

ABSTRACT
The fusion of autophagosomes and endosomes/lysosomes, also called autophagosome maturation, ensures the degradation of autophagic cargoes. It is an important regulatory step of the macroautophagy/autophagy process. STX17 is the key autophagosomal SNARE protein that mediates autophagosome maturation. Here, we report that the acetylation of STX17 regulates its SNARE activity and autophagic degradation. The histone acetyltransferase CREBBP/CBP and the deacetylase HDAC2 specifically regulate the acetylation of STX17. In response to cell starvation and MTORC1 inhibition, the inactivation of CREBBP leads to the deacetylation of STX17 at its SNARE domain. This deacetylation promotes the interaction between STX17 and SNAP29 and the formation of the STX17-SNAP29-VAMP8 SNARE complex with no effect on the recruitment of STX17 to autophagosomal membranes. Deacetylation of STX17 also enhances the interaction between STX17 and the tethering complex HOPS, thereby further promoting autophagosome-lysosome fusion. Our study suggests a mechanism by which acetylation regulates the late-stage of autophagy, and possibly other STX17-related intracellular membrane fusion events.
Abbreviations: ACTB: actin beta; CREBBP/CBP: CREB binding protein; Ctrl: control; GFP: green fluorescent protein; GST: glutathione S-transferase; HDAC: histone deacetylase; HOPS: homotypic fusion and protein sorting complex; KO: knockout; LAMP1/2: lysosomal associated membrane protein 1/2; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MEFs: mouse embryonic fibroblasts; MS: mass spectrometry; MTORC1: mechanistic target of rapamycin kinase complex 1; NAM: nicotinamide; PtdIns3K: phosphatidylinositol 3-kinase; RFP: red fluorescent protein; SNAP29: synaptosome associated protein 29; SNARE: soluble N-ethylamide-sensitive factor attachment protein receptor; SQSTM1/p62: sequestosome 1; STX17: syntaxin 17; TSA: trichostatin A; TSC1/2: TSC complex subunit 1/2; VAMP8: vesicle associated membrane protein 8; WT: wild type.
KEYWORDS: Acetylation, autophagosome maturation, CREBBP, HDAC2, HOPS, STX17
Introduction
Macroautophagy (hereafter called autophagy) protects the cell homeostasis by engulfing cytoplasmic components, including protein aggregates and unwanted/damaged organelles, into double-membraned phagophores, which mature into autophagosomes, subsequently delivering the cargo to the lysosomes for degradation [1,2]. The whole process of autophagy can be generally divided into four stages: autophagy initiation, autophagosome formation, autophagosome maturation, and lysosomal degradation [3,4]. Whereas the former two stages involve a series of kinases or kinase complexes and autophagy-related proteins [5,6], which act to sense homeostatic changes and facilitate the biogenesis of autophagosomes, the latter two stages are regulated by a set of membrane-associated proteins and acidic hydrolases that mediate the membrane fusion between autophagosomes and lysosomes and to maintain the digestive activity of lysosomes. The sequential coordination of these stages ensures effective autophagic degradation and continuous autophagy flux.
At present, in comparison to our understanding of autophagy initiation and autophagosome formation, the mechanisms regulating autophagosome maturation, especially in higher eukaryotes, remain largely unknown. Accumulating evidence supports a role for the homotypic fusion and vacuole protein sorting (HOPS) complex as the tether for autophagosome-lysosome fusion [7,8]. Also, it was recently shown that the de-O-GlcNAcylated form of the Golgi stacking protein GORASP2/GRASP55 (golgi reassembly stacking protein 2) can localize to autophagosomes/lysosomes and facilitate autophagosome maturation by interacting with autophagosomal MAP1LC3/LC3 (microtubule associated protein 1 light chain 3)-II and lysosomal LAMP2 (lysosomal associated membrane protein 2) [9]. STX17 (syntaxin 17) is the major autophagosomal soluble N-ethylmaleimide-sensitive protein receptor (SNARE) protein in mammals and Drosophila [10,11]. After arriving at the autophagosomes, STX17 serves as the Qa SNARE, which recruits and cooperates with the Qbc-SNARE SNAP29 (synaptosome associated protein 29) from the cytosol. Together, they form the SNARE complex along with the lysosomal R-SNARE VAMP8 (vesicle associated membrane protein 8), thereby mediating the fusion of autophagosomes with the lysosomes [10]. ATG14 (autophagy-related 14), an autophagy-specific component of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, which directly binds to the t-SNARE complex and primes it for VAMP8 interaction [12]. In addition, RUBCNL, a component of the UVRAG (UV radiation resistance associated)-PtdIns3K subcomplex, directs the PtdIns3K and the HOPS complex to autophagosomes by directly interacting with STX17 [13]. In metazoans, the small GTPases RAB7A and RAB2A coordinately promote autophagosome-lysosome fusion through the interaction with the HOPS complex [14–16].
