

Abstract
Purpose of the Review
Capillary malformations (CM), the most common type of vascular malformation, are caused by a somatic mosaic mutation in GNAQ, which encodes the Gαq subunit of heterotrimeric G-proteins. How the single amino acid change – predicted to activate Gαq - causes CM is not known but recent advances are helping to unravel the mechanisms.
Recent Findings
The GNAQ R183Q mutation is present in endothelial cells isolated from skin and brain CMs, but also in brain tissue underlying the CM, raising questions about the origin of CM-causing cells. Insights from computational analyses shed light on the mechanisms of constitutive activation and new basic science shows Gαq plays roles in sensing shear stress and in regulating cerebral blood flow.
Keywords: Vascular anomalies, capillary malformations, Sturge-Weber syndrome, GNAQ mutation, G-protein signaling
Summary
Several studies confirm the GNAQ R183Q mutation in 90% of non-syndromic and Sturge-Weber syndrome (SWS) CMs. The mutation is enriched in endothelial cells and blood vessels isolated from skin, brain and choroidal CMs but whether the mutation resides in other cell types must be determined. Further, the mechanisms by which the R183Q mutation alters microvascular architecture and blood flow must be uncovered to develop new treatment strategies for SWS in particular, a devastating disease for which there is no cure.
Introduction
Capillary malformation (CM) is a slow flow vascular malformation composed of enlarged capillaries and venules with thickened perivascular cell coverage[1]. The affected vessels show abnormalities such as bulging, sprouting, discontinuities in the endothelium, uneven perivascular coverage and blood stasis (Figure 1). Cutaneous CMs, often called port wine stains (PWS) or port wine birthmarks occur in 3/1000 infants; they can darken and form nodules over time, and in 55–70% of cases soft tissue overgrowth is observed, especially if the CM is located on the lip or cheek[1,2]. Vessel enlargement and wall thickness were measured in infant PWS compared to adjacent normal skin[3], which revealed a bimodal distribution of PWS vessels with increased circumferences. PWS vessels in the 25–200μm range showed a right shift towards larger circumference and a second peak of very large vessels were seen in PWS but not in adjacent normal skin.
Figure 1.

Leptomeningeal blood vessels, stained for endothelium with Ulex Europaeus Agglutinin I (red) and nuclei with Hoechst (blue). Left: Sturge-Weber brain. Right: normal brain, with leptomeningeal vessels in a sulcus seen on the left side of the image, adjacent to a molecular layer and dentate gyrus on the right. Scale bars: 50 μm.
In Sturge-Weber syndrome (SWS), a rare neurocutaneous disorder that occurs in 1/20,000 – 1/50,000 infants, CMs in the ipsilateral leptomeninges and ocular choroid occur along with PWS; see Images in Clinical Medicine[4]. Although a diagnosis of SWS is difficult to make before one year of age, the location of the PWS on a newborn is a strong predictor. A PWS located on the forehead, the midline of the face or in a hemifacial distribution increases risk of SWS to 20–50%[5-7]. Waelchli and colleagues noted the association of where PWS are located with embryonic vascular development of the face rather than trigeminal nerve involvement, a long-held notion[5]. Children with SWS are at high risk for seizures, stroke-like episodes and cognitive delays, which are thought to be due to the vascular stasis and poor perfusion in the cortex beneath the leptomeningeal CM. Further, ipsilateral CM can occur in the choroid plexus, leading to glaucoma, retinal detachment and choroidal bleeding. There is a pressing need to accurately diagnose infants at high risk for SWS so that preventative therapies may be considered. T1 weighted magnetic resonance imaging with gadolinium contrast is currently the standard neuroimaging procedure but it is not ideal. A new MRI-based approach that compares quantitative apparent diffusion coefficient (ADC) maps to an atlas of ADC maps from age-matched controls shows promise as a safer method to identify infants at high risk of SWS seizures[8].
