

Abstract
A recent discovery of a cooperative catalysis comprising a silver salt and an acid led to a dramatic improvement in the way glycosyl halides are glycosidated. Excellent yields have been achieved, but the stereoselectivity achieved with 2-O-benzylated donors was poor. Reported herein is our first attempt to refine the stereoselectivity of the cooperatively catalyzed galactosylation reaction. Careful optimization of the reaction conditions along with studying effects of the remote protecting groups led to excellent stereocontrol of α-galactosylation of a variety of glycosyl acceptors with differentially protected galactosyl donors.
Graphical Abstract

INTRODUCTION
Construction of the biologically relevant glycans has been a great challenge since the first chemical glycosidations of glycosyl halides performed by Michael,1 Koenigs–Knorr,2 and Fischer.3 A rigorous study of chemical glycosylation gave straightforward access to many 1,2-trans glycosidic linkages that can be reliably achieved with the aid of a neighboring participating ester group at C-2.4–7 Conversely, there is no universal method for installing 1,2-cis glycosidic linkages.8 Early work by Helferich, Zemplen, and others has enhanced our understanding of the synthesis of 1,2-cis glycosides with glycosyl halides as donors.9–12 However, these studies did not lead to practical application in the synthesis of complex oligosaccharides. Since then, many improvements for the construction of 1,2-cis glycosides with glycosyl halides have emerged: employing bases as acid scavengers,2,13–16 alcoholysis of glycosyl halides with or without halide additives,17–19 halide-ion catalyzed glycosylation,20–22 conversion of anomeric halides to positively charged leaving groups,23,24 β- to α-glycoside anomerization,25–27 in situ generation and glycosylation of glycosyl bromides from thioglycosides,28 bromine-promoted glycosylation,29,30 etc.
Due to their general instability, low reactivity profile, and the requirement for excess toxic reagents for their activation, glycosyl halides have been superseded by other glycosyl donors at the forefront of modern oligosaccharide synthesis. In recent years, imidates, thioglycosides, and phosphates were found to be more advantageous.8 Many current methods employed for 1,2-cis glycosylation rely on these glycosyl donors in combination with other effects including steric factors,31,32 remote participation,33–37 chiral auxiliaries,38–41 H-bond-mediated aglycone delivery (HAD),42 modification of catalysts,43–45 or glycosylation modulators.46,47 In spite of these significant advancements, completely stereocontrolled 1,2-cis glycosylation remains a notable challenge in chemical synthesis.8
Recently, glycosyl chlorides have been progressively gaining interest after Ye48 and Jacobsen49 reported organocatalyzed activation for the α- and β-stereoselective glycosidation, respectively. A new method for highly stereoselective 1,2-cis glycosylation with glycosyl bromides has also been recently reported.16 However, glycosidation of glycosyl halides may be very slow, even at elevated temperatures. Recently, our group has introduced a cooperative catalysis approach for the activation of glycosyl halides.50 An acid and a silver salt applied together have changed the fate of glycosyl halides by leading to a drastically reduced glycosylation reaction time to as low as 2 min and a dramatic increase in the yields of glycosides.51 However, our previous cooperatively catalyzed reactions were nonstereoselective, and reported herein is our first attempt to investigate 1,2-cis α-stereoselective galactosylation.
RESULTS AND DISCUSSION
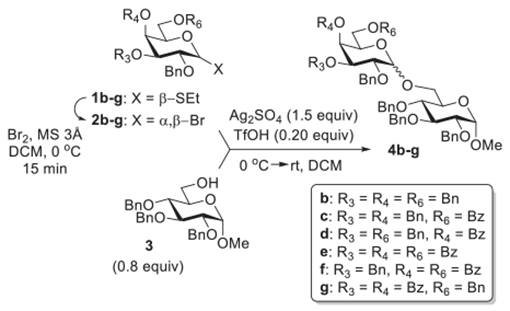
In the effort to investigate 1,2-cis stereoselective glycosylation we chose to focus on galactosyl bromide donors. One of the driving forces for this selection is the fact that the HAD method that gives excellent syn-selectivity in application to β-mannosides52 and α-glucosides53 cannot be applied to α-galactosides because all remote substituents are pointing upward (anti). Another motivation to investigate α-galactosylation was due to the very unexpected β-stereoselectivity observed in our recent work with galactosyl bromides.50 Glycosidation of per-benzoylated bromide 2a generated from the corresponding thioglycoside 1a with glycosyl acceptor 354 provided disaccharide 4a with complete β-stereoselectivity due to the participation of a 2-O-benzoyl neighboring group (Table 1, entry 1). This glycosylation was performed in the presence of Ag2O (3.0 equiv) and TMSOTf (0.25 equiv).50 However, when galactosyl bromide 2b generated from per-benzylated thioglycoside precursor 1b, was coupled with glycosyl acceptor 3, rather unexpectedly disaccharide 4b was obtained in 96% yield with a nearly complete β-stereoselectivity (α/β = 1:20, entry 2). This glycosylation was performed in the presence of Ag2O (3.0 equiv) and TMSOTf (0.10 equiv).50 With a general idea of developing universal reaction conditions that would be applicable to all kinds of substrates, we repeated glycosidation of 2b in the presence of Ag2O (3.0 equiv) using the larger amount of TMSOTf (0.25 equiv). Strikingly, increasing the amount of TMSOTf resulted in a complete loss of stereoselectivity and a significant reduction of the yield of 4b that dropped to 71% (entry 3).
Table 1.
Comparative Glycosidation of Galactosyl Bromides 2a–d and Optimization of the Reaction Conditions
 | |||
|---|---|---|---|
| Entry | Donor | Conditions | Product, yield, α/β |
| 150 | 2a | Ag2O (3.0 equiv), TMSOTf (0.25 equiv), 0 °C→rt, 10 min | 4a, 99%, β-only |
| 250 | 2b | Ag2O (3.0 equiv), TMSOTf (0.10 equiv), 0 °C→rt, 15 min | 4b, 96%, 1:20 |
| 3 | 2b | Ag2O (3.0 equiv), TMSOTf (0.25 equiv), 0 °C→rt, 15 min | 4b, 71%, 1.0:1 |
| 4 | 2c | Ag2O (3.0 equiv), TMSOTf (0.25 equiv), 0 °C→rt, 1 h | 4c, 84%, 1:3.0 |
| 5 | 2d | Ag2O (3.0 equiv), TMSOTf (0.25 equiv), 0 °C→rt, 3 h | 4d, 80%, 2.9:1 |
| 6 | 2d | Ag2O (3.0 equiv), TfOH (0.15 equiv), −10 °C, 1.5 h | 4d, 72%, 5.0:1 |
| 7 | 2d | Ag2O (3.0 equiv), TfOH (0.15 equiv), −30 °C, 2 h | 4d, 82%, 11:1 |
| 8 | 2d | Ag2O (3.0 equiv), TfOH (0.20 equiv), −50 °C, 3 h | 4d, 67%, 11:1 |
| 9 | 2d | Ag2SO4 (1.0 equiv), TfOH (0.20 equiv), −30 °C → rt, 18 h | 4d, 95%, 11:1 |
| 10 | 2d | Ag2SO4 (1.5 equiv), TfOH (0.20 equiv), −10 °C, 1 h | 4d, 92%, 11.5:1 |
| 11 | 2d | Ag2SO4 (1.5 equiv), TfOH (0.20 equiv), 0 °C→rt, 1 h | 4d, 87%, 6.0:1 |
Methods for enhancing α-stereoselectivity of galactosylation exist. Among these, utilizing the remote protecting group participation appealed to us as a promising next step toward improving the stereoselectivity. Previous studies demonstrated that acyl groups at various remote positions may have a profound effect on the stereoselectivity of glycosylation.55–58 To understand the effects of remote substituents on cooperatively catalyzed galactosylations, we obtained thioglycosides 1c and 1d, equipped with a 6-OBz and 4-OBz substituent, respectively. The corresponding galactosyl bromides 2c and 2d were generated in situ and activated in the presence of Ag2O (3.0 equiv) and TMSOTf (0.25 equiv). Glycosidation of 6-OBz bromide 2c with acceptor 3 was largely disappointing because it produced disaccharide 4c in a good yield of 84% albeit preferential β-stereoselectivity (α/β = 1:3.0, entry 4). Encouragingly, galactosyl bromide 2d afforded disaccharide 4d56 in 80% yield with a marginal shift toward α-stereoselectivity (α/β = 2.9:1, entry 5). From these results, it was apparent that only the benzoyl group at the C-4 position offers a promising structural feature for the galactosyl donor favoring α-1,2-cis stereoselectivity.
To enhance the utility of this galactosylation we then endeavored to optimize the reaction conditions bearing the following considerations. We note that some reactions were accompanied by a competing silyl transfer to the glycosyl acceptor that explains compromised yields of disaccharides seen in a number of experiments. Hence, we replaced TMSOTf with trifluoromethanesulfonic acid (TfOH), which provided comparable results in our previous study,51 to avoid the silyl transfer side reaction. All previous reactions were conducted at 0 °C followed by slowly bringing the reaction temperature to ambient. Hence, we wondered if modifying the reaction temperature would offer a better means for the stereocontrol. After preliminary screening, we found that the reaction of galactosyl bromide 2d with glycosyl acceptor 3 in the presence of Ag2O (3.0 equiv) and TfOH (0.15 equiv) at −10 °C affords disaccharide 4d in 72% yield with a further improved α-stereoselectivity (α/β = 5.0:1, entry 6). A decrease in temperature to −30 °C produced disaccharide 4d in a respectable yield and even higher stereoselectivity (82%, α/β = 11:1, entry 7). Further cooling to −50 °C (or −70 °C) did not enhance the stereoselectivity but led to a decreased yield (67%, α/β = 11:1, entry 8).
