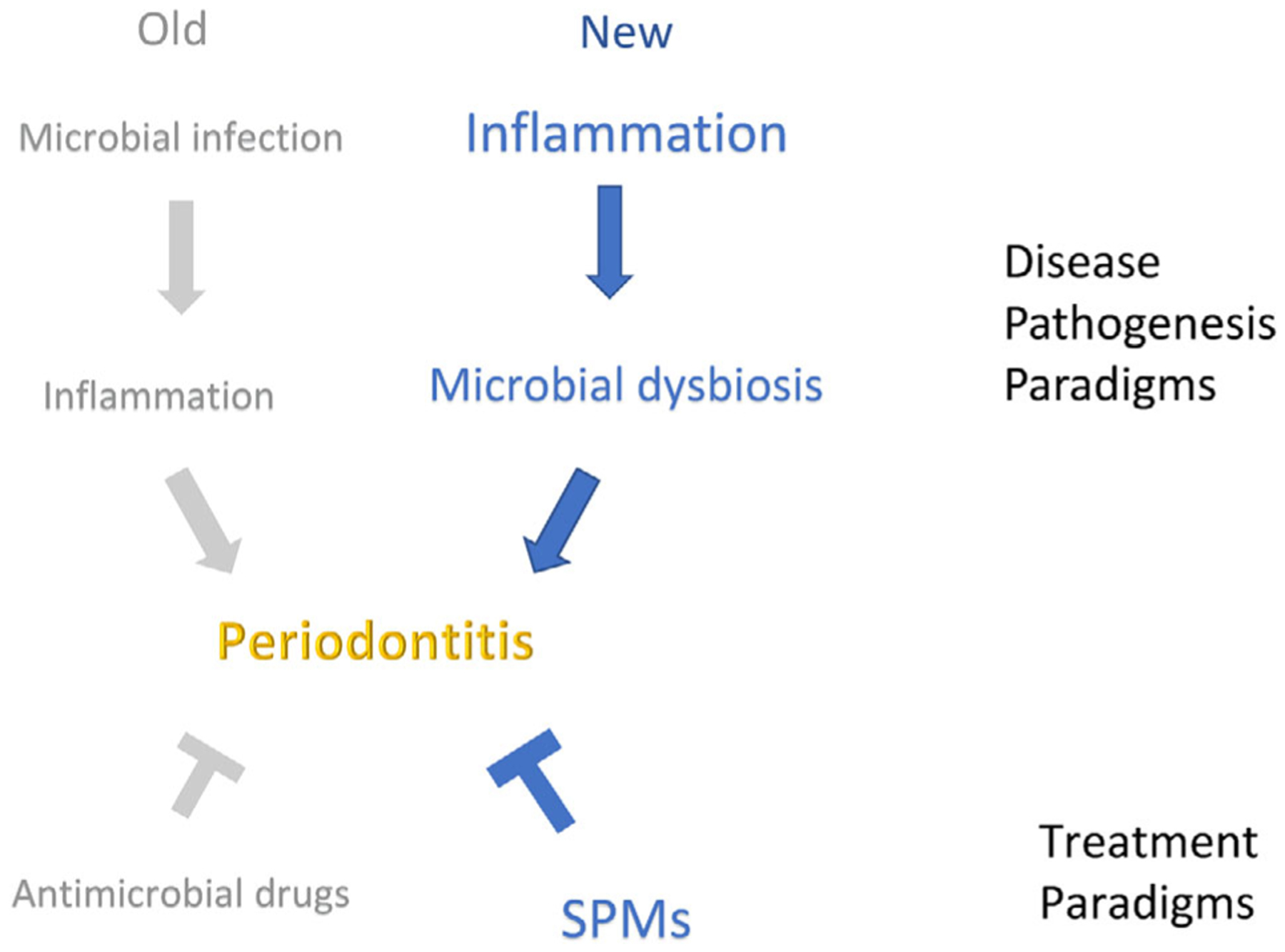
Paradigm shifts in understanding and treating periodontal disease. The paradigm shift is from the concept that one “catches” periodontitis from an outside source through the acquisition of specific pathogens to the concept of dysbiosis that is characterized by the overgrowth of certain segments of the endogenous microbiome. We have known for many years that microbial plaque accumulation on teeth, if not removed, results in gingival inflammation. We now understand that the gingival inflammation leads to changes in the bacterial composition by changing the growth environment and selecting for a different subset of organisms leading to the overgrowth of organisms that normally comprise a very small segment of the microbiome. Dysbiosis perpetuates and exacerbates inflammation. Why certain people develop a stable advanced gingivitis without periodontitis is still not quantifiable, but it is hypothesized to be related to the extent and severity of the inflammatory response, or a failure of the resolution response. [Credit: Nancy R. Gough, BioSerendipity, LLC, Elkridge, MD]
Fortunately, several companies are actively investigating resolution pharmacology. So, the translation of this concept into the clinic is already in progress.