The causal mutation in CM is firmly established as several laboratories have confirmed the landmark finding by Shirley and colleagues that identified the same somatic mutation in GNAQ – the arginine (R) at amino acid position 183 replaced by glutamine (Q) - in 90% on non-syndromic PWS and SWS brain cases [9-12]. Recently the R183Q mutation was identified in choroidal CM in SWS[13,14], linking the three sites – skin, brain and eye. The R183Q mutation was documented in 4 cases of a rare form of SWS that presents without skin or ocular involvement, called forme fruste or SWS type III[15]. In a small number of cases, the GNAQ mutation results in a different amino acid at R183 –e.g., glycine (G) or leucine (L)[11,12]. The loss of the arginine R is likely most important, as this reduces hydrogen bonding between Gαq and guanosine diphosphate (GDP), a critical bond needed for assembly of the GDP-Gαβγ “inactive” complex[16]. Thus, R183Q activates Gαq, which in turn activates phospholipase C-β (PLCβ)[17], (Figure 2).
Figure 2.
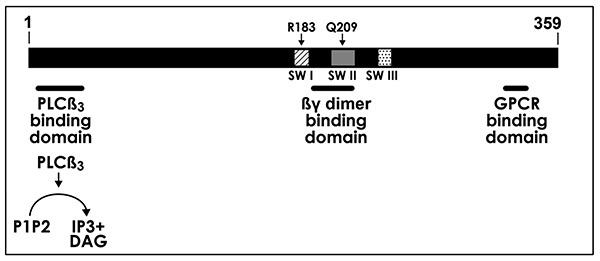
The human Gαq polypeptide is 359 amino acids in length. It contains three α-helical domains called switch I (SWI), switch II (SWII) and switch III (SWIII) shown as patterned boxes. The location of amino acid residues R183 in SWI and Q209 in SWII are shown. The amino-terminal domain of Gαq interacts with PLCβ3[33]; Gαq activates PLCβ3 to hydrolyze PIP2 into inositol 1, 4, 5 triphosphate (IP3) and diacylglycerol (DAG). The SWI region changes conformation upon receptor binding; the SWII region interacts with the βγ subunits of the heterotrimer G-protein; and putative G-protein coupled receptor (GPCR) binding is located in the C-terminal region.
As noted above, the R183Q is found in approximately 90% of PWS and SWS cases; this leaves approximately 10% unaccounted for. Efforts are underway to identify genetic alterations in R183Q-negative CMs as they will undoubtedly point to up and downstream regulators of Gαq that are needed for proper capillary morphogenesis. Three of eight CM cases without GNAQ mutations, all located on extremities, were found to have a R183C (cysteine) mutation in GNA11[18], which encodes Gα11, a Gαq homolog. Therefore, it is likely that the GNA11 R183C alters cellular function in a similar fashion to GNAQ R183Q. A much rarer form of CM, called CM-arteriovenous malformation (AVM) affects 1/100,000 individuals. The CMs are atypical: small, round, multifocal and fast flow, and associated with AVMs or arteriovenous fistulas. RASA1 loss-of-function mutations are found in approximately 50% of patients with CM-AVM [19], and recently, loss of function mutations in EPHB4, an upstream regulator of RASA1, were found in CM-AVM patients without RASA1 mutation. [20]. RASA1 inactivates Ras p21 by enhancing Ras-GTPase such that loss of RASA1 or EPHB4 function should result in chronic activation of the Ras/MAPK pathway. Mice genetically engineered to be mosaic for Rasa1 loss-of-function were found to develop abnormal cutaneous vessels, underscoring the critical role of Rasa1 and the Ras/MAPK pathway in vascular development[21].
Recent developments/advances –
Our understanding of how the GNAQ R183Q somatic activating mutation causes CM is emerging slowly. Here we will discuss recent findings in three areas that have made important progress and at the same time opened up new questions.