We then endeavored to change the source of the silver promoter and replaced silver(I) oxide with silver(I) sulfate (Ag2SO4). After preliminary screening, we noticed that the same stereoselectivity albeit with a much higher yield could be achieved in the presence of only 1.0 equiv of Ag2SO4, along with 0.20 equiv of TfOH. Thus, disaccharide 4d was produced in an excellent yield of 95% and high stereoselectivity (α/β = 11:1, entry 9). This reaction was much slower than the previous experiments. As a matter of fact, it was not proceeding at −30 °C during the initial 5 h. When the external cooling was removed, the reaction required an additional 13 h to complete. The subsequent experiment was performed with a larger amount of Ag2SO4 (1.50 equiv) and at higher reaction temperature (−10 °C). As a result, disaccharide 4d was produced in only 1 h in an excellent yield of 92% and with a similar stereoselectivity (α/β = 11.5:1, entry 10). A further increase in the starting reaction temperature to 0 °C followed by warming to room temperature produced disaccharide 4d in a similar yield, but the stereoselectivity dropped (87%, α/β = 6.0:1, entry 11). Nevertheless, we chose this latter experiment as the benchmark for our subsequent study. This choice was driven by experimental convenience as opposed to low temperature experiments that are more tedious to set up and maintain.
Under the established reaction conditions, with the addition Ag2SO4 (1.5 equiv) and TfOH (0.20 equiv) and a starting reaction temperature of 0 °C which was allowed to warm to room temperature, whereas 4-OBz donor 4d showed good stereoselectivity (α/β = 6.0:1, Table 2, entry 1), galactosyl bromides 2b and 2c failed. Thus, per-OBn bromide 2b and 6-OBz bromide 2c produced the respective disaccharides 4b and 4c in good yields albeit with preferential β-stereoselectivity (α/β = 1/1.2–2.7, entries 2 and 3). These results reinforce our previous assumption that the 4-OBz group is essential for achieving preferential α-stereoselectivity. To elaborate on this, we obtained a further series of differentially polybenzoylated ethylthio galactosides 1e–g. The corresponding bromides were then generated in situ followed by their glycosidation with acceptor 3. Galactosyl bromide donor 2e equipped with three benzoyl substituents at C-3, -4, and -6 provided disaccharide 4e in 97% yield with complete α-stereoselectivity (entry 4). Although galactosyl bromide 2e is equipped with the electronically superdisarming protecting group pattern,59 the reaction readily completed within 1 h. Glycosidation of 4,6-OBz galactosyl bromide 2f was faster, and the corresponding disaccharide 4f was smoothly produced in only 30 min in 93% yield and with nearly complete stereoselectivity (α/β = 33/1, entry 5). Glycosidation of 3,4-OBz galactosyl bromide 2g was slower (2 h), but the corresponding disaccharide 4g was smoothly produced in 96% yield with complete α-stereoselectivity (entry 6).
Table 2.
Comparative Glycosidation of Galactosyl Bromides 2b–g under Optimized Reaction Conditions
 | |||
|---|---|---|---|
| Entry | Donor | Time | Product, yield, α/β |
| 1 | 2d (4-OBz) | 1 h | 4d, 87%, 6.0:1 |
| 2 | 2b (all-OBn) | 1.5 h | 4b, 87%, 1:1.2 |
| 3 | 2c (6-OBz) | 1 h | 4c, 75%, 1:2.7 |
| 4 | 2e (3,4,6-OBz) | 1 h | 4e, 97%, α-only |
| 5 | 2f (4,6-OBz) | 30 min | 4f, 93%, 33:1 |
| 6 | 2g (3,4-OBz) | 2 h | 4g, 96%, α-only |
Excellent yields and α-stereoselectivity achieved with galactosyl bromide donors 2e–g and primary glucosyl acceptor 3 prompted us to broaden the scope of the cooperatively catalyzed 1,2-cis glycosylation. For this study, we selected dibenzoylated galactosyl bromides 2f and 2g due to the ease of their preparation from the corresponding cyclic acetal/ketal-protected precursors. We also chose a series of glycosyl acceptors including secondary alcohols 5–754 and primary alcohols of differential reactivity, highly reactive diacetone galactose 8, and electronically deactivated compound 960 (Figure 1). Results for reactions between glycosyl donors 2f and 2g and different glycosyl acceptors 5–9 are listed in Table 3. Glycosylation between donor 2f and 2-OH acceptor 5 was very efficient in the presence of Ag2SO4 (1.5 equiv) and TfOH (0.20 equiv) at 0 °C to rt. As a result, disaccharide 10 was obtained in 3 h in 91% yield and with complete α-stereoselectivity (entry 1). Glycosidation of donor 2f with the less reactive 3- and 4-OH acceptors 6 and 7 was slower, and to enhance the utility we increased the amount of TfOH to 0.40 equiv. As a result, disaccharides 11 and 12 were obtained in 6 h in 73% and 49% yield, respectively, with complete α-stereoselectivity in both cases (entries 2 and 3).
Figure 1.

Standard glycosyl acceptors 5–9 for broadening the scope of α-galactosylation
Table 3.
Expanding the Scope of Glycosidation of Donors 2f and 2g with Acceptors 5–9
 |
Glycosidations of donor 2f with differently protected primary acceptors were performed using 0.20 equiv of TfOH. Under these reaction conditions, diacetone galactose acceptor 8 gave disaccharide 1358 in 68% yield and good α-selectivity (α/β = 11/1, entry 4). Glycosylation of a much less reactive benzoylated 6-OH acceptor 9 with donor 2f gave disaccharide 14 in 87% yield and with complete α-stereoselectivity (entry 5). A practically identical trend was observed during glycosylations with 3,4-OBz protected bromide 2g (Table 3). All glycosylations were rather swift (2–6 h), and the respective disaccharide derivatives 15–19 were produced in good yields of 65–97% with practically complete stereoselectivity in all cases (α/β > 25/1 to α-only, entries 6–10).
In conclusion, combined effects of the cooperative catalysis and remote participation of benzoyl groups led us to develop a powerful new method for stereocontrolled α-galactosylation. This method has the following four (conservatively estimated) advantages in comparison with the known methods. First, the ease of the synthesis of glycosyl donors and a rapid in situ conversion to the corresponding bromides. Second, no specialized protecting groups are required; only altering positions of common benzyl and benzoyl groups provide high to exclusive α-stereoselectivity. Third, the reaction duration is relatively short, and the reaction can be conducted at ambient temperature without losing stereoselectivity. Fourth, promoter systems are stable, not environment sensitive, and commonly available. Further development of the methodology and its application to the stereoselective synthesis of biologically relevant molecules are currently underway in our laboratory.
EXPERIMENTAL SECTION
General.
The reactions were performed using commercially available reagents. CH2Cl2 used for reactions was distilled from CaH2 prior to the application. Other ACS-grade solvents used for reactions were purified and dried according to standard procedures. Prior to the initial application, silver salts were coevaporated with toluene (×2), dried in vacuo in the dark. Column chromatography was performed on silica gel 60 (70–230 mesh); reactions were monitored using Thin-Layer Chromatography (TLC) on Kieselgel 60 F254. TLC was examined under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40 °C. Molecular sieves (3 Å) used for reactions were crushed and activated under vacuum for 8 h at 390 °C in the first instance, and then for 2–3 h at 390 °C directly prior to application. Optical rotations were measured by a Jasco P2000 polarimeter. 1H and 13C NMR spectra were recorded in CDCl3 at 300 and 75 MHz, respectively. 1H NMR was calibrated to tetramethylsilane (TMS, δH = 0 ppm) in CDCl3. 13C NMR was calibrated to the central signal of CDCl3 δc = 77.00 ppm) in CDCl3. HRMS analysis was performed using an Agilent 6230 ESI TOF LC/MS mass spectrometer.
Synthesis of Thioglycoside Donors.
Ethyl 2,3,4,6-Tetra-O-benzoyl-1-thio-β-d-galactopyranoside (1a).
1a was synthesized as previously reported,61 and its analytical data were in accordance with those reported previously.61
Ethyl 2,3,4,6-Tetra-O-benzyl-1-thio-β-d-galactopyranoside (1b).
1b was synthesized as previously reported,28 and its analytical data were in accordance with those reported previously.28
Ethyl 6-O-Benzoyl-2,3,4-tri-O-benzyl-1-thio-β-d-galactopyranoside (1c).
A solution of ethyl 2,3,4-tri-O-benzyl-1-thio-β-d-galactopyranoside (20,62 2.34 g, 4.73 mmol) in pyridine (40 mL) was cooled to 0 °C. Benzoyl chloride (0.82 mL, 7.09 mmol) was added dropwise followed by the addition of 4-dimethylaminopyridine (DMAP, 115.6 mg, 0.95 mmol), the external cooling was removed, and the resulting mixture was stirred under argon for 16 h at rt. Afterward, the reaction was quenched with MeOH (~10 mL), the volatiles were removed under reduced pressure, and the residue was coevaporated with toluene (4 × 10 mL). The resulting residue was dissolved in CH2Cl2 (~40 mL) and washed with 1 N aq. HCl (15 mL), sat. aq. NaHCO3 (15 mL), and water (2 × 15 mL). The organic phase was separated, dried with MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane 10% gradient elution) to afford the title compound as a white amorphous solid (2.25 g, 3.75 mmol, 80% yield). Analytical data for 1c: Rf = 0.65 (EtOAc/hexanes, 3/7, v/v); [α]23D −13.2 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 7.99–7.88 (m, 2H, aromatic), 7.62–7.50 (m, 1H, aromatic), 7.48–7.15 (m, 17H, aromatic), 5.01 (d, 1H, 2J = 11.7 Hz, CHPh), 4.90 (d, 1H, 2J = 10.2 Hz, CHPh), 4.77 (dd, 4H, 4 × CHPh), 4.54–4.42 (m, 2H, J1,2 = 9.7, J6a,6b = 11.2 Hz, H-1, 6a), 4.31 (dd, 1H, H-6b), 3.93–3.82 (m, 2H, J2,3 = 9.3 Hz, H-2, 4), 3.70 (m, 1H, J5,6a = 6.9, J5,6b = 6.0 Hz, H-5), 3.61 (dd, 1H, J3,4 = 2.8 Hz, H-3), 2.85–2.63 (m, 2H, SCH2CH3), 1.29 (t, 3H, SCH2CH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ, 166.1, 138.2, 138.1 (×2), 133.1 129.7, 129.6 (×3), 128.4 (×2), 128.3 (×5), 128.2 (×2), 127.8, 127.7 (×2), 127.6 (×3), 85.3, 84.1, 78.4, 75.9, 75.8, 74.3, 73.2 (×2), 63.5, 24.9, 15.1 ppm; HR-FAB MS [C36H38O6SNa]+ calcd for 621.2281, found 621.2290.
