

Abstract
Release of neuropeptides from dense core vesicles (DCVs) is essential for neuromodulation. Compared with the release of small neurotransmitters, much less is known about the mechanisms and proteins contributing to neuropeptide release. By optogenetics, behavioral analysis, electrophysiology, electron microscopy, and live imaging, we show that synapsin SNN-1 is required for cAMP-dependent neuropeptide release in Caenorhabditis elegans hermaphrodite cholinergic motor neurons. In synapsin mutants, behaviors induced by the photoactivated adenylyl cyclase bPAC, which we previously showed to depend on ACh and neuropeptides (Steuer Costa et al., 2017), are altered as in animals with reduced cAMP. Synapsin mutants have slight alterations in synaptic vesicle (SV) distribution; however, a defect in SV mobilization was apparent after channelrhodopsin-based photostimulation. DCVs were largely affected in snn-1 mutants: DCVs were ∼30% reduced in synaptic terminals, and their contents not released following bPAC stimulation. Imaging axonal DCV trafficking, also in genome-engineered mutants in the serine-9 protein kinase A phosphorylation site, showed that synapsin captures DCVs at synapses, making them available for release. SNN-1 colocalized with immobile, captured DCVs. In synapsin deletion mutants, DCVs were more mobile and less likely to be caught at release sites, and in nonphosphorylatable SNN-1B(S9A) mutants, DCVs traffic less and accumulate, likely by enhanced SNN-1 dependent tethering. Our work establishes synapsin as a key mediator of neuropeptide release.
SIGNIFICANCE STATEMENT Little is known about mechanisms that regulate how neuropeptide-containing dense core vesicles (DCVs) traffic along the axon, how neuropeptide release is orchestrated, and where it occurs. We found that one of the longest known synaptic proteins, required for the regulation of synaptic vesicles and their storage in nerve terminals, synapsin, is also essential for neuropeptide release. By electrophysiology, imaging, and electron microscopy in Caenorhabditis elegans, we show that synapsin regulates this process by tethering the DCVs to the cytoskeleton in axonal regions where neuropeptides are to be released. Without synapsin, DCVs cannot be captured at the release sites and, consequently, cannot fuse with the membrane, and neuropeptides are not released. We suggest that synapsin fulfills this role also in vertebrates, including humans.
Keywords: electron microscopy, imaging, neuropeptides, optogenetics, synapsin, synaptic transmission
Introduction
Neurotransmitters are released from synaptic vesicles (SVs) (Jahn and Fasshauer, 2012; Sudhof, 2013). SVs are stored in the reserve pool (RP), tethered by the phosphoprotein synapsin (Benfenati et al., 1989; Rizzoli and Betz, 2005; Cesca et al., 2010). SVs are mobilized from the RP by synapsin phosphorylation through protein kinase A (PKA) and CaMKII (Kuromi and Kidokoro, 2000; Menegon et al., 2006; Milovanovic et al., 2018), translocate to the plasma membrane (PM), and are docked and primed, forming the readily releasable pool (RRP). Following Ca2+ influx into the terminal, SVs fuse with the PM. Ca2+ can also induce secretion of neuropeptides from dense core vesicles (DCVs), via protein kinase C signaling (Park et al., 2006; Sieburth et al., 2007; Xue and Wu, 2010). Also, cAMP and PKA affect DCV fusion, through an unknown target (Zhou et al., 2007; Wang and Sieburth, 2013). Among synaptic cAMP/PKA targets are Exchange Protein Activated by Cyclic AMP (EPAC), which binds cAMP to regulate SV release (Zhong and Zucker, 2005; Cheung et al., 2006; Gekel and Neher, 2008), synapsin, tomosyn, Rim1, ryanodine receptor, cysteine string protein, snapin, complexin, and SNAP-25 (Hosaka et al., 1999; Evans et al., 2001; Lonart et al., 2003; Rodriguez et al., 2003; Nagy et al., 2004; Thakur et al., 2004; Baba et al., 2005; Gracheva et al., 2006; McEwen et al., 2006; Cho et al., 2015). How PKA phosphorylation of these proteins affects transmission, and whether they also affect DCV fusion, is only partially understood.
Neuropeptides orchestrate numerous processes in the nervous system (Taghert and Nitabach, 2012; Graebner et al., 2015; Oranth et al., 2018; Steuer Costa et al., 2019). They are synthesized as precursors, packaged in DCVs, and processed during axonal transport (Hoover et al., 2014). In Drosophila, DCVs constantly traffic anterogradely and retrogradely between synapses, and capture events make them available for release (Wong et al., 2012). Capture is regulated in an activity-dependent fashion, involving FMRP in flies (Cavolo et al., 2016), as well as synaptotagmin-4. The latter was involved in regulating the interaction of DCV cargo with kinesin and dynein; thus, capture may depend on a tug-of-war between these anterograde and retrograde microtubule motors (Bharat et al., 2017). Scaffold proteins α-liprin and TANC2 are required for DCV capture at postsynaptic spines; they, as well as calmodulin, interact with actin to modulate kinesins (Stucchi et al., 2018). However, another study showed that actin was dispensable for activity-dependent DCV capture (Cavolo et al., 2016). In Caenorhabditis elegans, sentryn, α-liprin, and the SAD kinase regulate anterograde versus retrograde DCV traffic (Morrison et al., 2018).
Neuropeptide release requires the Ca2+-dependent activator protein for secretion (CAPS/UNC-31) (Rupnik et al., 2000; Charlie et al., 2006b). In C. elegans cholinergic motor neurons, optogenetic cAMP increase (induced by Beggiatoa photoactivated adenylyl cyclase [bPAC]), caused DCV fusion as well as ACh release (Steuer Costa et al., 2017). This involved SV mobilization from the RP, increased frequency of miniature postsynaptic current (mPSC) events, and increased mPSC amplitudes. The latter required neuropeptides, released from cholinergic neurons, acting in an autocrine fashion to induce filling of SVs via the vAChT (UNC-17).
The single C. elegans synapsin (SNN-1) resembles vertebrate synapsin II (Benfenati et al., 1989; Gitler et al., 2008). PKA phosphorylation of synapsin mobilizes SVs by releasing their mutual and cytoskeletal anchoring (Johnson et al., 1972; Hosaka et al., 1999; Menegon et al., 2006). Synapsin contains a C-terminal intrinsic disordered region that can form nonmembranous organelles by liquid-liquid phase separation, regulated by activity and CaMKII phosphorylation (Milovanovic et al., 2018). While vertebrate synapsin clearly interacts with SVs, much less was found on DCVs (Navone et al., 1984). Thus, synapsin appears unlikely to affect DCV cycling and release. Yet, here we identify C. elegans synapsin as a mediator of cAMP/PKA effects on transmission, DCV trafficking, and fusion. snn-1 mutants release no neuropeptides, and they exhibit reduced evoked SV release. SVs distribute abnormally, in line with synapsin organizing the RP, and they do not show the bPAC evoked, neuropeptide-dependent increase in diameter and content. DCV numbers are reduced and distribute aberrantly in snn-1 synaptic terminals. The SNN-1 protein colocalizes with immobile DCVs at likely release sites. DCV trafficking is aberrant in snn-1 deletion mutants, and synapses appear unable to capture DCVs to retain them for release. In SNN-1(S9A) animals, with nonphosphorylatable SNN-1B, DCVs show largely reduced motility, possibly because of enhanced tethering.
Materials and Methods
Strains and genetics
C. elegans strains were cultivated using standard methods on nematode growth medium (NGM) and fed Escherichia coli strain OP50-1 (Brenner, 1974). For all behavioral and electrophysiology experiments, animals were in lite-1(ce314) background (lacking the intrinsic photophobic response) (Edwards et al., 2008), for all bPAC EM work, the wt background was used. Strains used or generated are as follows: snn-1(tm2557), KG1034: ceIs33[rab-3::Tpde-4d(+) cDNA], KG1180: lite-1(ce314), RB830: epac-1(ok655), TR2171: unc-68(r1162), ZX460: zxIs6[punc-17::ChR2(H134R)::yfp;lin-15+], ZX1460: zxIs53[punc-17::bPAC::YFP, pmyo-2::mCherry], ZX1461: ceIs33[rab-3::Tpde-4d(+) cDNA]; zxIs53[punc-17::bPAC::YFP, pmyo-2::mCherry], ZX1555: nuIs183[Punc-129::NLP-21::Venus; myo-2::NLS::GFP], ZX1569: lite-1(ce314); zxIs53[punc-17::bPAC::YFP; pmyo-2::mCherry], ZX1570: lite-1(ce314); unc-31(n1304); zxIs53[punc-17::bPAC::YFP; pmyo-2::mCherry], ZX1815: snn-1(tm2557); zxIs6[punc-17::ChR2(H134R)::yfp;lin-15+], ZX1816: lite-1(ce314); snn-1(tm2557); zxIs6[punc-17::ChR2(H134R)::yfp;lin-15+], ZX1867: epac-1(ok655); zxIs53[punc-17::bPAC::YFP; pmyo-2::mCherry], ZX1868: snn-1(tm2557); lite-1(ce314), ZX1870: unc-31(n1304); zxIs53[punc-17::bPAC::YFP; pmyo-2::mCherry], ZX1871: snn-1(tm2557); lite-1(ce314); zxIs53[punc-17::bPAC::YFP; pmyo-2::mCherry], ZX1990: lite-1(ce314); zxIs53[punc-17::bPAC::YFP; pmyo-2::mCherry] nuIs183[Punc-129::NLP-21::Venus; myo-2::NLS::GFP], ZX1991: snn-1(tm2557); lite-1(ce314); zxIs53[punc-17::bPAC::YFP; pmyo-2::mCherry] nuIs183[punc-129::NLP-21::Venus; pmyo-2::NLS::GFP], ZX1992: unc-68(r1162), zxIs53[punc-17::bPAC::YFP; pmyo-2::mCherry], ZX2002: lite-1(ce314); zxIs6[punc-17::ChR2(H134R)::yfp, lin-15+], ZX2073: snn-1(S9A); lite-1(ce314); zxIs53[punc-17::bPAC::YFP; pmyo-2::mCherry], ZX2379: snn-1(S9E); lite-1(ce314); zxIs53[punc-17::bPAC::YFP; pmyo-2::mCherry], ZX2553: snn-1(tm2557); nuIs183[punc-129::NLP-21::Venus; pmyo-2::NLS::GFP], ZX2555: snn-1(S9E); nuIs183[punc-129::NLP-21::Venus; pmyo-2::NLS::GFP], ZX2559: snn-1(S9A); nuIs183[punc-129::NLP-21::Venus; pmyo-2::NLS::GFP], ZX2912: vjEx94[punc-129::SNN-1::mCherry, pmyo-2::NLS::mCherry]; nuIs183[punc-129::NLP-21::Venus; pmyo-2::NLS::GFP].
Molecular biology and CRISPR/Cas9 genome editing
The construct for the bPAC of Beggiatoa sp. was previously described (Steuer Costa et al., 2017). The snn-1(S9A) and snn-1(S9E) point mutations were engineered by CRISPR/Cas9 genome editing.
Generation of transgenic animals
Generation of transgenic C. elegans expressing punc-17::bPAC::YFP; pmyo-2::mCherry (strain ZX1460) was previously described (Steuer Costa et al., 2017). This transgene, zxIs53, was used for crossing into the mutant alleles tested in this work.
Behavioral assays
Locomotion parameters on NGM (see Fig. 1) were acquired with a previously described single worm tracker (Stirman et al., 2011) with the following modifications: First, a mechanical shutter (Sutter Instrument) was placed between projector and microscope and synchronized to the light stimulation. Second, a band pass filter (650 ± 25 nm) was inserted in the background light path. These modifications ensured an ambient light power, measured between 200 and 1000 nm, <20 nW/mm2 during tracking in “dark.” Light power was measured with a power meter (PM100, Thorlabs) at the focal plane while the sensor was placed at the expected worm's position. All animals were kept in darkness during growth. Young adults were transferred individually on plain NGM plates under red light (>600 nm) in a dark room and kept for 15 min in the dark before transfer to the tracker. Light stimulation protocol was 15 s in darkness, 25 s in 70 µW/mm2 470/10 nm light, and 15 s darkness. Tracks were automatically filtered to exclude data points from erroneously evaluated movie frames with a custom workflow in KNIME (Desktop version 2.10, www.KNIME.com) (Preisach et al., 2008; Warr, 2012). Our constraints were that animals do not move faster than 1.25 mm/s and their length does not show a discrepancy >25% to the mean first 5 s of the video. Videos were excluded from analysis when >15% of the data points had to be discarded by our constraints. Behavior data passed the Shapiro-Wilk normality test. Bending angle analysis during crawling was defined by the mean deviation from 180° from 11 equally interspaced 3-point angles defined by 13 points along the spine of the animal. Behavior data clustering was performed on the absolute change compared with the mean value before light using dynamic time warping distances and the hclust function with method “average” in R (version 3.2.1, libraries dtw, ape, gplots; http://www.R-project.org/).