1). Cellular origin –
PWS and SWS brain tissue specimens are mosaic for the R183Q mutation, evident from mutant allelic frequencies that range from 1–18%[9]. Endothelial cells isolated from skin and brain CM tissue showed increased mutant allelic frequencies compared to whole tissue indicating an endothelial enrichment and localization[12,22,23]. Hematopoietic and perivascular cell fractions were devoid of mutant cells, but not the stromal fraction, which showed mutant allelic frequencies as high as 8–9% in SWS brain [22]. Thus, there are unidentified cells that contain the GNAQ R183Q mutation. In a new study, investigators separated SWS brain tissue blocks into leptomeningeal CM and adjacent brain and tested for the GNAQ R183Q mutation. They found mutant alleles (0.16–2.9%) in the CM-adjacent brain tissue, showing that mutant cells reside in areas outside of the vascular malformation [24]. The authors suggest that R183Q mutant cells outside of the CM may be responsible for the brain abnormalities in SWS. It is also conceivable that the GNAQ R183Q mutation is “silent” in some cells and in certain locations due to microenvironmental factors such hemodynamic shear forces and/or paracrine signals from surrounding cells, and as such would have no functional impact. Such hypotheses may be tested when animal models with cell-specific and mosaic mutant alleles are developed. In summary, a hypothesis has emerged that the GNAQ R183Q mutation occurs in a progenitor cell that can give rise to multiple lineages, including the endothelial lineage, and that descendent cells can migrate to different locations in the developing embryo. Consistent with this, one report suggests the mutation arises in a mesodermal progenitor cell based on detection of GNAQ R183Q in blood samples from SWS patients using an ultra-sensitive droplet digital PCR technique [25].
2). Mutant Gαq signaling –
There is very little known mechanistically about how the GNAQ R183Q mutation affects signaling in endothelial cells, a cell that contains the mutant allele in PWS and SWS CM lesions. The mutation has been expressed in immortalized cells such as the human embryonic kidney cell line 293T and selected pathways analyzed based on what is known about the GNAQ Q209L mutation that drives uveal melanoma[9]. In 293T cells, GNAQ R183Q expression elicits a slight increase in phosphorylation of the mitogen-activated protein kinase (MAPK) target ERK compared to expression of GNAQ Q209L which increases phosphorylation of several targets (ERK, P38 and JNK) strongly. Endothelial-localized phosphorylated ERK has been reported in SWS brain vessels supporting the activation seen in 293T cells[26].
More investigation is needed to identify the Gαq signaling pathways impacted by the R183Q mutation, the duration and magnitude of the signaling, and further which of the perturbed pathways disrupt capillary morphogenesis, blood flow and neurovascular coupling. While the GNAQ Q209L mutation provides a template, a recent computational analysis predicts that the R183Q mutation drives activation of distinct downstream targets[16]. Briefly, the loss of arginine (R) at amino acid position 183 abolishes hydrogen bonding between the arginine and GDP, which effectively destabilizes the inactive GDP-bound Gαq and favors the active guanosine-triphosphate (GTP)-bound form. The R183Q mutation also affects the conformation of the switch 1 domain of Gαq which may alter interaction with the ADP-ribosylation factor 6 (ARF6), an immediate downstream effector that controls endocytosis of activated Gαq complexes[27]. In contrast, the Q209L mutation impairs reassociation of the Gαq subunit with the Gβ subunit which is needed to reform the inactive heterotrimeric complex and shut off signaling (Figure 2). The Q209L mutation also reduces hydrogen bonding between Q209 and negative regulators of G-protein signaling (RGS) 2 and 8. In summary, the differential effects of R183Q and Q209L on GDP binding affinity and heterotrimerization respectively may impact downstream signaling dynamics. New compounds such as the cyclic depsipeptide FR9000359 that specifically inhibit GDP/GTP exchange on Gαq and thereby favor the inactive GDP-bound Gαβγ heterotrimer[28] may prove to useful in sorting out relevant pathways affected by the R183Q mutation.