ACKNOWLEDGMENTS
This paper is based on a presentation given by the author at the Day of Celebration for Steven Offenbacher. The symposium was hosted in October 2019 by the University of North Carolina Adams School of Dentistry and supported by Colgate-Palmolive Company and the American Academy of Periodontology. Colgate-Palmolive Company provided generous support for publication of this Journal of Periodontology supplement but had no involvement with the content. The initial draft of this manuscript was developed by Nancy R. Gough, PhD (a medical writer with BioSerendipity, LLC, Elkridge, MD) based on content provided solely by Dr. Van Dyke. The final manuscript submitted was under the sole control of the author. This article is supported in part by USPHS Grant DE025020 from the National Institute of Dental and Craniofacial Research.
DISCLOSURES
Dr. Van Dyke is inventor on several granted and pending licensed and unlicensed patents awarded to the Forsyth Institute that are subject to consulting fees and royalty payments. He is a founder of Nocendra, Inc. and AIAH Inc. He also holds grant awards from the NIDCR and several corporations in the area of inflammatory disease prevention and treatment, including periodontal diseases.
REFERENCES
- 1.Offenbacher S, Odle BM, Gray RC, Van Dyke TE. Crevicular fluid prostaglandin E levels as a measure of the periodontal disease status of adult and juvenile periodontitis patients. J Periodontal Res. 1984;19(1):1–13. 10.1111/j.1600-0765.1984.tb01190.x [DOI] [PubMed] [Google Scholar]
- 2.Offenbacher S, Odle BM, Van Dyke TE. The use of crevicular fluid prostaglandin E2 levels as a predictor of periodontal attachment loss. J Periodontal Res. 1986;21(2):101–112. 10.1111/j.1600-0765.1986.tb01443.x [DOI] [PubMed] [Google Scholar]
- 3.Offenbacher S, Heasman PA, Collins JG. Modulation of host PGE2 secretion as a determinant of periodontal disease expression. J Periodontol. 1993;64(suppl 5S):432–444. 10.1902/jop.1993.64.5s.432 [DOI] [PubMed] [Google Scholar]
- 4.Van Dyke TE, Lester MA, Shapira L. The Role of the host response in periodontal disease progression: implications for future treatment strategies. J Periodontol. 1993;64(suppl 8S):792–806. 10.1902/jop.1993.64.8s.792 [DOI] [PubMed] [Google Scholar]
- 5.Hajishengallis G, Liang S, Payne MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10(5):497–506. 10.1016/j.chom.2011.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10(10):717–725. 10.1038/nrmicro2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanner AC Jr, Kent R, Kanasi E, et al. Clinical characteristics and microbiota of progressing slight chronic periodontitis in adults. J Clin Periodontol. 2007;34(11):917–930. 10.1111/j.1600-051X.2007.01126.x [DOI] [PubMed] [Google Scholar]
- 8.Hasturk H, Kantarci A, Goguet-Surmenian E, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. 2007;179(10):7021–7029. 10.4049/jimmunol.179.10.7021 [DOI] [PubMed] [Google Scholar]
- 9.Lee CT, Teles R, Kantarci A, et al. Resolvin E1 reverses experimental periodontitis and dysbiosis. J Immunol. 2016;197(7):2796–2806. 10.4049/jimmunol.1600859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yost S, Duran-Pinedo AE, Teles R, Krishnan K, Frias-Lopez J. Functional signatures of oral dysbiosis during periodontitis progression revealed by microbial metatranscriptome analysis [published correction appears in Genome Med. 2015;7(1):111]. Genome Med. 2015;7(1):27. Published April 27, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yost S, Duran-Pinedo AE, Teles R, Krishnan K, Frias-Lopez J. Erratum to: functional signatures of oral dysbiosis during periodontitis progression revealed by microbial metatranscriptome analysis. Genome Med. 2015;7(1):111. 10.1186/s13073-015-0231-6. Published October 27, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nibali L, Ready DR, Parkar M, et al. Gene polymorphisms and the prevalence of key periodontal pathogens. J Dent Res. 2007;86(5):416–420. 10.1177/154405910708600505 [DOI] [PubMed] [Google Scholar]