Ethyl 4-O-Benzoyl-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (1d).
A solution of ethyl 2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (21,63 1.50 g, 3.03 mmol) in pyridine (20 mL) was cooled to 0 °C. Benzoyl chloride (0.53 mL, 4.55 mmol) was added dropwise followed by addition of DMAP (74.5 mg, 0.61 mmol), the external cooling was removed, and the resulting mixture was stirred under argon for 16 h at rt. Afterward, the reaction was quenched with MeOH (~5 mL), the volatiles were removed under reduced pressure, and the residue was coevaporated with toluene (3 × 5 mL). The resulting residue was dissolved in CH2Cl2 (~20 mL) and washed with 1 N aq. HCl (8 mL), sat. aq. NaHCO3 (8 mL), and water (2 × 8 mL). The organic phase was separated, dried with MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane 10% gradient elution) to afford the title compound as a white amorphous solid (1.63 g, 2.73 mmol, 90% yield). Analytical data for 1d: Rf = 0.60 (EtOAc/hexanes, 3/7, v/v); [α]23D + 13.0 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 8.16–8.05 (m, 2H, aromatic), 7.59–7.14 (m, 18H, aromatic), 5.90 (br. d, 1H, H-4), 4.77 (dd, 2H, 2J = 10.2 Hz, CH2Ph), 4.70 (dd, 2H, 2J = 11.4 Hz, CH2Ph), 4.54 (d, 1H, J1,2 = 9.1 Hz, H-1), 4.47 (dd, 2H, 2J = 11.7 Hz, CH2Ph), 3.84 (m, 1H, J5,6a = J5,6b = 6.5 Hz, H-5), 3.72 (dd, 1H, J2,3 = 3.1 Hz, H-2), 3.71–3.59 (m, 2H, J6a,6b = 9.4 Hz, H-3, 6a), 3.53 (dd, 1H, H-6b), 2.79 (m, 2H, SCH2CH3), 1.34 (t, 3H, SCH2CH3) ppm; 13C NMR (CDCl3 at 75 MHz): δ 165.6, 137.9, 137.6, 137.4, 133.1, 129.9 (×2), 129.7, 128.4 (×4), 128.3 (×5), 128.2, 128.0 (×2), 127.8 (×2), 127.7 (×2), 127.6, 85.3, 81.0, 77.6, 75.9, 75.8, 73.6, 71.6, 68.2, 67.3, 24.8, 15.0 ppm; HR-FAB MS [C36H38O6SNa]+ calcd for 621.2297, found 621.2290.
Ethyl 3,4,6-Tri-O-benzoyl-2-O-benzyl-1-thio-β-d-galactopyranoside (1e).
A solution of ethyl 2-O-benzyl-1-thio-β-d-galactopyranoside (22,64 1.17 g, 3.72 mmol) in pyridine (20 mL) was cooled to 0 °C. Benzoyl chloride (2.59 mL, 22.33 mmol, 6.0 equiv) was added dropwise, followed by the addition of DMAP (90.4 mg, 0.74 mmol, 0.2 equiv). The external cooling was removed, and the resulting mixture was stirred under argon for 16 h at rt. Afterward, the reaction was quenched with MeOH (~8 mL), the volatiles were removed under reduced pressure, and the residue was coevaporated with toluene (3 × 5 mL). The resulting residue was dissolved in CH2Cl2 (~30 mL) and washed with 1 N aq. HCl (10 mL), sat. aq. NaHCO3 (10 mL), and water (2 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane 10% gradient elution) to afford the title compound as a white amorphous solid (2.28 g, 3.64 mmol, 98%). Analytical data for 1e: Rf = 0.70 (EtOAc/hexanes, 3/7 v/v); [α]23D + 103.3 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 8.03 (m, 4H, aromatic), 7.81 (d, 2H, aromatic), 7.71–7.37 (m, 7H, aromatic), 7.38–7.07 (m, 7H, aromatic), 5.93 (br d, 1H, H-4), 5.48 (dd, 1H, J3,4 = 3.2 Hz, H-3), 4.85 (d, 1H, 2J = 10.6 Hz, CHPh), 4.75 (d, 1H, J1,2 = 9.7 Hz, H-1), 4.67–4.57 (m, 2H, 2J = 11.0, J5,6a = 6.3 Hz, H-5, CHPh), 4.35 (dd, 1H, J6a,6b = 11.4 Hz, H-6a), 4.23 (dd, 1H, H-6b), 3.95 (dd, 1H, J2,3 = 9.6 Hz, H-2), 2.96–2.72 (m, 2H, SCH2CH3), 1.37 (t, 3H, SCH2CH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 166.0, 165.4, 165.3, 137.2, 133.5, 133.2, 133.1, 129.9 (×2), 129.7 (×2), 129.6 (×2), 129.4, 129.3, 129.2, 128.5, 128.4 (×2), 128.3, 128.2 (×5), 127.8, 85.7, 77.2, 76.2, 75.6, 74.5 (×2), 68.7, 62.2, 25.4, 15.2 ppm; HR-FAB MS [C36H34O8S + Na]+ calcd for 649.1867, found 649.1878.
Ethyl 4,6-Di-O-benzoyl-2,3-di-O-benzyl-1-thio-β-d-galactopyranoside (1f).
A solution of ethyl 2,3-di-O-benzyl-1-thio-β-d-galactopyranoside (23,63 1.06 g, 2.62 mmol) in pyridine (20 mL) was cooled to 0 °C. Benzoyl chloride (0.91 mL, 7.86 mmol, 3.0 equiv) was added dropwise, followed by the addition of DMAP (64.0 mg, 0.52 mmol, 0.2 equiv). The external cooling was removed, and the resulting mixture was stirred under argon for 16 h at rt. Afterward, the reaction was quenched with MeOH (~8 mL), the volatiles were removed under reduced pressure, and the residue was coevaporated with toluene. The resulting residue was dissolved in CH2Cl2 (~20 mL) and washed with 1 N aq. HCl (10 mL), sat. aq. NaHCO3 (10 mL), and water (2 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane 10% gradient elution) to afford the title compound as a white amorphous solid (1.43 g, 2.33 mmol, 90%). Analytical data for 1f: Rf = 0.75 (EtOAc/hexanes, 3/7, v/v); [α]23D +10.8 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 8.20–8.08 (m, 2H, aromatic), 8.03 (dd, 2H, aromatic), 7.58 (m, 2H, aromatic), 7.53–7.16 (m, 14H, aromatic), 5.90 (dd, 1H, H-4), 4.86 (d, 1H, 2J = 11.3 Hz, CHPh), 4.79 (dd, 2H, 2J = 10.1 Hz, CH2Ph), 4.62–4.52 (m, 3H, J6a,6b = 10.6 Hz, H-1, 6a, CHPh), 4.36 (dd, 1H, H-6b), 4.02 (m, 1H, J5,6a = 7.2, J5,6b = 6.3 Hz, H-5), 3.73 (m, 2H, J2,3 = 9.1, J3,4 = 2.8 Hz, H-2, 3), 2.94–2.63 (m, 2H, SCH2CH3), 1.34 (t, 3H, SCH2CH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 166.1, 165.7, 137.9, 137.5, 133.3 133.2, 130.0 (×3), 129.7 (×3), 129.5 (×2), 128.5, 128.4 (×2), 128.3 (×5), 128.0 (×2), 127.8, 127.7, 85.4, 80.9, 77.6, 75.9, 74.6, 71.9, 67.3, 62.8, 25.0, 15.1 ppm; HR-FAB MS [C36H36O7S + Na]+ calcd for 635.2074, found 635.2068.
Ethyl 3,4-Di-O-benzoyl-2,6-di-O-benzyl-1-thio-β-d-galactopyranoside (1g).
A solution of ethyl 2,6-di-O-benzyl-1-thio-β-d-galactopyranoside (24,65 3.10 g, 7.66 mmol) in pyridine (50 mL) was cooled to 0 °C. Benzoyl chloride (2.67 mL, 22.9 mmol, 3.0 equiv) was added dropwise, followed by the addition of DMAP (187 mg, 1.53 mmol, 0.2 equiv). The external cooling was removed, and the resulting mixture was stirred under argon for 16 h at rt. Afterward, the reaction was quenched with MeOH (~10 mL), the volatiles were removed under reduced pressure, and the residue was coevaporated with toluene. The resulting residue was dissolved in CH2Cl2 (~40 mL) and washed with 1 N aq. HCl (15 mL), sat. aq. NaHCO3 (15 mL), and water (2 × 15 mL). The organic phase was separated, dried with MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane 10% gradient elution) to afford the title compound as a clear syrup (4.22 g, 6.89 mmol, 90%). Analytical data for 1g: Rf = 0.75 (EtOAc/hexanes, 3/7, v/v); [α]23D +126.0 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 8.06–7.93 (m, 2H, aromatic), 7.86–7.74 (m, 2H, aromatic), 7.61 (d, 1H, aromatic), 7.53–7.42 (m, 3H, aromatic), 7.37–7.05 (m, 12H, aromatic), 5.88 (d, 1H, H-4), 5.43 (dd, 1H, J3,4 = 3.4 Hz, H-3), 4.83 (d, 1H, 2J = 10.6 Hz, CHPh), 4.69 (d, 1H, J1,2 = 9.7 Hz, H-1), 4.61 (d, 1H, 2J = 10.6 Hz, CHPh), 4.51 (d, 1H, 2J = 11.8 Hz, CHPh), 4.40 (d, 1H, 2J = 11.8 Hz, CHPh), 4.04 (m, 1H, J5,6a = 6.2 Hz, H-5), 3.89 (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.72–3.48 (m, 2H, H-6a, 6b), 2.93–2.76 (m, 2H, SCH2CH3), 1.38 (t, 3H, SCH2CH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.4 (×2), 137.5, 137.3, 133.3, 133.0, 129.8 (×3), 129.6 (×3), 129.5, 128.4 (×2), 128.3 (×2), 128.2 (×5), 127.7 (×3), 127.6, 85.7, 76.4, 76.0, 75.5, 74.7, 73.5, 68.9, 68.0, 25.3, 15.0 ppm; HR-FAB MS [C36H36O7S + Na]+ calcd for 635.2074, found 635.2078.