3). Cellular Effects –
Recent studies have shown that Gαq is involved in endothelial cell sensing of shear stress imposed by blood flow. Human umbilical artery endothelial cells exposed to either laminar or disturbed-flow shear stress activated the mechanosensor Piezo1, the G-protein coupled receptor (GPCR) P2Y and Gαq but then diverged [29]. Under laminar flow, endothelial nitric oxide synthase (eNOS) was upregulated whereas disturbed flow triggered the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and integrinα5, activating pro-inflammatory pathways. It is unclear how and at what point the different pathways diverge given the same initial signaling via Gαq but it is intriguing to speculate that the R183Q mutation in Gαq may disrupt the endothelial cell’s ability to distinguish between laminar and disturbed flow. Others showed that Gαq, being anchored in the lipid bilayer, can be activated solely by shear stress and independently from GPCR signaling[30], which further points to a central role for Gαq in sensing flow. Based on these new findings, one might speculate that capillary malformations form because the R183Q mutation in GNAQ alters the ability of Gαq to sense and integrate shear stress signals. One may further speculate that the R183Q mutation uniquely impacts Gαq sensing in the capillary-venule microenvironment but may have a lesser or no impact on mutant endothelial cells in large arteries or veins. That is, R183Q mutant endothelial cells residing in large vessels may not have the same impact on vessel architecture.
Recent studies have also connected Gαq to cerebral blood flow, which is known to be altered in the brains of patients with SWS. Normally, local neuronal activity is relayed from brain capillary endothelial cells to upstream to vasoactive feeding arterioles, which vasodilate to increase local blood supply to meet the demand for oxygen. Harraz and colleagues show that Gαq is part of a molecular switch critical for this process based on its ability to reduce phosphatidylinositol 4,5 biphosphate (PIP2) via its activation of phospholipase-Cβ (Figure 2). A Gαq-stimulated drop in PIP2 blocks the capillary potassium channel Kir2.1 and at the same time relieves inhibition of the calcium/sodium channel TRPV4 [31,32]. In effect, the drop in PIP2 acts to reset capillary-initiated electrical signaling, it reduces blood flow and prepares the cells to respond to subsequent neuronal signals. Constitutively active R183Q Gαq would likely disrupt this neurovascular coupling mechanism, a speculation that is consistent with the poor regional cerebral perfusion and chronic ischemia often seen in the brain of infants with SWS.
Conclusion –
At present, PWS and SWS are medically managed but there is no cure. The discovery of the GNAQ R183Q mutation in ~90% of cases provides a focal point but the questions that arise are immense. In what cell does the missense mutation occur and how do its progeny distribute to the primary locations of CM in SWS. Does the mutation impart cell autonomous or non-cell autonomous dysfunctions, or both? What are critical downstream effectors that cause the brain, ocular and skin abnormalies and can they be blocked specifically and without harm? Continued basic science – including the development of in vitro and in vivo models, computational studies, genomics and drug screens are essential in order to accelerate discovery and translate them to new treatments for SWS.
Key Bullet Points.
The GNAQ R183Q mutation is found in 90% of patients affected by CM and is enriched in endothelial cells, but the originating cell in which the mutation occurs is still unknown.
Both R183Q and the GNAQ Q209L mutation in uveal melanoma are “activating” but insights from G-protein structural biology indicate the precise mechanisms by which the individual mutations cause overactivation may differ, leading to differences in downstream signaling.
Recent work has linked Gαq signaling to regional blood flow regulation, raising the question of whether and how the GNAQ R183Q mutation affects neurovascular coupling in SWS.
Acknowledgements –
We thank Sanda Alexandrescu, M.D., Department of Pathology, Boston Children’s Hospital, for her expertise on brain histological sections.
Financial Support and Sponsorship –
Supported by the National Institutes of Health award number HL127030 (J.B.) and the Sturge-Weber Foundation (C.B.)
Footnotes
Conflicts of Interest – The authors have no conflicts of interest to disclose.
