

Abstract
Poly(ADP-ribose)polymerase 1 (PARP1), a widely explored anticancer drug target, plays an important role in single-strand DNA break repair processes. High-throughput virtual screening (HTVS) of the Maybridge small molecule library using the PARP1-benzimidazole-4-carboxamide crystal structure and pharmacophore model led to the identification of eleven compounds. These compounds were evaluated using recombinant PARP1 enzyme assay that resulted in the acquisition of three PARP1 inhibitors: 3 (IC50 = 12 μM), 4 (IC50 = 5.8 μM), and 10 (IC50 = 0.88 μM). Compound 4 (2,3-dihydro-1,4-benzodioxine-5-carboxamide) was selected as a lead and it was subjected to further chemical modifications, involving analogue synthesis and scaffold hopping. These efforts led to the identification of (Z)-2-(4-hydroxybenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxamide (49, IC50 = 0.082 μM) as the most potent inhibitor of PARP1 from the series.
Keywords: virtual screening; 1,4-benzodioxine; 1,4-benzoxazin-3-one; PARP1; Knoevenagel condensation
Introduction
Poly(ADP-ribose) polymerase 1 (PARP1) is the most abundant (~106 molecules/cell) member of the PARP family of proteins. It is a chromatin-bound nuclear protein known to facilitate DNA repair processes by catalyzing the poly(ADP)ribosylation (PARylation) of itself (automodification) or on an array of DNA damage repair-associated target proteins (heteromodification) such as topoisomerase I/II, p53, histones, DNA polymerases and DNA ligases [1]. Activation of PARP1 plays a critical role in mediating DNA single-strand break repair in cancer cells, thereby providing them survival advantages. PARP1 inhibitors are therefore being considered clinically as potentiators of chemo- and radio-therapy through suppression of the base excision repair (BER) pathway [2,3]. In addition, some of the FDA approved PARP1 inhibitors (e.g., olaparib, niraparib, rucaparib and talazoparib) exhibited single-agent cytotoxic effect on tumors with germline mutations in BRCA1- or BRCA2-dependent DNA double-stranded repair mechanisms (homologous recombination, HR) [4,5]. However, the rapid emergence of restored HR pathway function has led to the limited use of existing PARP1 inhibitors [6–8]. Thus, development of new classes of PARP1 inhibitors is highly desirable, and is considered a promising strategy for anticancer drug development.
In our continuing effort to identify novel PARP1 inhibitors [9,10], we conducted a structure- and pharmacophore-based virtual screening of a Maybridge database comprised of a refined 15,621 small molecule compound library; this initial refinement of the library was generated by applying six pre-defined filtering parameters (Figure 1) to the original 54,836 compound library. Among the 868 virtual hits identified this way, 11 were selected as potential inhibitors of PARP1 enzyme by consideration of novelty, diversity, and lead optimization potential. Further in vitro evaluation of these compounds in PARP1 enzyme assay led to the identification of three lead compounds 3 (IC50 = 12 μM), 4 (IC50 = 5.8 μM), and 10 (IC50 = 0.88 μM). Since compound 3 had the weakest PARP1 inhibition and compound 10 belonged to the phthalazinone scaffold, already present in clinically used PARP-inhibitors (e.g., olaparib and talazoparib), we selected 2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (compound 4, Figure 1) as a tractable lead for further SAR optimization. Using analogue synthesis and scaffold hopping strategies on compound 4, we obtained a modestly potent compound 49, (Z)-2-(4-hydroxybenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxamide (IC50 = 0.082 μM, Figure 1) that exhibited a high potential as a lead compound in future anticancer drug development studies targeting PARP1 enzyme.
Figure 1.

A schematic representation of the high-throughput virtual screening protocol and subsequent hit optimization that led to the identification of the most active compound 49.
Results and Discussion
Structure-based Virtual Screening
Initially, 54,836 compounds from Maybridge library were subjected to a ligand preparation protocol using six pre-defined parameters such as inclusion of molecular weight filter, controlling ionization, exclusion of tautomers, restricting stereoisomers to one per ligand, number of aromatic rings (1–3) and that all ligands must possess a primary or secondary carboxamide group as depicted in Figure 1. This generated a refined library of 15,621 compounds. These compounds were subsequently subjected to a high-throughput virtual screening (HTVS) protocol using the complex of the catalytic domain of human PARP1 enzyme and the benzimidazole-4-carboxamide ligand (RCSB Protein Data Bank (PDB) ID: 2RCW). The HTVS with Glide v5.5 docking (Schrodinger LLC, New York, NY) involved constraints such as (1) applying a −7.0 Glide score cutoff to eliminate ligands which have a higher Glide score and (2) employing a hydrogen bond constraint to eliminate the ligands which do not interact with the backbone of Gly863. These efforts led to narrowing of the potential PARP1 inhibitory leads to 900 candidates for pharmacophore-based screening.
Pharmacophore-based Virtual Screening
PHASE algorithm [11] was employed to quantitatively correlate the chemical structures of selected known PARP1 inhibitors to their biological activity for generating a valid pharmacophore model. A training set of 23 known PARP1 inhibitors [12] was chosen for this activity, with their experimental pIC50 values ranging from 0.568 to 2.853 and seven compounds were employed as the test set to validate the accuracy of the pharmacophore models (Table S1, supporting information). Compounds with pIC50 values greater than 1.5 were deemed active for the development of pharmacophore models. A pharmacophore site point generation protocol containing the default variant list: acceptor (A), donor (D), hydrophobic (H), negative (N), positive (P), and aromatic ring (R) along with a diversity of conformers for the training set molecules was selected to generate pharmacophore hypothesis models. Four top-scored pharmacophore hypotheses containing a combination of ADR variants were thus generated (Table S2, supporting information), exhibiting comparable scores for survival, site, vector, volume, and number of active sites. Visual inspection led to elimination of pharmacophore models AADRR.1 and ADRRR.5 due to the incorrect orientation of a vector, thereby preventing the models to form a key hydrogen bonding interaction between carboxamide group of mapped ligands and carbonyl oxygen of Gly863 (Figure S1, supporting information). The pharmacophore models AADRR.2 and ADRRR.7 were then subjected to atom-based 3D QSAR analysis which provided comparable results for the two models (Table S3, supporting information). Further results of overlapping pharmacophore models on an active PARP1 inhibitor revealed that ADRRR.7 could not identify the key intramolecular hydrogen bonding interaction between carboxamide group and the heteroatom located on the adjacent ring of the ligand (Figure S1, supporting information). Therefore, AADRR.2 was selected for further analysis, and the previously obtained 900 ligands were screened against the AADRR.2 pharmacophore model. During this process, all ligands were required to contain at least three (A2, D3 and R9) of the five features of the AADRR.2 model. This step resulted in 868 ligands, from which 11 compounds were selected for empirical testing in biochemical PARP1 enzyme assay based on their ability to form hydrogen bonding interaction between the carboxamide and Gly863/Ser904, novelty, diversity, and the potential for further optimization (Figure 1)[13].
PARP1 Inhibition Data
Ten of the eleven selected compounds (1–10) were obtained from Maybridge Chemicals Ltd (Tintagel, United Kingdom). Compound 11 was purchased from Matrix Scientific (Columbia, SC). These compounds were then tested for in vitro inhibition of PARP1 enzyme activity as described in our earlier reports [9,14]. A previously characterized PARP1 inhibitor, 8-amino-2-methylquinazolinone, developed in our laboratory and 3-aminobenzamide, recommended in the assay kit, were included as internal positive controls [14]. The preliminary screening was performed at a single concentration of 10 μM that resulted in three inhibitory classes of compounds: with high, moderate, and low PARP1 inhibitory activity (Table 1). Based on these results, the three best compounds (3, 4 and 10) were selected for further analysis using dose-response relationship. The IC50 values of these inhibitors thus obtained were 12 μM, 5.8 μM and 0.88 μM for 3, 4 and 10 respectively (Table 1). Based on the potency and novelty aspects described earlier, compound 4 was selected for further investigation. To that end, the binding model of compound 4 at the active site of PARP1 (Figure S2, supporting information) was generated to facilitate further lead optimization via extension of substituents at the C2- or C3-positions of the dioxine ring or by employing a scaffold hopping strategy.
Table 1.
Structures and PARP1 inhibitory activities of virtual screening hits 1-11
| Compound | Vendor Number | Structure | PARP1 IC50 (μM)a, b |
|---|---|---|---|
| 1 | S14246 |  |
>10 |
| 2 | AC40100 |  |
>10 |
| 3 | NRB02674 (R/S) |  |
12 ± 1.6 |
| 4 | MO00789 |  |
5.8 ± 0.10 |
| 5 | JFD03569 (R/S) |  |
~10 |
| 6 | GK03180 |  |
~10 |
| 7 | GK02796 |  |
>10 |
| 8 | GK00357 |  |
>10 |
| 9 | BTB08875 |  |
>10 |
| 10 | BTB02780 |  |
0.88 ± 0.090 |
| 11 | 003736 |  |
>10 |
3-Aminobenzamide sample included in the assay kit was used as an internal standard, and showed a consistent result as reported (60 – 80% inhibition at 50 μM).
All compounds were assayed at a single concentration of 10 μM in triplicates. Compounds with <40% inhibition at 10 μM were denoted as PARP1 IC50 >10 μM. Compounds showing 40 – 60% inhibition at 10 μM were considered to have PARP1 IC50 ~10 μM. Compounds showing >60% inhibition at 10 μM were further evaluated to obtain PARP1 IC50s.
Chemistry
To conduct SAR around compound 4, it was first synthesized according to Scheme 1. The 2,3-dihydroxybenzoic acid 12 was esterified using concentrated sulfuric acid in refluxing methanol to obtain 13. Methyl ester 13 was alkylated with 1,2-dibromoethane in the presence of potassium carbonate to afford cyclized ester 14. The ester group in 14 was hydrolyzed using lithium hydroxide. Resulting acid was obtained through basic and acidic work up and converted to carboxamide 4 using the mixed-anhydride method. Target compounds with C7-nitro (15) and C8-nitro (16) were obtained via a nitration reaction involving a mixture of nitric acid and trifluoroacetic acid. Corresponding amino derivatives 17 and 18 were obtained using catalytic hydrogenation reaction conditions [15].
Scheme 1.

(a) MeOH, concentrated H2SO4, reflux, 12 h, 85%; (b) 1,2-dibromoethane, K2CO3, DMF, reflux, 12 h, 60%; (c) (i) LiOH, H2O, THF, rt, 8–12 h; (ii) isobutyl chloroformate, N-methylmorpholine, THF, 0 °C, 30 min; (iii) NH3 in MeOH, rt, 60 min, 55%; (d) HNO3, CF3COOH, 0 °C, 6 h, 22% (15:16 = 40:60); (e) Pd/C, H2, MeOH, rt, 8–10 h, 60–70%.
To avoid the complexity of introducing racemates during installation of substituents at prochiral centers C2 or C3 of the dioxine ring, we reacted intermediate 13 with (2S)-glycidyl tosylate and (2R)-glycidyl tosylate, respectively, to obtain esters 19a/19b (40:60 ratio) (Scheme 2) and 24a/24b (40:60 ratio) (Scheme 3) [16]. Without further purification, these methyl esters were converted to their corresponding amides 20a/20b and 25a/25b via hydrolysis followed by amide formation using mixed-anhydride method. The primary alcohol group of these amides was subjected to mesylation by treating with methanesulfonyl chloride in the presence of triethylamine to obtain 21a/21b and 26a/26b. Displacement of the mesylate group in 21a/21b and 26a/26b by N-methylpiperazine or pyrimidinyl-piperazine yielded 22a/22b, 23a/23b, 27a/27b, and 28a/28b as shown in Schemes 2 and 3.
