

SUMMARY
Cancer cells function as primary architects of the tumor microenvironment. However, the molecular features of cancer cells that govern stromal cell phenotypes remain unclear. Here, we show that cancer-associated fibroblast (CAF) heterogeneity is driven by lung adenocarcinoma (LUAD) cells at either end of the epithelial-to-mesenchymal transition (EMT) spectrum. LUAD cells that have high expression of the EMT-activating transcription factor ZEB1 reprogram CAFs through a ZEB1-dependent secretory program and direct CAFs to the tips of invasive projections through a ZEB1-driven CAF repulsion process. The EMT, in turn, sensitizes LUAD cells to pro-metastatic signals from CAFs. Thus, CAFs respond to contextual cues from LUAD cells to promote metastasis.
In brief
Bota-Rabassedas et al. show that EMT in lung adenocarcinoma cells activates a secretory process that governs CAF heterogeneity and, in turn, sensitizes lung adenocarcinoma cells to pro-metastatic signals from CAFs. Thus, EMT positions lung adenocarcinoma cells at the apex of a signaling hierarchy in the tumor microenvironment.
Graphical Abstract
INTRODUCTION
Accumulation of a densely fibrotic, immunosuppressive tumor stroma facilitates metastasis and is correlated with a worse clinical outcome in advanced epithelial malignancies (Kalluri, 2016; Werb and Lu, 2015). Cancer-associated fibroblasts (CAFs) are phenotypically heterogeneous mesenchymal cells that originate from diverse cell types and are a major source of cytokines and extracellular matrix molecules that enhance tumor cell-invasive activity, recruit vasculature, and suppress anti-tumor immunity (Gascard and Tlsty, 2016; Kalluri, 2016). In line with these findings, single-cell RNA sequencing studies have identified functional differences between CAFs and have shown that extracellular matrix production and immunoregulatory functions segregate to distinct CAF populations (Bartoschek et al., 2018; Elyada et al., 2019). Pharmacologic and genetic approaches to target CAFs in preclinical models result in widely disparate effects that range from tumor suppression to tumor promotion (Feig et al., 2013; Kraman et al., 2010; Loeffler et al., 2006; Olive et al., 2009; Özdemir et al., 2014; Provenzano et al., 2012; Su et al., 2018). Clearly, the molecular underpinnings of CAF heterogeneity must be better understood before CAF-targeting approaches can be tested in cancer patients.
There is a growing appreciation that cancer cells function as primary architects of the tumor microenvironment (Li and Stanger, 2019; Prager et al., 2019). Oncogenic mutations and epigenetic events in cancer cells upregulate immunoregulatory molecules and activate cytokine secretion, leading to the creation of an immunosuppressive milieu marked by the presence of pro-tumorigenic lymphoid and myeloid cell populations and reductions in anti-tumor CD8+ T cells (Li et al., 2018; Spranger and Gajewski, 2018). Cancer stem cells, which have the capacity to self-renew and undergo a reversible epithelial-to-mesenchymal transition (EMT), are capable of immune evasion and creating and maintaining tumor-promoting myeloid and T cell populations in the tumor microenvironment (Prager et al., 2019; Zhou et al., 2015). In lung adenocarcinoma (LUAD) patients, EMT features are correlated with advanced disease and worse clinical outcome (Larsen et al., 2016). Based on this conceptual framework, here, we postulated that contextual cues from LUAD cells govern CAF heterogeneity.
RESULTS
CAF heterogeneity in LUAD
To test our hypothesis, we studied CAF heterogeneity in KrasLA1 mice, which develop LUAD from somatic activation of a latent KrasG12D allele (Johnson et al., 2001). KrasLA1 mice harbor a Thy-1+ CAF population (hereafter called tCAFs) that, in co-culture with KrasLA1-derived LUAD cells, generates a cytokine-rich collagenous matrix and promotes LUAD cell invasion (Pankova et al., 2016; Roybal et al., 2011). We isolated tCAFs from KrasLA1 mice (Figure 1A) and subjected them to multiple annealing and dC-tailing-based quantitative single-cell RNA sequencing (MATQ-seq), a highly sensitive single-cell RNA-seq assay (Sheng et al., 2017). After excluding non-fibroblastic cells based on expression levels of cell-type-specific markers (Table S1), we identified two distinct tCAF clusters (Figure 1B) that were distinguishable based on 980 differentially expressed genes (Table S2). Quality control metrics showed that clustering was not related to batch-to-batch variability or sequencing depth (Figure S1). By gene set enrichment analysis (GSEA), cluster 1 was enriched in, among other terms, “EMT,” “inflammatory response,” “hypoxia,” “glycolysis,” “myogenesis,” and “angiogenesis” (Figure 1C). These terms are features of activated fibroblasts (Kalluri, 2016). In contrast, cluster 2 was enriched in “Notch signalling” and “PI3K/AKT/mTOR signalling” (Figure 1C). Thus, tCAFs contained two distinct subpopulations.
Figure 1. LUAD cells shape tCAF heterogeneity.
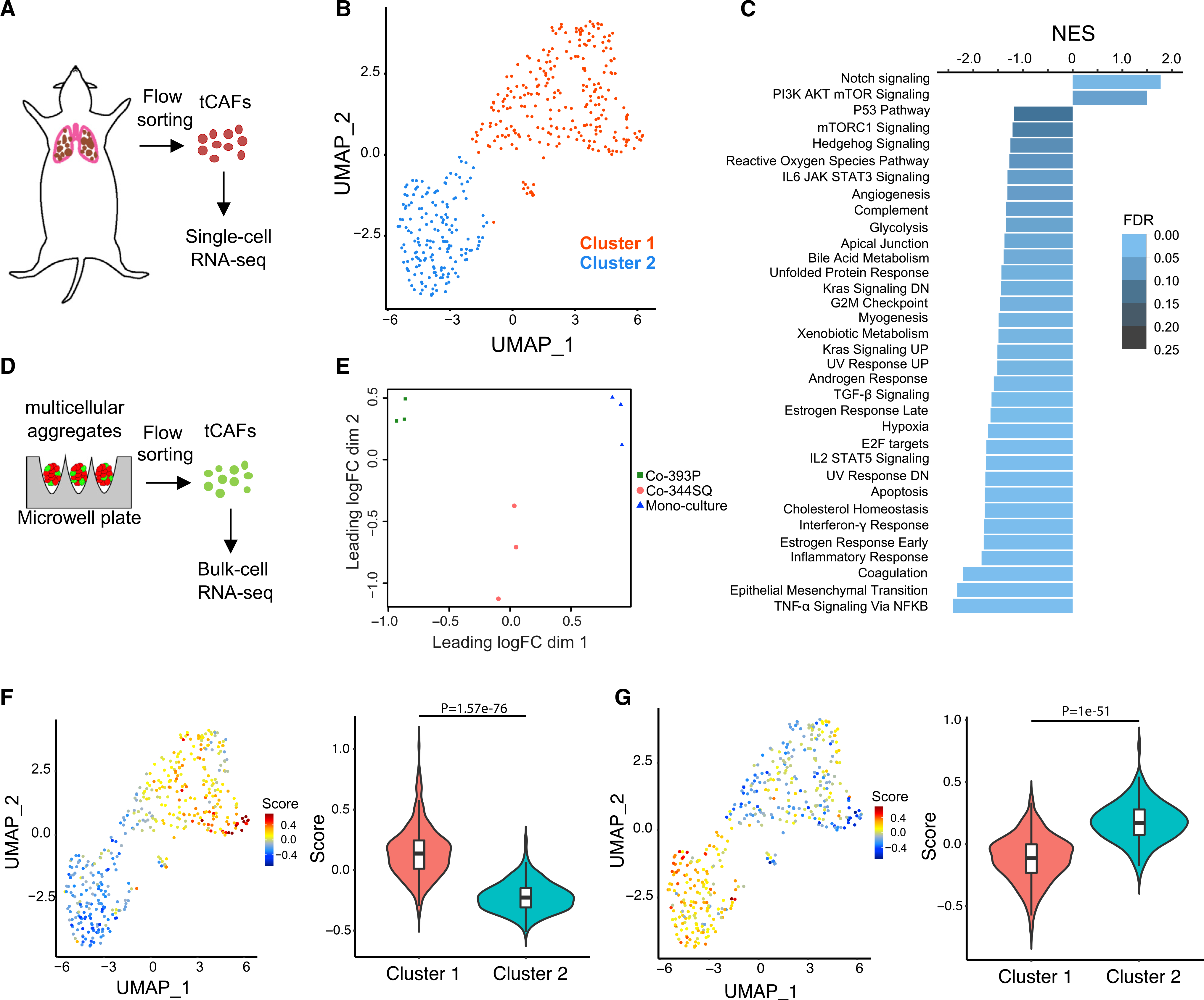
(A) tCAFs isolated by flow sorting from lung tissues in KrasLA1 mice were subjected to single-cell RNA sequencing.
(B) Principal-component analysis (PCA) plot of murine tCAFs (n = 428) subjected to single-cell RNA sequencing.
(C) GSEA of upregulated genes in tCAFs clusters. Normalized enrichment scores (NESs) represented by bar length. False discovery rate (FDR) values color-coded.
(D) Multicellular aggregates generated in microwell plates were incubated for 48 h and subjected to flow sorting to isolate tCAFs for bulk-cell RNA sequencing. n = 3 biological replicates per condition.
(E) PCA plot of tCAFs from triplicate multicellular aggregate preparations (dots) subjected to bulk-cell RNA sequencing.
(F and G) Correlations between single-cell RNA-sequencing data and expression signatures from tCAFs co-cultured with 344SQ cells (F) or 393P cells (G) are depicted with correlation plots (left) and violin plots (right).