References
* of special interest (4), ** of outstanding interest (4)
- 1.Mulliken JB: Capillary Malformations, Hyperkeratotic Stains, Telangiectasias, and Miscellaneous Vascular Blots. In Vascular Anomalies: Hemangioma and Malformations, edn Second edition /. Edited by Mulliken JB, Burrows PE, Fishman SJ: Oxford University Press; 2013:xviii, 1118 pages. [Google Scholar]
- 2.Greene AK, Taber SF, Ball KL, Padwa BL, Mulliken JB: Sturge-Weber syndrome: soft-tissue and skeletal overgrowth. The Journal of craniofacial surgery 2009, 20 Suppl 1:617–621. [DOI] [PubMed] [Google Scholar]
- 3.Tan W, Wang J, Zhou F, Gao L, Yin R, Liu H, Sukanthanag A, Wang G, Mihm MC Jr., Chen DB, et al. : Coexistence of Eph receptor B1 and ephrin B2 in port-wine stain endothelial progenitor cells contributes to clinicopathological vasculature dilatation. Br J Dermatol 2017, 177:1601–1611.*This study reveals the abnormal phenotype of CM-affected blood vessels in PWS compared to normal skin. The authors found increased vessel circumferance in PWS, thick and thin-walled vessels and a loss of distinct arterial or venous identity.
- 4.Desai S, Glasier C: Sturge-Weber Syndrome. N Engl J Med 2017, 377:e11. [DOI] [PubMed] [Google Scholar]
- 5.Waelchli R, Aylett SE, Robinson K, Chong WK, Martinez AE, Kinsler VA: New vascular classification of port-wine stains: improving prediction of Sturge-Weber risk. Br J Dermatol 2014, 171:861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zallmann M, Leventer RJ, Mackay MT, Ditchfield M, Bekhor PS, Su JC: Screening for Sturge-Weber syndrome: A state-of-the-art review. Pediatr Dermatol 2018, 35:30–42. [DOI] [PubMed] [Google Scholar]
- 7.Dutkiewicz AS, Ezzedine K, Mazereeuw-Hautier J, Lacour JP, Barbarot S, Vabres P, Miquel J, Balguerie X, Martin L, Boralevi F, et al. : A prospective study of risk for Sturge-Weber syndrome in children with upper facial port-wine stain. Journal of the American Academy of Dermatology 2015, 72:473–480. [DOI] [PubMed] [Google Scholar]
- 8.Pinto ALR, Ou Y, Sahin M, Grant PE: Quantitative Apparent Diffusion Coefficient Mapping May Predict Seizure Onset in Children With Sturge-Weber Syndrome. Pediatr Neurol 2018, 84:32–38.*Identifying children with facial PWS who will develop seizures is a major clinical challenge. The authors used contrast agent-free diffusion MRI scans with apparent diffusion coefficient mapping to identified presymptomatic brain abnormalities in seven infants who later developed seizures. Larger studies are needed to determine if this method can serve as a biomarker for likely onset of seizures.
- 9.Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J: Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. The New England journal of medicine 2013, 368:1971–1979.**This landmark paper identified the somatic mosaic mutation in Gαq, GNAQ R183Q (c.548G>A), in 90% of cases with non-syndromic and Sturge-Weber syndrome capillary malformations. This discovery provided an essential molecular foothold for basic, translational and clinical scientists to tackle PWS and SWS.