Scheme 2.

(a) (2S)-glycidyl tosylate, K2CO3, DMF, 40 °C, 30 h, 20–30%; (b) (i) LiOH, H2O, THF, rt, 10 h; (ii) isobutyl chloroformate, N-methylmorpholine, THF, 0 °C, 15–25 min; (iii) NH3 in MeOH, rt, 1 h, 60–70%; (c) methanesulfonyl chloride, triethylamine, DCM, rt, 10–12 h, 50–55%, (d) N-methylpiperazine (for 22a and 22b), 1-(2-pyrimidinyl)piperazine (for 23a and 23b), K2CO3, DCM, 0 °C to rt, 6–8 h, 20–33%.
Scheme 3.

(a) (2R)-glycidyl tosylate, K2CO3, DMF, 40 °C, 30 h, 20–30%; (b) (i) LiOH, H2O, THF, rt, 10 h; (ii) isobutyl chloroformate, N-methylmorpholine, THF, 0 °C, 15–25 min; (iii) NH3 in MeOH, rt, 1 h, 60–70%; (c) methanesulfonyl chloride, triethylamine, DCM, rt, 10–12 h, 50–55%, (d) N-methylpiperazine (for 27a and 27b), 1-(2-pyrimidinyl)piperazine (for 28a and 28b), K2CO3, DCM, 0 °C to rt, 6–8 h, 18–31%.
With the expectation that scaffold hopping could lead to more potent PARP1 inhibitors, a series of analogues with different core structures were also prepared. Compounds 31 and 32 were first prepared to evaluate the influence of varying dioxine ring size on PARP1 inhibition (Scheme 4). Thus, intermediate 13 was reacted with the dibromomethane or 1,3-dibromopropane, respectively, to obtain 29 and 30 [17]. Esters 29 and 30 were then converted to target carboxamides 31 and 32 following the similar method used for preparing the carboxamide 4 [15].
Scheme 4.

(a) dibromomethane (for 29), 1,3-dibromopropane (for 30), K2CO3, DMF, reflux, 8–12 h, 55–70%; (b) (i) LiOH, H2O, THF, rt, 8–12 h; (ii) corresponding acid, isobutyl chloroformate, N-methylmorpholine, THF, 0 °C, 15–30 min; (iii) NH3 in MeOH, rt, 50–60 min, 60–70%.
To obtain new scaffold hopping analogues, target compounds 44-50 were designed and synthesized by following a synthetic route outlined in Scheme 5. Commercially available acid 33 was initially converted to a methyl ester 34, and the phenolic hydroxyl group in 34 was subjected to O-alkylation with appropriate ethyl α-bromoacetate that afforded diesters 35-38 [18]. Reductive cyclization of 35-38 with iron and acetic acid afforded intermediates 39-42. The cyclic amide group in ester 39 was reduced to 43 using NaBH4 in the presence of boron trifluoride-diethyl etherate complex [19]. Target compounds 44-48 were synthesized according to the procedure used for the synthesis of 4. Finally, target compounds 49 and 50 were prepared via Knoevenagel condensation reaction conditions using 45 and corresponding benzaldehydes. It is noted that the Knoevenagel condensation reaction produced 49 and 50 in <10% yields. This is attributed to the weak acidic nature of the C2-methylene protons. To increase the amount of product formation, we conducted the reaction using following modifications: (a) conventional reflux of starting reagents in pyridine containing catalytic amounts of the piperidine, (b) microwave irradiation at 120 °C in a 1:1 ratio of acetic anhydride and triethylamine, and (c) microwave irradiation at 120 °C in pyridine containing catalytic amounts of the piperidine. However, the modified conditions did not result in improved product yields.
Scheme 5.

(a) MeOH, conc. H2SO4, reflux, 12 h, 88%; (b) appropriate ethyl α-bromoacetate, K2CO3, DMF, rt, 8 h, 69–84%; (c) Fe, acetic acid, reflux, 4 h, 69–78%; (d) BF3-Et2O, NaBH4, THF, 5 °C, 2 h, 23%; (e) (i) LiOH, H2O, THF, rt, 10 h; (ii) isobutyl chloroformate, N-methylmorpholine, THF, 0 °C, 20 min; (iii) NH3 in MeOH, rt, 30 min, 30–60%; (f) 45, 4-hydroxybenzaldehyde (for 49), 4-carboxybenzaldehyde (for 50), Et3N/Ac2O (1/1), reflux, 12 h, 8–10%.
Structural Elucidation of Regioisomers 21a and 21b by HMBC Experiments
Regioisomerism assignments were performed based on the NMR analyses of compounds 21b and 21a, as illustrated in Figures 2 and 3 respectively. While 1-bond 1H-13C heteronuclear single quantum coherence (HSQC) spectrum showed similar patterns for the two regioisomers (Figures S4 and S5, supporting information), the 3-bond long range 1H-13C coupling experiment (HMBC = Heteronuclear Multiple Bond Correlation) not only allowed for assignment of quaternary carbons, but also showed the differences between the two regioisomers. In the 1D proton spectrum H6 and H4 appear as doublets whereas H5 appears as a triplet. Assignment of H6 was confirmed by the 3-bond coupling to the carbonyl carbon in the HMBC spectra (peak C in Figures 2 and 3). Identifying H6 allowed for the identification of quaternary carbon, C2 (peak B in Figures 2 and 3) through its 3-bond coupling, while quaternary carbon, C3 was identified by its 3-bond coupling to H5 (peak E in Figures 2 and 3). Unequivocal assignment of C2 and C3 was utilized to find if C8 or C9 were substituted with a functional group. In the expanded HMBC spectra of 21b shown in Figure 2, C3 showed 3-bond coupling to a pair of methylene protons (peaks F and G). This was possible only if there was no substituent on C9. On the other hand, in the expanded HMBC spectra of 21a shown in Figure 3, C2 showed 3-bond coupling to a pair of methylene protons (peaks F and G) clearly indicating that C8 was unsubstituted. The regiochemistry of the hydroxymethyl, N-methylpiperazine and pyrimidinyl-piperazine substituted compounds was also assigned based on the fact that they shared the same intermediates such as 19a/19b and 24a/24b.
Figure 2.

1H-13C HMBC spectra of 21b regioisomer: Crosspeaks identified as A, B, and C are 3-bond couplings from H6 to C4, C2, and C7 respectively. Crosspeaks identified as D and E are 3-bond couplings from H5 to C1 and C3. Crosspeaks identified as F and G are 3-bond couplings from the C9 methylene protons to C3 confirming that the C9 carbon is unsubstituted.
Figure 3.

1H-13C HMBC spectra of 21a regioisomer: Crosspeaks identified as A, B, and C are 3-bond couplings from H6 to C4, C2, and C7 respectively. Crosspeaks identified as D and E are 3-bond couplings from H5 to C1 and C3. Crosspeaks identified as F and G are 3-bond couplings from the C8 methylene protons to C2 confirming that the C8 carbon is unsubstituted.
Structure-Activity Relationship
Initial structure- and pharmacophore-based virtual screening studies led to the identification of 11 compounds that were categorized into three classes: with high, moderate, or low PARP1 inhibition (Table 1). Based on novelty of structure and ease of synthesis, we selected compound 4 (IC50 = 5.8 μM) as our lead for further optimization. PARP-inhibitory positive controls (olaparib and veliparib) were used for comparison. Table 2 summarizes the installation of small to large substitutions at various positions on the bicyclic ring system of 4 and their resulting effects on activities. To obtain a direct comparison of activities between optimized analogues and lead compound 4, percentage inhibition against PARP1 by target compounds at 5 μM was determined. The compounds showing >60% inhibition were evaluated for their IC50 values. Introduction of nitro (15 and 16) or amino (17 and 18) groups on the phenyl portion of the benzodioxine ring led to a dramatic loss of activity. Further we wanted to explore the effect of various polar substituents on the dioxine portion of lead compound 4. Synthesis of compounds with such substitutions will generate a stereogenic center for each modification. To avoid the complexity involved in the purification and identification of active enantiomers, a synthetic route that would simplify both chemistry and SAR interpretation was necessary. As a result, we optimized the synthesis using two homochiral glycidyl tosylates as the starting materials. This assisted us in achieving the synthesis of the pure enantiomers with a desirable purity and additionally, the synthesis gave rise to two regioisomers for each enantiomer. The compound library thus generated, comprising 20a-23a, 20b-23b, 25a-28a, and 25b-28b, was screened in vitro against PARP1 enzyme (see Table 2).
Table 2.
PARP1 inhibitory activities of target compounds 15-18, 20-23, and 25-28
| Compd | Structure | PARP1 IC50 (μM)a,b | Compd | Structure | PARP1 IC50 (μM)a,b |
|---|---|---|---|---|---|
| 4 |  |
5.8 ± 0.10 | 26a |  |
>5 |
| 15 |  |
>5 | 26b |  |
1.2 ± 0.21 |
| 16 |  |
>5 | 22a |  |
>5 |
| 17 |  |
>5 | 22b |  |
>5 |
| 18 |  |
>5 | 27a |  |
>5 |
| 20a |  |
>5 | 27b |  |
>5 |
| 20b |  |
>5 | 23a |  |
>5 |
| 25a |  |
6.9 ± 0.95 | 23b |  |
>5 |
| 25b |  |
7.8 ± 2.7 | 28a |  |
>5 |
| 21a |  |
>5 | 28b |  |
>5 |
| 21b |  |
0.69 ± 0.27 | |||
| Olac | - | 0.0012 ± 0.00020 | Velid | - | 0.0015 ± 0.00020 |
3-Aminobenzamide sample included in the assay kit was used as an internal standard, and showed a consistent result as reported (60 – 80% inhibition at 50 μM).
All compounds were assayed at a single concentration of 5 μM in triplicates. Compounds with <40% inhibition at 5 μM were denoted as PARP1 IC50 >5 μM. Compounds showing >60% inhibition at 5 μM were further evaluated to obtain PARP1 IC50s.
olaparib;
veliparib
While analogues with hydroxymethyl extension, 25a (with C2-substitution, IC50 = 6.9 μM) and 25b (with C3-substitution, IC50 = 7.8 μM) displayed comparable activities as that of lead compound 4, the modifications leading to compounds 20a and 20b proved detrimental. Enantiomerically pure hydroxymethyl regioisomers were then converted to respective methanesulfonates 21a, 21b, 26a and 26b with the hypothesis that they would capture additional hydrogen bonding interactions in the adenosine-binding pocket (ABP) of PARP1 active site. Interestingly, C3-substituted enantiomers 21b (IC50 = 0.69 μM) and 26b (IC50 = 1.2 μM) exhibited ~8-fold and ~5-fold improvement in activity, compared to the unsubstituted lead 4 (IC50 = 5.8 μM), while analogues 21a and 26a with C2-mesylate demonstrated a loss of PARP1 inhibition. Compound 21b also showed ~10-fold improved activity over hydroxymethyl precursor 25a. From these results, it was evident that methanesulfonyl extensions at the C3-position were more favored regardless of its stereochemistry, compared to those at the C2-position. In conclusion, compound 21b with (S)-configuration was the most potent amongst these four analogues.
