

Abstract
The endothelial Protein C receptor (EPCR) is essential for the anticoagulant and cytoprotective functions of the protein C (PC) system. Selected variants of the malaria parasite protein, Plasmodium falciparum Erythrocyte Membrane Protein 1 (PfEMP1) associated with severe malaria, including cerebral malaria, specifically target EPCR on vascular endothelial cells.
Here, we examine the cellular response to PfEMP1 engagement to elucidate its role in malaria pathogenesis. Binding of the CIDRα1.1 domain of PfEMP1 to EPCR obstructed activated PC (APC) binding to EPCR and induced a loss of cellular EPCR functions. CIDRα1.1 severely impaired endothelial PC activation and effectively blocked APC-mediated activation of protease-activated receptor-1 (PAR1) and associated barrier protective effects of APC on endothelial cells. A soluble EPCR variant (E86A-sEPCR) bound CIDRα1.1 with high affinity and did not interfere with (A)PC binding to cellular EPCR. E86A-sEPCR used as a decoy to capture PfEMP1, permitted normal PC activation on endothelial cells normal barrier protective effects of APC, and greatly reduced cytoadhesion of infected erythrocytes to brain endothelial cells.
These data imply important contributions of PfEMP1-induced protein C pathway defects in the pathogenesis of severe malaria. Furthermore, the E86A-sEPCR decoy provides a proof-of-principle strategy for the development of novel adjunct therapies for severe malaria.
Keywords: Malaria, Endothelial cells, Endothelial Protein C Receptor, PfEMP1
Introduction
The clinical outcome of malaria due to Plasmodium falciparum infection ranges from no or minor symptoms to severe malaria, a life threatening disease with complications such as severe anemia, respiratory distress, and cerebral malaria. Of the different manifestations of severe malaria, cerebral malaria is most recognizable clinically and anatomically. Even with the latest antimalarial and supportive treatments available, cerebral malaria is fatal in around 15% of the cases, and survivors often suffer long-term neurological impairments (1, 2). P. falciparum infected erythrocytes (IE) express parasite-encoded proteins on the surface of the erythrocyte cell membrane that mediate adhesion to the vascular endothelium thereby allowing the parasites to avoid splenic clearance and an opportunity to mature and multiply inside the IE. The sequestration of IE provoke a strong and focused inflammatory response caused by alterations in the microenvironment and disruption of local blood flow that result in dysfunction of the endothelium, vascular leakage, edema, and may ultimately manifest in coma and death (3, 4).
Parasite sequestration is mediated by the P. falciparum Erythrocyte Membrane Protein 1 (PfEMP1) protein family, encoded by the var genes. A subset of PfEMP1s characterized by carrying a Cysteine-rich Inter Domain Region (CIDR) subtype alpha 1 (CIDRα1) domain has been associated with the development of severe malaria (5) . Recently, the endothelial protein C receptor (EPCR) was identified as the receptor for CIDRα1 domains, and EPCR-binding by parasites isolates from patients was associated with severe malaria symptoms of the donor (6). Thus it seems parasites has evolved to each carry a repertoire of three major distinct PfEMP1 groups conferring discrete receptor specificities and ensuing clinical outcomes: The group A or A-like PfEMP1 variants carrying CIDRα1 domains binding to EPCR and linked to development of severe including cerebral malaria symptoms; the group B and C PfEMP1 variants carrying CIDRα2-6 domains binding to CD36 and linked to uncomplicated malaria(7).
EPCR is best known for its multiple roles in the PC system but also acts as a vascular receptor for (activated) factor VII(a) (8-12). The anticoagulant and cytoprotective functions of the PC system are essential for the regulation of coagulation, vascular inflammation, and endothelial permeability (10, 13). EPCR facilitates the activation of PC by recruiting PC to the endothelial cell surface thereby enhancing its activation by the thrombin-thrombomodulin complex (12-14). Activated PC (APC) mediates potent anticoagulant activities as well as direct cytoprotective effects on cells that are independent of its anticoagulant activities (10, 13). The anticoagulant activities of APC involve the proteolytic inactivation of coagulation factors Va and VIIIa that are required for efficient thrombin generation (13, 15). Cytoprotective effects of APC include anti-apoptotic and anti-inflammatory activities, beneficial alterations of gene expression profiles and protection of endothelial and vascular barrier functions (13, 16). EPCR is required for APC’s cytoprotective effects and according to the current paradigm, EPCR facilitates APC-mediated non-canonical activation of protease activated receptor (PAR) 1 and PAR3 that result biased PAR signaling and activation of cytoprotective intracellular signaling pathways (10, 17, 18). Multiple in vivo disease and injury models demonstrate important beneficial contribution of APC’s activities both when APC is generated endogenously or administered therapeutically. Notably, APC cytoprotective activities provide important neuroprotective effects that include protection of the blood brain barrier as well as anti-inflammatory and anti-apoptotic activities within the neurovascular unit, and thus, these activities of APC might be directly relevant to the complications associated with cerebral malaria (19, 20).
The role of EPCR in cerebral malaria pathogenesis is unresolved; loss of EPCR staining has been reported at sites of sequestering IE in the brain, along with increased fibrin deposits (21). Furthermore, increased systemic plasma levels of TNFα and thrombin-antithrombin III (TAT) complexes as well as decreased levels of PC have been reported during P. falciparum infection (22). Collectively, these observations indicate that cerebral malaria infection results in procoagulant and proinflammatory vascular responses that are consistent with acquired insufficiencies of the protein C system. Here, we show that insufficiencies of the protein C system are inflicted by binding of the malaria protein PfEMP1 to EPCR on endothelial cells in vitro. Our data conclude that CIDRα1 induced a loss of EPCR function causing major defects in PC activation and in the ability of APC to convey cytoprotective effects. In addition, a PfEMP1 decoy strategy was developed using a non-(A)PC-binding soluble EPCR variant that effectively allowed normal endothelial EPCR function permitting normal PC activation and cytoprotective effects of APC in presence of CIDRα1.
Methods
Proteins
The recombinant EPCR-binding CIDRα1.1 domain derived from FCR3 IT4VAR20 and the non-EPCR-binding control domains CIDRα3.5 derived from FCR3 IT4VAR15 and CIDRa3.1 derived from Dd2 Dd2VAR01 were produced using the baculovirus expression system as described previously (6). PC, APC, and biotinylated-APC were prepared as described (23, 24) (ref 24 submitted for publication; see accompanying pdf copy). Soluble wild-type (wt) EPCR (sEPCR) and E86A-sEPCR were produced in HEK-293 cells and purified as described (25).
Cell Culture
EA.hy926 cells were maintained in complete medium (high-glucose DMEM supplemented with 2 mM L-glutamine, 1% penicillin-streptomycin, and 10% fetal calf serum). EPCR knock-down EA.hy926 cells were maintained in complete medium supplemented with 0.5 μg/ml puromycin. Human brain microvascular endothelial cells (HBMECs), isolated as described previously (26), were maintained in fibronectin (R&D Systems) coated flasks in endothelial growth medium (ScienCell).
Binding assays
APC binding to sEPCR and to EA.hy926 cells was performed as described (24). Binding of CIDR to soluble EPCR was determined in MaxiSorp plates (Nunc) coated with wt-sEPCR or E86A-sEPCR (10 μg/ml) in coating buffer (15 mM Na2CO3, 35 mM NaHCO3, 0.02% NaN3, pH 9.5) overnight at 4°C, blocked with Tris buffered saline (TBS; 50 mM Tris, 150 mM NaCl, pH 7.4) containing 3% bovine serum albumin (Sigma-Aldrich) for two hours at room temperature, and incubated which recombinant CIDR domain containing a V5-tag. Binding of CIDR domains was determined with a HRP-conjugated anti-V5 antibody (ICL) and 1-step Ultra-TMB (3,3′.5,5′-tetramethylbenzidine, Pierce).
