

Abstract
Various species of the intestinal microbiota have been associated with the development of colorectal cancer (CRC)1,2, yet a direct role of bacteria in the occurrence of oncogenic mutations has not been established. Escherichia coli can carry the pathogenicity island pks, which encodes a set of enzymes that synthesize colibactin3. This compound is believed to alkylate DNA on adenine residues4,5 and induces double strand breaks in cultured cells3. Here, we expose human intestinal organoids to genotoxic pks+ Escherichia coli by repeated luminal injection over a period of 5 months. Whole genome sequencing of clonal organoids before and after this exposure reveals a distinct mutational signature, absent from organoids injected with isogenic pks-mutant bacteria. The same mutational signature is detected in a subset of 5876 human cancer genomes from two independent cohorts, predominantly in CRC. Our study describes a distinct mutational signature in CRC and implies that the underlying mutational process directly results from past exposure to bacteria carrying the colibactin-producing pks pathogenicity island.
The intestinal microbiome has long been suggested to be involved in colorectal cancer (CRC) tumorigenesis1,2. Various bacterial species are reportedly enriched in stool and biopsies of CRC patients6–9, including genotoxic strains of Escherichia coli (E. coli)3,6,10,11. The genome of these genotoxic E. coli harbors a 50 kb hybrid polyketide-nonribosomal peptide synthase operon (pks, also referred to as clb) responsible for the production of the genotoxin colibactin. pks+ E. coli are present in a significant fraction of individuals (~20% healthy individuals, ~40% inflammatory bowel disease, ~60% familial adenomatous polyposis and CRC)6,10,11. pks+ E. coli induce - amongst others - interstrand crosslinks (ICLs) and double strand breaks (DSBs) in epithelial cell lines3,10–12 and in gnotobiotic mouse models of CRC, in which they can also contribute to tumorigenesis6,10,11. Recently, two studies have reported colibactin-adenine adducts, which are formed in mammalian cells exposed to pks+ E. coli4,5. While the chemistry of colibactin’s interaction with DNA is thus well-established, the outcome of this process in terms of recognizable mutations remains to be determined. Recent advances in sequencing technologies and the application of novel mathematical approaches allow classification of somatic mutational patterns. Stratton and colleagues have pioneered a mutational signature analysis which includes the bases immediately 5′ and 3′ to the single base substitution (SBS), and a number of different contexts characterizing insertions and deletions (indels)13,14. More than 50 mutational signatures have thus been defined in cancers. For some, the underlying causes (e.g. tobacco smoke, UV light, specific genetic DNA repair defects) are known13,15,16. However, for many the underlying etiology remains unclear. Human intestinal organoids, established from primary crypt stem cells17, have been useful to identify underlying causes of mutational signatures18: After being exposed to a specific mutational agent in culture, the organoids can be subcloned and analyzed by Whole Genome Sequencing (WGS) to reveal the consequent mutational signature16,19,20.
In order to define the mutagenic characteristics of pks+ E. coli, we developed a co-culture protocol in which a pks+ E. coli strain (originally derived from a CRC biopsy21) was microinjected into the lumen of clonal human intestinal organoids22 (Fig. 1a, b). An isogenic clbQ knock-out strain, incapable of producing active colibactin21,23, served as negative control. Both bacterial strains were viable for at least 3 days in co-culture and followed similar growth dynamics (Fig. 1c). DSBs and ICLs, visualized by γH2AX and FANCD2 immunofluorescence, were induced specifically in epithelial cells exposed to pks+ E. coli (Fig. 1d, e, Extended Data Fig. 1a), confirming that pks+ E. coli induced DNA damage in our model. This co-culture induced no significant viability difference between organoids exposed to pks+ and pksΔclbQ E. coli, although there was a modest decrease when compared to the dye-only injected organoids (Extended Data Fig. 1b, c).We then performed repeated injections (with pks+ E. coli, pksΔclbQ E. coli or dye-only) into single cell-derived organoids, in order to achieve long-term exposure over a period of 5 months. Subsequently, sub-clonal organoids were established from individual cells extracted from the exposed organoids. For each condition, three subclones were subjected to WGS (Fig. 2a). We also subjected the original clonal cultures to WGS to subtract the somatic mutations that were already present before co-culture. Organoids exposed to pks+ E. coli presented increased SBS levels compared to pksΔclbQ, with a bias towards T>N substitutions (Fig. 2b). These T>N substitutions occurred preferentially at ATA, ATT and TTT (of which the middle base is mutated). From this, we defined a pks-specific single base substitution signature (SBS-pks; Fig. 2c). This mutational signature was not observed in organoids exposed to pksΔclbQ E. coli or dye (Fig. 2b, c, Extended Data Fig. 2a–c), proving this to be a direct consequence of the pks+ E. coli exposure. Furthermore, exposure to pks+ E. coli induced a characteristic small indel signature (ID-pks), which was characterized by single T deletions at T homopolymers (Fig. 2d, e, Extended Data Fig. 2d–f). SBS-pks and ID-pks were replicated in an independent human intestinal organoid line (Extended Data Fig. 3a–d; SBS cosine similarity = 0.77; ID cosine similarity = 0.93) and with a clbQ-knockout E.coli strain recomplemented with the clbQ locus (pksΔclbQ:clbQ) (Extended Data Fig. 3e–h; SBS cosine similarity = 0.95; ID cosine similarity = 0.95).
Figure 1. Co-culture of healthy human intestinal organoids with genotoxic E. coli induces DNA damage.
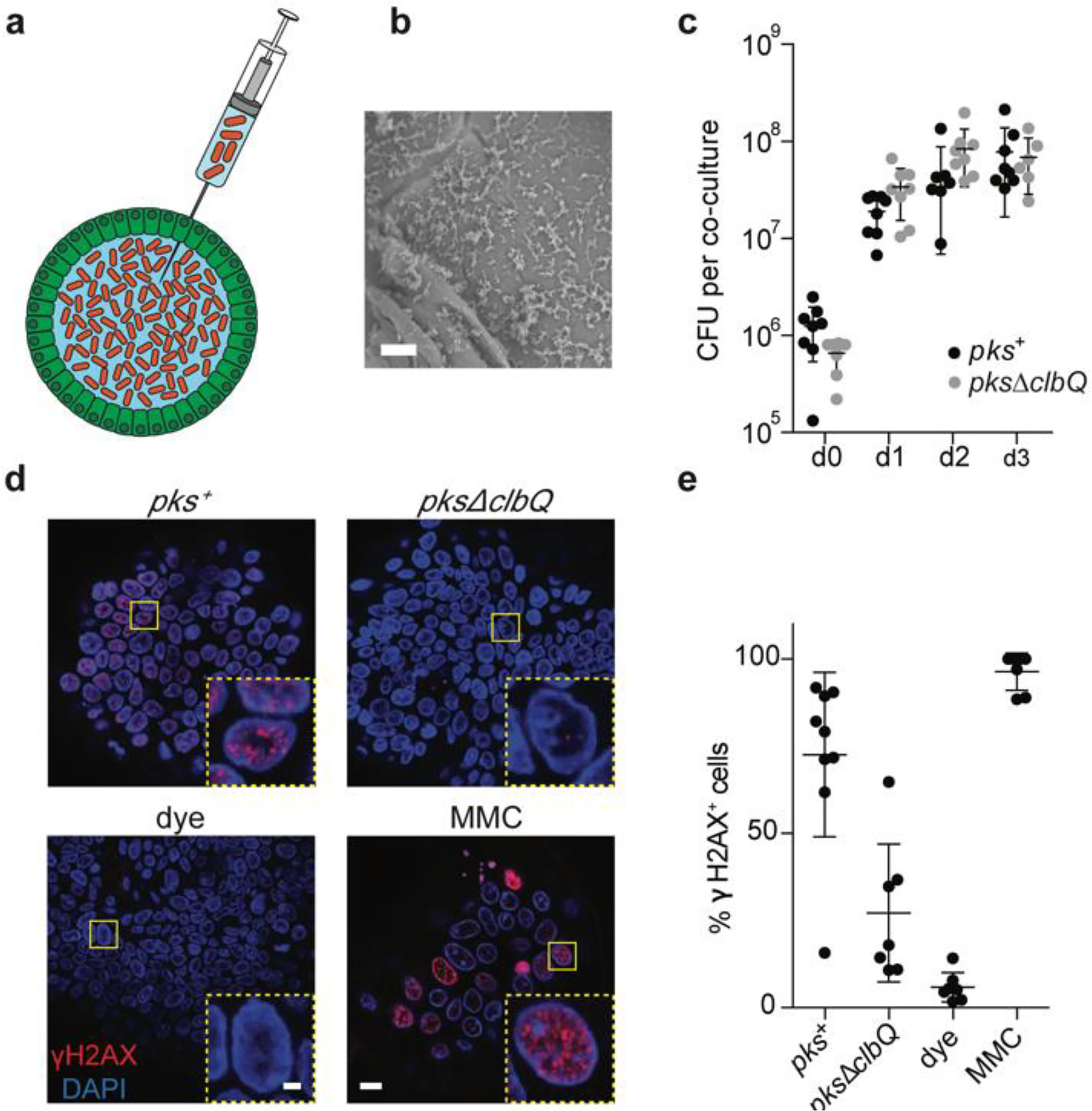
a, Schematic representation of genotoxic E. coli microinjection into the lumen of human intestinal organoids. b, Scanning electron microscopy image illustrating direct contact between organoid apical side and pks+ E. coli after 24h co-culture. Scale bar = 10 μm. c, Bacterial load of pks+ or pksΔclbQ at 0, 1, 2 and 3 days after co-culture establishment (n = 8 co-cultures per condition and timepoint, except pks+ d2 (n = 7) and pksΔclbQ d3 (n = 6)). CFU, colony forming units. Center line indicates mean, error bars represent SD. d, Representative images of DNA damage induction after 1 day of co-culture, measured by γH2AX immunofluorescence. One organoid is shown per image with one nucleus in the inset. Yellow boxes indicate inset area. Scale bars represent 10 μm (large image) and 2 μm (inset). e, Quantification of (d): Percentage of nuclei positive for γH2AX foci in pks+ (n= 9 organoids), pksΔclbQ (n=7 organoids), dye (n=7 organoids) and mitomycin C (MMC) (n=7 organoids) after 1 day of co-culture. Center line indicates mean, error bars represent SD.
Figure 2. Long-term co-culture of pks+ E. coli induces SBS-pks and ID-pks mutational signatures.