- 10.Nakashima M, Miyajima M, Sugano H, Iimura Y, Kato M, Tsurusaki Y, Miyake N, Saitsu H, Arai H, Matsumoto N: The somatic GNAQ mutation c.548G>A (p.R183Q) is consistently found in Sturge-Weber syndrome. J Hum Genet 2014, 59:691–693. [DOI] [PubMed] [Google Scholar]
- 11.Frigerio A, Wright K, Wooderchak-Donahue W, Tan OT, Margraf R, Stevenson DA, Grimmer JF, Bayrak-Toydemir P: Genetic Variants Associated with Port-Wine Stains. PLoS One 2015, 10:e0133158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Couto JA, Huang L, Vivero MP, Kamitaki N, Maclellan RA, Mulliken JB, Bischoff J, Warman ML, Greene AK: Endothelial Cells from Capillary Malformations are Enriched for Somatic GNAQ Mutations. Plastic and reconstructive surgery 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bichsel CA, Goss J, Alomari M, Alexandrescu S, Robb R, Smith LE, Hochman M, Greene A, Bischoff J: Association of Somatic GNAQ Mutation With Capillary Malformations in a Case of Choroidal Hemangioma. JAMA Ophthalmol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francis JH, Milman T, Grossniklaus H, Albert D, Folberg R, Levitin G, Coupland S, Catalanotti F, Rabady D, Kandoth C, et al. : GNAQ Mutations in Diffuse and Solitary Choroidal Hemangiomas. Ophthalmology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hildebrand MS, Harvey AS, Malone S, Damiano JA, Do H, Ye Z, McQuillan L, Maixner W, Kalnins R, Nolan B, et al. : Somatic GNAQ mutation in the forme fruste of Sturge-Weber syndrome. Neurol Genet 2018, 4:e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martins L, Giovani PA, Reboucas PD, Brasil DM, Haiter Neto F, Coletta RD, Machado RA, Puppin-Rontani RM, Nociti FH Jr, Kantovitz KR: Computational analysis for GNAQ mutations: New insights on the molecular etiology of Sturge-Weber syndrome. J Mol Graph Model 2017, 76:429–440.**This study used computational and structural biology methods to predict functional differences between the GNAQ R183Q mutation associated with CM and the Q209 mutation found in 90% of uveal melanoma. While R183Q affects the GDP/GTP exchange in Gαq, Q209 impedes the re-association of Gαq with the Gβγ subunits. Both are needed to return Gαq signaling to the off state, but alterations caused by each mutation likely impact the precise downstream pathways that are altered.
- 17.Litosch I: Decoding Galphaq signaling. Life Sci 2016, 152:99–106. [DOI] [PubMed] [Google Scholar]
- 18.Couto JA, Ayturk UM, Konczyk DJ, Goss JA, Huang AY, Hann S, Reeve JL, Liang MG, Bischoff J, Warman ML, et al. : A somatic GNA11 mutation is associated with extremity capillary malformation and overgrowth. Angiogenesis 2017, 20:303–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, Vanwijck R, Vikkula M: Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet 2003, 73:1240–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amyere M, Revencu N, Helaers R, Pairet E, Baselga E, Cordisco M, Chung W, Dubois J, Lacour JP, Martorell L, et al. : Germline Loss-of-Function Mutations in EPHB4 Cause a Second Form of Capillary Malformation-Arteriovenous Malformation (CM-AVM2) Deregulating RAS-MAPK Signaling. Circulation 2017, 136:1037–1048. [DOI] [PubMed] [Google Scholar]
- 21.Henkemeyer M, Rossi DJ, Holmyard DP, Puri MC, Mbamalu G, Harpal K, Shih TS, Jacks T, Pawson T: Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 1995, 377:695–701. [DOI] [PubMed] [Google Scholar]
- 22.Huang L, Couto JA, Pinto A, Alexandrescu S, Madsen JR, Greene AK, Sahin M, Bischoff J: Somatic GNAQ Mutation is Enriched in Brain Endothelial Cells in Sturge-Weber Syndrome. Pediatr Neurol 2017, 67:59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan W, Nadora DM, Gao L, Wang G, Mihm MC Jr., Nelson JS: The somatic GNAQ mutation (R183Q) is primarily located within the blood vessels of port wine stains. J Am Acad Dermatol 2016, 74:380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sundaram SK, Michelhaugh SK, Klinger NV, Kupsky WJ, Sood S, Chugani HT, Mittal S, Juhasz C: GNAQ Mutation in the Venous Vascular Malformation and Underlying Brain Tissue in Sturge-Weber Syndrome. Neuropediatrics 2017, 48:385–389.**The GNAQ R183Q mutation was detected in brain tissue outside of leptomeningeal CMs, although at a lower mutant allelic frequency. This finding raises the possibility that mutant cells are not be confined to the endothelial/vascular compartment and highlights the need to thoroughly investigate the cellular origin of the GNAQ mutant cells to fully understand their impact on CM progression and brain function.
