

Abstract
Purpose
Autosomal recessive bestrophinopathy (ARB) and vitelliform macular dystrophy (VMD) are distinct phenotypes, typically inherited through recessive and dominant patterns, respectively. Recessively inherited VMD (arVMD) has been reported, suggesting that dominant and recessive BEST1-related retinopathies represent a single disease spectrum. This study compares adVMD, arVMD, and ARB to determine whether a continuum exists and to define clinical and genetic features to aid diagnosis and management.
Methods
One arVMD patient and nine ARB patients underwent standard ophthalmic examination, imaging, electrophysiology, and genetic assessments. A meta-analysis of reported BEST1 variants was compiled, and clinical parameters were analyzed with regard to inheritance and phenotype.
Results
Among 10 patients with biallelic BEST1 variants, three novel ARB variants (p.Asp118Ala, p.Leu224Gln, p.Val273del) were discovered. A patient with homozygous p.Glu35Lys was clinically unique, presenting with VMD, including hyperautofluorescence extending beyond the macula, peripheral punctate lesions, and shortened axial-length. A tritan-axis color vision deficit was seen in three of six (50%) of ARB patients. Attempts to distinguish recessively-inherited ARB and dominantly-inherited VMD genotypically, by variant frequency and residue location, did not yield significant differences. Literature meta-analysis with principle component analysis of clinical features demonstrated a spectrum of disease with arVMD falling between adVMD and ARB.
Conclusions
This study suggests that arVMD is part of a continuum of autosomal recessive and dominant BEST1-related retinopathies. Detailed clinical and molecular assessments of this cohort and the literature are corroborated by unsupervised analysis, highlighting the overlapping heterogeneity among BEST1-associated clinical diagnoses. Tritan-axis color vision deficit is a previously unreported finding associated with ARB.
Keywords: BEST1, bestrophinopathy, genetic diseases
Bestrophin 1 (BEST1) is a 585 amino-acid calcium-activated Cl− channel1–4 localized to the basolateral membrane of retinal pigment epithelium (RPE).5 Although its exact role in RPE physiological function is unknown, mutations in the BEST1 protein lead to a collection of retinopathies: Best vitelliform macular dystrophy (VMD) (OMIM-153700), autosomal dominant vitreoretinochoroidopathy (ADVIRC) and microcornea, rod-cone dystrophy, cataract, and posterior staphyloma syndrome (MRCS) (OMIM-193220), retinitis pigmentosa (RP) (OMIM-613914), and autosomal recessive bestrophinopathy (ARB) (OMIM-611809). VMD and ARB are the most commonly diagnosed BEST1-related retinopathies. The manifestation of each disease, although recognizable, present along a continuum. Overlap in these diseases suggests a similar pathogenic mechanism despite labeling as distinct clinical entities.
The VMD phenotype consists of a prominent raised central macular lesion that undergoes morphological changes classified into six stages: previtelliform, vitelliform, vitelliruptive, pseudohypopion, atrophic, and cicatricial.6–8 The best corrected visual acuity (BCVA) is mildly decreased to 20/50 on average (range 20/20 to 20/200), until later stages, where natural progression and complication create a steep decline. Patients often have mild hyperopia and an increase in fundus autofluorescence.7,9 Neurosensory separation starts in the vitelliform stage, and electrophysiology changes show a moderately decreased electro-oculography (EOG) Arden ratio of less than 1.5 with frequently normal full-field electroretinogram (ffERG).6
ARB displays macular changes resembling the vitelliruptive, atrophic, and cicatricial stages of VMD. The phenotype additionally encompasses extramacular punctate deposits, intraretinal and subretinal fluid (SRF) accumulation, punctate or diffuse fundus hyperautofluorescence, hyperopia, short axial-length, central visual field loss, severely decreased Arden ratio (≤1.0) on EOG, and reduced photopic and scotopic ffERG.10–12 Amblyopia and angle closure glaucoma can co-occur.12,13
As such, VMD and ARB macular phenotypes may overlap depending on the stage, distinguishable by the typically normal ffERG in VMD. Both phenotypes display a propensity for SRF accumulation, RPE-photoreceptor separation, disruption of photoreceptor outer segment shedding11 and lipofuscin-like deposits between the neurosensory retina and RPE.2,14 These contribute to the reduction in central vision in both diseases. Although the EOG Arden ratio is often severely impacted in ARB, there is significant overlap in the observed Arden ratio between both diseases. Additionally, EOG changes are difficult to interpret in the setting of an abnormal ffERG, and many institutions lack access to electroretinography equipment. Thus an alternate metric to distinguish between ARB and VMD would be beneficial for the purposes of diagnosis and treatment. Previous reports have noted that the heterogeneity of VMD and ARB make classification difficult.15–17 Diagnosis is complicated by cases of VMD that show atypical extramacular involvement with ffERG abnormalities.18–22
Familial inheritance pattern and genetic testing are frequently used to differentiate between the two diseases in cases where the phenotype is ambiguous either due to extramacular findings or ffERG responses. Although VMD and ARB are typically associated with dominant and recessive inheritance patterns respectively, several cases of recessively inherited VMD phenotype have been reported and may be more common than previously thought.19,23–30 To avoid confusion of multiple inheritance patterns associated with similarly named diseases, the traditional, collective VMD phenotype will be subdivided into autosomal recessive (arVMD) and autosomal dominant (adVMD) classifications, whereas ARB will refer to the distinct recessive disease.
The overlap in presentation between the two disease states, particularly in instances of arVMD, prompts the need for better diagnostic classification because both the inheritance and phenotype may be used to inform prognosis and treatments. In this work, we explore the clinical manifestations of arVMD and ARB, as well as possible opportunities to better classify each disease through genotype and clinical data.
Methods
Clinical Examination
Patients were enrolled in an institutional review board (IRB)–approved protocol for ophthalmic and genetic evaluations (NCT02471287) at the National Institutes of Health between 2012 and 2019; families F and H were examined at the University of Kentucky. Investigations were conducted in accordance with the Declaration of Helsinki after obtaining informed consent. Examination comprised Snellen BCVA, refraction, intraocular pressure (IOP), Ishihara or Farnsworth Panel-D15 color vision assessment, dilated fundoscopy, axial-length measurement, Goldmann Visual Field (GVF), color and autofluorescence (FAF) fundus imaging (Topcon, Tokyo, Japan), and optical coherence tomography (OCT) (Carl Zeiss Meditec AG, Dublin, CA, USA; and Spectralis; Heidelberg Engineering, Heidelberg, Germany). Electrophysiological testing (ffERG and EOG) was performed according to International Society for Clinical Electrophysiology of Vision31–34 standards using a commercial electrophysiology system (LKC, Gaithersburg, MD, USA). Two additional siblings and an unrelated third case were examined at the University of Kentucky Ophthalmic Genetics Service. Similar evaluation was conducted except with a different ERG system (Diagnosys LLC, Lowell, MA, USA). Records and tests were reviewed according to an IRB-approved study protocol conducted in accordance with the Declaration of Helsinki (IRB number: 61133).