To assess whether clusters 1 and 2 are generated by contextual cues from LUAD cells, we co-cultured tCAFs with LUAD cells derived from KrasLA1;Trp53R172H mice (Gibbons et al., 2009). These murine LUAD cell lines are positioned at either end of the EMT spectrum and display variable metastatic properties (high or low) following injection into syngeneic, immunocompetent mice (Gibbons et al., 2009). Highly metastatic LUAD cell lines undergo a reversible EMT through mutual antagonism between the EMT-activating transcription factor zinc finger E-box binding homeobox 1 (ZEB1) and microRNA-200 (miR-200) family members (Ahn et al., 2012; Gibbons et al., 2009; Tan et al., 2017, 2018; Yang et al., 2011). We generated 80-cell aggregates containing tCAFs alone or tCAFs and LUAD cells (30:50 ratio) that are mesenchymal and highly metastatic (344SQ cells) or epithelial and poorly metastatic (393P cells). After 48 h, tCAFs were isolated from the multicellular aggregates by flow sorting and subjected to bulk-cell RNA-seq (Figure 1D). tCAFs acquired distinct transcriptomic features in co-culture with 344SQ cells or 393P cells (Figure 1E; Table S2). A gene expression signature derived from 344SQ-co-cultured tCAFs was more highly correlated with cluster 1 than cluster 2 (Figure 1F), whereas a gene expression signature derived from 393P-co-cultured CAFs was more highly correlated with cluster 2 than cluster 1 (Figure 1G). Thus, clusters 1 and 2 were distinguishable based on signatures tCAFs acquired in co-culture with LUAD cells at either end of the EMT spectrum.
LUAD cells at either end of the EMT spectrum govern tCAF heterogeneity
Based on GSEA of upregulated genes, the terms “TGF-β signaling,” “EMT,” “angiogenesis,” and “glycolysis” were higher in co-cultured than mono-cultured tCAFs (Figures 2A, left bar graph and S2A). Comparisons between tCAFs co-cultured with 344SQ cells or 393P cells revealed sharp differences; among other terms, “hypoxia,” “interferon-γ,” “TNF-α signaling via NF-κB,” “IL-2 STAT5 signaling,” and “inflammatory response” were higher in 344SQ-co-cultured tCAFs, whereas “angiogenesis,” “EMT,” “myogenesis,” “Notch signaling,” and “Wnt β-catenin signalling” were higher in 393P-co-cultured tCAFs (Figure 2A, center bar graph), which led us to postulate that the EMT state of LUAD cells influences the phenotypic properties of tCAFs. To test this hypothesis, we generated multicellular aggregates containing tCAFs and 344SQ cells that have been subjected to small hairpin RNA (shRNA)-mediated ZEB1 depletion, which induces a shift toward an epithelial state (Tan et al., 2017), and found that GSEA hallmarks upregulated in tCAFs co-cultured with ZEB1-deficient 344SQ cells overlap with those upregulated in 393P-co-cultured tCAFs (Figure 2A, center and right bar graphs), which supported our hypothesis.
Figure 2. LUAD cells at either end of the EMT spectrum shape tCAF heterogeneity.
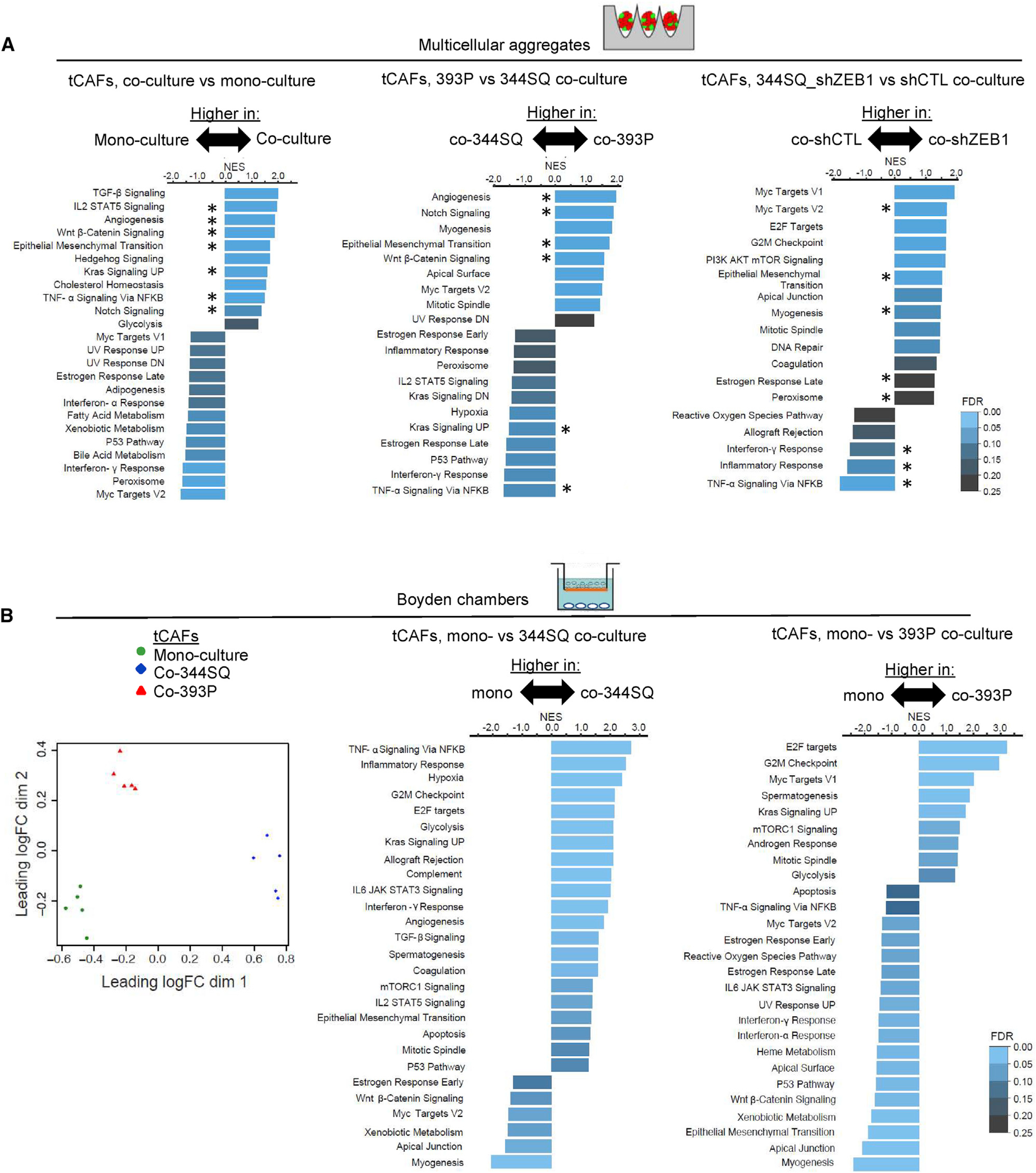
(A) GSEA of upregulated genes in tCAFs that were mono- or co-cultured (left), co-cultured with 344SQ cells or 393P cells (center), or co-cultured with ZEB1-deficient (shZEB1) or ZEB1-replete (control shRNA [shCTL]) 344SQ cells (right) in multicellular aggregates. NES represented by bar length. FDR values color-coded. Hallmarks shared between co-cultures are indicated (asterisks).
(B) PCA plot of tCAFs from Boyden chamber preparations (dots) subjected to bulk-cell RNA sequencing. GSEA of upregulated genes in tCAFs that were mono-cultured (mono-) or co-cultured (co-) with 393P cells or 344SQ cells in Boyden chambers. NES represented by bar length. FDR values color-coded. n = 5 biological replicates per condition.
In multicellular aggregates, cells can communicate through direct contact or soluble factor exchange. Both means of communication are thought to drive the outgrowth of myofibroblastic and pro-inflammatory CAFs in pancreatic cancer (Biffi et al., 2019; Dror et al., 2016; Mitra et al., 2012). To address the role of soluble factor exchange, we introduced tCAFs and LUAD cells (344SQ or 393P) into Boyden chambers separated by a porous filter. Based on bulk-cell RNA-seq, tCAFs acquired distinct transcriptomic features in co-culture with 344SQ cells and 393P cells (Figures 2B and S2B; Table S2). Concordance between the GSEA hallmarks that tCAFs acquired in Boyden chambers (Figure 2B) and multicellular aggregates (Figure S2A) was higher in tCAFs co-cultured with 344SQ cells than 393P cells (55% versus 26%, respectively), which suggests that soluble factor exchange played a more central role in tCAF reprogramming by 344SQ cells than by 393P cells.
EMT sensitizes LUAD cells to pro-metastatic signals from tCAFs
To assess how tCAF reprogramming influences LUAD progression, we generated orthotopic LUADs by intra-thoracic injection of LUAD cells alone (344SQ or 393P, 106 per mouse) or in combination with tCAFs (5 × 105 of each) into syngeneic, immunocompetent mice (Figure 3A) and found that tCAFs enhanced the metastatic activity of orthotopic tumors generated by 344SQ cells but not 393P cells (Figures 3B and 3C). Based on measurements of primary tumor sizes (Figure 3B), tCAF-induced metastatic activity was not related to increased 344SQ tumor growth. To test the hypothesis that EMT sensitizes LUAD cells to pro-metastatic signals from tCAFs, we repeated the experiment on ZEB1-deficient 344SQ cells and found that tCAFs did not enhance metastasis under those conditions (Figure 3D), which supported our hypothesis.
Figure 3. EMT sensitizes LUAD cells to pro-metastatic signals from tCAFs.