Analysis of CIDR binding to EA.hy926 cells was determined in an on-cell western (OCW) assay using wt and EPCR-knock down (EPCRKD) EA.hy926 cells (24). Cells were grown in black clear bottom 96-well plates (Corning) and were incubated on ice with CIDRα1.1 in HMM2 (Hanks’ balanced salt solution (GIBCO) supplemented with 1.3 mM CaCl2, 0.6 mM MgCl2, and 0.1% BSA(essential globulin and protease free, Sigma)) for one hour, fixed with 4% methanol-free paraformaldehyde (Pierce), and blocked with Odyssey Blocking buffer (OBB; Licor). Binding of CIDR domains was determined with 3 μg/ml goat anti-V5 antibody (ICL) and 1:600 IRDye CW800 donkey anti-rabbit IgG (Licor). DRAQ5 (Biostatus) diluted 1:100,000 was used for cell number normalization. Fluorescent signals were analyzed on the Odyssey (Licor) using Image Studio Software v2.0 and normalized to vehicle-treated controls.
Functional assays
PC activation was determined on EA.hy926 cells in 10 mM HEPES buffer (pH 7.4) supplemented with 150 mM NaCl, 3 mM CaCl2, 0.6 mM MgCl2, and 0.1% PEG 6000. PC (50 nM, Baxter) and 0.02 U/mL (0.17 nM) thrombin (ERL) were incubated with CIDRα1.1, CIDRα3.1 (50 nM) or the EPCR-blocking antibody RCR-252 (27) (20 μg/ml) with or without E86A-sEPCR. After 45 min, thrombin was inhibited by 0.2 μM lepirudin (Schering AG) and APC generation was determined using 0.4 mM S-2366 (Chromogenix). Substrate conversion was measured kinetically at 405 nm in an iEMS Reader (Labsystems).
PAR1 cleavage by APC was examined with an OCW assay essentially as previously described (18). Briefly, confluent EA.hy926 cells were incubated with 100 nM APC in the presence or absence of 100 nM CIDRα1.1 and/or 400 nM E86A-sEPCR for three hours at 37°C. After fixation and blocking, cells were stained with 10 μg/ml mouse anti-PAR1 ATAP2 antibody for one hour at room temperature. IRDye CW800 donkey anti-mouse IgG (Licor) diluted 1:600 was used for detection and DRAQ5 diluted 1:100,000 for cell number normalization.
Endothelial cell-layer permeability was measured in real time using the iCelligence system (ACEA) that determines changes in transendothelial electric resistance (TER) by electric cell-substrate impedance sensing (ECIS). EA.hy926 cells were grown on integrated electronic sensors to confluence, after which new medium containing vehicle, CIDRα1.1, CIDRα1.1 and/or E86A-sEPCR was added. After one hour, APC (50 nM) was added and incubated for an additional 3 hours. Barrier disruption was induced with 2 nM thrombin and compared to untreated cells. Barrier disruption by thrombin was set to 100%.
Parasite culture
Plasmodium falciparum laboratory strain FCR3 expressing the IT4VAR20 PfEMP1 (6) were maintained in blood type O+ with 0.125 μg/ml Albumax II (Invitrogen), 25 mM sodium bicarbonate (Lonza), 2 mM L-glutamine (Sigma-Aldrich), and 0.125 μg/ml gentamycin (Lonza) supplemented RPMI-1640 HEPES medium. The parasites were maintained in a mixture of 5% CO2 and 2% O2. Routine antibody and protein selections i were done to ensure proper parasite phenotype and QPCR was used to assess var gene transcription as previously described (6).
Infected erythrocyte cell adhesion assay
Adhesion assays were done as described (6). Briefly, infected red blood cells in the ring stage expressing IT4VAR20 were grown with tritiated hypoxanthine (3H) (Amersham; 8.75 MBq/ml packed erythrocytes) until maturing into the trophozoite stage. Trophozoite and schizont stage parasites were purified using magnetic cell sorting (Miltenyi Biotec) in RPMI-1640 supplemented with 2% FCS. The concentration was adjusted to 1.25x107 blood cells/ml. HBMECs were grown to confluence in endothelial cell medium (ScienCell) on flat-bottomed 96-well plates (Nunc) pre-coated with 2 μg/cm2 fibronectin (R&D Systems). Late stage IEs (250.000/well) were added to HBMECs along with various concentrations of sEPCR or E86A-sEPCR. After one hour at 37°C, the plates were washed using the Biomek 2000 (Beckman Coulter). Radioactive material was harvested using a Filtermate Harvester (PerkinElmer) and 50 μl scintillation liquid was added before radioactivity was measured as counts per minute using a Topcount NXT (PerkinElmer). Counts per minute in wells with HBMECs and parasite IE but without sEPCR or E86A-sEPCR were considered to represent 100% adhesion. The amounts of EPCR on a layer of confluent HBMECs in 8 well chambered glass slides (Thermo Scientific). Parasite medium or 50μl MACS purified parasites at a concentration of 1.25x107 blood cells/ml were added to wells, which was then left to incubate for various time intervals. After the incubations, the chambers were washed upside down for 30 minutes in warm PBS. The slides were subsequently fixed with 4% PFA. Staining was done with polyclonal goat anti-EPCR antibodies (R&D), rabbit anti-goat FITC (Invitrogen), and DAPI. Coverslips were mounted onto the slides using FluorSave (VWR). Images of the cells were acquired using a Nikon TE 2000-E confocal microscope with 60x oil immersion objective lens (DIC), and fluorescence intensity was quantified using FIJI (28).
Statistical analysis
Data was analyzed using the Student’s t-test or ANOVA with a multiple comparison test as appropriate. P-values <0.05 were considered statistically significant. Statistical analyses were performed using GraphPad Prism software, v5.04 (GraphPad).
Results
Inhibition of APC binding to EPCR by CIDRα1.1
To characterize the effects on EPCR function of the specific binding between the PfEMP1 malaria protein and EPCR, the interaction of CIDRα1 with EPCR was studied in more detail. The recombinant CIDRα1.1 subtype domain derived from the IT4VAR20 PfEMP1 bound to sEPCR in a purified system with an apparent Kd of 4.3 nM (Figure 1A). The CIDRα1.1 protein inhibited the binding of APC to sEPCR, with 50% inhibition at 12.5 nM CIDRα1.1 and complete inhibition of APC binding at 50 nM CIDRα1.1 (Figure 1B). This confirmed data from a previous study that showed reduced PC binding to sEPCR in the presence of CIDRα1 (6). To determine the effect of CIDRα1.1 on APC binding to cellular EPCR, CIDRα1.1 binding to endogenous EPCR expressed on EA.hy926 cells was examined. A concentration-dependent binding of CIDRα1.1 to the surface of wt-EA.hy926 cells was observed with an apparent Kd of 8 nM (Figure 1C). In contrast, no significant CIDRα1.1 binding was observed on EPCRKD EA.hy926 cells that express <20% EPCR (Figure 1C). This indicated that EPCR was the main receptor responsible for CIDRα1.1 binding on EA.hy926 cells. Consistent with the effects of CIDRα1.1 on sEPCR, APC binding to endothelial EPCR was inhibited in the presence of CIDRα1.1 with 50% inhibition of APC binding at around 33 nM CIDRα1.1 and complete inhibition at ~100 nM CIDRα1.1 (Figure 1D).
Figure 1. The CIDRα1.1 domain of PfEMP1 binds EPCR and inhibits APC binding.
