

Abstract
Benzodiazepines are the primary treatment option for organophosphate (OP)-induced status epilepticus (SE), but these antiseizure drugs (ASDs) lose efficacy as treatment is delayed. In the event of a mass civilian or military exposure, significant treatment delays are likely. New ASDs that combat benzodiazepine-resistant, OP-induced SE are critically needed, particularly if they can be efficacious after a long treatment delay. This study evaluated the efficacy of the Kv7 channel modulator, retigabine, as a novel therapy for OP-induced SE. Adult, male rats were exposed to soman or diisopropyl fluorophosphate (DFP) to elicit SE and monitored by electroencephalogram (EEG) recording. Retigabine was administered alone or adjunctive to midazolam (MDZ) at delays of 20- or 40-min in the soman model, and 60-min in the DFP model. Following EEG recordings, rats were euthanized and brain tissue was collected for Fluoro-Jade B (FJB) staining to quantify neuronal death. In the DFP model, MDZ + 15 mg/kg retigabine suppressed seizure activity and was neuroprotective. In the soman model, MDZ + 30 mg/kg retigabine suppressed seizures at 20- and 40-min delays. Without MDZ, 15 mg/kg retigabine provided partial antiseizure and neuroprotectant efficacy in the DFP model, while 30 mg/kg without MDZ failed to attenuate soman-induced SE. At 60 mg/kg, retigabine without MDZ strongly reduced seizure activity and neuronal degeneration against soman-induce SE. This study demonstrates the antiseizure and neuroprotective efficacy of retigabine against OP-induced SE. Our data suggest retigabine could be a useful adjunct to standard-of-care and has potential for use in the absence of MDZ.
Keywords: nerve agent, soman, diisopropyl fluorophosphate, Fluoro-Jade B, electroencephalogram, rat
Introduction
Organophosphates (OP) are potent toxic chemicals that pose a widespread danger to public health. OP pesticides are used around the globe for agricultural purposes, and each year 3 million people are poisoned resulting in an estimated 300,000 deaths (Robb & Baker, 2020). They are also a leading cause of global suicide accounting for up to 20% of all cases (WHO, 2019). In addition to OP pesticides, OP nerve agents pose significant health threats to both military and civilian personnel. OP nerve agents have been used in warfare as recently as the ongoing Syrian conflict (Costanzi et al., 2018; Clarke & Weir, 2019), and terrorist attacks by the cult Aum Shinriyko in Japan where the OP nerve agent sarin was used in the poisoning of over 5000 civilians (Yanagisawa et al., 2006).
OP compounds produce their deadly effects through the irreversible inhibition of the enzyme acetylcholinesterase (AChE) (Costanzi et al., 2018). AChE is found widely throughout the central and peripheral nervous systems and is crucial for the regulation of neuronal function through the breakdown of acetylcholine (ACh) within cholinergic synapses. If AChE is inhibited, excess ACh floods post-synaptic targets throughout the body (McDonough & Shih, 1997; Shih & McDonough, 1997). This “cholinergic crisis” symptomatically manifests as vomiting, hypersecretions, miosis, fasciculations, paralysis, status epilepticus (SE) and lethal respiratory and circulatory depression (King & Aaron, 2015).
OP intoxication is typically treated with a polytherapy regimen of the cholinergic antagonist atropine, an AChE reactivator oxime (such as 2-PAM), and an antiseizure benzodiazepine (McDonough & Shih, 1997). Termination of OP-induced SE is a critical component of treatment of OP poisoning. As little as 20 min of uncontrolled SE can produce neuropathological consequences, and neuronal damage can result in permanent cognitive dysfunction (Petras, 1994; McDonough & Shih, 1997; McDonough et al., 1998; Chen, 2012; Myhrer et al., 2018; Wu et al., 2018). Benzodiazepines are usually the first line of antiseizure treatment for SE and are most effective when administered immediately upon convulsive symptoms. However, as treatment times are delayed, benzodiazepines rapidly lose their effectiveness (Walton & Treiman, 1988; Towne et al., 1994; Lowenstein & Alldredge, 1998; Rice & DeLorenzo, 1999; Shih et al., 1999; Jones et al., 2002; McDonough et al., 2010; Jackson et al., 2019). For instance, following exposure to the nerve agent soman, diazepam loses efficacy when administered after a 40-min treatment delay (Shih et al., 1999). Unfortunately, given the time-dependent decrease in benzodiazepine antiseizure efficacy, novel approaches to treat OP-induced SE are critically needed. The need is especially pronounced in the event of high consequence, mass casualty public health chemical emergencies (and a major focus of the United States government)(Yeung et al., 2020). In such scenarios, significant delays in post-exposure medical treatments are anticipated as the immediate availability of first responders and medical products are likely to be constrained.
Two recent studies have demonstrated that M-type potassium channel openers (flupirtine and retigabine) may be effective adjunctive therapy to standard-of-care treatments in models of OP-exposure (Zhang et al., 2017; Gore et al., 2020). M-type (KCNQ/Kv7) potassium channels play an important role in regulating neuronal resting membrane potential, action potential (AP) threshold, and AP firing (Peters et al., 2005; Shah et al., 2008; Brown & Passmore, 2009), and are inactivated by cholinergic signaling through the muscarinic acetylcholine receptor (Selyanko et al., 2000; Brown & Passmore, 2009). Here, we present data demonstrating the effect of retigabine, alone or in conjunction with midazolam, as an antiseizure drug (ASD) in two rat models of OP-induced SE at treatment delays that normally render benzodiazepines ineffective.
Materials and Methods
Drugs
Retigabine was purchased from Axon Medchem (Reston, VA, cat#:1525) and dissolved in 100% PEG200. Retigabine was formulated at concentrations ranging from 5–20 mg/ml and administered intraperitoneally (IP) at doses ranging from 2–60 mg/kg.
In the DFP model, DFP was acquired from Battelle Memorial Institute (Columbus, OH) (Hess et al., 2016) and aliquoted at a concentration of 1 mg/100 μl (saline vehicle) for storage at −80°C. Atropine methyl nitrate (AMN) was purchased from Spectrum Chemicals (New Brunswick, NJ) and formulated at 4 mg/ml in saline vehicle). Pyridostigmine bromide and 2-PAM were purchased from Sigma-Aldrich (St. Louis, MO) and formulated at 0.052 mg/ml and 50 mg/ml respectively, in saline vehicle. MDZ was purchased pre-compounded at 5 mg/ml in saline from Akorn Pharmaceuticals (Vernon Hills, IL).
In the soman model, all of the following drugs were dissolved in sterile saline. HI-6 was acquired from Kalexsyn Medicinal Chemistry (Kalamazoo, MI) and formulated at 250 mg/ml. AMN was acquired from Sigma-Aldrich (St. Louis, MO) and formulated at 4 mg/ml. Soman was synthesized by the US Army Combat Capabilities Development Command Chemical Biological Center (Aberdeen Proving Ground, MD) and diluted to a final concentration of 360 μg/ml. Atropine sulfate was synthesized by Sparhawk laboratories (Lenexa, KS) and formulated at 0.9 mg/ml. Atropine sulfate was admixed with 2-PAM synthesized by Baxter Healthcare Corporation (Deerfield, IL) and formulated at 50 mg/ml. Midazolam was purchased pre-formulated at a concentration of 5 mg/ml from Akorn Pharmaceuticals (Vernon Hills, IL).
Animals
In both models of OP-induced SE, male Sprague-Dawley rats were purchased from Charles River (Wilmington, MA). In the DFP model, rats were ordered at a pre-surgery weight of 50–75 grams whereas in the soman model rats were ordered at a pre-surgery weight of 250–300 grams. All rats were housed in individual cages in a temperature- and humidity-controlled vivarium that was on a 12-hr light/dark cycle with ad libitum access to food and water except during experimental procedures in the soman model.
EEG Implant Surgery
All surgical procedures used in these experiments were reviewed and approved by the Institutional Animal Care and Use Committees (IACUC) at the United States Army Medical Research Institute of Chemical Defense and the University of Utah. All procedures for these experiments were conducted in accordance with the principles stated in the Guide for the Care and Use of Laboratory Animals, and the Animal Welfare Act of 1966 (P. L. 89–544), as amended. In both models, surgical implantation of cortical electroencephalogram (EEG) electrodes occurred 5–7 days prior to OP-exposure.
In the DFP model, rats were pretreated with dexamethasone (2.0 mg/ kg, SC) before being anesthetized with 3% isoflurane and placed in a stereotaxic instrument. Anesthesia was maintained at 1–5% for the duration of the procedure. After positioning, a longitudinal midline incision was made in the scalp, followed by lateral retraction to expose the skull. Next, six 500-μm holes were drilled through the skull, and small screws were placed in three of the holes for anchoring the headset. Then two 2–3 mm long bipolar recording electrodes (MS333-3-B, Plastics One, Roanoke, VA) were placed in two of the remaining holes on the right side of the midline, and a ground electrode was positioned through a hole on the left side of the skull. The headset was then dental cemented into place. Finally, the wound was closed with sutures, penicillin (0.2ml, SQ, 300,000 IU), 1.2 ml lactated ringer’s solution (SC), and topical lidocaine was administered and the rats were returned to their home cages for recovery. Buprenorphine (0.05 mg/kg, SQ) was also administered at the end of surgery, and then twice per day for 1 day post-surgery.
In the soman model, rats were administered the analgesics meloxicam (subcutaneous (SC), 1 mg/kg, at least 15 min before surgery), and bupivacaine (intradermal, 2.5 mg/ml, 0.1 ml total volume, at start of surgery). Rats were anesthetized with isoflurane (3–5% for induction followed by 1–3% for maintenance) and surgical plane was determined by the rat’s inability to respond to painful stimuli. To implant the EEG electrodes, a midline incision was made to expose the underlying skull. Next, hand drilled, bilateral burr holes were made above the parietal cortices as well as the cerebellum for a reference electrode. Stainless steel screw electrodes were then placed into the burr holes and connected to a recording headset. The completed EEG recording headset was secured in place by glass ionomer cement and the surgical incision was closed with sutures. Throughout the surgery, the rat’s heart rate, temperature, and oxygen saturation were monitored. At the conclusion of the surgery and recovery from anesthesia, rats were returned to their home cages.