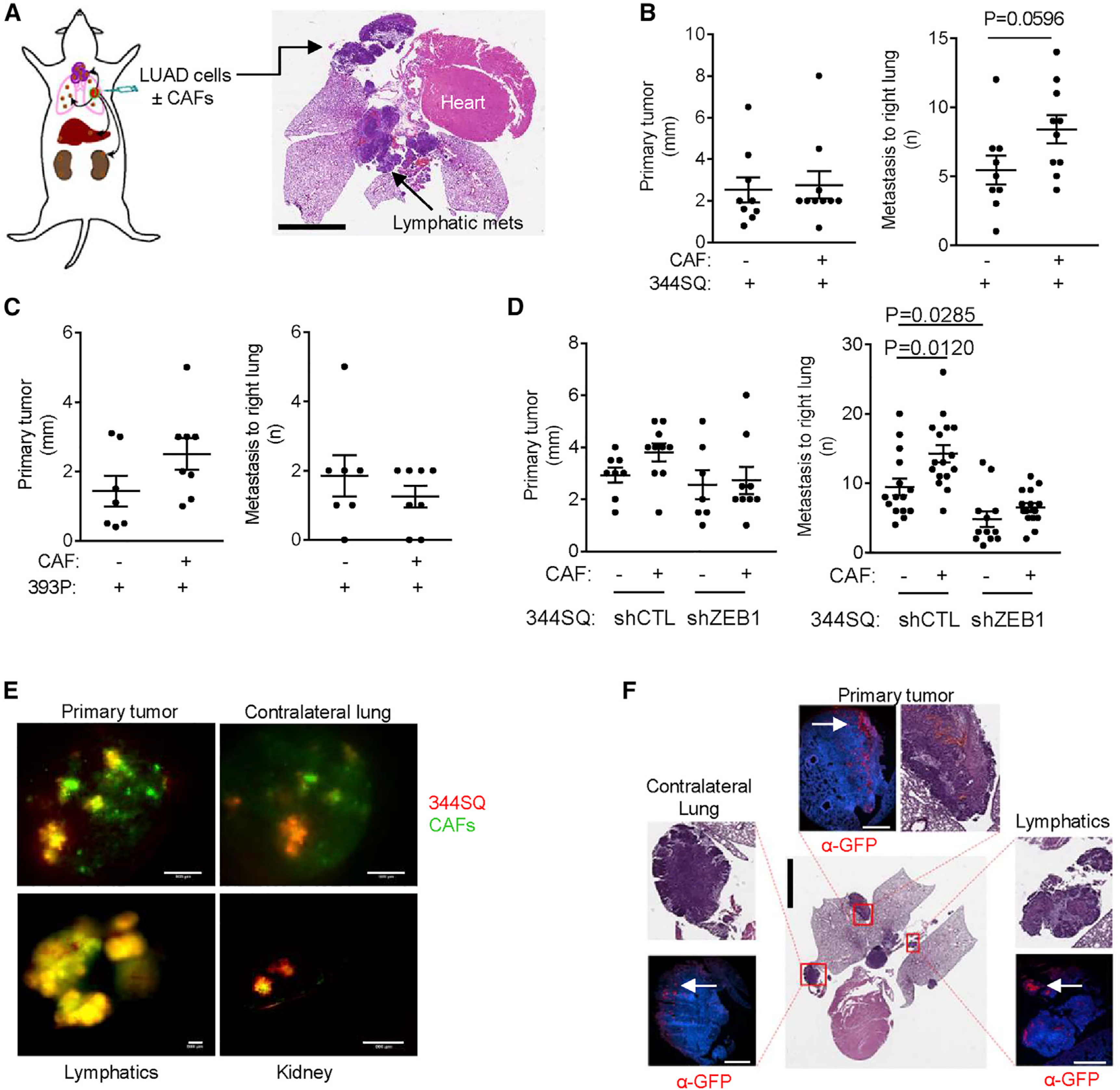
(A) Orthotopic LUADs generated by intra-thoracic injection of LUAD cells alone or in combination with tCAFs in syngeneic, immunocompetent mice. Arrows point to sites of primary tumor and mediastinal lymph node metastasis. Scale bar, 4 mm.
(B–D) Primary tumor diameters and numbers of contralateral lung metastases per mouse (dots) injected with 344SQ cells (B), 393P cells (C), or ZEB1-deficient (shZEB1) or ZEB1-replete (shCTL) 344SQ cells (D) alone (−) or in combination with tCAFs (+). n = at least 8 mice per condition. p values, 2-sided t test, 1-way ANOVA test.
(E) Merged fluorescence micrographs of fluorescently tagged 344SQ cells (red) and tCAFs (green) in primary tumor and metastases to contralateral lung, mediastinal nodes, and kidney. Intact tissues imaged immediately after resection. Scale bars, 500 μm. n = 2 mice.
(F) Micrographs of primary tumor and metastases to contralateral lung and mediastinal nodes in a mouse co-injected with fluorescently tagged 344SQ cells and tCAFs. Boxed regions are shown at higher magnifications (insets). Consecutive sections stained with hematoxylin and eosin or fluorescently tagged anti-GFP antibody (α-GFP) to identify co-injected tCAFs (arrows). Nuclei counterstained with DAPI (blue). Scale bars, 4 mm (central image), 500 μm (fluorescent micrographs). n = 2 mice.
High ZEB1 levels in LUAD cells drive the formation of tCAF-led invasive structures
Notably, the co-injected tCAFs were present in regional lymphatics and distant metastatic deposits (Figures 3E and 3F), which led us to reason that tCAFs and LUAD cells have the capacity to generate organized, invasive structures. To examine this possibility, we performed live-cell imaging on multicellular aggregates in 3-dimensional (3D) collagen gels (Figure 4A) and identified invasive projections containing a leading tCAF and LUAD cells following collectively behind (Figure 4B; Video S1). By time course studies, tCAF-led invasive structure formation was initiated by a tCAF that migrated radially to the aggregate periphery, exited the aggregate, and was followed by a LUAD cell that maintained contact with the leading tCAF (Figure 4C; Video S2). Treatment with a matrix metalloproteinase inhibitor at a dose that does not inhibit cell proliferation (Figure S3) reduced the numbers of invasive projections (Figure 4D), suggesting that invasion required proteolytic activity. These findings recapitulate features of “leader-follower” structures reported by Gaggioli et al. (2007). tCAF-led invasive projections were significantly more numerous in aggregates containing mesenchymal than epithelial LUAD cell lines (Figure 4E). In gain- and loss-of-function studies, ZEB1 enhanced tCAF-led invasive structure formation (Figures 4F and 4G). Based on reports that ZEB1 promotes LUAD metastasis by relieving key mediators from miR-dependent silencing (Ahn et al., 2012; Gibbons et al., 2009; Tan et al., 2017, 2018; Yang et al., 2011), we ectopically expressed ZEB1-silenced miRs (miR-200, miR-206, miR-148a, or miR-181) in 344SQ cells and found that the numbers of tCAF-led invasive structures sharply decreased (Figure 4H). Thus, miR silencing in LUAD cells plays a key role in tCAF-led invasive structure formation.
Figure 4. ZEB1 drives tCAF-led invasive structure formation.
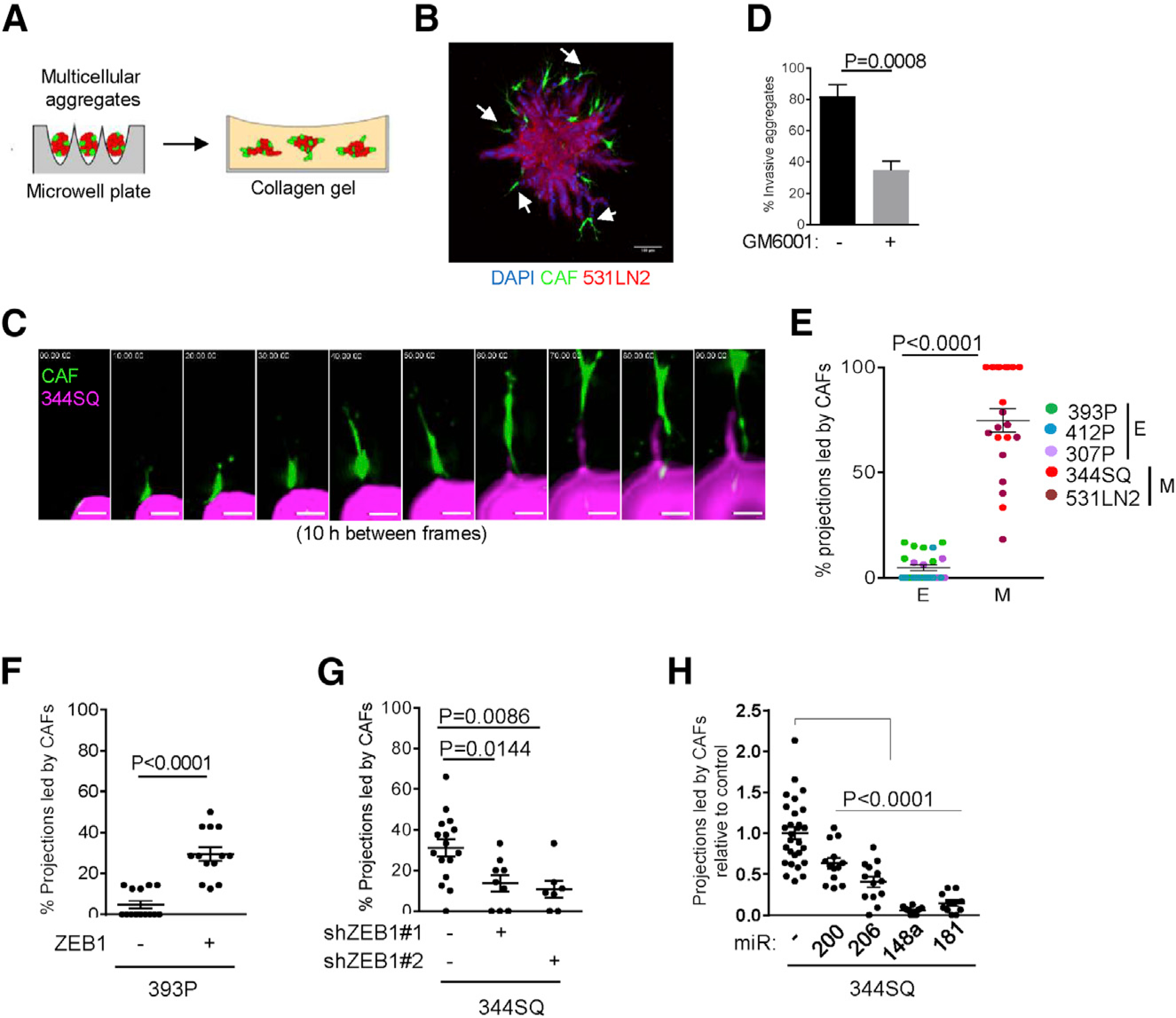
(A) Multicellular aggregates generated in microwell plates were transferred to collagen gels for imaging of invasive structure formation.
(B) Confocal micrograph of a multicellular aggregate containing tCAF-led invasive structures (arrows). Scale bar, 100 μm.
(C) Montage of confocal micrographs from live-cell imaging of a multicellular aggregate. tCAF (green), 344SQ cells (magenta). Scale bars, 15 μm.
(D) Percentage of aggregates that develop invasive projections in the presence or absence of protease inhibitor GM6001 (20 μM). n = at least 25 aggregates per condition. p values, Fisher’s exact test.
(E) Percentages of total projections led by tCAFs per multicellular aggregate (dots). n = at least 6 aggregates per cell line. Epithelial (E) or mesenchymal (M) LUAD cell lines. p values, 2-sided t test.