Genetic Component
BEST1 variants (NM_004183) were analyzed using the SIFT,35 PolyPhen,36 and CADD37 in silico tools for pathogenesis estimation. Sequence alignment using Clustal Omega was conducted on the following accession numbers: NP_004174.1 (Homo sapiens), NP_001270510.1 (Macaca fascicularis), NP_001073714.1 (Bos taurus), NP_001091014.1 (Canis lupus familiaris), NP_036043.2 (Mus musculus), XP_008158536.1 (Eptesicus fuscus), XP_421055.2 (Gallus gallus), XP_689098.3 (Danio rerio), NP_652603.1 (Drosophila melanogaster).
Meta-Analysis of Previously Published Patient Data
A list of mutations in the BEST1 gene were compiled from papers reported in HGMD and Association for Vision Research and Ophthalmology reports related to arVMD, adVMD and ARB until July 2019 (See Supplementary Tables S1 and S2 for a list of mutations). ARB reports were supplemented by searching “Autosomal Recessive Bestrophinopathy” in the PubMed directory. Reports detailing clinical updates of previously reported patients were excluded to avoid repeat sampling. Available patient data were recorded, including age, sex, BCVA, refraction, axial-length, EOG, photopic and scotopic ffERG, genotype, phenotype, and inheritance. Missense, nonsense, frameshift, and indels were categorized according to phenotype, inheritance, and residue and visualized using Protter.38 Patient data were then reduced to variants showing at least three separate reports for both VMD and ARB, to test the hypothesis that associated clinical data among recurrent instances of each variant were different between disease states. This data was manipulated in R using the basic stats and infer packages after testing for normality and equal variance and employing non-parametric tests when applicable. Comparisons were made using ANOVA followed by TukeyHSD. Principle component analysis (PCA) was accomplished using available data and groups stratified by inheritance and phenotype. The frequency of BEST1 variants was obtained from gnomAD and cross-referenced against inheritance and phenotype.
Results
Clinical Results arVMD Case
A six-year-old female was referred after a failed school vision screening one year prior to presentation. Her parents reported no significant vision issues. BCVA was measured at 20/32 OD and 20/80 OS. She had mild, nonspecific color vision deficits on the Farnsworth-D15 panel.
Fundus exam and OCT imaging indicated bilateral, large vitelliform lesions with SRF and mild pigmentary changes within the deposit, normal vessels, and punctate yellow lesions in the far periphery (Fig. 1A). EOG Arden ratio was unilaterally reduced and the ffERG was essentially normal except delayed photopic response (See Supplementary Table S3). Based on the clinical appearance, VMD was diagnosed.
Figure 1.
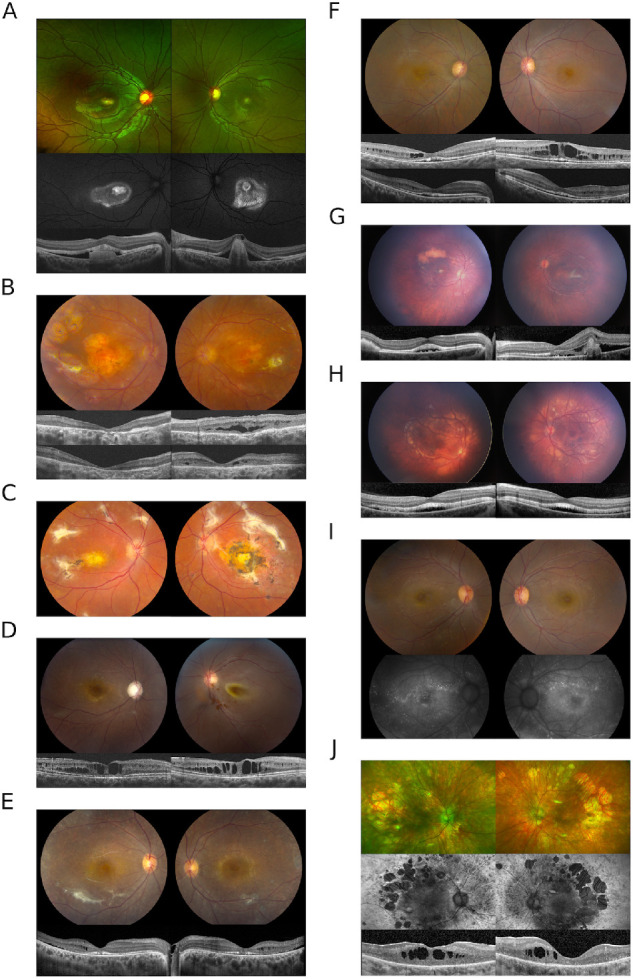
(A) Bilateral subfoveal lesions are present in the proband from family A. OCT imaging reveals subretinal material OD and fibrotic appearing pigment epithelial detachment OS with subretinal lucency separating the neurosensory retina from the RPE OU. Mild pigmentary changes are seen on color imaging and a ring of hyperautofluorescence extends beyond the subfoveal lesion. Punctate lesions (not shown) were observed in the far peripheral retina. (B) Chorioretinal atrophy accompanied by scarring and scattered punctate depositions throughout the peripheral retina in the proband from family B. OCT images at presentation (upper) and after CAI treatment (lower) show a partial resolution of intraretinal fluid in the left eye. (C) The fundus image of the sibling to proband B. (D) Macular cystic changes and optic nerve cupping in the right eye of proband C; retinal detachment in the left eye. The OCT images depict the right eye at two different time points, at presentation (left) and after one year (right). (E) Color fundus images from patient D show diffuse retinal atrophy with white dots and subretinal fibrosis. Intraretinal fluid, SRF, and disruption of the IS/OS juction can be seen on OCT. (F) Cystic macular changes in patient E on fundus images and OCT. The top row of OCT images shows cystic changes at presentation and the lower row corresponds to resolution with CAIs. (G) Fundus photos of proband F show focal yellow deposits in the macula with a superior large yellow conglomerate of subretinal material OD. The left eye has a perifoveal subretinal fibrosis. The disease in both eyes does not extend beyond the vascular arcades. OCT images show subretinal fluid at foveal center in the right eye and the subretinal fibrosis with subretinal fluid on the left eye. (H) Photos show more extensive disease than the sibling in (G) as the yellow deposits extend beyond the vascular arcades including the nasal retina. The corresponding OCTs show a shallow but extensive area of subretinal fluid. (I) Proband G had small scattered hyperfluorescent lesions on FAF. (J) Widespread retinal atrophy with hyperfluorescence in the periphery and patchy mid-peripheral loss of signal with macular hypofluorescence.