Lysine acetylation is one of the major post-translational modifications that play an important mechanistic role in the regulation of multiple cellular functions. The function of histone acetylation in gene transcription is well known. However, proteomic studies have revealed that acetylation is involved in the regulation of a great number of non-histone proteins in cell metabolism, and acetylation also targets macromolecular complexes [17,18]. As a constitutive and adaptive catabolic mechanism, autophagy can be rapidly initiated by cells to cope with stress conditions, including nutrient starvation and energy deprivation [19,20]. Accumulating pieces of evidence have indicated that under these conditions, acetylation or deacetylation of the core proteins of the autophagy machinery is essential to their autophagic function [21–24].
Meanwhile, the activity of the acetyltransferases or deacetylases for these proteins can also be directly regulated by post-translational modifications and their interactions with other proteins [25–27], in addition to the changes in their coenzyme levels [28,29]. Acetylation modifications have also been demonstrated to regulate early stages of autophagy, including autophagy initiation and autophagosome formation, by targeting a number of autophagy-related proteins [21–24]. Recently, protein acetylation was also reported to be involved in autophagosome maturation [30–33]. In this study, we show that STX17 can be modified by acetylation at its SNARE domain, which is specifically controlled by the histone acetyltransferase CREBBP/CBP (CREB binding protein) and the deacetylase HDAC2 (histone deacetylase 2). During autophagy, the deacetylation of STX17, caused by the inactivation of CREBBP, enables STX17 to interact with SNAP29 and HOPS. This interaction promotes the formation of the STX17-SNAP29-VAMP8 SNARE complex and the autophagosomal recruitment of HOPS, which eventually leads to the fusion of autophagosomes with the lysosomes.
Results
STX17 is deacetylated at K219 and K223 during autophagy
To identify potential modifications by the acetylation of proteins in the late-stage of autophagy, we primarily focused on STX17 because it is an evolutionarily conserved autophagosomal SNARE protein that plays a key role in mediating autophagosome-lysosome fusion. Using a specific anti-acetyl-lysine antibody, we detected the acetylation of STX17 in fed HEK293 cells (Figure 1(A)). Interestingly, the induction of autophagy in cells by starvation or treatment with torin1, an inhibitor of MTORC1, led to the deacetylation of STX17 (Figure 1(A)). We then treated cells with TSA, the broad-spectrum inhibitor of HDAC family deacetylases, and NAM, the inhibitor of the SIRT family deacetylases, and found that only TSA specifically promoted STX17 acetylation (Figure 1(B) and S1A). In addition, in mouse embryonic fibroblasts (MEFs), the deletion of Tsc1 or Tsc2, two upstream negative regulators of MTORC1, significantly increased STX17 acetylation (Figure 1(C)). Therefore, these results suggest that STX17 is an acetylated protein and undergoes deacetylation during starvation- or MTORC1 inhibition-induced autophagy.
Figure 1.