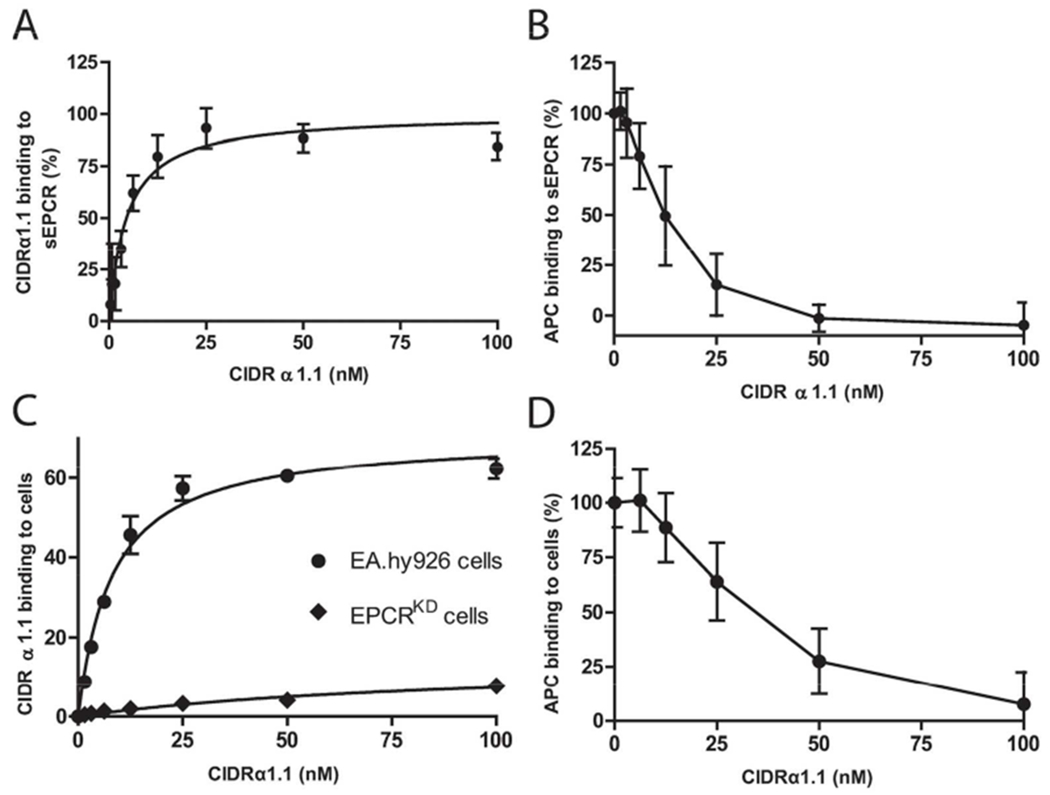
A) Solid phase binding assay of the CIDRα1.1 domain to sEPCR. The Bmax of individual experiments was set to 100%. B) Binding APC to sEPCR in the presence of CIDRα1.1. CIDRα1.1 was incubated with sEPCR for 30 min before addition of APC (50 nM). APC binding in the absence of CIDRα1.1 was considered to represent 100% binding. C) Binding of CIDRα1.1 to endothelial EPCR on EA.hy926 and EPCR knockdown (EPCRKD) EA.hy926 cells expressing <20% EPCR analyzed by on cell western. CIDRα1.1 binding was expressed in arbitrary fluorescence units (FL) reported by the Odyssey (K. Counts). D) APC binding (50 nM) to EA.hy926 endothelial cells in presence of CIDRα1.1. APC binding in the absence of CIDRα1.1 was considered to represent 100% binding. All data points represent the mean ± SD (n=3).
Inhibition of EPCR function by CIDRα1.1
To investigate the effect of CIDRα1.1 on stimulation of PC activation by EPCR, thrombin-mediated PC activation was determined on EA.hy926 cells, such that the required thrombomodulin and EPCR were provided by the cells. PC activation on EA.hy926 cells was inhibited >75% by CIDRα1.1 (Figure 2A). The non EPCR-binding CIDRα3.5 domain from PfEMP1 IT4VAR15 (6), did not inhibit PC activation, indicating that the inhibition was specific for the EPCR-binding CIDRα1.1 domain. Antibody RCR-252, previously shown to block PC binding to EPCR (27), was used to ascertain the fraction of EPCR-dependent PC activation and suggested that EPCR’s ability to enhance PC activation was completely blocked by CIDRα1.1 (Figure 2A).
Figure 2. CIDRα1.1 inhibits cellular functions of EPCR.
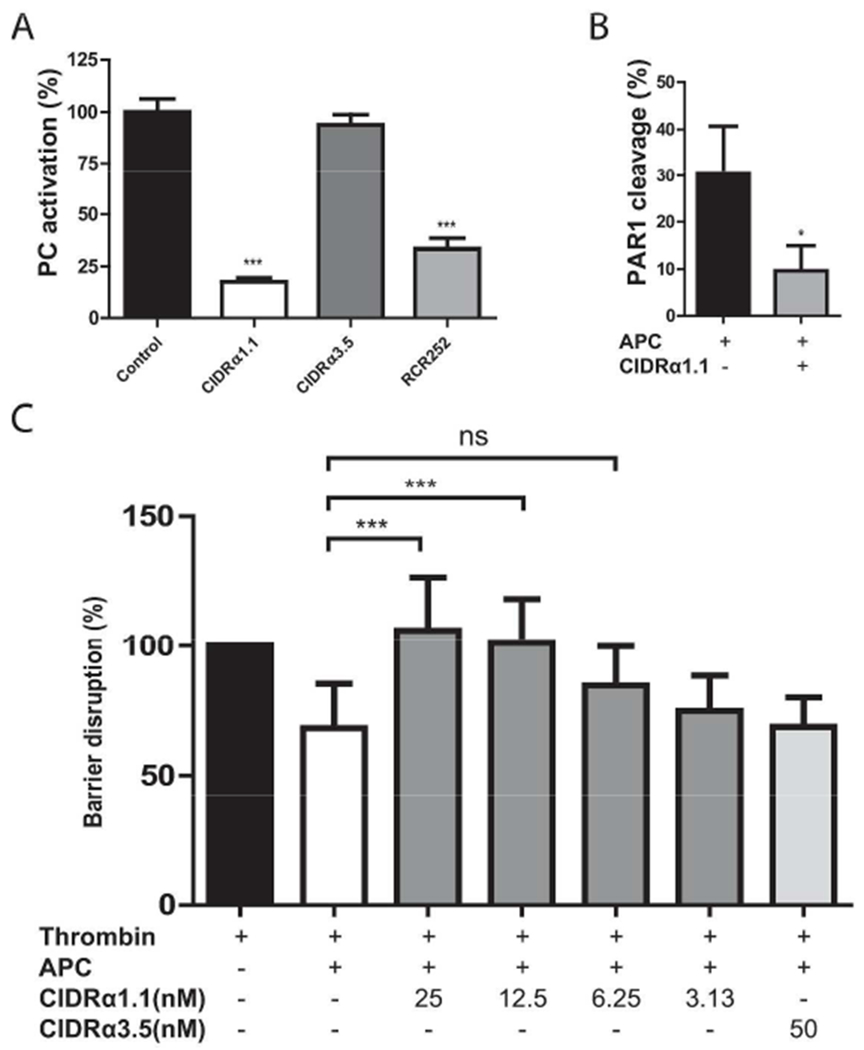
A) The effect of CIDRα1.1 on EPCR-dependent enhancement of PC activation. Thrombin-mediated PC activation was determined on EA.hy926 cells in presence of EPCR-binding CIDRα1.1 (50 nM), non-EPCR-binding CIDRα3.5 (50 nM), and the EPCR blocking antibody RCR-252 (20 μg/ml). PC activation in the absence of CIDR or antibody was set to 100%. B) The effect of CIDRα1.1 on EPCR-dependent PAR1 cleavage by APC. APC (100 nM)-mediated PAR1 cleavage as a percentage of the total endogenously expressed PAR1 on the surface of EA.hy926 cells was determined in the presence and absence of CIDRα1.1 (100 nM). C) The effect of CIDRα1.1 on EPCR-dependent protection of endothelial barrier function by APC. APC (50 nM)-mediated protection of thrombin (2 nM)-induced barrier disruption was determined on confluent EA.hy926 cell layers in the presence of EPCR-binding CIDRα1.1 and non-EPCR-binding CIDRα3.1 control. The reduction of impedance induced by thrombin was set as 100% barrier disruption. All data points represent the mean ± SD (A, B: n=3, C: n=6-8). P-values were calculated using: A) and C) 1-way Anova and Dunnett’s Multiple Comparison Test, and for B) Student’s t-test. * denotes p<0.05, *** denotes p<0.001.