Next-generation sequencing genetic analysis of 325 retinal degeneration associated genes was positive for a homozygous mutation in BEST1, c.103G>A (p.Glu35Lys), which has been previously reported39–41 and predicted to be damaging by Sift, PolyPhen, and CADD.
Clinical Results ARB Cases
Nine additional patients with autosomal recessive inheritance of biallelic pathogenic BEST1 variants were assessed (See supplemental patient narratives). In contrast to the arVMD case, most ARB cases presented to clinic in their second or third decade with reports of vision loss starting as early as age three (range 3–31). At initial presentation, four patients had visual acuity of 20/100 or better in each eye (range 20/20 to 20/800). Associated ocular conditions included strabismus, keratoconus, cataract, corneal scarring, cystoid macular edema (CME), shallow anterior chambers, decreased axial-length, central scotoma, and failed color vision along the triton axis (Table 1). A high incidence of glaucoma was observed with four of nine having undergone laser peripheral iridotomy or trabeculectomy and displaying glaucomatous cup-to-disk changes.
Table 1.
Clinical Data for Bestrophinopathy Patients
| Family: Member (Sex) | Allele 1/2 (AA Change) | Age at Exam (Onset) | Initial BCVA | Refraction | EOG Arden Ratio | Color Vision Score (axis) | Axial Length (ACD, mm) | IOP (mm Hg) | C/D | GVF | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A: III,1 (F) | c.103G>A (p.Glu35Lys) | 6 (5) | OD | 20/32 | +1.00 +1.50 × 82 | 1.8 | 11/15 (D15, nonspecific) | 22.17 | 15 | 0.40 | NA |
| c.103G>A (p.Glu35Lys) | OS | 20/80 | +1.00 +1.75 × 90 | 0.88 | 13/15 (D15, nonspecific) | 22.46 | 15 | 0.40 | NA | ||
| B: III,1 (F) | c.422G>A (p.R141H) | 44 (3) | OD | 20/320 | +0.75 +2.50 × 180 | 1.64 | 1/15 (D15, Tritan) | 21.35 | 11 | 0.40 | Paracentral scotoma, constricted I1e, off-center I4e |
| c.830C>T (p.T277M) | OS | 20/320 | +3.25 +2.25 × 13 | Unable | 1/15 (D15, Tritan) | 21.11 | 11 | 0.50 | Paracentral scotoma, absent I1e and I2e, and off-center I4e | ||
| B: III,2 (F) | c.422G>A (p.R141H) | 39 (4) | OD | 20/200 | +2.00 +1.23 × 112 | NA | 3/15 (D15, Tritan) | 21.40 | 15 | 0.20 | Dense central scotoma, absent I1e |
| c.830C>T (p.T277M) | OS | 20/640 | +2.75 +1.25 × 32 | NA | 2/15 (D15, Tritan) | 21.73 | 16 | 0.20 | Dense central scotoma, absent I1e | ||
| C: II,2 (F) | c.671T>A (p.L224Q) | 24 (12) | OD | 20/125 | −2.00 Sph | NA | NA | NA | 15 | 1.00 | Central scotoma with constriction of I3e, I4e, III4e, and IV4e |
| c.353A>C (p.D118A) | OS | 20/800 | Sph Plano | NA | NA | NA | 17 | 0.30 | Central scotoma with absent I3e and I4e, constricted III4e and IV4e | ||
| D: IV,2 (F) | c.418C>G (p.L140V) | 32 (30) | OD | 20/80 | −0.75 +1.75 × 5 | 1.12 | 12/16 (Ishihara) | 21.11 | 40 (22*) | 0.60 | Central scotoma constriction of I1e, I2e, and I4e |
| c.418C>G (p.L140V) | OS | 20/100 | −0.25 +1.50 × 10 | 1.11 | 11/16 (Ishihara) | 20.84 | 15 | 0.50 | Central scotoma constriction of I1e, I2e, and I4e | ||
| E: IV,5 (F) | c.816_818delTGT (p.V273del) | 26 (4) | OD | 20/50 | −1.25 +1.75 × 101 | 1.14 | 12/15 (D15, Tritan) | 21.31 (2.15) | 20 | 0.60 | Absent I1e and constricted I4e |
| c.816_818delTGT (p.V273del) | OS | 20/40 | −0.50 +1.25 × 94 | 1.21 | 9/15 (D15, Tritan) | 21.14 (2.47) | 13 | 0.60 | Constricted I1e | ||
| F: III,1 (M) | c.122T>C (p.Leu41Pro) | 9 (9) | OD | 20/20 | +0.50 −0.50 × 168 | NA | 9/15 (D15, nonspecific) | 22.4 (3.44) | 21 | 0.30 | NA |
| c.422G>A (p.Arg141His) | OS | 20/50 | +0.50 −0.25 × 47 | NA | 13/15 (D15, nonspecific) | 22.8 (3.17) | 20 | 0.30 | NA | ||
| F: III,2 (F) | c.122T>C (p.Leu41Pro) | 10 (10) | OD | 20/25 | +3.75 −0.75 × 147 | NA | 11/15 (D15, nonspecific) | 21.05 (3.00) | 17 | 0.30 | NA |
| c.422G>A (p.Arg141His) | OS | 20/20 | +3.50 −0.75 × 38 | NA | 15/15 | 21.04 (2.92) | 16 | 0.30 | NA | ||
| G: II,1 (M) | c.604C>T (p.Arg202Trp) | 23 (23) | OD | 20/320 | −4.50 +2.25 × 97 | NA | NA | NA | 14 | NA | Constricted I4e, I1e absent |
| c.604C>T (p.Arg202Trp) | OS | 20/200 | −4.50 +0.50 × 170 | NA | NA | NA | 25 | NA | Constricted I4e, I1e absent | ||
| H: II,1 (F) | c.122T>C (p.Leu41Pro) | 60 (31) | OD | 20/300 | +10.50 sph | NA | NA | NA | 17 | 0.30 | Central scotoma without constriction |
| c.122T>C (p.Leu41Pro) | OS | 20/150 | +10.50 sph | NA | NA | NA | 14 | 0.30 | Normal in all isopters |
AA amino acid, ACD anterior chamber depth, C/D cup-to-disk ratio, GVF Goldmann visual field, NA not assessed.
IOP tested after laser peripheral iridotomy treatment for acute angle closure episode.