- 25.Uchiyama Y, Nakashima M, Watanabe S, Miyajima M, Taguri M, Miyatake S, Miyake N, Saitsu H, Mishima H, Kinoshita A, et al. : Ultra-sensitive droplet digital PCR for detecting a low-prevalence somatic GNAQ mutation in Sturge-Weber syndrome. Sci Rep 2016, 6:22985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wellman RJ, Cho SB, Singh P, Tune M, Pardo CA, Comi AM, Workgroup BS-WsP: Galphaq and hyper-phosphorylated ERK expression in Sturge-Weber syndrome leptomeningeal blood vessel endothelial cells. Vasc Med 2018:1358863X18786068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoo JH, Shi DS, Grossmann AH, Sorensen LK, Tong Z, Mleynek TM, Rogers A, Zhu W, Richards JR, Winter JM, et al. : ARF6 Is an Actionable Node that Orchestrates Oncogenic GNAQ Signaling in Uveal Melanoma. Cancer Cell 2016, 29:889–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onken MD, Makepeace CM, Kaltenbronn KM, Kanai SM, Todd TD, Wang S, Broekelmann TJ, Rao PK, Cooper JA, Blumer KJ: Targeting nucleotide exchange to inhibit constitutively active G protein alpha subunits in cancer cells. Sci Signal 2018, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albarran-Juarez J, Iring A, Wang S, Joseph S, Grimm M, Strilic B, Wettschureck N, Althoff TF, Offermanns S: Piezo1 and Gq/G11 promote endothelial inflammation depending on flow pattern and integrin activation. J Exp Med 2018, 215:2655–2672.*This paper identifies Gq-protein signaling as integral part in shear sensing in endothelial cells, and demonstrates that while laminar and disturbed flow both signal through Gαq, they activate different downstream targets.
- 30.Dela Paz NG, Melchior B, Frangos JA: Shear stress induces Galphaq/11 activation independently of G protein-coupled receptor activation in endothelial cells. Am J Physiol Cell Physiol 2017, 312:C428–C437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harraz OF, Longden TA, Dabertrand F, Hill-Eubanks D, Nelson MT: Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. Proc Natl Acad Sci U S A 2018, 115:E3569–E3577.**Gαq activates phospholipase Cβ, which hydrolyzes PIP2 into diacylglycerol and IP3. This study shows that PIP2 depletion caused by activated GqPCR signaling affects an electrical signal relay from brain capillary endothelial cells to small arteries. The brain endothelial-localized R183Q mutation in Gαq may disrupt this neurovascular regulation, and thereby contribute to poor blood flow and chronic ischemia observed in brains of children with SWS
- 32.Harraz OF, Longden TA, Hill-Eubanks D, Nelson MT: PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. Elife 2018, 7.**This follow up study underscores the integral role Gαq signaling plays in neurovascular coupling by its affect on PIP2 levels. Neuronal activity sensed by GqPCR signaling decreases PIP2 levels, which in turn modulates ion-channel activation and ultimately local perfusion. Further work is needed to determine if the constitutive activation of Gαq in brain endothelial cells - as a result of the GNAQ R183Q mutation - disrupts regulation of PIP2 levels.
- 33.Lyon AM, Dutta S, Boguth CA, Skiniotis G, Tesmer JJ: Full-length Galpha(q)-phospholipase C-beta3 structure reveals interfaces of the C-terminal coiled-coil domain. Nat Struct Mol Biol 2013, 20:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]