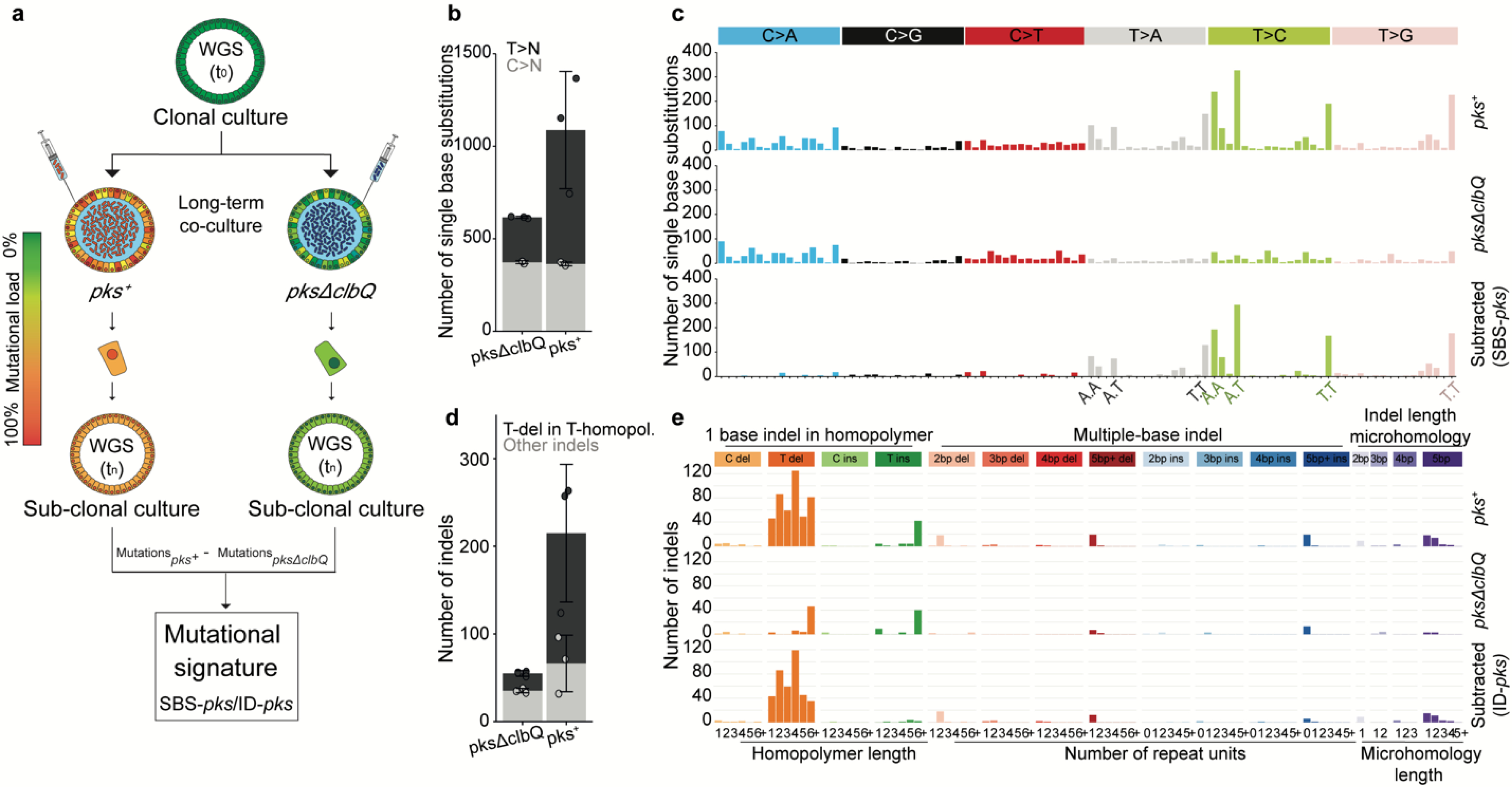
a, Schematic representation of the experimental setup. b, The number of single base substitutions (SBS) that accumulated in organoids co-cultured with either pks+ or pksΔclbQ E. coli (n = 3 clones). Box height indicates mean number of events, error bars represent SD. c, SBS 96-trinucleotide mutational spectra in organoids exposed to either pks+ (top) or pksΔclbQ (middle) E. coli. The bottom panel depicts the SBS-pks signature, which was defined by subtracting pksΔclbQ from pks+ SBS mutations. d, The number of small insertions and deletions (indels) that accumulated in organoids co-cultured either with pks+ or pksΔclbQ E. coli (n = 3 clones). Box height indicates mean number of events, error bars represent SD. e, Indel mutational spectra observed in organoids exposed to either pks+ (top) or pksΔclbQ (middle) E. coli. The bottom panel depicts the ID-pks signature, which was defined by subtracting pksΔclbQ from pks+ indel mutations.
Next, we asked if the SBS-pks and ID-pks mutations were characterized by other recurrent patterns. First, the assessed DNA stretch was extended beyond the nucleotide triplet. This uncovered the preferred presence of an adenine residue 3bp upstream to the mutated SBS-pks T>N site (Fig. 3a). Similarly, mutations that contributed to the ID-pks signature in poly-T stretches showed an enrichment of adenines immediately upstream of the affected poly-T stretch (Fig. 3b). Intriguingly, the lengths of the adenine stretch and the T-homopolymer were inversely correlated, consistently resulting in a combined length of 5 or more A/T nucleotides (Extended Data Fig. 4a). While SBS-pks and ID-pks are the predominant mutational outcomes of colibactin exposure, we also observed longer deletions at sites containing the ID-pks motif in organoids treated with pks+ E. coli (Fig. 3c). Additionally, the SBS-pks signature exhibited a striking transcriptional strand bias (Fig. 3d, e). We speculate that these observations reflect preferential repair of alkylated adenosines on the transcribed strand by transcription-coupled nucleotide excision repair. These features clearly distinguish the pks signature from published signatures of alkylating agents or other factors19.
Figure 3. Consensus motifs and extended features of SBS-pks and ID-pks mutational signatures.
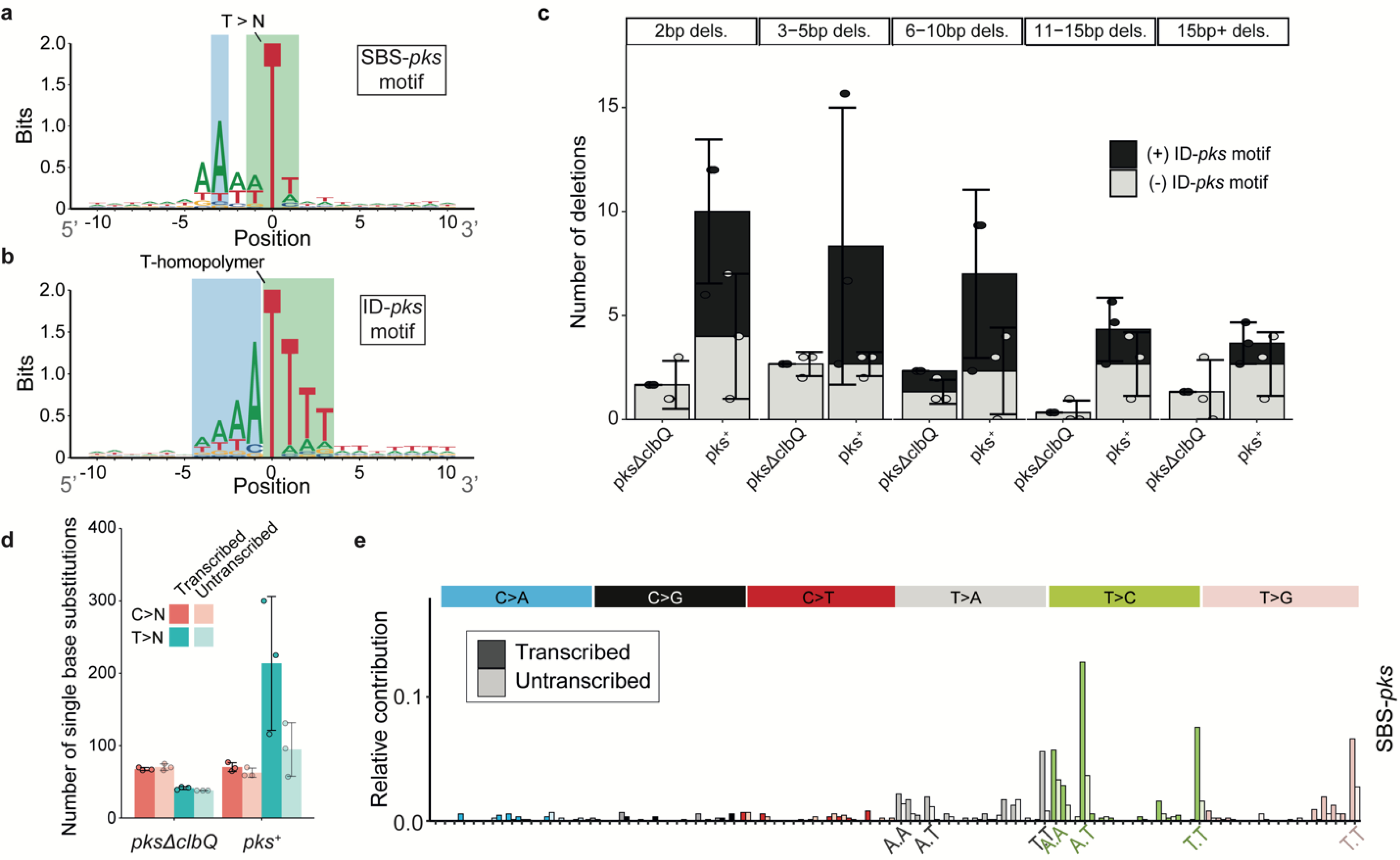
a, 2-bit representation of the extended sequence context of T>N mutations observed in organoids exposed to pks+ E. coli. Sequence directionality indicated in grey. Green: Highlighted T>N trinucleotide sequence; Blue: Highlighted A-enriched position characteristic of the SBS-pks mutations. b, 2-bit representation of the extended sequence context of single T-deletions in T-homopolymers observed in organoids exposed to pks+ E. coli. Sequence directionality indicated in grey. Green: Highlighted T-homopolymer with deleted T; Blue: Highlighted characteristic poly-A stretch. c, Mean occurrence of < 1 base pair deletions in pks+ or pksΔclbQ exposed organoids. Black bars correspond to deletions matching the ID-pks extended motifs; Grey bars correspond to deletions where no ID-pks motif is observed. Box height indicates mean number of events, error bars represent SD (n = 3 clones). d, Transcriptional strand-bias of T>N and C>N mutations occurring in organoids exposed to pks+ E. coli and pksΔclbQ E. coli. Pink: C>N; Blue: T>N; Dark color: Transcribed strand; Bright color: Untranscribed strand. Box height indicates mean number of events, error bars represent SD (n = 3 clones). e, Transcriptional strand bias of the 96-trinucleotide SBS-pks mutational signature. Color: Transcribed strand; White: Untranscribed strand.