Stargardt's macular dystrophy was a common referral diagnosis. The retinal picture of these patients consisted of yellow or white deposits in the mid-periphery accompanied by frequent subretinal fibrosis, chorioretinal atrophy, CME, and SRF (Figs. 1B–J). Pigment clumping with RPE changes was also observed in three patients, and hyperautofluorescence was visualized on FAF extending into the periphery. All ARB patients tested with EOG demonstrated a severely reduced Arden ratio. A reduced scotopic with delayed photopic response was also observed on ffERG (Supplementary Table S3).
Clinical Molecular Genetics
Genetic analysis of affected individuals, with absent family history (Fig. 2A.), returned several novel variants in the BEST1 protein. Variants c.353A>C, c.671T>A, c.816_818delTGT (p.Asp118Ala, p.Leu224Gln, p.Val273del) are predicted to be damaging by Sift, PolyPhen, and CADD, while frequencies were not listed in gnomAD (Table 2). The p.Asp118Ala variant is located at a phylogenetically conserved residue (Fig. 2B) in a large cytoplasmic domain (91-230). Other mutations in this area have been linked to adVMD (p.Phe113Leu19 and p.Glu119Gln42) and ARB (p.Thr124Met43). The p.Leu224Gln variant is located in the same cytoplasmic domain near the third transmembrane domain (TMD) (231-253) and shows similar conservation. Other variants around this residue have been linked to adVMD (p.Leu224Phe,44 p.Leu224Met,45 p.Gly222Glu,46 p.Tyr227Cys)44. The c.816_818delTGT variant is located in the fourth TMD (269-291) with nearby mutations leading to ARB (p.Val273Met,43 p.Pro274Arg,47 p.Thr277Met48) and adVMD (p.Pro274Arg, p.Val275Ile, p.Phe276Leu).19
Figure 2.

(A) The patient pedigrees demonstrate recessive inheritance. Arrows show probands from each family. (B) Clustal Omega sequence alignment of BEST1 shows highly conserved residues. The mutations examined in this study are highlighted with the bottom row indicating (*) a single, fully conserved residue, (†) a conserved residue with strongly similar properties, and (‡) a conserved residue with weakly similar properties.
Table 2.
Frequency of Variants and in Silico Predictions of Mutation Effects
| BEST1 Mutation | Amino Acid Change | Previously Reported | Gnomad Frequency | Sub Pop. Frequency (Origin) | SIFT (Score) | PolyPhen (Score, Sensitivity, Specificity) | CADD Score (PHREDD) |
|---|---|---|---|---|---|---|---|
| c.103G>A | p.Glu35Lys | Reported (Tian 2017, Stone 2017) | Not listed | Not listed | Damaging (0.01) | PD (1.000, 0.00, 1.00) | 2.24 (21.7) |
| c.125T>C | p.Leu41Pro | Reported (Kramer 2003, Zhao 2012, Burguess 2018) | 5/251264 | 0.0000615 (African) | Tolerated (0.13) | PD (0.966, 0.78, 0.95) | 1.16 (14.4) |
| c.353A>C | p.Asp118Ala | Novel | Not listed | Not listed | Damaging (0.02) | PD (1.000, 0.00, 1.00) | 4.16 (29.8) |
| c.418C>G | p.Leu140Val | Reported (Davidson 2009) | 5/150144 | 0.0002202 (South Asian) | Damaging (0.03) | PD (0.998, 0.27, 0.99) | 2.63 (22.8) |
| c.422G>A | p.Arg141His | Reported (White 2000, Schatz 2010, Kinnick 2011, Stone 2017, ...) | 96/181002 | 0.003275 (Finnish) | Damaging (0.00) | PD (1.000, 0.00, 1.00) | 4.42 (33) |
| c.604C>T | p.Arg202Trp | Davidson 2011 | 4/251348 | 0.0000578 (Latino) | Damaging (0.03) | PD (1.000, 0.00, 1.00) | 3.82 (26.4) |
| c.671T>A | p.Leu224Gln | Novel | Not listed | Not listed | Damaging (0.00) | PD (1.000, 0.00, 1.00) | 3.93 (27.3) |
| c.816_818delTGT | p.Val273del | Novel | Not listed | Not listed | Unable | Unable | Unable |
| c.830C>T | p.Thr277Met | Reported (Zaneveld 2015, Luo 2018, Zhong 2017, Tian 2017) | 3/251476 | 0.00003266 (South Asian) | Damaging (0.00) | PD (1.000, 0.00, 1.00) | 3.41 (24.8) |
Probably damaging (PD). PHREDD scaled C-score of 20 to 30 indicate the top 1% of all single nucleotide variants that are likely to cause disease and PHREDD scaled C-score of 30 to 40 indicate the top 0.1% of all single nucleotide variants (SNVs) that are likely to cause disease.
The c.422G>A and c.830C>T variants (p.R141H and p.T277M respectively) have been reported in ARB prior to this study.10,13,15,39,43,48,49 Similarly, the R141H variant has been associated with VMD in both dominant and recessive patterns.21,25,29,50 The L41P variant causes adVMD in the heterozygous state51 and ARB in the compound heterozygous state.10,52,53 Last, the missense mutation p.Leu140Val has been linked with autosomal recessive RP.16
Genetic Meta-Analysis
Given the recessive inheritance of VMD and ARB in this cohort and overlap in inheritance modes commonly associated with each phenotype in the literature, we investigated variant characteristics associated with phenotypic presentation and inheritance patterns. First, we examined the general population frequency and cDNA location of variants associated with phenotype (Fig. 3A) and inheritance patterns (Fig. 3B). There was no statistically significant difference in allele frequencies associated with different phenotype presentations or inheritance patterns (P = 0.808, 0.458, respectively; Fig. 3B). This suggests that there is not a protein “hotspot” that informs development of macular lesions versus global retinopathy.
Figure 3.

Log frequency distribution of known cDNA mutations in BEST1 associated with (A) VMD or ARB phenotypes and (B) Dominant or Recessive Inheritance patterns. (C) Overlap of mutated residues associated with VMD and ARB. (D) Overlap of recessively inherited VMD and ARB
We then analyzed BEST1 variants by functional domains (Figs. 3C, 3D). Although VMD mutations cluster around the four intracellular and transmembrane domains, ARB mutations show considerable variability in spatial organization. Still, BEST1 disease-associated alleles are skewed toward the N-terminus. Variants causing missense mutations or protein length alterations (frameshift, nonsense, or in-frame deletions) were not distributed differently. It was noted that 19% (35/180) of BEST1 disease-associated residues have been reported with both ARB and VMD phenotypes (Fig. 3C), and 16% (16/100) of residues overlap between arVMD and ARB (Fig. 3D). The implication is that BEST1-residues, when mutated, can be associated with either phenotype or inheritance pattern, but different phenotypes do not localize to separate protein domains. Alternately, the overlap suggests that some cases of BEST1-related retinopathy follow a semidominant inheritance pattern with incomplete penetrance, where one allele is sufficient to cause disease, and two alleles cause more severe disease. The idea that arVMD constitutes its own distinct disease state has not yet been explored but is also possible.