Next, the effects of CIDRα1.1 binding to EPCR on APC’s cellular functions were investigated by analyzing EPCR-dependent cleavage of PAR1 by APC on endothelial cells. APC cleaves PAR1 at non-canonical Arg46 in contrast to canonical cleavage by thrombin at Arg41. Since the epitope of the anti-PAR1 antibody ATAP2 is lost upon PAR1 cleavage at Arg46 by APC but is retained when PAR1 is cleaved by thrombin at Arg41, binding of ATAP2 to cells was used to measure PAR1 cleavage by APC (18). Addition of APC to EA.hy926 cells resulted in 30% PAR1 cleavage. CIDRα1.1 reduced PAR1 cleavage 3-fold to 10 %, suggesting that CIDRα1.1 blocks EPCR-facilitated PAR1 cleavage by APC (Figure 2B). To characterize the functional consequences of diminished PAR1 cleavage by APC in the presence of CIDRα1.1, protection of thrombin-induced endothelial cell barrier disruption was studied. The protective effect of APC on thrombin-induced barrier disruption were dose-dependently inhibited by CIDRα1.1 and APC protective effects were absent in the presence of 12.5 nM CIDRα1.1 (Figure 2C). The CD36-binding CIDRα3.1 domain had no effect on APC’s ability to inhibit thrombin-induced barrier disruption. Thus, CIDRα1.1 inhibited the EPCR-dependent functions of the protein C system that include inhibition of PC activation and inhibition of APC’s cytoprotective effects on endothelial cells.
E86A-sEPCR allows APC binding to EPCR in the presence of CIDRα1.1
It remains unclear whether inhibition of APC binding to EPCR by CIDRα1.1 is caused by steric hindrance or by overlap of the CIDRα1.1 and APC binding sites on EPCR. To obtain additional insights, binding of CIDRα1.1 to E86A-sEPCR was tested, since residue E86 is essential for APC binding to EPCR (17, 29, 30). Remarkably, CIDRα1.1 bound to E86A-sEPCR with an apparent Kd of 4.4 nM (Figure 3A), similar to that of wt-sEPCR (Figures 1A). Thus, E86 is essential for APC binding but was dispensable for CIDRα1.1 binding, indicating qualitatively difference between the binding sites of CIDRα1.1 and APC on EPCR. This provided the opportunity to use E86A-sEPCR as a decoy strategy for CIDRα1.1. E86A-sEPCR dose-dependently enabled APC binding to sEPCR in the presence of CIDRα1.1 (50 nM) (Figure 3B) with around. 50% of maximum APC binding at ~25 nM E86A-sEPCR and ~85% of maximum APC binding at 100 nM E86A.
Figure 3. CIDRα1.1 binds similarly to wild type sEPCR and mutated E86A-sEPCR.
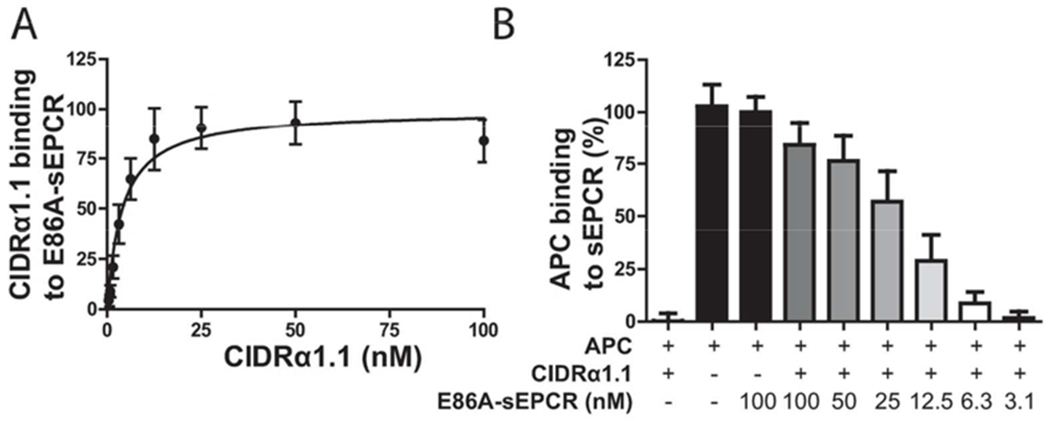
A) Solid phase binding assay of the CIDRα1.1 domain to E86A-sEPCR. The Bmax of individual experiments was set to 100%. B) Binding APC to sEPCR in the presence of CIDRα1.1 and E86A-sEPCR. CIDRα1.1 (50 nM) was incubated with E86A-sEPCR and sEPCR for 30 min before addition of APC (50 nM). APC binding in the absence of CIDRα1.1 was considered to represent 100% binding. All data points represent the mean ± SD (n=3).
E86A-sEPCR prevents cytoadhesion of infected erythrocytes to brain microvascular endothelial cells
To determine whether an E86A-sEPCR decoy strategy potentially would affect IE sequestration, the direct effect of E86A-sEPCR on reductions of IE binding to endothelial cells was determined. Erythrocytes infected with the laboratory parasite FCR3 express endogenous PfEMP1-IT4VAR20 containing CIDRα1.1 and show potent adhesion to primary brain microvascular endothelial cells (HBMEC) (6). When E86A-sEPCR was added to the primary brain endothelial cells together with the IT4VAR20-expressing IEs, it effectively reduced_-the adhesion of with an IC50 ~30 nM and a dose-response curve that was indistinguishable from that of wt-sEPCR (Figure 4). Endothelial cell incubation with EPCR-bound parasites did not affect the general EPCR expression levels (data not shown) as observed in brain microvasculature in vivo (21), supporting the interpretation that the parasite binding was inhibited by the E86A-EPCR.
Figure 4. E86A-sEPCR prevents cytoadhesion of P. falciparum-infected erythrocytes to primary brain endothelial cells.
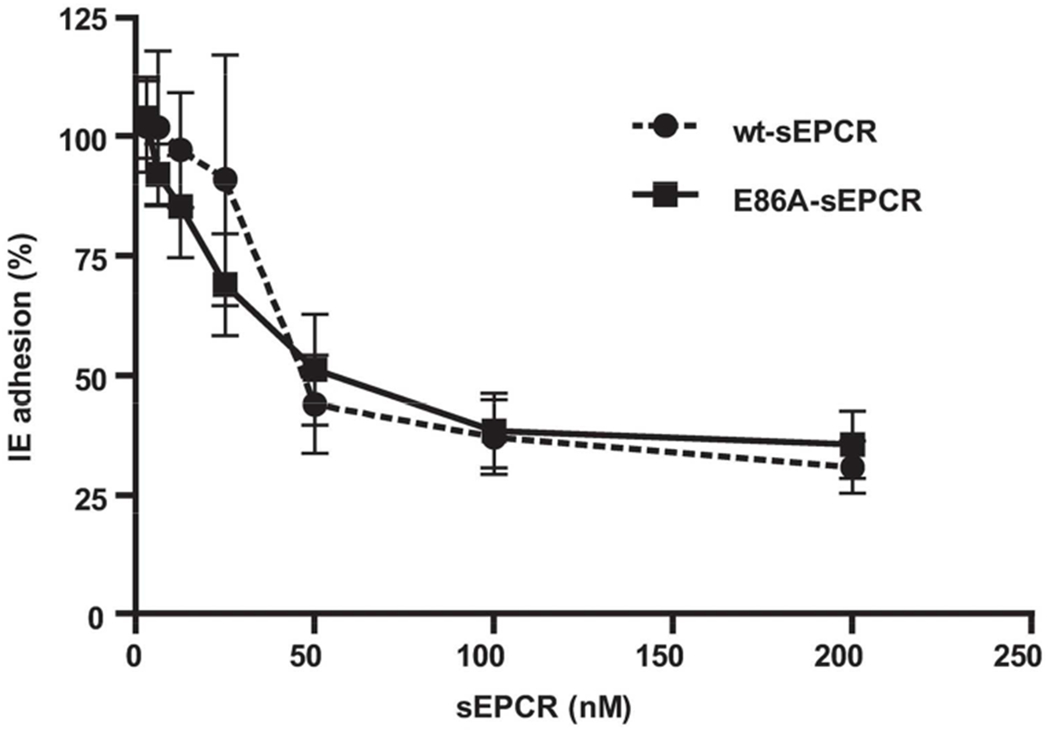
Erythrocytes were infected with the P. falciparum laboratory strain FCR3 expressing the IT4VAR20 PfEMP1 that contains the CIDRα1.1 domain. Infected erythrocytes (IE) were allowed to adhere to primary microvascular brain endothelial cells for 1 hour in the presence of various concentrations of wt-sEPCR or E86A-sEPCR. Adhesion proportion of parasite-IE in the absence of sEPCR following a standardized wash was considered to represent 100% adhesion. All data points represent the mean ± SD (n=3).
