

Abstract
Microglia, the resident immune cells of the CNS, have emerged as key regulators of neural precursor cell activity in the adult brain. However, the microglia-derived factors that mediate these effects remain largely unknown. In the present study, we investigated a role for microglial brain-derived neurotrophic factor (BDNF), a neurotrophic factor with well known effects on neuronal survival and plasticity. Surprisingly, we found that selective genetic ablation of BDNF from microglia increased the production of newborn neurons under both physiological and inflammatory conditions (e.g., LPS-induced infection and traumatic brain injury). Genetic ablation of BDNF from microglia otherwise also interfered with self-renewal/proliferation, reducing their overall density. In conclusion, we identify microglial BDNF as an important factor regulating microglia population dynamics and states, which in turn influences neurogenesis under both homeostatic and pathologic conditions.
SIGNIFICANCE STATEMENT (1) Microglial BDNF contributes to self-renewal and density of microglia in the brain. (2) Selective ablation of BDNF in microglia stimulates neural precursor proliferation. (3) Loss of microglial BDNF augments working memory following traumatic brain injury. (4) Benefits of repopulating microglia on brain injury are not mediated via microglial BDNF.
Keywords: cognition, glia, learning and memory, neurogenesis, neuroinflammation, repopulating microglia
Introduction
Microglia are myeloid cells that reside in the CNS and serve a number of critical immune and nonimmune functions (Hanisch and Kettenmann, 2007; Salter and Stevens, 2017). Specifically, in response to infection or injury, microglia undergo a characteristic phenotypic transformation (i.e., adopting a less-ramified morphology), and contribute to the removal of debris/pathogens and mounting of an inflammatory response through recruitment of peripheral immune cells (Kettenmann et al., 2011; Wolf et al., 2017). Microglia also shape brain function under homeostatic conditions by supporting myelination and by modifying neuronal connectivity (Stevens et al., 2007; Wake et al., 2009; Paolicelli, et al., 2011; Schafer et al., 2012; Vasek et al., 2016; Lehrman et al., 2018; Li and Barres, 2018; Weinhard et al., 2018). Parkhurst et al. (2013) demonstrated that microglia regulate synapse formation and elimination during motor learning, with microglial brain-derived neurotrophic factor (BDNF; Bennett et al., 2016; Gyoneva et al., 2019) proposed as the main molecular driver of these effects.
BDNF is an important and potent trophic factor for neuronal survival, including that of adult-born granule cells within the hippocampus (Ghosh et al., 1994; Wang et al., 2015). Here, neuronally derived BDNF positively regulates hippocampal neurogenesis, supporting the differentiation of immature neurons into mature granule neurons and promoting dendritogenesis (Chan et al., 2008; Waterhouse et al., 2012; Wang et al., 2015). Astrocytic BDNF expression also supports hippocampal neurogenesis, aiding the survival of immature neurons (Quesseveur et al., 2013). A putative contribution of microglial BDNF to these processes has, however, remained unexplored.
The role of microglia as regulators of adult hippocampal neurogenesis is now well established (Butovsky et al., 2006; Sierra et al., 2010; Diaz-Aparicio et al., 2020; for review, see Ekdahl, 2012; Sierra et al., 2014; Sato, 2015; Araki et al., 2020), although the mechanisms underlying their effects have remained mostly unclear. By removing microglia from hippocampal neurosphere cultures, we ourselves previously showed that microglia stimulate neural precursor cell proliferation in response to exercise, but suppress the activity of these cells during aging (Vukovic et al., 2012). Our more recent work additionally uncovered a unique microglia phenotype that can be induced through the turnover of these cells and supports functional neurogenesis (i.e., increased precursor activity, survival, and integration of newly generated cells) following brain injury (Willis et al., 2020). Microglia thus regulate adult neurogenesis across a range of settings but the precise molecular mechanisms, including any potential role of BDNF therein, remain to be unraveled.
In this study, we used Cx3cr1creERT2 x Bdnf flox/flox mice to selectively ablate BDNF from microglia in the adult mouse brain (Parkhurst et al., 2013) under both control and pathologic (e.g., inflammatory) conditions, subsequently examining the effects of this cell-specific ablation on spatial learning and memory, microglia behavior, neural precursor cell proliferation, and the survival and morphology of adult-born neurons.
Materials and Methods
Experimental model and subject details.
Cx3cr1creERT2 (Yona et al., 2013) and Bdnf flox/flox mice (Rios et al., 2001) were obtained from The Jackson Laboratory. The crossed mouse line was maintained as being heterozygous for the Cx3cr1creERT2 allele and homozygous for the floxed Bdnf allele. Wild-type C57BL/J6 mice were sourced from the Animal Resource Center. Cx3cr1CreERT2 x iDTR mice were maintained in-house and were heterozygous for the Cx3cr1creERT2 allele and homozygous for the iDTR allele; only female mice were used in this study. All animals were kept in individually ventilated cages (two to five mice/cage) on a 12 h light/dark cycle. Food and water were provided ad libitum. All experimental procedures were approved by The University of Queensland Animal Ethics Committee, conducted in accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes, and with adherence to the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (Kilkenny et al., 2010).
Tamoxifen preparation and delivery.
Tamoxifen (TAM) stock solution (25 mg/ml; Sigma-Aldrich) was freshly prepared on a daily basis in corn oil (CO; Sigma-Aldrich). Mice were gavaged with either 180 mg/kg tamoxifen or an equal volume of corn oil as a control for 5 consecutive days. All mice were 7–8 weeks old at the time of gavaging.
BDNF ELISA.
Cx3cr1creERT2 x Bdnf flox/flox mice were deeply anesthetized with sodium pentobarbital 6 d after oral gavage with tamoxifen or corn oil (see above), followed by transcardial perfusion with 0.1 m PBS. The hippocampus was then dissected from the brain, lysed [1 mm sodium orthovanadate, 1 mm phenylmethylsufonyl fluoride, 1 μm batimastat (MedChem), 20 mm sodium fluoride, 1% chlorine (Roche), and 1% Phos-STOP (Sigma-Aldrich)] and then spun at 12,000 g for 30 min at 4°C. Supernatants were aliquoted and stored at −80°C. Protein content for each sample was determined using a Bradford assay (BIO-RAD), and BDNF concentrations were measured using a sandwich ELISA (Thermo Fisher Scientific) as per the manufacturer instructions.
Microglia depletion and repopulation.
For PLX5622-mediated microglial depletion and/or repopulation experiments, mice were placed on a chow diet with PLX5622 incorporated at 1200 ppm (provided by Plexxikon via Research Diets) as specified. Control mice received standard mouse chow only (Research Diets). PLX5622 was used under the permissions of a material transfer agreement from Plexxikon. For genetic microglia depletion, Cx3cr1CreERT2 x iDTR mice at 3–4 weeks of age were orally gavaged with tamoxifen [12.5 mg/g body weight (BW)] or corn oil, once daily for 5 d. Exposure to tamoxifen induces CX3CR1-positive (CX3CR1pos) cells to express diphtheria toxin receptor (DTR) in Cx3cr1CreERT2 x iDTR mice, but not corn oil controls. After the fifth and final treatment with tamoxifen or corn oil, mice were left to rest for at least 4 weeks. Because microglia are self-renewing and have a slow turnover rate compared with circulating CX3CR1pos cells, only microglia continue to express DTR after this rest period (Parkhurst et al., 2013). As a result, microglia can be selectively depleted by administration of DT (30 ng/g BW; Sigma-Aldrich) via intraperitoneal injection once daily for 3 d. Both the genetic and pharmacological approach lead to very efficient depletion of microglia (Willis and Vukovic, 2020), with the genetic model offering the greatest specificity in that it is not hampered by potential peripheral effects of PLX5622 (Lei et al., 2020).
In vivo labeling of newborn cells.
5-Ethynyl-2′-deoxyuridine (EdU; Sigma-Aldrich) and 5-bromo-2'-deoxyuridine (BrdU; Sigma-Aldrich) were dissolved in 0.1 m phosphate buffer [23 mg/ml Na2HPO4, 5.93 mg/ml NaH2PO4. 2H2O, and 0.007% NaOH (w/v); Sigma-Aldrich] to concentrations of 16.4 and 20 mg/ml, respectively, and stored at −20°C until further use. Proliferating cells were labeled via intraperitoneal injection of EdU (82.4 mg/kg, i.p.) and/or BrdU (100 mg/kg, i.p.).
Tissue preparation for immunohistochemistry.
Mice were deeply anaesthetized using an injection of sodium pentobarbital (1.6 g/kg, i.p.; Verbac), followed by transcardial perfusion with 20 ml of ice-cold PBS and 30 ml of formalin [10% (w/v); Sigma-Aldrich]. Brains were dissected and postfixed in 10% formalin for 24 h. Brains were then cryoprotected in 30% (w/v) sucrose in PBS with 0.01% (w/v) sodium azide for 2 d at 4°C or 24 h at room temperature. Coronal brain sections (40 μm thick) were cut using a sliding microtome (Leica) and serially collected in six-well plates. Sections were stored in PBS with 0.01% (w/v) sodium azide at 4°C until further processing.
Immunofluorescent staining.
Brain sections were blocked in 5% (v/v) normal goat serum (NGS) with 0.3% (v/v) Triton X-100 (Sigma-Aldrich) in PBS, before being incubated overnight at 4°C with the following primary antibodies: rabbit anti-IBA1 (1:1000; Wako), rabbit anti-TBR2 (1:500; Abcam), rabbit anti-BDNF (1:500; Abcam; Chaaya et al., 2019), rat anti-BrdU (1:1000; Accurate Chemical and Scientific Corporation), rabbit anti-SOX2 antibody (1:500; Abcam), guinea pig anti-DCX (1:1000; Millipore), rabbit anti-GFAP (1:200; Abcam), rat anti-GFAP (1:1000; Millipore), chicken anti-NeuN (1:1000; Abcam), rat anti-CD11B (1:1000; Abcam), rat anti-complement 3 (1:500; Clone 11H9, Hycult Biotech), rat anti-CD68 (1:1000; BIO-RAD), and/or TMEM119 (1:500; Abcam) diluted in 3% (v/v) NGS with 0.1% (v/v) Triton X-100 in PBS. The following day, sections were washed three times in PBS for 5 min and incubated in 3% (v/v) NGS with 0.1% (v/v) Triton X-100 in PBS with an appropriate combination of the following secondary antibodies: goat anti-rabbit Alexa Fluor 488 (1:2000; Thermo Fisher Scientific), goat anti-rabbit Alexa Fluor 568 (1:1000; Thermo Fisher Scientific), goat anti-rabbit Alexa Fluor 647 (1:500 Thermo Fisher Scientific), goat anti-rat Alexa Fluor 568 (1:1000; Thermo Fisher Scientific), and/or goat anti-guinea pig Alexa Fluor 488 (1:2000; Thermo Fisher Scientific). Sections were then washed in PBS three times for 5 min, with the nuclear stain DAPI (1:1000) added with the last round of washing. Next, sections were mounted onto glass slides (Menzel Glaser) with Vectashield H-100 Mounting Medium (Vector Laboratories) and sealed by placing a glass coverslip over sections (Menzel Glaser) and coating the edges of the coverslip with clear nail polish.
For BrdU, BDNF, and SOX2 staining, an initial antigen retrieval step was conducted before incubation with blocking buffer. For BrdU staining, sections were incubated in 1 m HCl (Millipore) for 20 min at 45°C, followed by 0.1 m boric acid (Millipore) incubation at room temperature for 10 min. For SOX2 and BDNF staining, sections were incubated in sodium citrate buffer (10 mm tri-sodium citrate dihydrate; ChemSupply Australia) and 0.05% (v/v) Tween-20 (ChemSupply Australia) in double distilled H2O for 15 min at 95°C. Sections were then washed in PBS three times for 5 min before continuing with the above generic staining protocol.
To detect EdU, sections were stained using the Click-iT Plus EdU Alexa Fluor 647 or Alexa Fluor 488 Imaging Kit (Thermo Fisher Scientific) as per the manufacturer protocol, before proceeding with the generic staining protocol detailed. Any antigen retrieval step was always done before EdU staining.
Imaging and cell quantification.
For EdUpos, DCXpos, and TBR2pos cell counts, five 40 μm sections of the dorsal dentate gyrus were quantified per animal, starting at anteroposterior (AP) −1.5 mm from bregma (one in six series, 200 μm apart). Cell counts were performed live using StereoInvestigator software (version 2017.01.1; MicroBrightfield Bioscience) and 40× 0.75 numerical aperture (NA) air-objective on an AxioImager Z2 microscope (Carl Zeiss) with an ORCA-R2 digital charge-coupled device camera (Hamamatsu). Cell counts were expressed per volume unit (in cubic millimeters) of the granule cell layer (GCL) and presented as the mean from the five sections that were analyzed.
To quantify radial glia-like SOX2posEdUposGFAPradial cells, images were acquired using a spinning-disk confocal microscope and processed as described above. Cells were identified by their presence within the subgranular zone (SGZ), a single radial GFAPpos process extending directly from the SGZ into the GCL, and colocalization of GFAP with SOX2 and EdU.