Next, we sought to understand the nature of residues causing VMD or ARB by analyzing only reported cases with the same residue changes to discern relevant clinical differences that can be used in diagnosis and treatment. Additionally, VMD was split into dominant and recessive groupings to distinguish the influence of inheritance on clinical presentation. A total of 176 patients were analyzed: 73 adVMD, 83 ARB, 18 arVMD, and two with semidominant inherited bestrophiopathy. These cases carried mutations affecting residues 13, 25, 141, 195, 218, 225, and 312 that are commonly mutated in each disease. When comparing dominant to recessive inheritance there was a statistically different mean age at onset, BCVA, and EOG Arden ratio (Figs. 4A–4C). Principle component analysis also showed a separation between patients expressing different phenotypes and inheritance patterns (Fig. 4D). Interestingly, VMD patients who displayed a recessive mode of inheritance clustered between the dominantly inherited VMD cases and the ARB cases, indicating that recessive VMD represents an overlapping disorder. The first principle component (PC) was primarily composed of BCVA and the EOG Arden ratio, which explained 40.9% of the variance, and the second PC primarily comprised the reported age at onset. Autosomal recessive bestrophinopathy patients primarily clustered toward worse VA, lower EOG, and younger age at onset.
Figure 4.

Histograms with overlaid violin plots describing the distribution of (A) BCVA, (B) EOG Arden ratio, and (C) presentation age for VMD and ARB patients. Black cross bars indicate means. (D) PCA of ARB and VMD patients. PC1 is primarily composed of VA and EOG while PC2 is primarily composed of age.
Discussion
In this report, we attempted to differentiate unique features of adVMD from arVMD and ARB by comparing the clinical findings of patients with arVMD with recessively inherited ARB while simultaneously conducting a meta-analysis of previous reported BEST1 mutations. The clinical picture of arVMD was consistent with previous reports: a central vitelliform lesion with SRF, moderately reduced BCVA, and largely spared ERG. Family history was negative in arVMD. Clinically, we noticed that color vision deficits were more common in ARB individuals and that cystic macular changes were responsive to CAI therapy. We also identified three novel mutations displaying a characteristic ARB phenotype: reduced central visual acuity, extramacular lesions, absent EOG, and abnormal ERG. We were also better able to classify individuals based on their clinical metrics. Last, we identified arVMD as a biallelic vitelliform expression of BEST1-related retinopathy that appears along the VMD-Bestrophinopathy spectrum.
A high incidence of glaucoma associated with ARB10,13 was observed in this cohort. Four of the patients were diagnosed with narrow angle glaucoma and displayed above-average IOP and increased cup-to-disc ratio changes. Anterior segment abnormalities are likely the underlying factor contributing to the high incidence of glaucoma in ARB.54 Most ARB patients assessed demonstrated shortened axial length, with patient E also displaying decreased anterior chamber depth. As ARB patients age, growth of the lens can crowd an already narrow angle, and IOP control may be challenging even in patients with previous laser peripheral iridotomy. In these cases, treating physicians performing filtering procedures should be aware of the increased risk of aqueous misdirection. Curiously, the arVMD patient's axial-length was short but to a lesser extent than in ARB. There were also no signs of glaucoma, suggesting variable expression of recessive BEST1 variants leading to anterior segment dysgenesis and glaucoma. Together, this information hints at the role of BEST1 in ocular development that variably impacts the incidence of angle closure glaucoma in recessively but not dominantly inherited disease.
Six patients displayed intraretinal fluid accumulation with cystic changes, additionally, the arVMD patient had SRF accumulation (Fig. 1). CAIs have been attempted before in ARB with CME, yielding variable response.15,47,55 Crowley et al.55 reported bilateral anatomical changes with a unilateral increase in visual acuity, whereas another study reported minimal effect.47 Boon et al.15 noted a dose dependent reduction in the accumulation of fluid that reappeared after CAI taper and subsequently disappeared after returning to the initial dose, similar to proband B. Several factors likely influence the chances of successful treatment: age at onset, severity, duration of edema and pharmacogenomic differences in populations.15 In this cohort, three ARB patients were treated with topical CAIs. Patient A was not treated because fluid accumulation had not yet caused visual dysfunction. The retinal microstructure normalized in the probands from families B and E. No change was noted in patient C over one year. Only patient E experienced an improvement in visual acuity. The proband of family F when treated with Bevacizumab showed mild SRF reduction with stable vision. Probands G and H were not followed up long enough to note changes. The nature of the intraretinal and subretinal fluid and response to CAIs suggests widespread RPE dysfunction. SRF accumulation can also be secondary to capillary leak caused by occult choroidal neovascularization (CNV) identified in up to 36% of patients, which explains the positive response to anti-VEGF.56 Similar treatment strategies have been used successfully in adVMD complicated by CNV.57,58 Clinicians should interrogate these eyes for CNV and consider anti-VEGF agents if present. However, this is likely not a primary treatment for the underlying disease.
Color vision is one diagnostic measure that has not been extensively discussed in ARB patients. Six patients tested for color vision failed, and three ARB cases (50%) tested using the Panel-D15 test demonstrated Tritan axis deficits. Interestingly, Boon et al.15 found that patients were also deficient in the blue-yellow axis along with one individual experiencing red-green confusion. One potential explanation is the relative abundance of BEST1 in the extramacular regions14,59 that when decreased to a critical level in recessive diseases is able to affect a more widespread area compared with the macular restriction of adVMD. Rods are also known to assist tritan axis hue discrimination, which is impaired in rod photoreceptor degeneration.60 A recent study found that ARB patient's rod function was severely impacted, while their cones were preserved.61 It is possible that the null effect of ARB mutations negatively impacts rods more than that seen in adVMD, resulting in a clinically significant effect that might be used to distinguish ARB from adVMD, beyond extensive ERG testing. This defect could develop later in the disease course and instead correlate with severe macular dysfunction, explaining the absence of a specific axis deficit in family F. Color vision testing should be investigated in future studies to determine if it can be used to further classify ARB, arVMD and adVMD.