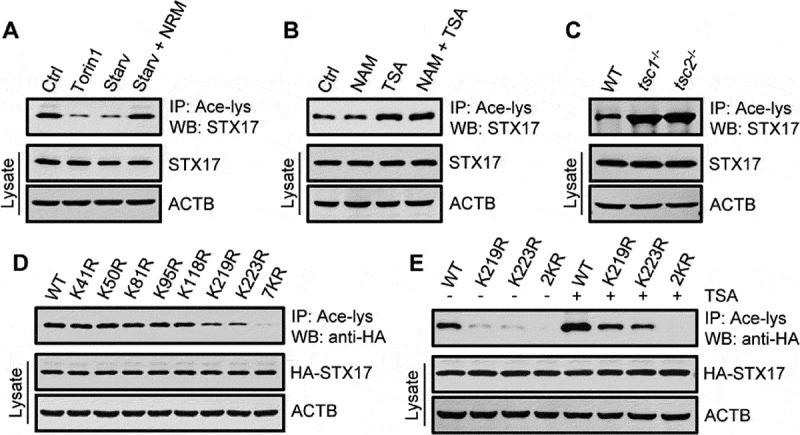
STX17 is deacetylated at K219 and K223 during autophagy. (A) Acetylation of STX17 in HEK293 cells with or without cell starvation or torin1 treatment. Acetylated proteins were immunoprecipitated from cell lysates with an anti-acetyl-lysine antibody (Ace-lys), and the immunoprecipitates were analyzed by western blot using anti-STX17. NRM: nutrient-rich medium. (B) STX17 acetylation in HEK293 cells treated with or without NAM and/or TSA. (C) STX17 acetylation in MEFs with or without the deletion of Tsc1 or Tsc2. (D) Acetylation of HA-STX17 (WT or lysine replacement mutants) expressed in HEK293T cells. 7KR, all seven Lys residues were replaced by Arg. (E) Acetylation of HA-STX17 (WT or lysine replacement mutants) in HEK293T cells treated with or without TSA. 2KR, Lys219, and Lys223 were replaced by Arg
To map the acetylation sites on STX17, STX17 protein immunopurified from TSA-treated cells was subjected to mass spectrometry (MS). Seven lysine residues located in the N-terminal region and SNARE domain were suggested (Figure S1B and C). We then created single-point mutants of STX17 by individually replacing each of the seven lysines with arginine via site-directed mutagenesis, and checked their acetylation levels in transfected cells. These changes revealed that K219 and K223, located in the SNARE domain of STX17, are the main acetylated residues (Figure 1(D,E)). In addition, TSA treatment increased the acetylation of K219R and K223R mutants to a similar degree, while the K219R-K223R double mutant (STX17-2KR) hardly responded to TSA treatment (Figure 1(E)).
STX17 is acetylated by CREBBP
To search for the acetyltransferase of STX17, we overexpressed the most common cell metabolism-related acetyltransferases in HEK293T cell lines, stably expressing HA-STX17. We found that only CREBBP overexpression increased the acetylation of STX17, and the effect was highly specific because overexpression of EP300/p300, a paralog of CREBBP, had no effect (Figure 2(A)). In addition, treatment of these cells with the specific EP300-CREBBP activator CTB or the inhibitor C646 correspondingly stimulated or suppressed STX17 acetylation (Figure 2(B)). Interestingly, knockdown of CREBBP, but not EP300, significantly reduced STX17 acetylation (Figure 2(C)). Furthermore, re-introduction of WT CREBBP, but not the acetyltransferase-defective CREBBP [34], into CREBBP-KO HEK293 cells restored STX17 acetylation (Figure 2(D)).
Figure 2.
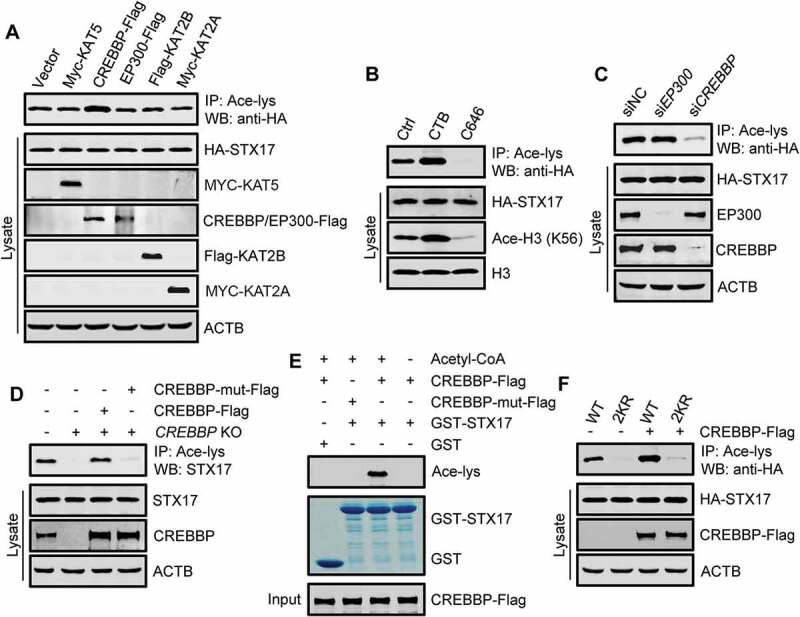
STX17 is acetylated by CREBBP. (A) Acetylation of HA-STX17 in HEK293T cells stably expressing HA-STX17 and overexpressing the indicated individual histone acetyltransferases. The anti-acetyl-lysine antibody was used for immunoprecipitation, and the precipitates were analyzed using anti-HA. (B) HA-STX17 acetylation in HEK293T cells stably expressing HA-STX17 after treatment with the CREBBP activator CTB or the CREBBP inhibitor C646. H3: histone H3; Ace-H3 (K56): histone H3 acetylated at