We then assessed if the experimentally deduced SBS-pks and ID-pks signatures occur in human tumors by interrogating WGS data from a Dutch collection of 3668 solid cancer metastases24. The mutations a cancer cell has acquired at its primary site will be preserved even in metastases, so that these provide a view on the entire mutational history of a tumor. We first performed non-negative matrix factorization (NMF) on genome-wide mutation data obtained from 496 CRC metastases in this collection. Encouragingly, this unbiased approach identified an SBS signature that highly resembled SBS-pks (cosine similarity = 0.95; Extended Data Fig. 5a, b). We then determined the contribution of SBS-pks and ID-pks to the mutations of each sample in the cohort. This analysis revealed a strong enrichment of the two pks signatures in CRC-derived metastases when compared to all other cancer types (Fisher’s exact test p-value < 0.0001, Extended Data Table 1), as is displayed for SBS-pks in Figure 4a and for ID-pks in Figure 4b. We noted 7.5% SBS-pks, 8.8% ID-pks and 6.25% SBS/ID-pks high samples when applying a cutoff contribution value at 0.05 (Extended Data Table 1, Fig. 4c). As expected, the SBS-pks and ID-pks signatures were positively correlated in this metastasis dataset (R2 = 0.46 (all samples); R2 = 0.70 (CRC-only); Fig. 4c), in line with their co-occurrence in our in vitro data set. The longer deletions at ID-pks sites were also found to co-occur with SBS-pks and ID-pks (Fig. 4e, f). Additionally, we evaluated the levels of the SBS-pks or ID-pks mutational signatures in an independent cohort, generated in the framework of the Genomics England 100,000 Genomes Project. This dataset is comprised of WGS data from 2208 CRC tumors, predominantly of primary origin. SBS-pks and ID-pks were enriched in 5.0% and 4.4% of patients respectively, while 44 samples were high in both SBS-pks and ID-pks (Fig. 4d). The relative contribution of both pks-signatures correlated with an R2 of 0.35 (Fig. 4d).
Figure 4. SBS-pks and ID-pks mutational signatures are present in a subset of CRC samples from 2 independent cohorts.
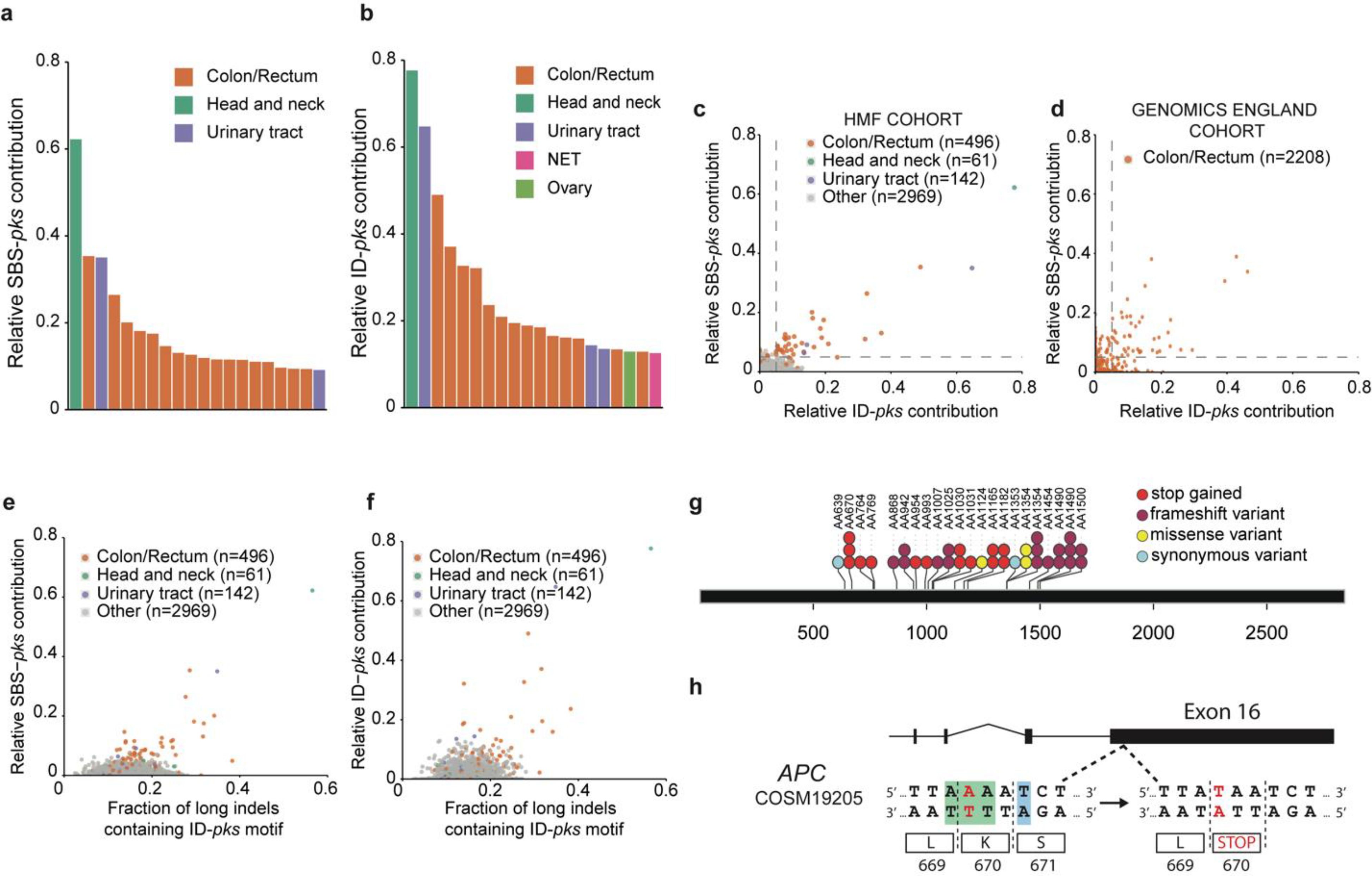
a, Top 20 out of 3668 metastases from the HMF cohort, ranked by the fraction of single base substitutions attributed to SBS-pks. CRC metastases (in orange) are enriched. Colors indicate tissue of origin. b, Top 20 out of 3668 metastases from HMF cohort. Samples are ranked by the fraction of indels attributed to ID-pks. CRC metastases (in orange) are also here enriched. NET indicates neuroendocrine tumor. Colors indicate tissue of origin. c, Scatterplot of fraction of single base substitutions and indels attributed to SBS-pks and ID-pks in 3668 metastases. Each dot represents one metastasis. Samples high for both SBS-pks and ID-pks (> 5% contribution, dashed lines) are enriched in CRC (orange). SBS-pks and ID-pks are correlated (R2 = 0.46; only CRC, R2 = 0.7). Colors indicate tissue of origin. d, Scatterplot of SBS-pks and ID-pks contribution in 2208 CRC tumor samples, predominantly of primary origin, from the Genomics England cohort. SBS-pks and ID-pks are correlated (R2 = 0.35). Each dot represents one primary tumor sample. Dashed lines delimitate samples with high SBS-pks or ID-pks contribution (> 5%). e, Scatterplot of SBS-pks and > 1 bp indels with ID-pks pattern in the HMF cohort. Colors indicate tissue of origin. f, Scatterplot of ID-pks and >1 bp indels with ID-pks pattern in the HMF cohort. Colors indicate tissue of origin. g, Exonic APC driver mutations found in the IntOGen collection matching the colibactin target SBS-pks or ID-pks motifs. h, Schematic representation of a driver mutation in APC causing a premature stop codon matching the SBS-pks motif, found in the IntOGen collection and in two independent SBS/ID-pks high patients from the HMF cohort.
Finally, we also investigated to what extent the pks signatures can cause oncogenic mutations. To this end, we investigated the most common driver mutations found in 7 CRC patient cohorts25 for hits matching the extended SBS-pks or ID-pks target motifs (Fig. 3a, b). This analysis revealed that 112 out of 4,712 (2.4%) CRC driver mutations matched the colibactin target motif (Supplementary Table 1). APC, the most commonly mutated gene in CRC, contained the highest number of mutations matching SBS-pks or ID-pks target sites, with 52 out of 983 driver mutations (5.3%) matching the motifs (Fig. 4g). We then explored the mutations of the 31 SBS/ID-pks high CRC metastases from the HMF cohort for putative driver mutations matching the extended motif. In total, this approach detected 209 changes in protein coding sequences (displayed in Supplementary Table 2). Remarkably, an identical APC driver mutation matching the SBS-pks motif was found in two independent donors (Fig. 4h).
While this study was in revision, an article26 was published describing the derivation of mutational signatures from healthy human colon crypts. Stratton c.s. note the co-occurrence of two mutational signatures in subsets of crypts from some of the subjects. These signatures were termed SBS-A and ID-A. The authors derived hierarchical lineages of the sequenced crypts, which allowed them to conclude that the -unknown- mutagenic agent was active only during early childhood. Intriguingly, SBS-A and ID-A closely match SBS-pks and ID-pks, respectively. Our data imply that pks+ E. coli is the mutagenic agent that is causative to the SBS-A and ID-A signatures observed in healthy crypts. We assessed if the SBS-pks mutational signature contributed early to the mutational load of metastatic samples from the Dutch cohort by evaluating their levels separately in clonal (pre-metastasis) or non-clonal (post-metastasis) mutations. The accumulation of SBS-pks and ID-pks at the primary tumor site or even earlier was substantiated by the abundant presence of SBS-pks in clonal mutations in the cohort (Extended Data Fig. 5c). In addition to CRCs, one head and neck- and three urinary tract-derived tumors from this cohort also displayed a clear SBS-pks and ID-pks signature (Fig. 4c). Both tissues have been described as sites of E. coli infection27–29. This rare occurrence of the pks signatures in non-CRC tumors was substantiated by a preprint report30 of signatures closely resembling SBS-pks and ID-pks in an oral squamous cell carcinoma patient.
The distinct motifs at sites of colibactin-induced mutations may serve as a starting point for deeper investigations into the underlying processes. Evidence is accumulating that colibactin forms interstrand crosslinks between two adenosines4,5,12, and our data imply a distance of 3–4 bases between these adenosines. These crosslinks formed by a bulky DNA adduct could be resolved in different ways, including induction of DSBs, Nucleotide Excision Repair or translesion synthesis, which in turn could result in various mutational outcomes. While our study unveils single base substitutions and deletions as a mutational consequence, the underlying mechanisms will need to be elucidated in more detailed DNA-repair studies.