Classification of ARB and VMD patients by phenotype and inheritance was attempted using common clinical parameters. Given the results of the PCA (Fig. 4D), algorithmic separation of ARB and VMD phenotypes should be feasible given additional clinical parameters such as refraction, ERG, and axial-length data. Such a program would be instrumental in determining likely candidates for gene therapy that has shown amelioration of disease in recessively inherited models of ARB.61 Gene therapy is theorized to have more success in recessively inherited disease, particularly in cases of gene supplementation for null phenotypes.62 However, many ARB patients contain alleles that are known to cause VMD in the homozygous state (Fig. 3C). Furthermore, the heterogeneity in bestrophinopathies has led to variable response to gene augmentation efforts; only some adVMD variants (N296H and R218C) show a recovery of chloride conductance by dilution of the mutant allele, while another variant, A146K required gene editing to silence the dominant-negative effect.62
In a further attempt to differentiate alleles associated with separate phenotypes, the frequency of these alleles was examined, but no distinction between phenotypes was achieved. A significant overlap in disease alleles occurs between ARB and VMD. It appears that there is too much heterogeneity, particularly in VMD, to reliably delineate variants that lead to a particular phenotype. In genetic supplementation, it is uncertain if complete cure could be achieved or if a dominant-negative or semi-dominant phenotype would result given the genotype-phenotype heterogeneity. Thus correctly classifying variants by inheritance and phenotype could highlight which variants are likely to respond to treatment. Defining the arVMD classification is also relevant when considering genetic testing, since the possibility of recessive disease in a patient with VMD influences pre-test genetic counseling. Similarly, the posttest counseling may also differ given the prognosis that arVMD may well progress to ARB, whereas adVMD has a different course and separate risks.
The genetics and inheritance of these diseases are complicated by the variable penetrance associated with VMD.44,46,63–66 Although VMD is typically associated with dominant inheritance, recessive cases have also been reported and are likely more common than previously thought.19,23–30 Indeed, multiple previous reports of p.Glu35Lys are all associated with autosomal recessive inheritance, without known parent carrier phenotypes.39,40,41 A major limitation to our study is that parents were not routinely clinically examined in the absence of reported symptoms or a concerning vision history. In rare cases, pathogenic BEST1 variants are inherited in a semidominant fashion. MacDonald et al.50 found that in a compound heterozygous state, two siblings manifested the VMD phenotype, whereas the heterozygous parents displayed more mild maculopathy. Schatz et al.21 found that homozygous or compound heterozygous VMD mutations produce a severe VMD phenotype that extends into the extramacular region. In another case, a son with biallelic BEST1 variants was diagnosed with ARB, whereas his heterozygous father was diagnosed with adVMD.17 The result of biallelic adVMD mutations can resemble the ARB phenotype often associated with recessive inheritance because of the extramacular involvement. Still, these cases are often classified as atypical adVMD because a large macular lesion is usually observed.19,20,22 Additionally, reports have cast doubt on whether BEST1-related RP is a clinical entity, because it may represent part of the ARB phenotypic spectrum.67 Collectively, these reports and our observations demonstrate that BEST1 mutations are associated with multiple inheritance patterns and a spectrum of phenotypes that overlap and are difficult to differentiate. These findings also support routine clinical examination of unaffected carriers of pathogenic variants associated with ARB.
In conclusion, this study suggests that arVMD is a clinical entity along the VMD-bestrophinopathy spectrum based on the combination of autosomal recessive inheritance and vitelliform endophenotype. As discussed previously, biallelic inheritance of BEST1 variants appears to predispose individuals to extramacular lesions akin to what is seen in ARB, yet the presence of tritan axis color vision deficits in our cohort is unique to ARB. In addition, our patient with arVMD has a reduced axial-length, similar to ARB but not adVMD. Longitudinal follow-up will reveal whether arVMD progresses to typical ARB over time. Further support for this idea stems from examination of the younger sibling of family F who shares characteristics of both adVMD (partial vitelliform endophenotype, preserved acuity, and normal axial length) and ARB (nonspecific color vision changes and diffuse ERG deficits). From both the literature review meta-analyses of visual parameters and the unsupervised analysis of multiple parameters simultaneously (PCA graph), arVMD presents as an intermediate rather than distinct group between ARB and adVMD, particularly with respect to PC1 that largely represents age and degree of visual impairment (Fig. 4D). Because algorithmic separation could not be achieved, one conclusion would be that these diseases lay along a continuum. If VMD and ARB are, in fact, a continuum, this would invoke higher rates of incomplete penetrance for variants associated with presumed autosomal recessive disease, compared to variants in families with observed autosomal dominant inheritance with high penetrance rates. Clinical assessment of known and obligate BEST1-ARB carriers for subclinical phenotypes will be critical to distinguish whether these cause semidominant or truly autosomal recessive disease. However, there are differences in macular endophenotypes, ERG findings, axial-length, and glaucoma risk among these conditions, which have permitted clinical categorization in the literature. This also introduces the notion that arVMD can progress to ARB over time; our arVMD patient has not been observed long-term to determine if the phenotype will appear more like ARB in later years. Alternatively, arVMD may be a distinct subgroup of disease rather than part of a continuum. A better understanding of this phenomenon will provide implications for future treatment, particularly in the application of gene therapy.
Supplementary Material
Acknowledgments
The authors thank Amy Turriff and Delphine Blain for genetic counseling, including pedigree analysis and pretest and posttest patient counseling.
Supported by the NEI-NIH Intramural Research Program.