For quantification of IBA1pos microglia, five consecutive sections of the dorsal dentate gyrus starting at approximately AP −1.5 mm were imaged on a spinning-disk confocal microscope (Innovative Instruments Inc.; with W1 spinning-disk module, Yokogawa), using a 20× 1.2 NA air-objective, a Hamamatsu Flash 4.0 sCMOS camera, and running SlideBook software (version 6.0.16; Innovative Instruments Inc.). For quantification of microglia in the dentate gyrus, a 16 bit 3D montage of the full 40 μm physical depth of the section was acquired with a z-interval of 1.2 μm and compressed into a maximum z-projection. Cell counts were quantified using Fiji software (Schindelin et al., 2012) by cropping a region of interest (ROI) around the dentate gyrus and manually counting cells within the ROI using the Cell Counter plugin. Cell counts were normalized to the 3D area of the ROI, and the mean cell count across five sections was calculated for each mouse.
For all traumatic brain injury (TBI) experiments (see Materials and Methods below), cell counts were performed live using StereoInvestigator software (MBF Bioscience) using an AxioImager Z2 microscope (Zeiss) and an ORCA-R2 digital charge-coupled device camera (Hamamatsu), as previously described (Willis et al., 2020). Cell counts in TBI samples were recorded as the average from four consecutive sections of the dentate gyrus, located at bregma −1.50, −1.70, −1.94, and −2.18 mm. DCXpos, DCXposBrdUpos, TBR2pos, and TBR2posEdUpos cells were counted within the granule cell layer and expressed per volume unit (in cubic millimeters) of the analyzed region. IBA1pos, IBA1posBrdUpos, and GFAPpos cell numbers were also counted in the dentate gyri of these mice. For quantification of IBA1pos cells in the motor cortex and CA1 region, cells were counted as above using StereoInvestigator software and were recorded as the average from four brain sections that were located at bregma −1.50, −1.70, −1.94, and −2.18 mm.
Analysis of immunofluorescence.
Analysis of BDNF colocalization, BDNF median fluorescence intensity (MFI), and C3 MFI within GFAPpos astrocytes was performed as previously described (Willis et al., 2020). Briefly, images were acquired using an inverted Diskovery spinning disk confocal microscope (Spectral Applied Research) with a Zyla sCMOS camera at 20× magnification using a CFI Plan Aprochromat VC 20×/NA 0.75/working distance 1.0 mm objective. Using Imaris software, BDNF MFI, the extent of colocalization of BDNF with CD11bpos microglia, GFAPpos astrocytes or NeuNpos neurons, and also C3 MFI within GFAPpos astrocytes in the dentate gyrus was calculated from four consecutive sections, located at bregma −1.50, −1.70, −1.94, and −2.18 mm/mouse. All tissue sections were stained in one batch, with the same imaging threshold and exposure time to ensure consistency for image analysis.
Retrovirus intrahippocampal injection.
To visualize dendrites and dendritic spines in adult-born neurons, proliferating cells in the SGZ were infected with a Moloney murine leukemia virus-based retroviral vector expressing tdTomato (∼106 colony-forming units/ml; Clontech), delivered to the dentate gyrus by stereotaxic injection. Briefly, 6-week-old Cx3cr1creERT2 x Bdnf flox mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg BW) and xylazine (8 mg/kg BW; Ilium), and placed into a stereotaxic frame. Fur was shaved from the head, alcohol was used to sterilize the skin, a midline incision was made in the skin using a scalpel, and the skin was retracted. A hole was drilled through the skull using a dental drill to allow for bilateral injection at the following coordinates [AP, −2.18 mm; mediolateral (ML), ±1.5 mm; dorsoventral (DV), −2.0 mm]. Virus (2 μl) was loaded into a pulled glass capillary (catalog #GC150F-7.5, Harvard Apparatus) and lowered into the brain to the above coordinates at a rate of 1 mm/min. Over a 5 min period, 2 μl of virus was injected using a Picospritzer III (Parker Hannifin). The glass capillary was then left in place for 5 min before being drawn up 0.5 mm in 0.1 mm increments over 3 min and then 2 mm continuously at a rate of 1 mm/min. The incision in the skin was glued closed using Vetbond (3M). To prevent infection, prophylactic injections of Baytril (5 mg/kg BW, s.c.; Bayer) were administered, along with an injection of Metacam (2 mg/kg BW, s.c.; Boehringer) to provide pain relief. Mice were allowed to recover on a heat pad to maintain core body temperature at 36–37°C.
Dendritic morphology and spine density analysis.
To analyze dendritic morphology and spine density, three consecutive sections of the dorsal dentate gyrus starting at approximately AP −1.70 mm were imaged on a spinning-disk confocal. Dendritic arbors of tdTomatopos cells in the GCL were acquired using a 60× 1.2 NA oil-immersion objective, imaging the full extent of the selected neuron with a z-interval of 0.5 μm. For dendritic spines, images were acquired using a 100× 1.8 NA oil-immersion objective with a z-interval of 0.1 μm. Dendritic morphology was analyzed from maximum z-projections using Neurolucida 360 software (version 11.03; MBF Bioscience). Briefly, dendrites were traced using the user-guided function, and with Sholl analysis performed with a 1 μm interval between radii. In addition, the number of branches per cell, the sum of dendritic length, and the mean distance to the nearest neighboring dendritic terminal were quantified to assess dendrite morphology. Two neurons/section, totaling six neurons/mouse were analyzed. Spine density was quantified from maximum z-projections by manually counting tdTomatopos protrusions from dendrites using Fiji software. A total of 800–1000 μm of dendrite length was analyzed per mouse.
Stereotaxic injection of LPS.
Lipopolysaccharide (LPS; Escherichia coli O111:B4; Sigma-Aldrich) was prepared by dissolving it in 0.9% (w/v) sterile saline to a concentration of 3 mg/ml and stored at −80°C. LPS was defrosted and stored on ice before injection. Eleven- to 12-week-old Cx3cr1creERT2 x Bdnf flox mice were anesthetized with Zoletil (40 mg/kg BW; Virbac) and xylazine (8 mg/kg BW; Ilium), administered via intraperitoneal injection. Fur was shaved from the head, Betadine was used to sterilize the skin, a midline incision was made in the skin using a scalpel, and the skin was retracted. Mice were placed into a stereotaxic frame, and a craniotomy was performed by drilling through the skull with a dental drill. LPS (1 μl) or a vehicle control of saline was loaded into a pulled glass capillary (catalog #GC150F-7.5, Harvard Apparatus) and was lowered into the brain at a rate of 1 mm/min at the following coordinates: AP, −2.18 mm; ML, +1.5 mm; DV, −2.0 mm. LPS was injected using a Picospritzer III (Parker Hannifin), with the first 0.5 μl injected over 3 min, followed by a 2 min rest period, and then the remaining 0.5 μl was injected over 3 min. The glass capillary was then left in place for 5 min before being drawn up 0.5 mm in 0.1 mm increments over 3 min and then 2 mm continuously at a rate of 1 mm/min. The wound site was disinfected with Betadine, closed, and sealed using Vetbond. Hartmann's solution with buprenorphine (0.25 mg/kg BW; Provet) was administered to mice subcutaneously for analgesia. Mice were then allowed to recover on a heat pad to maintain core body temperature at 36–37°C. Note that antibiotics were not administered to these mice to avoid any possible suppression of the inflammatory response to LPS.
Controlled cortical impact model of traumatic brain injury.
Mice were subjected to either moderate controlled cortical impact (CCI) or sham surgery as previously described (Willis et al., 2020). Briefly, mice were anesthetized with Zoletil (40 mg/kg BW; Virbac) and xylazine (8 mg/kg BW; Ilium), administered via intraperitoneal injection, and placed into a stereotaxic frame. Using sterile procedures, a midline incision was made, the skin retracted, and a 4 mm craniotomy performed midway between the lambdoid and bregma sutures, and laterally midway between the central suture and temporalis muscle. The skull cap was carefully removed, taking care not to damage the underlying meninges. The tip of the 3 mm impactor piston was angled and kept perpendicular to the exposed cortical surface. The parameters for the CCI were as follows: impact speed, 3.5 m/s; deformation depth, 1.0 mm; duration, 400 ms. After CCI, the incision was sutured, and the mice were allowed to recover on a heat pad to maintain core body temperature at 36–37°C. Shortly after injury, mice were subcutaneously injected with Hartmann's solution with buprenorphine (0.25 mg/kg BW; Provet). Sham mice underwent the same surgical operation but without CCI.
Real-time quantitative PCR.
Mice were killed using an intraperitoneal injection of sodium pentobarbital (1.6 g/kg BW) before being transcardially perfused with 20 ml of 0.1 m PBS, pH 7.4. Hippocampi were dissected from the brain and snap frozen in liquid nitrogen, then stored at −80°C. RNA was extracted and purified using TRIzol according to manufacturer instructions (Thermo Fisher Scientific), and RNA quality was assessed using a NanoDrop technology (Thermo Fisher Scientific). RNA was converted to cDNA using the High Capacity cDNA Reverse Transcription Kit according to manufacturer instructions (Thermo Fisher Scientific). Real-time quantitative PCRs (qPCRs) were conducted using Applied Biosystems 7500 Real-Time PCR systems (Thermo Fisher Scientific) with the following SYBR-Green probes (Sigma-Aldrich) used to amplify the gene of interest: Ccl2 (forward: 5′-GTGCTTGAGGTGGTTGTGGA-3′; reverse: 5′-GCTGACCCCAAGAAGGAATG-3′); Csf-1 (forward: 5′-AACACCCCCAATGCTAACGC-3′; reverse: 5′-ACACAGGCCTTGTTCTGCTC-3′); Hprt (forward: 5′-CCCAGCGTCGTGATTAGCG-3′; reverse: 5′-GCCACAATGTGATGGCCTCC-3′); IL1-β (forward: 5′-CAACCAACAAGTGATATTCTCCATG-3′; reverse: 5′-GATCCACACTCTCCAGCTGCA-3′); IL6 (forward: 5′-TAGTCCTTCCTACCCCAATTTCC-3′; reverse: 5′-TTGGTCCTTAGCCACTCCTTC-3′); IL-10 (forward: 5′-GGTGAGAAGCTGAAGACCCTC-3′; reverse: 5′-GCCTTGTAGACACCTTGGTCTT-3′); Serpina3n (forward: 5′-GCTGAAGTACACAGGAAATG-3′; reverse: 5′-ATCTATCATCCTGGGTTTCAG-3′); and Tnfα (forward: 5′-CATCTTCTCAAAATTCGAGTGACAA-3′; reverse: 5′-TGGGAGTAGACAAGGTACAACCC-3′). Relative gene expression to the geometric mean of reference gene hypoxanthine guanine phosphoribosyl transferase (Hprt) was determined using the following formula: 2-ΔCT, where ΔCT = CT(target gene) – CT(Hprt). Data represent the fold change in gene expression compared with that of corn oil-treated mice that received control surgery, being either a stereotaxic injection of saline into the dentate gyrus or a sham surgery for CCI.
Active place avoidance.
To assess working spatial learning and memory, mice were tested in the active place avoidance (APA) behavioral paradigm, as previously described by Willis et al. (2017). Before behavioral testing, mice were habituated to the experimenter by placing mice on the experimenter's arm for 1 min/d for 7 d. Mice were then habituated to the APA arena by placing them in the rotating arena for 20 min, during which the mice could freely explore with the shock zone turned off. The APA arena consisted of a rotating (1 rpm) elevated platform with a grid metal floor, fenced by a 32-cm-high clear Perspex circular boundary, with black and white A3 size visual cues placed on the adjacent four surrounding walls; each cue had a unique pattern. One day after habituation, mice were tested in the APA paradigm for 20 min, during which they had to avoid a shock zone occupying a 60° area of the circular arena. The position of the shock zone was stationary in relation to the room. Mice were tracked by an overhead camera linked to Tracker Analysis software (Bio-Signal Group), with entry into the shock zone leading to the delivery of a brief footshock (0.5 mA, 500 ms duration, 60 Hz, at 1.5 s intervals). Mice were placed into the arena, directly opposite to the shock zone location. Working short-term spatial learning and memory was assessed by analyzing the number of entries into the shock zone and the maximum duration of time that the shock zone was avoided in 5 min intervals over the 20 min trial, using Track Analysis software (Bio-Signal Group) for analysis. Longer-term spatial learning and memory abilities were assessed by testing across 5 consecutive days.
Magnetic resonance imaging.