K56. (C) HA-STX17 acetylation in HEK293T cells stably expressing HA-STX17 after incubation with siRNA against EP300 or CREBBP. (D) Acetylation of endogenous STX17 in CREBBP-KO HEK293 cells transfected with or without Flag-tagged WT CREBBP or acetyltransferase-defective CREBBP mutant. The anti-acetyl-lysine antibody was used for immunoprecipitation, and the precipitates were analyzed using anti-STX17. CREBBP-mut-Flag: acetyltransferase-defective CREBBP mutant. (E) In vitro STX17 acetylation assay using purified GST-STX17 and Flag-tagged WT CREBBP or the acetyltransferase-defective CREBBP mutant immunoprecipitated from HEK293T cells. Acetyl-CoA: acetyl-coenzyme A. CREBBP-mut-Flag: acetyltransferase-defective CREBBP mutant. (F) Acetylation of HA-STX17-WT or HA-STX17-2KR in HEK293T cells with or without CREBBP-Flag transfection
Like EP300, CREBBP shuttles between the nucleus and the cytoplasm, and several cytoplasmic proteins have been shown to interact with CREBBP [35,36]. To determine whether STX17 is a direct substrate of CREBBP, we carried out in vitro acetylation assays by incubating purified recombinant GST-STX17 with CREBBP-Flag immunoprecipitated from HEK293T cells. In the presence of acetyl-coenzyme A, GST-STX17 was clearly acetylated by WT CREBBP, but not by the acetyltransferase-defective CREBBP (Figure 2(E)).
Finally, to verify that K219 and K223 sites are the targets of CREBBP, we examined the acetylation of STX17-2KR mutant in cells overexpressing CREBBP-Flag. As expected, the acetylation of STX17-2KR did not respond to the overexpression of CREBBP-Flag, while the acetylation of STX17-WT was greatly promoted by the expression of CREBBP (Figure 2(F)). Together, these results suggest that CREBBP specifically mediates the acetylation of STX17 at K219 and K223.
HDAC2 is a deacetylase of STX17
To identify the deacetylase of STX17, we were guided by our earlier finding that TSA, but not NAM, stimulated STX17 acetylation in cells. We screened members of the HDAC family of deacetylases, focusing on the five, which may positively regulate autophagy [37,38]. By expressing each of these HDACs in HEK293T cell lines stably expressing HA-STX17, we found only HDAC2 markedly deacetylated STX17 (Figure 3(A)). Accordingly, knocking down HDAC2 enhanced STX17 acetylation (Figure 3(B)), and the enhancement was abolished by re-expressing WT HDAC2, but not the deacetylase-defective HDAC2, in the knockdown cells [39,40] (Figure 3(C) and S2A). In addition, co-immunoprecipitation assays detected a strong association of HA-STX17 with HDAC2 but not HDAC1 (Figure 3(D)). Moreover, recombinant GST-HDAC2 purified from E. coli specifically pulled down endogenous STX17 from cell lysates (Figure 3(E)). Finally, we found that knockdown of HDAC2 failed to increase the acetylation of STX17-2KR as it strongly increased the acetylation of STX17-WT (Figure 3(F)). Therefore, these results suggest that HDAC2 is the deacetylase of STX17 and might mediate the deacetylation of STX17 at K219 and K223.
Figure 3.