Figure 1.
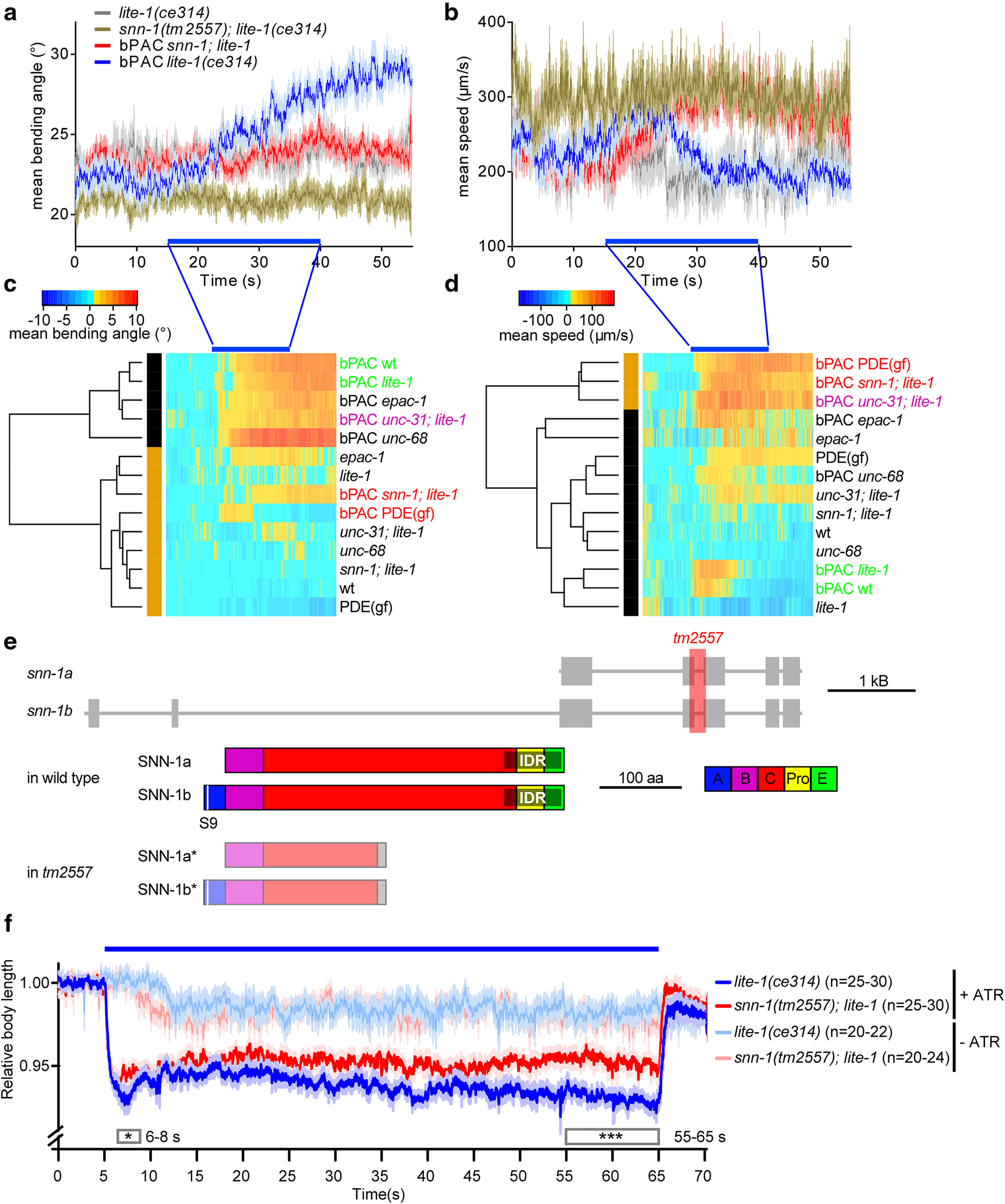
Behavioral phenotypes induced by bPAC and ChR2 photostimulation in cholinergic neurons uncover synapsin as a target of cAMP increase and as a mediator of evoked release. bPAC was expressed in cholinergic neurons, and light effects on locomotion behavior were analyzed after video microscopy of individual animals. a, Mean (±SEM) bending angles (n ≥ 29), measured for 11 angles defined by 13 points along the body or (b) velocity of animals before, during, and after blue light stimulation (blue bar). Animals of the indicated genotypes were assessed. Bending angles (c) and velocities (d) for the indicated genotypes and transgenes were normalized to the mean before light stimulation and compared by dynamic cluster analysis. Two clusters are observed for both behaviors: one with wt animals (green), and another with a gain-of-function phosphodiesterase (PDE(gf)) and snn-1 mutants (red). e, C. elegans snn-1 gene on chromosome IV (boxes represent exons; lines represent introns), and protein structure of the a and b splice variants. Domains A, B, C, Pro (proline-rich), E, named by homology to the mammalian isoforms (Sudhof et al., 1989). Domains A and E are required for SV and synapsin oligomerization, whereas domain C interacts with the SV membrane. The intrinsic disordered region (gray shade) mediating phase separation of synapsin and SVs (Milovanovic et al., 2018) was annotated based on the Predictor of Natural Disordered Regions (www.pondr.com). S9, main PKA phosphorylation site. f, Body contraction in response to cholinergic photostimulation (ChR2). Mean (±SEM) normalized body length, number of animals, genotype as indicated. +/− ATR indicates incubation in all-trans retinal, the obligate ChR2 cofactor. Blue bar represents photostimulation. Boxes represent periods for which datasets were statistically significantly different. ***p < 0.001; *p < 0.05; two-way ANOVA and Bonferroni correction.
Behavior was also recorded (see Fig. 6) using a multiworm tracking device (Swierczek et al., 2011). A set of red LEDs was used for back-illumination, to avoid bPAC preactivation; 60-100 age-synchronized adult animals were collected in M9 buffer, spread onto a NGM plate, and kept in the dark for at least 20 min before tracking. The light stimulation protocol was 15 s dark, 25 s in 70 µW/mm2 470/10 nm light, followed again by 15 s dark. Tracks were extracted using Choreography. Bending angle was defined by the mean deviation from 180° from four equally interspaced angles defined by 11 points along the object's skeleton. A custom MATLAB (The MathWorks R2020b) script (kindly provided by Ichiro Aoki) was used to organize Choreography output files into summary statistics. Data were plotted and analyzed in GraphPad Prism version 8.0.2.
Figure 6.
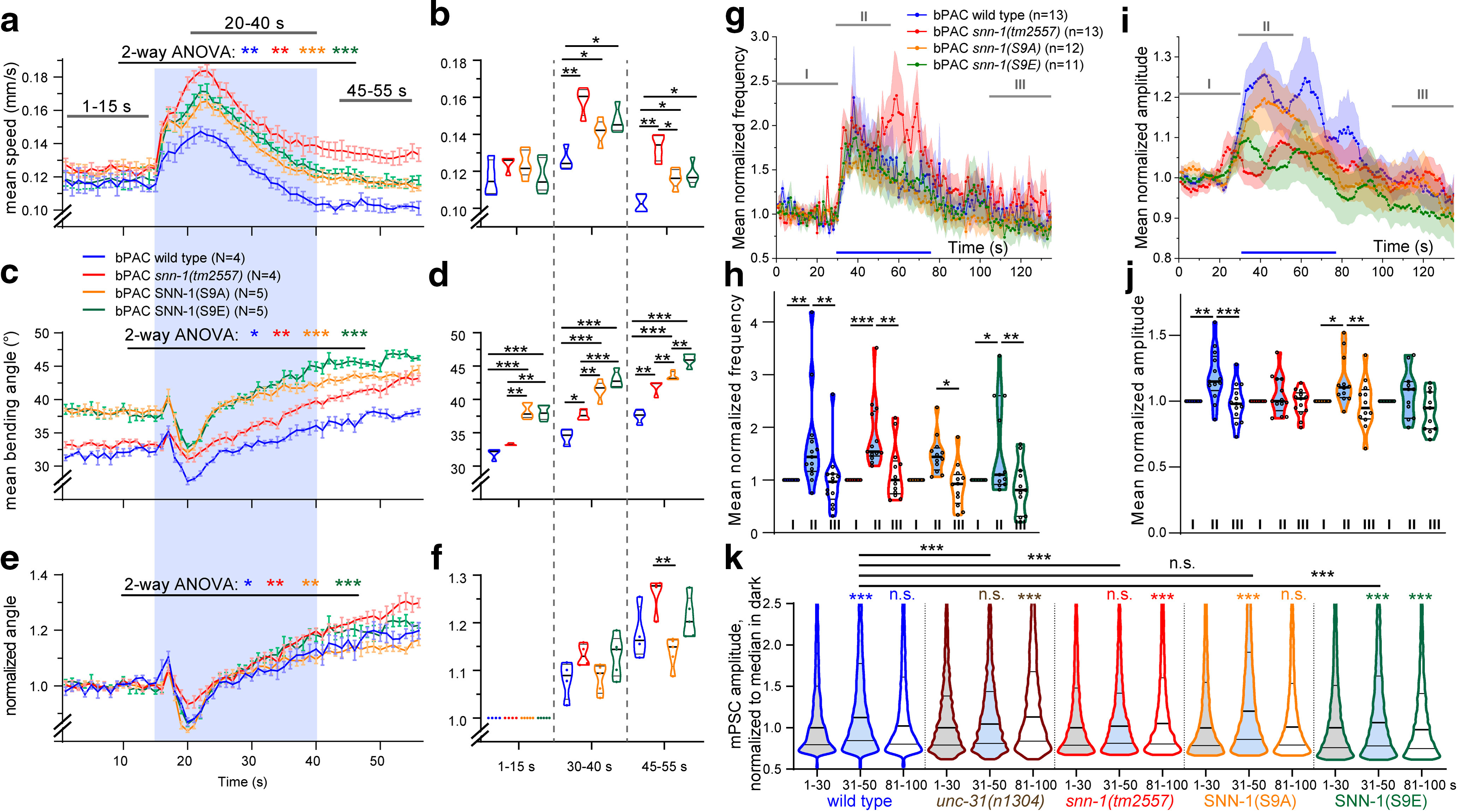
SNN-1B serine 9 mutations affect behaviors and postsynaptic currents induced by bPAC stimulation. a-f, Multiworm tracking (Swierczek et al., 2011) analysis of SNN-1B S9A and S9E mutants, as well as snn-1 and wt animals (N = 4 or 5 replicates, n = 60-100 animals each). S9A and S9E mutations affect bPAC induced changes in crawling velocity (a,b) and bending angles (c–f; e, f, normalized to initial value) compared with wt and snn-1(tm2557). a, c, e, Mean (±SEM). b, d, f, Velocities and bending angles in the indicated time windows shown as median and 25/75 quartiles. Blue shade in a, c, e represents illumination period. g, h, Mean, normalized mPSC events per second before (I), during (II), and after (III) presynaptic bPAC stimulation in cholinergic neurons, in the indicated genotypes. Blue bar represents blue light stimulation period. i, j, Mean (±SEM) normalized mPSC amplitudes in 1 s bins, before, during, and after photoactivation. k, Frequency distribution analysis of mPSC amplitudes in different time periods before, during, and after bPAC photostimulation, as indicated. Datasets represent 3223-13,348 single events. A shift to generally higher amplitudes is apparent for wt, S9A, and S9E animals, but no distinct populations of higher mPSC amplitudes emerge. *p < 0.05; **p < 0.01; ***p < 0.001; paired and unpaired t test (b,d,f) or two-way ANOVA (a,c,e,h,j) with Bonferroni post-test. g, i, Some data are replotted from Figure 2.
Contraction assay
The body length was determined as previously reported (Erbguth et al., 2012). Animals were kept in the dark on seeded NGM plates with or without all-trans retinal (Liewald et al., 2008). Single animals were transferred to empty NGM plates and kept for 15 min before recording. Worms were illuminated with blue light 1.4 mW/mm2 from a 50 W HBO mercury lamp, filtered through a GFP excitation filter, under a 10× objective in a Zeiss Axiovert 40 microscope (Carl Zeiss). The length values were normalized to the mean value before illumination 0-5 s. All values <80% were excluded to avoid measurement errors (i.e., when the animals roll up). Data were plotted and analyzed in GraphPad Prism 8.0.2.