Ex vivo magnetic resonance imaging (MRI) was performed as previously described (Willis et al., 2020). Briefly, perfusion-fixed brains were kept in skull, but with the ventral side opened up to facilitate the entry of contrast agent [0.2% Magnevist (gadopentetate dimeglumine); Bayer Healthcare) for 4 d at 4°C. Samples were then immersed in perfluoropolyether solution (Fomblin, Solvay Solexis), and high-resolution images were acquired using a Bruker 16.4 T small animal MRI system with a vertical wide bore, equipped with a Micro2.5 gradient set and a 15 mm linear saw coil (M2M imaging). T1/T2 imaging was performed using Paravision software (version 6.0.1) and a gradient echo fast low angle shot (FLASH) MRI sequence, with TR = 40 ms, TE = 10 ms, and flip angle = 30°. Images were acquired using a 20 × 12.7 × 9 mm field of view with 400 × 256 × 180 matrix to produce 180 slices with 50 μm slice resolution. T1/T2 FLASH scans were masked using ITK-SNAP software to remove skull fragments, and brains were mapped to the Australian Mouse Brain Mapping Consortium (AMBMC) template using FMRIB linear and nonlinear image registration tools FLIRT and FNIRT. ITK-SNAP software was then used to manually check and correct ROIs (i.e., the ipsilateral cortex and ipsilateral dorsal hippocampus), following which the ROI 3D volume measures were recorded.
Quantification and statistical analysis.
GraphPad Prism software (version 7.02) was used for all data visualization and statistical analysis. For Student's t test, a Welch's correction was used if the SD of groups differed by more than two times. For the analysis of experiments with more than two conditions, one-way or two-way ANOVA with Bonferroni post hoc test was used as appropriate. For behavioral experiments, two-way ANOVA with repeated measures and Bonferroni post hoc test were used. All tests were unpaired and two-tailed unless specified otherwise in text. All experiments were performed in a blinded fashion and with randomization. Data are presented as the mean ± SEM, with data points in bar graphs representing individual animals. Statistical significance was set at p < 0.05.
Results
Microglial BDNF deficiency reduces microglial proliferation
To ablate BDNF from microglia, Cx3cr1creERT2 x Bdnf flox/flox mice were gavaged with TAM, while controls received corn oil CO (vehicle) only (Fig. 1A). Immunostaining experiments confirmed the selective loss of BDNF from CD11bpos microglia (Fig. 1B,C) but not other cells (i.e., astrocytes and neurons) in the hippocampus of tamoxifen-treated mice (Fig. 1D,E). Similar observations were made for the cortex (data not shown). Total BDNF levels were otherwise unchanged compared with controls (Fig. 1F,G), reflecting the relatively small contribution of microglia to overall BDNF levels in the brain with only ∼5% of the staining localizing to these cells (Fig. 1B; data not shown).
Figure 1.
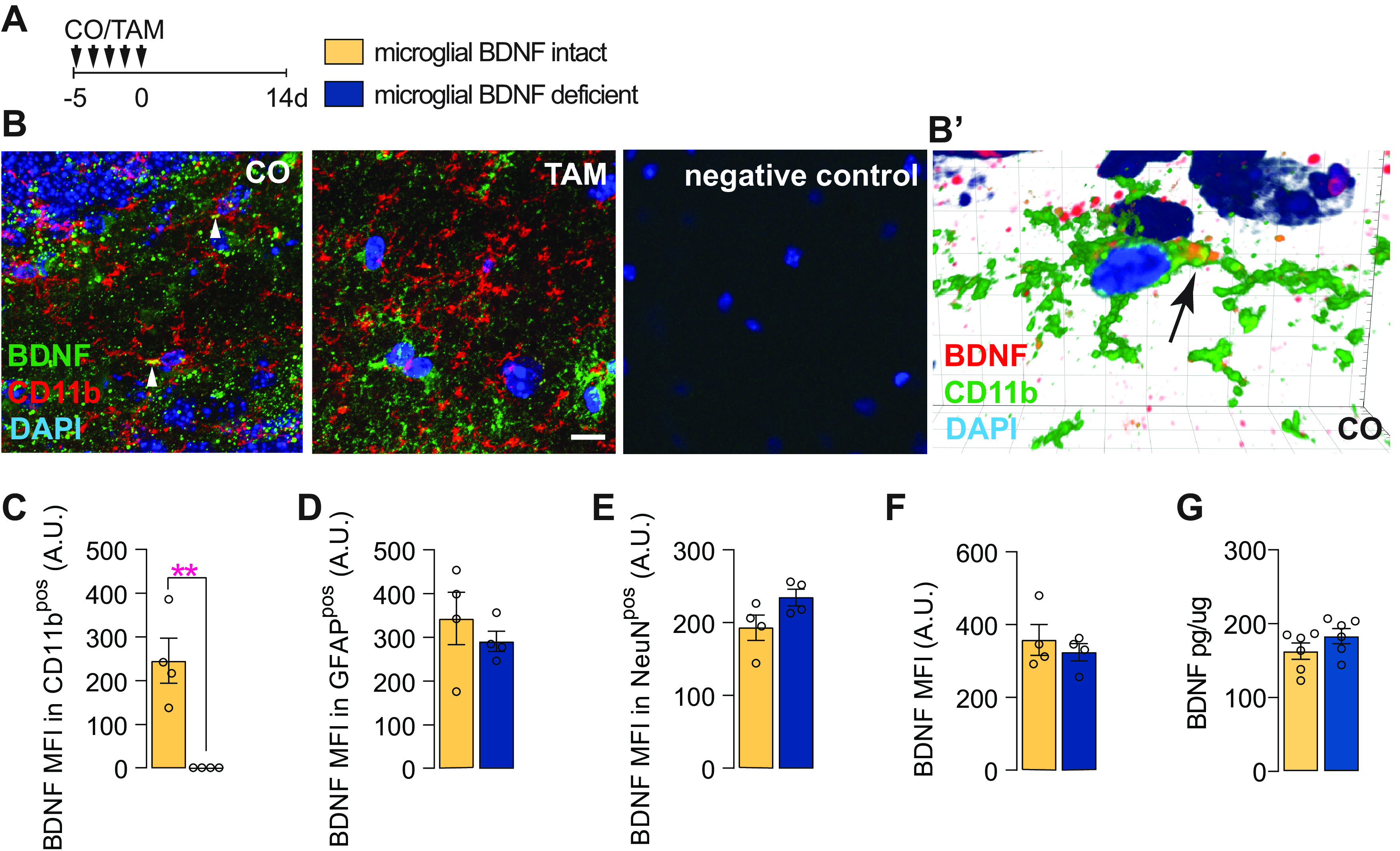
BDNF ablation in microglia in Cx3cr1creERT2 x Bdnf flox/flox mice. A, Experimental timeline for microglial BDNF ablation in Cx3cr1creERT2 x Bdnf flox/flox mice. B, Confocal images showing BDNF immunostaining in Cx3cr1creERT2 x Bdnf flox/flox mice treated with CO or TAM; arrows indicate expression of BDNF in CD11bpos microglia. B′, Imaris 3D reconstruction showing BDNF in CD11bpos brain microglia in Cx3cr1creERT2 x Bdnf flox/flox mice treated with corn oil. C, Quantification of BDNF MFI within CD11bpos microglia (t(6) = 4.74, p = 0.0032). D, Quantification of BDNF MFI within GFAPpos cells (t(6) = 0.45, p = 0.45). E, Quantification of BDNF MFI within NeuNpos cells (t(6) = 2.01, p = 0.092). F, Quantification of total BDNF MFI within the hippocampus (t(6) = 0.69, p = 0.51). G, ELISA quantification of BDNF protein levels in the hippocampus of Cx3cr1creERT2 x Bdnf flox/flox mice 6 d after tamoxifen-induced microglial BDNF deletion (t(10) = 1.34, p = 0.19). Data represent the mean ± SEM. **p < 0.01. Statistical tests: unpaired Student's t test (C–G). A.U., Arbitrary units. Small circles represent individual mice.
Pulse-chase studies with BrdU, delivered once daily for 4 d followed by killing 3 d later (Fig. 2A), revealed that microglial BDNF ablation significantly reduced both the total (11% reduction) and proliferating (66% reduction) number of IBA1pos microglia in the dentate gyrus (Fig. 2B–D). Similar changes were observed in the cortex where both microglial proliferation (CO, 338.30 ± 27.39 cells/mm3; vs TAM, 190.2 ± 3.767 cells/mm3; p = 0.0007; t(8) = 5.36, n = 5) and overall microglia density (CO, 6762 ± 547.4 cells/mm3; vs TAM, 5435 ± 107.6 cells/mm3; p = 0.045; t(8) = 2.38, n = 5) were reduced with BDNF deletion. The observed reductions in IBA1pos cell numbers persisted in the longer term after the selective ablation of BDNF from microglia (Fig. 2E,F), and they were specific as the treatment of wild-type mice with tamoxifen did not alter the number of IBA1pos microglia (Fig. 2G,H). Together, these results show that the genetic ablation of BDNF from microglia reduces the overall density and proliferation of these cells.
Figure 2.
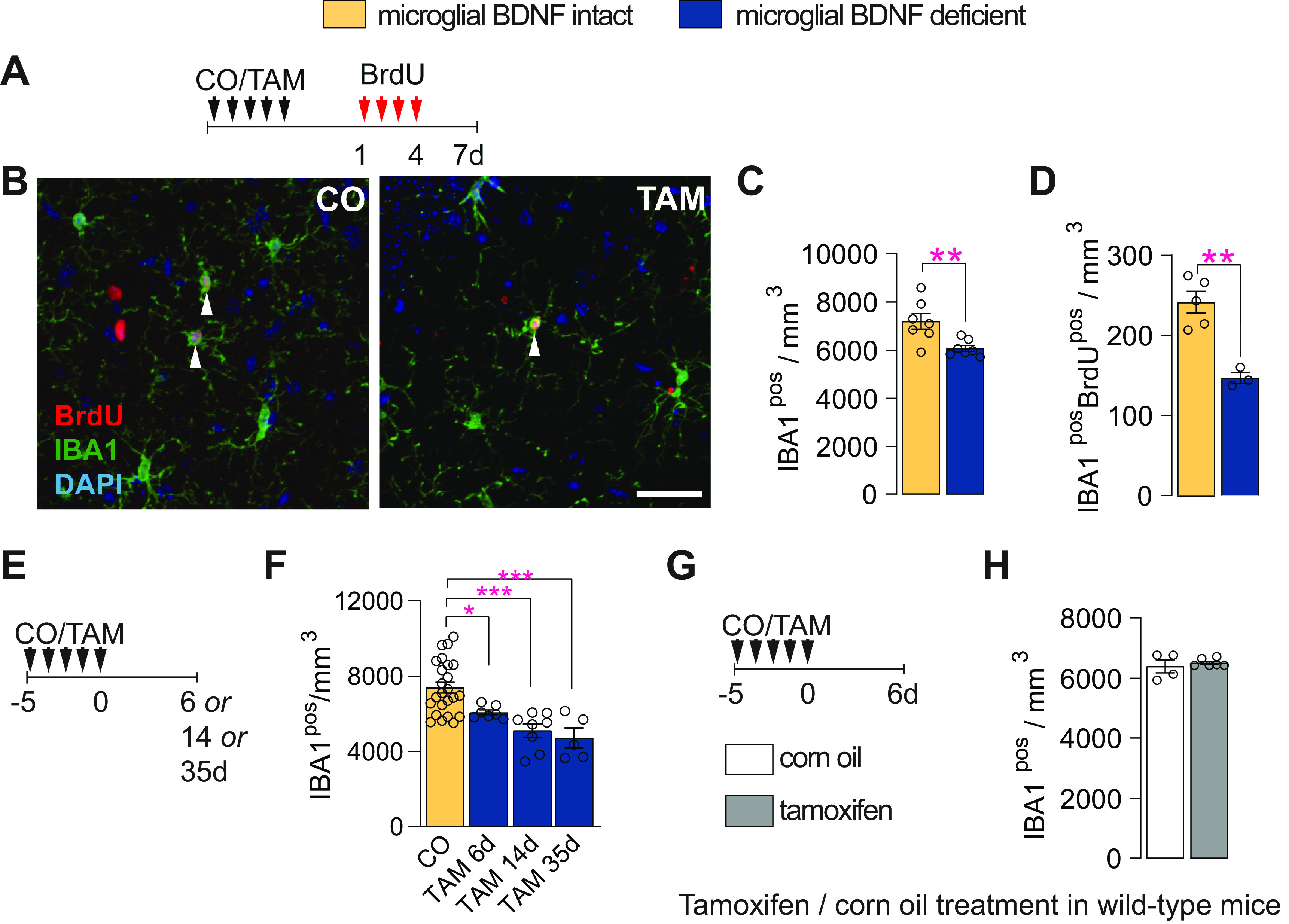
Microglial BDNF deficiency reduces microglial density and their proliferation. A, Experimental timeline for daily intraperitoneal BrdU injections in Cx3cr1creERT2 x Bdnf flox/flox mice, given 1–4 d after CO or TAM treatment, with mice killed 3 d after the last BrdU injection. B, Confocal images of IBA1pos and BrdUpos cells in the dentate gyrus of Cx3cr1creERT2 x Bdnf flox/flox mice. C, D, Quantification of total IBA1pos microglia (C; t(12) = 3.20, p = 0.0077) and newborn IBA1posBrdUpos microglia (D; t(8) = 6.78, p = 0. 0001) in the dentate gyrus. E, Experimental timeline for Cx3cr1creERT2 x Bdnf flox/flox mice treated with CO or TAM and killed 6, 14, or 35 d later. F, Quantification of IBA1pos microglia in the dentate gyrus at 6, 14, or 35 d post-corn oil or tamoxifen administration (F(3,40) = 11.74, p < 0.0001; CO vs TAM 6 d, p = 0.046; CO vs TAM 14 d, p = 0.0001; CO vs TAM 35 d, p = 0.0002). G, Experimental timeline for wild-type mice treated with either corn oil or tamoxifen once daily for 5 d and killed 6 d after the fifth and final treatment. H, Quantification of IBA1pos microglia in the dentate gyrus of wild-type mice (t = 0.65, df = 8, p = 0.53). Data represent the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. Statistical tests: unpaired Student's t test, C, D, H; one-way ANOVA with Bonferroni post hoc comparison test, F. Small circles represent individual mice.