In summary, we find that prolonged exposure of wild-type human organoids to genotoxic E. coli allows the extraction of a unique SBS and indel signature. As organoids do not model immune/inflammation effects or other microenvironmental factors, this provides evidence for immediate causality between colibactin and mutations in the host epithelial cells. The adenine-enriched target motif is in agreement with the proposed mode of action of colibactin’s ‘double-warhead’ attacking closely spaced adenine residues4,5,12. The pronounced sequence specificity reported here may inspire more detailed investigations on the interaction of colibactin with specific DNA contexts. As stated above, Stratton and colleagues26 likely describe SBS-pks and ID-pks mutational signatures of the same etiology in primary human colon crypts. This agrees with the notion that pks+ E. coli-induced mutagenesis indeed occurs in the healthy colon of individuals that harbor genotoxic E. coli strains31 and that such individuals may be at an increased risk of developing CRC. The small number of pks signature-positive urogenital and head-and-neck cancer cases suggests that pks+ bacteria act beyond the colon. Intriguingly, presence of the pks island in another strain of E. coli, Nissle 1917, is closely linked to its probiotic effect32. This strain has been investigated for decades for diverse disease indications33. Our data suggest that E. coli Nissle 1917 may induce the characteristic SBS/ID-pks mutational patterns. Future research should elucidate if this is the case in vitro, and in patients treated with pks+ bacterial strains. This study implies that detection and removal of pks+ E. coli, as well as re-evaluation of probiotic strains harboring the pks island, could decrease the risk of cancer in a large group of individuals.
METHODS
Human material and organoid cultures
Ethical approval was obtained from the ethics committees of the University Medical Center Utrecht, Hartwig Medical Foundation and Genomics England. Written informed consent was obtained from patients. All experiments and analyses were performed in compliance with relevant ethical regulations.
Organoid culture
Clonal organoid lines were derived and cultured as described previously16,17. In brief, wild type human intestinal organoids (clonal lines ASC-5a and ASC-6a, previously used in Blokzijl et al.,34) were cultured in domes of Cultrex Pathclear Reduced Growth Factor Basement Membrane Extract (BME) (3533–001, Amsbio) covered by medium containing Advanced DMEM/F12 (Gibco), 1x B27, 1x Glutamax, 10 mmol/L HEPES, 100 U/mL Penicillin-Streptomycin (all Thermo-Fisher), 1.25 mM N-acetylcysteine, 10 μM Nicotinamide, 10 μM p38 inhibitor SB202190 (all Sigma-Aldrich) and the following growth factors: 0.5 nM Wnt Surrogate-Fc Fusion Protein, 2% Noggin conditioned medium (both U-Protein Express), 20% Rspo1 conditioned medium (in-house), 50 ng/mL EGF (Peprotech), 0.5 μM A83–01, 1 μM PGE2 (both Tocris). For derivation of clonal lines, cells were FACS sorted and grown at a density of 50 cells/μl in BME. 10 μM ROCK inhibitor Y-27632 (Abmole, M1817) was added for the first week of growth. Upon reaching a size of >100 μm diameter, organoids were picked and transferred to one well per organoid. All organoid lines were regularly tested to rule out mycoplasma infection and authenticated using SNP profiling.
Organoid bacteria co-culture
The genotoxic pks+ E. coli strain was previously isolated from a CRC patient and isogenic pksΔclbQ knock out and pksΔclbQ:clbQ recomplemented strains were generated based on this strain21. Bacteria were initially cultured in Advanced DMEM (Gibco) supplemented with Glutamax and HEPES to an O.D. of 0.4. They were then microinjected into the lumen of organoids as previously described22,35. Bacteria were injected at a multiplicity of infection of 1 together with 0.05% (w/v) FastGreen dye (Sigma) to allow tracking of injected organoids. At this point, 5 μg/mL of the non-permeant antibiotic Gentamicin were added to the media to prevent overgrowth of bacteria outside the organoid lumen. Cell viability was assessed as follows: Organoids were harvested after 1, 3 or 5 days (bacteria were removed by primocin treatment at day 3) of co-culture in cold DMEM (Gibco) and incubated in TrypLE Express (Gibco) at 37°C for 5 minutes with repeated mechanical shearing. Single cells were resuspended in DMEM with added DAPI, incubated on ice for at least 15 minutes and assessed for viability on a BD FACS Canto™. Cells positive for DAPI were considered dead, while cells maintaining DAPI exclusion were counted as viable. Bacterial growth kinetics were assessed by harvesting, organoid dissociation with 0.5% saponin for 10 minutes and re-plating of serial dilutions on LB plates. Colony forming units were quantified after overnight culture at 37°C. E. coli were killed with 1x Primocin (InvivoGen) after 3 days of co-culture, after which organoids were left to recover for 4 days before being passaged. When the organoids reached a cystic stage again (typically after 2–3 weeks), the injection cycle was repeated. This procedure was repeated 5 times (3 times for ASC Clone 6-a and the clbQ recomplementation experiment in ASC Clone 5-a) to nivellate injection heterogeneity and ensure accumulation of enough mutations for reliable signature detection.
Whole-mount organoid immunofluorescence, DNA damage quantification and scanning electron microscopy
Organoids co-cultured with pks+/pksΔclbQ E. coli21 were collected in Cell Recovery Solution (Corning) and incubated at 4°C for 30 minutes with regular shaking in order to free them from BME. For FANCD2 staining, organoids were pre-permeabilized with 0.2% Triton-X (Sigma) for 10 minutes at room temperature. Then, organoids were fixed in 4% formalin overnight at 4°C. Subsequently, organoids were permeabilized with 0.5% Triton-X (Sigma), 2% donkey serum (BioRad) in PBS for 30 minutes at 4°C and blocked with 0.1% Tween-20 (Sigma) and 2% donkey serum in PBS for 15 minutes at room temperature. Organoids were incubated with mouse anti-γH2AX (Millipore; clone JBW301; 1:1000 dilution) or rabbit anti-FANCD2 (affinity purified in Pace et al.36; 1mg/ml) primary antibody overnight at 4°C. Then, organoids were washed 4 times with PBS and incubated with either secondary goat anti-mouse AF-647 (Thermo Fisher, catalog number A-21235, 1:500 dilution) or goat anti-rabbit AF-488 (Life Technologies, catalog number A21206, 1:500 dilution) antibodies, respectively, for 3h at room temperature in the dark and washed again with PBS. Organoids were imaged using an SP8 confocal microscope (Leica). Fluorescent microscopic images of γH2AX foci were quantified as follows: Nuclei were classified as containing either 0 or one or more foci. The fraction of nuclei containing foci over all nuclei is displayed as one datapoint per organoid. Organoids co-cultured with bacteria for 24h were harvested as described above and processed for scanning electron microscopy as previously described35.
WGS and read alignment
For WGS, clonal and subclonal cultures were generated for each condition. From these clonal cultures DNA was isolated using the DNeasy Blood and Tissue Kit (Qiagen) using manufacturer’s instructions. Illumina DNA libraries were prepared using 50 ng of genomic DNA isolated from the (sub-)clonal cultures isolated using TruSeq DNA Nano kit. The parental ASC 5a clone was sequenced on a HiSeq XTEN instrument at 30x base coverage. All other samples were sequenced using an Illumina Novaseq 6000 with 30x base coverage. Reads were mapped against the human reference genome version GRCh37 by using Burrows-Wheeler Aligner37 (BWA) version v0.7.5 with settings bwa mem -c 100 -M. Sequences were marked for duplicates using Sambamba (v0.4.732) and realigned using GATK IndelRealigner (GATK version 3.4–46). The full description and source code of the pipeline is available at https://github.com/UMCUGenetics/IAP.
Mutation calling and filtration
Mutations were called using GATK Haplotypecaller (GATK version 3.4–46) and GATK Queue producing a multi-sample Vcf file20. The quality of the variants was evaluated usingGATK VariantFiltration v3.4–46 using the following settings: -snpFilterName SNP_LowQualityDepth -snpFilterExpression “QD < 2.0” -snpFilterName SNP_MappingQuality -snpFilterExpression “MQ < 40.0” -snpFilterName SNP_StrandBias -snpFilterExpression “FS > 60.0” -snpFilterName SNP_HaplotypeScoreHigh -snpFilterExpression “HaplotypeScore > 13.0” -snpFilterName SNP_MQRankSumLow -snpFilterExpression “MQRankSum < −12.5” -snpFilterName SNP_ReadPosRankSumLow -snpFilterExpression “ReadPosRankSum < −8.0” -snpFilterName SNP_HardToValidate -snpFilterExpression “MQ0 >= 4 && ((MQ0 / (1.0 * DP)) > 0.1)” -snpFilterName SNP_LowCoverage -snpFilterExpression “DP < 5” -snpFilterName SNP_VeryLowQual -snpFilterExpression “QUAL < 30” -snpFilterName SNP_LowQual -snpFilterExpression “QUAL >= 30.0 && QUAL < 50.0 “ -snpFilterName SNP_SOR -snpFilterExpression “SOR > 4.0” -cluster 3 -window 10 -indelType INDEL -indelType MIXED -indelFilterName INDEL_LowQualityDepth -indelFilterExpression “QD < 2.0” -indelFilterName INDEL_StrandBias -indelFilterExpression “FS > 200.0” -indelFilterName INDEL_ReadPosRankSumLow -indelFilterExpression “ReadPosRankSum < −20.0” -indelFilterName INDEL_HardToValidate -indelFilterExpression “MQ0 >= 4 && ((MQ0 / (1.0 * DP)) > 0.1)” -indelFilterName INDEL_LowCoverage -indelFilterExpression “DP < 5” -indelFilterName INDEL_VeryLowQual -indelFilterExpression “QUAL < 30.0” -indelFilterName INDEL_LowQual -indelFilterExpression “QUAL >= 30.0 && QUAL < 50.0” -indelFilterName INDEL_SOR -indelFilterExpression “SOR > 10.0.
Somatic single base substitution and indel filtering
To obtain high confidence catalogues of mutations induced during culture, we applied extensive filtering steps previously described by Jager et al.20. First, only variants obtained by GATK VariantFiltration with a GATK phred-scaled quality score ≥ 100 for single base substitutions and ≥ 250 for indels were selected. Subsequently, we only considered variants with at least 20x read coverage in control and sample. We additionally filtered base substitutions with a GATK genotype score (GQ) lower than 99 or 10 in WGS(tn) or WGS(t0), respectively. Indels were filtered when GQ scores were higher than 60 WGS(tn) or 10 in WGS(t0). All variants were filtered against the Single Nucleotide Polymorphism Database v137.b3730, from which SNPs present in the COSMICv76 database were excluded. To exclude recurrent sequencing artefacts, we excluded all variants variable in at least three individuals in a panel of bulk-sequenced mesenchymal stromal cells38. Next, all variants present at the start of co-culture (denominated WGS(t0) in Fig. 2a) were filtered from those detected in the clonal pks+ E. coli, pksΔclbQ E. coli co-cultures (denominated WGS(tn) in Fig. 2a) or dye culture. Indels were only selected when no called variants in WGS(t0) were present within 100bp of the indel and if not shared in WGS(t0). In addition, both indels and SNVs were filtered for the additional parameters: mapping quality (MQ) of at least 60 and a variant allele (VAF) of 0.3 or higher to exclude variants obtained during the clonal step. Finally, all multi-allelic variants were removed. Scripts used for filtering single base substitutions (SNVFIv1.2) and indels (INDELFIv1.5) are deposited on https://github.com/ToolsVanBox/.