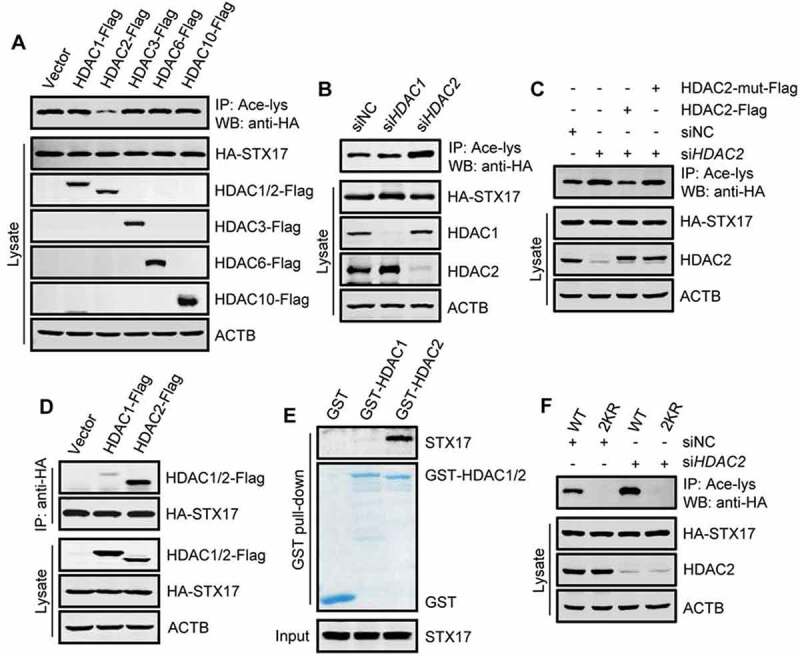
HDAC2 is a deacetylase for STX17. (A) Acetylation of HA-STX17 in HEK293T cells stably expressing HA-STX17 and overexpressing the indicated individual histone deacetylases. (B) HA-STX17 acetylation in HEK293T cells stably expressing HA-STX17 after incubation with siRNA against HDAC1 or HDAC2. (C) HEK293T cells stably expressing HA-STX17 were transfected with Flag-tagged WT HDAC2 or deacetylase-defective HDAC2 and incubated with HDAC2 siRNA. Acetylation of HA-STX17 was then analyzed. HDAC2-mut-Flag: deacetylase-defective HDAC2. (D) Co-precipitation of HDAC1 and HDAC2 with STX17. HA-STX17 was immunoprecipitated from HEK293T cells transfected with HDAC1-Flag or HDAC2-Flag, and the precipitates were analyzed using anti-Flag. (E) Pull-down assay of STX17-HDAC2 binding. Purified recombinant GST-HDAC1 or GST-HDAC2 was incubated with HEK293 cell lysates, and the bound STX17 was detected by western blot using anti-STX17. (F) Acetylation of HA-STX17-WT or HA-STX17-2KR in HEK293T cells incubated with or without HDAC2 siRNA
However, in cells under starvation or torin1 treatment, the interaction between STX17 and HDAC2 was barely changed (Figure S2B). In addition, fluorometric HDAC2 activity assays, with Boc-Lys (Ac)-AMC as a substrate, indicated that the activity of the endogenous HDAC2 was not altered by cell starvation or torin1 treatment, while it was dramatically inhibited by the TSA treatment (Figure S2C). Because it is known that CREBBP is inactivated in starved or torin1-treated cells due to MTORC1 inactivation [27], we concluded that the deacetylation of STX17 in these cells is mainly due to the inhibition of CREBBP.
Deacetylation of STX17 is required for autophagosome-lysosome fusion
To evaluate the physiological effects of STX17 acetylation, we first checked the localization of STX17 to the autophagosomal membranes where it displays a SNARE activity. Thus, we created acetylation-mimetic, lysine-to-glutamine mutations of STX17 at K219 and K223 (STX17-2KQ). Under normal culture conditions, cellular STX17 predominantly resides on the endoplasmic reticulum (ER) and the mitochondria [10]. Using an ER marker SEC61-GFP and a mitochondria marker Mito-GFP, we confirmed that acetylation does not change the intracellular distribution of STX17 (Figure S3A and B). When expressed in torin1-treated stable GFP-LC3 cells, both STX17-2KQ and STX17-2KR were normally recruited, like STX17-WT, to the autophagosomal membranes, as indicated by their colocalization with GFP-LC3 puncta (Figure S3C and D).