Disclosure: T.A. Pfister, None; W.M. Zein, None; C.A. Cukras, None; H.N. Sen, None; R.S. Maldonado, None; L.A. Huryn, None; R.B. Hufnagel, None
References
- 1. Yang T, Liu Q, Kloss B, et al.. Structure and selectivity in bestrophin ion channels. Science. 2014; 346: 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT.. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiol Rev. 2008; 88: 639–672. [DOI] [PubMed] [Google Scholar]
- 3. Milenkovic A, Brandl C, Milenkovic VM, et al.. Bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells. Proc Natl Acad Sci USA . 2015; 112: 2630–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moshfegh Y, Velez G, Li Y, Bassuk AG, Mahajan VB, Tsang SH.. Bestrophin1 mutations cause defective chloride conductance in patient stem cell-derived RPE. Hum Mol Genet . 2016; 25: 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marmorstein AD, Marmorstein LY, Rayborn M, Wang X, Hollyfield JG, Petrukhin K.. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc Natl Acad Sci USA. 2000; 97: 12758–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boon CJ, Klevering BJ, Leroy BP, Hoyng CB, Keunen JE, den Hollander AI. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009; 28: 187–205. [DOI] [PubMed] [Google Scholar]
- 7. Querques G, Zerbib J, Georges A, et al.. Multimodal analysis of the progression of Best vitelliform macular dystrophy. Mol Vis. 2014; 20: 575–592. [PMC free article] [PubMed] [Google Scholar]
- 8. Tsang SH, Sharma T.. Best vitelliform macular dystrophy. Adv Exp Med Biol. 2018; 1085: 157–158. [DOI] [PubMed] [Google Scholar]
- 9. Renner AB, Tillack H, Kraus H, et al.. Morphology and functional characteristics in adult vitelliform macular dystrophy. Retina. 2004; 24: 929–939. [DOI] [PubMed] [Google Scholar]
- 10. Burgess R, Millar ID, Leroy BP, et al.. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am J Hum Genet. 2008; 82: 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gerth C, Zawadzki RJ, Werner JS, Heon E.. Detailed analysis of retinal function and morphology in a patient with autosomal recessive bestrophinopathy (ARB). Doc Ophthalmol. 2009; 118: 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kalevar A, Chen JJ, McDonald HR, Fu AD.. Autosomal recessive bestrophinopathy: multimodal imaging update. Retin Cases Brief Rep. 2018; 12: 51–54. [DOI] [PubMed] [Google Scholar]
- 13. Zhong Y, Guo X, Xiao H, et al.. Flat anterior chamber after trabeculectomy in secondary angle-closure glaucoma with BEST1 gene mutation: case series. PLoS One. 2017; 12: e0169395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mullins RF, Kuehn MH, Faidley EA, Syed NA, Stone EM.. Differential macular and peripheral expression of bestrophin in human eyes and its implication for best disease. Invest Ophthalmol Vis Sci. 2007; 48: 3372–3380. [DOI] [PubMed] [Google Scholar]
- 15. Boon CJ, van den Born LI, Visser L, et al.. Autosomal recessive bestrophinopathy: differential diagnosis and treatment options. Ophthalmology. 2013; 120: 809–820. [DOI] [PubMed] [Google Scholar]
- 16. Davidson AE, Millar ID, Urquhart JE, et al.. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet. 2009; 85: 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakanishi A, Ueno S, Hayashi T, et al.. Clinical and genetic findings of autosomal recessive bestrophinopathy in Japanese cohort. Am J Ophthalmol. 2016; 168: 86–94. [DOI] [PubMed] [Google Scholar]
- 18. Boon CJ, Theelen T, Hoefsloot EH, et al.. Clinical and molecular genetic analysis of best vitelliform macular dystrophy. Retina. 2009; 29: 835–847. [DOI] [PubMed] [Google Scholar]
- 19. Kinnick TR, Mullins RF, Dev S, et al.. Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients. Retina. 2011; 31: 581–595. [DOI] [PubMed] [Google Scholar]
- 20. Peiretti E, Caminiti G, Forma G, et al.. A novel p.Asp304Gly mutation in BEST1 gene associated with atypical best vitelliform macular dystrophy and high intrafamilial variability. Retina. 2016; 36: 1733–1740. [DOI] [PubMed] [Google Scholar]
- 21. Schatz P, Bitner H, Sander B, et al.. Evaluation of macular structure and function by OCT and electrophysiology in patients with vitelliform macular dystrophy due to mutations in BEST1. Invest Ophthalmol Vis Sci. 2010; 51: 4754–4765. [DOI] [PubMed] [Google Scholar]
- 22. Schatz P, Klar J, Andreasson S, Ponjavic V, Dahl N.. Variant phenotype of Best vitelliform macular dystrophy associated with compound heterozygous mutations in VMD2. Ophthalmic Genet. 2006; 27: 51–56. [DOI] [PubMed] [Google Scholar]
- 23. Bitner H, Mizrahi-Meissonnier L, Griefner G, Erdinest I, Sharon D, Banin E.. A homozygous frameshift mutation in BEST1 causes the classical form of Best disease in an autosomal recessive mode. Invest Ophthalmol Vis Sci. 2011; 52: 5332–5338. [DOI] [PubMed] [Google Scholar]
- 24. Cascavilla ML, Querques G, Stenirri S, Parodi MB, Querques L, Bandello F.. Unilateral vitelliform phenotype in autosomal recessive bestrophinopathy. Ophthalmic Res. 2012; 48: 146–150. [DOI] [PubMed] [Google Scholar]
- 25. Iannaccone A, Kerr NC, Kinnick TR, Calzada JI, Stone EM.. Autosomal recessive best vitelliform macular dystrophy: report of a family and management of early-onset neovascular complications. Arch Ophthalmol. 2011; 129: 211–217. [DOI] [PubMed] [Google Scholar]
- 26. Lacassagne E, Dhuez A, Rigaudière F, et al.. Phenotypic variability in a French family with a novel mutation in the BEST1 gene causing multifocal best vitelliform macular dystrophy. Mol Vis. 2011; 17: 309–322. [PMC free article] [PubMed] [Google Scholar]
- 27. Sodi A, Menchini F, Manitto MP, et al.. Ocular phenotypes associated with biallelic mutations in BEST1 in Italian patients. Mol Vis. 2011; 17: 3078–3087. [PMC free article] [PubMed] [Google Scholar]
- 28. Tian R, Yang G, Wang J, Chen Y.. Screening for BEST1 gene mutations in Chinese patients with bestrophinopathy. Mol Vis. 2014; 20: 1594–1604. [PMC free article] [PubMed] [Google Scholar]
- 29. Wittström E, Ekvall S, Schatz P, Bondeson ML, Ponjavic V, Andréasson S.. Morphological and functional changes in multifocal vitelliform retinopathy and biallelic mutations in BEST1. Ophthalmic Genet. 2011; 32: 83–96. [DOI] [PubMed] [Google Scholar]
- 30. Piñeiro-Gallego T, Alvarez M, Pereiro I, et al.. Clinical evaluation of two consanguineous families with homozygous mutations in BEST1. Mol Vis. 2011; 17: 1607–1617. [PMC free article] [PubMed] [Google Scholar]
- 31. McCulloch DL, Marmor MF, Brigell MG, et al.. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015; 130: 1–12. [DOI] [PubMed] [Google Scholar]
- 32. Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M.. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol. 2009; 118: 69–77. [DOI] [PubMed] [Google Scholar]
- 33. Constable PA, Bach M, Frishman LJ, Jeffrey GB, Robson AG.. ISCEV standard for clinical electro-oculography (2017 update). Doc Ophthalmol. 2017; 134: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marmor MF, Brigell MG, McCulloch DL, et al.. ISCEV standard for clinical electro-oculography (2010 update). Doc Ophthalmol. 2011; 122: 1–7. [DOI] [PubMed] [Google Scholar]