Mutational profile analysis
In order to extract mutational signatures from the high-quality mutational catalogues after filtering, we used the R package “MutationalPatterns” to obtain 96-trinucleotide single base substitution and indel subcategory counts for each clonally cultured sample39 (Extended Data Fig. 1a, d). In order to obtain the additional mutational effects induced by pks+ E. coli (SBS and ID) we pooled mutation numbers for each culture condition (pksΔclbQ and pks+), and subtracted mutational counts of pksΔclbQ from pks+ (Fig. 2c, e, Extended Data Fig 2b, d). For the clones exposed to pksΔclbQ:clbQ, we subtracted relative levels of the pksΔclbQ mutations in the same organoid line. This enabled us to correct for the background of mutations induced by pksΔclbQ E. coli and injection dye. To determine transcriptional strand bias of mutations induced during pks+ E. coli exposure, we selected all single base substitutions within gene bodies and checked whether the mutated C or T was located on the transcribed or non-transcribed strand. We defined the transcribed area of the genome as all protein coding genes based on Ensembl v75 (GCRh37)40 and included introns and untranslated regions. The extended sequence context around mutation sites was analyzed and displayed using an in-house script (“extended_sequence_context.R”). 2-bit sequence motifs were generated using the R package “ggseqlogo”. Cosine similarities between indel and single-base substitution profiles were calculated using the function ‘cos_sim_matrix’ from the MutationalPatterns package.
Analysis of clonal mutations in the SBS/ID-pks high CRC tumors
From the 31 SBS/ID-pks high CRC tumors clonal and subclonal single base substitutions were defined to contain a purity/ploidy adjusted allele-fraction (PURPLE_AF) of < 0.4 or > 0.2, respectively41. Signature re-fitting on both fractions was performed with the same signatures as described above for the initial re-fitting of the HMF cohort.
Analysis of >1bp deletions matching pks-motif
For each > 1 bp T-deletion observed in organoid clones or the HMF cohort, the sequence of the deleted bases and 5 base-pair flanking regions was retrieved using the R function “getSeq” from the package “BSgenome”. Retrieved sequences were examined for the presence of a 5 base-pair motif matching the pks-motifs identified (Extended Data Fig. 4a) “AAAAT”, “AAATT, “AATTT” or “ATTTT”. Sequences containing one or more matches with the motifs were marked as positive for containing the motif.
NMF extraction of signatures from HMF Colorectal cancer cohort
In order to identify SBS-pks in an unbiased manner, signature extraction was performed on all 496 samples from colorectal primary tumors present in the HMF metastatic cancer database24. All variants containing the ‘PASS’ flag were used for analysis. Signature extraction was performed using non-negative matrix factorization (NMF), using the R package “MutationalPatterns” function “extract_signatures” with the following settings: rank = 17, nrun = 200. The cosine similarity of the extracted signature matching SBS-pks was re-fitted to the COSMIC SigProfiler signatures and SBS-pks was determined as described above to determine similarity (Extended Data Fig. 5a, b).
Signature re-fitting on HMF cohort
Mutation catalogues containing somatic variants processed according to Priestley et al, 2019 were obtained from the HMF. All variants containing the ‘PASS’ flag in the HMF dataset were selected. Single base trinucleotide and indel subcategory counts were extracted using the R package “MutationalPatterns” and in house-written R scripts respectively. In order to determine the contribution of SBS-pks and ID-pks to these mutational catalogues, we re-fitted the COSMIC SigProfiler mutational SBS and ID signatures v3 (https://cancer.sanger.ac.uk/cosmic/signatures/), in combination with SBS-pks and ID-pks, to the mutational catalogues using the MutationalPatterns function “fit_to_signatures”. Signatures marked as possible sequencing artefacts were excluded from the re-fitting. Cutoff values for high SBS-pks and ID-pks levels were manually set at 5%, each. Numbers of SBS/ID-pks positive samples were compared between CRC and other cancer types by Fisher’s exact test (two-tailed).
Mutation calling and filtration (Genomics England cohort)
As part of the Genomics England 100,000 Genomes Project (main programme version 7)42 standard pipeline, 2208 CRC genomes were sequenced on the Illumina HiSeq X platform. Reads were aligned to the human genome (GRCh38) using the Illumina iSAAC aligner 03.16.02.143. Mutations were called using Strelka and filtered in accordance with the HMF dataset24.
Before examining somatic mutations for the pks mutational signature, mutation calls were first subjected to additional filtering steps similar to those previously described24. All calls present in the matched normal sample were removed. The calls were split into high and low confidence genomic regions according to lists available at ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/NA12878_HG001/NISTv3.3.1/GRCh38/. Somatic mutation calls in high confidence regions were passed with a somatic score (QSI or QSS) of 10, whilst calls in low confidence regions were passed with a score of 20. A pool of 200 normal samples was constructed, and any calls present in three or more normal samples were removed. Any groups of single nucleotide variants within 2bp were considered to be miss-called multiple nucleotide variants and were removed. Finally, all calls had to pass the Strelka “PASS” filter. Mutational signatures were then analysed as described above for the HMF cohort.
Detection of pks-signature mutations in protein coding regions
Mutations were extracted from the 31 SBS/ID-pks high CRC-samples. Exonic regions were defined as all autosomal exonic regions reported in Ensembl v75 (GCRh37)40. All extracted CRC mutations were filtered for localization in exonic regions using the Bioconductor packages “GenomicRanges”44 and “BSgenome”. In a second filtering step, the sequence context of mutations was required to match the following criteria:
For SBS-pks: T>N mutation, A or T directly upstream and downstream, A 3 bases upstream. For ID-pks: Single T deletion, A directly upstream, a stretch of an A homopolymer followed by a T polymer with combined length of at least 5 nucleotides, but no stretch exceeding 10 nucleotides in length. Mutations passing both filter steps were further filtered for presence of a predicted “HIGH” or “MODERATE” score in the transcript with highest impact score according to the reported SnpEff annotation.
To assess the mutagenic impact of pks, we obtained all mutations from the 50 highest mutated genes in CRC from IntOGen25, release 2019.11.12. Mutations were filtered matching the pks motif according to the sequence criteria stated above apart from the predicted impact score. Mutations in APC were plotted using the R package “rtrackViewer”, using only exonic mutations.
Extended Data
Extended Data Fig. 1. Co-culture with genotoxic pks+ E. coli induces DNA interstrand crosslinks in healthy human intestinal organoids.
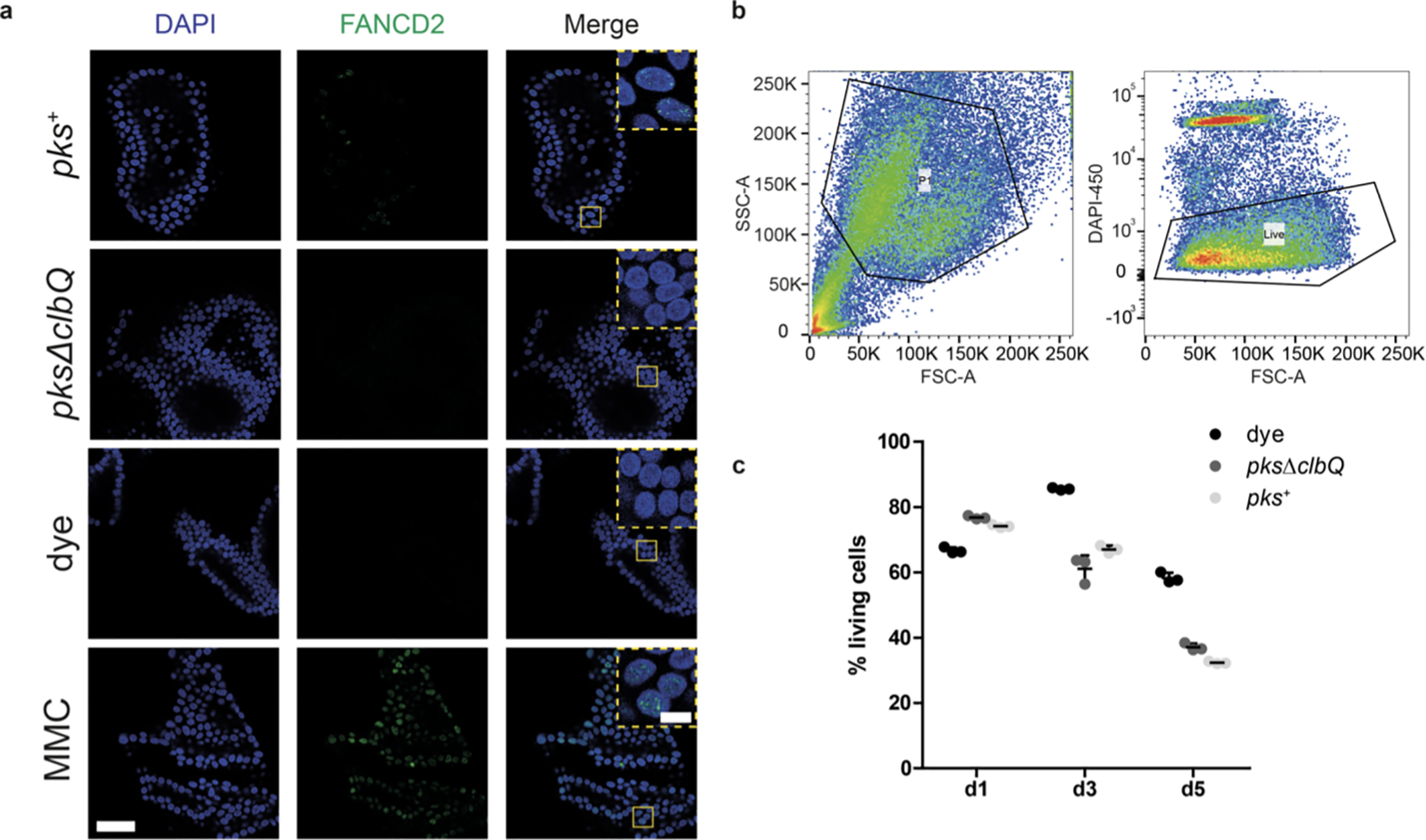
a, Representative images (out of n = 5 organoids per group) of DNA interstrand crosslink formation after 1d of co-culture, measured by FANCD2 immunofluorescence (green). Nuclei were stained with DAPI (blue). Yellow boxes represent inset area. Scale bars represent 50 μm (large image) and 10 μm (inset). Experiment was repeated independently twice with similar results. b, Gating strategy to select epithelial cells (left) and to quantify viable cells (right). c, Viability of intestinal organoid cells after 1, 3 and 5 days of co-culture (n = 3 technical replicates) (bacteria eliminated after 3 days of co-culture). Points are independent replicates, center line indicates mean, error bars represent SD.