DFP Exposure
On the day of DFP exposure, rats were weighed (150–225 g, estimated ~7 weeks old) and placed into individual Plexiglass® EEG recording chambers. Implanted EEG electrodes were connected to spring covered cables (Plastics One, Roanoke, VA) to begin the recording. The experimental protocol used a delayed treatment rodent model of DFP exposure to cause OP-induced SE (Pouliot et al., 2016). Recordings began with 60-min of baseline EEG with pyridostigmine bromide (0.026 mg/kg) administered intramuscularly (IM) 30-min into this recording period. After baseline EEG was recorded, DFP (5.5 mg/kg, SC) was administered. AMN (2 mg/kg, IM) and 2-PAM (25 mg/kg, IM) were given 1 min after DFP injection to decrease mortality caused by OP-induced peripheral toxicity and to ensure that a majority of rats survived throughout the entirety of the experiment. After the administration of DFP, EEG recordings were directly observed and the time of electrographic seizure initiation was noted. Initiation was determined by the appearance of repetitive spikes and sharp waves with an amplitude greater than twice that of the baseline EEG and having a sustained duration of more than 10 s. At 60-min after the start of ictal activity, rats were administered MDZ (1.78 mg/kg, IM) in conjunction with either retigabine (2–30 mg/kg, IP from 5 mg/ml solution) or PEG200 vehicle. EEG recordings persisted for 24 hr following treatment at which time rats were euthanized and brain tissue was collected for histopathology.
Soman Exposure
On the day of soman exposure, rats were weighed (284–460 g, estimated ~12 weeks old), placed into individual Plexiglass® EEG recording chambers and connected via headset tether to the EEG recording system. Baseline EEG was recorded for 60 min, with an HI-6 pretreatment (IP) administered 30 min into the recording period. Following the baseline EEG recording, the OP nerve agent soman was administered (SC) at a dose of 180 μg/kg, an amount that elicited EEG seizure activity in 100% of rats studied. One minute after the administration of soman, AMN was given (IM) at a dose of 2 mg/kg. AMN, in conjunction with HI-6 pretreatment, was given to protect against systemic nerve agent toxicity, thus increasing the number of rats that survived until the onset of neurological symptoms. OP-induced SE initiated at an average latency of 4 min and 27 s (SEM = ± 14 s) after soman administration based on the same criteria described for the DFP model. At 20- or 40-min after the onset of SE, rats were administered 0.45 mg/kg atropine sulfate admixed with 25 mg/kg 2-PAM (IM), 1.8 mg/kg MDZ (IM), and either PEG200 vehicle or retigabine (IP). In some experiments, MDZ was not administered to test the stand-alone efficacy of retigabine. EEG recordings were continued for 4 hr following treatment, at which time drug efficacy was evaluated. If rats remained in SE, treatment was considered a failure and rats were euthanized so brain tissue could be collected for histopathology. If a rat’s EEG recordings revealed no ictal activity, they were returned to their home cage. Twenty-four hours after soman exposure, these rats were again attached to the recording system and EEG was recorded for 30 min to determine if SE termination persisted. Following this recording session, rats were euthanized and brain tissue was collected for histopathology.
EEG Recording and Analysis
In the DFP model, EEG signals were recorded using AcqKnowledge software (BioPac Systems, Inc. Santa Barbara, CA), amplified using EEG100 amplifiers (high-pass filter 1 Hz, low-pass filter 100 Hz, notch filter 60 Hz, 5000x gain) and digitized at 500 Hz with a MP150 analog-to-digital converter. In the soman model, EEG signals were recorded using Spike2 software (Cambridge Electronic Design Limited, Cambridge, England). EEG signal was amplified with 1902 amplifiers and digitized using a Micro1401 data acquisition interface (both from Cambridge Electronic Design Limited, Cambridge, England). EEG data channels were sampled at 512 Hz and then digitally filtered with a 0.3 Hz high-pass filter, a 100 Hz low-pass filter and a 60 Hz notch filter,
In both models, two previously described algorithms were used to analyze the intensity of electrographic SE from the collected EEG following the administration of soman or DFP. Gamma (20–60 Hz) power spectral density along with spike rate were calculated using custom Python-based software developed at the University of Utah (White et al., 2006; Lehmkuhle et al., 2009). Changes in mean power and spike rate frequency during SE were calculated by subtracting baseline values of these variables that were measured during a 10-min time bin prior to the administration of DFP or soman from values determined during sequential, 50% overlapping bins (5-min bin size in soman model, 15-min bin size in DFP model) during SE. If an accurate baseline determination could not be made due to excessive artifact or lack of sufficient data for comparison, a rat’s EEG data was removed from the study. In addition, in the soman model the raw EEG was examined by two experienced researchers and each animal was consensually rated as having the SE terminated or not terminated, based on the overall appearance of the EEG record (McDonough et al., 2010; Jackson et al., 2019). To be rated as having SE terminated, all spiking and/or rhythmic waves had to stop and the EEG remain free of ictal activity. This allowed for an estimate of the latency for SE control for the different experimental treatments.
Tissue Preparation and Histopathology
Upon completion of EEG studies in the DFP model, rats were deeply anesthetized with isoflurane and perfused with phosphate-buffered saline. Following exsanguination, tissue was perfused with 10% formalin and fixed brains were removed and placed into a 30% sucrose solution for cryoprotection. Brains were then flash frozen and coronally sliced (40 μm) between Bregma coordinates −1.8 to −6.3 mm (Paxinos, 2007). The brain sections were slide-mounted and subsequently stained for FJB using previously described techniques (Schmued & Hopkins, 2000). Stained brain slices were imaged using a Hamamatsu Nanozoomer 2.0 HT (Olympus) and FJB positive neurons were quantified as previously described (Johnstone et al., 2019; Spampanato et al., 2019; Barker et al., 2020). Brain regions of interest were selected based on previous research demonstrating that they play a role in initiating, propagating, and/or maintaining seizure activity (Gale, 1992; Myhrer, 2007; Myhrer et al., 2008; Apland et al., 2010; Skovira et al., 2010; Skovira et al., 2012).
Following the completion of EEG recordings in the soman model, rats were deeply anesthetized with sodium pentobarbital and perfused transcardially with phosphate-buffered saline. Once fully exsanguinated, rats were then perfused with 10% formalin to fix tissue. When perfusion was completed, the rat’s brain was removed and stored in formalin until they could be embedded in paraffin and sectioned coronally (5 μm slices). Brain sections in the area 3.24 mm posterior to Bregma were slide-mounted and stained for FJB positive neurons as previously described (Schmued & Hopkins, 2000). Once stained, brain sections were imaged with an Olympus VS120-L100-W microscope using VS-ASW software (Olympus Corporation, Tokyo, Japan). Areas of interest from each scanned brain section were cropped out using FIJI software using the dimensions previously described (Johnstone et al., 2019). Counting in the hippocampus included all FJB positive neurons within the anatomical boundaries of the structure. FJB positive cell counting was performed by treatment-blinded laboratory technicians.
Statistics
EEG and histopathology data were analyzed using GraphPad Prism 7 software (Graphpad Software, San Diego, California). For EEG analysis, values for change in power and spike frequency relative to baseline were compared hourly for retigabine- and vehicle-treated groups using an unpaired t-test or one-way ANOVA. For histopathology data, the mean number of FJB positive neurons per brain region was calculated for retigabine- and vehicle-treated rats and compared using an unpaired t-test or one-way ANOVA. For all statistical analyses, p<0.05 was considered to be statistically significant.
Results
MDZ + retigabine (15 mg/kg) provides prolonged antiseizure efficacy and neuroprotection in a DFP model of OP-induced SE
In the DFP model, MDZ was administered in conjunction with retigabine (2 or 15 mg/kg) or PEG200 vehicle 60 min after the initiation of SE. Rats (n=21) were treated with MDZ + 15 mg/kg retigabine and of these, 2 died following treatment (p>0.05, Fisher’s exact test; survival, MDZ + RTG vs. MDZ + vehicle, Table 1). The rapid and robust antiseizure efficacy of MDZ + 15 mg/kg retigabine compared to standard-of-care MDZ + PEG200 vehicle can be easily seen in the raw, compressed EEG traces (Figure 1A), and this suppression of SE was consistent for the group of rats treated similarly as can be seen in the averaged, quantitative EEG analysis (Figure 1B, lines are group average and shaded areas are 95% confidence). Compared to MDZ + PEG200 controls (n = 18), rats that received MDZ + 15 mg/kg retigabine demonstrated significant reductions in gamma power at 1–18 hr following the administration of treatment (Figure 1B1). Similar results on the efficacy of retigabine were seen in mean spike-frequency analysis, with MDZ + 15 mg/kg retigabine-treated rats showing significant reductions at 1–20 hr post-treatment compared to MDZ + PEG200 controls (Figure 1B2). No significant enhancement of the MDZ effects were seen when 2 mg/kg retigabine was co-administered (Figure 1B1 and 1B2).