Electrophysiology
Recordings from dissected body wall muscle cells were conducted as described previously (Liewald et al., 2008). Animals were immobilized with Histoacryl glue (B. Braun Surgical), and a lateral incision was made to access neuromuscular junctions (NMJs) along the anterior ventral nerve cord. The basement membrane overlying body wall muscles was enzymatically removed by incubation in 0.5 mg/ml collagenase for 10 s (C5138, Sigma Aldrich). Integrity of body wall muscle cells and nerve cord was visually examined via DIC microscopy. Recordings from body wall muscles were acquired in whole-cell patch-clamp mode at room temperature (20°C-22°C) using an EPC-10 amplifier equipped with Patchmaster software (HEKA). The head stage was connected to a standard HEKA pipette holder for fire-polished borosilicate pipettes (1B100F-4, Worcester Polytechnic Institute) of 4-9 mΩ resistance. The extracellular bath solution consisted of 150 mm NaCl, 5 mm KCl, 5 mm CaCl2, 1 mm MgCl2, 10 mm glucose, 5 mm sucrose, and 15 mm HEPES, pH 7.3, with NaOH, ∼330 mOsm. The internal/patch pipette solution consisted of K-gluconate 115 mm, KCl 25 mm, CaCl2 0.1 mm, MgCl2 5 mm, BAPTA 1 mm, HEPES 10 mm, Na2ATP 5 mm, Na2GTP 0.5 mm, cAMP 0.5 mm, and cGMP 0.5 mm, pH 7.2, with KOH, ∼320 mOsm. Light activation was performed using an LED lamp (KSL-70, Rapp OptoElectronic; 470 nm, 8 mW/mm2) and controlled by an EPC 10 amplifier and Patchmaster software (HEKA). mPSC analysis was done by Mini Analysis software (Synaptosoft, version 6.0.7). Amplitude and mean number of mPSC events per second were analyzed during the following time bins: 30 s before illumination, the first 25 s of illumination, and the last 30 s after illumination. Subsequent analysis and graphing were performed using Patchmaster, and Origin (Originlabs).
Electron microscopy
Young adult animals were used for HPF fixation, based on methods previously described (Weimer, 2006). Briefly, ∼10-20 worms were loaded into a 100 µm deep aluminum planchette (Microscopy Services) filled with E. coli OP50, covered with a 0.16 mm sapphire disk and a 0.4 mm spacer ring (Engineering office M. Wohlwend) for subsequent photostimulation. To prevent preactivation of bPAC, all manipulations were done under low-light conditions or under red light. For light stimulation experiments, worms were continuously illuminated with a ∼470 nm blue LED (3 mW/mm2) for 30 s, followed by high-pressure freezing at −180°C under 2100 bar pressure in a Bal-Tec HPM010 or an HPM100 machine (Leica Microsystems). After freezing, specimens were transferred under liquid nitrogen into a Reichert AFS machine (Leica Microsystems) for freeze substitution. Tannic acid (0.1% in dry acetone) fixative was used to incubate samples at −90°C for 100 h. Then, a process of washing was performed to substitute with acetone, followed by an incubation of 2% OsO4 for 39.5 h (in dry acetone) while slowly increasing temperature up to room temperature. Afterwards, Epoxy resin (Agar Scientific, AGAR 100 Premix kit hard) embedding process was executed with increasing concentration from 50% to 90% at room temperature and 100% at 60°C over 48 h. Cross sections were cut at a thickness of 40 nm, transferred on formvar-covered copper slot grids, and counterstained in 2.5% aqueous uranyl acetate for 4 min, followed by distilled water wash. Then, grids were carried onto Reynolds lead citrate solution for 2 min in a CO2-free chamber and then washed in distilled water again. Images of regions in the ventral nerve cord were taken with a Erlangshen ES500W CCD camera (Gatan) in a Philips CM12 operated at 80 kV. Images were tagged in ImageJ (version 1.47, National Institutes of Health) for synapse perimeter, SVs, docked SVs, DCVs, large vesicle (LVs), and dense projection (DP), scored blind for each condition. ImageJ ROIs were stored for position, area, circumference, and largest diameter, and then quantified and automatically analyzed by custom software written in R, called by a KNIME workflow. The diameters of synapses from each stimulation condition varied in size; thus, each value for the number of docked SVs was normalized to the mean perimeter and represents the number of docked SVs along a membrane whose perimeter is 1548 nm in a profile. The other organelles are represented as the numbers of SVs, DCVs, or LVs in a typical synaptic profile of 164 100 nm2. 3D reconstructions of serial sections were performed using “Reconstruct” (Fiala, 2005), as described previously (Kittelmann et al., 2013; Steuer Costa et al., 2017).
Fluorescence imaging
For coelomocyte imaging, animals were either kept in dark or illuminated for 15 min with a 470 nm LED, 350 µW/mm2 at 20°C. Images were acquired up to 30 min after illumination. Image acquisition was performed with a Zeiss Cell Observer SD with an α Plan-Apochromoat 100×/1.46 Oil DIC (UV) objective (Carl Zeiss), 488 nm excitation laser at 40% power, and a Rolera EM-C2 with EM gain of 100, full resolution, and 100 ms exposure time. Images were exported as 16 bit .tif files. In ImageJ (version 1.47, National Institutes of Health), ROIs were traced for the anterior coelomocyte and a background area outside of the animal. Similarly, cell bodies and processes in the ventral nerve cord were analyzed for fluorescence. Mean intensity of these ROIs was exported, and fluorescence intensity was corrected for background intensity before normalization to the NLP-21::Venus marker strain in the dark (Sieburth et al., 2007); Venus is an enhanced YFP variant.
Analysis of trafficking of NLP-21::Venus-containing DCVs
z stacks of fluorescence images, enclosing the dorsal nerve cord, were acquired on a Zeiss Cell Observer Spinning Disk confocal microscope with a Plan-Apochromat 63×/1.40 Oil DIC M27 objective (Carl Zeiss), 488 nm excitation laser at 20% power, and a Rolera EM-C2 EMCCD camera with EM gain of 100, full resolution, and 70 ms exposure time. Live animals were immobilized on a 10% agarose pad (in M9 buffer) supplemented with a drop of 20 mm tetramisole, under a coverslip. To generate kymographs, we used Fiji (Schindelin et al., 2012) and the Velocity Measurement Tool (http://dev.mri.cnrs.fr/projects/imagej-macros/wiki/Velocity_Measurement_Tool). A segmented line was drawn along the nerve cord process (i.e., the path of moving particles) and used to obtain line scan kymographs. Trajectories of single DCVs for each kymograph were traced and analyzed for anterograde or retrograde trafficking, and velocity, by using the respective features of the macro. Further analysis was done using Microsoft Excel.
SNN-1::mCherry and NLP-21::Venus colocalization was analyzed on a Zeiss Cell Observer Spinning Disk confocal microscope (Carl Zeiss) with 488 nm excitation laser at 20% power, 561 nm excitation laser at 30% power, and a Rolera EM-C2 EMCCD camera with EM gain of 100, full resolution, and 70 ms exposure time. z stacks of images were taken and z-projected in Fiji.
Fluorescence analysis of DCVs after photo-bleaching
Single z plane images were acquired on a Zeiss LSM 780 confocal microscope (Carl Zeiss) for photo-bleaching experiments. Photo-bleaching was performed using an Argon laser power set to 100% 514 nm output. Immediately after photo-bleaching, a 400-frame image stream was acquired at 2 volumes per second. Kymographs and motion analyses were done as described above.
Experimental design and statistical analysis
Experiments/data analyses, particularly for EM analyses, were performed blind to the experimenter. Data are given as mean ± SEM. Significance between datasets after two-tailed Student's t test or after ANOVA is given as p value, the latter after Bonferroni's multiple comparison test, or Tukey's post hoc test. For some of the analyses of DCV trafficking, no normal distribution of the data was observed; thus, Kruskal–Wallis tests were used, with Dunn's post-tests. Data were analyzed and plotted in GraphPad Prism (GraphPad Software, version 5.01), or R (version 3.2.1, libraries dtw, ape, gplots; http://www.R-project.org/), or OriginPro 2015G (OriginLab).
For empirical cumulative distribution functions, the sum of SV linear distances to the DP was divided by the profile area, to represent the distribution of SVs relative to the DP in a profile area-independent manner. These values were plotted as an empirical cumulative distribution function to show the distribution of SVs across the profiles in one group. Equivalency of distributions was analyzed by a Kolmogorov–Smirnov test between groups. We report the empirical distribution functions as originating from different distribution functions when the Kolmogorov–Smirnov test p value is <0.05. We conclude that the distribution of SVs relative to the DP is changed across groups that fulfill this statement.
Results
Synapsin mediates the effects of cAMP in synaptic augmentation
As we found previously, bPAC stimulation in cholinergic neurons of C. elegans (thus generating cAMP acutely) increased locomotion speed and body bending angles, through a mechanism involving PKA signaling, neuropeptide release, and neuropeptide-mediated increased filling of cholinergic SVs (Steuer Costa et al., 2017). However, the synaptic target(s) of PKA that mediate these effects were as yet unknown. To identify such proteins, we assessed candidate synaptic cAMP- and PKA-target mutants for altered bPAC-evoked behavioral parameters, using video microscopy. We tested a modulator of synaptic strength (EPAC-1) (Kaneko and Takahashi, 2004), and the PKA targets UNC-68 (ryanodine receptor, which affects synaptic mPSC frequency and amplitude) (Q. Liu et al., 2005), as well as synapsin SNN-1. As controls, we included unc-31 (CAPS) mutants (lacking neuropeptide release) and pde-4d (PDE(gf)) animals with constitutively active phosphodiesterase and thus reduced cAMP levels (Charlie et al., 2006a).
Data were acquired for individual animals and averaged to estimate differences (i.e., changes of locomotion bending angles or speed) relative to before illumination (Fig. 1a,b). During 25 s photostimulation, bending angles increased progressively and stayed elevated afterward (they return to baseline after ∼90 s) (Steuer Costa et al., 2017). Locomotion speed showed a transient increase; while animals continue exaggerated movement, as their body bending increases, their net speed goes down. Since some genotypes had very different basal values, we normalized the data and used dynamic cluster analysis to compare them and identify potential mechanisms underlying the effects on the induced behaviors. Yet, since locomotion behavior is influenced by multiple circuits beyond the motor neurons, as well as muscle, it is unlikely that single molecular pathways can be connected to distinct behavioral aspects. Thus, we rather used the behavioral analysis to pinpoint interesting genes for further analysis. We used wt or lite-1 background to account for photophobic responses to blue light (Edwards et al., 2008). epac-1, unc-68, and unc-31 mutants, but not snn-1 and PDE(gf) animals, showed the progressive increase of bending angles, such as wt (Fig. 1a,c). bPAC-stimulated wt animals moderately increased their speed, which was similar in unc-68 and epac-1 mutants; however, a much larger speed increase was observed for unc-31, PDE(gf), and snn-1(tm2557) mutants (Fig. 1b,d). snn-1(tm2557) mutants were the only animals that mimicked PDE(gf) mutants in both behaviors, differing from wt, and they phenocopied unc-31/CAPS mutants (lacking neuropeptide release) in speed increase. We therefore analyzed snn-1 mutants in more detail.
The snn-1(tm2557) allele truncates the 3'-half of the gene, likely eliminating its expression (Fig. 1e). Even if some expression remains, only the N-terminal half, including the PKA phosphorylation site I (serine 9), would be produced, while truncation of the central C domain, interacting with SVs and actin, should abolish synapsin function. To assess whether snn-1 mutants have a general defect in cholinergic transmission, these neurons were stimulated using channelrhodopsin-2 (ChR2). During stimulation, ACh is released and the body shrinks because of evoked muscle contraction, with characteristic differences depending on the type of mutant (Liewald et al., 2008). snn-1 mutants contracted significantly less than controls (Fig. 1f). This may indicate a reduced release rate, but could also reflect reduced SV filling state, as we previously observed in unc-31 mutants lacking neuropeptide release (Steuer Costa et al., 2017). It does not indicate a strong synaptic defect, though, because such mutants (e.g., lacking synaptobrevin or UNC-13) contract stronger than wild type, because of a homeostatic increase of postsynaptic excitability (Liewald et al., 2008).
Synapsin is required for neuropeptide release
First, we assessed bPAC-induced neuropeptide release. Exocytosis of neuropeptide precursors fused to a fluorescent protein can be detected in so-called scavenger cells (coelomocytes). These kidney-like cells “clean up” the body fluid by endocytosis. Coelomocytes become fluorescent when Venus-tagged NLP-21 neuropeptides are expressed in, and released from, motor neurons (NLP-21 is expressed intrinsically in these cells; Fig. 2a) (Sieburth et al., 2007). bPAC stimulation induced NLP-21::Venus release, and was abolished in unc-31/CAPS mutants, as we showed previously (Steuer Costa et al., 2017). Release was also abolished in snn-1(tm2557) mutants (Fig. 2b,c), despite a significant enrichment of NLP-21::Venus in neuronal cell bodies and processes, compared with wt (Fig. 2d,e). The latter likely results from an inability to release neuropeptides.