Microglial BDNF deficiency transiently stimulates neural precursor proliferation
Having established an effect on microglia proliferation, we next investigated whether the selective ablation of BDNF from these cells also affected hippocampal neurogenesis. To label proliferating cells in the neurogenic niche (i.e., SGZ of the hippocampus), all mice were injected with EdU, 5 d after oral gavage with CO or TAM, and then killed 25 h later (Fig. 3A). In doing so, we found that microglial BDNF ablation significantly increased EdUposIBA1neg cell numbers in the SGZ (Fig. 3B,C). Costaining with more selective stem/progenitor cell markers revealed that the intermediate neuronal precursor pool, which expresses the transcription factor TBR2 (Hodge et al., 2012), was significantly expanded (Fig. 3D) and more active (Fig. 3E) in mice with microglial BDNF ablation. Treatment of wild-type mice with tamoxifen did not alter the number of EdUposIBA1neg and/or TBR2pos cells (Fig. 3F–H). It should be noted that the stimulatory effect of microglial BDNF deletion on TBR2pos precursor cell proliferation appeared otherwise transient under these experimental conditions and, unlike that for microglia themselves (Fig. 2F), was not present longer term, that is, at 30 d post-tamoxifen treatment (CO, 3459 ± 335.70 cells/mm3; vs TAM, 3263 ± 366.3 cells/mm3; p = 0.72, t(8) = 0.37, n = 4–6).
Figure 3.
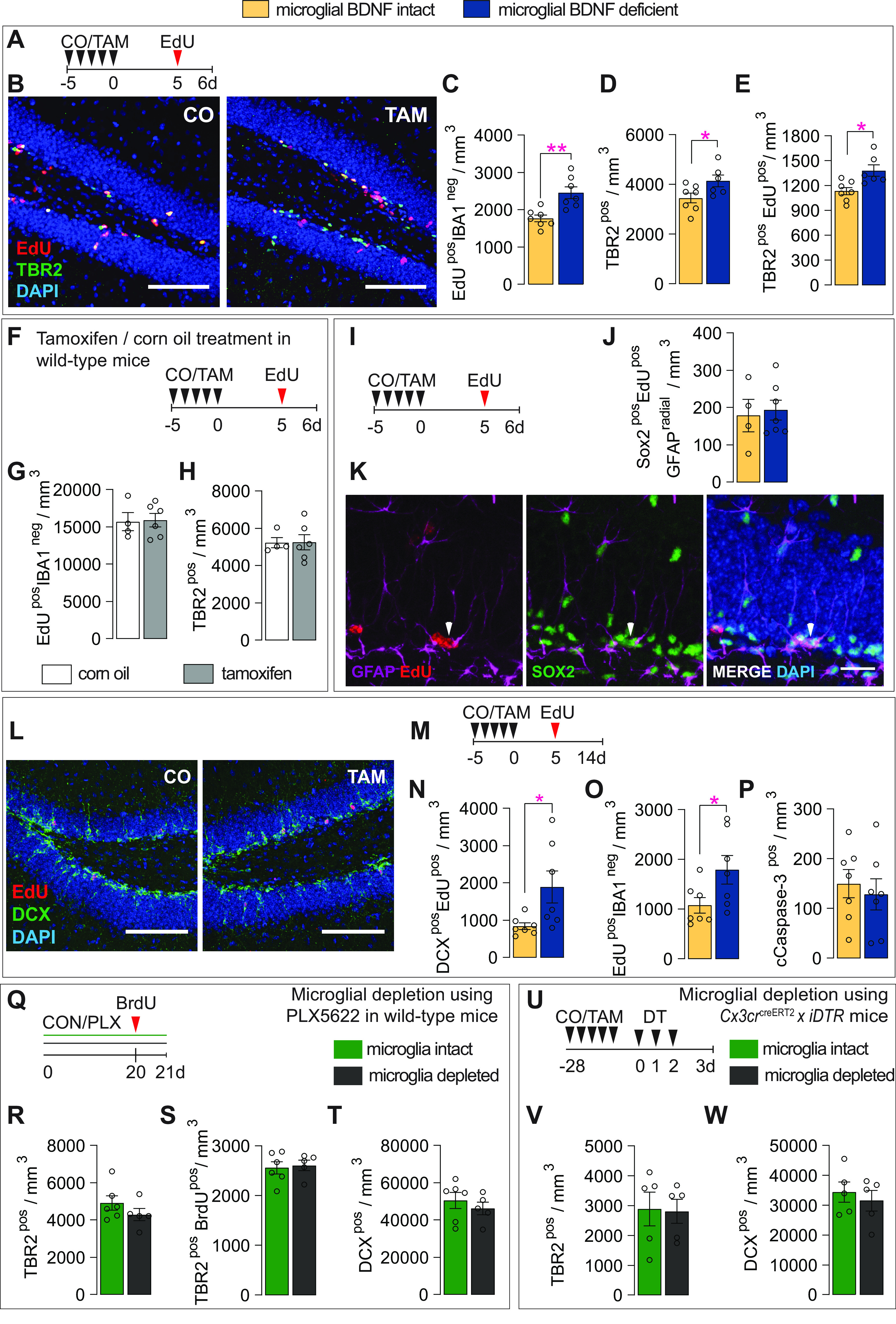
Microglial BDNF deficiency acutely stimulates neural precursor proliferation. A, Experimental timeline for intraperitoneal injection of EdU in control (CO) and microglial BDNF ablated (TAM) Cx3cr1creERT2 x Bdnf flox/flox mice. B, Representative images of proliferating EdUpos cells and TBR2pos neural precursors in the dentate gyrus of CO- and TAM-treated Cx3cr1creERT2 x Bdnf flox/flox mice. Scale bar, 100 μm. C, Quantification of EdUposIBA1neg proliferating cells (t(11) = 3.86, n = 6) 6 d after microglial BDNF ablation in Cx3cr1creERT2 x Bdnf flox/flox mice. D, E, Total TBR2pos (D: t(11) = 2.21, p = 0.049) and proliferating TBR2posEdUpos (E: t(11) = 3.06, p = 0.011) neural precursor in the SGZ. F, Experimental timeline for wild-type mice killed 6 d after CO or TAM treatment. G, Quantification of the density of EdUpos cells in the SGZ of wild-type mice (t(8) = 0.90, n = 4–6). H, Quantification of the density of TBR2pos neural precursors in the SGZ of wild-type mice (t(8) = 0.96, n = 4–6). I, Experimental timeline for intraperitoneal EdU injection into Cx3cr1creERT2 x Bdnf flox/flox mice. J, Quantification of SOX2posEdUposGFAPpos neural stem cell-like cells (t(9) = 0.32, n = 4–7). K, Representative images of neural precursor cells (indicated by arrow) identified as SOX2posEdUpos cells with a single radial GFAPpos process penetrating the SGZ. Scale bar, 20 μm. L, Representative images of the number of DCXposEdUpos immature neurons in the SGZ of CO- and TAM-treated Cx3cr1creERT2 x Bdnf flox/flox mice. Scale bar, 100 μm. M, Experimental timeline for Cx3cr1creERT2 x Bdnf flox/flox mice killed 9 d after intraperitoneal EdU injection. N, Quantification of newly born DCXposEdUpos immature neurons (t(12) = 2.39, n = 7, p = 0.034). O, Quantification of newborn surviving EdUposIBA1neg cells in Cx3cr1creERT2 x Bdnf flox/flox mice, killed 9 d after EdU injection (t(12) = 2.21, p = 0.048). P, Quantification of cleaved Caspase3pos (cCaspase-3pos) cells in dentate gyrus of Cx3cr1creERT2 x Bdnf flox/flox mice (t(12) = 0.50, p = 0.63). Q, Experimental timeline for microglial depletion induced by continuous PLX5622 treatment in wild-type mice. R, S, TBR2pos (R: t(9) = 1.20, p = 0.26, n = 5–6) and proliferating TBR2posBrdUpos (S: t(9) = 0.30 p = 0.77, n = 5–6) neural precursor in the SGZ of wild-type mice treated with PLX5622. T, Quantification of the density of DCXpos immature neurons in the SGZ of wild-type mice treated with PLX5622 (t(9) = 0.75, p = 0.48 n = 5–6). U, Experimental timeline for microglial depletion in Cx3cr1creERT2 x iDTR mice. V, W, Quantification of TBR2pos intermediate neuronal progenitors (T: t(8) = 0.11, p = 0.91, n = 5) and DCXpos immature neurons (U: t(5) = 0.59, p = 0.57, n = 5) in the dentate gyrus of Cx3Cr1creERT2 x iDTR mice 1 d post DT administration. Data represent the mean ± SEM. *p < 0.05, **p < 0.01. Statistical tests: unpaired Student's t test. Small circles represent individual mice.
Having established an acute stimulatory effect of microglial BDNF ablation on the TBR2pos precursor population, we next analyzed the number of SOX2pos cells in the SGZ that had a single radial GFAPpos process penetrating into the granule cell layer (Fig. 3I–K, arrowhead); these neural stem cell-like cells are known to give rise to TBR2pos neural precursors. However, in contrast to their progeny, the activity of these SOX2posGFAPpos cells was not altered by microglial BDNF ablation (Fig. 3J). We then tested whether expansion of the TRB2pos neural precursor pool in the absence of microglial BDNF also led to an increase in the number of immature adult-born granule cells; these immature neurons were identified by their expression of the microtubule-associated protein doublecortin (DCX; Rao and Shetty, 2004; Fig. 3L). For this, Cx3cr1creERT2 x Bdnf flox/flox mice were injected with EdU on day 5 and killed 9 d later, allowing for fate tracking of a wave of TBR2pos cells as they differentiate into immature neurons (Fig. 3M). Microglial BDNF ablation did indeed result in a significant increase in the absolute number of DCXposEdUpos cells (Fig. 3N). Consistent with the relatively short experimental timeline and also the fact that these EdU-labeled cells represent only a small proportion of the total pool of immature neurons, we found no difference in overall DCXpos cell numbers with microglial BDNF ablation (mice killed 6 d after corn oil/tamoxifen administration; CO = 61,036 ± 2731 cells/mm3; TAM = 51,559 ± 3796 cells/mm3; p = 0.70, t = 0.40, df = 10, n = 5–7/group). The proportion of DCXpos cells within the total EdUpos population was also not different between conditions (CO, 80.15 ± 4.24; vs TAM, 84.65 ± 2.76; p = 0.38; t(13) = 0.9, n = 7–8), suggesting that the observed increase in DCXposEdUpos cell numbers in the absence of microglial BDNF resulted from an expanded TBR2pos reservoir rather than from augmented differentiation of these cells into neurons. Consistent with this, the relative reduction in IBA1negEdUpos cells between 1 and 9 d postinjection (Fig. 3, compare C, O) was not different between conditions (i.e., in the presence or absence of microglial BDNF; p = 0.42, t(12) = 0.84), and there was also no difference in cleaved caspase-3 staining (apoptotic cells; Fig. 3P).
Having previously observed that the removal of microglia from hippocampal cell preparations of naive sedentary mice does not change neurosphere formation in vitro (Vukovic et al., 2012), we theorized that deleting BDNF from microglia likely changes their phenotype (because of a lack of autocrine BDNF signaling) to one that transiently stimulates neurogenesis. Consistent with this premise, in vivo depletion of microglia through both pharmacologic (Fig. 3Q–T; 78% IBA1pos cell depletion) and genetic (Fig. 3U–W; 97% IBA1pos cell depletion) approaches—and with it thus also microglia-derived BDNF—did not change the rate of adult neurogenesis in the hippocampus. Together, the above findings demonstrate that the ablation of BDNF from microglia stimulates TBRpos precursors, most likely through a change in microglial phenotype.