Table 1:
Summary of treatment mortality.
| Treatment | Animal’s Treated | 4 hr Post-treatment Mortality (n) | 24 hr Post-treatment Mortality (n) | Seizure Termination (n) |
|---|---|---|---|---|
| MDZ + PEG200 | 19 | 1 (5%) | 0 | N.E. |
| MDZ + 2 mg/kg Retigabine | 16 | 0 | 0 | N.E. |
| MDZ + 15 mg/kg Retigabine | 21 | 1 (5%) | 1 (5%) | N.E. |
| MDZ alone | 18 | 0 | 0 | N.E. |
| 15 mg/kg Retigabine | 19 | 1 (5%) | 0 | N.E. |
| 30 mg/kg Retigabine | 7 | 4 (57%) | 3 (43%) | N.E. |
| Treatment | Animal’s Treated | 4 hr Post-treatment Mortality (n) | No Seizure Termination (n) | Seizure Termination (n) |
| MDZ + PEG200 @ 20 Min | 17 | 1 (6%) | 16 (94%) | 0 (0%) |
| MDZ + 15 mg/kg Retigabine @ 20 Min | 14 | 8 (57%) | 6 (43%) | 0 (0%) |
| MDZ + 30 mg/kg Retigabine @ 20 Min | 12 | 3 (25%) | 1 (8%) | 8 (67%) |
| 30 mg/kg Retigabine @ 20 Min | 8 | 1 (12.5%) | 6 (75%) | 1 (12.5%) |
| 60 mg/kg Retigabine @ 20 Min | 14 | 3 (21%) | 1 (7%) | 10 (71%) |
| MDZ + PEG200 @ 40 Min | 11 | 2 (18%) | 9 (82%) | 0 (0%) |
| MDZ + 30 mg/kg Retigabine @ 40 Min | 15 | 3 (20%) | 3(20%) | 9 (60%) |
Figure 1: Retigabine adjunct to standard-of-care therapy provides dose-dependent enhancement of antiseizure and neuroprotective effects in a delayed treatment model of DFP exposure.
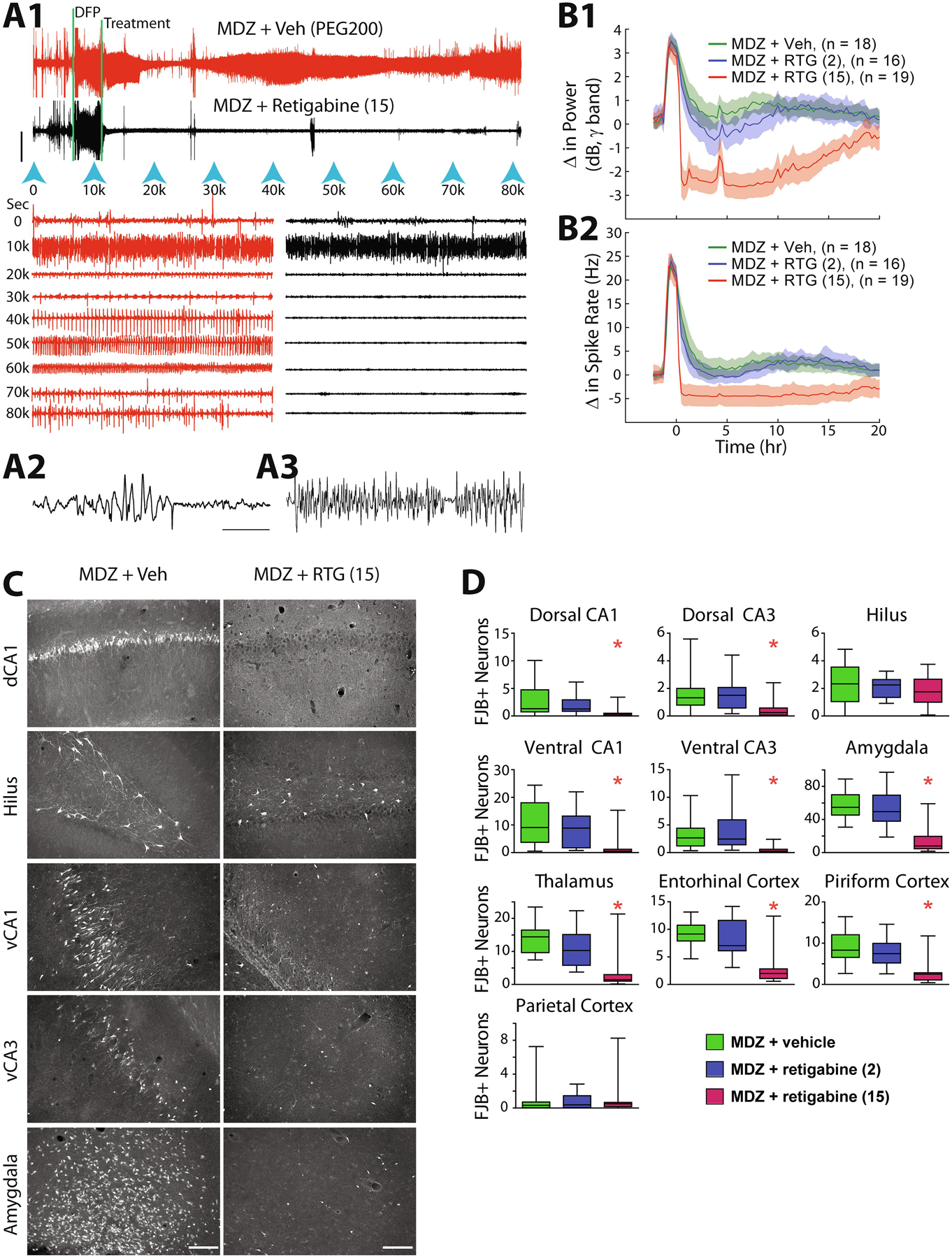
(A1) Compressed traces represent the continuous EEG recorded for more than 22 hr total in two rats exposed to DFP (first vertical bar). Treatment time is indicated by the second vertical bar and represents the time the rat on the top (red trace) received MDZ (1.78 mg/kg, IM) + vehicle (PEG200, IP) and the bottom rat (black trace) received MDZ (1.78 mg/kg, IM) + 15 mg/kg retigabine (IP). Blue arrowheads indicate the time in seconds and correspond to the expanded traces below (1-min duration). In both the compressed and expanded traces, clear transition into SE following DFP can be seen prior to treatment, followed by the complete suppression of SE in the MDZ + retigabine treated rat compared to the transient suppression seen in the MDZ + vehicle rat. Calibration bar = 1 mV. (A2) Further expanded EEG trace demonstrating movement artifact compared to (A3) untreated seizure activity during DFP-induced SE. Calibration bar = 2 s. (B1) Automated calculation of change in power in the gamma band (20–60 Hz) and (B2) change in the mean spike rate (Hz) are presented for the cohort of rats treated with either MDZ + vehicle (green lines), MDZ + 2 mg/kg retigabine (blue lines) and MDZ + 15 mg/kg retigabine (red lines). Shaded regions represent 95% confidence intervals. Treatment with MDZ + 15 mg/kg retigabine was significantly different from both other groups for 19–20 hr (p < 0.05, ANOVA, Games Howell post hoc test). (C) Fluoro-Jade B labeled dying neurons at 24 hr after SE can be seen in the representative image from MDZ + vehicle treated rat (left) compared to MDZ + 15 mg/kg retigabine treated rat (right) for dCA1, hilus, vCA1 vCA3 and amygdala. (D) The average number of dead neurons for the cohort of treated rats are represented for each brain region sampled for MDZ + vehicle (green), MDZ + 2 mg/kg retigabine (blue) and MDZ + 15 mg/kg retigabine (red). Asterisks indicate significantly different values for the MDZ + 15 mg/kg retigabine compared to MDZ + vehicle treatment (p < 0.05, ANOVA, Tukey’s multiple comparison test).
In addition to being a strong antiseizure adjunct to midazolam, retigabine also provided significant neuroprotection against neuronal death in the DFP model. Compared to brains from MDZ + PEG200 treated rats, FJB staining from rats treated with MDZ + 15 mg/kg retigabine (n = 19) demonstrated significant reductions in the number of fluorescent neurons in all examined regions, except the hilus and parietal cortex (Figure 1C and 1D). These data from the DFP model demonstrate that retigabine is a potent antiseizure and neuroprotective drug when combined as adjunctive therapy with MDZ for DFP-induced SE.
MDZ + retigabine at 30 mg/kg, but not 15 mg/kg, provides antiseizure efficacy in a soman model of OP-induced SE
To expand on the results obtained from the DFP model, MDZ + retigabine was tested in the soman model of OP-induced SE. MDZ + 15 mg/kg retigabine was administered 20 min after the initiation of SE. A total of 14 rats received MDZ + 15 mg/kg retigabine and of these rats, 8 died at latencies ranging from 5.0–270.5 min. None of the surviving 6 rats demonstrated SE termination (Figure 2A & B; Table 1). Of the 8 rats that died, none of these rats demonstrated SE termination leading up to their death. In the control group, 17 rats were treated with MDZ + PEG200 at a 20-min treatment delay. Of these 17 rats, 1 died at a latency of 71.0 min without demonstrating SE termination and 16 survived to the 4-hr endpoint, but none of these rats demonstrated SE termination at any time point (Figure 2A & B; Table 1).