Synthesis of Disaccharides.
A General Procedure for Bromination of Thioglycosides followed by Glycosylation.
A mixture containing a thioglycoside precursor (0.047–0.050 mmol), which was dried in vacuo for 45 min, and activated molecular sieves (3 Å, 90 mg) in freshly distilled CH2Cl2 (1.0 mL) was stirred under argon for 30 min at rt. The resulting mixture was cooled to 0 °C, Br2 (0.062–0.065 mmol, 1.3 equiv) was added, and the reaction mixture was stirred under argon for 15 min at 0 °C. Afterward, the volatiles were removed under reduced pressure, and the residue was dried in vacuo for 30 min. A silver salt (1.50 equiv) and a glycosyl acceptor (0.038–0.040 mmol, 0.80 equiv) were added, and the resulting mixture was dried in vacuo for 1.5 h. Freshly distilled CH2Cl2 (1.0 mL) was added, and the resulting mixture was stirred under argon for 10 min at rt. The mixture was cooled to the temperature specified in tables, TMSOTf or TfOH (0.20–0.40 equiv) was added, and the resulting mixture was stirred under argon for the time and temperature specified in tables. Afterward, the solids were filtered off through a pad of Celite and rinsed successively with CH2Cl2. The combined filtrate (~25 mL) was washed with water (2 × 10 mL). The organic phase was separated, dried with magnesium sulfate, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to yield a corresponding disaccharide derivative in a yield and with a stereoselectivity specified in Tables 1–3 and below.
Methyl 6-O-(2,3,4,6-Tetra-O-benzoyl-β-d-galactopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (4a).
4a was obtained as described previously,50 and its analytical data were consistent with those reported previously.66
Methyl 6-O-(2,3,4,6-Tetra-O-benzyl-α/β-d-galactopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (4b).
4b was obtained as a colorless amorphous solid from galactosyl bromide donor 2b and acceptor 3 by the general glycosylation method (35 mg, 0.035 mmol, 87% yield, α/β = 1:1.2). The analytical data for 4b were consistent with those reported previously.67
Methyl 6-O-(6-O-Benzoyl-2,3,4-tri-O-benzyl-α/β-d-galactopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (4c).
4c was obtained as a colorless foam from galactosyl bromide donor 2c and acceptor 3 by the general glycosylation method (34 mg, 0.034 mmol, 75% yield, α/β = 1:2.7). Analytical data for 4c: Rf = 0.50 (EtOAc/hexane, 3/7, v/v); Selected 1H NMR data for α-4c (CDCl3 at 300 MHz): δ 4.94 (d, H-1′), 4.49 (d, H-1), 4.05 (dd, H-2′), 3.89 (dd, H-3), 3.22 (dd, H-2) ppm; Selected 1H NMR data for β-4c (CDCl3 at 300 MHz): δ 4.58 (d, J1,2 = 3.5 Hz, H-1), 4.33 (d, H-1′), 3.98 (dd, H-3) 3.90 (dd, H-2′), 3.52 (dd, H-3′), 3.48 (dd, H-2) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 166.0 (×2), 138.8, 138.6, 138.5, 138.3, 138.2, 138.1 (×2), 133.1, 133.0, 129.9, 129.7, 129.5 (×2), 128.4, 128.3 (×2), 128.2 (×2), 128.1, 127.9 (×2), 127.8, 127.7, 127.6 (×2), 127.5, 127.4, 104.1, 97.8, 97.5, 97.4, 82.2, 81.9, 80.0, 79.7, 79.1, 78.2, 78.0, 77.2, 75.6, 75.1, 74.9, 74.8, 74.5, 74.4, 73.3 (×2), 73.2, 73.1, 72.7, 72.0, 70.0, 69.8, 68.6, 66.2, 64.0, 63.1, 55.1, 54.9 ppm; HR-FAB MS [C62H64O12 + Na]+ calcd for 1023.4290, found 1023.4312.
Methyl 6-O-(4-O-Benzyl-2,3,6-tri-O-benzyl-α/β-d-galactopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (4d).
4d was obtained from thioglycoside donor 2d and acceptor 3 by the general glycosylation method (35 mg, 0.035 mmol, 87% yield, α/β = 6.0:1). The analytical data for 4d were consistent with those reported previously.56
Methyl 6-O-(3,4,6-Tri-O-benzoyl-2-O-benzyl-α-d-galactopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (4e).
4e was obtained from galactosyl bromide donor 2e and acceptor 3 by the general glycosylation method (38 mg, 0.037 mmol, 97% yield). Analytical data for 4e: Rf = 0.30 (EtOAc/hexane, 3/10, v/v); [α]23D +117.8 (c = 1.0 CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 7.94 (m, 4H, aromatic), 7.80 (d, 2H, aromatic), 7.66–7.05 (m, 29H, aromatic), 5.88 (d, 1H, H-4′, 5.72 (dd, 1H, J3′,4’ = 3.2 Hz, H-3′), 5.16 (d, 1H, J1′,2’ = 3.3 Hz, H-1′), 4.94 (2 d, 2H, 2J = 10.9, 2J = 11.2 Hz, 2 × CHPh), 4.81 (d, 1H, 2J = 10.9 Hz, CHPh), 4.71 (d, 1H, 2J = 12.2 Hz, CHPh), 4.65–4.54 (m, 5H, J1,2 = 3.4 Hz, H-1, 4 × CHPh), 4.53–4.36 (m, 2H, J5′,6a’ = 4.1, J6a’,6b’ = 10.2 Hz, H-5′, 6a′), 4.29 (dd, 1H, H-6b′), 4.11 (dd, 1H, J2′,3′ = 10.4 Hz, H-2′), 3.98 (dd, 1H, J3,4 = 9.2 Hz, H-3), 3.89–3.71 (m, 3H, J5,6a = 6.8, J6a,6b = 12.4 Hz, H-5, 6a, 6b), 3.55–3.28 (m, 5H, J2,3 = 3.4 Hz, H-2, 4, OCH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.9, 165.4 (×2), 138.7, 138.3, 138.0, 137.7, 133.3, 133.1, 133.0, 129.7 (×2), 129.6 (×5), 129.3, 128.4 (×9), 128.3 (×5), 128.1 (×2), 127.9 (×3), 127.8 (×3), 127.7 (×3), 127.6, 127.5, 97.7, 97.2, 82.0, 79.8, 77.8, 75.6, 75.0, 73.2, 73.0, 72.3, 70.1, 70.0, 69.5, 66.8, 66.1, 62.7, 55.1 ppm; HR-FAB MS [C62H60O14 + Na]+ calcd for 1051.3875, found 1051.3890.
Methyl 6-O-(4,6-Di-O-benzoyl-2,3-di-O-benzyl-α-d-galactopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (4f).
4f was obtained from galactosyl bromide donor 2f and acceptor 3 by the general glycosylation method (36 mg, 0.035 mmol, 93% yield, α/β = 33:1). Analytical data for α-4f: Rf = 0.55 (EtOAc/hexane, 3/7, v/v); [α]23D +80.4 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 8.10–7.91 (m, 4H, aromatic), 7.64–7.49 (m, 2H, aromatic), 7.47–7.10 (m, 29H, aromatic), 5.80 (br d, 1H, H-4′), 5.03 (d, 1H, J1′,2’ = 3.4 Hz, H-1′), 4.95 (d, 1H, 2J = 10.9 Hz, CHPh), 4.86–4.74 (m, 4H, 2J = 12.3 Hz, 4 × CHPh), 4.69 (d, 1H, 2J = 12.0 Hz, CHPh), 4.67–4.61 (dd, 2H, 2J = 11.9 Hz, CH2Ph), 4.59 (d, 1H, CHPh), 4.52 (dd, 2H, J1,2 = 3.4, 2J = 12.0 Hz, H-1, CHPh), 4.41–4.28 (m, 3H, H-5′, 6a′, 6b′), 4.03, (dd, 1H, J3′,4’ = 2.9 Hz, H-3′), 3.98–3.87 (m, 2H, J2′,3′ = 10.0 Hz, J3,4 = 9.2 Hz, H-2′, 3), 3.83–3.69 (m, 3H, H-5, 6a, 6b), 3.38 (dd 1H, H-4), 3.32–3.19 (m, 4H, J2,3 = 9.5 Hz, H-2, OCH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.9, 165.7, 138.7, 138.4, 138.3, 138.1, 137.9, 133.1 (×2), 129.9 (×2), 129.7, 129.6, 129.5 (×2) 128.4 (×7), 128.3, (×5), 128.2 (×4), 127.9 (×3), 127.8 (×3), 127.7 (×2), 127.6 (×3), 127.5, 127.4, 97.5 (×2), 82.0, 79.9, 78.0, 75.6, 75.1, 75.0, 74.8, 73.1, 72.9, 71.6, 70.0, 68.6, 67.1, 66.1, 63.2, 54.9 ppm; HR-FAB MS [C62H62O13 + Na]+ calcd for 1037.4083, found 1037.4094.
Methyl 6-O-(3,4-Di-O-benzoyl-2,6-di-O-benzyl-α-d-galactopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (4g).