Figure 2.

Synapsin is required for cAMP-induced neuropeptide release. a, NLP-21::Venus neuropeptides are released from cholinergic neurons and endocytosed by coelomocytes. b, Representative images of coelomocytes in bPAC, snn-1, and snn-1;bPAC animals, without and with photostimulation. False color representation: scale bar, 10 µm. c, Fluorescence quantification in coelomocytes, normalized to fluorescence level in nonstimulated wt. Data are median and 25/75 quartiles (thick and thin lines), min to max. Fluorescence (d) and quantification (e) of NLP-21::Venus in cell bodies and processes of cholinergic neurons, in wt and snn-1(tm2557) mutants. Number of animals indicated. c, e, Data are mean ± SEM. **p < 0.01; ***p < 0.001; two-way or one-way ANOVA with Tukey test or Bonferroni correction.
Previously, we showed that bPAC stimulation increases SV release rate and mPSC amplitude in patch-clamp recordings: The amplitude increase resulted from neuropeptide release, impacting on filling of SVs with ACh, and mutation of unc-31 abolished the mPSC amplitude increase (Steuer Costa et al., 2017). In snn-1(tm2557) mutants, the mean basal mPSC rate was slightly reduced (Fig. 3a,b), although not significantly different from wt (Fig. 3d). bPAC stimulation increased the mPSC rate in wt, and likewise in snn-1(tm2557) animals (Fig. 3a,b,d). Yet, unlike wt (and like unc-31), snn-1 animals showed no increased mPSC amplitudes (Fig. 3c,e), suggesting that synapsin is required for neuropeptide release.
Figure 3.
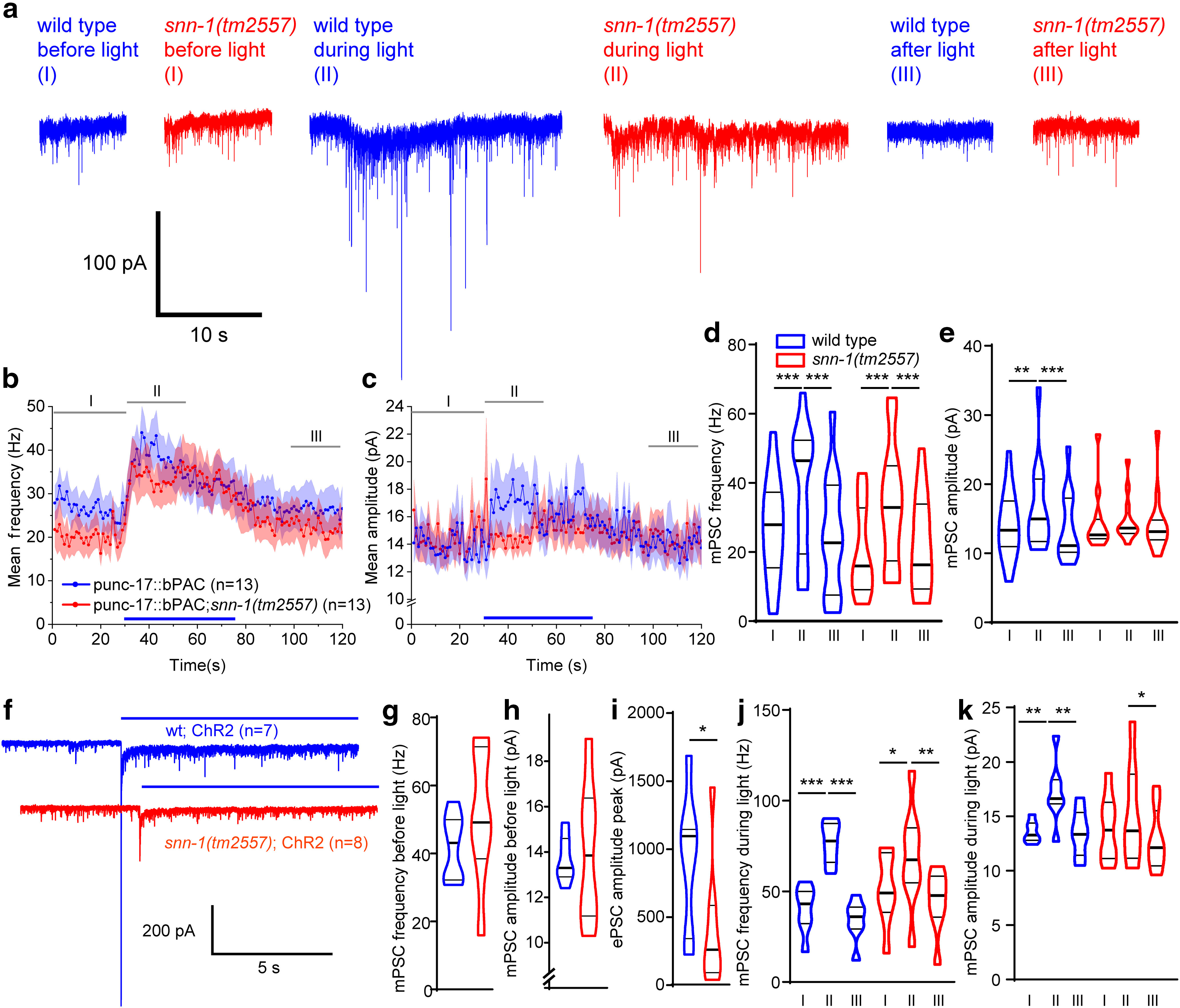
Postsynaptic currents at the NMJ after presynaptic bPAC and ChR2 stimulation uncover SNN-1 function in SV release and cAMP-dependent amplitude increase. a, Postsynaptic currents (voltage clamp of body wall muscle cells) before (I), during (II), and after (III) presynaptic bPAC stimulation in cholinergic neurons, in wt (blue) or snn-1(tm2557) background (red). b, Mean (±SEM) mPSC events per second and (c) mPSC amplitudes, in 1 s bins, before, during, and after photoactivation (blue bar), in animals expressing bPAC in wt and snn-1 mutants. d, e, Group data from b, c, averaged in the indicated time windows I, II, and III. f, Postsynaptic currents after ChR2 stimulation (blue bar), in wt and snn-1(tm2557). g, h, Mean (±SEM) mPSC frequency and amplitude are not different before stimulation. i, ChR2 photostimulation-evoked PSCs are reduced in snn-1(tm2557) animals. j, k, mPSC rate and amplitudes before, during, and after the light stimulus, averaged in the same time windows as in a–c. d, e, j, k, *p < 0.05; **p < 0.01; ***p < 0.001; one-way repeated-measures ANOVA with Bonferroni correction.
The lack of bPAC induced neuropeptides and thus the reduced filling state of ACh vesicles in snn-1 mutants may also underlie the reduced body contraction we observed in response to cholinergic ChR2 stimulation (Fig. 1f). However, synapsin's role in organizing the RP makes it likely that a defect in synaptic transmission per se contributes to this finding. We thus recorded evoked PSCs following ChR2 stimulation of cholinergic neurons (Fig. 3f–k). snn-1 mutants had normal mPSC frequency and amplitude before the light stimulus. However, photo-evoked peak currents were significantly smaller in snn-1 animals (mean evoked amplitude was ∼25% of wt; Fig. 3i). Also, the rate of evoked PSCs was much more increased in wt than in snn-1 mutants (Fig. 3j). During the stimulation phase, mPSC amplitude increased for wt, but not for snn-1 (Fig. 3k). Thus, snn-1 mutants release fewer SVs on depolarization, in line with a function of SNN-1 in providing SVs for docking, and also for ongoing acute release, when SVs must be efficiently mobilized from the RP.
Synapsin mutants have normal SV numbers, but reduced DCV numbers at synapses
To assess the role of SNN-1 in synaptic transmission, and specifically in neuropeptide release, we looked at the synaptic ultrastructure. High-pressure-freeze electron microscopy (Kittelmann et al., 2013; Steuer Costa et al., 2017; Yu et al., 2018) can be used to analyze the content and distribution of synaptic organelles in the terminals of cholinergic motor neurons, including SVs, docked SVs, DCVs, and large vesicles (LVs), which are endosomes formed after activity-induced SV release (Fig. 4a–c).
Figure 4.
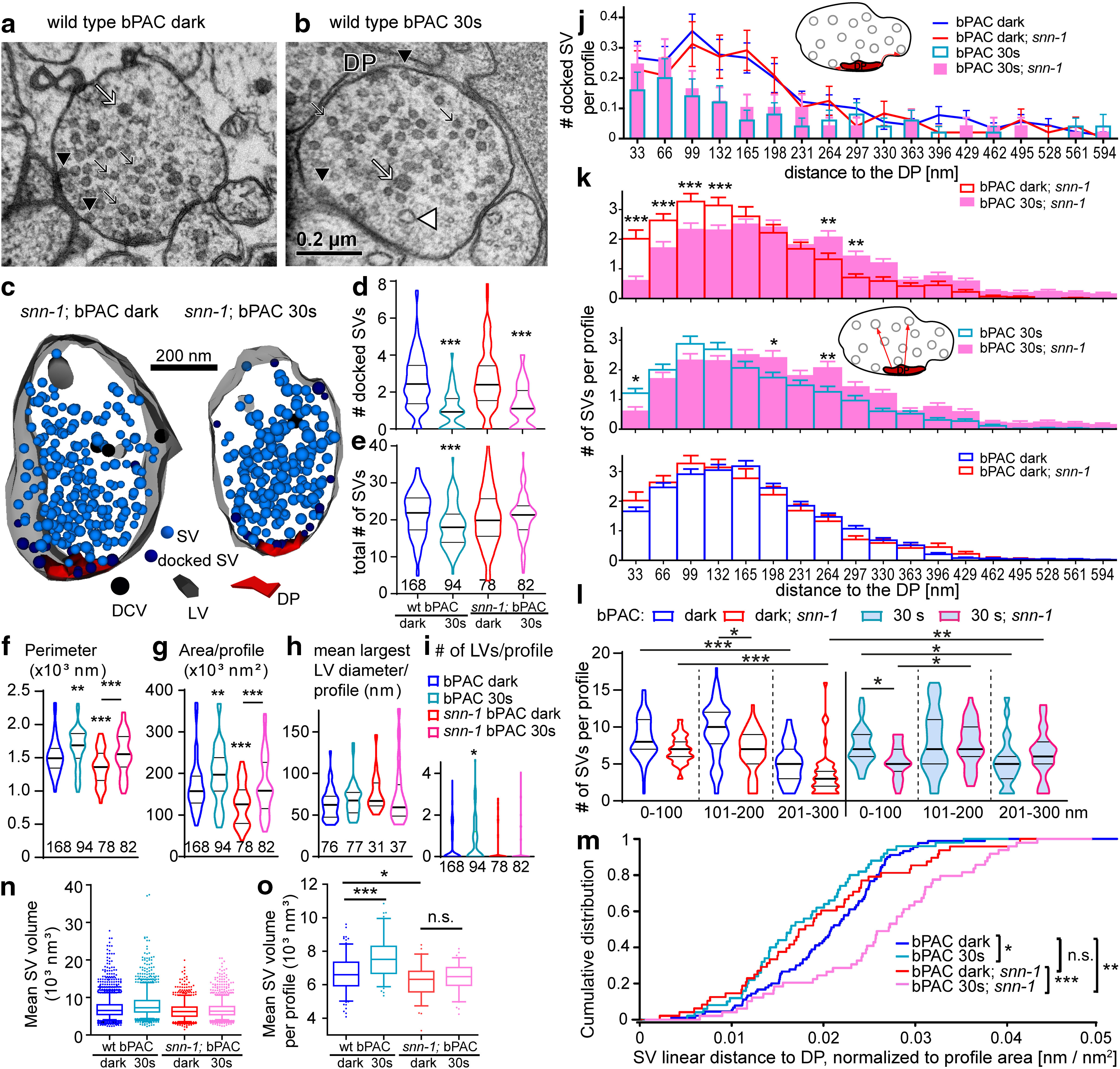
Electron microscopy analysis of the NMJ reveals reduced SV release, altered SV redistribution after stimulation, and smaller SV diameter in snn-1(tm2557) mutants. a, Thin section of a wt cholinergic neuron synaptic terminal. The DP is indicated. Black arrows indicate SVs. Black arrowheads indicate docked SVs. White arrow indicates DCV. White arrowhead indicates stimulation-induced endosomal LV. b, Same as in a, but after 30 s blue light stimulation. c, 3D reconstruction of snn-1;bPAC synapses, dark control (left) and 30 s photostimulated (right). Synaptic structures as in a, b are indicated. d, Number of docked SVs and (e) total SVs per profile, normalized to perimeter or area, respectively. Data shown as median and 25/75 quartiles (thick and thin lines), min to max. Number of sections analyzed is indicated. f, g, Mean size characteristics of synapses analyzed by EM in this work. Mean perimeter and area per profile, per condition and genotype, as indicated. h, i, Quantification of endocytic LVs, induced by bPAC stimulation. Largest diameter of individual LVs observed (h), and mean LV number per synaptic profile (i). j, Binned distribution of docked SVs per profile, at indicated distances (along the PM) to DP, in wt or snn-1(tm2557) before and after 30 s stimulation. k, Binned distribution of SVs in the RP in snn-1 versus wt, before and after 30 s stimulation, at indicated linear distances to the DP (analyzed in 33 nm bins). l, Summary analysis of SVs per profile in 100 nm bins of linear distances to DP, as indicated. m, Empirical cumulative distribution plot of summed distances of RP SVs to DP per profile. n, Volume of SVs (n = 4067, 2079, 1400, and 1888), for the indicated genotypes and conditions of bPAC stimulated synapses. SV inner diameter was measured and used to calculate the volume. Also shown are means, medians, interquartile range. Whiskers represent 5-95 percentile. All statistical comparisons (ANOVA with Bonferroni correction) were highly significant because of high n values. o, SV volumes, as in i, were averaged per profile, for profiles with ≥10 SVs (n = 168, 94, 78, and 82). Data are mean ± SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; one-way ANOVA with Tukey's multiple comparison test in d–i, k, o; Kruskal–Wallis test with Dunn's multiple comparison in l; Bonferroni correction in n; and Kolmogorov–Smirnov test in m.