We, therefore, focused our study on the effect of STX17 acetylation on the maturation of autophagosomes, although STX17 may also be involved in autophagy initiation [41]. We generated an STX17-KO HEK293 cell line using the CRISPR-Cas9 system (Figure 4(A)). Compared with the WT HEK293 cells, we observed in these cells a dramatic increase in the number of LC3 puncta (Figure 4(A)), and elevated cellular LC3-II and SQSTM1 levels (Figure 4(B) and S3E) in the presence or absence of torin1. This result suggested the accumulation of immature autophagosomes and decreased autophagic degradation. Interestingly, the transfection into the STX17-KO cells of STX17-WT or STX17-2KR, but not STX17-2KQ, neutralized the effects of STX17 knockout (Figure 4(B) and S3E). Under torin1 treatment, the colocalization of GFP-LC3 with LAMP1-RFP was significantly decreased in STX17-KO cells, and the expression of STX17-WT or STX17-2KR reversed this change, but not STX17-2KQ (Figure 4(C) and S3F). Under the same conditions, while most of the puncta of the re-introduced HA-STX17-WT or HA-STX17-2KR were LAMP1-RFP-positive, only a small portion of HA-STX17-2KQ colocalized with LAMP1-RFP (Figure S3G). Meanwhile, the ratio of GFP-LC3 puncta positive for HA-STX17-WT or the mutants was very close to that of the WT HEK293 cells (Figure S3H). Furthermore, we monitored the autophagosome maturation using the Cherry-GFP-LC3 reporter. GFP fluorescence is rapidly quenched in the acidic lysosomal environment, whereas Cherry fluorescence remains; therefore, yellow (GFP+ and Cherry+) LC3 puncta represent autophagosomes, while red LC3 puncta (GFP− and Cherry+) indicate autolysosomes. After torin1 treatment, we observed a significant increase in the proportion of yellow puncta in STX17-KO cells compared to WT HEK293 cells, and this increase was abolished by re-expression of the STX17-WT or STX17-2KR, but not STX17-2KQ (Figure 4(D) and S3I). Meanwhile, most yellow LC3 puncta were positive for HA-STX17 (WT, 2KR, and 2KQ) (Figure 4(D) and S3J), but none of the red LC3 puncta were positive for STX17. These data further supported the acetylation-independent recruitment of STX17 to the autophagosomal membranes and confirmed the quick disassociation of STX17 from the membrane of the autolysosomes [42]. Because the lack of STX17 does not cause significant changes in the number and acidification of the lysosomes [7], these results strongly suggested that STX17 acetylation has an inhibitory effect on autophagosome maturation.
Figure 4.
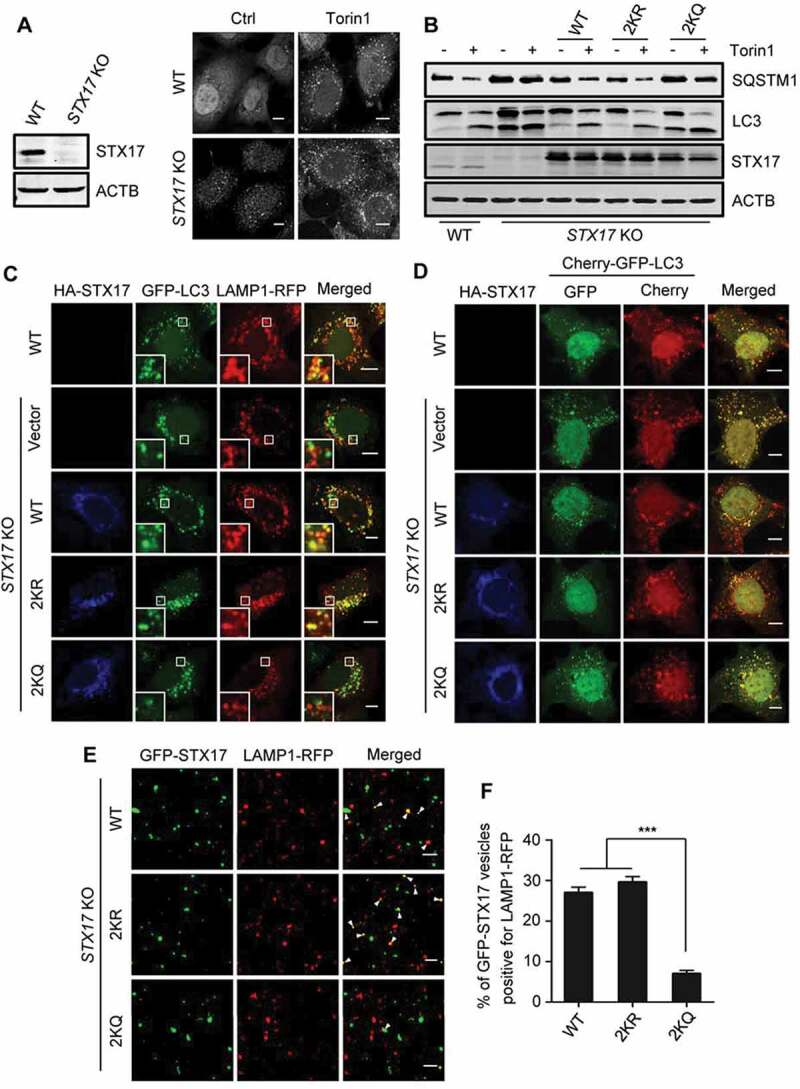
The requirement of STX17 deacetylation in autophagosome-lysosome fusion. (A) Formation of autophagosomes in STX17-KO HEK293 cells treated with or without torin1. The cells were immunostained with anti-LC3. The expression level of STX17 in STX17-KO cells is also shown. (B) SQSTM1 and LC3-II levels in STX17-KO cells. The cells were transfected with HA-STX17 (WT or mutants) with or without torin1 treatment. (C) Colocalization of LAMP1-RFP with GFP-LC3 in torin1-treated WT or STX17-KO HEK293 cells. The cells were transfected with HA-STX17 (WT or mutants). Scale bars: 10 µm. (D) WT or STX17-KO HEK293 cells expressing Cherry-GFP-LC3 were transfected with HA-STX17 (WT or mutants) and treated with torin1 for 12 h. The cells were then imaged by confocal microscopy. Scale bars: 10 µm. (E) Confocal microscopy analysis of the in vitro autophagosome-lysosome fusion assay. Arrowheads indicate co-localized GFP-STX17-positive autophagosomes and LAMP1-RFP-positive lysosomes. Scale bars: 5 µm. (F) Quantification of the data shown in (E). Data are presented as mean ± SEM; n = 30. ***P < 0.001