4g was obtained from galactosyl bromide donor 2g and acceptor 3 by the general glycosylation method (37 mg, 0.036 mmol, 96% yield). Analytical data for 4g: Rf = 0.60 (EtOAc/hexane, 3/7, v/v); [α]23D + 132.5 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 7.88 (d, 2H, aromatic), 7.80 (d, 2H, aromatic), 7.59 (dd, 1H, aromatic), 7.54–7.10 (m, 30H, aromatic), 5.83 (d, 1H, H-4′), 5.70 (dd, 1H, J3′,4’ = 2.9 Hz, H-3′), 5.19 (d, 1H, J1′,2’ = 3.4 Hz, H-1′), 4.96 (dd, 2H, 2J = 11.0, Hz, CH2Ph), 4.83 (d, 1H, 2J = 11.0 Hz, CHPh), 4.71 (2 d, 2H, 2J = 11.2, 2J = 12.3 Hz, 2 × CHPh), 4.59 (m, 4H, H-1, 3 × CHPh,), 4.46 (d, 1H, 2J = 11.9 Hz, CHPh), 4.40–4.31 (m, 2H, H-5′, CHPh), 4.13–4.02 (dd, 1H, J2′,3′ = 10.4 Hz, H-2′), 3.99 (dd, 1H, J3,4 = 9.3 Hz, H-3), 3.92–3.75 (m, 3H, H-5, 6a, 6b), 3.63 (dd, 1H, H-4), 3.58–3.30 (m, 6H, J2,3 = 9.3 Hz, H-2, 6a’, 6b’, OCH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.4 (×2), 138.7, 138.3, 138.1, 137.8, 137.6, 133.1, 132.8, 129.7 (×3), 129.6 (×3), 128.4 (×7), 128.3 (×3), 128.2 (×3), 128.1, 128.0, 127.9 (×4), 127.8 (×4), 127.7, 127.6 (×2), 127.5 (×4), 97.9, 97.4, 82.0, 79.8, 77.7, 75.6, 75.0, 73.3, 73.2, 72.1, 70.4, 70.2, 69.8, 68.3, 67.6, 66.1, 55.1 ppm; HR-FAB MS [C62H62O13 + Na]+ calcd for 1037.4083, found 1037.4096.
Methyl 2-O-(4,6-Di-O-benzoyl-2,3-di-O-benzyl-α-d-galactopyranosyl)-3,4,6-tri-O-benzyl-α-d-glucopyranoside (10).
10 was obtained from galactosyl bromide donor 2f and acceptor 5 by the general glycosylation method (35 mg, 0.034 mmol, 91% yield). Analytical data for 10: Rf = 0.50 (EtOAc/hexane, 3/7, v/v); [α]23D +81.9 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 8.09–7.89 (m, 4H, aromatic), 7.60–7.52 (m, 1H, aromatic), 7.47–7.03 (m, 30H, aromatic), 5.33 (d, 1H, H-4′), 4.96 (d, 1H, J1′,2’ = 3.7 Hz, H-1′), 4.92 (d, 1H, J1,2 = 3.0 Hz, H-1), 4.74 (m, 6H, 2J = 11.1, 2J = 12.4 Hz, 6 × CHPh), 4.60–4.43 (m, 4H, 4 × CHPh), 4.39 (dd, 1H, J5′,6a’ = 3.5, J5′,6b’ = 8.1 Hz, H-5′), 4.22–4.02 (m, 2H, J3,4 = 9.2 Hz, H-3, 6a′), 4.00–3.88 (m, 3H, J3′,4’ = 2.9 Hz, H-2, 3′, 6b′), 3.88–3.73 (m, 3H, H-2′, 5, 6a), 3.69 (m, 1H, H-6b), 3.60 (dd, 1H, H-4), 3.47 (s, 3H, OCH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.8, 165.6, 138.7, 138.5, 138.0, 137.9, 137.8, 133.1, 132.9, 129.8 (×5), 129.7, 129.6, 128.4 (×9), 128.3 (×3), 128.2 (×2), 128.0 (×2), 127.9 (×2), 127.8, (×2), 127.7 (×2), 127.6, 127.5, 127.2 (×2), 96.3, 94.9, 80.7, 78.4, 77.2, 76.0, 75.6, 74.9, 74.6, 74.1, 73.6, 73.3, 71.8, 70.1, 68.5 (×2), 66.9, 63.3, 55.0, 53.4 ppm; HR-FAB MS [C62H62O13 + Na]+ calcd for 1037.4083, found 1037.4096.
Methyl 3-O-(4,6-Di-O-benzoyl-2,3-di-O-benzyl-α-d-galactopyranosyl)-2,4,6-tri-O-benzyl-α-d-glucopyranoside (11).
11 was obtained from galactosyl bromide donor 2f and acceptor 6 by the general glycosylation method (28 mg, 0.028 mmol, 73% yield). Analytical data for 11: Rf = 0.40 (EtOAc/hexane, 3/7, v/v); [α]23D +85.7 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 8.13–8.00 (m, 2H, aromatic), 8.00–7.88 (m, 2H, aromatic), 7.55 (m, 2H, aromatic), 7.41 (m, 5H, aromatic), 7.35–7.10 (m, 18H, aromatic), 7.10–6.90 (m, 6H, aromatic), 5.58 (br d, 2H, J1′,2’ = 3.4 Hz, H-1′, 4′), 5.00, (d, 1H, 2J = 11.7 Hz, CHPh), 4.84 (m, 2H, J5′,6a’ = 7.6, J5′,6b’ = 6.8, 2J = 10.9 Hz, H-5′, CHPh), 4.78–4.67 (m, 2H, J1,2 = 3.4, 2J = 11.6 Hz, H-1, CHPh), 4.65–4.38 (m, 7H, 7 × CHPh), 4.33 (dd, 1H, J3,4 = 9.0 Hz, H-3), 4.22 (dd, 1H, J6a’,6b’ = 12.0 Hz, H-6a′), 4.13 (dd, 1H, J3′,4’ = 3.0 Hz, H-3′), 4.00 (dd, 1H, H-6b′), 3.90 (dd, 1H, J2′,3′ = 10.2 Hz, H-2′), 3.83 (dd, 1H, J4,5 = 9.2 Hz, H-4), 3.78–3.54 (m, 4H, H-2, 5, 6a, 6b), 3.31 (s, 3H, OCH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 166.2, 165.7, 138.3, 137.9, 137.7 (×2), 137.6, 133.0, 132.7, 130.0 (×3), 129.8 (×2), 129.7, 128.4 (×9), 128.3 (×5), 128.2 (×2), 128.1 (×3), 128.0 (×3), 127.8, 127.6, 127.5, 127.4, 127.2, 126.7 (×3), 97.8, 97.3, 78.8, 78.7, 76.5, 74.8, 74.0, 73.9, 73.4, 72.7, 71.7, 69.7, 68.8, 68.2, 66.6, 63.8, 55.0 ppm; HR-FAB MS [C62H62O13 + Na]+ calcd for 1037.4083, found 1037.4096.
Methyl 4-O-(4,6-Di-O-benzoyl-2,3-di-O-benzyl-α-d-galactopyranosyl)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (12).
12 was obtained from galactosyl bromide donor 2f and acceptor 7 by the general glycosylation method (19 mg, 0.019 mmol, 49% yield). Analytical data for 12: Rf = 0.45 (EtOAc/hexane, 3/7, v/v); [α]23D +34.6 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 7.99 (m, 4H, aromatic), 7.57 (m, 2H, aromatic), 7.49–7.05 (m, 29H, aromatic), 5.70 (br d, 2H, J1′,2’ = 3.2 Hz, H-1′, 4′), 5.00–4.64 (m, 5H, 2J = 11.8 Hz, 5 × CHPh), 4.61–4.47 (m, 4H, J1,2 = 3.2 Hz, H-1, 3 × CHPh), 4.43 (2 d, 2H, 2J = 9.1, 2J = 11.6 Hz, 2 × CHPh), 4.37–4.27 (m, 1H, H-6a′), 4.22–3.97 (m, 3H, J5′,6a’ = 5.3 Hz, H-3, 5′, 6a’), 3.96–3.79 (m, 4H, J2′,3′ = 10.2 Hz, H-2′, 3′, 4, 5), 3.64 (dd, 2H, J6a,6b = 10.0 Hz, H-6a, 6b), 3.52 (dd, 1H, J2,3 = 9.4 Hz, H-2), 3.39 (s, 3H, OCH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.9, 165.7, 139.0, 137.9 (×2), 137.8, 133.1 (×2), 129.8 (×2), 129.6 (×5), 129.0, 128.4, (×9), 128.3, 128.1 (×5), 127.9 (×3), 127.8, 127.7, 127.6 (×2), 127.5, 127.4, 127.0, 126.7, 126.5, 125.2, 97.6, 97.5, 81.7, 79.9, 76.1, 74.2, 73.7, 73.6, 73.3, 73.1, 71.9, 69.3 (×2), 69.2, 68.3, 67.4, 63.1, 55.1 ppm; HR-FAB MS [C62H62O13 + Na]+ calcd for 1037.4083, found 1037.4096.
6-O-(4,6-Di-O-benzoyl-2,3-di-O-benzyl-α/β-d-galactopyranosyl)-1,2:3,4-di-O-isopropylidene-α-d-galactopyranose (13).
13 was obtained from galactosyl bromide donor 2f and acceptor 8 by the general glycosylation method (21 mg, 0.026 mmol, 68% yield, α/β = 11.0:1). The analytical data for 13 were consistent with those reported previously.58
Methyl 2,3,4-Tri-O-benzoyl-6-O-(4,6-di-O-benzoyl-2,3-di-O-benzyl-α-d-galactopyranosyl)-α-d-glucopyranoside (14).
14 was obtained from galactosyl bromide donor 2f and acceptor 9 by the general glycosylation method (35 mg, 0.033 mmol, 87% yield). Analytical data for 14: Rf = 0.40 (EtOAc/hexane, 3/7, v/v); [α]23D +100.31 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 8.14–7.77 (m, 10H, aromatic), 7.66–7.14 (m, 25H, aromatic), 6.05 (dd, 1H, J3,4 = 9.8 Hz, H-3), 5.85 (d, 1H, H-4′), 5.24 (dd, 1H, H-4), 5.13 (d, 1H, J1,2 = 3.7 Hz, H-1), 4.94 (dd, 1H, J2,3 = 9.9 Hz, H-2), 4.89–4.80 (m, 3H, J1′,2’ = 3.5 Hz, H-1′, 2 × CHPh), 4.67 (d, 2H, 2J = 11.2 Hz, 2 × CHPh), 4.47–4.36 (m, 2H, J5’,6b’ = 8.5 Hz, H-5′, 6a′), 4.30–4.20 (m, 2H, H-5, 6b′), 4.12 (dd, 1H, J3′,4’ = 2.8 Hz, H-3′), 3.99–3.84 (m, 2H, J2′,3′ = 10.0, J6a,6b = 10.6 Hz, H-2′, 6a), 3.58 (d, 1H, H-6b), 3.28 (s, 3H, OCH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.9, 165.7, 165.6, 165.5, 165.3, 138.2, 137.8, 133.4, 133.2 (×2), 133.0, 129.9 (×4), 129.8, 129.7 (×4), 129.6, 129.5 (×2), 129.1, 129.0, 128.7, 128.5 (×2), 128.4 (×3), 128.3 (×2), 128.2 (×6), 128.1 (×3), 127.8, (×3), 127.7, 127.5, 97.4, 96.4, 75.3, 74.6, 73.4, 71.9, 71.6, 70.4, 69.3, 68.7, 68.1, 67.3, 66.0, 63.3, 55.2 ppm; HR-FAB MS [C62H56O16 + Na]+ calcd for 1079.3461, found 1079.3471.