First, we analyzed SV abundance. bPAC stimulation strongly reduces the number of docked SVs, as well as overall SV numbers throughout the terminal (i.e., the sum of RRP and RP) in wt animals (Steuer Costa et al., 2017). No obvious difference in SV numbers was observed between unstimulated wt and snn-1(tm2557) mutants (Fig. 4d,e). This is surprising given that mammalian synapses lacking synapsin show largely reduced SV content (Gitler et al., 2004; Milovanovic et al., 2018), but is in line with our finding of unaltered spontaneous SV release rate (Fig. 3b,d). Yet, on ChR2 stimulation, snn-1 mutants demonstrated significantly reduced SV release (Fig. 3f,i). Since docked SVs were similarly reduced in wt and snn-1 mutants after photostimulation (Fig. 4d), and since no significant reduction of the RP was observed in snn-1 animals, unlike in wt (Fig. 4e), the reduced ePSC rate must reflect a defect of the snn-1 animals to mobilize SVs from the RP. Compared with wt synapses, snn-1 synapses were smaller (Fig. 4f,g) and did not accumulate endocytic LVs on bPAC stimulation (Fig. 4h,i), probably as reduced SV release necessitated less SV recycling. Nonetheless, snn-1 synapses increased their size in response to 30 s bPAC stimulation, as did wt.
Second, we analyzed the distribution of docked SVs, relative to the DP (central to the active zone). This was similarly altered by bPAC stimulation in snn-1 and wt (Fig. 4j). Synapsin therefore does not directly affect SV priming or exocytosis. Analysis of the RP distribution (Fig. 4k–m) showed that depending on which “spheres” of SVs were analyzed in different distances to the DP, some regions showed different SV content in wt versus snn-1 mutants in unstimulated animals (100-200 nm; Fig. 4l). Upon bPAC photostimulation, SVs in snn-1 mutants were more depleted near the DP. If SVs approach the active zone by tethering to the DP, and then are docked and distributed laterally, depletion of SVs near the DP may reflect a deficit in refilling of the RRP, again supporting a role of synapsin in mobilizing the RP.
Third, we analyzed SV size. We previously found that bPAC stimulation, cAMP and neuropeptide signaling regulate SV loading with ACh, and thus SV diameter, and this was abolished in unc-31/CAPS mutants (Steuer Costa et al., 2017). Also, snn-1 mutants had significantly smaller SVs, which did not increase their size on bPAC stimulation (Fig. 4n,o). Thus, in line with our findings of abolished neuropeptide release, snn-1 mutants also do not upregulate SV filling.
Fourth, we analyzed the distribution and fate of DCVs in snn-1(tm2557) synapses before and after bPAC stimulation. DCVs were significantly reduced in snn-1 mutants, compared with wt, under both conditions, in sections containing the DP (Fig. 5a,b), and in sections neighboring the DP (to ∼240 nm axial DP distance; Fig. 5c,d,f). We also assessed the radial distribution of DCVs relative to the DP. DCVs were most abundant in 150-250 nm distance to the DP, peaking at ∼233 nm (Fig. 5d,e). Although bPAC stimulation caused DCV depletion in the 150 nm bin (Fig. 5e), overall distribution and abundance were similar to without stimulation, peaking at 200-230 nm radial distance to the DP in wt. However, in snn-1(tm2557) mutants, DCVs peaked at 250-350 nm radial distance, with no marked further depletion on bPAC stimulation. The reduction of DCVs in snn-1 mutants indicates that DCVs are delivered to snn-1 terminals but may not be efficiently tethered there to be eventually released. Thus, they may be “lost” to the neuronal process and cell body (Fig. 2d).
Figure 5.

DCVs are largely depleted in snn-1(tm2557) synapses and distribute differently compared with wt. a, Sections analyzed by HPF-EM either contain the DP or are flanking the region containing the DP. b, DCVs per profile containing the DP plus two flanking sections in snn-1(tm2557) compared with wt, without and with 30 s photostimulation. Data shown as median and 25/75 quartiles (thick and thin lines), min to max. c, Same as in b, but in DP-flanking sections. d, Abundance of DCVs was analyzed either radially within a section, in distinct distances to the DP, or along the axon. e, Abundance of DCVs in distinct radial distances of DCVs relative to DP, quantified in 50 nm bins in untreated and 30 s stimulated wt and snn-1(tm2557) synapses. f, DCV abundance in sections containing the DP, or in sections of the indicated axial distance to the DP. b, c, e, f, Data are mean ± SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; one-way ANOVA with Tukey correction.
As reported previously (Hammarlund et al., 2008), DCVs did not cluster at active zones, but distributed evenly along the axon. DCVs in sections flanking the DP out to 240 nm (Fig. 5f) did not exhibit any obvious peak, yet they were depleted in snn-1 synapses (also out to 800 nm; see Fig. 7). In sum, snn-1 mutants have reduced DCV numbers in synapses, which are not released in response to bPAC stimulation.
Figure 7.
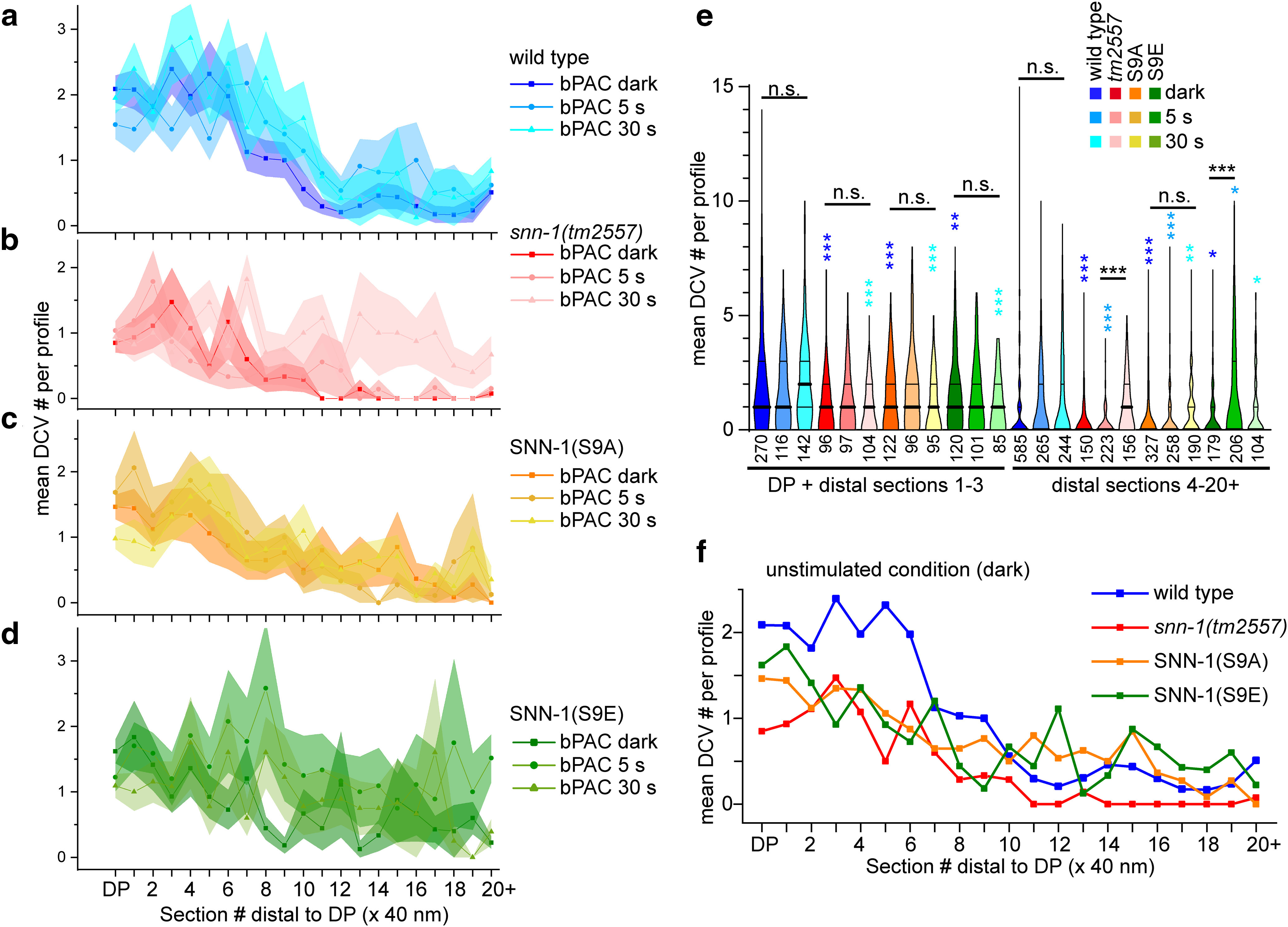
SNN-1B serine 9 mutations affect DCV distribution in sections containing DPs and flanking sections following bPAC stimulation. DCV distribution and abundance were analyzed in the DP-containing and DP-flanking sections, out to 800 nm (in 20 consecutive 40 nm sections) in wt (a), snn-1(tm2557) (b), SNN-1B S9A (c), and S9E (d) knock-in mutants, in unstimulated, as well as 5 and 30 s bPAC photostimulated animals. Shown are mean ± SEM DCV number per section. e, DCV numbers were binned in the indicated sections at and near the DP (DP + flanking sections 1-3) or in the distal section (sections 4-20). Data are median and 25/75 percentiles (thick and thin lines), min to max. n = number of sections is indicated in e. *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA with Bonferroni's multiple comparison test. Different blue colored asterisks indicate significance to the respective wt controls. f, Same as in a–d, but comparing mean DCV distribution in indicated genotypes in the nonstimulated condition. a–d, SEM not shown for clarity.
Synapsin S9A and S9E phosphorylation site mutants show behavioral and electrophysiological defects
cAMP augments SV release and induces neuropeptide release (Steuer Costa et al., 2017), and here we show that this is facilitated by synapsin. Despite effects on distribution and mobilization, the bPAC-induced increase of SV release was not abolished in snn-1(tm2557) mutants, which may retain the N-terminal half of the protein (Fig. 1e). PKA-mediated phosphorylation of serine 9 reduces the affinity for actin, and was suggested to regulate SV mobilization (Hosaka et al., 1999; Cesca et al., 2010). To explore this in C. elegans and to assess the potential function of S9 in synapsin SNN-1B function, we obtained point mutations by CRISPR/Cas9-mediated genome editing: S9A abolishes phosphorylation and mimics a constitutively dephosphorylated state, and S9E mimics constitutively phosphorylated synapsin.