Loss of microglial BDNF does not alter the dendritic morphology of adult-born hippocampal neurons
As microglia have been demonstrated to shape neuronal connectivity in both development and adulthood (Paolicelli et al., 2011; Parkhurst et al., 2013), we next investigated whether microglial BDNF normally supports the development of dendrites and/or spine density on emerging adult-born neurons. To address this question, a retrovirus expressing the fluorescent reporter tdTomato was delivered into the dentate gyrus, labeling a wave of adult-born neurons that enabled the selective visualization of these cells as they matured and integrated into local circuitry (Fig. 4A,B). Mice were administered tamoxifen 5–7 d after retroviral injection to ablate microglial BDNF and then left for 24 d before being killed (Fig. 4A). Detailed examination of the morphology of these adult-born neurons revealed that the loss of microglial BDNF did not change the total dendritic length, number of branches, mean distance of dendritic terminals to their nearest neighbor, or overall dendritic complexity of adult-born neurons (Fig. 4C–F). Dendritic spine/protrusion density on adult-born tdTomatopos hippocampal granule cells was similarly unaltered following ablation of microglial BDNF (Fig. 4G,H), and there was also no difference in the total number of tdTomatopos neurons between groups (CO, 4076 ± 625.8; vs TAM, 4221 ± 460.2 cells/mm3; p = 0.81, t(6) = 0.25, n = 4). Collectively, these experiments suggest that microglial BDNF deficiency does not alter the dendritic morphology of adult-born hippocampal neurons, and they also corroborate our earlier conclusions that neuronal differentiation, maturation, and survival/integration is not affected under these conditions.
Figure 4.

Microglial BDNF deficiency does not influence the dendritic morphology of adult-born hippocampal neurons. A, Experimental timeline for tdTomato retrovirus intrahippocampal injection into Cx3cr1creERT2 x Bdnf flox/flox mice followed by either tamoxifen or corn oil gavage. B, Confocal images of dendritic morphology of tdTomato-positive retrovirus-labeled adult-born hippocampal neurons. Scale bar, 50 μm. C, D, Quantification of the total length of dendrites (t(9) = 0.92, n = 5–6; C) and the number of branches (t(9) = 0.34, n = 5–6; D). E, Quantification of the mean distance to the nearest neighboring dendritic terminal (t(9) = 0.43, n = 5–6). F, Sholl analysis of the complexity of dendrites (F(1,9) = 0.44, n = 5–6). G, Quantification of the density of dendritic spines (t(6) = 0.43, n = 4). H, Representative images of dendritic spines. Scale bar, 10 μm. Data represent the mean ± SEM. Statistical tests: Student's t test, C–E, G; repeated-measures two-way ANOVA followed by Bonferroni post hoc comparison test, F. Small circles represent individual mice.
Microglial BDNF deficiency differentially influences newborn immature neuron survival and microglial proliferation following LPS injection
Having established that microglial BDNF ablation impacted the proliferation of these cells and also hippocampal neurogenesis, we next sought to corroborate these results in mice treated with LPS; this immunologic challenge is a well established model for inducing microgliosis and also is known for its disruptive effects on hippocampal neurogenesis (Ekdahl et al., 2003; Monje et al., 2003). When injecting LPS into the hippocampus of Cx3cr1creERT2 x Bdnf flox/flox mice and then killing these animals 7 d later, we indeed observed significant reductions in both the total (DCXpos; 50%) and proliferated (DCXposBrdUpos; 81%) number of immature neurons compared with saline-injected controls (Fig. 5A–C). Similar to earlier experiments under unchallenged conditions (Fig. 3), ablation of microglial BDNF significantly increased the number of newly generated DCXposBrdUpos cells in response to LPS challenge (an approximately twofold increase; CO plus LPS, 451.58 ± 84.43; vs TAM plus LPS, 916.5 ± 130.70 cells/mm3; Fig. 5B,D). However, as these cells represent only a small proportion (<10%) of the total pool of immature neurons, overall DCXpos cell numbers were not significantly changed (Fig. 5B,C). Otherwise, there was also no difference in total DCXpos cell numbers between saline-injected mice with and without microglial BDNF deletion (CO plus saline, 26,606 ± 1347 cells/mm3; vs TAM plus saline, 25,264 ± 2871 cells/mm3; p = 0.66, t(9) = 0.45, n = 5), although a significant increase in the number of newly generated DCXposBrdUpos cells was again observed here under conditions where microglial BDNF was ablated (1.26-fold increase; CO plus saline: 1908 ± 75.66 cells/mm3 versus TAM plus saline: 2413 ± 159 cells/mm3; p = 0.014, t(9) = 3.01, n = 5).
Figure 5.

Microglial BDNF deficiency supports newborn immature neuron survival and reduces microglial proliferation following LPS infection. A, Experimental timeline for intraperitoneal BrdU injection following intrahippocampal LPS administration in Cx3cr1creERT2 x Bdnf flox/flox mice. B, Representative images of DCXpos and DCXposBrdUpos immature neurons in the dentate gyrus. Scale bar, 50 μm. Dotted lines indicated the GCL. C, Quantification of DCXpos immature neurons 7 d after intrahippocampal LPS injection (F(2,19) = 18.98, p < 0.0001). D, Quantification of newly born DCXposBrdUpos immature neurons that were born 1–4 d after LPS stimulation and survive until day 7 post-LPS (F(2,19) = 47.39, p < 0.0001). E, Representative confocal images of IBA1posBrdUpos microglia in the dentate gyrus. Scale bar, 50 μm. F, Quantification of IBA1pos microglia in the dentate gyrus (F(2,24) = 76.99, p < 0.0001). G, Quantification of IBA1posBrdUpos microglia in the dentate gyrus (F(2,19) = 11.87, p = 0.0005). H, Quantification of IBA1pos mean fluorescence intensity (F(2,15) = 28.31, p < 0.0001). I, Representative confocal images of Tmem119pos, CD68pos microglia in the dentate gyrus. J, K, Quantification of TMEM119pos (J: F(2,16) = 3.73, p = 0.047) and CD68pos (K: F(2,16) = 2.17, p = 0.15) mean fluorescence intensity. L, Experimental timeline for Cx3cr1creERT2 x Bdnf flox/flox killed 6 h after intrahippocampal LPS administration. M, qPCR data for select genes associated with the neuroinflammatory response to LPS infection in the ipsilateral hippocampus of Cx3cr1creERT2 x Bdnf flox/flox mice 6 h after saline or LPS injection. Expression is relative to the Hprt housekeeping gene and presented as the “fold change” from saline-injected corn oil-gavaged vehicle controls (Ccl2: F(2,10) = 11.70, p = 0.0024; Csf-1: F(2,10) = 12.56, p = 0.0019; IL-1β: F(2,10) = 77.99, p < 0.0001; IL-6: F(2,10) = 4.87, p = 0.033; IL-10: F(2,10) = 3.88, p = 0.035; TNF-α: F(2,10) = 5.08, p = 0.030, n = 3–5/group). Data represent the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Statistical tests: one-way ANOVA followed by Bonferroni post hoc comparison; asterisks indicate post hoc results (C, D, F–H, J, K, M). Small circles represent individual mice. A.U., Arbitrary units.
When assessing microglial responses in these mice, we found that both microglia proliferation (IBA1posBrdUpos cells) and overall microglia density (IBA1pos cells) were significantly increased following LPS challenge (Fig. 5E–G). Importantly, this LPS-induced microgliosis was significantly blunted under conditions where BDNF was deleted from these cells, as is evident from a reduction in overall microglia numbers (Fig. 5F) and microglial proliferation (Fig. 5G); both readouts were also significantly reduced in saline-injected mice with microglial BDNF ablation (IBA1pos: CO plus saline, 7335 ± 232 cells/mm3; vs TAM plus saline, 6230 ± 136.9 cells/mm3; p = 0.0008, t(19) = 4.00, n = 5; IBA1posBrdUpos: CO plus saline, 329.2 ± 69.91 cells/mm3; vs TAM plus saline, 78.97 ± 20.01 cells/mm3; p = 0.018, t(7) = 3.08, n = 5). The MFI of IBA1 staining was also attenuated with microglial BDNF ablation (Fig. 5H), suggesting lessened microglia activation in response to LPS injection. Although the staining intensity was much lower (>10-fold), a reduced IBA1 MFI was similarly observed in saline-injected control mice with microglial BDNF deletion [CO plus saline, 42.95 ± 4.92 (n = 5); vs TAM plus saline, 25.98 ± 1.40 (n = 5); p = 0.035, t(5) = 2.86]. Further examination showed that LPS stimulation also increased other markers of microglial activation, including the lysosomal protein CD68 (Fig. 5I,J; surrogate marker of phagocytic activity) and TMEM119 (Fig. 5K). However, in contrast to IBA1, microglial BDNF did not significantly alter the LPS-induced changes in either CD68 or TMEM119 MFI (Fig. 5I–K). Last, we examined the expression of cytokines typically induced by LPS stimulation, including Ccl2, Csf-1, IL-1β, IL-6, IL-10, and TNFα. All of these were indeed found to be upregulated 6 h after intrahippocampal LPS administration, and without any influence thereon of microglial BDNF ablation (Fig. 5L,M). Together, these data reveal that microglial BDNF deficiency augments the production and/or survival of newly generated immature neurons under neuroinflammatory conditions, while reducing microglia activation.
Microglial BDNF deficiency differentially influences neurogenesis, cognition, and microglia proliferation in traumatic brain injury
Last, we investigated the effect of microglial BDNF deficiency in a well defined model of TBI (i.e., unilateral controlled cortical impact). Here, Cx3cr1creERT2 x BDNF flox/flox mice were administered either corn oil (vehicle) or tamoxifen at least 4 weeks prior TBI (Fig. 6A); this gap in time avoids any unintended effects of tamoxifen on myeloid cells that may become recruited to the injury site and ensures the turnover of short-lived monocytes while leaving the longer-lived microglia BDNF deficient at the time of surgery (Parkhurst et al., 2013). Overall, TBI led to a significant loss of DCXpos immature neurons in the ipsilateral hippocampus at 12 d postinjury compared with sham-operated controls (63% loss; Fig. 6B). This TBI-induced loss of DCXpos cells was attenuated in mice with microglial BDNF ablation (an approximately twofold increase; Fig. 6B). The number of immature neurons that were born shortly after TBI (1–3 d postsurgery) and survived up to 12 d postinjury (i.e., DCXposBrdUpos cells) was also significantly greater in the absence of microglial BDNF (an approximately twofold increase; Fig. 5C). No significant differences in total DCXpos cell numbers were otherwise observed between sham-operated mice with and without microglial BDNF (CO sham, 27,644 ± 761.4; TAM sham, 24,891 ± 1683 cells/mm3; t(11) = 1.41, p = 0.19), although an increase in the number of newly generated immature neurons (i.e., DCXposBrdUpos cells) was once again also observed here with microglial BDNF ablation (CO sham, 227.5 ± 62.98; vs TAM sham, 406.5 ± 43.56 cells/mm3; t(11) = 2.39, p = 0.036, n = 6–7).
Figure 6.