Methyl 2-O-(3,4-Di-O-benzoyl-2,6-di-O-benzyl-α-d-galactopyranosyl)-3,4,6-tri-O-benzyl-α-d-glucopyranoside (15).
15 was obtained from galactosyl bromide donor 2g and acceptor 5 by the general glycosylation method (36 mg, 0.035 mmol, 93% yield). Analytical data for 15: Rf = 0.45 (EtOAc/hexane, 3/7, v/v); [α]23D +209.2 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 7.93–7.79 (m, 4H, aromatic), 7.64–7.06 (m, 31H, aromatic), 5.85 (dd, 1H, J3′,4’ = 3.3 Hz, H-3′), 5.62 (br d, 1H, H-4′), 5.13 (d, 1H, J1′,2’ = 3.5 Hz, H-1′), 5.04–4.82 (m, 4H, J1,2 = 3.4, 2J = 11.1 Hz, H-1, 3 × CHPh), 4.73 (d, 1H, 2J = 12.4 Hz, CHPh), 4.63 (d, 2H, 2J = 12.2 Hz, 2 × CHPh), 4.53 (m, 3H, J5′,6a’ = 6.8, J5′,6b’ = 5.3, 2J = 12.0 Hz, H-5′, CH2Ph), 4.21–4.06 (m, 4H, J2′,3′ = 10.5 Hz, H-2′, 3, CH2Ph), 3.93 (dd, 1H, J2,3 = 9.9 Hz, H-2), 3.86–3.56 (m, 4H, H-4, 5, 6a, 6b), 3.46 (s, 3H, OCH3), 3.38 (dd, 1H, J6a’,6b’ = 10.4 Hz, H-6a′), 3.17 (dd, 1H, H-6b’) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.4, 165.3, 138.2, 138.0 (×2), 137.8, 133.1, 132.8, 129.8, 129.7, 129.6 (×2), 128.5, (×3), 128.4 (×8), 128.3 (×2), 128.2 (×4), 128.0, 127.9 (×5), 127.8, (×2), 127.7 (×4), 127.3 (×3), 127.2, 96.4, 94.8, 80.6, 78.2, 75.0, 74.7, 73.5, 73.0, 72.7, 72.5, 70.4, 70.2, 69.8, 68.4, 68.3, 67.0, 55.0 ppm; HR-FAB MS [C62H62O13 + Na]+ calcd for 1037.4190, found 1037.4095.
Methyl 3-O-(3,4-Di-O-benzoyl-2,6-di-O-benzyl-α-d-galactopyranosyl)-2,4,6-tri-O-benzyl-α-d-glucopyranoside (16).
16 was obtained from galactosyl bromide donor 2g and acceptor 6 by the general glycosylation method (27 mg, 0.027 mmol, 70% yield). Analytical data for 16: Rf = 0.50 (EtOAc/hexane, 3/7, v/v); [α]23D + 114.4 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 7.84 (dd, 4H, aromatic), 7.64–6.90 (m, 31H, aromatic), 5.92 (dd, 1H, J3′,4’ = 3.2 Hz, H-3′), 5.74 (dd, 2H, J1′,2’ = 3.3 Hz, H-1′, 4′), 5.03–4.85 (m, 2H, J5′,6a’ = 6.1, J5′,6b’ = 6.7, 2J = 11.8 Hz, H-5′, CHPh), 4.78 (d, 1H, 2J = 11.3 Hz, CHPh), 4.68–4.42 (m, 3H, J1,2 = 3.7 Hz, H-1, CH2Ph), 4.52–4.40 (m, 4H, 4 × CHPh), 4.38–4.17 (m, 3H, J3,4 = 9.1, 2J = 12.0, Hz, H-3, CH2Ph), 4.12 (dd, 1H, J2′,3′ = 10.7 Hz, H-2′), 3.88–3.57 (m, 5H, H-2, 4, 5, 6a, 6b), 3.50 (dd, 1H, J6a’,6b’ = 10.1 Hz, H-6a′), 3.34 (d, 1H, H-6b′), 3.31 (s, 3H, OCH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.5, 165.4, 138.4, 138.0, 137.8, 137.7, 137.2, 133.0, 132.8, 129.9, 129.8, 129.7 (×2), 129.6 (×2), 128.7 (×2), 128.6 (×2), 128.3 (×5), 128.2 (×4), 128.1 (×4), 128.0 (×4), 127.8 (×2), 127.7 (×2), 127.6, 127.5, 127.2, 127.1, 126.6, 97.7, 97.5, 78.9, 78.6, 77.2, 76.1, 73.5 (×2), 73.4, 73.2, 72.8, 70.5, 70.3, 69.6, 68.3, 67.1, 55.0 ppm; HR-FAB MS [C62H62O13 + Na]+ calcd for 1037.4083, found 1037.4094.
Methyl 4-O-(3,4-Di-O-benzoyl-2,6-di-O-benzyl-α-d-galactopyranosyl)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (17).
17 was obtained from galactosyl bromide donor 2g and acceptor 7 by the general glycosylation method (25 mg, 0.025 mmol, 65% yield). Analytical data for 17: Rf = 0.45 (EtOAc/hexanes, 3/7, v/v); [α]23D +114.0 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 7.90–7.83 (m, 2H, aromatic), 7.80–7.72 (m, 2H, aromatic), 7.63–7.56 (m, 1H, aromatic), 7.51–7.38 (m, 3H, aromatic), 7.35–7.23 (m, 18H, aromatic), 7.17–6.94 (m, 9H, aromatic), 5.84 (br. d, 2H, J1′,2’ = 3.6 Hz, H-1′, 4′), 5.70 (dd, 1H, J3′,4’ = 2.4 Hz, H-3′), 5.05 (d, 1H, 2J = 11.9 Hz, CHPh), 4.84 (d, 1H, 2J = 11.9 Hz, CH2Ph), 4.74–4.38 (m, 7H, J1,2 = 3.6 Hz, H-1, 6 × CHPh), 4.32–4.22 (m, 2H, 2J = 12.6 Hz, H-5′, CHPh), 4.14–4.07 (m, 3H, H-3, 4, CHPh), 4.01 (dd, 1H, J2′,3′ = 10.6 Hz, H-2′), 3.97–3.85 (m, 2H, J5,6b = 9.2 Hz, H-5, 6a), 3.70 (dd, 1H, H-6b), 3.60 (dd, 1H, J2,3 = 9.3 Hz, H-2), 3.45–3.29 (m, 5H, H-6a′, 6b′, OCH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.5, 165.3, 139.2, 138.4, 138.2, 138.0, 137.9, 137.8, 137.7, 137.5, 137.3, 137.2, 133.0, 132.8, 129.7, 129.6, 128.7, 128.5, 128.3 (×2), 128.2, 128.1 (×2), 128.0 (×2), 127.8 (×3), 127.7, 127.6, 127.5, 127.4, 127.3, 127.2, 127.1, 126.9, 126.6, 126.5, 97.7, 97.6, 97.3, 81.8, 80.1, 78.9, 78.7, 76.1, 74.2, 73.4, 73.2, 73.0, 72.8, 70.5, 70.3, 70.1, 69.6, 69.3, 69.0, 68.3, 68.1, 67.8, 67.1, 55.1 ppm; HR-FAB MS [C62H62O13 + Na]+ calcd for 1037.4083, found 1037.4095.
6-O-(3,4-Di-O-benzoyl-2,6-di-O-benzyl-α-d-galactopyranosyl)-1,2:3,4-di-O-isopropylidene-α-d-glucopyranose (18).
18 was obtained from galactosyl bromide donor 2g and acceptor 8 by the general glycosylation method (23 mg, 0.028 mmol, 75% yield). Analytical data for 18: Rf = 0.45 (EtOAc/hexane, 3/7, v/v); [α]23D +58.6 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 7.90 (d, 2H, aromatic), 7.82 (d, 2H, aromatic), 7.64–7.56 (m, 1H, aromatic), 7.53–7.39 (m, 3H, aromatic), 7.36–7.11 (m, 12H, aromatic), 5.88 (d, 1H, H-4′), 5.74 (dd, 1H, J3′,4’ = 2.8 Hz, H-3′), 5.54 (d, 1H, J1,2 = 5.0 Hz, H-1), 5.18 (d, 1H, J1′,2’ = 3.5 Hz, H-1′), 4.70–4.57 (m, 3H, J3,4 = 8.0, Hz, H-3, 5′, CHPh), 4.54–4.37 (m, 4H, 2J = 12.0 Hz, H-4, 3 × CHPh), 4.32 (dd, 1H, J2,3 = 2.3 Hz, H-2), 4.14–4.05 (m, 2H, J2′,3′ = 10.5, J5,6a = 6.0, J5,6b = 8.0 Hz, H-2′, 5), 3.92 (dd, 1H, J6a,6b = 10.0 Hz, H-6a), 3.87–3.75 (m, 1H, H-6b), 3.62–3.48 (m, 2H, H-6a’, 6b′), 1.61, 1.46, 1.37, 1.24 (4 s, 12H, 4 × CH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.4 (×2), 137.8, 137.6, 133.1, 132.8, 129.8, 129.7 (×3), 129.6 (×2), 128.4 (×2), 128.2 (×6), 128.1, 127.6 (×4), 127.5, 109.0, 108.7, 97.7, 96.2, 77.2, 73.4, 73.3, 72.1, 70.6 (×2), 70.5, 70.2, 69.8, 68.1, 67.6, 66.7, 65.9, 26.0, 24.9, 24.5 ppm; HR-FAB MS [C46H50O13 + Na]+ calcd for 833.3144, found 833.3152.