To further assess the importance of methanesulfonyl group, we developed a compound library where this group was displaced with N-methylpiperazinyl extension resulting in compounds, 22a, 22b, 27a, and 27b (see Table 2). This modification was anticipated to capture ionic and/or hydrogen bonding interactions within the ABP of PARP1 active site; however, these analogues showed a significant decrease (<20% inhibition at 5 μM) in the activity profile compared to the lead compound 4 (IC50 = 5.8 μM). Furthermore, replacement of the methyl group in N-methylpiperazine with a 2-pyrimidinyl extension (analogues 23a, 23b, 28a and 28b) also exhibited a significant loss of activity (<20% inhibition at 5 μM). These results together suggested that bulky extensions at the C2- or C3-positions, were perhaps pointing toward a sterically hindered region in the active site of PARP1.
Finally, to further improve the inhibitory potency of benzodioxine lead compound 4, the scaffold hopping strategy was implemented. We therefore explored the effect of modifying the size of the fused ring system and studied its effect on inhibitory potency (Table 3). With the intention of securing compounds with enhanced potency, target compounds that exhibited >60% inhibition at 2.5 μM concentration were prompted for PARP1 IC50 determination. The synthesized benzodioxole compound 31 exhibited an IC50 value of ≥2.5 μM (literature IC50 = 5.3 μM [20] while benzodioxepine 32 showed significantly decreased activity (15% inhibition at 2.5 μM). This data suggested that the six-membered ring was perhaps the optimum size ring for PARP1 inhibitory activity. To evaluate the contribution of the oxygen atom in the dioxine ring, we replaced one of the oxygen atoms (H-bond acceptor) in compound 4 with bioisosteric –NH– (H-bond donor) group. Our hypothesis, based on structural evidence, was that this modification could significantly improve PARP1 inhibition through favorable water-mediated hydrogen bonding interaction with the catalytically crucial Glu988 residue in the nicotinamide binding pocket. To our surprise, 1,4-benzoxazine derivative 44 (IC50 = 11 μM) was ~2-fold less potent than 4. However, converting a moderately basic ring amine in 44 to a neutral cyclic carboxamide led to 45 that exhibited 15-fold enhanced activity (IC50 = 0.75 μM) in comparison to 44. This improvement in potency may be attributed to a water-mediated hydrogen bonding interaction of 45 with Glu988 residue. Analogue 45 was considered as a refined lead compound and subsequent substitutions were performed at its C2-position to enhance hydrophobic/hydrogen bonding interactions. A phenyl extension at C2-position was thus introduced as a racemate to capture hydrophobic interactions, which resulted in decreased activity (46, IC50 ~ 2.5 μM). Electron-donating (methoxy) or electron-withdrawing (bromo) substitutions at the para-position of the C2-phenyl led to racemic mixtures 47 (methoxy) and 48 (bromo). Compound 47 (IC50 = 0.80 μM) displayed a comparable activity whereas 48 (IC50 > 2.5 μM) showed decreased activity with respect to 45. Since benzylidene substitutions in our previously published work proved successful in generating potent analogues, [9,15] we decided to introduce a rigid substitution such as para-substituted benzylidene moiety at the C2-position. This led to the synthesis of two compounds with further gain in potency, e.g., compounds 49 (IC50 = 0.082 μM) and 50 (IC50 = 0.42 μM). The pattern in which various structural modifications of the benzodioxine and benzoxazinone scaffolds favor or disfavor PARP1 inhibition is depicted in Figure 4.
Table 3.
PARP1 inhibitory activities of Scaffold Hopping analogues
| Compound | Structure | PARP1 IC50 (μM)a,b |
|---|---|---|
| 4 |  |
5.8 ± 0.10 |
| 31 |  |
>2.5 (5.3)c |
| 32 |  |
>2.5 |
| 44 |  |
11 ± 1.9 |
| 45 |  |
0.75 ± 0.090 |
| 46 |  |
~2.5 |
| 47 |  |
0.80 ± 0.15 |
| 48 |  |
>2.5 |
| 49 |  |
0.082 ± 0.045d |
| 50 |  |
0.42 ± 0.046d |
| Olae | - | 0.0012 ± 0.00020 |
| Velif | - | 0.0015 ± 0.00020 |
3-Aminobenzamide sample included in the assay kit was used as an internal standard, and showed a consistent result as reported (60 – 80% inhibition at 50 μM).
All compounds were assayed at a single concentration of 2.5 μM in triplicates. Compounds with <40% inhibition at 2.5 μM were denoted as PARP1 IC50 >2.5 μM. Compounds showing 40 – 60% inhibition at 2.5 μM were considered to have PARP1 IC50 ~2.5 μM. Compounds showing >60% inhibition at 2.5 μM were further evaluated to obtain PARP1 IC50.
Literature IC50 value;
Upon observing >70% inhibition at 0.5 μM in PARP1 enzyme assay, IC50 values of compounds 49, 50, olaparib, and veliparib were determined by chemiluminescence assay (BPS Bioscience, San Diego, CA);
olaparib;
veliparib
Figure 4.

SAR summary of the 1,4-benzodioxine and 3-oxo-2,3-dihydrobenzoxazine scaffolds.
Because PARP-isoform selectivity is a key to obtain clinically useful new compounds to treat cancers, we conducted screening of the most potent compound 49 along with positive controls against seven clinically investigated PARP-isoforms (e.g., PARP2, PARP3, TNKS1 (PARP5A), TNKS2 (PARP5B), PARP8, PARP10, and PARP14 (Figure 5 and Table S5). Compound 49 (PARP1 IC50 = 0.082 μM) showed complete inhibition of PARP2 at 0.5 μM concentration, which is not surprising due to a highly conserved catalytic domain (>69% homology) being shared by both PARP1 and PARP2.[21] Interestingly, 49 also substantially inhibited PARP3, TNKS1, and TNKS2, which are involved in cancer progression.[22,23] Conversely, 49 did not interfere with other PARP-isoforms such as PARP8, PARP10 or PARP14. Further investigation on PARP-isoform screening with low testing concentrations will be conducted based on the progress of future structure optimization.
Figure 5. Percentage inhibition data of 49 against seven PARP-isoforms.

The reported data is based on the two independent experiments, and each experiment was performed in duplicate. Olaparib for PARP2, PARP3, PARP8, and PARP10, XAV- 939 for TNKS1 and TNKS2 and rucaparib for PARP14 were used as the positive controls (%inhibition data of the positive controls are reported in supporting information, Table S5).
To elucidate the plausible binding interactions of the most potent compound 49 at the active site of PARP1, we docked compound 49 using the Glide docking tool and co-crystal structure of full length PARP1-dihydrobenzofuran-7-carboxamide (Figure 6) [15]. In the binding model, the pharmacophore carboxamide group of 49 captures three key hydrogen bonding interactions with the side chain of Ser904 and the backbone atoms of Gly863, similar to what we observed for lead 4 (Figure S2, supporting information). Furthermore, a productive intramolecular hydrogen bond between the amide proton and the oxazinone ring oxygen atom was also evident. The phenyl ring of the benzoxazinone moiety formed a π-π stacking interaction with the side chain of Tyr907. The 4-hydroxybenzylidene moiety extended toward the ABP of the PARP1 active site while forming a hydrogen bond with the side chain of Ser864. Additionally, the phenolic hydroxyl group of 49 could form an electrostatic contact with the side chain of a key residue Asn868 due to the proximity between the two groups [9]. The benzoxazinone ring –NH- is found located near Glu988, which suggests a plausible water-mediated hydrogen bonding. Future studies will explore the effect of a wide range of substituents on the benzylidene ring to extend the molecule further into the ABP of PARP1 enzyme. These efforts will be facilitated by optimizing the reaction conditions for the Knoevenagel condensation, which is the low-yielding final step to produce target compounds.
Figure 6.
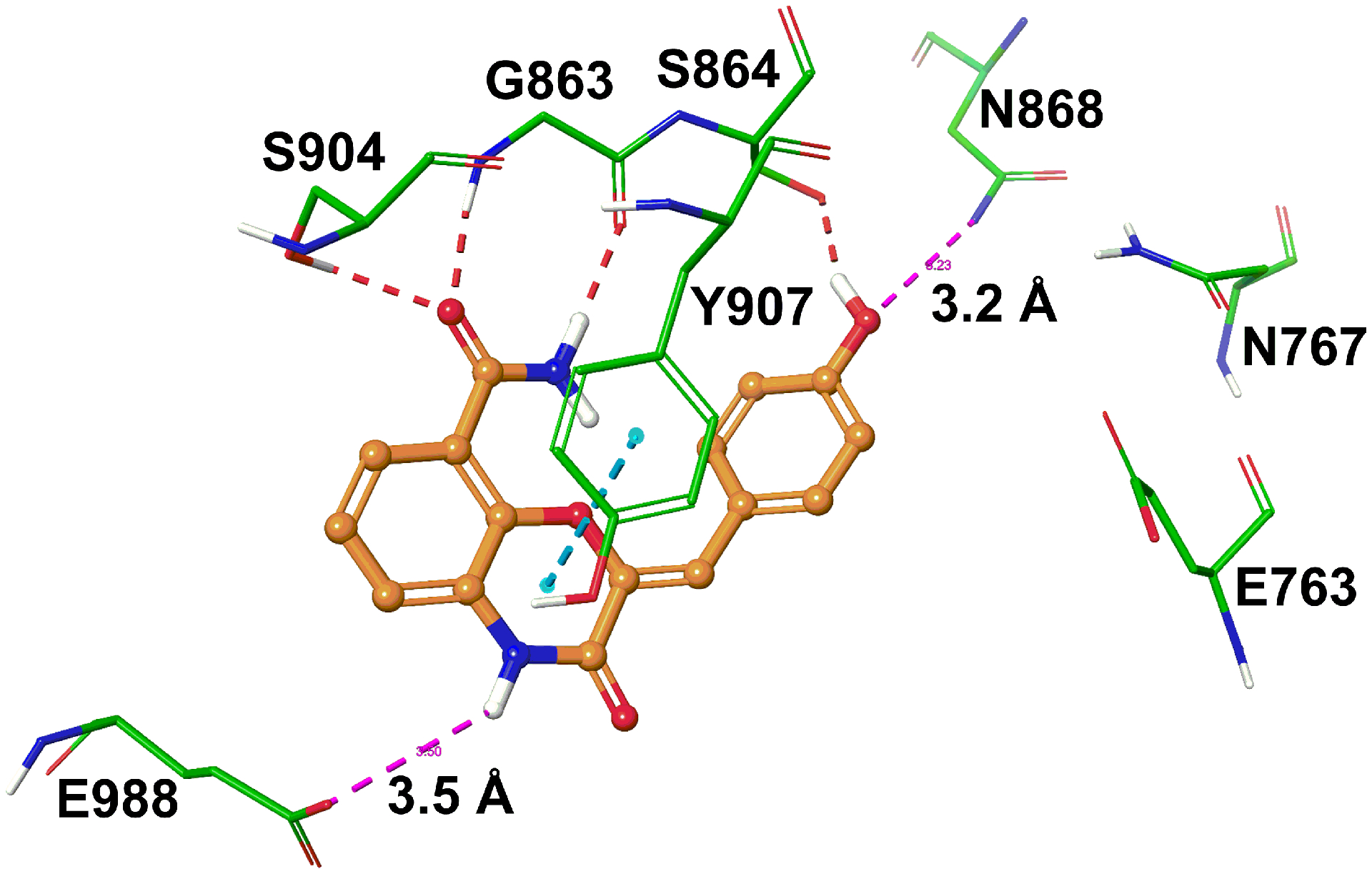
A binding model of 49 in the active site of PARP1. Key amino acids are depicted in stick model with the atoms colored as carbon – green, hydrogen – white, nitrogen – blue, and oxygen – red. The ligand is shown in ball and stick model with the same color scheme as above except carbon atoms are represented in orange. The broken red, pink and cyan lines indicate hydrogen bonding interaction, potential electrostatic contacts and π-π interaction, respectively.