The E86A-sEPCR decoy enables cellular functions of EPCR in the presence of CIDRα1.1
To determine the functional efficacy of the E86A-sEPCR decoy, PC activation was determined in the presence of CIDRα1.1 and E86A-sEPCR. CIDRα1.1 inhibited PC activation and E86A-sEPCR overcame the inhibitory effect of CIDRα1.1 (Figure 5A). PC activation was normalized by 50 nM E86A-sEPCR, whereas 5 nM E86A-sEPCR already resulted in a significant improved PC activation in the presence of CIDRα1.1. The functional efficacy of the E86A-sEPCR decoy to allow APC’s cellular effects in presence of CIDRα1 was analyzed by APC-mediated PAR1 cleavage and by APC’s ability to protect against thrombin-induced endothelial barrier disruption. The inhibitory effect of CIDRα1.1 on cleavage of PAR1 by APC was greatly diminished by E86A-sEPCR. E86A-sEPCR increased APC-mediated PAR1 cleavage 4-fold in the presence of CIDRα1.1 (Figure 5B). Similarly, E86A-sEPCR fully enabled APC’s barrier protective effects in the presence of CIDRα1.1 (figure 5C). Although E86A-sEPCR did have a small but not significant effect on the barrier protective effects of APC for reasons not currently understood, E86A-sEPCR by itself showed no effects on barrier integrity (data not shown). In summary, E86A-sEPCR greatly improved cellular functionality of EPCR in the presence of CIDRα1.1 and allowed endothelial PC activation and normal APC’ cellular activity with no significant evidence for direct interference of E86A-sEPCR in these activities.
Figure 5. E86A-sEPCR allows PC activation and APC endothelial barrier protective effects in the presence of CIDRα1.1.
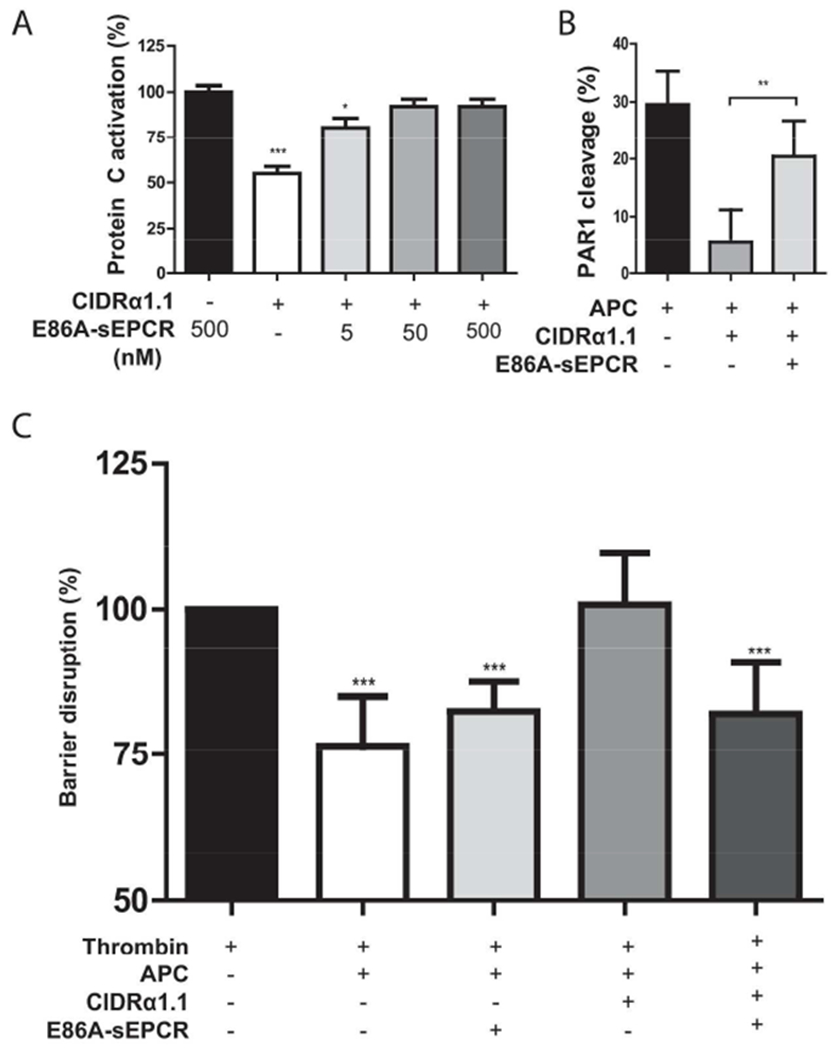
A) The effect of E86A-sEPCR on CIDRα1.1-mediated inhibition of EPCR-dependent enhancement of PC activation. Thrombin-mediated PC activation was determined on EA.hy926 cells in presence of EPCR-binding CIDRα1.1 (50 nM) and E86A-sEPCR. PC activation in the absence of CIDRα1.1 but in the presence of E86A-sEPCR (500 nM) was set to 100%. B) The effect of E86A-sEPCR on CIDRα1.1-mediated inhibition of EPCR-dependent PAR1 cleavage by APC. APC (100 nM)-mediated PAR1 cleavage of endogenous expressed PAR1 on the surface of EA.hy926 cells was determined in the presence and absence of CIDRα1.1 (100 nM) and/or E86A-sEPCR (400 nM). C) The effect of E86A-sEPCR on CIDRα1.1-mediated inhibition of EPCR-dependent protection of endothelial barrier function by APC. APC (25 nM)-mediated protection of thrombin (2 nM)-induced barrier disruption was determined on confluent EA.hy926 cell layers in the presence and absence of EPCR-binding CIDRα1.1 (12.5 nM) and/or E86A-sEPCR (75 nM). The reduction of impedance induced by thrombin was set as 100% barrier disruption. All data points represent the mean ± SD (n=3). P-values were calculated using: A) and C) 1-way ANOVA and Dunnett’s Multiple Comparison Test, B) Student’s t-test. * denotes p<0.05, ** denotes p<0.005, *** denotes p<0.001.
Discussion
Cerebral malaria is generally accepted to be the result of the interplay between the sequestration of parasites in the microvasculature, coagulation, and inflammation (4). The identification of EPCR as a PfEMP1 binding partner for parasites associated with severe childhood malaria prompts the question whether the engagement of EPCR by PfEMP1 directly contributes to the pathogenesis of severe P. falciprum malaria in addition to the complications associated with parasite sequestration (6). PfEMP1 molecules are unique to malaria parasites infecting humans and the great African apes (31). The lack of an animal model mimicking parasite host interactions in play during precipitation of cerebral malaria in humans represents a major hurdle in investigating cerebral malaria. The murine P. berghei ANKA model of cerebral malaria does not epitomize the sequestration of parasites in the microvasculature of the brain comparable to human cerebral malaria. Thus, this model is unable to account for the pathogenic role of parasite cytoadhesion (32).