Methyl 2,3,4-Tri-O-benzoyl-6-O-(3,4-di-O-benzoyl-2,6-di-O-benzyl-α-d-galactopyranosyl)-α-d-glucopyranoside (19).
19 was obtained from galactosyl bromide donor 2g and acceptor 9 by the general glycosylation method (39 mg, 0.037 mmol, 97% yield). Analytical data for 19: Rf = 0.35 (EtOAc/hexane, 3/7, v/v); [α]23D +154.7 (c = 1.0, CHCl3); 1H NMR (CDCl3 at 300 MHz): δ 8.11–7.75 (m, 10H, aromatic), 7.67–7.09 (m, 25H, aromatic), 6.18 (dd, 1H, J3,4 = 8.7 Hz, H-3), 5.87 (br d, 1H, H-4′), 5.77 (dd, 1H, J3′,4’ = 3.0 Hz, H-3′), 5.52 (dd, 1H, H-4), 5.27 (dd, 2H, J1,2 = 3.4, J2,3 = 10.8 Hz, H-1, 2), 4.98 (d, 1H, J1′,2′ = 3.4 Hz, H-1′), 4.76–4.28 (m, 6H, J5,6a = 7.3, J5,6b = 9.1, 2J = 12.4 Hz, H-5, 5′, 4 × CHPh), 4.11 (dd, 1H, J2′,3′ = 10.4 Hz, H-2′), 3.96 (dd, 1H, J6a,6b = 10.9 Hz, H-6a), 3.65 (d, 1H, H-6b), 3.57–3.39 (m, 5H, H-6a′, 6b′ OCH3) ppm; 13C{1H} NMR (CDCl3 at 75 MHz): δ 165.7 (×2), 165.4, 165.3 (×2), 137.8, 137.6, 133.4, 133.3, 133.1, 133.0, 132.8, 129.9 (×5), 129.8, 129.7 (×2), 129.6, (×5), 129.1, 129.0, 128.8, 128.3 (×4), 128.2 (×8), 128.1 (×2), 127.9, 127.7, 127.5 (×2), 127.4, 97.4, 96.7, 73.5, 73.2, 72.8, 72.1, 70.4, 70.2, 69.8, 69.6, 68.5, 68.0, 67.6, 66.7, 55.6 ppm; HR-FAB MS [C62H56O16 + Na]+ calcd for 1079.3461, found 1079.3471.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by awards from the NIGMS (GM111835) and the NSF (CHE-1058112). We thank Dr. Rensheng Luo (UM—St. Louis) for help with aquiring spectral data using an NMR spectrometer that was purchased thanks to the NSF (Award CHE-0959360).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c01279.
Characterization data for all new compounds (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.0c01279
The authors declare no competing financial interest.
DEDICATION
In loving memory of Kyle Dewayne Robert Hill and Lester Gene Shadrick.
Contributor Information
Melanie Shadrick, Department of Chemistry and Biochemistry, University of Missouri—St. Louis, St. Louis, Missouri 63121, United States.
Yashapal Singh, Department of Chemistry and Biochemistry, University of Missouri—St. Louis, St. Louis, Missouri 63121, United States.
Alexei V. Demchenko, Department of Chemistry and Biochemistry, University of Missouri—St. Louis, St. Louis, Missouri 63121, United States.
REFERENCES
- (1).Michael A On the synthesis of helicin and phenolglucoside. Am. Chem. J 1879, 1, 305–312. [Google Scholar]
- (2).Koenigs W; Knorr E Über einige derivate des traubenzuckers und der galactose. Ber. Dtsch. Chem. Ges 1901, 34, 957–981. [Google Scholar]
- (3).Fischer E; Armstrong EF Over isomers of the aceto-halogen derivative of grape sugar and the synthesis of the glucosides. Ber. Dtsch. Chem. Ges 1901, 34, 2885–2900. [Google Scholar]
- (4).Helferich B; Bohn E; Winkler S Unsaturated derivatives of gentiobiose and cellobiose. Ber. Dtsch. Chem. Ges. B 1930, 63B, 989–998. [Google Scholar]
- (5).Hudson C Relations between rotatory power and structure in the sugar group. XXV. The ring structures of various monosaccharides. J. Am. Chem. Soc 1930, 52, 1680–1700. [DOI] [PubMed] [Google Scholar]
- (6).Isbell H The structures of the acetylmethylmannosides. J. Am. Chem. Soc 1930, 52, 5298–5298. [Google Scholar]
- (7).Perlin A Formation of diastereomeric orthoacetates of d-mannose: N.M.R. spectral evidence. Can. J. Chem 1963, 41, 399–406. [Google Scholar]
- (8).Nigudkar SS; Demchenko AV Stereocontrolled 1,2-cis glycosylation as the driving force of progress in synthetic carbohydrate chemistry. Chem. Sci 2015, 6, 2687–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Helferich B; Bredereck H Zuckersynthesen. VIII. Liebigs Ann. Chem 1928, 465, 166–184. [Google Scholar]
- (10).Zemplén G; Pacsu E Über die Verseifung acetylierter Zucker und verwandter Substanzen. Ber. Dtsch. Chem. Ges. B 1929, 62, 1613–1614. [Google Scholar]
- (11).Zemplen G; Gerecs A Action of mercury salts on acetohalogenosugars. IV. Direct preparation of alkyl biosides of the α-series. Ber. Dtsch. Chem. Ges. B 1930, 63B, 2720–2729. [Google Scholar]
- (12).Helferich B; Zirner J Synthesis of tetra-O-acetyhexoses with a free 2-hydroxyl group. Synthesis of disaccharides. Chem. Ber 1962, 95, 2604–2611. [Google Scholar]
- (13).Fischer E; von Mechel L Synthesis of phenyl glucosides. Ber. Dtsch. Chem. Ges 1916, 49, 2813–2820. [Google Scholar]
- (14).Helferich B; Doppstadt A; Gottschlich A Synthesen von Glykosiden im homogenen Medium. Naturwissenschaften 1953, 40, 441–442. [Google Scholar]
- (15).Lemieux R; Morgan A The mechanism for the formation of 1, 2-cis-pyridine nucleosides from 1, 2-cis-acetohalogenosugars. A novel rearrangement. J. Am. Chem. Soc 1963, 85, 1889–1890. [Google Scholar]
- (16).Yu F; Li J; DeMent PM; Tu Y-J; Schlegel HB; Nguyen HM Phenanthroline-Catalyzed Stereoretentive Glycosylations. Angew. Chem., Int. Ed 2019, 58, 6957–6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ishikawa T; Fletcher HG The synthesis and solvolysis of some d-glucopyranosyl bromides having a benzyl group at C-2. J. Org. Chem 1969, 34, 563–571. [DOI] [PubMed] [Google Scholar]
- (18).Frechet JM; Schuerch C Solid-phase synthesis of oligosaccharides. II. Steric control by C-6 substituents in glucoside syntheses. J. Am. Chem. Soc 1972, 94, 604–609. [Google Scholar]
- (19).Kronzer FJ; Schuerch C The methanolysis of some derivatives of 2,3,4-tri-O-benzyl-J-d-glucopyranosyl bromide in the presence and absence of silver salts. Carbohydr. Res 1973, 27, 379–390. [Google Scholar]
- (20).Lemieux RU; Hendriks KB; Stick RV; James K Halide ion catalyzed glycosylation reactions. Syntheses of -linked disaccharides. J. Am. Chem. Soc 1975, 97, 4056–4062 and references therein. [Google Scholar]
- (21).Lemieux RU; Driguez H The chemical synthesis of 2-O-(-L-fucopyranosyl)-3-O-(-d-galactopyranosyl)-d-galactose. The terminal structure of blood-group B antigenic determinant. J. Am. Chem. Soc 1975, 97, 4069–4075. [DOI] [PubMed] [Google Scholar]
- (22).Lemieux R; Driguez H Chemical synthesis of 2-acetamido-2-deoxy-4-O-(alpha-L-fucopyranosyl)-3-O-(beta-D-galactopyranosyl)-D-glucose. Lewis a blood-group antigenic determinant. J. Am. Chem. Soc 1975, 97, 4063–4069. [DOI] [PubMed] [Google Scholar]
- (23).West AC; Schuerch C The reverse anomeric effect and the synthesis of -glycosides. J. Am. Chem. Soc 1973, 95, 1333–1335. [Google Scholar]
- (24).Kronzer FJ; Schuerch C The use of 2,3,4,6-tetra-O-benzyl-o-D-glycopyranosyl iodides in –glycoside synthesis. Carbohydr. Res 1974, 34, 71–78 and references therein. [Google Scholar]
- (25).Higashi K; Nakayama K; Uoto K; Shioya E; Kusama T A novel glycoside anomerization catalyzed by trimethylsilyl bromide and zinc bromide in combination. Chem. Pharm. Bull 1991, 39, 590–592. [Google Scholar]
- (26).Higashi K; Nakayama K; Shioya E; Kusama T Direct transformation of O-glycosides into other O-glycosides using trimethylsilyl bromide and zinc bromide. Chem. Pharm. Bull 1992, 40, 1042–1043. [Google Scholar]
- (27).Nakayama K; Higashi K; Soga T; Uoto K; Kusama T Zinc triflate-promoted glycosidation: synthesis of lipid A disaccharide intermediates. Chem. Pharm. Bull 1992, 40, 2909–2912. [DOI] [PubMed] [Google Scholar]
- (28).Kihlberg JO; Leigh DA; Bundle DR The in situ activation of thioglycosides with bromine: an improved glycosylation method. J. Org. Chem 1990, 55, 2860–2863. [Google Scholar]
- (29).Kaeothip S; Yasomanee JP; Demchenko AV Glycosidation of thioglycosides in the presence of bromine: mechanism, reactivity, and stereoselectivity. J. Org. Chem 2012, 77, 291–299. [DOI] [PubMed] [Google Scholar]
- (30).Panza M; Civera M; Yasomanee JP; Belvisi L; Demchenko AV Bromine-promoted glycosidation of conformationally superarmed thioglycosides. Chem. - Eur. J 2019, 25, 11831–11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Imamura A; Ando H; Korogi S; Tanabe G; Muraoka O; Ishida H; Kiso M Di-tert-butylsilylene (DTBS) group-directed α-selective galactosylation unaffected by C-2 participating functionalities. Tetrahedron Lett. 2003, 44, 6725–6728. [Google Scholar]