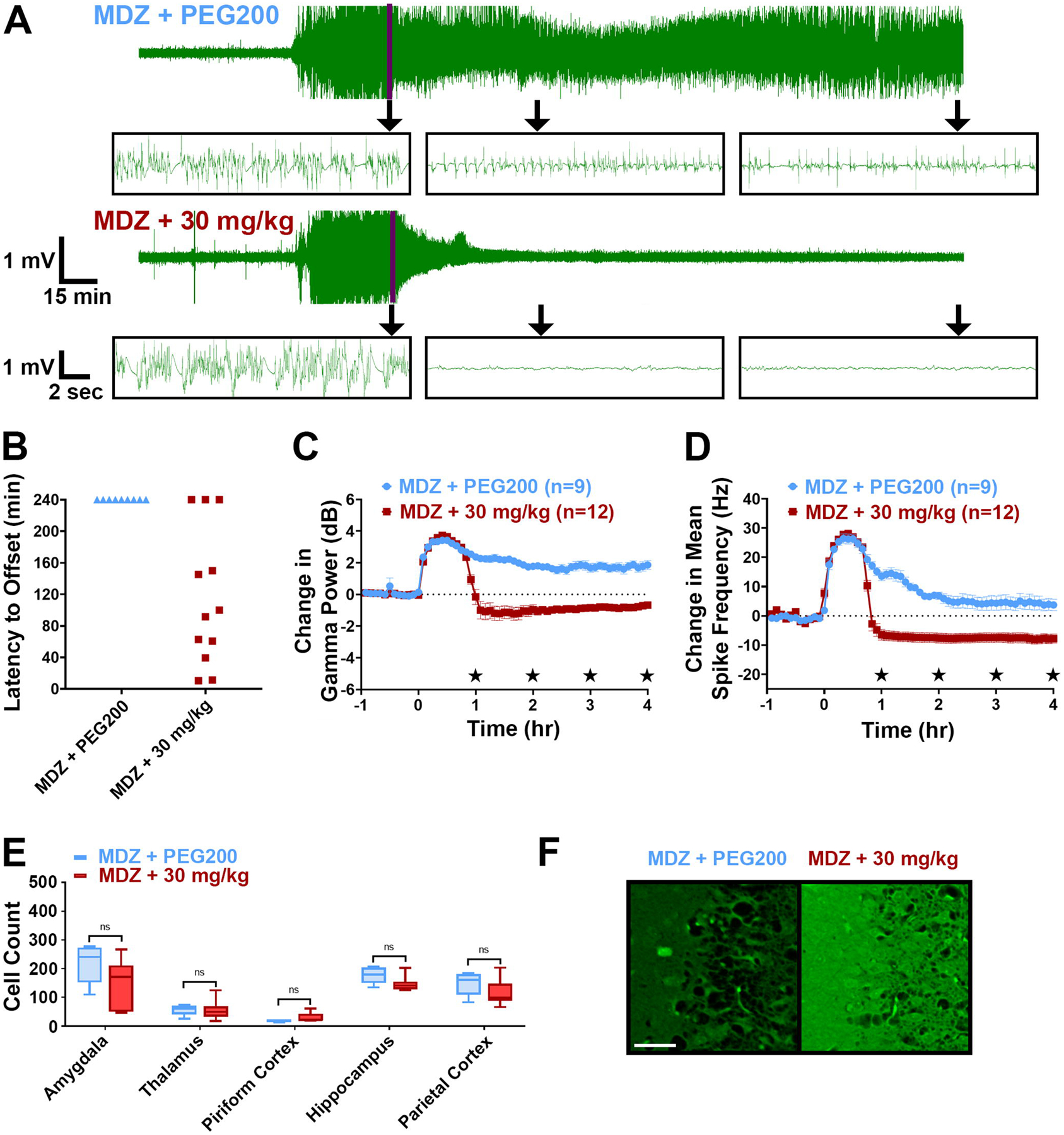
Figure 2: 30 mg/kg, but not 15 mg/kg, retigabine is an effective antiseizure adjunct therapy when administered at a 20-min delay in the soman model of OP-induced SE.

(A) Representative EEG traces from rats that received MDZ + PEG200, MDZ + 15 mg/kg retigabine, and MDZ + 30 mg/kg retigabine. Purple line marks time of treatment, which occurred 20-min after SE onset. Inset boxes show representative expanded EEG traces from each treatment group at: time of treatment (Tx), 1 hr post-treatment, and 4 hr post-treatment. (B) Latency to SE termination (min) for each treatment group. Data points at the 240-min mark represent rats that failed to demonstrate SE termination. (C) Change in EEG gamma power relative to baseline for each treatment group. Significant differences at each hour time point are noted on the y axis. (D) Change in mean spike frequency relative to baseline for each treatment group. Significant differences at each hour time point are noted on the y axis. (E) Average Fluoro-Jade B count for each brain region of interest from each treatment group. (F) Representative Fluoro-Jade B staining from the piriform cortex of each treatment group. Scale bar represents 50 μm. Significance values: %p<0.05 MDZ +15 mg/kg retigabine vs. MDZ + PEG200; *p<0.05 MDZ + 30 mg/kg retigabine vs. MDZ + PEG200; #p<0.05 MDZ + 30 mg/kg retigabine vs. MDZ + 15 mg/kg retigabine, One-way ANOVA, Tukey’s multiple comparison test. EEG data represents average ± 95% confidence intervals.
EEG analysis further revealed the lack of efficacy of MDZ + 15 mg/kg retigabine. Similar to MDZ + PEG200 controls, rats that received MDZ + 15 mg/kg retigabine failed to show significant reductions in gamma power or spike rate at any experimental time point (Figure 2C & D). MDZ + 15 mg/kg retigabine was also a poor neuroprotective treatment regimen in the soman model. Significant reductions in FJB positive neurons were not seen in any of the observed brain regions of rats given MDZ + 15 mg/kg retigabine (n = 6 brains) compared to MDZ + PEG200 controls (n = 16 brains, Figure 2E & F). Thus, retigabine at 15 mg/kg showed no efficacy in this protocol.
In response to the lack of efficacy of MDZ + 15 mg/kg retigabine in the soman model, we hypothesized that a higher dose of retigabine may be needed to alleviate nerve agent-induced seizures. As a result, MDZ + 30 mg/kg retigabine was tested against soman-induced SE. A total of 12 rats received MDZ + 30 mg/kg retigabine and of these rats, 3 died at latencies ranging from 20.3 – 240.0 min post-treatment with 1 of these rats showing a brief period of SE cessation for 25.4 min. Of the remaining 9 rats that survived, 8 demonstrated SE termination at latencies ranging from approximately 5.0 – 116.5 min (average = 43.8, median = 18.4 min) (Figure 2A & B; Table 1) that persisted until the end of the 4-hr experimental paradigm. Of these 8 rats that were seizure free at the end of the 4-hr EEG recording, 1 was perfused early by error (post-hoc analysis revealed animal was SE free), 4 were seizure free at the 24-hr recording session and 3 died before or during the end of the 24-hr recording period. EEG analysis further revealed the robust antiseizure efficacy of MDZ + 30 mg/kg retigabine. Compared to MDZ + PEG200 controls and MDZ + 15 mg/kg retigabine-treated rats, rats that received MDZ + 30 mg/kg retigabine demonstrated significant reductions in gamma power and mean spike frequency at 1 – 4 hr post-treatment (Figure 2C & D). Despite being a strong antiseizure treatment, MDZ + 30 mg/kg retigabine provided only minor neuroprotection in the soman model. Compared to MDZ + PEG200 controls (n = 16), brain sections from rats treated with MDZ + 30 mg/kg retigabine (n = 6) demonstrated significant reductions in FJB positive neurons in only the amygdala and thalamus (Figure 2E & F). No significant reductions in FJB staining were seen in the piriform cortex, hippocampus or parietal cortex of rats treated with MDZ + 30 mg/kg retigabine. These data reveal that MDZ + 30 mg/kg retigabine had significant antiseizure effects against soman-induced SE but provided minimal reductions in FJB staining in the observed brain regions.
MDZ + 30 mg/kg retigabine retains antiseizure efficacy at a 40-min treatment delay in the soman model of OP-induced SE
To determine if MDZ + 30 mg/kg retigabine showed antiseizure efficacy at longer treatment delays, these treatments were administered 40 min after the onset of soman-induced SE. Previous research has shown that administering MDZ at this treatment delay terminates nerve agent-induced status epilepticus in <1% of animals (Jackson et al., 2019). A total of 15 rats received MDZ + 30 mg/kg retigabine at a 40-min post-SE initiation delay. Of these rats, 3 died at latencies ranging from 9.5 – 240.0 min post-treatment with 1 of these rats showing termination of SE at time of death. Of the remaining 12 rats that survived, 9 demonstrated SE termination at latencies ranging from 10.5 – 150.1 min post-treatment (average = 74.6, median = 62.8) (Figure 3A & B; Table 1). Of the 9 rats that demonstrated SE termination, only 1 rat returned to having seizures during the 4-hr recording after an offset period of 103.4 min, and this rat was perfused at the 4-hr time point. Of the 8 rats that remained SE free at the end of the 4-hr recording, 5 died before or during the 24-hr EEG recording period, 2 remained out of SE at the 24-hr recording period and 1 rat returned to SE. In the control group, 11 rats were treated with MDZ + PEG200 at a 40-min treatment delay. Of these 11 rats, 2 died at latencies of 139.0 and 217.7 min and neither of these rats demonstrated SE termination before their deaths. Of the 9 rats that survived until the 4-hr endpoint, none showed SE termination at any time point of the experiment (Figure 3A & B; Table 1).
Figure 3: MDZ + 30 mg/kg retigabine retains its antiseizure efficacy at a treatment delay of 40-min post-SE onset.
(A) Representative EEG traces from rats that received MDZ + PEG200 and MDZ + 30 mg/kg retigabine. Purple line marks time of treatment, which occurred 40-min after SE onset. Inset boxes show representative expanded EEG traces from each treatment group at: time of treatment (Tx), 1 hr post-treatment, and 4 hr post-treatment. (B) Latency to SE termination (min) for each treatment group. Data points at the 240-min mark represent rats that failed to demonstrate SE termination. (C) Change in EEG gamma power relative to baseline for each treatment group. Significant differences at each hour time point are noted on the y axis. (D) Change in mean spike frequency relative to baseline for each treatment group. Significant differences at each hour time point are noted on the y axis. (E) Average Fluoro-Jade B count for each brain region of interest from each treatment group. (F) Representative Fluoro-Jade B staining from the piriform cortex of each treatment group. Scale bar represents 50 μm. Significance values: *p<0.05 MDZ + 30 mg/kg retigabine vs. MDZ + PEG200, Student’s t-test. EEG data represents average ± 95% confidence intervals.