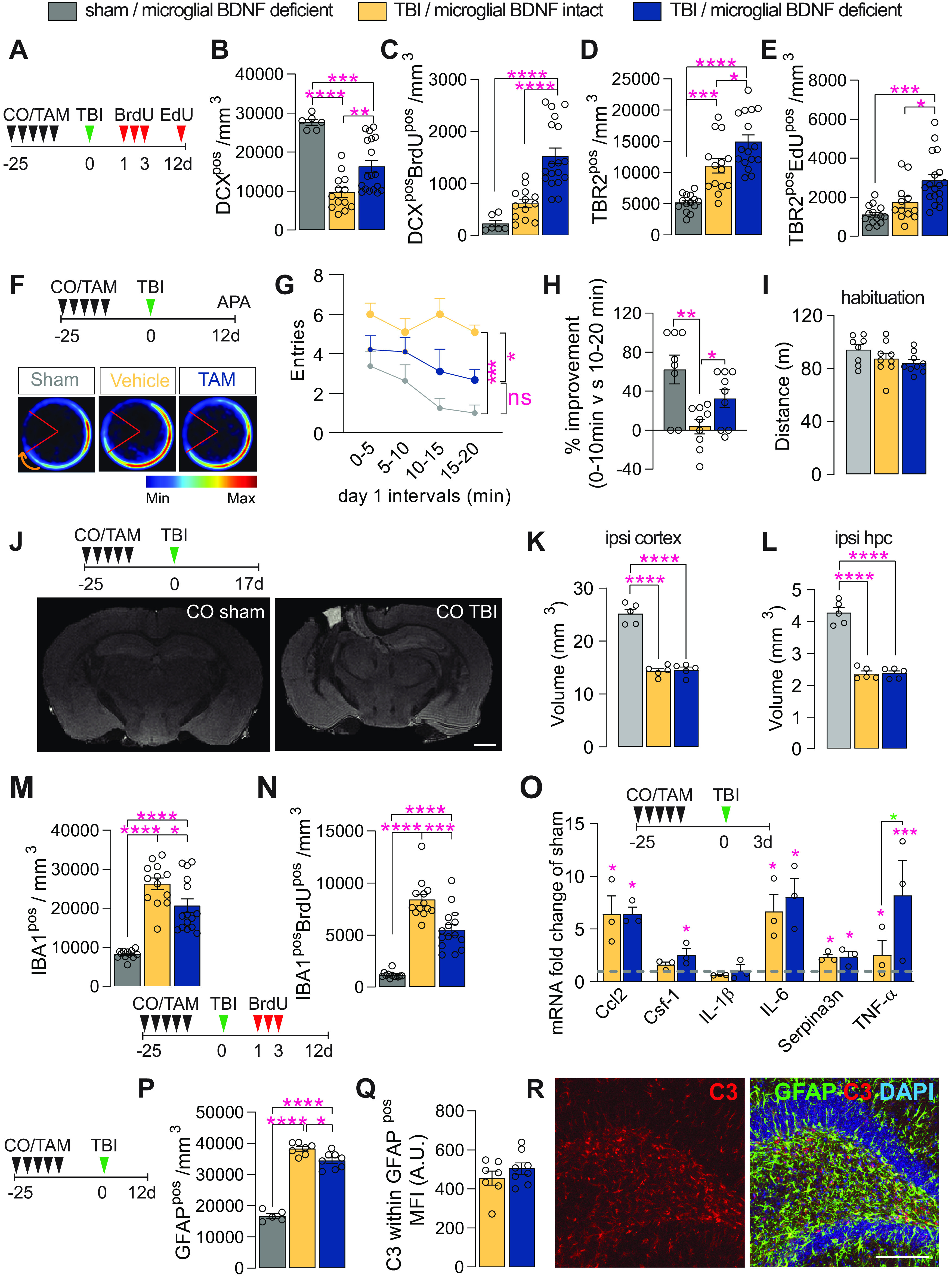
Microglial BDNF deficiency stimulates hippocampal neurogenesis and has a modest effect on short-term learning and memory, while reducing microglial proliferation following traumatic brain injury. A, Experimental timeline for intraperitoneal injection of BrdU and EdU in Cx3cr1creERT2 x Bdnf flox/flox mice following sham or TBI surgery. B, Quantification of total DCXpos immature neurons at 12 d postinjury (F(2,34) = 22.55, p < 0.0001). C, Quantification of immature neurons (i.e., DCXposBrdUpos) born after TBI that survive until day 12 postinjury (F(2,34) = 22.36, p < 0.0001). D, E, Quantification of total (D: F(2,41) = 25.95, p < 0.0001) and proliferating (E: F(2,41) = 12.34, p < 0.0001; TBR2posEDUpos) TBR2pos neural precursors at 12 d postinjury. F, Experimental timeline for microglial BDNF ablation in Cx3cr1creERT2 x Bdnf flox/flox mice followed by TBI surgery and APA testing. Merged heatmaps showing the mean proportion of time spent in a particular location during day 1 of APA testing. G, Within-session APA performance of sham and TBI mice with and without microglial BDNF ablation on day 1 of testing (F(2,23) = 13.04, p = 0.0002). Asterisks indicate post hoc results for the 15–20 min interval between groups. H, Percentage improvement in APA within-session performance for individual mice, as assessed by the change in the number of shock zone entries between the first and the last 10 min of the 20 min session (F(2,23) = 7.45, p = 0.0032). I, Distance traveled during habituation trial of APA testing (F(2,23) = 2.09, p = 0.15). J, Top, Experimental timeline for microglial BDNF ablation in Cx3cr1creERT2 x Bdnf flox/flox mice and ex vivo MRI 17 d after TBI surgery. Bottom, T1/T2 FLASH images of sham-operated and TBI mice. K, L, T1/T2 ex vivo MRI measurements of total spared tissue volumes for the ipsilateral (ipsi) cortex (K: F(2,12) = 94.21, p < 0.0001), and ipsilateral hippocampus (L: F(2,12) = 92.63, p < 0.0001), in both sham and TBI mice with microglial BDNF intact or ablated. M, Quantification of IBA1pos microglia in the dentate gyrus 12 d post-TBI (F(2,37) = 38.82, p < 0.0001). N, Quantification of IBA1posBrdUpos newborn microglia born on days 1–3 post-TBI and surviving until day 12 post-TBI (F(2,37) = 61.86, p < 0.0001). O, qPCR data for select genes associated with TB1-related immune processes in the ipsilateral hippocampus of Cx3cr1creERT2 x Bdnf flox/flox mice with intact versus ablated microglial BDNF 3 d after TBI. Expression is relative to the Hprt housekeeping gene and presented as the “fold change” from sham-operated controls (dotted gray line; Ccl2: F(2,7) = 11.65, p = 0.0059; Csf-1: F(2,7) = 5.93, p = 0.031; IL-1β: F(2,7) = 0.54, p = 0.61; IL-6: F(2,7) = 10.30, p = 0.0082; Serpina3n: F(2,7) = 7.23, p = 0.020; TNF-α: F(2,7) = 33.03, p = 0.0003; t test analysis for TNF-α: t(7) = 3.99, p = 0.016 (blue asterisk). P, Quantification of GFAPpos astrocytes in the dentate gyrus (F(2,17) = 153.6, p < 0.0001). Q, Quantification of C3 MFI within GFAPpos astrocytes in the ipsilateral dentate gyrus of Cx3cr1creERT2 x Bdnf flox/flox mice after TBI surgery (t(13) = 1.07, p = 0.31). R, Representative confocal images of GFAPposC3pos astrocytes in the dentate gyrus. Data represent the mean ± SEM. *p < 0.05, **p < 0.01 ***p < 0.001, ****p < 0.0001. Statistical tests: one-way ANOVA, B–E, G, I, K–P, Q; repeated-measures two-way ANOVA, H; followed by Bonferroni post hoc comparison test for all; asterisks indicate ANOVA post hoc result; unpaired Student's t test, Q. n = 6–18/group (B–E), n = 8–10/group (G–I), n = 5/group (K, L), n = 12–15 (M, N), n = 3/group (O), n = 7–8/group (P, Q). Circles represent individual mice. A.U., arbitrary units.
Consistent with our previous observations (Willis et al., 2020), a significant increase in the overall size and activity of the TBR2pos neural progenitor pool was observed in response to TBI, which was further augmented by microglial BDNF ablation (34–76% increase; Fig. 6D,E). Similar effects were seen in sham-operated controls where microglial BDNF ablation also resulted in significant increases in both total and proliferating TBR2pos intermediate neuronal progenitor cell numbers (TBR2pos CO sham, 5183 ± 373.5 cells/mm3; vs TBR2pos TAM sham, 6385 ± 308.1 cells/mm3; p = 0.041, t(20) = 2.19, n = 8–14; and TBR2posEdUpos CO sham, 1104 ± 127.5 cells/mm3; vs TAM sham, 1544 ± 83.05 cells/mm3; p = 0.033, t(19) = 2.30, n = 7–14).
We next investigated whether the observed increase in newly proliferated DCXpos cells in the absence of microglial BDNF could improve working memory and spatial learning following TBI as we previously demonstrated that immature neurons positively contribute to performance in such hippocampus-dependent tasks (Vukovic et al., 2013). Cognitive abilities of sham and TBI mice with and without microglial BDNF were tested in APA, a robust behavioral task that requires animals to navigate a rotating turntable and continuously use visual cues in their surroundings to avoid being moved into a stationary shock zone (Vukovic et al., 2013; Willis et al., 2017). As expected, sham-operated mice quickly learned to avoid the shock zone within a single session, as evident from their improved performance (i.e., reduction in shock zone entries) during a 20 min APA test (Fig. 6F–H); this “within-session” learning reflects their short-term working memory (Willis et al., 2017). Vehicle-treated TBI mice are not able to learn this task, making significantly more mistakes (greater number of shock zone entries) than their sham-operated counterparts (Fig. 6G) and failing to show any within-session learning over time (Fig. 6H). On the other hand, TBI mice with microglial BDNF deficiency clearly displayed some learning, resulting in a significantly better APA performance (Fig. 6G,H). Consistent with our recent observations (Willis et al., 2020), the rescue and/or increase in DCXpos cell numbers that was observed here in the absence of microglial BDNF thus appears to increase the cognitive flexibility of TBI mice. Last, assessment of the distance that was traveled by each mouse during APA habituation (i.e., when shock zone turned off) demonstrated no differences between groups (Fig. 6I), ruling out differences in general locomotor activity between groups as a factor of influence. Ex vivo MR imaging of in-skull samples otherwise showed that there were also no differences in lesion size, with the volume of spared cortical tissue being the same in TBI mice with and without microglial BDNF (Fig. 6J,K); hippocampal volumes were also not different between these two groups (Fig. 6L).
Having established neurogenic and behavioral benefits, we next examined how microglial BDNF ablation impacted on the responses of these cells to TBI. Consistent with previous observations under both homeostatic conditions and following LPS challenge, microglial BDNF ablation significantly reduced both the total number (27% decrease) and proliferating number (BrdUpos; 34% decrease) of IBA1pos microglia in the ipsilateral dentate gyrus (Fig. 6M,N). Similar observations were made in sham-operated mice where microglial BDNF ablation also significantly reduced both the total number (CO sham, 8211 ± 325.6 IBA1pos cells/mm3; vs TAM sham, 7007 ± 243.2 IBA1pos cells/mm3; p = 0.040, t(15) = 2.25, n = 5–12) and proliferating number (CO sham, 1164 ± 90.02 IBA1posBrdUpos cells/mm3; vs TAM sham, 723.2 ± 174.5 IBA1posBrdUpos cells/mm3; p = 0.026, t(15) = 2.48, n = 5–12) of microglia.
Given the consistent effects of microglial BDNF on the proliferation and overall density of these cells, including after brain insult, we examined last whether there were any associated changes in the expression of inflammatory genes and/or astrocyte reactivity in the ipsilateral hippocampus of TBI mice. Consistent with the LPS experiments, microglial BDNF deletion did not impact on TBI-associated increases in the expression of select inflammatory chemokines/cytokines, with the exception of TNFα (Fig. 6O); microglial BDNF deficiency also did not change the expression of the A1 astrocyte gene Serpina3n. A modest reduction in astrogliosis was observed in the hippocampi of TBI mice with microglial BDNF deletion (Fig. 6P), but without an overt change in C3 expression (A1 astrocyte marker) by these cells (Fig. 6Q,R). Together, we found that microglial BDNF deficiency augments hippocampal neurogenesis after TBI, attenuating the deficits in working memory induced by such insult. Injury-induced gliosis was also reduced under conditions where BDNF was absent from microglia.
Microglial BDNF deficiency does not alter the beneficial effects of repopulating microglia on attenuating cognitive deficits and hippocampal degeneration after TBI
We recently reported the discovery of a neuroprotective and proregenerative microglia phenotype that can be induced through the experimental turnover of these cells (Willis et al., 2020). Specifically, we showed that repopulating microglia augment both neuronal survival and neurogenesis post-TBI in an IL-6-dependent manner. With previous in vitro work indicating that such positive IL-6 effects may require BDNF (Murphy et al., 2001), we directly probed a possible mechanistic involvement and/or contribution of microglial BDNF to the beneficial effects of repopulating microglia in our final set of experiments. For this, corn oil-treated (i.e., vehicle controls) and tamoxifen-treated Cx3cr1creERT2 x BDNF flox/flox mice were placed on a 3 week PLX5622-containing diet to induce microglia depletion/repopulation at the time of TBI (Fig. 7A). When testing these mice in APA, the previously established benefits of repopulating microglia for improved spatial learning and memory after TBI were clearly evident (Fig. 7B,C). However, no influence of microglial BDNF therein was observed.
Figure 7.