- 35. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC.. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012; 40: 452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adzhubei IA, Schmidt S, Peshkin L, et al.. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kircher M, Whitten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014; 46: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Omasits U, Ahrens CH, Müller S, Wollscheid B.. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 2014; 30: 884–886. [DOI] [PubMed] [Google Scholar]
- 39. Tian L, Sun T, Zhang X, Peng X, Li Y.. Screening of BEST1 gene in a chinese cohort with Best vitelliform macular dystrophy or autosomal recessive bestrophinopathy. Invest Ophthalmol Vis Sci. 2017; 58: 3366–3375. [DOI] [PubMed] [Google Scholar]
- 40. Stone EM, Andorf JL, Whitmore SS, et al.. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017; 124: 1314–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Habibi I, Falfoul Y, Todorova MG, et al.. Clinical and genetic findings of autosomal recessive bestrophinopathy (ARB). Genes. 2019; 10: 953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meunier I, Senechal A, Dhaenens CM, et al.. Systematic screening of BEST1 and PRPH2 in juvenile and adult vitelliform macular dystrophies: a rationale for molecular analysis. Ophthalmology. 2011; 118: 1130–1136. [DOI] [PubMed] [Google Scholar]
- 43. Luo J, Lin M, Guo X, et al.. Novel BEST1 mutations and special clinical characteristics of autosomal recessive bestrophinopathy in Chinese patients. Acta Ophthalmol. 2019; 97: 247–259. [DOI] [PubMed] [Google Scholar]
- 44. Lotery AJ, Munier FL, Fishman GA, et al.. Allelic variation in the VMD2 gene in best disease and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000; 41: 1291–1296. [PubMed] [Google Scholar]
- 45. White K, Marquardt A, Weber BH.. VMD2 mutations in vitelliform macular dystrophy (Best disease) and other maculopathies. Hum Mutat. 2000; 15: 301–308. [DOI] [PubMed] [Google Scholar]
- 46. Marchant D, Yu K, Bigot K, et al., New VMD2 gene mutations identified in patients affected by Best vitelliform macular dystrophy. J Med Genet. 2007; 44: e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fung AT, Yzer S, Goldberg N, et al.. New Best1 mutations in autosomal recessive bestrophinopathy. Retina. 2015; 35: 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zaneveld J, Siddiqui S, Li H, et al.. Comprehensive analysis of patients with Stargardt macular dystrophy reveals new genotype-phenotype correlations and unexpected diagnostic revisions. Genet Med. 2015; 17: 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Borman AD, Davidson AE, O'Sullivan J, et al.. Childhood-onset autosomal recessive bestrophinopathy. Arch Ophthalmol. 2011; 129: 1088–1093. [DOI] [PubMed] [Google Scholar]
- 50. MacDonald IM, Gudiseva HV, Villanueva A, Greve M, Caruso R, Ayyagari R.. Phenotype and genotype of patients with autosomal recessive bestrophinopathy. Ophthalmic Genet. 2012; 33: 123–129. [DOI] [PubMed] [Google Scholar]
- 51. Kramer F, Mohr N, Kellner U, Rudolph G, Webber BH.. Ten novel mutations in VMD2 associated with Best macular dystrophy (BMD). Hum Mutat . 2003; 22: 418. [DOI] [PubMed] [Google Scholar]
- 52. Zhao L, Grob S, Cory R, et al.. A novel compound heterozygous mutation in the BEST1 gene causes autosomal recessive Best vitelliform macular dystrophy. Eye (Lond.) . 2012; 26: 866–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davidson AE, Millar ID, Burgess R, et al.. Functional characterization of bestrophin-1 missense mutations associated with autosomal recessive bestrophinopathy. Invest Ophthalmol Vis Sci. 2011; 52: 3730–3736. [DOI] [PubMed] [Google Scholar]
- 54. Jansson RW, Berland S, Bredrup C, Austeng D, Andréasson S, Wittström E.. Biallelic mutations in the BEST1 gene: additional families with autosomal recessive bestrophinopathy. Ophthalmic Genet. 2016; 37: 183–193. [DOI] [PubMed] [Google Scholar]
- 55. Crowley C, Paterson R, Lamey T, et al.. Autosomal recessive bestrophinopathy associated with angle-closure glaucoma. Doc Ophthalmol. 2014; 129: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Parodi MB, Romano F, Cicinelli MV, et al.. Retinal vascular impairment in best vitelliform macular dystrophy assessed by means of optical coherence tomography angiography. Am J Ophthalmol. 2018; 187: 61–70. [DOI] [PubMed] [Google Scholar]
- 57. Hori K, Ishida S, Inoue M, et al.. Treatment of cystoid macular edema with oral acetazolamide in a patient with BEST vitelliform macular dystrophy. Retina. 2004; 3: 481–482. [DOI] [PubMed] [Google Scholar]
- 58. Leu J, Schrage NF, Degenring RF.. Choroidal neovascularisation secondary to Best's disease in a 13-year-old boy treated by intravitreal bevacizumab. Graefes Arch Clin Exp Ophthalmol. 2007; 245: 1723–1725. [DOI] [PubMed] [Google Scholar]
- 59. Whitmore SS, Wagner AH, DeLuca AP, et al.. Transcriptomic analysis across nasal, temporal, and macular regions of human neural retina and RPE/choroid by RNA-Seq. Exp Eye Res. 2014; 129: 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Knight R, Buck SL, Fowler GA, Nguyen A.. Rods affect S-cone discrimination on the Farnsworth-Munsell 100-hue test. Vision Res. 1998; 38: 3477–3481. [DOI] [PubMed] [Google Scholar]
- 61. Guziewicz KE, Cideciyan AV, Beltran WA, et al.. Gene therapy corrects a diffuse retina-wide microdetachment modulated by light exposure. Proc Natl Acad Sci USA. 2018; 115: 2839–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sinha D, Steyer B, Shahi PK, et al.. Human IPSC modeling reveals mutation-specific responses to gene therapy in a genotypically diverse dominant maculopathy. Am J Hum Genet . 2020; 170: 278–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Arora R, Khan K, Kasilian ML, et al.. Unilateral BEST1-associated retinopathy. Am J Ophthalmol. 2016; 169: 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wabbels B, Preising MN, Kretschmann U, Demmler A, Lorenz B.. Genotype-phenotype correlation and longitudinal course in ten families with Best vitelliform macular dystrophy. Graefes Arch Clin Exp Ophthalmol. 2006; 244: 1455–1466. [DOI] [PubMed] [Google Scholar]
- 65. Seddon JM, Sharma S, Chong S, Hutchinson A, Allikmets R, Adelman RA.. Phenotype and genotype correlations in two best families. Ophthalmology. 2003; 110: 1724–1731. [DOI] [PubMed] [Google Scholar]
- 66. Renner AB, Tillack H, Kraus H, et al.. Late onset is common in best macular dystrophy associated with VMD2 gene mutations. Ophthalmology. 2005; 112: 586–592. [DOI] [PubMed] [Google Scholar]
- 67. Shah M, Broadgate S, Shanks M, et al.. Association of Clinical and Genetic Heterogeneity with BEST1 Sequence Variations. JAMA Ophthalmol . 2020; 138: 644–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.