First, we assessed their behavioral phenotypes (Fig. 6a–f). To facilitate these measurements, we used a multi-worm tracking system (Swierczek et al., 2011) instead of the analyses we had performed on single animals (Stirman et al., 2011) as shown in Figure 1. Upon photostimulation, velocity transiently increased for all genotypes tested but was generally higher for all mutants and most pronounced for snn-1(tm2557), while both S9A and S9E animals crawled at an intermediate velocity (Fig. 6a,b). These elevated speeds were slowly decreasing, likely because animals continued increasing their bending angles in response to bPAC stimulation (Fig. 6c–f): Bending angles were generally higher for S9A and S9E mutants. Upon illumination, all strains showed a biphasic behavior: A brief increase in bending was followed by a transient drop, after which bending angles increased again and kept doing so even after the light stimulus ended. This indicates a strong and persistent cAMP generation that causes sustained (PKA-dependent) effects that are enhanced by the absence of SNN-1. S9A and S9E mutants behaved similarly. They had an intermediate effect on locomotion velocity increase, and thus did not affect synapsin function as severely as the deletion mutant. However, they showed the highest basal bending angles, and most pronounced increases therein. The phosphomimetic and phospho-deficient mutations showed no obvious opposing differences in their behaviors, although S9A animals increased bending significantly less.
To address possible differences between S9A and S9E mutants in more detail, we used electrophysiology. Both mutants had essentially the same relative increase in mPSC rate during bPAC stimulation as wt (Fig. 6g,h), and as snn-1(tm2557) mutants. The deletion mutant had a prolonged increase in mPSC rates compared with the other genotypes. With respect to mPSC amplitudes, the S9A mutant showed an increase, like wt, while S9E, just as the snn-1(tm2557) mutant, showed no amplitude increase during bPAC stimulation (Fig. 6i,j). This may indicate that SNN-1B(S9E) animals do not release neuropeptides, which could be because of an inability to tether DCVs and to capture them near synaptic release sites.
To analyze whether there was a general increase in mPSC amplitude in wt, and/or whether there may be different populations of SVs filled to a different extent with transmitter, we performed a frequency distribution analysis of mPSC amplitudes (Fig. 6k). We did not observe any distinct populations of amplitudes, which equally distributed over a wide range, but with a clear maximum between 10 and 20 pA. We observed that, in wt, a general increase in amplitudes occurred during the light stimulus and returned to basal levels after the light was turned off. In snn-1(tm2557) animals, this increase was not observed, as was the case for unc-31 mutants (data included here was reanalyzed from Steuer Costa et al., 2017). Interestingly, unc-31(n1304) as well as snn-1(tm2557) animals exhibited a significant difference (increase for unc-31) in mPSC amplitudes after the end of the light stimulus. For S9A animals, we observed an increase as in wt, and also S9E animals showed a significant increase during light; however, this increase was significantly smaller than the one observed in wt. After light off, S9E animals even showed a drop in mPSC amplitudes below basal levels.
SNN-1B(S9B), but not S9A mutants, resemble snn-1(tm2557) deletion mutants in their DCV distribution after bPAC stimulation
We further explored the ability of SNN-1B(S9) mutants to tether DCVs by HPF-EM. Therefore, the axial distribution of DCVs in the S9A and S9E mutants was analyzed up to 800 nm from the DP/synapse region (Fig. 7a–d). All mutants showed significantly less DCVs than wt, both without and with bPAC stimulation (Fig. 7e,f). In addition, following photostimulation, we found a significant enrichment of DCVs at sites distal to the synaptic terminal/DP region in the snn-1(tm2557) and the S9E mutants, whereas S9A and wt did not increase DCV numbers distal to the DP (Fig. 7a–e). This could indicate that the tm2557 and S9E mutants, either because of the lack of SNN-1 or because of the inability to capture DCVs with a constitutively “phosphorylated” S9 residue, are unable to accumulate DCVs at synapses. Instead, the few DCVs present there were even further depleted, while DCVs accumulated outside synapses, in line with NLP-21::Venus imaging (Fig. 2d).
Without SNN-1, DCVs cannot be retained at release sites, while they accumulate and become immobilized in axonal processes in SNN-1(S9A) mutants
Last, we analyzed the dynamic properties of DCVs (i.e., their trafficking in cholinergic motor axons). We used time-lapse imaging of NLP-21::Venus-containing DCVs, expressed in the dorsally innervating A-type (DA) class of motor neurons (Fig. 8a; Extended Data Movie 1). Dorsal axonal processes were imaged as confocal stacks (4 volumes/s), and maximum-projected. Line scans were used to generate kymographs, for analysis of anterograde and retrograde traffic of DCVs, as well as their stationary accumulations (Fig. 8b). The stationary fluorescent particles may represent DCV release sites, near which DCVs are captured and made available for fusion. Distinct capture, as well as release events could be observed (Fig. 8b). We compared wt, snn-1(tm2557), S9A, and S9E mutants. Mobile particles moved with mostly uniform velocity, although S9A and S9E mutants showed significantly slower retrograde traffic (Fig. 8c). Overall, a similar fraction of the particles observed in each kymograph were stationary; thus, the mutations did not cause a loss or gain of release sites (Fig. 8d). However, when we assessed the overall movement for each group of animals, wt showed the highest accumulated distance covered by the mobile particles, whereas all snn-1 mutants showed significantly less motility, with the most prominent reduction for the S9A mutant, which may constitutively tether DCVs (Fig. 8e). To score for general DCV abundance, the overall fluorescence along the axon was measured. Here, S9A animals showed a significant (>twofold) increase of DCVs, compared with wt (Fig. 8f), where axon and dendrite showed similar fluorescence levels. Thus, S9A causes an accumulation of DCVs in axons, probably in between synapses (compare EM analysis, near synapses; Fig. 7).
Figure 8.
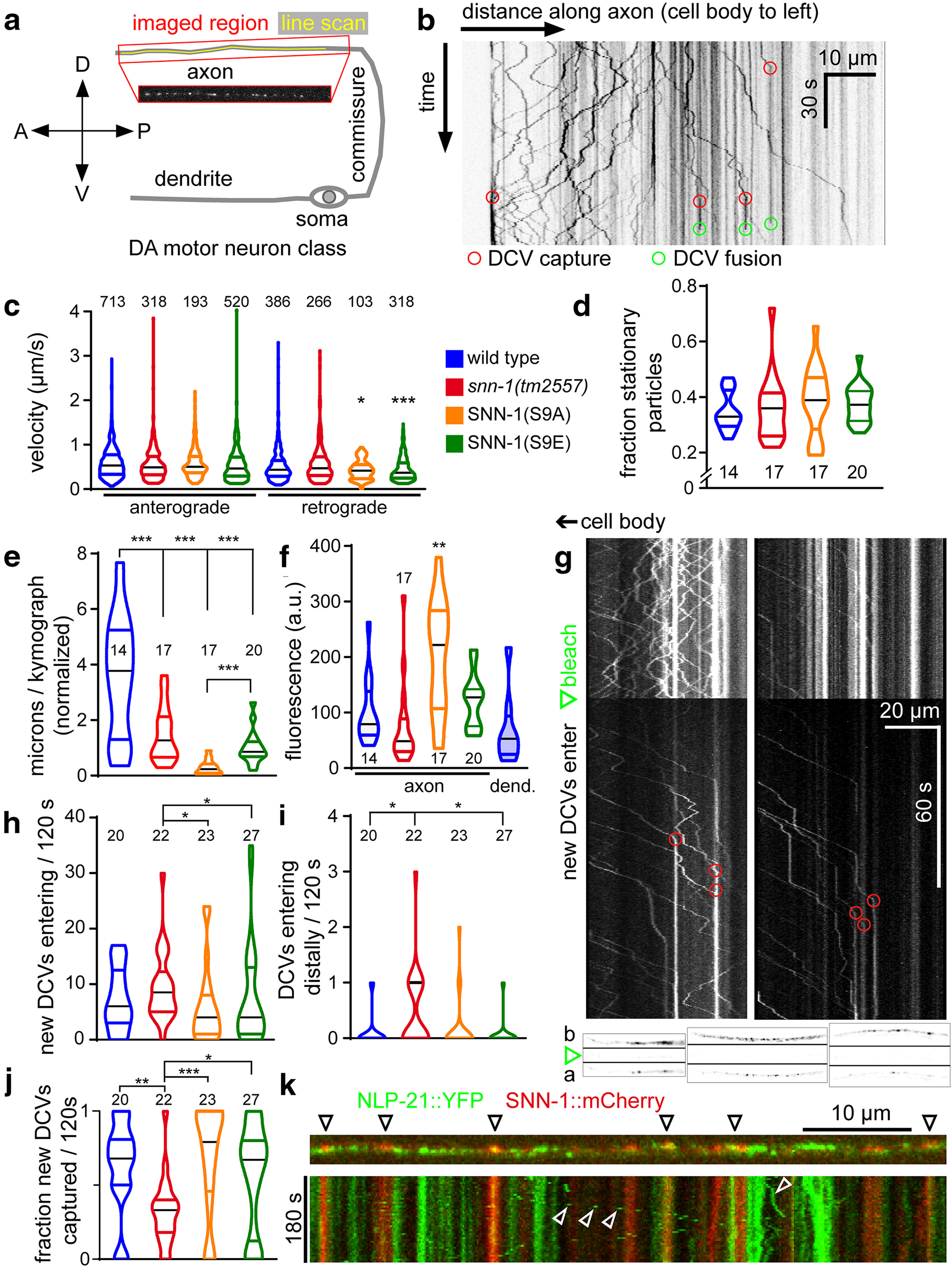
DCV trafficking in axons of DA-type motor neurons is affected in synapsin mutants. DCVs labeled with the NLP-21::Venus neuropeptide precursor protein were analyzed in axonal processes of DA motor neurons. a, Schematic showing the region analyzed and location of the line scan used for kymograph analysis. b, Example kymograph, 80 µm along the axon pointing away from the cell body (anterograde traffic is to the right), and covering 105 s in the time domain (down). Numerous DCVs traffic, some appear static. Capture events (red circles) occur when moving particles becomes static. DCV fusion events (green circles; disappearance of DCV fluorescence) are evident. c, DCV velocity (anterograde and retrograde, as indicated) was analyzed as increments of straight movement, between observable changes in velocity, or phases of no movement. Number of analyzed increments as well as genotypes are indicated. d, Fraction of stationary particles per genotype. e, Overall movement was assessed by summing up the individual distances of each moving increment in axial direction. f, The density of the fluorescence in the analyzed axonal segments, as well as in wt dendrites, was compared. g, To assess individual DCV traffic, a section of the axon was photo-bleached after 60 s (green arrow), and observation continued for 120 s. DCVs entering the bleached region are eventually captured (red circles). Shown are two examples. Bottom insets, Three examples of axons before (b), right after (green arrow), and 120 s after bleach (a; top to bottom). h, The incidence of new DCVs entering the bleached area was counted. i, Some DCVs entered the bleached area from the distal region of the axon. j, Fraction of new DCVs being captured. k, SNN-1::mCherry was imaged along with NLP-21::Venus in the same axon (top). Bottom, Time-lapse analysis, distance, and time indicated. Few DCVs are seen to move (open white arrows), whereas others are stationary. SNN-1 puncta are only stationary; some of them colocalize with stationary DCVs. *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA with Bonferroni correction (c-f) and Kruskal–Wallis multiple comparisons analysis with Dunn's test (h–j). c–f, h–j, Data are frequency distribution, with median (black) and 25-75 percentile. d–f, h–j, The number of analyzed animals is indicated.
Trafficking of NLP-21::Venus-containing DCVs in the dendrite of a DA type motor neuron.
Last, we wanted to obtain more information about mobile DCVs and the propensity of DCV capture in the axon. Because such events are often obscured by the fluorescence of DCVs already present in that segment, we eliminated existing axonal fluorescent signals by photo-bleaching (Fig. 8g). New DCVs could be observed to enter the bleached segment of the axon and were eventually captured. Capture sites coincided with regions where high fluorescence was detected prebleach, indicating that these are preferred DCV release sites (Fig. 8g, bottom insets). We then counted the newly delivered DCVs (i.e., not photo-bleached, Fig. 8h; some DCVs entered the bleached area from the distal end, Fig. 8i), as well as the fraction of newly arriving DCVs that were captured (Fig. 8j). Significantly more DCVs were entering in snn-1(tm2557) animals, and in particular, also from the distal side. This indicates that, in the absence of synapsin, DCVs cycle within the axon along microtubules, as they are not captured. Capture events were largely reduced in snn-1(tm2557) animals. The S9 mutants were not significantly different from wt; however, S9E mutants had a very high variability in trafficking DCVs, and S9A mutants showed a slightly higher capture rate, in line with the dephosphorylated state being more “sticky” for DCVs. Last, we assessed where, relative to DCVs, the synapsin protein is located (Fig. 8k). SNN-1::mCherry was observed in puncta along the axon, which sometimes coincided with DCV fluorescence (NLP-21::Venus). Time-lapse analyses showed that SNN-1 puncta were exclusively static, whereas NLP-21 puncta were mobile or static. Thus, SNN-1 does not traffic along with DCVs; however, it may be bound to cytoskeletal elements, or the SV cluster, and there provide a “sticky” environment for DCVs. Thus, dual-labeled sites may be capture sites where DCVs interact with SNN-1.