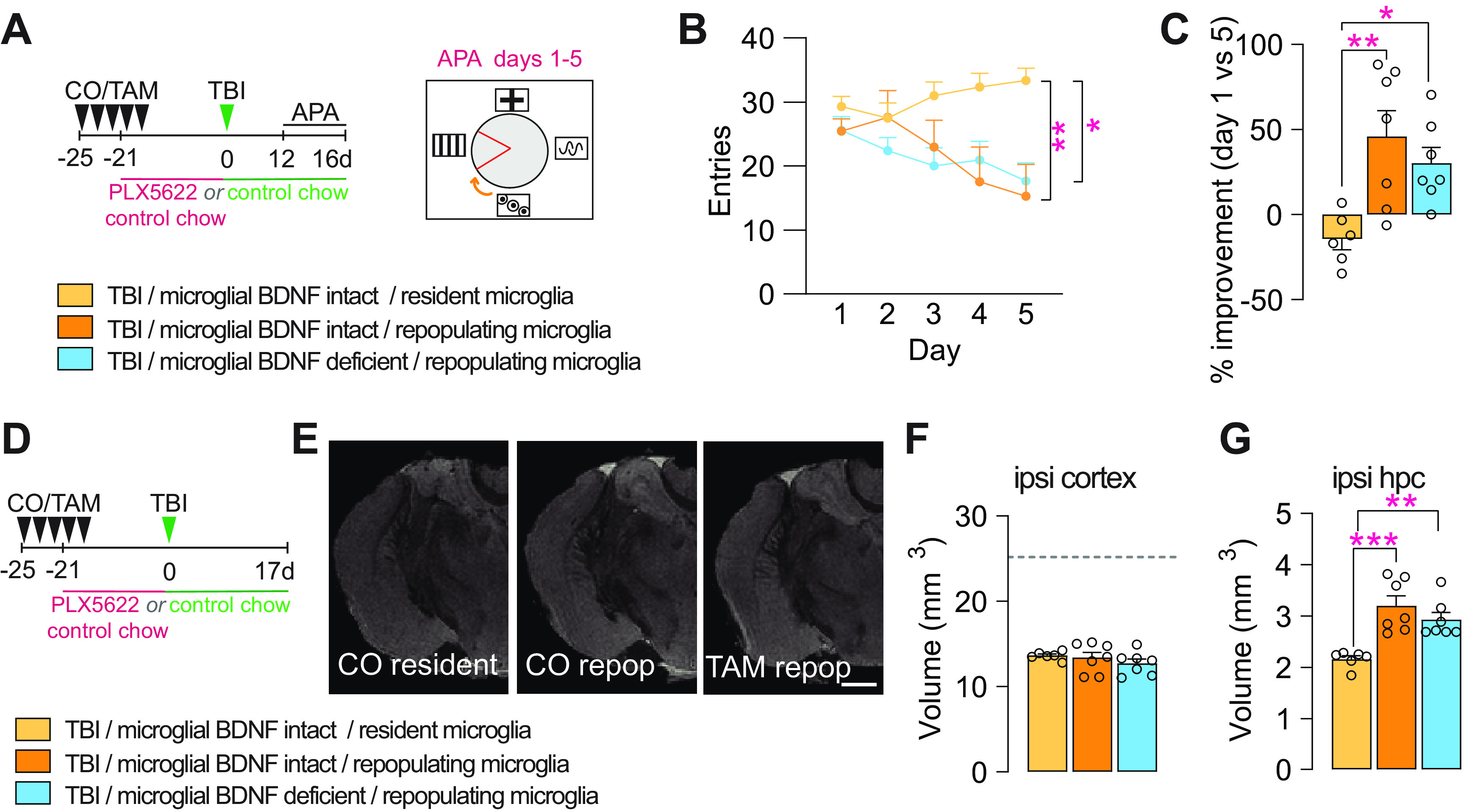
Microglial BDNF deficiency does not alter the beneficial effects of repopulating microglia on attenuating cognitive deficits and hippocampal degeneration induced by traumatic brain injury. A, Experimental timeline for microglial depletion and repopulation using transient PLX5622 treatment in control (CO) and microglial BDNF-ablated (TAM) Cx3cr1creERT2 x Bdnf flox/flox mice and subsequent APA testing after TBI. B, Shock zone entries for TB1 mice with resident or repopulating microglia in conditions with microglial BDNF intact or deficient (F(2,85) = 12.58, p < 0.0001); asterisks indicate post hoc results for testing day 5. C, Percentage improvement in APA performance, assessed by the change in entries on testing day 5 versus day 1 for individual mice (F(2,17) = 7.93, p = 0.0049). D, Experimental timeline for microglial repopulation experiments with transient PLX5622 treatment; mice were killed 17 d post-TBI. E, T1/T2 ex vivo MRI snapshots of Cx3cr1creERT2 x Bdnf flox/flox mice with microglial BDNF intact (CO) and microglial BDNF ablated (TAM), and in the presence of resident (control chow) versus repopulating microglia. F, G, T1/T2 ex vivo MRI measurement of total (spared) tissue volumes for the ipsilateral cortex (F: F(2,17) = 0.82, p = 0.46) and ipsilateral hippocampus (G: F(2,17) = 12.53, p = 0.0005) after TBI, with microglial BDNF intact (CO) or ablated (TAM), and under resident or repopulating microglia conditions; dotted line represents sham-operated mice from Figure 6K. Data represent the mean ± SEM. *p < 0.05, **p < 0.01 ***p < 0.001. Statistic tests: repeated-measures two-way ANOVA, B; one-way ANOVA, C, F, G; followed by Bonferroni post hoc comparison for all; asterisks indicate ANOVA post hoc result. A–G, n = 6–7/group. Circles represent individual mice. repoP, Repopulation; ipsi, ipsilateral; hpc, hippocampus.
Ex vivo in-skull MR imaging otherwise revealed no differences in injury severity/lesion size, with the volume of spared cortical tissue being the same across the experimental groups (Fig. 7D–F). Microglial repopulation did attenuate secondary degeneration of the ipsilateral hippocampus, but no effect of microglial BDNF thereon was observed (Fig. 7G). In summary, these experiments confirm that repopulating microglia attenuate cognitive deficits and hippocampal degeneration in this mouse model of TBI; they further show that microglial BDNF is dispensable in this.
Discussion
Microglial density in the noninflamed brain is maintained at a constant level by a balance of proliferation and apoptosis (Askew et al., 2017). The most well known regulator of microglial density is the CSF-1/CSF-1R axis, in which the neuronally derived ligands CSF-1 and IL-34 activate microglial CSF-1R to stimulate the survival and/or proliferation of these cells (Greter et al., 2012; Nandi et al., 2012; Elmore et al., 2014; Guan et al., 2016). In recent times, we and others have harnessed this dependency of microglia on CSF-1R signaling to deplete and/or induce the turnover of these cells in the adult brain using both genetic approaches and small-molecule inhibitors of CSF-1R signaling, such as PLX5622 (Rice et al., 2017; O'Neil et al., 2018; Willis et al., 2020). Here, we show that cell-specific ablation of BDNF from microglia negatively regulates their proliferation and overall density under both homeostatic and challenged conditions (infection and injury), while increasing adult neurogenesis.
Microglial BDNF contributes to self-regulation of proliferation and density
Loss of microglial BDNF reduced the proliferation of these cells to one-third of their normal cycling activity under homeostatic conditions, resulting in a 16% reduction in overall microglial density within 6 d and a progressive further loss beyond this time point (i.e., 35% reduction 5 weeks post-microglial BDNF depletion). We suggest that microglial BDNF acts very locally and in an autocrine manner considering that the lack of it leaves total BDNF levels in the brain unchanged (Parkhurst et al., 2013). Consistent with this, several previous studies have demonstrated that microglia express the BDNF receptor TrkB, both in vivo (Spencer-Segal et al., 2011; Khan et al., 2015) and in vitro (Nakajima et al., 1998; Mizoguchi et al., 2009). It has also been shown that the addition of BDNF to both primary microglia and the immortalized microglial BV2 cell line stimulates their proliferation (Zhang et al., 2003; Gomes et al., 2013). Mechanistically, these effects of BDNF are thought to be mediated via signaling events downstream of TrkB that cause the degradation of the kinase Iκβ and subsequent activation of the transcription factor nuclear factor-κβ, which is a well known positive regulator of microglial proliferation (Nakajima et al., 1998). Indeed, sequestering BDNF with a neutralizing antibody in N9 microglial-like cell cultures attenuates LPS-induced proliferation of microglia (Gomes et al., 2013). A positive autocrine BDNF feedback loop has otherwise been suggested where ATP-mediated microglial activation induces BDNF expression and/or release (Coull et al., 2005; Zhang et al., 2014). However, as there are various cellular sources of BDNF in the brain, the significance of its expression by microglia in vivo had remained mostly obscure. We note that Parkhurst et al. (2013) reported no change in IBA1pos microglia numbers in the cortex and CA1 region of Cx3cr1creERT2 x Bdnf flox/flox mice; however, this was based on comparing mice that were either homozygous or heterozygous for the Bdnf flox allele, and we posit that dose-dependency effects account for this seeming discrepancy between our studies. Indeed, a comparison to WT mice, as performed here, convincingly demonstrates that microglial BDNF contributes to regulating the proliferation and density of these cells within the adult mammalian brain in an autocrine manner.
Loss of microglial BDNF stimulates adult neurogenesis in the dentate gyrus
In addition to the effects on microglial self-renewal, we also show an increased capacity of BDNF-deficient microglia to support the rate of adult neurogenesis in vivo, under homeostatic as well as injured and/or neuroinflammatory conditions. Previous studies have demonstrated that BDNF expression in the mouse brain is largely attributed to astrocytes and neurons (Zhang et al., 2014), and that BDNF from both these cell types supports hippocampal neurogenesis (Chan et al., 2008; Waterhouse et al., 2012; Quesseveur et al., 2013; Wang et al., 2015). Consistent with microglia being only a minor source, total BDNF protein levels were unaltered in the hippocampi of Cx3cr1creERT2 x Bdnf flox/flox mice. Yet, as shown, the selective ablation of BDNF from microglia had a very distinct stimulatory (as opposed to diminishing) effect on adult hippocampal neurogenesis. These findings are very similar to those of Groves et al. (2019), who reported that pharmacologic blockade of TrkB (i.e., the main signaling receptor for BDNF) increases neural precursor cell proliferation and also the number of adult-born neurons in the dentate gyrus. The present study thus identifies microglia as the putative cellular target via which these in vivo effects of TrkB antagonism may be mediated. Our microglia depletion experiments otherwise strongly suggest that the proneurogenic effects of BDNF-deficient microglia result from a change in phenotype rather than a loss of BDNF per se as the absence of these cells does not alter the normal rate of neurogenesis. This finding is consistent with previous observations that absence of microglia has a negligible influence on hippocampal stem/progenitor cell activity (Willis et al., 2020), thus arguing against a direct inhibitory role for microglial BDNF in adult neurogenesis. Last, while the effects of microglial BDNF depletion on adult neurogenesis were transient under control conditions, they were much more long term (>30 d) under inflammatory conditions, as induced by either LPS injection or brain injury. These findings strongly suggest that endogenous mechanisms that may be able to compensate for the loss of microglial BDNF under normal conditions (e.g., the CSF-1/CSF-1R axis) cannot sufficiently do so following a major physiological challenge. Future studies can now focus on defining the molecular changes within BDNF-deficient microglia that lead to increased neural precursor proliferation, most likely through direct interactions between these two cell types (Vukovic et al., 2012), and to also further delineate more subtle effects that BDNF-deficient microglia may have on progenitor cell differentiation.
In light of our recent findings that inducing microglia turnover similarly yields a phenotype that is supportive of immature neuron production/survival (Willis et al., 2020), we investigated whether microglial BDNF has any direct involvement in these established benefits of repopulating microglia. The present study shows, however, that microglia-derived BDNF is dispensable and/or not required for repopulating microglia to exert their neuroprotective effects, as the selective removal of BDNF from these cells does not annul their mitigating influence over secondary injury processes and the cognitive deficits induced by TBI. Similarly, considering that repopulating microglia mediate their beneficial effects by upregulating IL-6 expression within hippocampal granule cells (Willis et al., 2020), microglial BDNF cannot be the factor inducing this.
Microglial BDNF deficiency does not alter the morphology and/or integration of adult-born neurons in the dentate gyrus
Microglia have been demonstrated to shape neuronal connectivity, the number of synaptic elements and dendritic branches in both the developing and adult cerebral cortex (Paolicelli et al., 2011; Parkhurst et al., 2013; Wallace et al., 2020). Whether microglia also influence the maturation and/or integration of newly generated granule cells into existing hippocampal circuits throughout adult life remains poorly understood. Parkhurst et al. (2013) demonstrated that the ablation of microglial BDNF interfered with learning-induced plasticity in the motor cortex. We found no evidence that such a role for microglial BDNF also extends to the maturation and integration of newly generated granule neurons in the adult hippocampus, with no overt effect on behavior and the morphology and/or changes in dendritic spine numbers for these cells. We recognize, however, that our analysis did not include a comparison of spine morphology, motility, formation, and/or disappearance in real time with and without microglial BDNF; a subtler role in experience-induced synapse formation by and/or onto these newly generated granule cells can therefore not be fully excluded at the present time.
In summary, the present study reports the following two previously undescribed functions of microglial BDNF in vivo: the autocrine regulation of cell cycling activity; and paracrine effects on neuronal precursor cell activity under both homeostatic and injured/inflammatory conditions. Our findings pave the way for additional mechanistic studies as to how microglial BDNF influences the behavior and phenotype of these cells in vivo with regard to self-renewal and their interactions with neural precursors.
Footnotes
J.V. holds a Senior Medical Research Fellowship from the Sylvia and Charles Viertel Foundation and was also supported by both the Australian Research Council (Discovery Early Career Research Award 150101578) and the National Health and Medical Research Council (NHMRC; Project Grant 1124503). S.B.R.H. was supported by an Australian government Research Training Scholarship, a Queensland Brain Institute Student Top-Up Scholarship, and a UQ Career Development Scholarship Extension. E.F.W. was supported by the University of Queensland Senate Scholarship Herdsman Fellowship in Medical Science and a UQ Career Development Scholarship Extension. M.J.R. was supported by SpinalCure Australia, the Wings for Life Spinal Cord Research Foundation, and the NHMRC (Grants 1060538 and 1163835). Imaging was performed at the Advanced Microscopy Facilities of the Queensland Brain Institute, using Yokogawa and Diskovery spinning disk confocal microscopes, Imaris software, and the stereology microscope, the use of which was supported by the Australian Government through ARC (Australian Research Council) LIEF (Linkage Infrastructure, Equipment and Facilities) Grant LE100100074. Behavioral tests were performed at the Behavior and Surgical Facility of the Queensland Brain Institute. We thank Robert Sullivan for experimental assistance. We also thank the staff of The University of Queensland Biological Resources Facility for breeding and maintaining the animals in this study. In addition, we thank the National Imaging Facility at the Center for Advanced Imaging, University of Queensland for use of the facilities, and for scientific and technical assistance.