Therefore, in the absence of a rodent or other animal model for P. falciparum-induced severe malaria, the functional consequences of the interaction of EPCR with PfEMP1s associated with severe malaria can only be assessed in in vitro, using human endothelial cell assays for PC activation and cellular APC activities.
Our results demonstrate that the interaction of EPCR with the PfEMP1 CIDRα1.1 domain obstructed normal EPCR function thereby severely impairing endothelial PC activation and generation of APC (Figure 6A). In vivo murine models with modulated EPCR expression indicate the importance of functional EPCR for the efficient activation of PC. For instance, PC activation is decreased by 30% in EPCR heterozygous mice and by 95% in EPCR deficient mice (33, 34). Although, mice with <10% (Procrδ/δ) or no (Procr−/−) EPCR develop normally without overt thrombotic complications, responses to prothrombotic challenges in mice with dysfunctional EPCR are greatly exacerbated (33, 35-38). Also the antithrombotic activities of APC are well recognized clinically as its deficiency, either mild or severe, are associated with venous thrombosis or neonatal purpura fulminans, blindness, and mental retardation, respectively, and the APC cleavage site mutation in factor V (Arg506Gln, factor V Leiden) is the most commonly identifiable hereditary risk factor for venous thrombosis among Caucasians (13, 39). Thus, the procoagulant state observed in cerebral malaria and the deposition of fibrin at sites of IE sequestration in the brain is consistent with and likely exacerbated by the acquired insufficiency of the protein C system to generate APC due to obstruction of EPCR by the PfEMP1 protein (21, 22, 40).
Figure 6. Schematic model of acquired PC system deficiency due to IE binding to EPCR and restoration of cellular EPCR function by E86A-sEPCR.
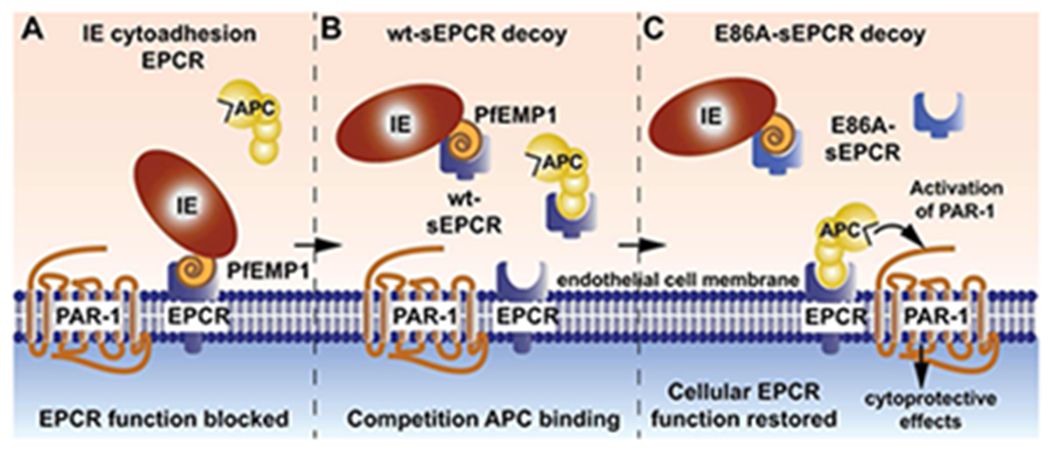
A) Binding of the CIDRα1 domain of PfEMP1 to EPCR prevents the interaction of PC and APC with EPCR resulting in a loss of cellular EPCR function. The inability of EPCR to promote protein C activation to generate APC (not shown) and to facilitate APC-mediated PAR1 activation to induce cytoprotective activities causes an acquired functional PC system deficiency. B) Although wt-sEPCR binds the CIDRα1 domain of PfEMP1 with high affinity and increased endogenous sEPCR levels have been implicated to protect against severe symptoms of P. falciparum infection, the competition of wt-sEPCR at high concentrations with APC binding to cellular EPCR may counteract the intended restoration PC and APC binding to cellular EPCR. C) The non-(A)PC binding sEPCR variant, E86A-sEPCR, overcomes the limitation of wt-sEPCR and provides a viable decoy strategy for PfEMP1 to enable cellular EPCR functions. Consequently, E86A-sEPCR prevents binding of the CIDRα1 domain of PfEMP1 to cellular EPCR and allows cellular EPCR function to promote protein C activation and to facilitate APC-mediated PAR1 activation to induce cytoprotective activities without competition for APC binding to cellular EPCR.
In addition to antithrombotic effects, APC also conveys EPCR-dependent cytoprotective effects on vascular endothelial cells that require activation of PAR1 and PAR3 (10, 16, 19, 20, 41). These cytoprotective effects of APC are essential for the observed beneficial effects of APC in multiple in vivo injury and disease models, including models for APC’s neuroprotective effects in ischemic stroke and APC-mediated mortality reduction in sepsis (19, 20, 41). CIDRα1.1 significantly reduced PAR1 cleavage by APC and thereby blunted APC’s endothelial barrier-protective effects. Thus, binding of the PfEMP1 parasite protein to EPCR severely impaired cytoprotective functions of APC. Mice with low expression levels of EPCR (<10%) fail to respond to APC therapy for mortality reduction in lipopolysaccharide (LPS)-induced sepsis and showed greatly increased susceptibility to LPS-induced death. Similarly, these mice show an exacerbated worsening of vascular permeability in the brain and other organs in response LPS, indicating that the EPCR-dependent endogenous cytoprotective PC pathway provides important contributions to maintain vascular integrity in response to inflammatory challenges (12, 37, 38, 41-43). Cerebral tissue-samples from children with cerebral malaria show significant depletions of vascular EPCR and thrombomodulin at sites of sequestering IE in the microvasculature, accompanied by fibrin deposits and petechial hemorrhagic lesions (21). Thus, although sequestered IEs also are found in other tissue of cerebral malaria patients, and the interaction with EPCR is likely affecting the PC system in the rest of the body as well, the brain may be more sensitive to the combination of rapid developing PC system defects accompanied by hypoxia and inflammation compared to other affected organ systems. In absence of currently available in vivo rodent models for in vivo proof-of-principle studies, our data support the new paradigm that acquired deficiency of the protein C system due to the obstruction of EPCR by cytoadherent parasites binding EPCR via PfEMP1, contributes directly to the pathogenesis of severe malaria.
Another line of investigation also supports the important role of EPCR in the pathogenesis of severe malaria. The non-synonymous SNP rs867186 in the PROCR gene resulting in a Ser219 to Gly substitution in the transmembrane domain of EPCR is associated with protection of Thai patients from severe malaria (44). Soluble EPCR reduces the adhesion of IE to the endothelial cells at physiologically relevant concentrations and the increased circulating plasma levels of sEPCR associated with rs867186 (Gly219-EPCR) likely provide an endogenous prophylactic decoy for sequestration of EPCR-binding IE to the microvasculature (6, 45, 46). This suggests potential applications of sEPCR as an adjunct therapy in patients with severe malaria. However, competition of sEPCR with (A)PC-binding to vascular EPCR can potentially limit the efficacy of the protein C system (Figure 6B) (46-49). Although physiologic levels of sEPCR in the circulation (0.1-0.3 μg/ml) are generally well tolerated without obvious deleterious effects, higher levels of sEPCR (3-8 μg/ml; 100-250 nM) significantly reduces PC activation and result in severe coagulation disturbance, raising serious concerns about the safety of wt-sEPCR therapeutic applications (34).