Extended Data Fig. 2. Genotoxic pks+ E. coli induce SBS-pks and ID-pks mutational signatures after long-term co-culture with wild-type intestinal organoids.
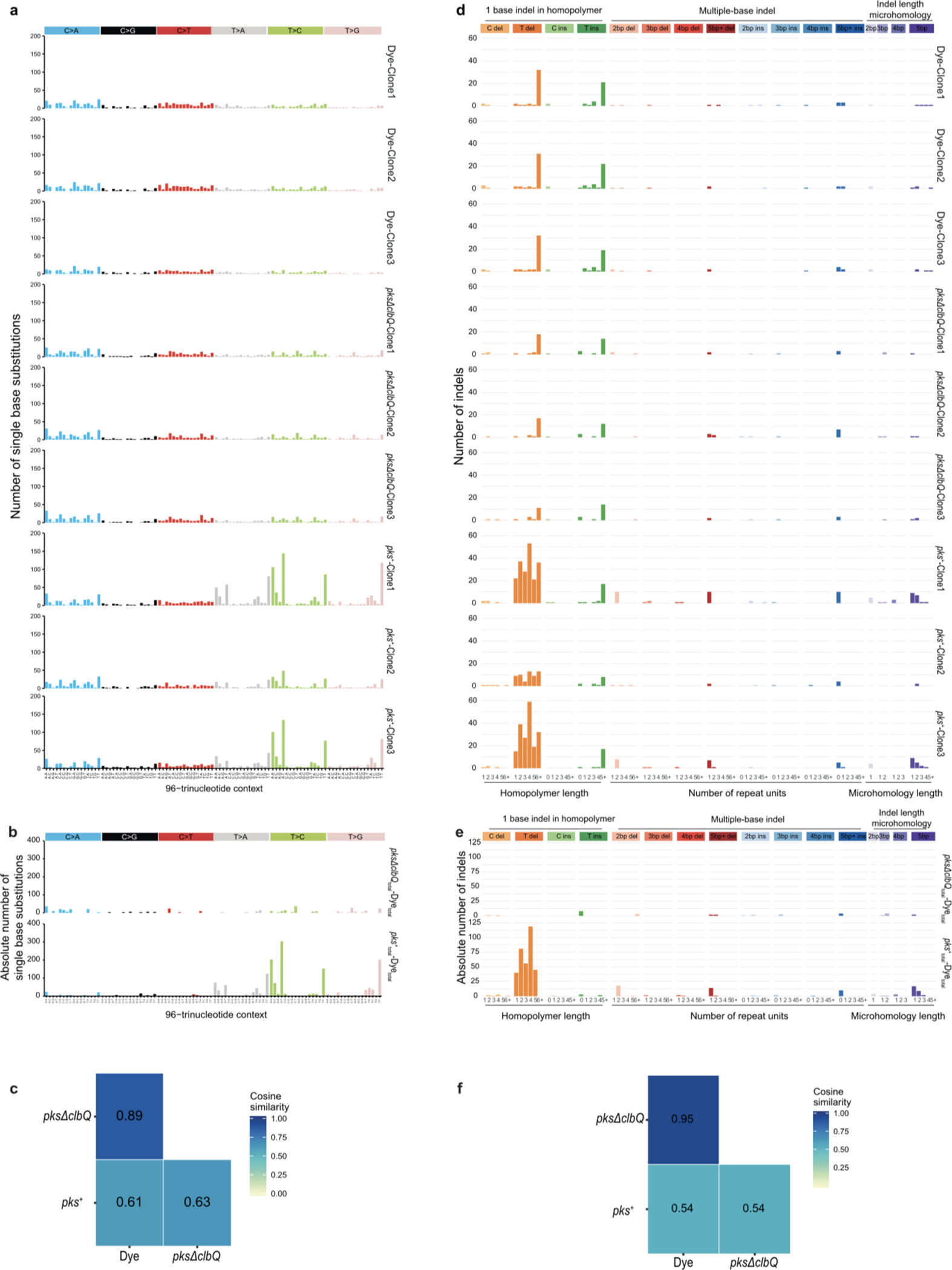
a, 96-trinucleotide mutational spectra of SBS in each of the 3 individual clones sequenced per condition. Top 3: dye; middle 3: pksΔclbQ E. coli; bottom 3: pks+ E. coli. b, Total 96-trinucleotide mutational spectra of pks+ and pksΔclbQ from which dye single base substitutions are subtracted. c, Heatmap depicting cosine similarity between dye, pks+ E. coli and pksΔclbQ E. coli 96-trinucleotide mutational profiles. d, Indel mutational spectra plots from each of the 3 individual clones sequenced per condition. Top 3: dye; middle 3: pksΔclbQ E. coli bottom 3: pks+ E. coli e, Total indel mutational spectra of values of pks+ E. coli and pksΔclbQ E. coli from which dye indels are subtracted. f, Heatmap depicting cosine similarity between dye, pks+ E. coli and pksΔclbQ E. coli indel mutational profiles.
Extended Data Fig. 3. Genotoxic pks+ E. coli and isogenic strain reconstituted with pksΔclbQ:clbQ induce SBS-pks and ID-pks mutational signatures after co-culture.
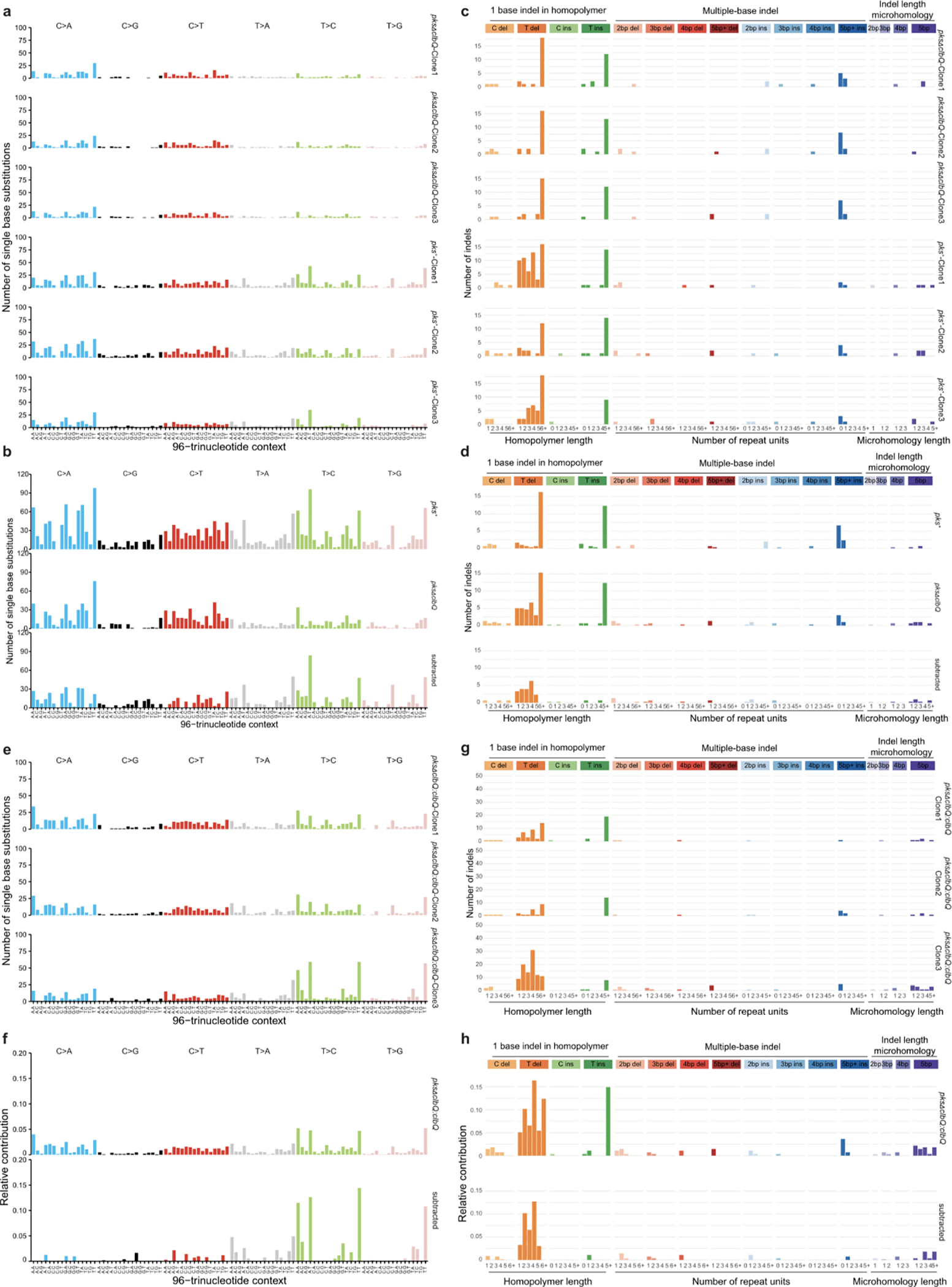
a, 96-trinucleotide mutational spectra of SBS in 3 individual clones from the independent human healthy intestinal organoid line ASC 6-a co-cultured for 3 rounds with pks+ or pksΔclbQ E. coli. b, Top: Total 96-trinucleotide mutational spectrum from the 3 clones from pks+ or pksΔclbQ E. coli shown in (a). Bottom: Resulting 96-trinucleotide mutational spectrum from ASC 6-a co-cultured with pks+ E. coli after the subtraction of background mutations from 3 parallel pksΔclbQ E. coli co-cultures (cosine similarity to SBS-pks = 0.77). c, Indel mutational spectra plots from the 3 independent ASC 6-a clones co-cultured for 3 rounds with pks+ or pksΔclbQ E. coli. d, Top: Total indel mutational spectrum from the 3 clones from pks+ or pksΔclbQ E. coli shown in (c). Bottom: Resulting indel mutational spectrum from the independent ASC 6-a co-cultured with pks+ E. coli after the subtraction of background mutations from 3 parallel pksΔclbQ E. coli co-cultures (cosine similarity to ID-pks = 0.93). e, 96-trinucleotide mutational spectrum from 3 individual clones of the ASC 5-a line co-cultured for 3 rounds with the isogenic recomplemented strain pksΔclbQ:clbQ. f, Top: Total 96-trinucleotide mutational spectrum from the 3 clones from pksΔclbQ:clbQ shown in (e). Bottom: Resulting mutational spectrum after subtracting pksΔclbQ background (cosine similarity to SBS-pks = 0.95). g, Indel mutational spectrum from 3 individual clones of the ASC 5-a line co-cultured for 3 rounds with the isogenic recomplemented strain pksΔclbQ:clbQ. h, Top: Total indel mutational spectrum from the 3 clones from pksΔclbQ:clbQ shown in (e). Bottom: Resulting mutational spectrum after subtracting pksΔclbQ background (cosine similarity to ID-pks = 0.95).