The authors declare no competing financial interests.
References
- Araki T, Ikegaya Y, Koyama R (2020) The effects of microglia- and astrocyte-derived factors on neurogenesis in health and disease. Eur J Neurosci. Advance online publication. Retrieved September 13, 2020. doi: 10.1111/ejn.14969. 10.1111/ejn.14969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, Tipton T, Chapman MA, Riecken K, Beccari S, Sierra A, Molnár Z, Cragg MS, Garaschuk O, Perry VH, Gomez-Nicola D (2017) Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep 18:391–405. 10.1016/j.celrep.2016.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, Weissman IL, Chang EF, Li G, Grant GA, Hayden Gephart MG, Barres BA (2016) New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 113:E1738–E1746. 10.1073/pnas.1525528113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M (2006) Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis or oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci 31:149–160. 10.1016/j.mcn.2005.10.006 [DOI] [PubMed] [Google Scholar]
- Chaaya N, Jacques A, Belmer A, Beecher K, Ali SA, Chehrehasa F, Battle AR, Johnson LR, Bartlett SE (2019) Contextual fear conditioning alter microglia number and morphology in the rat dorsal hippocampus. Front Cell Neurosci 13:214. 10.3389/fncel.2019.00214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JP, Cordeira J, Calderon GA, Iyer LK, Rios M (2008) Depletion of central BDNF in mice impedes terminal differentiation of new granule neurons in the adult hippocampus. Mol Cell Neurosci 39:372–383. 10.1016/j.mcn.2008.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JAM, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438:1017–1021. 10.1038/nature04223 [DOI] [PubMed] [Google Scholar]
- Diaz-Aparicio I, Paris I, Sierra-Torre V, Plaza-Zabala A, Rodríguez-Iglesias N, Márquez-Ropero M, Beccari S, Huguet P, Abiega O, Alberdi E, Matute C, Bernales I, Schulz A, Otrokocsi L, Sperlagh B, Happonen KE, Lemke G, Maletic-Savatic M, Valero J, Sierra A (2020) Microglia actively remodel adult hippocampal neurogenesis through the phagocytosis secretome. J Neurosci 40:1453–1482. 10.1523/JNEUROSCI.0993-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl CT (2012) Microglial activation—tuning and pruning adult neurogenesis. Front Pharmacol 3:41. 10.3389/fphar.2012.00041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A 100:13632–13637. 10.1073/pnas.2234031100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82:380–397. 10.1016/j.neuron.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME (1994) Requirement for BDNF in activity-dependent survival of cortical neurons. Science 263:1618–1623. 10.1126/science.7907431 [DOI] [PubMed] [Google Scholar]
- Gomes C, Ferreira R, George J, Sanches R, Rodrigues DI, Gonçalves N, Cunha RA (2013) Activation of microglial cells triggers a release of brain-derived neurotrophic factor (BDNF) inducing their proliferation in an adenosine A2A receptor-dependent manner: a 2A receptor blockade prevents BDNF release and proliferation of microglia. J Neuroinflammation 10:16. 10.1186/1742-2094-10-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M, Kündig TM, Frei K, Ginhoux F, Merad M, Becher B (2012) Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 37:1050–1060. 10.1016/j.immuni.2012.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves N, O'Keeffe I, Lee W, Toft A, Blackmore D, Bandhavkar S, Coulson Elizabeth J, Bartlett PF, Jhaveri DJ (2019) Blockade of TrkB but not p75 NTR activates a subpopulation of quiescent neural precursor cells and enhances neurogenesis in the adult mouse hippocampus. Develop Neurobiol 79:868–879. 10.1002/dneu.22729 [DOI] [PubMed] [Google Scholar]
- Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, Abboud-Werner SL, Lanier LL, Lomvardas S, Basbaum AI (2016) Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci 19:94–101. 10.1038/nn.4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoneva S, Hosur R, Gosselin D, Zhang B, Ouyang Z, Cotleur AC, Peterson M, Allaire N, Challa R, Cullen P, Roberts C, Miao K, Reynolds TL, Glass CK, Burkly L, Ransohoff RM (2019) Cx3cr1-deficient microglia exhibit a premature aging transcriptome. Life Sci Alliance 2:e201900453. 10.26508/lsa.201900453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–1394. 10.1038/nn1997 [DOI] [PubMed] [Google Scholar]
- Hodge RD, Nelson BR, Kahoud RJ, Yang R, Mussar KE, Reiner SL, Hevner RF (2012) Tbr2 is essential for hippocampal lineage progression from neural stem cells to intermediate progenitors and neurons. J Neurosci 32:6275–6287. 10.1101/cshperspect.a018812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch U-K, Noda M, Verkhratsky A (2011) Physiology of Microglia. Physiol Rev 91:461–553. 10.1152/physrev.00011.2010 [DOI] [PubMed] [Google Scholar]
- Khan N, Gordon R, Woodruff TM, Smith MT (2015) Antiallodynic effects of alpha lipoic acid in an optimized RR-EAE mouse model of MS-neuropathic pain are accompanied by attenuation of upregulated BDNF-TrkB-ERK signaling in the dorsal horn of the spinal cord. Pharmacol Res Perspect 3:e00137. 10.1002/prp2.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010) Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8:e1000412. 10.1371/journal.pbio.1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, Walker AJ, Heller MD, Umemori H, Chen C, Stevens B (2018) CD47 protects synapses from excess microglia-mediated pruning during development. Neuron 100:120–134. 10.1016/j.neuron.2018.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei F, Cui N, Zhou C, Chodosh J, Vavvas DG, Paschalis EI (2020) CSF1R inhibition by a small-molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proc Natl Acad Sci U S A 117:23336–23338. 10.1073/pnas.1922788117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Barres BA (2018) Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18:225–242. 10.1038/nri.2017.125 [DOI] [PubMed] [Google Scholar]
- Mizoguchi Y, Monji A, Kato T, Seki Y, Gotoh L, Horikawa H, Suzuki SO, Iwaki T, Yonaha M, Hashioka S, Kanba S (2009) Brain-derived neurotrophic factor induces sustained elevation of intracellular Ca2+ in rodent microglia. J Immunol 183:7778–7786. 10.4049/jimmunol.0901326 [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD (2003) Inflammatory blockade restores adult hippocampal neurogenesis. Science 302:1760–1765. 10.1126/science.1088417 [DOI] [PubMed] [Google Scholar]
- Murphy PG, Borthwick LA, Altares M, Gauldie J, Kaplan D, Richardson PM (2001) Reciprocal actions of interleukin-6 and brain-derived neurotrophic factor on rat and mouse primary sensory neurons. Eur J Neurosci 12:1891–1899. 10.1046/j.1460-9568.2000.00074.x [DOI] [PubMed] [Google Scholar]
- Nakajima K, Kikuchi Y, Ikoma E, Honda S, Ishikawa M, Liu Y, Kohsaka S (1998) Neurotrophins regulate the function of cultured microglia. Glia 24:272–289. 10.1002/(SICI)1098-1136(199811)24:3<272::AID-GLIA2>3.0.CO;2-4 [DOI] [PubMed] [Google Scholar]
- Nandi S, Gokhan S, Dai XM, Wei S, Enikolopov G, Lin H, Mehler MF, Stanley ER (2012) The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev Biol 367:100–113. 10.1016/j.ydbio.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil SM, Witcher KG, McKim DB, Godbout JP (2018) Forced turnover of aged microglia induces an intermediate phenotype but does not rebalance CNS environmental cues driving priming to immune challenge. Acta Neuropathol Commun 6:129–149. 10.1186/s40478-018-0636-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–1458. 10.1126/science.1202529 [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155:1596–1609. 10.1016/j.cell.2013.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesseveur G, David DJ, Gaillard MC, Pla P, Wu MV, Nguyen HT, Nicolas V, Auregan G, David I, Dranovsky A, Hantraye P, Hen R, Gardier AM, Déglon N, Guiard BP (2013) BDNF overexpression in mouse hippocampal astrocytes promotes local neurogenesis and elicits anxiolytic-like activities. Transl Psychiatry 3:e253. 10.1038/tp.2013.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Shetty AK (2004) Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci 19:234–246. [DOI] [PubMed] [Google Scholar]
- Rice RA, Pham J, Lee RJ, Najafi AR, West BL, Green KN (2017) Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia 65:931–944. 10.1002/glia.23135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R (2001) Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol 15:1748–1757. 10.1210/mend.15.10.0706 [DOI] [PubMed] [Google Scholar]
- Salter MW, Stevens B (2017) Microglia emerge as central players in brain disease. Nat Med 23:1018–1027. 10.1038/nm.4397 [DOI] [PubMed] [Google Scholar]
- Sato K (2015) Effects of microglia on neurogenesis. Glia 63:1394–1405. 10.1002/glia.22858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74:691–705. 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M (2010) Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 8:483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Beccari S, Diaz-Aparicio I, Encinas JM, Comeau S, Tremblay M (2014) Surveillance, phagocytosis, and inflammation: how never-resting microglia influence adult hippocampal neurogenesis. Neural Plast 2014:610343. 10.1155/2014/610343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer-Segal JL, Waters EM, Bath KG, Chao MV, McEwen BS, Milner TA (2011) Distribution of phosphorylated TrkB receptor in the mouse hippocampal formation depends on sex and estrous cycle stage. J Neurosci 31:6780–6790. 10.1523/JNEUROSCI.0910-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SWM, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178. 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez-Torres C, Frouin A, Wilton DK, Funk K, DeMasters BK, Jiang X, Bowen JR, Mennerick S, Robinson JK, Garbow JR, Tyler KL, Suthar MS, Schmidt RE, et al. (2016) A complement-microglia axis drives synapse loss during virus-induced memory impairment. Nature 534:538–543. 10.1038/nature18283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukovic J, Colditz MJ, Blackmore DG, Ruitenberg MJ, Bartlett PF (2012) Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J Neurosci 32:6435–6443. 10.1523/JNEUROSCI.5925-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukovic J, Borlikova GG, Ruitenberg MJ, Robinson GJ, Sullivan RKP, Walker TL, Bartlett PF (2013) Immature doublecortin-positive hippocampal neurons are important for learning but not for remembering. J Neurosci 33:6603–6613. 10.1523/JNEUROSCI.3064-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29:3974–3980. 10.1523/JNEUROSCI.4363-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace J, Lord J, Dissing-Olesen L, Stevens B, Murthy VN (2020) Microglial depletion disrupts normal functional development of adult-born neurons in the olfactory bulb. eLife 9:e50531. 10.7554/eLife.50531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Chang X, She L, Xu D, Huang W, Poo MM (2015) Autocrine action of BDNF on dendrite development of adult-born hippocampal neurons. J Neurosci 35:8384–8393. 10.1523/JNEUROSCI.4682-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse EG, An JJ, Orefice LL, Baydyuk M, Liao GY, Zheng K, Lu B, Xu B (2012) BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. J Neurosci 32:14318–14330. 10.1523/JNEUROSCI.0709-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, Exiga M, Vadisiute A, Raggioli A, Schertel A, Schwab Y, Gross CT (2018) Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun 9:1228. 10.1038/s41467-018-03566-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis EF, Vukovic J (2020) Protocol for brain-wide or region-specific microglia depletion and repopulation in adult mice. STAR Protoc 1:100211. 10.1016/j.xpro.2020.100211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis EF, Bartlett PF, Vukovic J (2017) Protocol for short- and longer-term spatial learning and memory in mice. Front Behav Neurosci 11:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis EF, MacDonald KPA, Nguyen QH, Garrido AL, Gillespie ER, Harley SBR, Bartlett PF, Schroder WA, Yates AG, Anthony DC, Rose-John S, Ruitenberg MJ, Vukovic J (2020) Repopulating microglia promote brain repair in an IL-6-dependent manner. Cell 180:833–846. 10.1016/j.cell.2020.02.013 [DOI] [PubMed] [Google Scholar]
- Wolf SA, Boddeke HW, Kettenmann H (2017) Microglia in Physiology and Disease. Annu Rev Physiol 79:619–643. 10.1146/annurev-physiol-022516-034406 [DOI] [PubMed] [Google Scholar]
- Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38:79–91. 10.1016/j.immuni.2013.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Geula C, Lu C, Koziel H, Hatcher LM, Roisen FJ (2003) Neurotrophins regulate proliferation and survival of two microglial cell lines in vitro. Exp Neurol 183:469–481. 10.1016/s0014-4886(03)00222-x [DOI] [PubMed] [Google Scholar]
- Zhang X, Zeng L, Yu T, Xu Y, Pu S, Du D, Jiang W (2014) Positive feedback loop of autocrine BDNF from microglia causes prolonged microglia activation. Cell Physiol Biochem 34:715–723. 10.1159/000363036 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch Nm, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu KQ (2014) An RNA-sequencing transcriptome and splicing dataset of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:119929–111947. [DOI] [PMC free article] [PubMed] [Google Scholar]