EEG analysis further demonstrated the antiseizure efficacy of MDZ + 30 mg/kg retigabine at a 40-min treatment delay. Compared to MDZ + PEG200 controls, rats that received MDZ + 30 mg/kg retigabine demonstrated significant reductions in both gamma power and mean spike frequency at 1–4 hr post-treatment (Figure 3C & D).
Despite the robust antiseizure efficacy of MDZ + 30 mg/kg retigabine at a 40-min treatment delay, no significant neuroprotection was provided by this treatment regimen. Compared to brain sections from rats given MDZ + PEG200, brain sections from rats given MDZ + 30 mg/kg retigabine at a 40-min treatment delay failed to demonstrate significant reductions in FJB staining in any of the examined brain regions (Figure 3E & F).
15 mg/kg retigabine without MDZ provides delayed antiseizure efficacy in the DFP model of OP-induced SE.
To determine if retigabine is an effective stand-alone ASD for OP-induced SE, retigabine treatment was tested head-to-head against MDZ in the DFP model. Rats (n = 19) were treated with 15 mg/kg retigabine alone and of these rats, 1 died compared to no deaths in the MDZ-alone group (n = 18; Table 1). Compared to the rapid efficacy provided by the combined MDZ + retigabine, retigabine monotherapy demonstrated delayed antiseizure efficacy when administered at a 60-min delay (Figure 4A). This effect was consistent for the group; however, treatment with retigabine alone caused significantly high levels of SE suppression as measured by both the change in gamma power (Figure 4B1) as well as the change in mean spike frequency (Figure 4B2). Compared to the combination treatment, retigabine alone reached peak effect at a later time (5 hr after treatment) and lasted for a shorter duration (7–9 hr). The diminished effect of monotherapy correlated with reduced neuroprotection, as can be seen in the raw images (Figure 4C), as well as the group-averaged bar graphs for each brain region (Figure 4D). Monotherapy treatment with retigabine did, however, still significantly reduce neuronal death in dorsal CA3, ventral CA1, thalamus, entorhinal cortex and piriform cortex (Figure 4D, Student’s t-Test; n = 18 MDZ alone; n = 18 retigabine 15 mg/kg alone).
Figure 4: Head-to-head comparison of retigabine and MDZ as stand-alone delayed- treatments for DFP-induced SE.
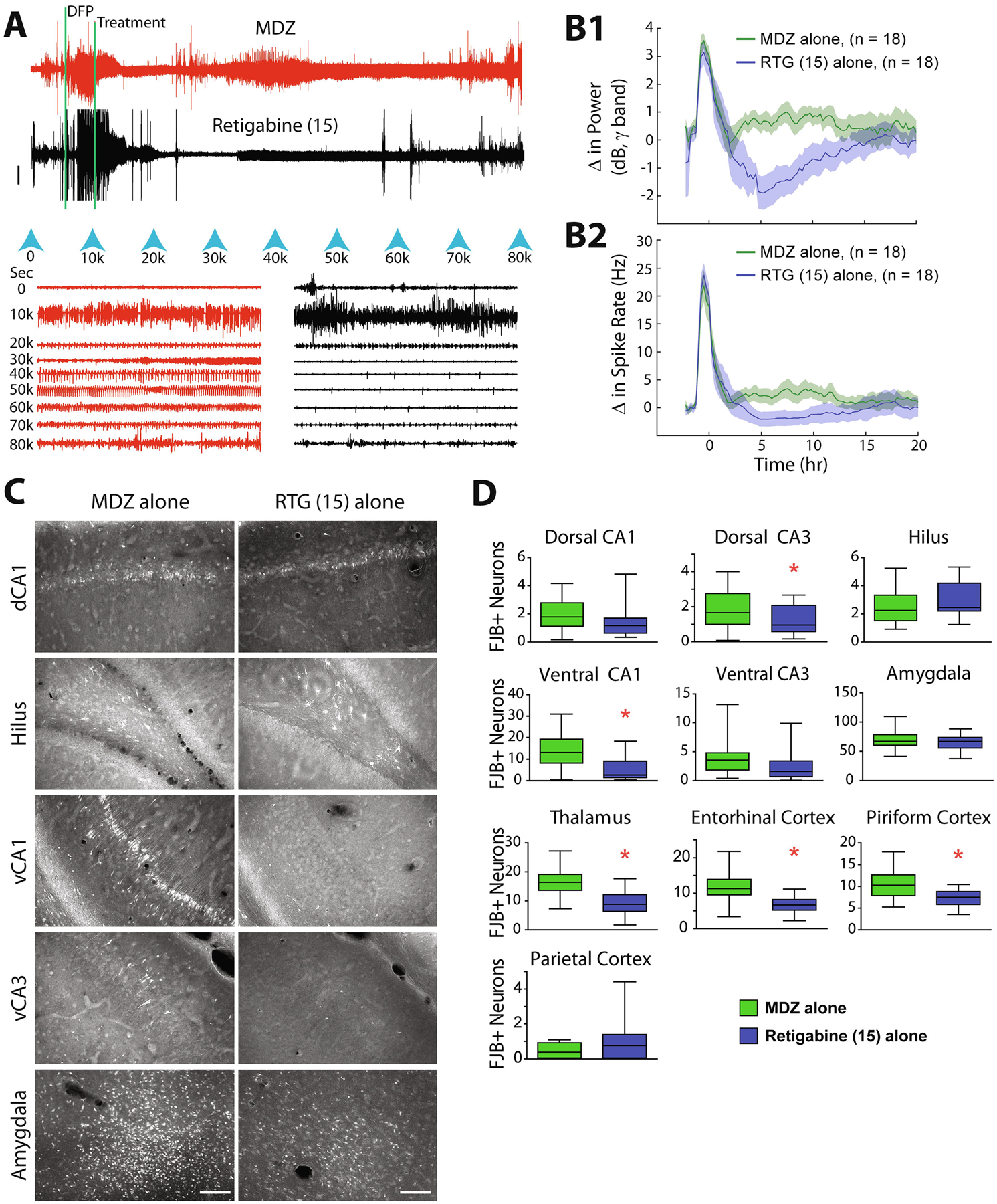
Data are presented as in Figure 1. (A) Raw, compressed EEG recordings demonstrate the difference between treatment with MDZ alone (+ standard antidotes, red trace, top) or retigabine alone (+ standard antidotes, black trace bottom). (B) Quantitative analysis of the group data demonstrates that treatment with retigabine alone is consistently and significantly superior to MDZ alone for time 4–11 hr (p < 0.05, Student’s t-test). (C) FJB labeled neurons from a rat treated with either MDZ alone or retigabine alone demonstrate the reduced neuronal death in vCA1 and vCA3 but no change in dCA1, hilus or amygdala. (D) Quantified neuronal death in each of the 10 brain regions analyzed are plotted for each group. Asterisks indicate significant reductions in rats treated with retigabine alone (p < 0.05, Student’s t-test).
To determine if a higher dose of retigabine without MDZ would provide improved antiseizure and neuroprotective effects against OP-induced SE, a 30-mg/kg dose of the drug was tested in the DFP model. Surprisingly, at the 30 mg/kg dose, 14/15 of the rats treated with retigabine died within 24 hr of treatment, with 5 out of 15 rats dying within the first 4 hr of treatment (Table 1). Blinded, qualitative assessment of the behavior at 4 hr after treatment revealed that the rats treated with a 30-mg/kg dose of retigabine appeared “over-sedated” with extremely slow breathing and discoloration of the exposed skin.
60 mg/kg retigabine without MDZ is an efficacious antiseizure and partial neuroprotectant in the soman model of OP-induced SE
In the soman model, 30 mg/kg retigabine without MDZ was not an effective antiseizure treatment. A total of eight rats received 30 mg/kg retigabine without MDZ. Of these rats one died without showing SE termination and one demonstrated seizure termination at a latency of 192.9 min (Figure 5A & B; Table 1). The single rat that demonstrated SE termination died overnight before the 24-h recording session. The remaining six rats that received 30 mg/kg retigabine did not demonstrate SE termination at any time point of the experiment (Figure 5A & B; Table 1). EEG analysis for rats treated with 30 mg/kg retigabine further revealed the lack of antiseizure efficacy when MDZ was not present. Compared to our previously referenced MDZ + PEG200 control group, rats treated with 30 mg/kg retigabine demonstrated no significant reductions in gamma power and only demonstrated a significant reduction in mean spike frequency at 1 hr (Figure 5C & D). Retigabine (30 mg/kg) without MDZ was also an ineffective neuroprotective treatment for soman-induced SE. No significant reductions in FJB positive neurons were seen in any of the observed brain regions of rats treated with 30 mg/kg retigabine (n=5) compared to MDZ + PEG200 controls (n=16) (Figure 5E & F).
Figure 5: 60 mg/kg, but not 30 mg/kg, retigabine alone, demonstrates improved antiseizure efficacy compared to MDZ+PEG200 for OP-induced SE when administered at a 20-min treatment delay.