Conclusion
In this study, PARP1 inhibitory hit compound 4 was first identified from a structure- and pharmacophore-based virtual screening protocol. Polar substitutions installed at the C2- or C3-positions of the benzodioxine ring of 4 led to analogues 21b and 26b with improved enzymatic inhibitory activities. Finally, utilization of the scaffold hopping strategy identified a refined lead compound 45 that was further optimized to generate a potent PARP1 inhibitor 49. Binding models of key compounds in the PARP1 active site allowed rationalization of the observed SAR trends. It is anticipated that compound 49 will serve as a promising lead compound for generation of selective PARP1 inhibitory agents with low nanomolar potency.
Experimental
High-throughput virtual screening
A detailed information regarding high-throughput virtual screening (ligand preparation, protein refinement, Glide docking procedure, and 3D pharmacophore modeling) can be found in supporting information.
Materials and methods
The selected 11 compounds from HTVS were obtained from Maybridge Chemicals Ltd. (Tintagel, United Kingdom) and Matrix Scientific (Columbia, SC). Starting materials for synthesis were received from the following vendors and used as received: Aldrich Chemical Co. (Milwaukee, WI), AK scientific (Union City, CA), Combi-Blocks Inc. (San Diego, CA), TCI America (Portland, OR), Gold Biotechnology (St. Louis, MO), Sigma-Aldrich (St. Louis, MO) and Alfa Aesar (Ward Hill, MA). The uniformity of all synthesized compounds was confirmed by thin layer chromatography (Agela Technologies). Thin layer chromatography (TLC) was employed to monitor the progress of reactions at 254 nm ultraviolet light. Bruker 400 UltrashieldTM spectrometer (1H at 400 MHz and 13C at 100 MHz) equipped with a z-axis gradient probe was used to record NMR experiments. Tetramethylsilane (TMS) was used as an internal standard, and chemical shift (δ) of 1H NMR was reported downfield of the TMS signal in parts per million (ppm). The 1H NMR data are presented as follows: chemical shift [multiplicity s (singlet), br (broad singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), and m (multiplet), number of protons, coupling constant]. 2D NMR experiments were carried out on Avance III NMR spectrometer with a 1H frequency of 500 MHz and 13C frequency of 125 MHz and equipped with an inverse probe. Samples were dissolved in DMSO-d6 or chloroform-d and experiments were carried out at room temperature. 1H and 13C chemical shift assignments were carried out with the aid of 1H-1H COSY, 1H-13C HSQC and 1H-13C HMBC experiments. Purification by flash chromatography was performed on Reveleris X2 flash chromatography system (BUCHI Corporation, New Castle, DE). High resolution mass spectra (HRMS) were collected for all unknown target compounds using a Waters Xevo G2-XS QToF mass spectrometer equipped with H-Class UPLC inlet and a LockSpray ESI source. Chiral HPLC analysis was performed using Agilent 1100 HPLC system and column (250 mm × 4.00 mm, serial no. 111156). Compounds were dissolved in acetonitrile and injected (10 μL) into Chirex column (phenomenex, Torrance, CA) with stationary phase as (S)-valine and (R)-1-(α-naphthyl)ethylamine. Optimum resolution of the enantiomers was achieved using isocratic mobile phases eluting at a flow rate of 1 mL/min. The elution was monitored at UV 254 nm in the form of major and minor peaks representing each enantiomer. Enantiomeric excess (%ee) was calculated based on the following equation, ee = ((R−S)/(R+S)) × 100%, where (R) and (S) denote individual optical isomers in the mixture and (R) + (S) = 1.
Synthetic procedures
General procedure to convert ester-containing intermediate to respective amides using mixed-anhydride method (A) [15].
Mixture of ester containing intermediate (2.0 mmol), lithium hydroxide (0.096 g, 4.0 mmol), and water (2 mL) in 10 mL of tetrahydrofuran was stirred at room temperature for 10 h. After removal of solvent under vacuum, the reaction mixture was diluted with ethyl acetate and extracted with water. The aqueous extract was then acidified using 2N aqueous hydrochloric acid solution followed by extraction with ethyl acetate (3 X 20 mL). The combined organic portion was dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to obtain the corresponding acid, which was subjected to the next step without further purification.
The acid was dissolved in 10 mL tetrahydrofuran, and the temperature was reduced to 0 °C under nitrogen atmosphere. After addition of isobutyl chloroformate (0.3 mL, 2.2 mmol) and N-methylmorpholine (0.24 mL, 2.2 mmol), the same condition was maintained until acid was completely converted to the desired mixed anhydride intermediate as monitored by TLC. Then a 2.0 mL solution of 7N ammonia in methanol was added, and the reaction mixture was stirred for 20 min at room temperature. After extraction with ethyl acetate and water, crude product was purified using flash chromatography with appropriate combination of ethyl acetate and hexanes.
Methyl 2,3-dihydroxybenzoate (13) [24].
To a solution of 2,3-dihydroxybenzoic acid (0.3 g, 1.9 mmol) in 20 mL of methanol, concentrated sulfuric acid (0.3 mL) was added at 5 °C. The reaction mixture was refluxed for 12 h, cooled to room temperature, and concentrated under vacuum. The resulted mass was dissolved in ethyl acetate and washed with sodium carbonate solution three times. The organic extract was dried over magnesium sulfate and concentrated under reduced pressure to obtain 13 as a white powder (yield: 0.278 g, 85%). 1 H NMR (400 MHz; DMSO-d6; TMS) δ 9.94 (s, 2H), 7.23 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 7.04 (dd, J = 7.9 Hz, 1.6 Hz, 1H), 6.75 (t, J = 7.9 Hz, 1H), 3.88 (s, 3H).
Methyl 2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylate (14) [17].
1,2-Dibromoethane (0.17 mL, 2.0 mmol) was added to a suspension of 13 (0.336 g, 2.0 mmol) and K2CO3 (0.304 g, 2.2 mmol) in 5 mL dimethylformamide. The reaction mixture was stirred under reflux for 10 h. When the starting material was completely consumed as indicated by TLC, the mixture was diluted with water and extracted with ethyl acetate. The organic portion was dried over anhydrous magnesium sulfate and evaporated under reduced pressure. The crude product was purified by flash chromatography (ethyl acetate:n-hexane 10:90) to obtain 14 as a white powder (yield: 0.272 g, 70%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.24 (d, J = 7.8 Hz, 1H), 7.05 (d, J = 7.8 Hz, 1H,), 6.87 (t, J = 8.1 Hz, 1H), 4.29 – 4.28 (m, 4H), 3.79 (s, 3H).
2,3-Dihydrobenzo[b][1,4]dioxine-5-carboxamide (4).
Compound 4 was prepared from 14 (0.288 g, 2.0 mmol) according to general procedure A as a white powder (yield: 0.186 g, 70%); mp 130–132 °C [lit. mp 130 °C] [25]. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.62 (s, 1H), 7.48 (s, 1H), 7.34 (dd, J = 7.9 Hz, 1.8 Hz, 1H), 6.98 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 6.87 (t, J = 7.9 Hz, 1H), 4.37 – 4.35 (m, 2H), 4.28 – 4.26 (m, 2H). 13C NMR (100 MHz; DMSO-d6; TMS): δ 165.87, 143.56, 141.91, 123.64, 122.27, 120.41, 119.53, 64.49, 63.55. HRMS (m/z): [M + H]+ calcd for C9H10NO3, 180.0655; found: 180.0634.
7-Nitro-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (15) [15].
A solution of 4 (0.540 g, 3.0 mmol) in 5 mL (65.3 mmol) of trifluoroacetic acid was cooled in an ice bath, and 1.2 mL (28.8 mL) nitric acid was added dropwise. The reaction mixture was stirred for 6 h, and then poured into ice water. The precipitate was filtered and the crude product was purified using flash chromatography and subsequently reprecipitated using ethyl acetate and hexane to obtain pure compound 15 as a yellow powder (yield: 0.061 g, 9%); 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.13 (d, J = 2.8 Hz, 1H), 7.86 (s, 1H), 7.82 (d, J = 2.8 Hz, 1H), 7.62 (s, 1H), 4.49 (d, J = 3.9 Hz, 2H), 4.38 (d, J = 4.0 Hz, 2H). 13C NMR (100 MHz; DMSO-d6; TMS): δ 164.00, 147.73, 143.85, 140.09, 124.13, 117.72, 113.92, 65.21, 63.65.
8-Nitro-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (16).
Compound 16 was isolated from the above-mentioned nitration reaction of 4 as yellow powder (yield: 0.088 g, 13%); 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.86 (s, 1H), 7.69 (d, J = 9.1 Hz, 1H), 7.62 (s, 1H), 7.07 (d, J = 9.1 Hz, 1H), 4.39 – 4.37 (m, 2H), 4.34 – 4.32 (m, 2H).
7-Amino-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (17) [15].
To a solution of 15 (0.448 g, 2.0 mmol) in 30 mL methanol, Pd/C (0.04 g) was added. The mixture was stirred under 4 atmosphere of hydrogen gas at room temperature for 8 h. After a halt in pressure drop, the suspension was filtered through celite pad. The filtrate was concentrated, diluted with ethyl acetate and washed with water. Ethyl acetate layer was dried over magnesium sulfate and evaporated under vacuum. The crude product was purified using flash chromatography (methanol:dichloromethane 5:95) to obtain 17 as a pale brown powder (yield: 0.233 g, 60%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.44 (s, 1H), 7.39 (s, 1H), 6.64 (d, J = 2.8 Hz, 1H), 6.22 (d, J = 2.8 Hz, 1H), 4.79 (s, 2H), 4.23 – 4.19 (m, 4H). 13C NMR (100 MHz; DMSO-d6; TMS): δ 166.08, 143.75, 142.22, 132.89, 123.05, 108.23, 105.29, 64.14, 63.83. HRMS (m/z): [M + H]+ calcd for C9H11N2O3, 195.0764; found: 195.0747.
8-Amino-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (18).
Compound 18 was prepared following the procedure used for synthesis of 17, using 16 (0.448 g, 2.0 mmol) as a starting material. Compound 18 (yield: 0.272 g, 70%) was isolated as a light brown powder. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.47 (s, 1H), 7.36 (s, 1H), 6.66 (d, J = 8.9 Hz, 1H), 6.20 (d, J = 8.9 Hz, 1H), 5.78 (s, 2H), 4.26 – 4.25 (m, 2H), 4.12 – 4.10 (m, 2H). HRMS (m/z): [M + H]+ calcd for C9H11N2O3, 195.0764; found: 195.0741.
Methyl (S)-2-(Hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylate (19a) [16].
Commercially available (2S)-glycidyl tosylate (0.456 g, 2.0 mmol) was added to a suspension of 13 (0.336 g, 2.0 mmol) and K2CO3 (0.304 g, 2.2 mmol) in 10 mL dimethylformamide. The mixture was stirred for 30 h at 40 °C, diluted with water and extracted with ethyl acetate. Ethyl acetate layer was dried over magnesium sulfate, and concentrated under reduced pressure. Flash chromatography (methanol:dichloromethane 5:95) was used to obtain 19a (yield: 0.089 g, 20%) as a white solid. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.36 (dd, J = 7.9 Hz, 1.6 Hz, 1H), 7.00 (dd, J = 8.1 Hz, 1.6 Hz, 1H), 6.81 (t, J = 7.9 Hz, 1H), 4.42 (dd, J = 11.6 Hz, 2.2 Hz, 1H), 4.28 – 4.23 (m, 1H), 4.15 – 4.05 (m, 4H), 3.84 (s, 3H).