Extended Data Fig. 4. Detailed sequence context for ID-pks and longer deletions by length.
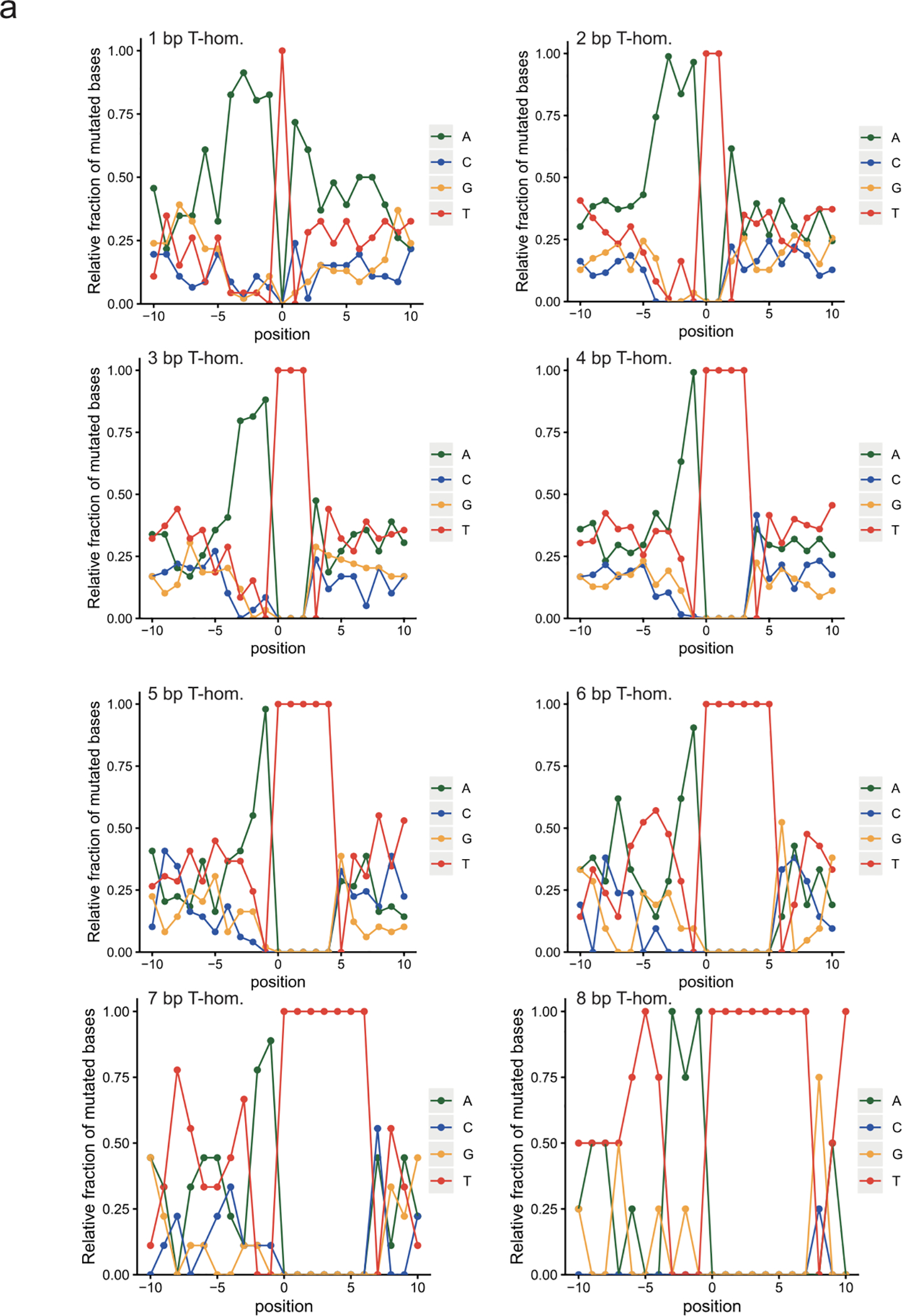
a, 10 base up- and downstream profile shows an upstream homopolymer of adenosines favoring induction of T-deletions. The length of the adenosine stretch decreases with increasing T-homopolymer length (1—8, top left to bottom right).
Extended Data Fig. 5. Signature extraction and clonal contribution of SBS-pks in CRC metastases.
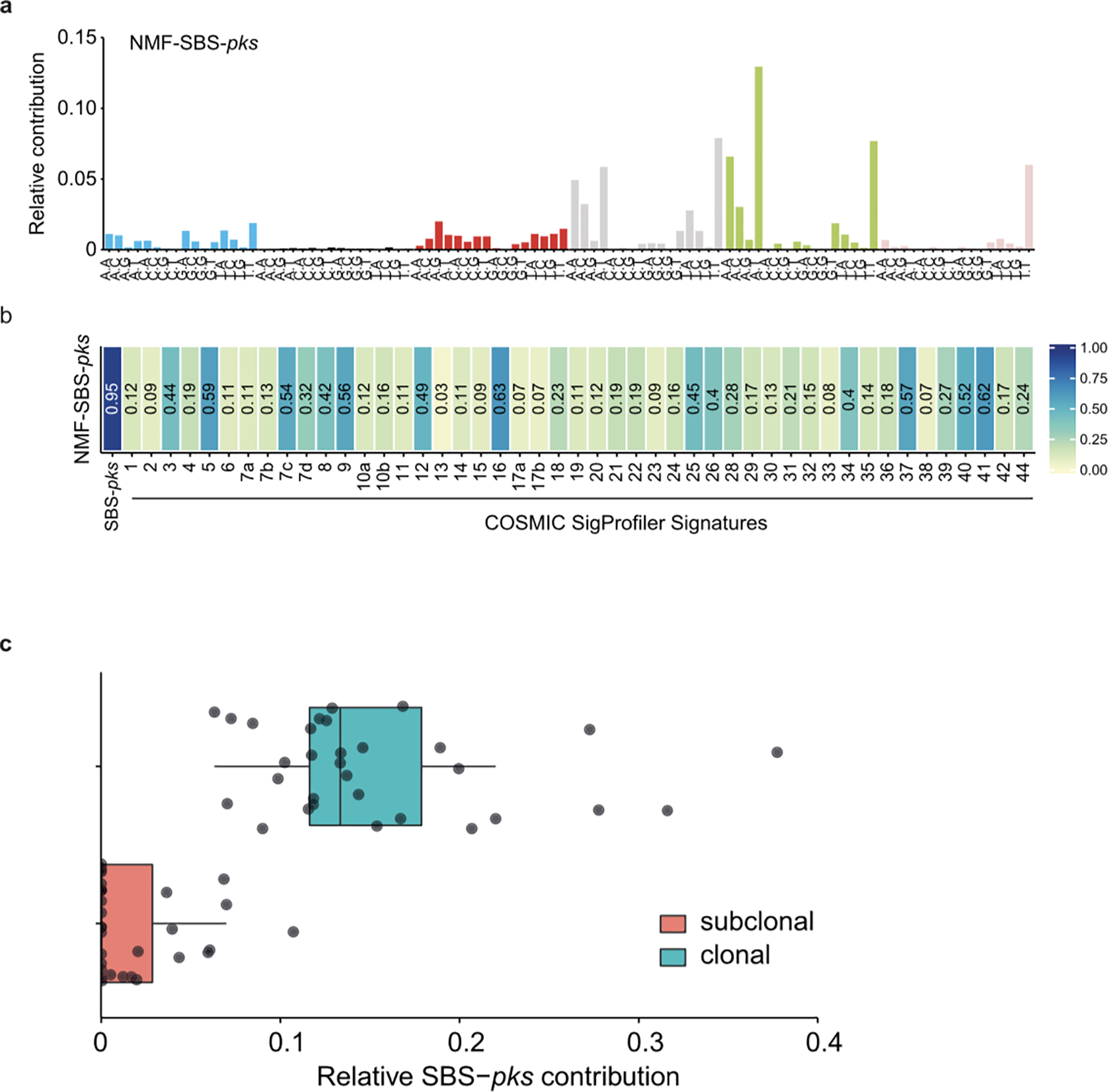
a, De-novo extracted NMF-SBS-pks signature by non-negative matrix factorization (NMF) on all 496 CRC metastases in the HMF dataset. b, Cosine similarity scores between the de-novo extracted SBS signature in (a) and COSMIC SigProfiler signatures, including our experimentally defined SBS-pks signature (left). c, Relative contribution of SBS-pks to clonal (corrected variant allele frequency > 0.4, blue bar) and subclonal fraction (corrected variant allele frequency < 0.2, red bar) of mutations in the 31 SBS/ID-pks high CRC metastases from the HMF cohort. Box indicates upper and lower quartiles with the center line indicating the mean. Box whiskers: largest value no further than 1.5 times the interquartile range extending from the box. Points indicate individual CRC metastases.
Extended Data Table 1|.
SBS-pks and ID-pks levels across tissue types.
| Primary Tumor Location | Total number | SBS-pks > 0.05 | ID-pks > 0.05 | SBS-pks > 0.05 & ID-pks > 0.05 |
|---|---|---|---|---|
| CRC | 496 | 37 (7,5%) | 44 (8,8%) | 31 (6,25%) |
| Head & Neck | 61 | 1 (1,6%) | 1 (1,6%) | 1 (1,6%) |
| Urinary Tract | 142 | 3 (2,1%) | 6 (4,2%) | 3 (2,1%) |
| Other | 2969 | 12 (0,4%) | 134 (4,5%) | 1 (0,03%) |
Sample numbers are displayed by primary tumor type per row. Numbers of samples with more than 5% contribution of either ID-pks, SBS-pks or both are shown; the proportion of positive samples per tissue is indicated in brackets.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Hoeijmakers J., Knipscheer P and Garaycoechea J. for discussions on DNA damage. We also thank Robinson P., and Vervier K., Lawley T., and Stratton M. for explorative analysis and discussions.