We found that the binding sites of CIDRα1.1 and APC on EPCR were qualitatively different since Glu86 was dispensable for CIDRα1.1 binding but is required for (A)PC binding to EPCR (17, 29, 30). This provided the opportunity to use E86A-sEPCR as a decoy strategy for CIDRα1, similar to previously decoy strategies employed for VEGF or factor Xa inhibitors (e.g. the VEGF-trap using a soluble VEGFR1 domains-antibody fusion to block VEGF interactions with its receptors, or the S195A-desGLA-factor Xa decoy to absorb the direct factor Xa inhibitors and restore coagulation) (50, 51). E86A-sEPCR inhibited the adhesion of IE to cellular EPCR and enabled the barrier-protective effects of APC in presence of CIDRα1. Since E86A-sEPCR is devoid of (A)PC binding and therefore does not compete for (A)PC binding to cellular EPCR (Figure 6C), and FVII(A) (9) it will likely be tolerated at higher levels than wt-sEPCR without interfering with the coagulation system. Our data suggest that sEPCR derivates like E86A-sEPCR would be able to inhibit circulating parasites from enganging EPCR, but due to the very low off rates of the PfEMP1-EPCR interaction (6), sEPCR derivates are unlikely to release bound parasites. This inability to release EPCR-bound parasites together with the finding that EPCR appear to be absent at sites of sequestered parasites in brains of patients (21) might limit ability of the decoy-strategy to restore APC functions at the lesions sites in the brain. Finally other interactions for sEPCR, such as the interaction with CD11b/CD18 (52, 53), should be clarified.
PfEMP1 are multi-domain molecules, and other PfEMP1-host receptor interactions may contribute to the parasite sequestration in severe malaria. However, given the association of EPCR-binding PfEMP1 variants with parasites causing severe childhood malaria and the potential adverse clinical effects of the PfEMP1-EPCR interaction suggested here, the potential of E86A-sEPCR as an adjunct therapy for patients in treatment for severe malaria deserves further investigations.
In summary, PfEMP1 variants with CIDRα1 domains and IE binding to EPCR have been associated with severe malaria (5, 6). The data presented here provides novel evidence suggesting that the obstruction of EPCR by IE sequestration contributes directly to the pathogenesis of severe malaria. PfEMP1 induced an acquired insufficiency of the protein C system to generate APC and to mediate cytoprotective effects of APC, thereby leaving the affected endothelium vulnerable to the vicious cycle of inflammation, thrombosis, and loss of vascular integrity caused by IE sequestration that is critical in the pathogenesis of severe malaria. Breaking this vicious cycle by targeting the cytoadhesion of IE during severe malaria could potentially reduce the mortally and neurological damages associated with the disease. A potential decoy strategy using E86A-sEPCR showed encouraging results in this respect as it allows the protein C system to function.
“What is known about this topic?”.
The pathogenesis of severe malaria is an interplay between coagulation, inflammation, and cytoadhesion.
Recently the CIDRα1 domain characteristic for the subset of the PfEMP1 proteins associated with severe malaria has been found to mediate cytoadhesion of infected erythrocytes by binding EPCR.
The interaction between EPCR and CIDRα1 blocks PC binding at protein level.
APC is involved in the haemostasis and endothelial barrier protection by cleaving PAR1.
Even with the best current treatment severe malaria often results in death of the patient highlighting the need for developing better adjuvant therapies.
“What does this paper add?
CIDRα1 interaction with EPCR on the cell surface blocks PC activation and APC mediated PAR1 cleavage resulting blocking APC mediated endothelial barrier protection, directly implicating the CIDRα1-EPCR interaction in the pathogenesis aside from enabling cytoadhesion.
Furthermore, we show proof of principle of a potential novel therapeutic strategy utilizing the mutant E86A-sEPCR as decoy, as it blocked infected erythrocyte binding to brains cells similarly to wt-sEPCR.
The E86A-sEPCR can effectively out-compete the endogenous cellular EPCR for CIDRα1 binding, thereby enabling C/APC binding and downstream effects.
Acknowledgements
We kindly thank Dr. Matthew K. Higgins (Oxford University, United Kingdom) for providing recombinant sEPCR, Baxter Healthcare Corporation (Westlake Village, CA) for the unrestricted gift of protein C (Ceprotin®), Dr. Kenji Fukudome (Saga Medical School, Saga, Japan) for the anti-EPCR antibody RCR-252, Dr. L. Brass (University of Pennsylvania, Philadelphia, PA) for the anti-PAR1 antibody ATAP2, and Dr C. J. S. Edgell (University of North Carolina, Chapel Hill, NC) for the EA.hy926 endothelial cells.
This work was supported by the Lundbeck Foundation, The Danish Council for Independent Research, Medical Sciences (the Sapere Aude programme DFF–4004-00624B (T.L.) and grant ID 1333-00220 (C.W.W.), Axel Muusfeldts Foundation, A.P. Møller Foundation, University of Copenhagen, the Ministerio de Economía y Competitividad (Inncorpora-PTQ), an American Heart Association Western States Affiliate postdoctoral fellowship (E.A.M.B.) and National Institutes of Health (NHLBI) grant HL104165 (L.O.M.).
Footnotes
Conflict of interest
The authors declare no conflict of interest
References
- 1.Carme B, Bouquety JC, Plassart H. Mortality and sequelae due to cerebral malaria in African children in Brazzaville, Congo. The American journal of tropical medicine and hygiene 1993; 48(2): 216–21. [DOI] [PubMed] [Google Scholar]
- 2.Birbeck GL, Molyneux ME, Kaplan PW, et al. Blantyre Malaria Project Epilepsy Study (BMPES) of neurological outcomes in retinopathy-positive paediatric cerebral malaria survivors: a prospective cohort study. Lancet neurology 2010; 9(12): 1173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moxon CA, Heyderman RS, Wassmer SC. Dysregulation of coagulation in cerebral malaria. Molecular and biochemical parasitology 2009; 166(2): 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Storm J, Craig AG. Pathogenesis of cerebral malaria--inflammation and cytoadherence. Frontiers in cellular and infection microbiology 2014; 4: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lavstsen T, Turner L, Saguti F, et al. Plasmodium falciparum erythrocyte membrane protein 1 domain cassettes 8 and 13 are associated with severe malaria in children. Proceedings of the National Academy of Sciences of the United States of America 2012; 109(26): E1791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner L, Lavstsen T, Berger SS, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature 2013; 498(7455): 502–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rowe JA, Claessens A, Corrigan RA, et al. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: molecular mechanisms and therapeutic implications. Expert reviews in molecular medicine 2009; 11: e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukudome K, Esmon CT. Identification, cloning, and regulation of a novel endothelial cell protein C/activated protein C receptor. The Journal of biological chemistry 1994; 269(42): 26486–91. [PubMed] [Google Scholar]
- 9.Lopez-Sagaseta J, Montes R, Puy C, et al. Binding of factor VIIa to the endothelial cell protein C receptor reduces its coagulant activity. Journal of thrombosis and haemostasis : JTH 2007; 5(9): 1817–24. [DOI] [PubMed] [Google Scholar]
- 10.Bouwens EA, Stavenuiter F, Mosnier LO. Mechanisms of anticoagulant and cytoprotective actions of the protein C pathway. Journal of thrombosis and haemostasis : JTH 2013; 11 Suppl 1: 242–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vatsyayan R, Kothari H, Mackman N, et al. Inactivation of Factor VIIa by Antithrombin In Vitro, Ex Vivo and In Vivo: Role of Tissue Factor and Endothelial Cell Protein C Receptor. PloS one 2014; 9(8): e103505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohan Rao LV, Esmon CT, Pendurthi UR. Endothelial cell protein C receptor: a multiliganded and multifunctional receptor. Blood 2014; 124(10): 1553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood 2007; 109(8): 3161–72. [DOI] [PubMed] [Google Scholar]
- 14.Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, et al. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proceedings of the National Academy of Sciences of the United States of America 1996; 93(19): 10212–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esmon CT. The protein C pathway. Chest 2003; 124(3 Suppl): 26S–32S. [DOI] [PubMed] [Google Scholar]