Our findings indicate that SNN-1 phosphorylation by PKA is required to mobilize DCVs, releasing them from the cytoskeleton. However, also the S9E mutation caused reduced DCV motility, as did the snn-1(tm2557) deletion allele; and while these two mutants did not release neuropeptides, the S9A animals did (based on the observed mPSC amplitude increase; Fig. 6j). Thus, synapsin S9 phosphorylation may affect multiple steps of DCV trafficking and recruitment to release sites, which is abolished in the deletion allele, and differently affected in S9E and S9A mutants.
Discussion
Here, we analyzed how synapsin, a known organizer of the SV cluster, functions in C. elegans cholinergic motor neurons, and particularly in neuropeptide release. In the snn-1(tm2557) deletion mutant, we observed some alteration in SV localization, a mild reduction of spontaneous and bPAC evoked SV release, but a profound defect on (ChR2)-evoked release. This argues for defective mobilization of SVs from the RP, in line with previous reports (Johnson et al., 1972; Benfenati et al., 1989; Hosaka et al., 1999; Menegon et al., 2006; Gitler et al., 2008). However, we also found a previously unknown role of synapsin in neuropeptide release: snn-1(tm2557) mutants, in response to bPAC-induced cAMP increase, showed evoked behavior different from wt, no neuropeptide release, less overall synaptic DCVs but no acute further reduction of DCVs in terminals, no increase in mPSC amplitudes, and no SV size increase (i.e., ACh loading). This is surprising, since in mammalian neurons, synapsin was not associated with DCVs (Navone et al., 1984; Pieribone et al., 1995).
SNN-1 is present throughout the axon in a punctate fashion, immobile, and in some puncta colocalizes with immobile DCVs, whereas mobile DCVs were not associated with SNN-1. Thus, the majority of synapsin may only transiently interact with DCVs, in line with EM in mammalian neurons, and we propose that this occurs at capture sites. In addition, synapsin clusters (or phase-separated areas) may provide a “sticky” substrate for DCVs, depending on its phosphorylation state, to slow down their free movement in the axon, thus promoting capture. On the other hand, wt SNN-1 also promoted DCV traffic. Possibly, as it organizes the SV cluster, it may prevent SVs from obstructing DCV motility. DCV abundance (reduced at synapses, increased in neurites and somata) and localization were affected in the absence of SNN-1, as well as in mutants of the phosphorylation site S9 in SNN-1B: Both abolished (S9A) or “constitutive” phosphorylation (S9E) caused reduced DCV numbers near the center of synapses, while S9A accumulated immobile DCVs in axons. The S9 site may thus affect DCV trafficking and function by regulating their clustering and/or their interaction with the cytoskeleton. At synapses, DCVs have to be “captured” from microtubule tracts. This is largely abolished in snn-1(tm2557) mutants and affected in S9E animals. However, DCVs also have to be released again to make them available for fusion (see model, Fig. 9). The right sequence of these differential dephosphorylation/phosphorylation events may be more affected by the synapsin S9E mutation rather than by S9A because DCVs have to be bound by synapsin to make them available at release site, and this may be ineffective in S9E mutants.
Figure 9.

Model. DCVs are delivered from the cell body and circulate in the neuronal process. Synapsin is required for DCV capture at synapses. In the absence of synapsin and in the S9E mutant, DCVs are not retained at synapses but accumulate between synapses and in the cell body, likely as they cannot be released. In the S9A mutant, DCVs are anchored tightly. Neuropeptide release, based on electrophysiology (see Fig. 6h), can still occur.
The lower number of DCVs in snn-1(tm2557) synapses showed an altered distribution on stimulation by bPAC, where they were depleted near DPs and accumulated distal to the synapse. In SNN-1B(S9A) animals, bPAC stimulation did not alter the DCV distribution at synapses. In S9E animals, as in tm2557, bPAC stimulation caused an increase of DCVs distal to the synapse. Possibly, activity causes a redistribution of DCVs, but no proper recruitment at release sites occurs; thus, DCVs may be “dropped” in these regions of the axon when no synapsin-based tethering of DCVs can occur. This supports two conclusions: (1) Contrary to previous publications that attributed “capture” merely to a tightly regulated balance of anterograde and retrograde molecular motors of the tubulin cytoskeleton (Bharat et al., 2017; Morrison et al., 2018; Stucchi et al., 2018), our data suggest that also a direct tethering of DCVs occurs by synapsin, as established for SVs, to keep them near release sites. (2) This process appears to depend on S9, where phosphorylation leads to release of the captured DCVs to enable their PM localization and fusion. According to the “conveyor belt” model of DCV delivery to synapses in Drosophila (i.e., by circulation in axons and terminals of motor neurons) (Wong et al., 2012), also DCVs in C. elegans traffic into the axon distally and return if they are not captured, possibly even back to the cell body. This is in line with our kymographs of NLP-21::Venus tagged DCVs.
The C. elegans genome encodes one synapsin gene, giving rise to two splice variants, SNN-1A, which lacks the N-terminal A domain and the S9 phosphosite, and SNN-1B, which includes the A domain (Fig. 1e). Our data indicate that the SNN-1B variant contributes to function in cholinergic neurons. Despite effects of the snn-1 deletion on synaptic SV distribution and particularly on SV mobilization from the RP, the bPAC-induced increase of SV release was only partially affected by SNN-1. However, the stronger, ChR2 (depolarization)-evoked SV fusion was largely affected. Synapsin binds to SV membranes through its central C domain, which is also responsible for dimerization and interaction with the actin cytoskeleton. Phosphorylation of S9 affects the affinity for actin and is responsible for PKA-dependent mobilization of SVs from the RP (Hosaka et al., 1999; Cesca et al., 2010). The snn-1(tm2557) deletion eliminates regions for SNN-1 interaction with SVs and actin, and affects dimerization (Esser et al., 1998). Since SV mobilization from the RP is defective in snn-1(tm2557), the SNN-1 protein may recruit additional proteins to the SV cluster that promote SV mobilization. The almost normal bPAC-induced increase of mPSC rate in snn-1(tm2557) mutants indicates that cAMP/PKA regulates SV release not only through SNN-1, but possibly also through targets that affect priming (e.g., tomosyn) (Baba et al., 2005; McEwen et al., 2006) or SNAP-25 (Nagy et al., 2004).
SNN-1 is required for DCV fusion. Discharged neuropeptides activate auto-receptors, increasing the ACh load of SVs (Steuer Costa et al., 2017), and this was absent in snn-1 mutants, as it was in unc-31/CAPS mutants. CAPS-dependent signaling in secretory granule filling was found in chromaffin cells of mice, as CAPS1 KOs were deficient in catecholamine loading (Speidel et al., 2005) and showed reduced DCV priming and release (Y. Liu et al., 2008). CAPS1 and CAPS2 were also shown to be required for SV priming in mice (Jockusch et al., 2007). This may be reflected in C. elegans, as unc-31 mutants lacked not only the cAMP-induced amplitude increase but also had a reduced mPSC rate (Steuer Costa et al., 2017). Unlike the findings of Charlie et al. (2006b), our data place UNC-31 downstream of cAMP/PKA effects on synapsin, and upstream of neuropeptide release in evoking increased SV quantal size. Thus, the unknown PKA target postulated by Zhou et al. (2007) likely is SNN-1. cAMP signaling enables synapses to undergo homeostatic changes, in response to altered NMJ signaling (e.g., to adapt to different locomotion regimens) within seconds to tens of seconds. In mammalian neurons, neuromodulators were shown to affect synapsin phosphorylation after 30 min through cAMP pathways, and to alter SV numbers (Patzke et al., 2019). Although the authors did not assess rapid effects after a few seconds of cAMP increase, the observed decrease in SV numbers indicates that cAMP/SNN-1 pathways of SV regulation are conserved during evolution. These authors also analyzed a number of mutations of synapsin found in human genetic diseases. Our work implies that these pathologies may, at least in part, be mediated at the level of neuropeptide signaling.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grants CRC1080-B2, GO1011/4-1/-2, and EXC115, and Goethe University. Some nematode strains were obtained from the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health National Center for Research Resources, and from the Japanese National Bioresource Project for the Experimental Animal Nematode C. elegans. We thank Kenneth Miller, Derek Sieburth and Joshua Kaplan for providing strains; Ichiro Aoki for MATLAB scripts; and Mona Hoeret and Franziska Baumbach for expert technical assistance.
The authors declare no competing financial interests.
References
- Baba T, Sakisaka T, Mochida S, Takai Y (2005) PKA-catalyzed phosphorylation of tomosyn and its implication in Ca2+-dependent exocytosis of neurotransmitter. J Cell Biol 170:1113–1125. 10.1083/jcb.200504055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Bahler M, Jahn R, Greengard P (1989) Interactions of synapsin I with small synaptic vesicles: distinct sites in synapsin I bind to vesicle phospholipids and vesicle proteins. J Cell Biol 108:1863–1872. 10.1083/jcb.108.5.1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat V, Siebrecht M, Burk K, Ahmed S, Reissner C, Kohansal-Nodehi M, Steubler V, Zweckstetter M, Ting JT, Dean C (2017) Capture of dense core vesicles at synapses by JNK-dependent phosphorylation of synaptotagmin-4. Cell Rep 21:2118–2133. 10.1016/j.celrep.2017.10.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavolo SL, Bulgari D, Deitcher DL, Levitan ES (2016) Activity induces Fmr1-sensitive synaptic capture of anterograde circulating neuropeptide vesicles. J Neurosci 36:11781–11787. 10.1523/JNEUROSCI.2212-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F (2010) The synapsins: key actors of synapse function and plasticity. Prog Neurobiol 91:313–348. 10.1016/j.pneurobio.2010.04.006 [DOI] [PubMed] [Google Scholar]
- Charlie NK, Thomure AM, Schade MA, Miller KG (2006a) The Dunce cAMP phosphodiesterase PDE-4 negatively regulates G alpha(s)-dependent and G alpha(s)-independent cAMP pools in the Caenorhabditis elegans synaptic signaling network. Genetics 173:111–130. 10.1534/genetics.105.054007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlie NK, Schade MA, Thomure AM, Miller KG (2006b) Presynaptic UNC-31 (CAPS) is required to activate the G alpha(s) pathway of the Caenorhabditis elegans synaptic signaling network. Genetics 172:943–961. 10.1534/genetics.105.049577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung U, Atwood HL, Zucker RS (2006) Presynaptic effectors contributing to cAMP-induced synaptic potentiation in Drosophila. J Neurobiol 66:273–280. 10.1002/neu.20218 [DOI] [PubMed] [Google Scholar]
- Cho RW, Buhl LK, Volfson D, Tran A, Li F, Akbergenova Y, Littleton JT (2015) Phosphorylation of complexin by PKA regulates activity-dependent spontaneous neurotransmitter release and structural synaptic plasticity. Neuron 88:749–761. 10.1016/j.neuron.2015.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Charlie NK, Milfort MC, Brown BS, Gravlin CN, Knecht JE, Miller KG (2008) A novel molecular solution for ultraviolet light detection in Caenorhabditis elegans. PLoS Biol 6:e198. 10.1371/journal.pbio.0060198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbguth K, Prigge M, Schneider F, Hegemann P, Gottschalk A (2012) Bimodal activation of different neuron classes with the spectrally red-shifted channelrhodopsin chimera C1V1 in Caenorhabditis elegans. PLoS One 7:e46827. 10.1371/journal.pone.0046827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser L, Wang CR, Hosaka M, Smagula CS, Sudhof TC, Deisenhofer J (1998) Synapsin I is structurally similar to ATP-utilizing enzymes. EMBO J 17:977–984. 10.1093/emboj/17.4.977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans GJ, Wilkinson MC, Graham ME, Turner KM, Chamberlain LH, Burgoyne RD, Morgan A (2001) Phosphorylation of cysteine string protein by protein kinase A: implications for the modulation of exocytosis. J Biol Chem 276:47877–47885. 10.1074/jbc.M108186200 [DOI] [PubMed] [Google Scholar]