Methyl (R)-3-(hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylate (19b).
From the same reaction of intermediate 13 (0.336 g, 2.0 mmol) and (2S)-glycidyl tosylate (0.456 g, 2.0 mmol), compound 19b (yield: 0.116 g, 26%) was isolated as a white solid. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.73 (dd, J = 7.9 Hz, 1.7 Hz, 1H), 7.17 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 7.02 (t, J = 8.0 Hz, 1H), 4.61 (dd, J = 15.2 Hz, 6.6 Hz, 1H), 4.41 – 4.34 (m, 2H), 4.03 – 3.91 (m, 3H), 3.83 (s, 3H).
(S)-2-(Hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (20a).
Compound 20a was prepared according to general procedure A, starting with 19a (0.448 g, 2.0 mmol). Purified compound 20a (yield: 0.293 g, 70%) was obtained as a white powder, mp 142–144 °C. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.54 (s, 2H), 7.27 (dd, J = 7.8 Hz, 1.7 Hz, 1H), 6.99 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 6.87 (t, J = 7.8 Hz, 1H), 5.10 (t, J = 5.7 Hz, 1H), 4.45 (dd, J = 9.3 Hz, 2.1 Hz, 1H), 4.21 – 4.16 (m, 1H), 4.11 – 4.06 (m, 1H), 3.69 – 3.58 (m, 2H). HRMS (m/z): [M + H]+ calcd for C10H12NO4, 210.0761; found: 210.0753. ee = 94%, (tR (major) = 10.42, tR (minor) = 9.77).
(R)-3-(Hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (20b).
Compound 20b was prepared according to general procedure A, starting with 19b (0.448 g, 2.0 mmol). Purified compound 20b (yield: 0.251 g, 60%) was obtained as a white powder, mp 144–146 °C. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.61 (s, 1H), 7.55 (s, 1H), 7.37 (dd, J = 7.8 Hz, 1.7 Hz, 1H), 7.01 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 6.88 (t, J = 7.9 Hz, 1H), 5.19 (t, J = 11.5 Hz, 1H), 4.36 (dd, J = 8.7 Hz, 2.6 Hz, 1H), 4.32 – 4.27 (m, 1H), 4.08 – 4.03 (m, 1H), 3.74 – 3.63 (m, 2H). HRMS (m/z): [M + H]+ calcd for C10H12NO4, 210.0761; found: 210.0744. ee = 93%, (tR (major) 7.43, tR (minor) = 9.01).
(R)-(5-Carbamoyl-2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl methanesulfonate (21a) [26].
Methanesulfonyl chloride (0.15 mL, 2.0 mmol) was added to a solution of 20a (0.518 g, 2.0 mmol) and triethylamine (0.3 mL, 2.2 mmol) in 20 mL dichloromethane at −10 °C. The mixture was stirred for 10 h at room temperature, and the reaction mixture was washed with water. Then the combined organic portion was dried over magnesium sulfate, and concentrated under vacuum. After purification with column chromatography, compound 21a (yield:0.356 g, 50%) was obtained as a white solid, mp 145–147 °C. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.56 (br, 2H), 7.29 (dd, J = 7.7 Hz, 1.5 Hz, 1H), 7.05 (dd, J = 7.9 Hz, 1.5 Hz, 1H), 6.92 (t, J = 7.9 Hz, 1H), 4.56 – 4.43 (m, 4H), 4.18 – 4.13 (m, 1H), 3.26 (s, 3H). HRMS (m/z): [M + H]+ calcd for C11H14NO6S, 288.0536; found: 288.0540. ee = 94%, (tR (major) 11.71, tR (minor) = 13.05).
(S)-(8-Carbamoyl-2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl methanesulfonate (21b).
Compound 21b was prepared following the procedure used for the synthesis of 21a, using 20b (0.287 g, 1.0 mmol) as a starting material. Purified compound 21b (yield: 0.217 g, 55%) was obtained as a white solid, mp 140–143 °C. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.64 (s, 1H), 7.47 (s, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.04 (d, J = 7.8 Hz, 1H), 6.92 (t, J = 7.8 Hz, 1H), 4.63 (d, J = 10.1 Hz 2H), 4.51 (dd, J = 11.6 Hz, 5.7 Hz, 1H), 4.44 (d, J = 11.1 Hz, 1H), 4.09 (dd, J = 11.2 Hz, 7.7 Hz 1H), 3.26 (s, 3H). HRMS (m/z): [M + H]+ calcd for C11H14NO6S, 288.0536; found: 288.0511. ee = 93%, (tR (major) 12.88, tR (minor) = 13.59).
(S)-2-((4-Methylpiperazin-1-yl)methyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (22a) [27].
A mixture of 21a (0.287 g, 1.0 mmol) and N-methylpiperazine (0.33 mL, 3.0 mmol) in ethanol was stirred for 10 h under reflux. Ethanol was then removed under vacuum. The concentrated mass was diluted with water, and extracted with ethyl acetate at potassium carbonate-assisted alkaline pH. After drying over magnesium sulfate, the organic layer was concentrated under reduced pressure and purified using preparative TLC (methanol:dichloromethane = 5: 95) to obtain compound 22a (yield: 0.058 g, 20%) as a white solid, mp 173–175 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.74 (dd, J = 7.8 Hz, 1.8 Hz, 1H), 7.52 (s, 1H), 7.04 (dd, J = 8.1 Hz, 1.8 Hz, 1H), 6.94 (t, J = 7.8 Hz, 1H), 5.76 (br, 1H), 4.50 (dd, J = 11.1 Hz, 2.3 Hz, 1H), 4.38 – 4.32 (m, 1H), 4.13 (dd, J = 11.3 Hz, 10.0 Hz, 1H), 2.76 – 2.45 (m, 10H), 2.30 (s, 3H). HRMS (m/z): [M + H]+ calcd for C15H22N3O3, 292.1656; found: 292.1685.
(R)-3-((4-Methylpiperazin-1-yl)methyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (22b).
Compound 22b was synthesized according to the procedure for compound 22a, starting with 21b (0.287 g, 1.0 mmol) and N-methylpiperazine (0.33 mL, 3.0 mmol). The mixture was purified using preparative TLC to obtain compound 22b (yield: 0.070 g, 24%) as a white solid, mp 185–187 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.76 (dd, J = 8.1 Hz, 1.9 Hz, 1H), 7.46 (s, 1H), 7.08 (dd, J = 8.1 Hz, 1.7 Hz, 1H), 6.97 (t, J = 8.0 Hz, 1H), 5.75 (br, 1H), 4.51 – 4.35 (m, 4H), 4.20 (dd, J = 8.5 Hz, 6.7 Hz, 1H), 3.50 (s, 4H), 2.39 – 2.30 (m, 7H). HRMS (m/z): [M + H]+ calcd for C15H22N3O3, 292.1656; found: 292.1629.
(S)-2-((4-(Pyrimidin-2-yl)piperazin-1-yl)methyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (23a).
Compound 23a was synthesized according to the procedure used to obtain compound 22a, starting with 21a (0.287 g, 1.0 mmol) and 1-(2-pyrimidinyl)piperazine (0.43 mL, 3.0 mmol). Compound 23a (yield: 0.117 g, 33%) was isolated as a white solid, mp 180–182 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.31 (d, J = 4.8 Hz, 2H), 7.76 (dd, J = 7.8 Hz, 1.9 Hz, 1H), 7.52 (s, 1H), 7.05 (dd, J = 8.1 Hz, 1.8 Hz, 1H), 6.96 (t, J = 7.9 Hz, 1H), 6.50 (t, J = 4.8 Hz, 1H), 5.73 (br, 1H), 4.56 (dd, J = 11.6 Hz, 2.5 Hz, 1H), 4.40 (t, J = 6.1 Hz, 1H), 4.56 (dd, J = 11.3 Hz, 9.5 Hz, 1H), 3.84 (t, J = 4.9 Hz, 4H), 2.80 – 2.57 (m, 6H). HRMS (m/z): [M + H]+ calcd for C18H22N5O3, 356.1717; found: 356.1736.
(R)-3-((4-(Pyrimidin-2-yl)piperazin-1-yl)methyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (23b).
Compound 23b was synthesized according to the procedure reported for compound 22a, starting with 21b (0.287 g, 1.0 mmol) and 1-(2-pyrimidinyl)piperazine (0.43 mL, 3.0 mmol). Compound 23b (yield: 0.135 g, 38%) was isolated as a white solid, mp 155–157 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.33 (d, J = 4.7 Hz, 2H), 7.76 (dd, J = 7.8 Hz, 1.8 Hz, 1H), 7.46 (s, 1H), 7.09 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 6.98 (t, J = 8.0 Hz, 1H), 6.54 (t, J = 4.8 Hz, 1H), 5.75 (br, 1H), 4.54 – 4.42 (m, 4H), 4.23 (dd, J = 12.0 Hz, 10.8 Hz, 1H), 3.83 (s, 4H), 3.54 (dd, J = 12.7 Hz, 6.3 Hz, 4H). HRMS (m/z): [M + H]+ calcd for C18H22N5O3, 356.1717; found: 356.1711.
Methyl (R)-2-(hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylate (24a).
Compound 24a was prepared by the procedure used for the synthesis of 19a, with 13 (0.336 g, 2.0 mmol) and (2R)-glycidyl tosylate (0.456 g, 2.0 mmol) as starting materials. Intermediate 24a (yield: 0.098 g, 22%) was isolated as a white powder. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.35 (dd, J = 7.9 Hz, 1.6 Hz, 1H), 7.00 (dd, J = 8.1 Hz, 1.6 Hz, 1H), 6.81 (t, J = 7.9 Hz, 1H), 4.42 (dd, J = 11.6 Hz, 2.1 Hz, 1H), 4.28 – 4.20 (m, 1H), 4.15 – 4.05 (m, 4H), 3.83 (s, 3H).
Methyl (S)-3-(hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylate (24b).
Compound 24b was isolated from the same reaction from which 24a was obtained. Crude product was purified to yield compound 24b (yield: 0.157 g, 35%) as a white solid. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.73 (dd, J = 7.9 Hz, 1.7 Hz, 1H), 7.17 (dd, J = 7.9 Hz, 1.7 Hz, 1H), 7.02 (t, J = 8.0 Hz, 1H), 4.61 (dd, J = 15.2 Hz, 6.6 Hz, 1H), 4.43 – 4.34 (m, 2H), 4.05 – 3.91 (m, 3H), 3.83 (s, 3H).
(R)-2-(Hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (25a).
Compound 25a was prepared according to general procedure A, starting with 24a (0.448 g, 2.0 mmol). Crude product was purified to yield compound 25a (yield: 0.251 g, 60%) as a white powder, mp 142–144 °C. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.54 (s, 2H), 7.27 (dd, J = 8.1 Hz, 1.7 Hz, 1H), 6.99 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 6.87 (t, J = 7.9 Hz, 1H), 5.10 (t, J = 5.7 Hz, 1H), 4.45 (dd, J = 9.3 Hz, 2.1 Hz, 1H), 4.21 – 4.16 (m, 1H), 4.11 – 4.06 (m, 1H), 3.69 – 3.58 (m, 2H). HRMS (m/z): [M + H]+ calcd for C10H12NO4, 210.0761; found: 210.0772. ee = 93%, (tR (major) = 9.75, tR (minor) = 10.42).
(S)-3-(Hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (25b).