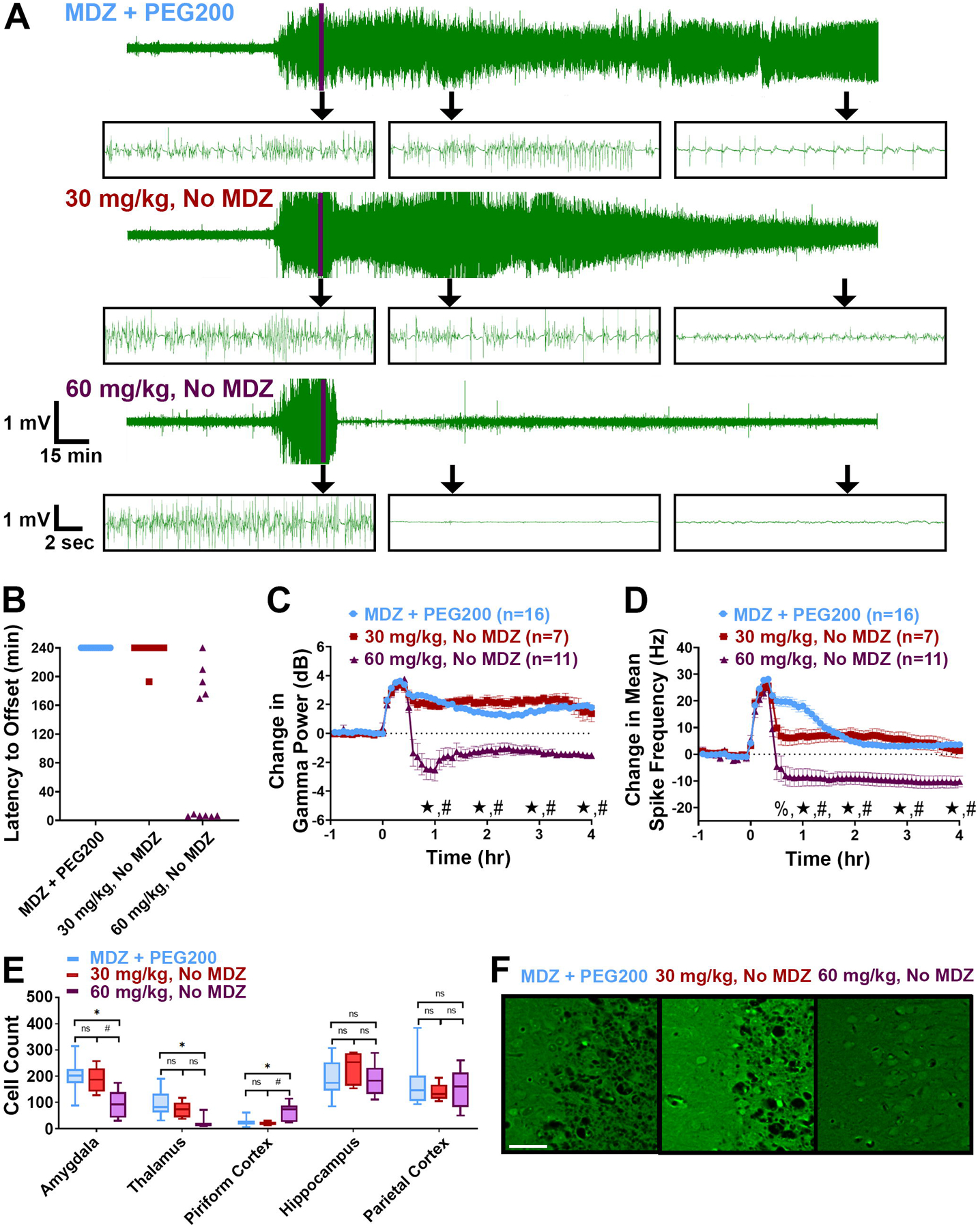
(A) Representative EEG traces from rats that received MDZ + PEG200, 30 mg/kg retigabine without MDZ, and 60 mg/kg retigabine without MDZ. Purple line marks time of treatment, which occurred 20 min after SE onset. Inset boxes show representative expanded EEG traces from each treatment group at: time of treatment (Tx), 1 hr post-treatment, and 4 hr post-treatment. (B) Latency to SE termination (min) for each treatment group. Data points at the 240-min mark represent rats that failed to demonstrate SE termination. (C) Change in EEG gamma power relative to baseline for each treatment group. Significant differences at each hour time point are noted on the y axis. (D) Change in mean spike frequency relative to baseline for each treatment group. Significant differences at each hour time point are noted on the y axis. (E) Average Fluoro-Jade B count for each brain region of interest from each treatment group. (F) Representative Fluoro-Jade B staining from the piriform cortex of each treatment group. Scale bar represents 50 μm. Significance values: %p<0.05 30 mg/kg retigabine, No MDZ vs. MDZ + PEG200; *p<0.05 60 mg/kg retigabine, No MDZ vs. MDZ + PEG200; #p<0.05 30 mg/kg retigabine, No MDZ vs. 60 mg/kg retigabine, No MDZ, One-way ANOVA, Tukey’s multiple comparison test. EEG data represents average ± 95% confidence intervals.
In response to the lack of antiseizure and neuroprotective efficacy provided by 30 mg/kg retigabine without MDZ in the soman model, the dose of retigabine was increased to 60 mg/kg. Rats (n = 14) were treated with 60-mg/kg retigabine without MDZ and of these rats, 3 died at latencies ranging from 52.6 to 211.1 min post-treatment with 1 of these rats showing SE termination at time of death. Of the remaining 11 rats, 1 failed to stop seizing and 10 demonstrated SE termination at latencies ranging from 5.3 – 209.8 min (average = 79.55, median = 6.52 min) (Figure 5A & B; Table 1). Of the 10 rats that showed SE termination, 4 returned to minor spiking EEG activity ranging from approximately 3–45 min post-SE termination. This spiking activity persisted for the duration of the recording in 2 rats and for 130 and 172 min in the remaining two rats. Of the 10 rats that showed periods of SE termination, 2 died overnight and 8 were seizure free during the 24-hr recording period. EEG analysis further demonstrated the antiseizure efficacy of 60 mg/kg retigabine without MDZ. Compared to MDZ + PEG200 controls and rats treated with 30 mg/kg, those that received 60 mg/kg retigabine without MDZ (n = 11) demonstrated significant reductions in gamma power and mean spike frequency at 1–4 hr post-SE initiation (Figure 5C & D).
In addition to being a strong ASD, 60 mg/kg retigabine also demonstrated some neuroprotective efficacy. Compared to MDZ + PEG200 controls (n = 16), rats treated with 60 mg/kg retigabine demonstrated significant reductions in FJB staining in the amygdala and thalamus (Figure 5E–F). These data from the soman model of OP-induced SE suggest that even in the absence of MDZ, a 60 mg/kg dose of retigabine is a promising antiseizure therapy for OP-induced SE.
Discussion
This study evaluated the efficacy of retigabine as either adjunctive or stand-alone therapy for the delayed treatment of DFP- or soman-induced SE. Our data demonstrate that retigabine is capable of reducing seizure activity and lessening neuronal death; however, its therapeutic effects were more pronounced when used as an adjunct to MDZ. Effective treatment required relatively high doses of retigabine, which revealed an unexpected lethality in the DFP model at a dose that was well-tolerated in the soman model. These data expand upon recent studies that have identified Kv7 as a druggable target for the treatment of OP- induced SE.
The antiseizure efficacy of retigabine in our two models of OP exposure adds further validation for the targeting of Kv7 potassium channels in SE. Retigabine has been shown to possess widespread antiseizure efficacy in models of electrical- and chemical-induced seizures (Rostock et al., 1996; Tober et al., 1996), and was an effective ASD in both rat and mouse genetic epilepsy models of audiogenic seizures (Dailey et al., 1995; Rostock et al., 1996). Regulation of Kv7 channel activity may be a particularly effective antiseizure approach for the treatment OP-induced SE where irreversible inhibition of AChE leads to a rapid increase in ACh at cholinergic synapses (McDonough & Shih, 1997; Shih & McDonough, 1997). This increase in ACh overstimulates muscarinic AChRs (mAChRs), which in turn directly inhibits neural Kv7 channels (Selyanko et al., 2000). This inhibition is believed to occur as a result of IP3 being formed from the hydrolysis and subsequent depletion of PIP2, a signaling molecule that plays a crucial role in Kv7 channels entering the open state (Brown et al., 2007; Brown & Passmore, 2009). By acting directly at Kv7 channels as a channel opener, retigabine may be directly restoring Kv7 channel activity; thereby overriding the effect of OP-induced mAChR overstimulation. Recent in vivo studies have supported targeting Kv7 channels for the treatment of OP-induced SE. Flupirtine in conjunction with diazepam was shown to be an effective antiseizure therapy for DFP-induced SE (Zhang et al., 2017) and retigabine itself was demonstrated to be an antiseizure and neuroprotectant adjunctive treatment for sarin-induced SE (Gore et al., 2020). Our work expanded on these studies by demonstrating that retigabine can be an effective ASD for OP-induced SE at delayed treatment times with and without co-administration of a benzodiazepine.
In addition to acting directly on Kv7 channels, both flupirtine and retigabine are known to act as positive allosteric modulators of GABA receptors at therapeutic or close-to-therapeutic doses (Gunthorpe et al., 2012; Klinger et al., 2012). Indeed, our data seem to demonstrate a synergistic effect with MDZ as the latency to seizure suppression was shortened when combination therapy was administered in the DFP model (see onset of effects in Figures 1B & 4B). Previous studies have suggested that retigabine and flupirtine may preferentially target δ-subunit containing, extrasynaptic GABAA receptors (van Rijn & Willems-van Bree, 2003; Klinger et al., 2015; Treven et al., 2015). These data are consistent with the effects of neurosteroids and enaminones, which have been shown to be robust ASDs in models of OP-induced SE (Althaus et al., 2017; Johnstone et al., 2019). While retigabine is not thought to reach a high enough concentration to act at GABAA receptors in healthy brains at the low chronic therapeutic dose of ~2 mg/kg, potential blood brain barrier (BBB) breakdown during ongoing SE and high treatment doses may allow for activity at these channels given the close effective concentration for GABAA receptor modulation and Kv7 activation (Gunthorpe et al., 2012; Gorter et al., 2015; Treven et al., 2015). Though speculative, BBB breakdown may also explain the differences in effective and lethal doses observed between the two studies, given the difference in timing of treatment: 30 mg/kg was lethal at 60-min of SE (DFP) when BBB was potentially disrupted, but 30 and 60 mg/kg were not lethal at either 20- and 40-min (soman) when BBB is potentially still intact (Gorter et al., 2015; Gorter et al., 2019; Mendes et al., 2019).