This publication and the underlying study have been made possible partly on the basis of the data that Hartwig Medical Foundation and the Center of Personalised Cancer Treatment (CPCT) have made available to the study.
This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support.
This work was supported by CRUK grant OPTIMISTICC [C10674/A27140], the Gravitation projects CancerGenomiCs.nl and the Netherlands Organ-on-Chip Initiative (024.003.001) from the Netherlands Organisation for Scientific Research (NWO) funded by the Ministry of Education, Culture and Science of the government of the Netherlands (C.P.M, J.P.), the Oncode Institute (partly financed by the Dutch Cancer Society) the European Research Council under ERC Advanced Grant Agreement no. 67013 (J.P., T.M, H.C.), a VIDI grant of the Netherlands Organisation for Scientific Research (NWO) (no. 016.Vidi.171.023) to R.v.B (A.R.H.) and NWO building blocks of life project: Cell dynamics within lung and intestinal organoids (737.016.009)(M.H.G).
This work was supported by the Netherlands Organ-on-Chip Initiative, an NWO Gravitation project (024.003.001) funded by the Ministry of Education, Culture and Science of the government of the Netherlands
Footnotes
DATA AVAILABILITY STATEMENT
Whole-genome sequence data has been deposited in the European Genome-phenome Archive (EGA; https://ega-archive.org); accession number EGAS00001003934. The data used from the Hartwig Medical Foundation and Genomics England databases consist of patient-level somatic variant data (annotated variant call data) and are considered privacy sensitive and available through access-controlled mechanisms.
Patient-level somatic variant and clinical data have been obtained from the Hartwig Medical Foundation under the data request number DR-084. Somatic variant and clinical data are freely available for academic use from the Hartwig Medical Foundation through standardized procedures. Privacy and publication policies, including co-authorship policies, can be retrieved from: https://www.hartwigmedicalfoundation.nl/en/data-policy/. Data request forms can be downloaded from https://www.hartwigmedicalfoundation.nl/en/applying-for-data/.
To gain access to the data, this data request form should be emailed to info@hartwigmedicalfoundation.nl., upon which it will be evaluated within 6 weeks by the HMF Scientific Council and an independent Data Access Board. When access is granted, the requested data become available through a download link provided by HMF.
Somatic variant data from the Genomics England dataset was analyzed within the Genomics England Research Environment secure data portal, under Research Registry project code RR87 and exported from the Research Environment following data transfer request 1000000003652 on 3rd December 2019.
- Your insitution has to sign the GeCIP Participation Agreement, which outlines the key principles that members of each institution must adhere to, including our Intellectual Property and Publication Policy.
- Submit your application using the relevant form found at the bottom of the page (https://www.genomicsengland.co.uk/join-a-gecip-domain/).
- The domain lead will review your application, and your institution will verify your identity for Genomics England and communicate confirmation directly to Genomics England.
- Your user account will be created.
- You will be sent an email containing a link to complete Information Governance training and sign the GeCIP Rules (https://www.genomicsengland.co.uk/wp-content/uploads/2019/07/GeCIP-Rules_29–08-2018.pdf). Completing the training and signing the GeCIP Rules are requirements for you to access the data. After you have completed the training and signed the rules, you will need to wait for your access to the Research Environment to be granted.
- This will generally take up to one working day. You will then receive an email letting you know your account has been given access to the environment, and instructions for logging in.
(for more detail, see: https://www.genomicsengland.co.uk/join-a-gecip-domain/)
Details of the data access agreement can be retrieved from: https://figshare.com/articles/GenomicEnglandProtocol_pdf/4530893/5. All requests will be evaluated by the Genomics England Access Review Committee taking into consideration patient data protection, compliance with legal and regulatory requirements, resource availability and facilitation of high quality research.
All analysis of the data must take place within the Genomics England Research Environment secure data portal, https://www.genomicsengland.co.uk/understanding-genomics/data/ and exported following approval of a data transfer request.
Regarding co-authorship, all publications using data generated as part of the Genomics England 100,000 Genomes Project must include the Genomics England Research Consortium as co-authors. The full publication policy is available at https://www.genomicsengland.co.uk/about-gecip/publications/.
All other data supporting the findings of this study are available from the corresponding author upon request.
CODE AVAILABILITY
All analysis scripts are available on https://github.com/ToolsVanBox/GenotoxicEcoli.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
GENOMICS ENGLAND RESEARCH CONSORTIUM AUTHOR LIST
Ambrose J. C. 1, Arumugam P.1, Baple E. L. 1, Bleda M. 1, Boardman-Pretty F. 1,2, Boissiere J. M. 1, Boustred C. R. 1, Brittain H.1, Caulfield M. J.1,2, Chan G. C. 1, Craig C. E. H. 1, Daugherty L. C. 1, de Burca A. 1, Devereau, A. 1, Elgar G. 1,2, Foulger R. E. 1, Fowler T. 1, Furió-Tarí P. 1, Hackett J. M. 1, Halai D. 1, Hamblin A.1, Henderson S.1,2, Holman J. E. 1, Hubbard T. J. P. 1, Ibáñez K.1,2, Jackson R. 1, Jones L. J. 1,2, Kasperaviciute D. 1,2, Kayikci M. 1, Lahnstein L. 1, Lawson K. 1, Leigh S. E. A. 1, Leong I. U. S. 1, Lopez F. J. 1, Maleady-Crowe F. 1, Mason J. 1, McDonagh E. M. 1,2, Moutsianas L. 1,2, Mueller M. 1,2, Murugaesu N. 1, Need A. C. 1,2, Odhams C. A. 1, Patch C. 1,2, Perez-Gil D. 1, Polychronopoulos D. 1, Pullinger J. 1, Rahim T. 1, Rendon A. 1, Riesgo-Ferreiro P.1, Rogers T. 1, Ryten M. 1, Savage K. 1, Sawant K. 1, Scott R. H. 1, Siddiq A. 1, Sieghart A. 1, Smedley D. 1,2, Smith K. R. 1,2, Sosinsky A. 1,2, Spooner W. 1, Stevens H. E. 1, Stuckey A. 1, Sultana R. 1, Thomas E. R. A. 1,2, Thompson S. R. 1, Tregidgo C. 1, Tucci A. 1,2, Walsh E. 1, Watters, S. A. 1, Welland M. J. 1, Williams E. 1, Witkowska K. 1,2, Wood S. M. 1,2, Zarowiecki M.1.
1. Genomics England, London, UK
2. William Harvey Research Institute, Queen Mary University of London, London, EC1M 6BQ, UK.
REFERENCES
- 1.Allen J & Sears CL Impact of the gut microbiome on the genome and epigenome of colon epithelial cells: contributions to colorectal cancer development. Genome Med. 11, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gagnaire A, Nadel B, Raoult D, Neefjes J & Gorvel J-P Collateral damage: insights into bacterial mechanisms that predispose host cells to cancer. Nat. Rev. Microbiol. 15, 109–128 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Nougayrède J-P et al. Escherichia coli Induces DNA Double-Strand Breaks in Eukaryotic Cells. Science 313, 848–851 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Wilson MR et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 363, eaar7785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xue M et al. Structure elucidation of colibactin and its DNA cross-links. Science 365, eaax2685 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dejea CM et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359, 592–597 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bullman S et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443–1448 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kostic AD et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14, 207–215 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wirbel J et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 25, 679 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buc E et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PloS One 8, e56964 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arthur JC et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 338, 120–123 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bossuet-Greif N et al. The Colibactin Genotoxin Generates DNA Interstrand Cross-Links in Infected Cells. mBio 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexandrov LB et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 578, 94–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nik-Zainal S et al. Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drost J et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 358, 234–238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato T et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Tuveson D & Clevers H Cancer modeling meets human organoid technology. Science 364, 952–955 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Kucab JE et al. A Compendium of Mutational Signatures of Environmental Agents. Cell 177, 821–836.e16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jager M et al. Measuring mutation accumulation in single human adult stem cells by whole-genome sequencing of organoid cultures. Nat. Protoc. 13, 59–78 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Cougnoux A et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 63, 1932–1942 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Bartfeld S et al. In Vitro Expansion of Human Gastric Epithelial Stem Cells and Their Responses to Bacterial Infection. Gastroenterology 148, 126–136.e6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z-R et al. Divergent biosynthesis yields a cytotoxic aminomalonate-containing precolibactin. Nat. Chem. Biol. 12, 773–775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Priestley P et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Perez A et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 10, 1081–1082 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee-Six H et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532–537 (2019). [DOI] [PubMed] [Google Scholar]
- 27.McLellan LK & Hunstad DA Urinary Tract Infection: Pathogenesis and Outlook. Trends Mol. Med. 22, 946–957 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zawadzki PJ et al. Identification of infectious microbiota from oral cavity environment of various population group patients as a preventive approach to human health risk factors. Ann. Agric. Environ. Med. 23, 566–569 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Banerjee S et al. Microbial Signatures Associated with Oropharyngeal and Oral Squamous Cell Carcinomas. Sci. Rep. 7, 4036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boot A et al. Mutational signature analysis of Asian OSCCs reveals novel mutational signature with exceptional sequence context specificity. bioRxiv 368753 (2018) doi: 10.1101/368753. [DOI] [Google Scholar]
- 31.Payros D et al. Maternally acquired genotoxic Escherichia coli alters offspring’s intestinal homeostasis. Gut Microbes 5, 313–512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olier M et al. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes 3, 501–509 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beimfohr C A Review of Research Conducted with Probiotic E. coli Marketed as Symbioflor. Int J Bacteriol 2016, 3535621 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
REFERENCES FROM METHODS SECTION
- 34.Blokzijl F et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heo I et al. Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nat. Microbiol. 3, 814–823 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pace P et al. FANCE: the link between Fanconi anaemia complex assembly and activity. EMBO J. 21, 3414–3423 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H & Durbin R Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osorio FG et al. Somatic Mutations Reveal Lineage Relationships and Age-Related Mutagenesis in Human Hematopoiesis. Cell Rep. 25, 2308–2316.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blokzijl F, Janssen R, Boxtel R. van & Cuppen E MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 10, 1–11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cunningham F et al. Ensembl 2015. Nucleic Acids Res. 43, D662–669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cameron DL et al. GRIDSS, PURPLE, LINX: Unscrambling the tumor genome via integrated analysis of structural variation and copy number. bioRxiv 781013 (2019) doi: 10.1101/781013. [DOI] [Google Scholar]
- 42.The National Genomics Research and Healthcare Knowledgebase | Genomics England. https://www.genomicsengland.co.uk/the-national-genomics-research-and-healthcare-knowledgebase/ (2017).
- 43.Raczy C et al. Isaac: ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinforma. Oxf. Engl. 29, 2041–2043 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Lawrence M et al. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 9, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.