Compound 25b was prepared according to general procedure A, starting with 24b (0.448 g, 2.0 mmol). Crude product was purified to yield compound 25b (yield: 0.293 g, 70%) as a white solid, mp 144–146 °C. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.61 (s, 1H), 7.55 (s, 1H), 7.37 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 7.01 (dd, J = 7.9 Hz, 1.7 Hz, 1H), 6.88 (t, J = 7.9 Hz, 1H), 5.19 (t, J = 5.8 Hz, 1H), 4.36 (dd, J = 8.7 Hz, 2.6 Hz, 1H), 4.32 – 4.27 (m, 1H), 4.08 – 4.03 (m, 1H), 3.74 – 3.63 (m, 2H). HRMS (m/z): [M + H]+ calcd for C10H12NO4, 210.0761; found: 210.0752. ee = 92%, (tR (major) 8.92, tR (minor) = 7.40).
(S)-(5-Carbamoyl-2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl methanesulfonate (26a).
Compound 26a was prepared by the procedure used for the synthesis of 21a using 25a (0.287 g, 1.0 mmol) as the starting material. Crude product was purified to yield compound 26a (yield: 0.197 g, 50%) as a white solid, mp 145–147 °C. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.56 (br, 2H), 7.29 (dd, J = 7.9 Hz, 1.5 Hz, 1H), 7.05 (dd, J = 7.9 Hz, 1.5 Hz, 1H), 6.92 (t, J = 7.8 Hz, 1H), 4.56–4.43 (m, 4H), 4.16 (dd, J = 11.5 Hz, 6.8 Hz, 1H), 3.26 (s, 3H). ee = 93%, (tR (major) 13.05, tR (minor) = 11.72).
(R)-(8-Carbamoyl-2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl methanesulfonate (26b).
Compound 26b was prepared by the procedure used for the synthesis of 21a using 25b (0.287 g, 1.0 mmol) as the starting material. Crude product was purified to yield compound 26b (yield: 0.217 g, 55%) as a white solid, mp 140–143 °C. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.64 (s, 1H), 7.47 (s, 1H), 7.36 (d, J = 7.9 Hz, 1H), 7.04 (d, J = 8.1 Hz, 1H), 6.92 (t, J = 7.7 Hz, 1H), 4.63 (d, J = 10.8 Hz, 2H), 4.53 – 4.49 (m, 1H), 4.44 (d, J = 11.9 Hz, 1H), 4.09 (dd, J = 10.7 Hz, 7.5 Hz, 1H), 3.26 (s, 3H). HRMS (m/z): [M + H]+ calcd for C11H14NO6S, 288.0536; found: 288.0517. ee = 92%, (tR (major) 13.59, tR (minor) = 12.88).
(R)-2-((4-Methylpiperazin-1-yl)methyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (27a).
Compound 27a was synthesized according to the procedure used for compound 22a using 26a (0.287 g, 1.0 mmol) and N-methylpiperazine (0.33 mL, 3.0 mmol) as the starting materials. Crude product was purified to yield compound 27a (yield: 0.058 g, 20%) as white solid, mp 173–175 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.74 (dd, J = 7.8 Hz, 1.8 Hz, 1H), 7.52 (s, 1H), 7.04 (dd, J = 8.1 Hz, 1.8 Hz, 1H), 6.94 (t, J = 7.8 Hz, 1H), 5.76 (br, 1H), 4.50 (dd, J = 11.1 Hz, 2.3 Hz, 1H), 4.38 – 4.32 (m, 1H), 4.13 (dd, J = 11.3 Hz, 10.0 Hz, 1H), 2.76 – 2.45 (m, 10H), 2.30 (s, 3H). HRMS (m/z): [M + H]+ calcd for C15H22N3O3, 292.1656; found: 292.1637.
(S)-3-((4-Methylpiperazin-1-yl)methyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (27b).
Compound 27b was synthesized according to the procedure reported for compound 22a using 26b (0.287 g, 1.0 mmol) and N-methylpiperazine (0.33 mL, 3.0 mmol) as the starting materials. Crude product was purified to yield compound 27b (yield: 0.058 g, 20%) as a white solid, mp 185–187 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.76 (dd, J = 8.1 Hz, 1.9 Hz, 1H), 7.46 (s, 1H), 7.08 (dd, J = 8.1 Hz, 1.7 Hz, 1H), 6.97 (t, J = 8.0 Hz, 1H), 5.75 (br, 1H), 4.51 – 4.35 (m, 4H), 4.20 (dd, J = 8.5 Hz, 6.7 Hz, 1H), 3.50 (s, 4H), 2.39 – 2.30 (m, 7H). HRMS (m/z): [M + H]+ calcd for C15H22N3O3, 292.1656; found: 292.1685.
(R)-2-((4-(Pyrimidin-2-yl)piperazin-1-yl)methyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (28a).
Compound 28a was synthesized according to the procedure used for 22a with 26a (0.287 g, 1.0 mmol) and 1-(2-pyrimidinyl)piperazine (0.43 mL, 3.0 mmol) as the starting materials. Crude product was purified to yield compound 28a (yield: 0.092 g, 26%) as a white solid, mp 180–182 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.31 (d, J = 4.8 Hz, 2H), 7.76 (dd, J = 7.8 Hz, 1.9 Hz, 1H), 7.52 (s, 1H), 7.05 (dd, J = 8.1 Hz, 1.8 Hz, 1H), 6.96 (t, J = 7.9 Hz, 1H), 6.50 (t, J = 4.8 Hz, 1H), 5.73 (br, 1H), 4.56 (dd, J = 11.6 Hz, 2.5 Hz, 1H), 4.40 (t, J = 6.1 Hz, 1H), 4.56 (dd, J = 11.3 Hz, 9.5 Hz, 1H), 3.84 (t, J = 4.9 Hz, 4H), 2.80 – 2.57 (m, 6H). HRMS (m/z): [M + H]+ calcd for C18H22N5O3, 356.1717; found: 356.1702.
(S)-3-((4-(Pyrimidin-2-yl)piperazin-1-yl)methyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (28b).
Compound 28b was synthesized according to the procedure for compound 22a with 26b (0.287 g, 1.0 mmol) and 1-(2-pyrimidinyl)piperazine (0.43 mL, 3.0 mmol) as the starting materials. Crude product was purified to yield compound 28b (yield: 0.110 g, 31%) as a white solid, mp 155–157 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.33 (d, J = 4.7 Hz, 2H), 7.76 (dd, J = 7.8 Hz, 1.8 Hz, 1H), 7.46 (s, 1H), 7.09 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 6.98 (t, J = 8.0 Hz, 1H), 6.54 (t, J = 4.8 Hz, 1H), 5.75 (br, 1H), 4.54 – 4.42 (m, 4H), 4.23 (dd, J = 12.0 Hz, 10.8 Hz, 1H), 3.83 (s, 4H), 3.54 (dd, J = 12.7 Hz, 6.3 Hz, 4H). HRMS (m/z): [M + H]+ calcd for C18H22N5O3, 356.1717; found: 356.1698.
Methyl benzo[d][1,3]dioxole-4-carboxylate (29).
Compound 29 was prepared in the same manner as 14, using 13 (0.336 g, 2.0 mmol) and dibromomethane (0.14 mL, 2.0 mmol) as starting materials. Crude product was purified to yield compound 29 (yield: 0.234 g, 65%) as a white powder. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.30 (dd, J = 8.1 Hz, 1.2 Hz, 1H), 7.16 (dd, J = 7.7 Hz, 1.2 Hz, 1H), 6.93 (t, J = 7.8 Hz, 1H), 6.15 (s, 2H,), 3.82 (s, 3H).
Methyl 3,4-dihydro-2H-benzo[b][1,4]dioxepine-6-carboxylate (30).
Compound 30 was prepared by the procedure for 14, using 13 (0.336 g, 2.0 mmol) and 1,3-dibromopropane (0.21 mL, 2.0 mmol) as starting materials. Crude product was purified to yield compound 30 (yield: 0.229 g, 55%) as a white powder. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.34 (dd, J = 7.8 Hz, 1.8 Hz, 1H), 7.08 (dd, J = 8.0 Hz, 1.8 Hz, 1H), 6.99 (t, J = 7.8 Hz, 1H), 4.28 (t, J = 5.3 Hz, 2H), 4.13 (t, J = 5.3 Hz, 2H), 2.15 – 2.09 (m, 2H), 3.77 (s, 3H).
Benzo[d][1,3]dioxole-4-carboxamide (31).
Compound 31 was prepared according to general procedure A, starting with 29 (0.360 g, 2.0 mmol). Crude product was purified to yield compound 31 (yield: 0.198 g, 60%) as a white solid. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.65 (s, 1H), 7.25 (d, J = 8.0 Hz, 1H), 7.16 (s, 1H), 7.07 (d, J = 7.7 Hz, 1H), 6.91 (t, J = 7.9 Hz, 1H), 6.13 (s, 2H). HRMS (m/z): [M + H]+ calcd for C8H8NO3, 166.0499; found: 166.0477.
3,4-Dihydro-2H-benzo[b][1,4]dioxepine-6-carboxamide (32).
Compound 32 was prepared according to general procedure A, using 30 (0.416 g, 2.0 mmol) as the starting material. Crude product was purified to yield compound 32 (yield: 0.232 g, 60%) as a white powder, mp 106–108 °C. 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.62 (s, 1H), 7.48 (s, 1H), 7.34 (dd, J = 7.9 Hz, 1.8 Hz, 1H), 7.08 (dd, J = 8.0 Hz, 1.8 Hz, 1H), 6.99 (t, J = 7.8 Hz, 1H), 4.23 (t, J = 5.5 Hz, 2H), 4.14 (t, J = 5.4 Hz, 2H), 2.16 – 2.10 (m, 2H). 13C NMR (100 MHz; DMSO-d6; TMS): δ 166.69, 151.75, 149.38, 128.67, 123.90, 123.70, 122.89, 70.89, 70.42, 31.15. HRMS (m/z): [M + H]+ calcd for C10H12NO3, 194.0812; found: 194.0799.
Methyl 2-hydroxy-3-nitrobenzoate (34).
Compound 34 was prepared by the procedure for 13 using commercially available 2-hydroxy-3-nitrobenzoic acid 33 (0.366 g, 2.0 mmol) as the starting material. Crude product was purified to yield compound 34 (yield: 0.347 g, 88%) as a white powder. 1H NMR (400 MHz; DMSO-d6; TMS) δ 11.48 (s, 1H), 8.18 (dd, J = 8.1 Hz, 1.7 Hz, 1H), 8.09 (dd, J = 7.9 Hz, 1.7 Hz, 1H), 7.11 (t, J = 8.1 Hz, 1H), 3.93 (s, 3H).
Methyl 2-(2-ethoxy-2-oxoethoxy)-3-nitrobenzoate (35) [18].
Ethyl bromoacetate (0.12 mL, 1.1 mmol) and K2CO3 (0.276 g, 2.0 mmol) were added to a solution of compound 34 (0.197 g, 1.0 mmol) in 10 mL dimethylformamide. After stirring for 8 h at room temperature, the mixture was diluted with ethyl acetate, and washed with water. Pure product 35 (yield: 0.212 g, 75%) was obtained by flash chromatography (methanol:dichloromethane 10:90) as a white solid. 1H NMR (400 MHz; chloroform-d) δ 8.10 (dd, J = 6.3 Hz, 1.6 Hz, 1H), 7.99 (dd, J = 8.0 Hz, 1.3 Hz, 1H), 7.36 (t, J = 8.1 Hz, 1H), 4.82 (s, 2H), 4.30 (q, J = 7.2 Hz, 2H), 3.94 (s, 3H), 1.32 (t, J = 7.1 Hz, 3H).
Methyl 2-(2-ethoxy-2-oxo-1-phenylethoxy)-3-nitrobenzoate (36).