(F–H) Percentages of total projections led by tCAFs per multicellular aggregate (dots). 393P cells stably transfected with ZEB1 (+) or empty vector (−). n = at least 13 aggregates per condition (F). 344SQ cells stably transfected with ZEB1 shRNA (+) or scrambled control shRNA (−). n = at least 7 aggregates per condition. (G) 344SQ cells stably transfected with miRs or empty vector (−). (H) n = at least 11 aggregates per condition.
p values, 2-sided t test, 1-way ANOVA test.
LUAD cells repel tCAFs through a ZEB1-driven secretory process
To assess the importance of tCAF motility in invasive structure formation, we subjected tCAFs to small interfering RNA (siRNA)-mediated depletion of the DDR2 collagen receptor (Figure 5A), which promotes CAF motility (Bayer et al., 2019), and found that tCAF migration to the aggregate periphery and tCAF-led invasive structure formation were reduced (Figures 5B and 5C). To determine whether tCAF motility was related to the EMT state of LUAD cells, we quantified radial tCAF movement in aggregates containing LUAD cells at either end of the EMT spectrum and found that movement was greater in aggregates containing mesenchymal than epithelial LUAD cells (Figures 5D and 5E; Video S3) and was impaired by shRNA-mediated ZEB1 depletion in 344SQ cells (Figure 5F). To determine whether high ZEB1 expression causes LUAD cells to repel tCAFs, we assessed the ability of tCAFs and LUAD cells to mix across a scratch wound and found that tCAFs mixed to a greater extent with epithelial than mesenchymal LUAD cells (Figure 6A; Videos S4 and S5). In gain- and loss-of-function studies, ZEB1 impaired LUAD cell mixing with tCAFs (Figures 6B and 6C).
Figure 5. Radial tCAF movement precedes tCAF-led invasive structure formation.
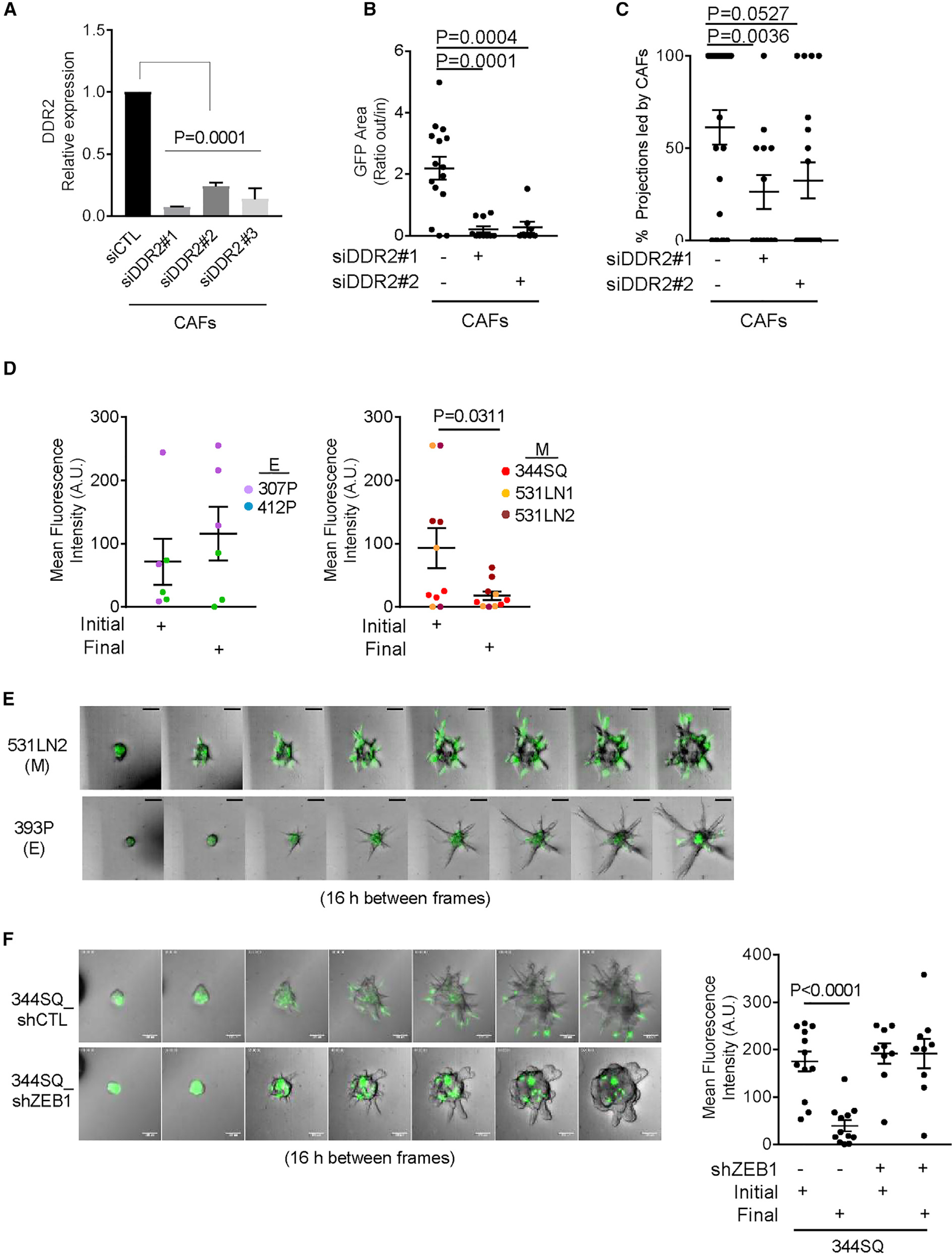
(A) Quantitative PCR analysis of DDR mRNA expression in tCAFs transfected with DDR2 or control (CTL) siRNAs. Values expressed relative to control siRNA (siCTL). n = 3 replicates per condition. p values, 1-way ANOVA test.
(B) Migratory properties of DDR2-deficient and DDR2-replete tCAFs in multicellular aggregates expressed as a ratio of extra:intra-aggregate GFP fluorescence per aggregate (dot). tCAFs transfected with siDDR2 (+) or scrambled control siRNA (siCTL) (−) are indicated. n = at least 8 aggregates per condition. p values, 1-way ANOVA test.
(C) Percentages of total projections led by tCAFs per multicellular aggregate (dots). tCAFs transfected with siDDR2 (+) or siCTL (−). n = at least 13 aggregates per condition. p values, 1-way ANOVA test.
(D) tCAFs in the center of aggregates (dots) containing epithelial (E) or mesenchymal (M) LUAD cell lines were quantified at T = 0 (initial) and 4d (final). n = 3 movies per cell line. Loss of fluorescence over time indicative of tCAF radial movement. LUAD cell lines color-coded. p values, 2-sided t test.
(E and F) Montages of confocal micrographs from live-cell imaging of multicellular aggregates. tCAFs (green). Scale bars, 100 μm. Scatterplot (F) shows GFP fluorescence at T = 0 (initial) and 4d (final) in the center of aggregate bodies (dots). Loss of fluorescence over time indicative of tCAF radial movement. n = at least 9 aggregates per condition.p values, 1-way ANOVA test.
Figure 6. LUAD cells repel tCAFs through a ZEB1-driven secretory program.

(A) Micrographs (far-left panels) of the leading front of a scratch wound containing tCAFs (green) and LUAD cells (red). Masks generated for each channel (2nd and 3rd panels) were overlaid (far-right panels). Scale bar, 100 μm. Mixing was quantified based on the overlapping area per field (dots) in scratch wounds containing epithelial (E) or mesenchymal (M) LUAD cell lines (color-coded). n = 3 biological replicates per cell line. p values, 2-sided t test.
(B and C) Mixing quantified in scratch wound assays containing tCAFs and 393P cells stably transfected with ZEB1 (+) or empty vector (−) (B) or 344SQ cells transfected with shZEB1 (+) or scrambled shRNA (−) (C). n = 3 biological replicates per condition. p values, 2-sided t test, 1-way ANOVA test.
(D) Quantitative PCR analysis of Itih2 mRNA levels in 393P cells and 344SQ cells (left bar graph) or 393P cells stably transfected with ZEB1 (+) or empty vector (−) (right bar graph). Values expressed relative to 393P cells and 393P_vector cells, respectively. n = 3 replicates per condition. p values, 2-sided t test.
(E) Western blot assay showing itih2 knockdown efficiency in 344SQ cells transiently transfected with control siRNA (NTC) or Itih2 siRNA (#1–3). β-Actin used as loading control. Results shown are representative of triplicate experiments.
(F) Micrographs of tCAFs (green) and siRNA-transfected 344SQ cells (red) at the leading fronts of scratch wounds. Scatterplot shows the overlapping area per field (dots). Scale bar, 100 μm. n = 3 replicates per condition. p values, 1-way ANOVA test.
(G) Schematic illustration of a working model.
Cancer cells secrete cytokines and extracellular matrix molecules that can attract or repulse cells in the tumor microenvironment (Lu et al., 2012). To identify factors secreted by 344SQ cells that mediate tCAF repulsion, we performed liquid chromatography-mass spectrometry on conditioned medium samples isolated from LUAD cells (344SQ or 393P) and tCAFs in mono-culture or co-culture. Of the 1,296 total proteins identified, numerous collagens, matricellular proteins, and proteases were present at higher levels in 344SQ cells (Figure S4), including inter-alpha-trypsin inhibitor heavy chain 2 (ITIH2), a secreted protein that stabilizes hyaluronan, which reduces the elasticity of tumor tissue and increases interstitial fluid pressure (Whatcott et al., 2011). ITIH2 mRNA levels were higher in 344SQ cells than 393P cells and were upregulated by ectopic ZEB1 expression in 393P cells (Figure 6D). tCAFs mixed to a greater extent with ITIH2-deficient than ITIH2-replete 344SQ cells (Figures 6E and 6F), which suggests that LUAD cells repel tCAFs through a ZEB1-dependent secretory process.