The dramatic mortality seen in rats given 30 mg/kg retigabine in the DFP, but not soman model, was surprising given previously published gait toxicity TD50 of approximately 94 mg/kg in mice (Kalappa et al., 2015). The difference in mortality between the two models at a 30-mg/kg retigabine dose could be the result of a few differences between the studies. In addition to differences in the time of treatment (already discussed above), another possible explanation could be different drug-drug interactions. In the DFP model, pyridostigmine bromide is given as a pre-treatment drug instead of the oxime HI-6, which is used in the soman model. Furthermore, 2-PAM is given 1 min after SE onset, compared to 20 min in the soman model. Additionally, in the soman model, atropine sulfate is administered at the time of antiseizure treatment whereas in the DFP model it is not administered at this time. Additional experiments will be necessary to determine why 30 mg/kg retigabine was fatal in the DFP model, but had antiseizure effects in the soman model. Despite numerous screens of other compounds using these models, we have not previously observed a similar dose-dependent difference in efficacy or mortality as observed in this study (Barker et al., 2020).
Retigabine has been reported to promote neuronal survival in models of neuronal injury including traumatic brain injury and spinal cord injury (Ebert et al., 2002; Boscia et al., 2006; Vigil et al., 2020; Wu et al., 2020). Our data in the DFP model, using retigabine as an adjunctive therapy to MDZ was consistent with these studies; however, a surprising finding of our study was the modest neuroprotection or lack of neuroprotection provided by MDZ + 30 mg/kg retigabine in the soman model at both the 20- and 40-min treatment delays. At both of these time points, MDZ + 30 mg/kg retigabine was a robust antiseizure therapy. The delay in seizure offset is one possible driving force behind this finding. When given at a 20-min treatment delay, MDZ + 30 mg/kg retigabine produced a median seizure offset time of 18.4-min, meaning SE lasted for approximately 38 min in these rats. In rats given MDZ + 30 mg/kg at a 40-min treatment delay, where no neuroprotection was observed, median offset time was even longer at 62.8 min. Both of these median SE durations far surpass the minimal time needed to see neuropathological evidence of neuronal death in rats (McDonough & Shih, 1997). In support of this idea, it’s important to note that rats that were given 60 mg/kg retigabine without MDZ showed the most widespread neuroprotection and the shortest median offset time.
Retigabine was once heralded for its first-in-class mechanism of action, and it was originally considered a significant advancement in the treatment of epilepsy; however, due to skin and retinal discoloration from prolonged, chronic use, it has since been discontinued. The majority (95%) of skin discoloration cases occurred after 2 yr of treatment, and retinal abnormalities were only seen in patients taking retigabine for a minimum of 3 yr (Clark et al., 2015). These safety issues would not be a significant concern if retigabine was administered acutely for SE, since side-effects from acute clinical trials were not sufficient to prevent FDA approval for use in humans. Our data suggest that a “one-off” treatment regimen for retigabine may be sufficient to terminate benzodiazepine-resistant SE; however, given the unexpected mortality observed, perhaps other more selective Kv7 channel openers should be considered (Roeloffs et al., 2008; Kalappa et al., 2015). Likewise, a lower dose of retigabine may be more efficacious and safer in humans (Gore et al., 2020).
In conclusion, we have described here, the antiseizure efficacy of retigabine in two models of benzodiazepine-resistant, OP-induced SE. Our data demonstrate that in models of delayed treatment, retigabine is an effective adjunct to current medical countermeasures and in soman-induced SE, retigabine may provide superior antiseizure efficacy compared to midazolam. Further exploration into the targeting of Kv7 channels for benzodiazepine-resistant SE may be highly useful, and retigabine and future analogues of the drug may have broad potential for the treatment of refractory and super-refractory SE.
Highlights.
The Kv7 channel agonist retigabine is a novel treatment option for OP-induced SE.
In two delayed-treatment rat models, retigabine with and without MDZ, significantly attenuated electrographic seizure activity.
The addition of retigabine to MDZ provided moderate neuroprotection measured as a reduction in Fluoro-Jade B labeled neurons.
Acknowledgements
This research was supported by two interagency agreements (AOD18013-001-00000 & AOD19020-001-00000) between the NIH Office of the Director (OD) and the U.S. Army Medical Research Institute of Chemical Defense under the oversight of the Chemical Countermeasures Research Program (CCRP) within the Office of Biodefense Research (OBRS) at the National Institute of Allergy and Infectious Diseases (NIAID/NIH). The views expressed are solely those of the authors and do not necessarily represent the official views of the CCRP, NIAID, NIH, HHS, USAMRICD, the Department of Army, Department of Defense, or the U.S. Government. Additionally, B. Barker, K. Berger, and C. Jackson were supported by appointments to the Postgraduate Research Participation Program at the U.S. Army Medical Research Institute of Chemical Defense administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and U.S. Army Medical Research and Development Command.
Disclosure of Conflicts of Interest
FED has received financial support in the form of consulting fees or grants from Neurona Therapeutics, Rugen Biomedical, and The Epilepsy Therapy Project. He has also received gifts (discounts on equipment) and consulting fees from - and has equity interest in - Epitel, Inc. and Cage Data Corp., which manufacture and sell Epoch™ recording devices, although these devices were not used in this research. The other authors do not have any conflicts of interest to disclose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Althaus AL, McCarren HS, Alqazzaz A, Jackson C, McDonough JH, Smith CD, Hoffman E, Hammond RS, Robichaud AJ & Doherty JJ (2017) The synthetic neuroactive steroid SGE-516 reduces status epilepticus and neuronal cell death in a rat model of soman intoxication. Epilepsy Behav, 68, 22–30. [DOI] [PubMed] [Google Scholar]
- Apland JP, Figueiredo TH, Qashu F, Aroniadou-Anderjaska V, Souza AP & Braga MF (2010) Higher susceptibility of the ventral versus the dorsal hippocampus and the posteroventral versus anterodorsal amygdala to soman-induced neuropathology. Neurotoxicology, 31, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker BS, Spampanato J, McCarren HS, Smolik M, Jackson CE, Hornung EN, Yeung DT, Dudek FE & McDonough JH (2020) Screening for Efficacious Anticonvulsants and Neuroprotectants in Delayed Treatment Models of Organophosphate-induced Status Epilepticus. Neuroscience, 425, 280–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscia F, Annunziato L & Taglialatela M (2006) Retigabine and flupirtine exert neuroprotective actions in organotypic hippocampal cultures. Neuropharmacology, 51, 283–294. [DOI] [PubMed] [Google Scholar]
- Brown DA, Hughes SA, Marsh SJ & Tinker A (2007) Regulation of M(Kv7.2/7.3) channels in neurons by PIP(2) and products of PIP(2) hydrolysis: significance for receptor-mediated inhibition. J Physiol, 582, 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA & Passmore GM (2009) Neural KCNQ (Kv7) channels. Br J Pharmacol, 156, 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y (2012) Organophosphate-induced brain damage: mechanisms, neuropsychiatric and neurological consequences, and potential therapeutic strategies. Neurotoxicology, 33, 391–400. [DOI] [PubMed] [Google Scholar]
- Clark S, Antell A & Kaufman K (2015) New antiepileptic medication linked to blue discoloration of the skin and eyes. Ther Adv Drug Saf, 6, 15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SA & Weir AGA (2019) UK resilience to a chemical incident. J R Army Med Corps. [DOI] [PubMed] [Google Scholar]
- Costanzi S, Machado JH & Mitchell M (2018) Nerve Agents: What They Are, How They Work, How to Counter Them. ACS Chem Neurosci, 9, 873–885. [DOI] [PubMed] [Google Scholar]
- Dailey JW, Cheong JH, Ko KH, Adams-Curtis LE & Jobe PC (1995) Anticonvulsant properties of D-20443 in genetically epilepsy-prone rats: prediction of clinical response. Neurosci Lett, 195, 77–80. [DOI] [PubMed] [Google Scholar]
- Ebert U, Brandt C & Loscher W (2002) Delayed sclerosis, neuroprotection, and limbic epileptogenesis after status epilepticus in the rat. Epilepsia, 43 Suppl 5, 86–95. [DOI] [PubMed] [Google Scholar]
- Gale K (1992) Subcortical structures and pathways involved in convulsive seizure generation. J Clin Neurophysiol, 9, 264–277. [DOI] [PubMed] [Google Scholar]
- Gore A, Neufeld-Cohen A, Egoz I, Baranes S, Gez R, Grauer E, Chapman S & Lazar S (2020) Efficacy of retigabine in ameliorating the brain insult following sarin exposure in the rat. Toxicol Appl Pharmacol, 395, 114963. [DOI] [PubMed] [Google Scholar]
- Gorter JA, Aronica E & van Vliet EA (2019) The Roof is Leaking and a Storm is Raging: Repairing the Blood-Brain Barrier in the Fight Against Epilepsy. Epilepsy Curr, 19, 177–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA & Aronica E (2015) Status epilepticus, blood-brain barrier disruption, inflammation, and epileptogenesis. Epilepsy Behav, 49, 13–16. [DOI] [PubMed] [Google Scholar]
- Gunthorpe MJ, Large CH & Sankar R (2012) The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia, 53, 412–424. [DOI] [PubMed] [Google Scholar]