- 16.Esmon CT. The endothelial protein C receptor. Current opinion in hematology 2006; 13(5): 382–5. [DOI] [PubMed] [Google Scholar]
- 17.Burnier L, Mosnier LO. Novel mechanisms for activated protein C cytoprotective activities involving noncanonical activation of protease-activated receptor 3. Blood 2013; 122(5): 807–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosnier LO, Sinha RK, Burnier L, et al. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 2012; 120(26): 5237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zlokovic BV, Griffin JH. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends in neurosciences 2011; 34(4): 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mosnier LO, Zlokovic BV, Griffin JH. Cytoprotective-selective activated protein C therapy for ischaemic stroke. Thrombosis and haemostasis 2014; 112(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moxon CA, Wassmer SC, Milner DA Jr., et al. Loss of endothelial protein C receptors links coagulation and inflammation to parasite sequestration in cerebral malaria in African children. Blood 2013; 122(5): 842–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hemmer CJ, Kern P, Holst FG, et al. Activation of the host response in human Plasmodium falciparum malaria: relation of parasitemia to tumor necrosis factor/cachectin, thrombin-antithrombin III, and protein C levels. The American journal of medicine 1991; 91(1): 37–44. [DOI] [PubMed] [Google Scholar]
- 23.Mosnier LO. Platelet factor 4 inhibits thrombomodulin-dependent activation of thrombin-activatable fibrinolysis inhibitor (TAFI) by thrombin. The Journal of biological chemistry 2011; 286(1): 502–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouwens EA, Stavenuiter F, Mosnier LO. Cell painting with an engineered membrane-anchoring endothelial protein c receptor enhances protein c anticoagulant and cytoprotective pathways. Submitted for publication 2014 2014. [Google Scholar]
- 25.Mosnier LO, Zampolli A, Kerschen EJ, et al. Hyperantithrombotic, noncytoprotective Glu149Ala-activated protein C mutant. Blood 2009; 113(23): 5970–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stins MF, Gilles F, Kim KS. Selective expression of adhesion molecules on human brain microvascular endothelial cells. Journal of neuroimmunology 1997; 76(1-2): 81–90. [DOI] [PubMed] [Google Scholar]
- 27.Ye X, Fukudome K, Tsuneyoshi N, et al. The endothelial cell protein C receptor (EPCR) functions as a primary receptor for protein C activation on endothelial cells in arteries, veins, and capillaries. Biochemical and biophysical research communications 1999; 259(3): 671–7. [DOI] [PubMed] [Google Scholar]
- 28.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nature methods 2012; 9(7): 676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liaw PC, Mather T, Oganesyan N, et al. Identification of the protein C/activated protein C binding sites on the endothelial cell protein C receptor. Implications for a novel mode of ligand recognition by a major histocompatibility complex class 1-type receptor. The Journal of biological chemistry 2001; 276(11): 8364–70. [DOI] [PubMed] [Google Scholar]
- 30.Preston RJ, Villegas-Mendez A, Sun YH, et al. Selective modulation of protein C affinity for EPCR and phospholipids by Gla domain mutation. The FEBS journal 2005; 272(1): 97–108. [DOI] [PubMed] [Google Scholar]
- 31.Otto TD, Rayner JC, Bohme U, et al. Genome sequencing of chimpanzee malaria parasites reveals possible pathways of adaptation to human hosts. Nature communications 2014; 5: 4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Craig AG, Grau GE, Janse C, et al. The role of animal models for research on severe malaria. PLoS pathogens 2012; 8(2): e1002401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W, Zheng X, Gu JM, et al. Extraembryonic expression of EPCR is essential for embryonic viability. Blood 2005; 106(8): 2716–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng X, Li W, Gu JM, et al. Effects of membrane and soluble EPCR on the hemostatic balance and endotoxemia in mice. Blood 2007; 109(3): 1003–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castellino FJ, Liang Z, Volkir SP, et al. Mice with a severe deficiency of the endothelial protein C receptor gene develop, survive, and reproduce normally, and do not present with enhanced arterial thrombosis after challenge. Thrombosis and haemostasis 2002; 88(3): 462–72. [PubMed] [Google Scholar]
- 36.Taylor FB Jr., Peer GT, Lockhart MS, et al. Endothelial cell protein C receptor plays an important role in protein C activation in vivo. Blood 2001; 97(6): 1685–8. [DOI] [PubMed] [Google Scholar]
- 37.Centelles MN, Puy C, Lopez-Sagaseta J, et al. Blocking endothelial protein C receptor (EPCR) accelerates thrombus development in vivo. Thrombosis and haemostasis 2010; 103(6): 1239–44. [DOI] [PubMed] [Google Scholar]
- 38.Tamayo I, Velasco SE, Puy C, et al. sPLA2-V impairs EPCR-dependent protein C activation and accelerates thrombosis in vivo. Journal of thrombosis and haemostasis : JTH 2014. [DOI] [PubMed] [Google Scholar]
- 39.Cushman M Inherited risk factors for venous thrombosis. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program 2005: 452–7. [DOI] [PubMed] [Google Scholar]
- 40.Clemens R, Pramoolsinsap C, Lorenz R, et al. Activation of the coagulation cascade in severe falciparum malaria through the intrinsic pathway. British journal of haematology 1994; 87(1): 100–5. [DOI] [PubMed] [Google Scholar]
- 41.Kerschen EJ, Fernandez JA, Cooley BC, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. The Journal of experimental medicine 2007; 204(10): 2439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor FB Jr., Stearns-Kurosawa DJ, Kurosawa S, et al. The endothelial cell protein C receptor aids in host defense against Escherichia coli sepsis. Blood 2000; 95(5): 1680–6. [PubMed] [Google Scholar]
- 43.von Drygalski A, Furlan-Freguia C, Ruf W, et al. Organ-specific protection against lipopolysaccharide-induced vascular leak is dependent on the endothelial protein C receptor. Arteriosclerosis, thrombosis, and vascular biology 2013; 33(4): 769–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naka I, Patarapotikul J, Hananantachai H, et al. Association of the endothelial protein C receptor (PROCR) rs867186-G allele with protection from severe malaria. Malaria journal 2014; 13(1): 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qu D, Wang Y, Song Y, et al. The Ser219-->Gly dimorphism of the endothelial protein C receptor contributes to the higher soluble protein levels observed in individuals with the A3 haplotype. Journal of thrombosis and haemostasis : JTH 2006; 4(1): 229–35. [DOI] [PubMed] [Google Scholar]
- 46.Saposnik B, Reny JL, Gaussem P, et al. A haplotype of the EPCR gene is associated with increased plasma levels of sEPCR and is a candidate risk factor for thrombosis. Blood 2004; 103(4): 1311–8. [DOI] [PubMed] [Google Scholar]
- 47.Fukudome K, Kurosawa S, Stearns-Kurosawa DJ, et al. The endothelial cell protein C receptor. Cell surface expression and direct ligand binding by the soluble receptor. The Journal of biological chemistry 1996; 271(29): 17491–8. [DOI] [PubMed] [Google Scholar]
- 48.Regan LM, Stearns-Kurosawa DJ, Kurosawa S, et al. The endothelial cell protein C receptor. Inhibition of activated protein C anticoagulant function without modulation of reaction with proteinase inhibitors. The Journal of biological chemistry 1996; 271(29): 17499–503. [DOI] [PubMed] [Google Scholar]
- 49.Liaw PC, Neuenschwander PF, Smirnov MD, et al. Mechanisms by which soluble endothelial cell protein C receptor modulates protein C and activated protein C function. The Journal of biological chemistry 2000; 275(8): 5447–52. [DOI] [PubMed] [Google Scholar]
- 50.Holash J, Davis S, Papadopoulos N, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(17): 11393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu G, DeGuzman FR, Hollenbach SJ, et al. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nature medicine 2013; 19(4): 446–51. [DOI] [PubMed] [Google Scholar]
- 52.Kurosawa S, Esmon CT, Stearns-Kurosawa DJ. The soluble endothelial protein C receptor binds to activated neutrophils: involvement of proteinase-3 and CD11b/CD18. Journal of immunology 2000; 165(8): 4697–703. [DOI] [PubMed] [Google Scholar]
- 53.Fink K, Busch HJ, Bourgeois N, et al. Mac-1 directly binds to the endothelial protein C-receptor: a link between the protein C anticoagulant pathway and inflammation? PloS one 2013; 8(2): e53103. [DOI] [PMC free article] [PubMed] [Google Scholar]