Compound 36 was prepared in a similar manner as 35 using compound 34 (0.197 g, 1.0 mmol) and commercially available ethyl 2-bromo-2-phenylacetate (0.267 g, 1.1 mmol) as the starting materials. Purification by column chromatography using ethyl acetate:hexane mixture as an eluent yielded compound 36 (yield: 0.280 g, 78%) as a yellow oil. 1H NMR (400 MHz, Chloroform-d) δ 7.98 (dd, J = 7.9 Hz, 1.7 Hz, 1H), 7.84 (dd, J = 8.0 Hz, 1.9 Hz, 1H), 7.46 – 7.37 (m, 2H), 7.37 – 7.28 (m, 3H), 7.22 (t, J = 8.0 Hz, 1H), 5.58 (s, 1H), 4.10 (q, J = 7.1 Hz, 2H), 3.79 (s, 3H), 1.10 (t, J = 7.1 Hz, 3H).
Methyl 2-(2-ethoxy-1-(4-methoxyphenyl)-2-oxoethoxy)-3-nitrobenzoate (37).
Compound 37 was prepared as for 35 using compound 34 (0.197 g, 1.0 mmol) and commercially available ethyl 2-bromo-2-(4-methoxyphenyl)acetate (0.301 g, 1.1 mmol) as the starting materials. Purification of crude product by column chromatography using ethyl acetate:hexane mixture as an eluent yielded compound 37 (yield: 0.269 g, 69%) as a dark yellow colored oil. 1H NMR (400 MHz, DMSO-d6) δ 8.05 (td, J = 8.0 Hz, 1.8 Hz, 2H), 7.42 (t, J = 8.0 Hz, 1H), 7.33 – 7.27 (m, 2H), 6.98 – 6.90 (m, 2H), 5.63 (s, 1H), 4.09 (q, J = 7.1 Hz, 2H), 3.81 (s, 3H), 3.76 (s, 3H), 1.08 (t, J = 7.1 Hz, 3H).
Methyl 2-(1-(4-bromophenyl)-2-ethoxy-2-oxoethoxy)-3-nitrobenzoate (38).
Compound 38 was prepared as for 35 using compound 34 (0.197 g, 1.0 mmol) and commercially available ethyl 2-bromo-2-(4-bromophenyl)acetate (0.354 g, 1.1 mmol) as the starting materials. Purification of crude product by column chromatography using ethyl acetate:hexane mixture as an eluent yielded compound 38 (yield: 0.368 g, 84%) as a brown colored oil. 1H NMR (400 MHz, DMSO-d6) δ 8.16 – 8.03 (m, 2H), 7.63 (d, J = 8.2 Hz, 2H), 7.46 (t, J = 7.9 Hz, 1H), 7.36 (d, J = 8.3 Hz, 2H), 5.74 (br, 1H), 4.15 – 4.01 (m, 2H), 3.77 (s, 3H), 1.07 (t, J = 7.1 Hz, 3H).
Methyl 3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxylate (39).
Iron (0.283 g) was added into a solution of 35 (0.283 g, 1.0 mmol) in 10 mL of acetic acid. After stirring for 4 h under reflux conditions, the reaction mixture was concentrated under vacuum and the crude product was directly loaded onto a silica gel column and purified using ethyl acetate:hexane mixture to obtain 39 (yield: 0.145 g, 70%) as a white solid. 1H NMR (400 MHz; DMSO-d6) δ 10.88 (s, 1H), 7.33 (dd, J = 7.8 Hz, 1.8 Hz, 1H), 7.09 (dd, J = 7.8 Hz, 1.7 Hz, 1H), 7.03 (t, J = 7.8 Hz, 1H), 4.65 (s, 2H), 3.80 (s, 3H).
Methyl 3-oxo-2-phenyl-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxylate (40).
Compound 40 was prepared as for 39 using intermediate 36 (0.359 g, 1.0 mmol). The resulting mixture was purified by column chromatography using ethyl acetate:hexane mixtures as an eluent to obtain compound 40 (yield: 0.221 g, 78%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.14 (s, 1H), 7.63 – 7.22 (m, 6H), 7.07 (dd, J = 23.9 Hz, 7.8 Hz, 2H), 5.91 (s, 1H), 3.80 (s, 3H).
Methyl 2-(4-methoxyphenyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxylate (41).
Compound 41 was prepared as for 39 using intermediate 37 (0.389 g, 1.0 mmol). The resulting mixture was purified by column chromatography using ethyl acetate:hexane mixtures as an eluent to yield compound 41 (yield: 0.216 g, 69%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.11 (s, 1H), 7.33 (d, J = 7.9 Hz, 3H), 7.16 – 6.98 (m, 2H), 6.92 (d, J = 8.4 Hz, 2H), 5.82 (s, 1H), 3.79 (s, 3H), 3.73 (s, 3H).
Methyl 2-(4-bromophenyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxylate (42).
Compound 42 was prepared as for 39 using intermediate 38 (0.438 g, 1.0 mmol). The resulting mixture was purified by column chromatography using ethyl acetate:hexane mixtures as an eluent to obtain compound 42 (yield: 0.257 g, 78%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.18 (s, 1H), 7.80 – 7.24 (m, 5H), 7.24 – 6.91 (m, 2H), 5.92 (s, 1H), 3.81 (s, 3H).
Methyl 3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxylate (43) [19].
A solution of 39 (0.414 g, 2.0 mmol) in 10 mL of tetrahydrofuran was treated with boron trifluoride diethyl etherate complex (0.52 mL, 4.2 mmol) in an ice bath, and NaBH4 (0.159 g, 4.2 mmol) was added after stirring for 30 min. 2M hydrochloric acid solution was used to quench the reaction, followed by extraction with ethyl acetate. The organic portion was dried over magnesium sulfate and concentrated under reduced pressure. The resulted crude product was purified using flash chromatography (ethyl acetate:hexane 30:70) to obtain 43 (yield: 0.088 g, 23%) as a white solid. 1H NMR (400 MHz; DMSO-d6) δ 6.70–6.63 (m, 2H), 6.48 (1H, dd, J = 5.2 Hz, 2.1 Hz, 1H), 4.56 (s, 1H), 4.14 (t, J = 4.4 Hz, 2H), 3.97 (s, 3H), 3.23 (t, J = 4.4 Hz, 2H).
3,4-Dihydro-2H-benzo[b][1,4]oxazine-8-carboxamide (44).
Compound 44 was prepared according to general procedure A, starting from 43 (0.354 g, 2.0 mmol) as a white powder (yield: 0.214 g, 60%), mp 165–168 °C. 1H NMR (400 MHz; DMSO-d6) δ 7.53 (s, 1H), 7.40 (s, 1H), 6.98 (dd, J = 7.9 Hz, 1.9 Hz, 1H), 6.70 (t, J = 7.7 Hz, 1H), 6.66 (dd, J = 7.8 Hz, 1.9 Hz, 1H), 5.95 (s, 1H), 4.23 (t, J = 4.4 Hz, 2H), 3.32–3.29 (m, 2H). HRMS (m/z): [M + H]+ calcd for C9H11N2O2, 179.0815; found: 179.0844.
3-Oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxamide (45).
Compound 45 was prepared according to general procedure A using 39 (0.207 g, 1.0 mmol) as the starting material. Purified product 45 (yield: 0.099 g, 52%) was obtained as a white solid, mp 290–292 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 7.58 (s, 2H), 7.35 (t, J = 4.8 Hz, 1H), 7.01 (d, J = 4.9 Hz, 2H), 4.68 (s, 2H). 13C NMR (100 MHz; DMSO-d6): δ 165.75, 164.84, 141.71, 128.08, 124.02, 123.47, 122.20, 118.34, 67.15. HRMS (m/z): [M + H]+ calcd for C9H9N2O3, 193.0608; found: 193.0582.
3-Oxo-2-phenyl-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxamide (46).
Compound 46 was prepared according to general procedure A using 40 (0.283 g, 1.0 mmol) as the starting material. Purified product 46 (yield: 0.097 g, 52%) was obtained as a white solid, mp 259–261 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.11 (s, 1H), 7.63 (br, 1H), 7.56 (br, 1H), 7.48 – 7.32 (m, 6H), 7.07 – 7.00 (m, 2H), 5.93 (s, 1H). HRMS (m/z): [M + H]+ calcd for C15H13N2O3, 269.0921; found: 269.0894.
2-(4-Methoxyphenyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxamide (47).
Compound 47 was prepared according to general procedure A using 41 (0.313 g, 1.0 mmol) as the starting material. Purified product 47 (yield: 0.116 g, 39%) was obtained as a white solid, mp 258–260 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 7.60 (br, 1H), 7.51 (br, 1H), 7.39 (dd, J = 7.0 Hz, 1.7 Hz, 1H), 7.34 (d, J = 8.6 Hz, 2H), 7.10 – 6.98 (m, 2H), 6.93 (d, J = 8.4 Hz, 2H), 5.86 (s, 1H), 3.74 (s, 3H). HRMS (m/z): [M + H]+ calcd for C16H15N2O4, 299.1026; found: 299.1004.
2-(4-Bromophenyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxamide (48).
Compound 48 was prepared according to general procedure A using 42 (0.362 g, 1.0 mmol) as the starting material. Purified product 48 (yield: 0.104 g, 30%) was obtained as a white solid, mp 255–258 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 7.70 – 7.49 (m, 4H), 7.46 – 7.29 (m, 3H), 7.04 (d, J = 5.1 Hz, 2H), 5.92 (s, 1H). HRMS (m/z): [M + H]+ calcd for C15H1279BrN2O3, 347.0026; found: 347.0007 and calcd for C15H1281BrN2O3, 349.0005; found: 349.0025.
(Z)-2-(4-Hydroxybenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxamide (49).
Compound 49 was prepared according to the reported procedure with modifications [28]. Compound 45 (0.192 g, 1.0 mmol) and 4-hydroxybenzaldehyde (0.183 g, 1.5 mmol) were added to a flask containing a 50:50 mixture of triethyl amine and acetic anhydride (10 mL:10 mL). The mixture was then refluxed for a period of 12 h and purified using preparative TLC to yield 49 (yield: 0.027 g, 8%) as a pale green solid, mp >310 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.07 (s, 1H), 9.81 (s, 1H), 7.94 (s, 1H), 7.83 (d, J = 8.5 Hz, 3H), 7.06 – 7.00 (m, 3H), 6.80 (d, J = 8.6 Hz, 2H), 6.72 (s, 1H). 13C NMR (100 MHz; DMSO-d6): δ 167.58, 157.97, 156.69, 138.97, 137.99, 132.16, 126.19, 125.49, 124.17, 122.90, 122.40, 116.65, 115.65, 112.13. HRMS (m/z): [M + H]+ calcd for C16H13N2O4, 297.0870; found: 297.0850.
(Z)-2-(4-Carboxybenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxamide (50).
Compound 50 was obtained according to the procedure used for the preparation of compound 49. Compound 45 (0.192 g, 1.0 mmol) and 4-carboxybenzaldehyde (0.225 g, 1.5 mmol) were employed to generate target compound 50 (yield: 0.031 g, 10%) as a dark yellow solid, mp >310 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.43 (s, 1H), 11.36 (s, 1H), 8.25 (d, J = 8.5 Hz, 1H), 7.98 – 7.91 (m, 4H), 7.81 (d, J = 8.3 Hz, 1H), 7.14 – 7.11 (m, 3H), 6.90 (s, 1H). 13C NMR (100 MHz; DMSO-d6): δ 164.36, 162.87, 161.89, 154.68, 152.75, 144.14, 140.80, 139.02, 131.94, 130.64, 130.55, 127.34, 125.93, 116.22, 116.01. HRMS (m/z): [M + H]+ calcd for C17H13N2O5, 325.2995; found: 325.3010.