DISCUSSION
Metastasis requires a high degree of tumor cell plasticity as manifested by the capacity to switch between migratory modes, evade immune surveillance, and maintain viability under stressful conditions (Egeblad et al., 2010; Odenthal et al., 2016). In one working hypothesis, EMT confers the invasive and stem-ness properties necessary for cancer cells to dissolve junctional complexes, intravasate into vascular structures, and colonize the metastatic niche (Chockley and Keshamouni, 2016; Krebs et al., 2017; Puisieux et al., 2014). The findings presented here support a paradigm that incorporates the tumor microenvironment into the regulatory network of EMT-activating transcription factors and a model in which cancer cells reside at the apex of a signaling hierarchy in the tumor microenvironment (Li and Stanger, 2019).
Reports on CAF heterogeneity have primarily used droplet-based single-cell RNA-seq technologies to interrogate large numbers of fibroblastic cells (Lambrechts et al., 2018). Here, we used a highly sensitive RNA sequencing platform to explore heterogeneity within an immunophenotypically defined (Thy-1+) CAF population. To deconvolve tCAF complexity, we probed single-cell RNA-seq data with expression signatures identified in co-cultured and mono-cultured tCAFs and found that tCAFs could be distinguished based on these signatures. Thus, comparative studies on defined CAF populations are feasible in the context of a highly sensitive single-cell RNA-seq method, and insight into the molecular underpinnings of tCAF complexity provided the reaction coordinates needed for complexity deconvolution.
The viability of circulating cancer cells is higher if they are incorporated into stromal fragments that provide an early growth advantage to cancer cells in the metastatic niche, which is the basis for the current belief that cancer cells bring their own “soil” to the metastatic niche (Duda et al., 2010). We found that tCAFs localized in the metastatic niche and generated tCAF-led invasive structures in response to ZEB1-driven repulsive forces from LUAD cells (Figure 6F), which builds on evidence that CAF-led invasive structures are stabilized by mechanically active heterotypic contacts that generate force and enable invasion (Labernadie et al., 2017). In line with our findings, EMT activates contact inhibition of locomotion to guide cell migration during embryonic development (Scarpa et al., 2015; Stramer and Mayor, 2017).
Collagen fibers secreted by CAFs create a stiff, fibrotic tumor stroma that promotes cancer cell invasion (Bayer et al., 2019; Corsa et al., 2016; Erdogan et al., 2017; Yamauchi et al., 2018). Selective antagonists of DDR2 and collagen-binding integrins (α1b1, α2b1, α10b1, α11b1) inhibit the pro-metastatic activity of CAFs (Grither and Longmore, 2018; Raab-Westphal et al., 2017). Based on the findings presented here, clinical trials designed to inhibit CAF motility through the use of selective inhibitors of collagen receptors are warranted as part of the ongoing efforts to develop CAF-targeting therapies (Kalluri, 2016; LeBleu and Kalluri, 2018).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jonathan M. Kurie (jkurie@mdanderson.org)
Materials availability
Mouse lines generated during this study are completely available upon request to the Lead Contact.
Data and code availability
The raw data of single cell RNA-seq and mini-bulk RNA-seq have been submitted to GEO: GSE166480. Sequencing depth and mapping can be found in Table S1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal husbandry and in vivo experiments
All mouse studies were approved by the Institutional Animal Care and Use Committee at The University of Texas MD Anderson Cancer Center. Mice received standard care and were euthanized according to the standards set forth by the Institutional Animal Care and Use Committee. To generate orthotopic lung tumors, 2–4 months old male littermates were randomly assigned to experimental groups. An incision was made in syngeneic, immunocompetent mice between the 2nd and 3rd ribs on the left side. Cells were directly injected into the left lung, and the wound was sutured. LUAD cells (RFP-tagged 344SQ, RFP-tagged 393P, 344SQ_shCTL or 344SQ_shZEB1) were injected alone (106 cells) or in combination with GFP-tagged tCAFs (5×105 of each cell type). Tissues were formalin-fixed and paraffin-embedded for histologic confirmation of tumor cells by hematoxylin and eosin staining and fluorescent antibody staining. Investigators were blinded to sample identities during analysis.
Cell lines
LUAD cells generated from KrasLA1/+;Tp53R172H/+ male mice (344SQ, 393P, 531LN1, 531LN2, 307P, 412P, and their transfected derivatives) (Gibbons et al., 2009) were cultured in RPMI 1640 containing 10% FBS. Cells were maintained at 37°C in an incubator with a humidified atmosphere containing 5% CO2. Cells were passaged every 2–3 days or when they reached 80% confluence. tCAFs isolated from KrasLA1 male mice were cultured in DMEM with 10% FBS, 1% Pyruvate, and 1% Penicillin-Streptomycin, were passaged every 3–5 days or upon reaching 75% confluence, and were used for a maximum of 5 passages after thawing. All co-culture experiments and paired mono-culture controls were performed using a mix of both cell culture mediums, at a 1:1 ratio. 393_RFP cells were generated using the vector pLVX_mCherry (puromycin selection); the mCherry coding sequence was amplified by PCR and inserted into a pLVX-puro vector (Clontech, Cat#632164). GFP-transfected tCAFs were generated with the vector EF-pLenti6.3-GFP (Blasticidin selection). The remaining stably transfected cells are described elsewhere: 344SQ_RFP (Padhye et al., 2019); 344SQ_shCtL, 344SQ_shZEB1, 344SQ_mir206, 344SQ_mir148a (Tan et al., 2017); 393P_vec, 393P_ZEB1, 344SQ_miR-181 (Tan et al., 2018), and 344SQ_miR-200 (Ahn et al., 2012). For co-cultureco-culture experiments, 50,000 tCAFs were seeded into the bottom chamber of a 0.4μm Boyden chamber with or without 80,000 LUAD cells in the upper chamber. After 48h, tCAFs were collected from the bottom chamber and subjected to RNA extraction. For transient transfection, cells were transfected with jetPRIME® (Polyplus, Cat#114–15) transfection reagent following the manufacturer’s suggested protocol.
METHOD DETAILS
Cell proliferation
Viable cell densities were quantified in sub-confluent monolayer culture conditions using water-soluble tetrazolium salt-1 (WST-1, Takara Cat#MK400) reagent as suggested by manufacturer’s instructions (Takara).
Fibroblast isolation for single-cell RNA sequencing analysis
To obtain fresh fibroblasts for single cell RNA-seq, we resected whole lung tissues from age-matched (10–12 months-old) KrasLA1 mice (for tCAF isolation). The lungs were immediately perfused with 2% FBS in Hank buffered salt solution (FBS-HBSS) and dispersed into single-cell suspensions by immersion in 3 μg/mL of collagenase (Worthington Biochemical; Cat#LS004216) and Dispase II (Roche; Cat#04942078001) on a gentleMACS Dissociator (miltenyibiotec; Cat# 130–093-235) using the lung tissue dissociation programs (Lung_01 and Lung_02). Dispersed cells were filtered (70 μm and 40 μm), centrifuged, washed with FBS-HBSS, and subjected to red blood cell lysis by adding RBC buffer (Biolegend; Cat#420301). The remaining cells were centrifuged, washed, and counted. To isolate immunophenotypically defined tCAF populations (Thy-1+ Epcam− CD45− Sca-1− CD31−) (Roybal et al., 2011), cells were stained for 45min on ice simultaneously with anti-Epcam-FITC (Biolegend; Cat#118210), anti-CD31-PE (Biolegend; Cat#102508), anti-CD45-PE (Biolegend; Cat# 103106), anti-Sca-1-PECy7 (Biolegend; Cat#108114), and anti-Thy1-APC (Abcam; Cat# ab25322). Control tubes containing 1×105 cells were stained with single antibodies or no antibody. Cells were subjected to flow cytometry (FACSAriaII instrument, BD FACSDiva 6.1.3 software) using a 75μm nozzle. Cells were gated with the following scheme: cells, singlets, live cells, negative for: FITC (EpCam), PE (CD31 and CD45), and PECy7 (sca-1), and positive for APC (Thy1). Live/dead marker (DAPI; Thermo Fischer Scientific; Cat#R37606 was added, and only live cells were placed individually in wells on a 96-well plate. The 96-well plate was kept at 4°C before and during sorting and contained the first enzymes of the RNA isolation protocol to minimize RNA degradation. Immediately after sorting, the plate was centrifuged for 30 s to mix the cells and enzymes. The remaining steps of the RNA sequencing protocol are described below.
RNA sequencing
We used a Bravo® Automated Liquid Handler to robotically perform MATQ-seq analysis (Sheng et al., 2017). To prevent evaporation during the reactions, we first added 10 μL of PCR-grade mineral oil (Sigma, Cat. No. M8662) to every well of a 96-well plate (Bio-rad, Cat. No. HSS9601). For cell lysis, 1 μL of lysis buffer containing 0.65 μL of 0.2% Triton X-100 ultrapure water (Thermo Fisher Scientific, 10777019), 0.2 μL of MATQ-seq primer mix (Sheng et al., 2017), 0.05 μL of dNTP (10mM each), 0.05 μL 0.1M DTT, and 0.05 μL RnaseOUT (Thermo Fisher Scientific, Cat. No. 10777019) were premixed and then added to each well using the Liquid Handler. The cell lysis plate was then briefly centrifuged. Individual tCAFs were flow sorted into 96-well plates. We lysed the cells with a 3-min incubation at 72°C on a 96-well PCR machine and then quickly transferred the plate to ice for 1 min. We then added 1 μL of reverse transcription mix containing 0.4 μL 5X FS buffer, 0.1 μL 0.1 M DTT, 0.05 μL RnaseOUT, 0.05 μL Superscript III (Thermo Fisher Scientific, Cat. No. 18080093), and 0.4 μL RNase-free H2O to each well of the plate using the Liquid Handler. We performed all the following steps of MATQ-seq using the robot and a 96-well PCR machine for temperature ramping according to the protocol previously described (Sheng et al., 2017) with minor changes in enzyme amount. We also redesigned the second strand primer to create a 3-base mismatch at the 3′ end of the primer for later library preparation. For mini-bulk cell RNA sequencing analysis of tCAFs isolated from multicellular aggregates, we used MATQ-seq as previously described (Sheng et al., 2017). As a quality control step, mapping rates from single-cell and bulk RNA sequencing were quantified (Table S1).