- (32).Imamura A; Ando H; Ishida H; Kiso M Di-tertbutylsilylene-Directed a-Selective Synthesis of 4-Methylumbelliferyl T-Antigen. Org. Lett 2005, 7, 4415–4418. [DOI] [PubMed] [Google Scholar]
- (33).Demchenko AV; Rousson E; Boons GJ Stereoselective 1,2-cis-galactosylation assisted by remote neighboring group participation and solvent effects. Tetrahedron Lett. 1999, 40, 6523–6526. [Google Scholar]
- (34).Kalikanda J; Li Z Study of the stereoselectivity of 2-azido-2-deoxygalactosyl donors: remote protecting group effects and temperature dependency. J. Org. Chem 2011, 76, 5207–5218. [DOI] [PubMed] [Google Scholar]
- (35).Komarova BS; Tsvetkov YE; Nifantiev NE Design of α-Selective Glycopyranosyl Donors Relying on Remote Anchimeric Assistance. Chem. Record 2016, 16, 488–506. [DOI] [PubMed] [Google Scholar]
- (36).Zhang Y; Zhou S; Wang X; Zhang H; Guo Z; Gao J A new method for α-specific glucosylation and its application to the one-pot synthesis of a branched α-glucan. Org. Chem. Front 2019, 6, 762–772. [Google Scholar]
- (37).Liu H; Hansen T; Zhou SY; Wen GE; Liu XX; Zhang QJ; Codee JDC; Schmidt RR; Sun JS. Dual-Participation Protecting Group Solves the Anomeric Stereocontrol Problems in Glycosylation Reactions. Org. Lett 2019, 21, 8713–8717. [DOI] [PubMed] [Google Scholar]
- (38).Kim JH; Yang H; Park J; Boons GJ A general strategy for stereoselective glycosylations. J. Am. Chem. Soc 2005, 127, 12090–12097. [DOI] [PubMed] [Google Scholar]
- (39).Kim JH; Yang H; Khot V; Whitfield D; Boons GJ Stereoselective glycosylations using (R)- or (S)-(ethoxycarbonyl)-benzyl chiral auxiliaries at C-2 of glycopyranosyl donors. Eur. J. Org. Chem 2006, 2006, 5007–5028. [Google Scholar]
- (40).Boltje TJ; Kim JH; Park J; Boons GJ Chiral-auxiliary-mediated 1,2-cis-glycosylations for the solid-supported synthesis of a biologically important branched α-glucan. Nat. Chem 2010, 2, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Mensink RA; Boltje TJ Advances in Stereoselective 1,2-cis Glycosylation using C-2 Auxiliaries. Chem. - Eur. J 2017, 23, 17637–17653. [DOI] [PubMed] [Google Scholar]
- (42).Yasomanee JP; Demchenko AV The effect of remote picolinyl and picoloyl substituents on the stereoselectivity of chemical glycosylation. J. Am. Chem. Soc 2012, 134, 20097–20102. [DOI] [PubMed] [Google Scholar]
- (43).Mensah EA; Yu F; Nguyen HM Nickel-catalyzed stereoselective glycosylation with C(2)-N-substituted benzylidene D-glucosamine and galactosamine trichloroacetimidates for the formation of 1,2-cis-2-amino glycosides. Applications to the synthesis of heparin disaccharides, GPI anchor pseudodisaccharides, and a-GalNAc. J. Am. Chem. Soc 2010, 132, 14288–14302. [DOI] [PubMed] [Google Scholar]
- (44).Sletten ET; Tu Y-J; Schlegel HB; Nguyen HM Are Brønsted Acids the True Promoter of Metal-Triflate-Catalyzed Glycosylations? A Mechanistic Probe into 1, 2-Cis-Aminoglycoside Formation by Nickel Triflate. ACS Catal 2019, 9, 2110–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bradshaw G; Colgan A; Allen N; Pongener I; Boland M; Ortin Y; McGarrigle E Stereoselective organocatalyzed glycosylations-thiouracil, thioureas and monothiophthalimide act as Brønsted acid catalysts at low loadings. Chem. Sci 2019, 10, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Park J; Kawatkar S; Kim JH; Boons GJ Stereoselective glycosylations of 2-azido-2-deoxy-glucosides using intermediate sulfonium ions. Org. Lett 2007, 9, 1959–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lu SR; Lai YH; Chen JH; Liu CY; Mong KK Dimethylformamide: an unusual glycosylation modulator. Angew. Chem., Int. Ed 2011, 50, 7315–7320. [DOI] [PubMed] [Google Scholar]
- (48).Sun L; Wu X; Xiong DC; Ye XS Stereoselective Koenigs-Knorr glycosylation catalyzed by urea. Angew. Chem., Int. Ed 2016, 55, 8041–8044. [DOI] [PubMed] [Google Scholar]
- (49).Park Y; Harper KC; Kuhl N; Kwan EE; Liu RY; Jacobsen EN Macrocyclic bis-thioureas catalyze stereospecific glycosylation reactions. Science 2017, 355, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Singh Y; Demchenko AV Koenigs-Knorr Glycosylation Reaction Catalyzed by Trimethylsilyl Trifluoromethanesulfonate. Chem. - Eur. J 2019, 25, 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Singh Y; Demchenko AV Defining the scope of the acid-catalyzed glycosidation of glycosyl bromides. Chem. - Eur. J 2020, 26, 1042–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Pistorio SG; Yasomanee JP; Demchenko AV Hydrogen bond-mediated aglycone delivery: focus on β-mannosylation. Org. Lett 2014, 16, 716–719. [DOI] [PubMed] [Google Scholar]
- (53).Yasomanee JP; Demchenko AV Hydrogen bond-mediated aglycone delivery: the synthesis of linear and branched α-glucans. Angew. Chem., Int. Ed 2014, 53, 10453–10456. [DOI] [PubMed] [Google Scholar]
- (54).Ranade SC; Kaeothip S; Demchenko AV Glycosyl alkoxythioimidates as complementary building blocks for chemical glycosylation. Org. Lett 2010, 12, 5628–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lourenço EC; Ventura MR The effect of electron withdrawing protecting groups at positions 4 and 6 on 1,2-cis galactosylation. Tetrahedron 2013, 69, 7090–7097. [Google Scholar]
- (56).Baek JY; Kwon H-W; Myung SJ; Park JJ; Kim MY; Rathwell DCK; Jeon HB; Seeberger PH; Kim KS Directing effect by remote electron-withdrawing protecting groups at O-3 or O-4 position of donors in glucosylations and galactosylations. Tetrahedron 2015, 71, 5315–5320. [Google Scholar]
- (57).Marianski M; Mucha E; Greis K; Moon S; Pardo A; Kirschbaum C; Thomas D; Meijer G; von Helden G; Gilmore K; Seeberger P; Pagel K Direct Evidence for Remote Participation in Galactose Building Blocks during Glycosylations Revealed by Cryogenic Vibrational Spectroscopy. Angew. Chem., Int. Ed 2020, 59, 6166–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Cheng YP; Chen HT; Lin CC A convenient and highly stereoselective approach for -galactosylation performed by galactopyranosyl dibenzyl phosphite with remote participating groups. Tetrahedron Lett. 2002, 43, 7721–7723. [Google Scholar]
- (59).Bandara MD; Yasomanee JP; Demchenko AV: Application of armed, disarmed, superarmed and superdisarmed building blocks in stereocontrolled glycosylation and expeditious oligosaccharide synthesis. In Selective Glycosylations: Synthetic Methods and Catalysts; Bennett CS, Ed.; Wiley: 2017; pp 29–58. [Google Scholar]
- (60).Zhang F; Zhang W; Zhang Y; Curran DP; Liu G Synthesis and Applications of a Light-Fluorous Glycosyl Donor. J. Org. Chem 2009, 74, 2594–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Dondoni A; Marra A; Scherrmann M-C; Casnati A; Sansone F; Ungaro R Synthesis and properties of O-glycosyl calix[4]arenes (calixsugars). Chem. - Eur. J 1997, 3, 1774–1782. [Google Scholar]
- (62).Yan S; Ding N; Zhang W; Wang P; Li Y; Li M An efficient and recyclable catalyst for the cleavage of tert-butyldiphenylsilyl ethers. Carbohydr. Res 2012, 354, 6–20. [DOI] [PubMed] [Google Scholar]
- (63).Garegg PJ; Kvarnstrom I; Niklasson A; Niklasson G; Svensson SCT Partial substitution of thioglycosides by phase transfer catalyzed benzoylation and benzylation. J. Carbohydr. Chem 1993, 12, 933–953. [Google Scholar]
- (64).Gao PC; Zhu SY; Cao H; Yang JS Total Synthesis of Marine Glycosphingolipid Vesparioside B. J. Am. Chem. Soc 2016, 138, 1684–1688. [DOI] [PubMed] [Google Scholar]
- (65).Agoston K; Kerekgyarto J; Hajko J; Batta G; Lefeber DJ; Kamerling JP; Vliegenthart JFG Synthesis of fragments of the glycocalyx glycan of the parasite Schistosoma mansoni. Chem. - Eur. J 2002, 8, 151–161. [DOI] [PubMed] [Google Scholar]
- (66).Codee JDC; Van den Bos LJ; Litjens REJN; Overkleeft HS; Van Boeckel CAA; Van Boom JH; Van der Marel GA Chemoselective glycosylations using sulfonium triflate activator systems. Tetrahedron 2004, 60, 1057–1064. [Google Scholar]
- (67).Vibhute AM; Dhaka A; Athiyarath V; Sureshan KM A versatile glycosylation strategy via Au (III) catalyzed activation of thioglycoside donors. Chem. Sci 2016, 7, 4259–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.