PARP1 enzyme assays and isoform profiling
Inhibitory activity against PARP1 was evaluated by in vitro PARP1 enzyme assay using the PARP Universal Chemiluminescent Assay Kit (Trevigen, Inc. Gaithersburg, MD) as described previously [9,15]. Compounds that showed more than 50% PARP1 inhibition at respective concentrations were further evaluated to obtain IC50 values. The IC50 values were determined from dose-response curves, which were plotted with absorbance against log of the concentrations (five concentration points for each compound), and obtained using regression wizard from Sigma Plot, version 10.0 (San Jose, CA).
The PARP1 inhibitory assays for compounds 49 and 50 were performed at BPS Bioscience using the BPS PARP1 Chemiluminescent Activity Assay Kit (Catalog #80551) using recently reported protocol [9]. Dose-response curves are depicted in supporting information Figures S3 and S4. The reported data, mean ± standard deviation (SD), are the averages from at least two independent dose-response curves, each carried out in triplicate. PARP-isoform screening for analogue 49 was performed at BPS Bioscience using the Chemiluminescent Activity Assay Kit (PARP2, catalog # 80502, PARP3, catalog # 80503, TNKS1, catalog # 80504, TNKS2, catalog # 80505, PARP8, catalog # 79076, PARP10, catalog # 80510 and PARP14, catalog # 80513) [9].
Supplementary Material
Acknowledgment
We are grateful to Dr. Leonard Barasa (St. John’s University) for his help during LC-MS analysis. We thank Dr. Suman Pathi for helpful discussions. We thank Dr. Brandon Fowler from the Department of Chemistry, Columbia University for his assistance in obtaining HR-MS data.
Funding:
This project was supported by the Department of Pharmaceutical Sciences of St. John’s University; St. John’s University Seed Grant No. 579-1110; and in part by the National Institute of General Medical Sciences (SC3GM131971) to T.T.T.
Footnotes
Conflicts of interest
TTT is a co-founder of Hysplex, LLC, with interests in PARP inhibitor development. The other authors declare no competing financial interest.
References
- [1].Langelier MF, Pascal JM, PARP-1 mechanism for coupling DNA damage detection to poly(ADP-ribose) synthesis, Curr. Opin. Struct. Biol 23(1) (2013) 134–143. DOI: 10.1016/j.sbi.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pratz KW, Rudek MA, Gojo I, Litzow MR, McDevitt MA, Ji J, Karnitz LM, Herman JG, Kinders RJ, Smith BD, Gore SD, Carraway HE, Showel MM, Gladstone DE, Levis MJ, Tsai HL, Rosner G, Chen A, Kaufmann SH, Karp JE, A phase I study of topotecan, carboplatin and the PARP inhibitor veliparib in acute leukemias, aggressive myeloproliferative neoplasms, and chronic myelomonocytic leukemia, Clin. Cancer Res 23(4) (2017) 899–907. DOI: 10.1158/1078-0432.ccr-16-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wahner Hendrickson AE, Menefee ME, Hartmann LC, Long HJ, Northfelt DW, Reid JM, Boakye-Agyeman F, Kayode O, Flatten KS, Harrell MI, Swisher EM, Poirer GG, Satele D, Allred J, Lensing JL, Chen A, Ji J, Zang Y, Erlichman C, Haluska P, Kaufmann SH, A phase I clinical trial of the poly(ADP-ribose) polymerase inhibitor veliparib and weekly topotecan in patients with solid tumors, Clin. Cancer Res 24(4) (2018) 744–752. DOI: 10.1158/1078-0432.ccr-17-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T, Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase, Nature 434(7035) (2005) 913–917. DOI: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- [5].Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A, Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy, Nature 434(7035) (2005) 917–921. DOI: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- [6].Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T, Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance, Cancer Res. 68(8) (2008) 2581–2586. DOI: 10.1158/0008-5472.can-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fojo T, Bates S, Mechanisms of resistance to PARP inhibitors-three and counting, Cancer Discov. 3(1) (2013) 20–23. DOI: 10.1158/2159-8290.cd-12-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ricks TK, Chiu HJ, Ison G, Kim G, McKee AE, Kluetz P, Pazdur R, Successes and challenges of PARP inhibitors in cancer therapy, Front. Oncol 5 (2015) 222. DOI: 10.3389/fonc.2015.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Velagapudi UK, Langelier MF, Delgado-Martin C, Diolaiti ME, Bakker S, Ashworth A, Patel BA, Shao X, Pascal JM, Talele TT, Design and synthesis of Poly(ADP-ribose) polymerase inhibitors: Impact of adenosine pocket-binding motif appendage to the 3-oxo-2,3-dihydrobenzofuran-7-carboxamide on potency and selectivity, J. Med. Chem 62(11) (2019) 5330–5357. DOI: 10.1021/acs.jmedchem.8b01709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zandarashvili L, Langelier MF, Velagapudi UK, Hancock MA, Steffen JD, Billur R, Hannan ZM, Wicks AJ, Krastev DB, Pettitt SJ, Lord CJ, Talele TT, Pascal JM, Black BE, Structural basis for allosteric PARP-1 retention on DNA breaks, Science (New York, N.Y.) 368(6486) (2020) DOI: 10.1126/science.aax6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA, PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results, J. Comput. Aided Mol. Des 20(10–11) (2006) 647–671. DOI: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- [12].Jones P, Altamura S, Boueres J, Ferrigno F, Fonsi M, Giomini C, Lamartina S, Monteagudo E, Ontoria JM, Orsale MV, Palumbi MC, Pesci S, Roscilli G, Scarpelli R, Schultz-Fademrecht C, Toniatti C, Rowley M, Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors, J. Med. Chem 52(22) (2009) 7170–7185. DOI: 10.1021/jm901188v. [DOI] [PubMed] [Google Scholar]
- [13].Ferraris DV, Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic, J. Med. Chem 53(12) (2010) 4561–4584. DOI: 10.1021/jm100012m. [DOI] [PubMed] [Google Scholar]
- [14].Kulkarni SS, Singh S, Shah JR, Low WK, Talele TT, Synthesis and SAR optimization of quinazolin-4(3H)-ones as poly(ADP-ribose)polymerase-1 inhibitors, Eur. J. Med. Chem 50 (2012) 264–273. DOI: 10.1016/j.ejmech.2012.02.001. [DOI] [PubMed] [Google Scholar]
- [15].Patel MR, Bhatt A, Steffen JD, Chergui A, Murai J, Pommier Y, Pascal JM, Trombetta LD, Fronczek FR, Talele TT, Discovery and structure-activity relationship of novel 2,3-dihydrobenzofuran-7-carboxamide and 2,3-dihydrobenzofuran-3(2H)-one-7-carboxamide derivatives as poly(ADP-ribose)polymerase-1 inhibitors, J. Med. Chem 57(13) (2014) 5579–5601. DOI: 10.1021/jm5002502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Takahashi B, Funami H, Iwaki T, Maruoka H, Shibata M, Koyama M, Nagahira A, Kamiide Y, Kanki S, Igawa Y, Muto T, Orally active ghrelin receptor inverse agonists and their actions on a rat obesity model, Bioorg. Med. Chem 23(15) (2015) 4792–4803. DOI: 10.1016/j.bmc.2015.05.047. [DOI] [PubMed] [Google Scholar]
- [17].Adam W, Peters EM, Peters K, Platsch H, Schmidt E, Von Schnering HG, Takayama K, Synthesis, thermal stability, and chemiluminescence properties of the dioxetanes derived from 1,4-dioxins, J. Org. Chem 49(21) (1984) 3920–3928. DOI: 10.1021/jo00195a008. [DOI] [Google Scholar]
- [18].Rajitha C, Dubey PK, Sunku V, Veeramaneni VR, Pal M, H3PO4/(CF3CO)2O mediated acylation followed by Hurd–Mori reaction: Synthesis of novel 1,2,3-thiadiazol derivatives, J. Het. Chem 50(3) (2013) 630–637. DOI: doi: 10.1002/jhet.1625. [DOI] [Google Scholar]
- [19].Kuroita, N. TM; Sano M; Kanzaki K; Inaba K; Kawakita T, Benzoxazines. II. Synthesis, conformational analysis, and structure--activity relationships of 3,4-dihydro-2H-1,4-benzoxazine-8-carboxamide derivatives as potent and long-acting serotonin-3 (5-HT3) receptor antagonists, Chem. Pharm. Bull 44(11) (1996) 2051–2060. DOI: 10.1248/cpb.44.2051. [DOI] [PubMed] [Google Scholar]
- [20].Griffin RJ, Calvert AH, Curtin NJ, Newell DR, Golding BT, Preparation of benzamide analogs as poly(ADP-ribose) polymerase inhibitors. PCT Int. Appl 1995, A1 1995091, pp 79. [Google Scholar]
- [21].Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Hoger T, Menissier-de Murcia J, de Murcia G, PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase, J. Biol. Chem 274(25) (1999) 17860–17868. DOI: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- [22].Rodriguez-Vargas JM, Nguekeu-Zebaze L, Dantzer F, PARP3 comes to light as a prime target in cancer therapy, Cell cycle (Georgetown, Tex.) 18(12) (2019) 1295–1301. DOI: 10.1080/15384101.2019.1617454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mizutani A, Yashiroda Y, Muramatsu Y, Yoshida H, Chikada T, Tsumura T, Okue M, Shirai F, Fukami T, Yoshida M, Seimiya H, RK-287107, a potent and specific tankyrase inhibitor, blocks colorectal cancer cell growth in a preclinical model, Cancer Sci. 109(12) (2018) 4003–4014. DOI: 10.1111/cas.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rubino MT, Maggi D, Laghezza A, Loiodice F, Tortorella P, Identification of novel matrix metalloproteinase inhibitors by screening of phenol fragments library, Archiv. der Pharmazie 344(9) (2011) 557–563. DOI: 10.1002/ardp.201000350. [DOI] [PubMed] [Google Scholar]
- [25].Heinrich T, Böttcher H, Prücher H, Gottschlich R, Ackermann K-A, van C Amsterdam, 1-(1-Phenethylpiperidin-4-yl)-1-phenylethanols as potent and highly selective 5-HT2A antagonists, ChemMedChem 1(2) (2006) 245–255. DOI: 10.1002/cmdc.200500023. [DOI] [PubMed] [Google Scholar]
- [26].Bolchi C, Fumagalli L, Moroni B, Pallavicini M, Valoti E, A short entry to enantiopure 2-substituted 1,4-benzodioxanes by efficient resolution methods, Tetrahedron: Asymmetry 14(23) (2003) 3779–3785. DOI: 10.1016/j.tetasy.2003.09.012. [DOI] [Google Scholar]
- [27].Costantino L, Gandolfi F, Sorbi C, Franchini S, Prezzavento O, Vittorio F, Ronsisvalle G, Leonardi A, Poggesi E, Brasili L, Synthesis and structure-activity relationships of 1-aralkyl-4-benzylpiperidine and 1-aralkyl-4-benzylpiperazine derivatives as potent sigma ligands, J. Med. Chem 48(1) (2005) 266–273. DOI: 10.1021/jm049433t. [DOI] [PubMed] [Google Scholar]
- [28].Honda T, Terao T, Aono H, Ban M, Synthesis of novel 1,4-benzoxazin-3-one derivatives as inhibitors against tyrosine kinases, Bioorg. Med. Chem 17(2) (2009) 699–708. DOI: 10.1016/j.bmc.2008.11.060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.