- 13.Nibali L, Fedele S, D’Aiuto F, Donos N. Interleukin-6 in oral diseases: a review. Oral Dis. 2012;18(3):236–243. 10.1111/j.1601-0825.2011.01867.x [DOI] [PubMed] [Google Scholar]
- 14.Morgan XC, Tickle TL, Sokol H, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79. 10.1186/gb-2012-13-9-r79. Published April 16, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franceschi F, Niccoli G, Ferrante G, et al. CagA antigen of Helicobacter pylori and coronary instability: insight from a clinico-pathological study and a meta-analysis of 4241 cases. Atherosclerosis. 2009;202(2):535–542. 10.1016/j.atherosclerosis.2008.04.051 [DOI] [PubMed] [Google Scholar]
- 16.Rajendhran J, Shankar M, Dinakaran V, Rathinavel A, Gunasekaran P. Contrasting circulating microbiome in cardiovascular disease patients and healthy individuals. Int J Cardiol. 2013;168(5):5118–5120. 10.1016/j.ijcard.2013.07.232 [DOI] [PubMed] [Google Scholar]
- 17.Giongo A, Gano KA, Crabb DB, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5(1):82–91. 10.1038/ismej.2010.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larsen N, Vogensen FK, van den Berg FW, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5(2):e9085. 10.1371/journal.pone.0009085. Published February 5, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Offenbacher S, Odle BM, Green MD, et al. Inhibition of human periodontal prostaglandin E2 synthesis with selected agents. Agents Actions. 1990;29(3–4):232–238. 10.1007/bf01966452 [DOI] [PubMed] [Google Scholar]
- 20.Paquette DW, Lawrence HP, McCombs GB, et al. Pharmacodynamic effects of ketoprofen on crevicular fluid prostanoids in adult periodontitis. J Clin Periodontol. 2000;27(8):558–566. 10.1034/j.1600-051x.2000.027008558.x [DOI] [PubMed] [Google Scholar]
- 21.Heasman PA, Offenbacher S, Collins JG, Edwards G, Seymour RA. Flurbiprofen in the prevention and treatment of experimental gingivitis. J Clin Periodontol. 1993;20(10):732–738. 10.1111/j.1600-051x.1993.tb00699.x [DOI] [PubMed] [Google Scholar]
- 22.Offenbacher S, Williams RC, Jeffcoat MK, et al. Effects of NSAIDs on beagle crevicular cyclooxygenase metabolites and periodontal bone loss. J Periodontal Res. 1992;27(3):207–213. 10.1111/j.1600-0765.1992.tb01670.x [DOI] [PubMed] [Google Scholar]
- 23.Petit C, Batool F, Bugueno IM, Schwinté P, Benkirane-Jessel N, Huck O. Contribution of statins towards periodontal treatment: a review. Mediators Inflamm. 2019;2019:6367402. 10.1155/2019/6367402. Published February 27, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8(5):349–361. 10.1038/nri2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40(3):315–327. 10.1016/j.immuni.2014.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dalli J, Serhan CN. Identification and structure elucidation of the pro-resolving mediators provides novel leads for resolution pharmacology. Br J Pharmacol. 2019;176(8):1024–1037. 10.1111/bph.14336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herrera BS, Hasturk H, Kantarci A, et al. Impact of resolvin E1 on murine neutrophil phagocytosis in type 2 diabetes. Infect Immun. 2015;83(2):792–801. 10.1128/IAI.02444-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damgaard C, Kantarci A, Holmstrup P, Hasturk H, Nielsen CH, Van Dyke TE. Porphyromonas gingivalis-induced production of reactive oxygen species, tumor necrosis factor-α, interleukin-6, CXCL8 and CCL2 by neutrophils from localized aggressive periodontitis and healthy donors: modulating actions of red blood cells and resolvin E1. J Periodontal Res. 2017;52(2):246–254. 10.1111/jre.12388 [DOI] [PMC free article] [PubMed] [Google Scholar]