To prepare libraries, we diluted 20 ng of amplified product into 10 μL for each cell. Samples were heated to 95°C for 30 s to denature the cDNA. We then added double strand conversion mix containing 0.6 μL 10 μM barcoded primer, 2 μL 10X Thermopol Buffer, 0.5 μL 10 mM each dNTP, 0.3 μL Bst 2.0 WarmStart® DNA Polymerase (NEB, Cat. No. M0538S), and 6.6 μL PCR grade water to each sample. Twenty cycles of 60°C 20 s, 65°C 30 s were performed to convert the cDNA into double strand and to add Illumina adaptor and barcode to the samples. 5 μL of 50mM EDTA was then added to each sample. We then pooled the samples into a single tube or several tubes according to the assigned barcode. Pooled libraries were purified with 1.2x AMPure XP beads (Beckman Coulter, Cat. No. A63880) into 20 μL of water. We then used the Nextera DNA library prep kit (Illumina, Cat. No. FC-121–1030) to tag 50 ng of the libraries according to the protocol provided with the kit. We purified the tagged libraries with 1.2x AMPure XP beads. We then amplified the libraries according to the Nextera protocol using Illumina sequencing adapters. A size selection was then performed using AMPure XP beads to select cDNA ranging from 250bp to 600 bp. We removed the majority of the ribosomal cDNA with a treatment of Duplex-specific Nuclease (Evrogen, Cat. No. EA003) using 100 ng of the library. We then purified the library using 0.9x AMPure XP and amplified the library using KAPA HIFI Hotstart Ready Mix (KAPA Biosystem, Cat. No. KK2601). Libraries were then diluted and sequenced on a Nextseq 500 machine, using the NextSeq 500/550 High Output Kit v2.5 (150 Cycles) or NextSeq 500/550 Mid Output Kit v2.5 (150 Cycles) (Illumina, Cat. No. 20024907 and 20024904).
Multicellular aggregates
Multicellular aggregates were created in a 24-well plate containing 1700 laser-ablated microwells per well as described (Albritton et al., 2016). The microwells were passivated with 0.05% pluronic acid (Sigma-Aldrich; Cat#p2443–250G) for 1 h prior to seeding the cells. For invasion assays in collagen gels, 85,000 LUAD cells alone or in combination with 51,000 tCAFs were seeded per well and cultured for 48 h to generate aggregates, each containing 50 LUAD cells alone or 50 LUAD cells and 30 tCAFs. tCAFs used to generate the aggregates originated from a common pool. Aggregates were mixed with rat tail collagen I (Collagen R solution 0.4%, Serva; Cat#47256.01) to generate collagen gels with defined concentrations (2mg/ml), volumes (200μl/gel), and aggregate densities (35 aggregates/ gel). The gels were allowed to polymerize upside-down on a glass-bottom 35mm plate (Mattek; Cat#P35G-1.5–14-C) or a glass-bottom 24-well plate (Mattek; Cat#P24G-1.5–13-F) at 37°C for 30 min. The aggregates were cultured for up to 5 days. For protease activity inhibition, aggregates were treated with GM6001 (20μM, MD Millipore; Cat#364206–1MG) in the collagen gel, for 3 days. Aggregates were fixed with 4% paraformaldehyde (Electron Microscopy Sciences; 15714-S) for 20 min at room temperature (RT) for confocal microscopy. For bulk-RNA sequencing studies, GFP-tagged tCAFs were isolated from multicellular aggregates (cultured for 48h) by disrupting the aggregates using a 301/2 gauge needle and isolating the tCAFs by flow sorting in the green channel (FACSAriaII instrument, BD FACSDiva 6.1.3 software) using a 75μm nozzle. Live/dead marker (DAPI, Thermo Fischer Scientific; Cat#R37606) was added. Live cells (at least 500 per replicate) were selected and subjected to RNA extraction protocol described elsewhere in this methods section.
Liquid chromatography-mass spectrometry of conditioned medium samples
For proteomic studies, conditioned medium samples were collected from microwells containing multicellular aggregates that had been in culture for 48 h. After collection, the conditioned medium samples were filtered, concentrated, and analyzed as previously described (Tan et al., 2020).
Microscopy
Images of fixed multicellular aggregates in 3D collagen gels were acquired with a NikonA1 confocal microscope, 10x objective. After collecting z-slices, the volume was rendered into 2-dimensional projections by maximum intensity projection algorithm (ImageJ; Schneider et al., 2012), and invasive structures were analyzed. An invasive projection was defined as at least one visible LUAD cell protruding out of the aggregate. A tCAF-led invasive structure was defined as a projection containing at least 1 tCAF at the tip. Invasive projections were manually counted. Results were determined from at least 10 multicellular aggregates per condition, at least two collagens gels per condition, and at least two independent batches of aggregates. When possible, investigators were blinded to sample identity when performing these quantifications.
Live-cell imaging was performed in an automated wide-field microscope (Nikon Eclipse TiE) with 20x objective. For 3-dimensional image stacks, Z stacks were projected by using a sum of the slices algorithm. Invasive projections were identified after thresholding to remove aggregate bodies (ImageJ). Diameters were measured after thresholding (ImageJ) to remove invasive projections from the live-cell images. Results were determined from at least 3 aggregates per condition, at least two gels per condition, and two independent batches of aggregates.
Cell-cell repulsion
Multicellular aggregates were generated, transferred to collagen gels, and subjected to time-lapse movies. Fluorescence intensity in the center of the aggregate was measured at initial (T = 0) and final (T = 4d) time points by performing radial profile analysis (ImageJ) on tCAFs in the green channel. Results represent values from at least 3 biological replicates per condition. For scratch-wound closure assays, 15,000 LUAD cells and 6,000 tCAFs were seeded on either side of a 35 mm culture-2-well insert glass-bottom dish (Ibidi; Cat#81176). tCAFs and LUAD cells were distinguishable on the basis of fluorescent tags that included GFP in tCAFs and RFP or CellTracker deep red dye (Fisher Scientific; Cat#C34565) in LUAD cells. Cells were stained with CellTracker dye for 30 min at RT at a concentration of 1.5μM prior to seeding. The cells were allowed to reach confluence in each chamber, at which time the insert was lifted to initiate cell migration and wound closure. Cells were imaged when the insert was lifted (initial time point, T = 0) and at the final time point (T = 4 d) using a confocal microscope (NikonA1, 10x objective). Leading edges of the migrating cells were imaged and analyzed for area of overlap using ImageJ. In brief, 16-bit raw images were converted to 8-bit, and a mask was generated for each channel, which comprised the total area of each cell type. The two channels were overlaid and converted to RGB. The overlapping area was measured and adjusted for the initial area of the chamber as described (Carmona-Fontaine et al., 2008). Results represent values from at least 4 images per replicate, 3 biological replicates per condition.
To quantify extrusion of DDR2-deficient and –replete tCAFs from multicellular aggregates, we quantified GFP outside and inside each aggregate body and used those values to calculate a ratio (outside/inside GFP area). For this purpose, z stack images of paraformaldehyde-fixed aggregates in collagen gels were acquired by confocal microscopy (10X objective). Z stacks were rendered into 2-dimensional images by using a maximum intensity projection algorithm (ImageJ), converted to 8-bit, thresholded, and segmented to generate masks of red and green channels, which were used to quantify LUAD cells and tCAFs, respectively, in the aggregate body. GFP areas within and outside the aggregate body mask were determined.
tCAF detection in distant metastases
For direct detection of fluorescently tagged cells, tumor-bearing organs were resected from mice bearing orthotopic LUAD, placed into a glass bottom dish, covered with 2% FBS in 1X phosphate-buffered saline, and imaged using a wide-field Nikon Eclipse TiE microscope (NIS-Elements Software, Nikon). As a second approach, 4 μm tissue slices from FFPE tumor-bearing lungs were stained with anti-GFP primary antibody and a fluorescently-labeled secondary antibody (anti-rabbit 568) and imaged with a wide-field Nikon Eclipse TiE microscope (NIS-Elements Software, Nikon), 10x objective.
Western blotting
Cells were lysed with RIPA buffer (Cell Signaling Technology) containing protease inhibitors (Cell Signaling Technology). Cell lysates were separated on a 4%–20% Bis-Tris gel, transferred to nitrocellulose membranes using Trans-Blot Turbo Transfer System (Bio-Rad), and then incubated with primary antibodies and HRP-conjugated secondary antibodies (Cell Signaling Technology). Protein bands were visualized with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). Antibodies against ITIH2 (Novus Biologicals, #NBP2–31750) and β-actin (Cell Signaling Technology, #4967) were purchased.
QUANTIFICATION AND STATISTICAL ANALYSIS
For MATQ data analysis, we first trimmed the unique molecular identifiers (UMI) sequence using seqtk (Shen et al., 2016). STAR (Dobin et al., 2013) was used to map the trimmed reads to the transcriptome. We annotated the mapped reads using HT-seq with genecode GRCm38 as a reference. We then parsed the reads annotation to the untrimmed reads and counted the gene expression using UMI. The expression level of every gene was then normalized to the total UMI numbers of the sample. To identify non-CAFs cells, we performed first PCA and clustering analysis by using all cells as an explorative analysis. A small proportion of cells (n = 32) were spreading out on the PCA plot and most of them were outliers. To investigate the underlying source of these outliers, we performed PCA analysis on these 32 cells and found that these cells are separated into three branches on the PCA. By looking into the genes on PC1 and PC2, we identified several cell lineage markers including epithelial markers (Cdh1, Krt7, Krt18, Krt19) and hematopoietic lineage markers (Laptm5, Hoxa10) expressed in these 32 cells. Additionally, we checked the expression of three markers (Cdh1, Hoxa10, Laptm5) across all cells. The cells with the highest expression level of each marker were the outlier cells. These observations suggested these outlier cells were potentially non-CAFs. Principal component analysis and clustering analysis were performed by using ‘Seurat” R package. Genes differentially expressed between tCAF clusters were identified by using ‘limma-trend’ function in ‘limma’ R package.
For bulk-cell RNA sequencing of mono-cultured and co-cultured tCAFs, STAR (Dobin et al., 2013) was used to map the reads to the transcriptome. Reads annotation was performed using HT-seq with genecode GRCm38 as a reference. Reads normalization was performed using EdgeR (Robinson et al., 2010). To identify significantly enriched pathways and functional hallmarks, gene set enrichment analyses were performed using GSEA 4.0.3 (Subramanian et al., 2005). Gene set shuffling was used for all bulk data. For differentially expressed gene analyses, EdgeR was used for tCAFs isolated from multicellular aggregates and limma-trend was used for tCAFs co-cultured with LUAD cells in Boyden chambers.