- Fiala JC (2005) Reconstruct: a free editor for serial section microscopy. J Microsc 218:52–61. 10.1111/j.1365-2818.2005.01466.x [DOI] [PubMed] [Google Scholar]
- Gekel I, Neher E (2008) Application of an Epac activator enhances neurotransmitter release at excitatory central synapses. J Neurosci 28:7991–8002. 10.1523/JNEUROSCI.0268-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Cheng Q, Greengard P, Augustine GJ (2008) Synapsin IIa controls the reserve pool of glutamatergic synaptic vesicles. J Neurosci 28:10835–10843. 10.1523/JNEUROSCI.0924-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Xu Y, Kao HT, Lin D, Lim S, Feng J, Greengard P, Augustine GJ (2004) Molecular determinants of synapsin targeting to presynaptic terminals. J Neurosci 24:3711–3720. 10.1523/JNEUROSCI.5225-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracheva EO, Burdina AO, Holgado AM, Berthelot-Grosjean M, Ackley BD, Hadwiger G, Nonet ML, Weimer RM, Richmond JE (2006) Tomosyn inhibits synaptic vesicle priming in Caenorhabditis elegans. PLoS Biol 4:e261. 10.1371/journal.pbio.0040261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graebner AK, Iyer M, Carter ME (2015) Understanding how discrete populations of hypothalamic neurons orchestrate complicated behavioral states. Front Syst Neurosci 9:111. 10.3389/fnsys.2015.00111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Watanabe S, Schuske K, Jorgensen EM (2008) CAPS and syntaxin dock dense core vesicles to the plasma membrane in neurons. J Cell Biol 180:483–491. 10.1083/jcb.200708018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover CM, Edwards SL, Yu SC, Kittelmann M, Richmond JE, Eimer S, Yorks RM, Miller KG (2014) A novel CaM kinase II pathway controls the location of neuropeptide release from Caenorhabditis elegans motor neurons. Genetics 196:745–765. 10.1534/genetics.113.158568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka M, Hammer RE, Sudhof TC (1999) A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron 24:377–387. 10.1016/s0896-6273(00)80851-x [DOI] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D (2012) Molecular machines governing exocytosis of synaptic vesicles. Nature 490:201–207. 10.1038/nature11320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockusch WJ, Speidel D, Sigler A, Sorensen JB, Varoqueaux F, Rhee JS, Brose N (2007) CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell 131:796–808. 10.1016/j.cell.2007.11.002 [DOI] [PubMed] [Google Scholar]
- Johnson EM, Ueda T, Maeno H, Greengard P (1972) Adenosine 3′,5-monophosphate-dependent phosphorylation of a specific protein in synaptic membrane fractions from rat cerebrum. J Biol Chem 247:5650–5652. [PubMed] [Google Scholar]
- Kaneko M, Takahashi T (2004) Presynaptic mechanism underlying cAMP-dependent synaptic potentiation. J Neurosci 24:5202–5208. 10.1523/JNEUROSCI.0999-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittelmann M, Liewald JF, Hegermann J, Schultheis C, Brauner M, Steuer Costa W, Wabnig S, Eimer S, Gottschalk A (2013) In vivo synaptic recovery following optogenetic hyperstimulation. Proc Natl Acad Sci USA 110:E3007–E3016. 10.1073/pnas.1305679110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y (2000) Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron 27:133–143. 10.1016/S0896-6273(00)00015-5 [DOI] [PubMed] [Google Scholar]
- Liewald JF, Brauner M, Stephens GJ, Bouhours M, Schultheis C, Zhen M, Gottschalk A (2008) Optogenetic analysis of synaptic function. Nat Methods 5:895–902. 10.1038/nmeth.1252 [DOI] [PubMed] [Google Scholar]
- Liu Q, Chen B, Yankova M, Morest DK, Maryon E, Hand AR, Nonet ML, Wang ZW (2005) Presynaptic ryanodine receptors are required for normal quantal size at the Caenorhabditis elegans neuromuscular junction. J Neurosci 25:6745–6754. 10.1523/JNEUROSCI.1730-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Schirra C, Stevens DR, Matti U, Speidel D, Hof D, Bruns D, Brose N, Rettig J (2008) CAPS facilitates filling of the rapidly releasable pool of large dense-core vesicles. J Neurosci 28:5594–5601. 10.1523/JNEUROSCI.5672-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudhof TC, Linden DJ (2003) Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell 115:49–60. 10.1016/s0092-8674(03)00727-x [DOI] [PubMed] [Google Scholar]
- McEwen JM, Madison JM, Dybbs M, Kaplan JM (2006) Antagonistic regulation of synaptic vesicle priming by tomosyn and UNC-13. Neuron 51:303–315. 10.1016/j.neuron.2006.06.025 [DOI] [PubMed] [Google Scholar]
- Menegon A, Bonanomi D, Albertinazzi C, Lotti F, Ferrari G, Kao HT, Benfenati F, Baldelli P, Valtorta F (2006) Protein kinase A-mediated synapsin I phosphorylation is a central modulator of Ca2+-dependent synaptic activity. J Neurosci 26:11670–11681. 10.1523/JNEUROSCI.3321-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milovanovic D, Wu Y, Bian X, De Camilli P (2018) A liquid phase of synapsin and lipid vesicles. Science 361:604–607. 10.1126/science.aat5671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison LM, Edwards SL, Manning L, Stec N, Richmond JE, Miller KG (2018) Sentryn and SAD kinase link the guided transport and capture of dense core vesicles in Caenorhabditis elegans. Genetics 210:925–946. 10.1534/genetics.118.300847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sorensen JB (2004) Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron 41:417–429. 10.1016/s0896-6273(04)00038-8 [DOI] [PubMed] [Google Scholar]
- Navone F, Greengard P, De Camilli P (1984) Synapsin I in nerve terminals: selective association with small synaptic vesicles. Science 226:1209–1211. 10.1126/science.6438799 [DOI] [PubMed] [Google Scholar]
- Oranth A, Schultheis C, Tolstenkov O, Erbguth K, Nagpal J, Hain D, Brauner M, Wabnig S, Steuer Costa W, McWhirter RD, Zels S, Palumbos S, Miller DM III, Beets I, Gottschalk A (2018) Food sensation modulates locomotion by dopamine and neuropeptide signaling in a distributed neuronal network. Neuron 100:1414–1428.e1410. 10.1016/j.neuron.2018.10.024 [DOI] [PubMed] [Google Scholar]
- Park YS, Hur EM, Choi BH, Kwak E, Jun DJ, Park SJ, Kim KT (2006) Involvement of protein kinase C-epsilon in activity-dependent potentiation of large dense-core vesicle exocytosis in chromaffin cells. J Neurosci 26:8999–9005. 10.1523/JNEUROSCI.2828-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzke C, Brockmann MM, Dai J, Gan KJ, Grauel MK, Fenske P, Liu Y, Acuna C, Rosenmund C, Sudhof TC (2019) Neuromodulator signaling bidirectionally controls vesicle numbers in human synapses. Cell 179:498–513.e422. 10.1016/j.cell.2019.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P (1995) Distinct pools of synaptic vesicles in neurotransmitter release. Nature 375:493–497. 10.1038/375493a0 [DOI] [PubMed] [Google Scholar]
- Preisach C, Burkhardt H, Schmidt-Thieme L, Decker R, eds (2008) Data analysis, machine learning and applications. Berlin: Springer. [Google Scholar]
- Rizzoli SO, Betz WJ (2005) Synaptic vesicle pools. Nat Rev Neurosci 6:57–69. 10.1038/nrn1583 [DOI] [PubMed] [Google Scholar]
- Rodriguez P, Bhogal MS, Colyer J (2003) Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem 278:38593–38600. 10.1074/jbc.C301180200 [DOI] [PubMed] [Google Scholar]
- Rupnik M, Kreft M, Sikdar SK, Grilc S, Romih R, Zupancic G, Martin TF, Zorec R (2000) Rapid regulated dense-core vesicle exocytosis requires the CAPS protein. Proc Natl Acad Sci USA 97:5627–5632. 10.1073/pnas.090359097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieburth D, Madison JM, Kaplan JM (2007) PKC-1 regulates secretion of neuropeptides. Nat Neurosci 10:49–57. 10.1038/nn1810 [DOI] [PubMed] [Google Scholar]
- Speidel D, Bruederle CE, Enk C, Voets T, Varoqueaux F, Reim K, Becherer U, Fornai F, Ruggieri S, Holighaus Y, Weihe E, Bruns D, Brose N, Rettig J (2005) CAPS1 regulates catecholamine loading of large dense-core vesicles. Neuron 46:75–88. 10.1016/j.neuron.2005.02.019 [DOI] [PubMed] [Google Scholar]
- Steuer Costa W, Yu SC, Liewald JF, Gottschalk A (2017) Fast cAMP modulation of neurotransmission via neuropeptide signals and vesicle loading. Curr Biol 27:495–507. 10.1016/j.cub.2016.12.055 [DOI] [PubMed] [Google Scholar]
- Steuer Costa W, Van der Auwera P, Glock C, Liewald JF, Bach M, Schuler C, Wabnig S, Oranth A, Masurat F, Bringmann H, Schoofs L, Stelzer EH, Fischer SC, Gottschalk A (2019) A GABAergic and peptidergic sleep neuron as a locomotion stop neuron with compartmentalized Ca2+ dynamics. Nat Commun 10:4095. 10.1038/s41467-019-12098-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirman JN, Crane MM, Husson SJ, Wabnig S, Schultheis C, Gottschalk A, Lu H (2011) Real-time multimodal optical control of neurons and muscles in freely behaving Caenorhabditis elegans. Nat Methods 8:153–158. 10.1038/nmeth.1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucchi R, Plucińska G, Hummel JJ, Zahavi EE, Guerra San Juan I, Klykov O, Scheltema RA, Altelaar AF, Hoogenraad CC (2018) Regulation of KIF1A-driven dense core vesicle transport: Ca(2+)/CaM controls DCV binding and Liprin-alpha/TANC2 recruits DCVs to postsynaptic sites. Cell Rep 24:685–700. 10.1016/j.celrep.2018.06.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC (2013) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80:675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, De Camilli P (1989) Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science 245:1474–1480. 10.1126/science.2506642 [DOI] [PubMed] [Google Scholar]
- Swierczek NA, Giles AC, Rankin CH, Kerr RA (2011) High-throughput behavioral analysis in C. elegans. Nat Methods 8:592–598. 10.1038/nmeth.1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghert PH, Nitabach MN (2012) Peptide neuromodulation in invertebrate model systems. Neuron 76:82–97. 10.1016/j.neuron.2012.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur P, Stevens DR, Sheng ZH, Rettig J (2004) Effects of PKA-mediated phosphorylation of Snapin on synaptic transmission in cultured hippocampal neurons. J Neurosci 24:6476–6481. 10.1523/JNEUROSCI.0590-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sieburth D (2013) PKA controls calcium influx into motor neurons during a rhythmic behavior. PLoS Genet 9:e1003831. 10.1371/journal.pgen.1003831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr WA (2012) Scientific workflow systems: pipeline Pilot and KNIME. J Comput Aided Mol Des 26:801–804. 10.1007/s10822-012-9577-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer RM (2006) Preservation of C. elegans tissue via high-pressure freezing and freeze-substitution for ultrastructural analysis and immunocytochemistry. Methods Mol Biol 351:203–221. 10.1385/1-59745-151-7:203 [DOI] [PubMed] [Google Scholar]
- Wong MY, Zhou C, Shakiryanova D, Lloyd TE, Deitcher DL, Levitan ES (2012) Neuropeptide delivery to synapses by long-range vesicle circulation and sporadic capture. Cell 148:1029–1038. 10.1016/j.cell.2011.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Wu LG (2010) Post-tetanic potentiation is caused by two signalling mechanisms affecting quantal size and quantal content. J Physiol 588:4987–4994. 10.1113/jphysiol.2010.196964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SC, Janosi B, Liewald JF, Wabnig S, Gottschalk A (2018) Endophilin A and B join forces with clathrin to mediate synaptic vesicle recycling in Caenorhabditis elegans. Front Mol Neurosci 11:196. 10.3389/fnmol.2018.00196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N, Zucker RS (2005) cAMP acts on exchange protein activated by cAMP/cAMP-regulated guanine nucleotide exchange protein to regulate transmitter release at the crayfish neuromuscular junction. J Neurosci 25:208–214. 10.1523/JNEUROSCI.3703-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou KM, Dong YM, Ge Q, Zhu D, Zhou W, Lin XG, Liang T, Wu ZX, Xu T (2007) PKA activation bypasses the requirement for UNC-31 in the docking of dense core vesicles from C. elegans neurons. Neuron 56:657–669. 10.1016/j.neuron.2007.09.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trafficking of NLP-21::Venus-containing DCVs in the dendrite of a DA type motor neuron.