We then performed in vitro autophagosome-lysosome fusion assays to further verify the effect of STX17 acetylation on autophagosome maturation [9,43,44]. GFP-STX17 and LAMP1-RFP were separately expressed in two groups of STX17-KO HEK293 cells. After treatment with torin1, the post-nuclear supernatants containing GFP-STX17-positive autophagosomes were incubated with the post-nuclear fraction containing LAMP1-RFP-positive lysosomes, in the presence of ATP. We found that the fusion activity of GFP-STX17-2KQ autophagosomes with LAMP1-RFP lysosomes was significantly lower than that of GFP-STX17-WT or GFP-STX17-2KR (Figure 4(E,F). Together, these results suggest that the deacetylation of STX17 at K219 and K223 is required for autophagosome-lysosome fusion.
To show that HDAC2 indeed regulates autophagosome maturation by regulating STX17 acetylation, we knocked down HDAC2 in HEK293 cells and monitored autophagosome maturation using Cherry-GFP-LC3 reporter. After torin1 treatment, compared with control siRNA-treated cells, autophagosome maturation in HDAC2 siRNA-treated cells was significantly inhibited, and the overexpression of STX17-WT or STX17-2KR reversed this inhibition, but not STX17-2KQ (Figure S4A and B). We also tested whether the downregulation of CREBBP enhances autophagic degradation and whether STX17-2KQ can potentially inhibit this effect. The deletion of CREBBP led to the degradation of intracellular SQSTM1 (Figure S4C). Knocking down STX17 in CREBBP-KO cells blocked SQSTM1 degradation, which was reversed by the expression of STX17-WT or STX17-2KR, but not STX17-2KQ (Figure S4C).
Deacetylation of STX17 promotes the formation of the STX17-SNAP29-VAMP8 SNARE complex
To explore the mechanism by which STX17 acetylation inhibits autophagosome maturation, we checked the formation of the STX17-SNAP29-VAMP8 SNARE complex. Immunoprecipitation from HEK293T cells of exogenously expressed HA-STX17-WT resulted in the co-precipitation of the endogenous SNAP29 and VAMP8 (Figure 5(A)). By comparison, more SNAP29 and VAMP8 co-precipitated with HA-STX17-2KR, and much lower levels co-precipitated with HA-STX17-2KQ (Figure 5(A)). In addition, when cell starvation or torin1 treatment augmented the association of SNAP29 and VAMP8 with HA-STX17-WT, they hardly affected the strong binding of SNAP29 and VAMP8 with HA-STX17-2KR, and the feeble binding with HA-STX17-2KQ (Figure 5(A)). We further validated these findings with in vitro pull-down assays using GST-STX17-WT, STX17-2KR, and STX17-2KQ proteins purified from E. coli. Compared with the recombinant GST-STX17-WT and GST-STX17-2KR, which efficiently pulled down SNAP29 and VAMP8, GST-STX17-2KQ pulled down only low levels of SNAP29 and VAMP8 from the cell lysates (Figure 5(B)). These results suggest that the acetylation of STX17 weakens its interaction with SNAP29 and VAMP8.
Figure 5.