To determine whether tCAFs were distinguishable based on gene expression signatures they acquire in co-culture with highly or poorly metastatic LUAD cells, we first performed differential expression analyses on bulk RNA sequencing data from tCAFs co-cultured with highly or poorly metastatic LUAD cells. The genes with FDR < 0.05 were selected for expression signatures of each condition. We calculated the average gene expression Z-score of each signature in each tCAF analyzed by single-cell RNA sequencing. The Z-scores were compared between clusters 1 and 2 by Student’s t test.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GFP | Novus Biologicals | Cat# NB600-308; RRID:AB_10003058 |
| Anti-Itih2 | Novus Biologicals | Cat# NBP2-31750 |
| Anti-β-Actin | Cell Signaling Technology | Clone 13E5, Cat# 4970; RRID:AB_2223172 |
| PE anti-mouse CD45 | Biolegend | Cat# 103106; RRID:AB_312971 |
| PE Rat IgG2a, κ Isotype Ctrl | Biolegend | Cat# 400508; RRID:AB_326530 |
| PE anti-mouse CD31 | Biolegend | Cat# 102508; RRID:AB_312915 |
| PE Rat IgG2b, kappa Isotype Ctrl | Biolegend | Cat# 400608; RRID:AB_326552 |
| Alexa Fluor® 488 anti-mouse CD326 (Ep-CAM) | Biolegend | Cat# 118210; RRID:AB_1134099 |
| Alexa Fluor® 488 Rat IgG2a, κ Isotype Ctrl | Biolegend | Clone RTK2758; Cat# 400525 |
| PE/Cy7 anti-mouse Ly-6A/E (Sca-1) | Biolegend | Cat# 108114; RRID:AB_493596 |
| PE/Cy7 Rat IgG2a, κ Isotype Ctrl | Biolegend | Cat# 400522; RRID:AB_326542 |
| CD90 / Thy1 antibody [G7] | Abcam | Cat# ab25322; RRID:AB_470438 |
| Rat IgG2b kappa Isotype Control (eB149/10H5), APC-eFluor 780 | Thermo Fisher Scientific | Cat# 47-4031-80; RRID:AB_1272021 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A10042; RRID:AB_2534017 |
| Alexa Fluor 568 Phalloidin | Thermo Fischer Scientific | Cat# A12380 |
| Bacterial and virus strains | ||
| Bacteria: Subcloning Efficiency DH5α Competent Cells | Thermo Fischer Scientific | Cat# 18265017 |
| Biological samples | ||
| Human Lung Adenocarcinoma tissue | UT MD Anderson Cancer Center | N/A |
| Human IPF tissue | The University of California San Francisco | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DAPI | Thermo Fischer Scientific | Cat# R37606 |
| Collagenase type I (CLSS-1 filtered) | Worthington Biochemical | Cat# LS004216 |
| Dispase II | Roche | Cat# 04942078001 |
| RBC buffer | Biolegend | Cat# 420301 |
| Puromycin | InvivoGene | Cat#ant-pr |
| Blasticidin | InvivoGene | Cat# ant-bl-1 |
| Matrigel | Corning | Cat# 356231 |
| CellTracker Deep Red Dye | Fisher Scientific | Cat# C34565 |
| Collagen R solution 0.4% | Serva | Cat# 47256.01 |
| Protease inhibitor GM6001 | MD Millipore | Cat# 364206-1MG |
| paraformaldehyde | Electron Microscopy Sciences | Cat# 15714-S |
| JetPRIME® transfection reagent | Polyplus | Cat# 114-15 |
| Pluronic F-127 | Sigma-Aldrich | Cat# p2443-250G |
| PCR-grade mineral oil | Sigma-Aldrich | Cat# No. M8662 |
| RnaseOUT | Thermo Fischer Scientific | Cat# 10777019 |
| Superscript III | Thermo Fischer Scientific | Cat# 18080093 |
| Bst 2.0 WarmStart® DNA Polymerase | NEB | Cat# M0538S |
| AMPure XP beads | Beckman Coulter | Cat#. A63880 |
| Duplex-specific Nuclease | Evrogen | Cat# EA003 |
| KAPA HIFI Hotstart Ready Mix | KAPA Biosystem | Cat# KK2601 |
| Critical commercial assays | ||
| RNeasy Mini Kit | QIAGEN | Cat# 74106 |
| qScript cDNA SuperMix | QuantaBio | Cat# 101414-106 |
| WST-1 | Takara | Cat# MK400 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fischer Scientific | Cat# 34096 |
| Nextera DNA library prep kit | Illumina | Cat# FC-121-1030 |
| NextSeq 500/550 High Output Kit v2.5 (150 Cycles) | Illumina | Cat# 20024907 |
| NextSeq 500/550 Mid Output Kit v2.5 (150 Cycles) | Illumina | Cat# 20024904 |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | NEB | Cat# E7490 |
| NEBNext Ultra II RNA library Prep Kit | NEB | Cat# E7770 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE166480 |
| NCBI Gene Expression Omnibus database | Tian et al., 2020 | GEO: GSE136904 |
| Mouse reference genome NCBI build 38, mm10 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/mouse |
| Experimental models: cell lines | ||
| Mouse: 344SQ, 393P, 531LN1, 531LN2, 307P, 412P, and their transfected derivatives | Gibbons et al., 2009 | N/A |
| Mouse: 344SQ_RFP | Padhye et al., 2019 | N/A |
| Mouse: 344SQ_shCtL, 344SQ_shZEB1, 344SQ_mir206, 344SQ_mir148a | Tan et al., 2017 | N/A |
| Mouse: 393P_vec, 393P_ZEB1, 344SQ_miR-181 | Tan et al., 2018 | N/A |
| Mouse: 344SQ_miR-200 | Ahn et al., 2012 | N/A |
| Mouse: CAFS-GFP | This paper | N/A |
| Mouse: 393_RFP | This paper | N/A |
| Experimental models: organisms/strains | ||
| Mouse: KrasLA1/+, 129/SV | The Jackson Laboratory | N/A |
| Oligonucleotides | ||
| siRNA against murine DDR2 | Sigma-Aldrich | SASI_Mm01_00106702 |
| siRNA against murine DDR2 | Sigma-Aldrich | SASI_Mm01_00106703 |
| siRNA against murine DDR2 | Sigma-Aldrich | SASI_Mm01_00106704 |
| siRNA against murine Itih2 | Sigma-Aldrich | SASI_Mm01_00067716 |
| siRNA against murine Itih2 | Sigma-Aldrich | SASI_Mm01_00067717 |
| siRNA against murine Itih2 | Sigma-Aldrich | SASI_Mm01_00067718 |
| Universal siRNA negative control #2 | Sigma-Aldrich | N/A |
| DDR2_F: TCATCCTGTGGAGGCAGTTCTG | Sigma-Aldrich | N/A |
| DDR2_R: CTGTTCACTTGGTGATGAGGAGC | Sigma-Aldrich | N/A |
| RLP32_F: GGAGAAGGTTCAAGGGCCAG | Sigma-Aldrich | N/A |
| RLP32_R: TGCTC CCATAACCGATGTTTG | Sigma-Aldrich | N/A |
| Itih2_F: ACCAGGACACATCCTCTCAGCT | Sigma-Aldrich | N/A |
| Itih2_R: CAGAACCTCCGAAGTAGTTGTGG | Sigma-Aldrich | N/A |
| MATQ-seq primer mix | Sheng et al., 2017 | N/A |
| Recombinant DNA | ||
| pLVX-puro | Clontech | Cat# 632164 |
| EF-pLenti6.3-GFP | Dr. Scott lab (BCM) | N/A |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| GraphPad Prism 7.03 | GraphPad | N/A |
| BD FACSDiva 6.1.3 software | BD Biosciences | N/A |
| NIS-Elements | Nikon | N/A |
| R Studio | R studio | https://www.r-project.org/ |
| GSEA 4.0.3 | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| MATLAB R2019a | The MathWorks, Inc. | https://www.mathworks.com/products/matlab.html |
| Cutadapt 1.18 | Martin, 2011 | https://github.com/marcelm/cutadapt/ |
| STAR 2.5.3a | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Samtools 1.7 | Li et al., 2009 | https://github.com/samtools/ |
| htseq-count 0.10.0 | Anders et al., 2015 | https://pypi.org/project/HTSeq/ |
| seqtk 1.2-r94 | Shen et al., 2016 | https://github.com/lh3/seqtk |
| gencode.vM10.annotation.gtf | N/A | https://www.gencodegenes.org/mouse/release_M10.html |
| edgeR 3.26.8 | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Other | ||
| Transwell Boyden chambers | Thermo Scientific | Cat# 141078 |
| Glass-bottom 35mm dishes | Mattek | Cat# P35G-1.5-14-C |
| Glass bottom 24-well plate | Mattek | Cat# P24G-1.5-13-F |
| Culture-insert 2-well, 35 mm plate | Ibidi | Cat# 81176 |
Highlights.
Epithelial-mesenchymal transition (EMT) of cancer cells governs CAF heterogeneity
EMT sensitizes cancer cells to pro-metastatic signals from CAFs
EMT initiates formation of CAF-led invasive structures
EMT triggers CAF repulsion
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grants R01 CA181184 (to J.M.K.), R01 CA2111125 (to J.M.K.), P30 CA125123 (to C.J.C.), R01 CA196205 (to G.D.L.); Department of Defense PROSPECT grant W81XWH-07–1-0306 (to I.I.W.); National Cancer Institute (NCI) Specialized Program of Research Excellence (SPORE) grant 1-P50-CA70907–01; CPRIT-MIRA grant RP160652 (to J.M.K.); and National Research Foundation of Korea (NRF) grant NRF-2020R1A5A2019210 (to Y.-H.A.). NCI P30 CA16672 Core grant supported flow cytometry. J.M.K. holds the Gloria Lupton Tennison Distinguished Professorship in Lung Cancer. C.Z. is supported by a McNair Scholarship. D.L.G. is an R. Lee Clark Fellow of the University of Texas MD Anderson Cancer Center, supported by the Jeane F Shelby Scholarship Fund. This project was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the CPRIT Core Facility Support Award (CPRIT-RP180672), the NIH (P30 CA125123 and S10 RR024574), and the expert assistance of Joel M. Sederstrom. The UTMB Mass Spectrometry Facility is supported in part by CPRIT grant RP190682 (to W.K.R.).