- Jackson C, Ardinger C, Winter KM, McDonough JH & McCarren HS (2019) Validating a model of benzodiazepine refractory nerve agent-induced status epilepticus by evaluating the anticonvulsant and neuroprotective effects of scopolamine, memantine, and phenobarbital. J Pharmacol Toxicol Methods, 97, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone TBC, McCarren HS, Spampanato J, Dudek FE, McDonough JH, Hogenkamp D & Gee KW (2019) Enaminone Modulators of Extrasynaptic alpha4beta3delta gamma-Aminobutyric AcidA Receptors Reverse Electrographic Status Epilepticus in the Rat After Acute Organophosphorus Poisoning. Front Pharmacol, 10, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DM, Esmaeil N, Maren S & Macdonald RL (2002) Characterization of pharmacoresistance to benzodiazepines in the rat Li-pilocarpine model of status epilepticus. Epilepsy Res, 50, 301–312. [DOI] [PubMed] [Google Scholar]
- Kalappa BI, Soh H, Duignan KM, Furuya T, Edwards S, Tzingounis AV & Tzounopoulos T (2015) Potent KCNQ2/3-specific channel activator suppresses in vivo epileptic activity and prevents the development of tinnitus. J Neurosci, 35, 8829–8842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AM & Aaron CK (2015) Organophosphate and carbamate poisoning. Emerg Med Clin North Am, 33, 133–151. [DOI] [PubMed] [Google Scholar]
- Klinger F, Bajric M, Salzer I, Dorostkar MM, Khan D, Pollak DD, Kubista H, Boehm S & Koenig X (2015) delta Subunit-containing GABAA receptors are preferred targets for the centrally acting analgesic flupirtine. Br J Pharmacol, 172, 4946–4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger F, Geier P, Dorostkar MM, Chandaka GK, Yousuf A, Salzer I, Kubista H & Boehm S (2012) Concomitant facilitation of GABAA receptors and KV7 channels by the non-opioid analgesic flupirtine. Br J Pharmacol, 166, 1631–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmkuhle MJ, Thomson KE, Scheerlinck P, Pouliot W, Greger B & Dudek FE (2009) A simple quantitative method for analyzing electrographic status epilepticus in rats. J Neurophysiol, 101, 1660–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH & Alldredge BK (1998) Status epilepticus. N Engl J Med, 338, 970–976. [DOI] [PubMed] [Google Scholar]
- McDonough JH Jr., Clark TR, Slone TW Jr., Zoeffel D, Brown K, Kim S & Smith CD (1998) Neural lesions in the rat and their relationship to EEG delta activity following seizures induced by the nerve agent soman. Neurotoxicology, 19, 381–391. [PubMed] [Google Scholar]
- McDonough JH Jr. & Shih TM (1997) Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci Biobehav Rev, 21, 559–579. [DOI] [PubMed] [Google Scholar]
- McDonough JH, McMonagle JD & Shih TM (2010) Time-dependent reduction in the anticonvulsant effectiveness of diazepam against soman-induced seizures in guinea pigs. Drug Chem Toxicol, 33, 279–283. [DOI] [PubMed] [Google Scholar]
- Mendes NF, Pansani AP, Carmanhaes ERF, Tange P, Meireles JV, Ochikubo M, Chagas JR, da Silva AV, Monteiro de Castro G & Le Sueur-Maluf L (2019) The Blood-Brain Barrier Breakdown During Acute Phase of the Pilocarpine Model of Epilepsy Is Dynamic and Time-Dependent. Front Neurol, 10, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhrer T (2007) Neuronal structures involved in the induction and propagation of seizures caused by nerve agents: implications for medical treatment. Toxicology, 239, 1–14. [DOI] [PubMed] [Google Scholar]
- Myhrer T, Enger S & Aas P (2008) Anticonvulsant efficacy of drugs with cholinergic and/or glutamatergic antagonism microinfused into area tempestas of rats exposed to soman. Neurochem Res, 33, 348–354. [DOI] [PubMed] [Google Scholar]
- Myhrer T, Mariussen E & Aas P (2018) Development of neuropathology following soman poisoning and medical countermeasures. Neurotoxicology, 65, 144–165. [DOI] [PubMed] [Google Scholar]
- Paxinos GW,C (2007) The Rat Brain in Stereotaxic Coordinates Sixth Edition. Elsevier Academic Press., 170. [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF & Isbrandt D (2005) Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci, 8, 51–60. [DOI] [PubMed] [Google Scholar]
- Petras JM (1994) Neurology and neuropathology of Soman-induced brain injury: an overview. J Exp Anal Behav, 61, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouliot W, Bealer SL, Roach B & Dudek FE (2016) A rodent model of human organophosphate exposure producing status epilepticus and neuropathology. Neurotoxicology, 56, 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AC & DeLorenzo RJ (1999) N-methyl-D-aspartate receptor activation regulates refractoriness of status epilepticus to diazepam. Neuroscience, 93, 117–123. [DOI] [PubMed] [Google Scholar]
- Robb EL & Baker MB (2020) Organophosphate Toxicity StatPearls, Treasure Island (FL). [PubMed] [Google Scholar]
- Roeloffs R, Wickenden AD, Crean C, Werness S, McNaughton-Smith G, Stables J, McNamara JO, Ghodadra N & Rigdon GC (2008) In vivo profile of ICA-27243 [N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide], a potent and selective KCNQ2/Q3 (Kv7.2/Kv7.3) activator in rodent anticonvulsant models. J Pharmacol Exp Ther, 326, 818–828. [DOI] [PubMed] [Google Scholar]
- Rostock A, Tober C, Rundfeldt C, Bartsch R, Engel J, Polymeropoulos EE, Kutscher B, Loscher W, Honack D, White HS & Wolf HH (1996) D-23129: a new anticonvulsant with a broad spectrum activity in animal models of epileptic seizures. Epilepsy Res, 23, 211–223. [DOI] [PubMed] [Google Scholar]
- Schmued LC & Hopkins KJ (2000) Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res, 874, 123–130. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Jentsch TJ & Brown DA (2000) Inhibition of KCNQ1–4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J Physiol, 522 Pt 3, 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC & Brown DA (2008) Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A, 105, 7869–7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih T, McDonough JH Jr. & Koplovitz I (1999) Anticonvulsants for soman-induced seizure activity. J Biomed Sci, 6, 86–96. [DOI] [PubMed] [Google Scholar]
- Shih TM & McDonough JH Jr. (1997) Neurochemical mechanisms in soman-induced seizures. J Appl Toxicol, 17, 255–264. [DOI] [PubMed] [Google Scholar]
- Skovira JW, McDonough JH & Shih TM (2010) Protection against sarin-induced seizures in rats by direct brain microinjection of scopolamine, midazolam or MK-801. J Mol Neurosci, 40, 56–62. [DOI] [PubMed] [Google Scholar]
- Skovira JW, Shih TM & McDonough JH (2012) Neuropharmacological specificity of brain structures involved in soman-induced seizures. Neurotoxicology, 33, 463–468. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Pouliot W, Bealer SL, Roach B & Dudek FE (2019) Antiseizure and neuroprotective effects of delayed treatment with midazolam in a rodent model of organophosphate exposure. Epilepsia, 60, 1387–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tober C, Rostock A, Rundfeldt C & Bartsch R (1996) D-23129: a potent anticonvulsant in the amygdala kindling model of complex partial seizures. Eur J Pharmacol, 303, 163–169. [DOI] [PubMed] [Google Scholar]
- Towne AR, Pellock JM, Ko D & DeLorenzo RJ (1994) Determinants of mortality in status epilepticus. Epilepsia, 35, 27–34. [DOI] [PubMed] [Google Scholar]
- Treven M, Koenig X, Assadpour E, Gantumur E, Meyer C, Hilber K, Boehm S & Kubista H (2015) The anticonvulsant retigabine is a subtype selective modulator of GABAA receptors. Epilepsia, 56, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn CM & Willems-van Bree E (2003) Synergy between retigabine and GABA in modulating the convulsant site of the GABAA receptor complex. Eur J Pharmacol, 464, 95–100. [DOI] [PubMed] [Google Scholar]
- Vigil FA, Bozdemir E, Bugay V, Chun SH, Hobbs M, Sanchez I, Hastings SD, Veraza RJ, Holstein DM, Sprague SM, C, M.C., Cavazos JE, Brenner R, Lechleiter JD & Shapiro MS (2020) Prevention of brain damage after traumatic brain injury by pharmacological enhancement of KCNQ (Kv7, “M-type”) K(+) currents in neurons. J Cereb Blood Flow Metab, 40, 1256–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton NY & Treiman DM (1988) Response of status epilepticus induced by lithium and pilocarpine to treatment with diazepam. Exp Neurol, 101, 267–275. [DOI] [PubMed] [Google Scholar]
- White AM, Williams PA, Ferraro DJ, Clark S, Kadam SD, Dudek FE & Staley KJ (2006) Efficient unsupervised algorithms for the detection of seizures in continuous EEG recordings from rats after brain injury. J Neurosci Methods, 152, 255–266. [DOI] [PubMed] [Google Scholar]
- WHO, W.H.O. (2019) Suicide https://www.who.int/news-room/fact-sheets/detail/suicide.
- Wu X, Kuruba R & Reddy DS (2018) Midazolam-Resistant Seizures and Brain Injury after Acute Intoxication of Diisopropylfluorophosphate, an Organophosphate Pesticide and Surrogate for Nerve Agents. J Pharmacol Exp Ther, 367, 302–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Li L, Xie F, Xu G, Dang D & Yang Q (2020) Enhancing KCNQ Channel Activity Improves Neurobehavioral Recovery after Spinal Cord Injury. J Pharmacol Exp Ther, 373, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa N, Morita H & Nakajima T (2006) Sarin experiences in Japan: acute toxicity and long-term effects. J Neurol Sci, 249, 76–85. [DOI] [PubMed] [Google Scholar]
- Yeung DT, Harper JR & Platoff GE Jr. (2020) The National Institutes of Health Chemical Countermeasures Research Program (NIH CCRP): A collaborative opportunity to develop effective and accessible chemical medical countermeasures for the American people. Drug Dev Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Todorovic MS, Williamson J & Kapur J (2017) Flupirtine and diazepam combination terminates established status epilepticus: results in three rodent models. Ann Clin Transl Neurol, 4, 888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]