DECLARATION OF INTERESTS
D.L.G. serves on scientific advisory committees for AstraZeneca, GlaxoSmithKline, Sanofi, and Janssen; provides consultation to Ribon Therapeutics; and receives research support from Janssen, Takeda, and AstraZeneca. I.I.W. serves on advisory boards for Genentech/Roche, Bristol-Myers Squibb, Medscape, Astra Zeneca/Medimmune, HTG Molecular, Merck, GlaxoSmithKline, and MSD and receives research support from Genentech, Oncoplex, HTG Molecular, DepArray, Merck, Bristol-Myers Squibb, Medimmune, Adaptive, Adaptimmune, EMD Serono, Pfizer, Takeda, Amgen, Karus, Johnson & Johnson, Bayer, 4D, Novartis, and Perkin-Elmer (Akoya). G.D.L. has received financial support from Pfizer-CTI. J.M.K. has received consulting fees from Halozyme. P.B. has received consulting fees from ExpertConnect.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109009.
REFERENCES
- Ahn YH, Gibbons DL, Chakravarti D, Creighton CJ, Rizvi ZH, Adams HP, Pertsemlidis A, Gregory PA, Wright JA, Goodall GJ, et al. (2012). ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J. Clin. Invest. 122, 3170–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albritton JL, Roybal JD, Paulsen SJ, Calafat N, Flores-Zaher JA, Farach-Carson MC, Gibbons DL, and Miller JS (2016). Ultrahigh-throughput Generation and Characterization of Cellular Aggregates in Laser-ablated Microwells of Poly(dimethylsiloxane). RSC Advances 6, 8980–8991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoschek M, Oskolkov N, Bocci M, Lövrot J, Larsson C, Sommarin M, Madsen CD, Lindgren D, Pekar G, Karlsson G, et al. (2018). Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 9, 5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SV, Grither WR, Brenot A, Hwang PY, Barcus CE, Ernst M, Pence P, Walter C, Pathak A, and Longmore GD (2019). DDR2 controls breast tumor stiffness and metastasis by regulating integrin mediated mechanotransduction in CAFs. eLife 8, e45508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, and Tuveson DA (2019). IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 9, 282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Fontaine C, Matthews HK, Kuriyama S, Moreno M, Dunn GA, Parsons M, Stern CD, and Mayor R (2008). Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature 456, 957–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chockley PJ, and Keshamouni VG (2016). Immunological Consequences of Epithelial-Mesenchymal Transition in Tumor Progression. J. Immunol. 197, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsa CA, Brenot A, Grither WR, Van Hove S, Loza AJ, Zhang K, Ponik SM, Liu Y, DeNardo DG, Eliceiri KW, et al. (2016). The Action of Discoidin Domain Receptor 2 in Basal Tumor Cells and Stromal Cancer-Associated Fibroblasts Is Critical for Breast Cancer Metastasis. Cell Rep. 15, 2510–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror S, Sander L, Schwartz H, Sheinboim D, Barzilai A, Dishon Y, Apcher S, Golan T, Greenberger S, Barshack I, et al. (2016). Melanoma miRNA trafficking controls tumour primary niche formation. Nat. Cell Biol. 18, 1006–1017. [DOI] [PubMed] [Google Scholar]
- Duda DG, Duyverman AM, Kohno M, Snuderl M, Steller EJ, Fukumura D, and Jain RK (2010). Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 107, 21677–21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Nakasone ES, and Werb Z (2010). Tumors as organs: complex tissues that interface with the entire organism. Dev. Cell 18, 884–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 9, 1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, Washington MK, Shi C, Franco OE, Weaver AM, et al. (2017). Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 216, 3799–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. (2013). Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 110, 20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, and Sahai E (2007). Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 9, 1392–1400. [DOI] [PubMed] [Google Scholar]
- Gascard P, and Tlsty TD (2016). Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 30, 1002–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, Thilaganathan N, Du L, Zhang Y, Pertsemlidis A, and Kurie JM (2009). Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 23, 2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grither WR, and Longmore GD (2018). Inhibition of tumor-microenvironment interaction and tumor invasion by small-molecule allosteric inhibitor of DDR2 extracellular domain. Proc. Natl. Acad. Sci. USA 115, E7786–E7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, and Jacks T (2001). Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410, 1111–1116. [DOI] [PubMed] [Google Scholar]
- Kalluri R (2016). The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598. [DOI] [PubMed] [Google Scholar]
- Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, and Fearon DT (2010). Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 330, 827–830. [DOI] [PubMed] [Google Scholar]
- Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D, Reichardt W, Bronsert P, et al. (2017). The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 19, 518–529. [DOI] [PubMed] [Google Scholar]
- Labernadie A, Kato T, Brugués A, Serra-Picamal X, Derzsi S, Arwert E, Weston A, González-Tarragó V, Elosegui-Artola A, Albertazzi L, et al. (2017). A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 19, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwé H, Pircher A, Van den Eynde K, et al. (2018). Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 24, 1277–1289. [DOI] [PubMed] [Google Scholar]
- Larsen JE, Nathan V, Osborne JK, Farrow RK, Deb D, Sullivan JP, Dospoy PD, Augustyn A, Hight SK, Sato M, et al. (2016). ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J. Clin. Invest. 126, 3219–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, and Kalluri R (2018). A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis. Model. Mech. 11. 10.1242/dmm.029447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, and Stanger BZ (2019). The tumor as organizer model. Science 363, 1038–1039. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Sub-group (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. (2018). Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 49, 178–193.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler M, Krüger JA, Niethammer AG, and Reisfeld RA (2006). Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Invest. 116, 1955–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Weaver VM, and Werb Z (2012). The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 196, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet. J. 17, 10–12. [Google Scholar]
- Mitra AK, Zillhardt M, Hua Y, Tiwari P, Murmann AE, Peter ME, and Lengyel E (2012). MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2, 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odenthal J, Takes R, and Friedl P (2016). Plasticity of tumor cell invasion: governance by growth factors and cytokines. Carcinogenesis 37, 1117–1128. [DOI] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhye A, Ungewiss C, Fradette JJ, Rodriguez BL, Albritton JL, Miller JS, and Gibbons DL (2019). A novel ex vivo tumor system identifies Srcmediated invasion and metastasis in mesenchymal tumor cells in non-small cell lung cancer. Sci. Rep. 9, 4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankova D, Chen Y, Terajima M, Schliekelman MJ, Baird BN, Fahrenholtz M, Sun L, Gill BJ, Vadakkan TJ, Kim MP, et al. (2016). Cancer-Associated Fibroblasts Induce a Collagen Cross-link Switch in Tumor Stroma. Mol. Cancer Res. 14, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager BC, Xie Q, Bao S, and Rich JN (2019). Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 24, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, and Hingorani SR (2012). Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puisieux A, Brabletz T, and Caramel J (2014). Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 16, 488–494. [DOI] [PubMed] [Google Scholar]
- Raab-Westphal S, Marshall JF, and Goodman SL (2017). Integrins as Therapeutic Targets: Successes and Cancers. Cancers (Basel) 9, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal JD, Zang Y, Ahn YH, Yang Y, Gibbons DL, Baird BN, Alvarez C, Thilaganathan N, Liu DD, Saintigny P, et al. (2011). miR-200 Inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol. Cancer Res. 9, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa E, Szabó A, Bibonne A, Theveneau E, Parsons M, and Mayor R (2015). Cadherin Switch during EMT in Neural Crest Cells Leads to Contact Inhibition of Locomotion via Repolarization of Forces. Dev. Cell 34, 421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Le S, Li Y, and Hu F (2016). SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 11, e0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng K, Cao W, Niu Y, Deng Q, and Zong C (2017). Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat. Methods 14, 267–270. [DOI] [PubMed] [Google Scholar]
- Spranger S, and Gajewski TF (2018). Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 18, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stramer B, and Mayor R (2017). Mechanisms and in vivo functions of contact inhibition of locomotion. Nat. Rev. Mol. Cell Biol. 18, 43–55. [DOI] [PubMed] [Google Scholar]
- Su S, Chen J, Yao H, Liu J, Yu S, Lao L, Wang M, Luo M, Xing Y, Chen F, et al. (2018). CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 172, 841–856.e16. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Banerjee P, Guo HF, Ireland S, Pankova D, Ahn YH, Nikolaidis IM, Liu X, Zhao Y, Xue Y, et al. (2017). Epithelial-to-mesenchymal transition drives a pro-metastatic Golgi compaction process through scaffolding protein PAQR11. J. Clin. Invest. 127, 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Banerjee P, Liu X, Yu J, Gibbons DL, Wu P, Scott KL, Diao L, Zheng X, Wang J, et al. (2018). The epithelial-to-mesenchymal transition activator ZEB1 initiates a prometastatic competing endogenous RNA network. J. Clin. Invest. 128, 3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Banerjee P, Pham EA, Rutaganira FUN, Basu K, Bota-Rabassedas N, Guo HF, Grzeskowiak CL, Liu X, Yu J, et al. (2020). PI4KIIIβ is a therapeutic target in chromosome 1q-amplified lung adenocarcinoma. Sci. Transl. Med. 12, eaax3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Lian R, Li Y, Liu C, Liang S, Li W, Tao T, Wu X, Ye Y, Yang X, et al. (2020). AKT-induced lncRNA VAL promotes EMT-independent metastasis through diminishing Trim16-dependent Vimentin degradation. Nat. Commun. 11, 5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werb Z, and Lu P (2015). The Role of Stroma in Tumor Development. Cancer J 21, 250–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whatcott CJ, Han H, Posner RG, Hostetter G, and Von Hoff DD (2011). Targeting the tumor microenvironment in cancer: why hyaluronidase deserves a second look. Cancer Discov. 1, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi M, Barker TH, Gibbons DL, and Kurie JM (2018). The fibrotic tumor stroma. J. Clin. Invest. 128, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ahn YH, Gibbons DL, Zang Y, Lin W, Thilaganathan N, Alvarez CA, Moreira DC, Creighton CJ, Gregory PA, et al. (2011). The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J. Clin. Invest. 121, 1373–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, et al. (2015). Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 17, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data of single cell RNA-seq and mini-bulk RNA-seq have been submitted to GEO: GSE166480. Sequencing depth and mapping can be found in Table S1.