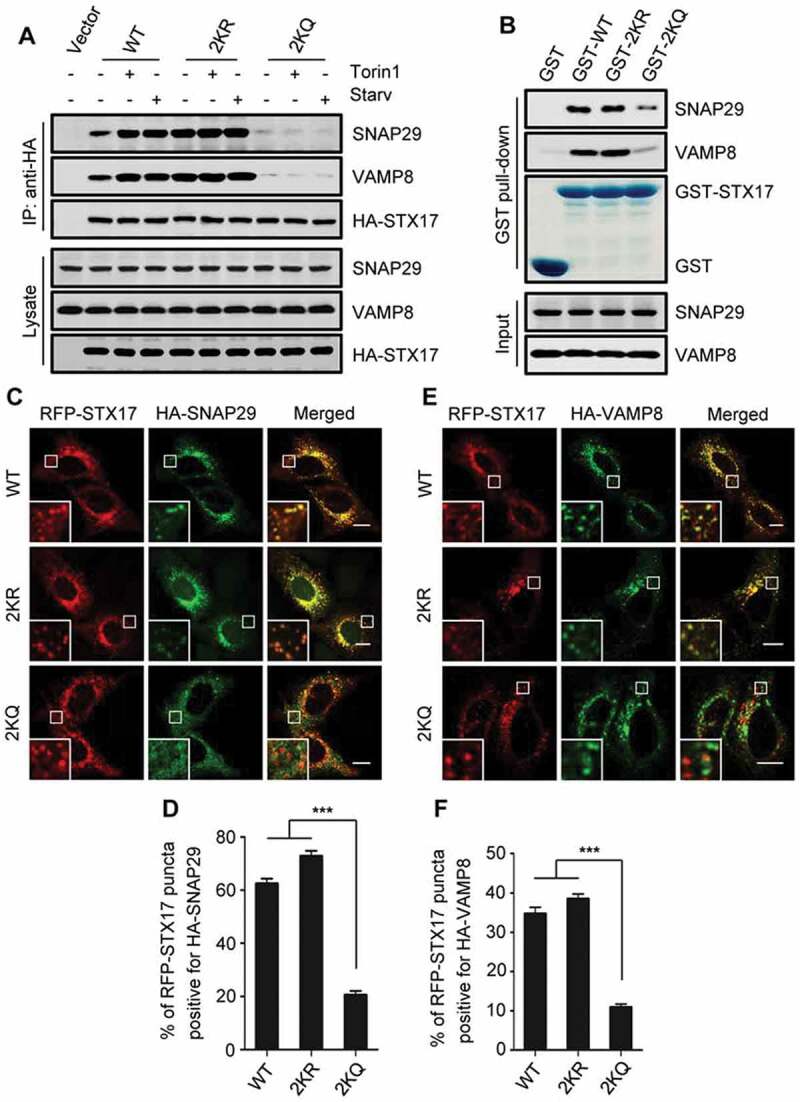
Acetylation of STX17 affects the formation of the STX17-SNAP29-VAMP8 SNARE complex. (A) Co-precipitation of endogenous SNAP29 and VAMP8 with HA-STX17. HA-STX17 (WT or mutants) was immunoprecipitated from HEK293T cells with anti-HA, and the precipitates were analyzed using anti-SNAP29 and anti-VAMP8. (B) Purified recombinant GST-STX17 (WT or mutants) was incubated with HEK293 cell lysates, and the bound SNAP29 and VAMP8 were detected by western blot using anti-SNAP29 and anti-VAMP8, respectively. (C-F) Colocalization of HA-SNAP29 with RFP-STX17 (C and D), and HA-VAMP8 with RFP-STX17 (E and F) in HEK293 cells. The cells were treated with torin1 and imaged by confocal microscopy. Scale bars: 10 µm. The quantitative data are shown as mean ± SEM; n = 30. ***P < 0.001
We then assessed the colocalization of STX17 with SNAP29 and VAMP8 in cells. In torin1-treated cells co-expressing RFP-STX17-WT or RFP-STX17-2KR, HA-SNAP29 formed many punctate structures, and most of them co-localized with RFP-STX17-WT puncta or RFP-STX17-2KR puncta (Figure 5(C,D)). In contrast, in cells expressing RFP-STX17-2KQ, few HA-SNAP29 puncta formed that merge with RFP-STX17-2KQ puncta (Figure 5(C,D)). Accordingly, in torin1-treated cells, the colocalization of HA-VAMP8 puncta with RFP-STX17-2KQ puncta was much lower than their colocalization with RFP-STX17-WT or RFP-STX17-2KR puncta (Figure 5(E,F)).
Together, these results suggest that the deacetylation of STX17 is essential for the membrane recruitment of SNAP29 and the subsequent formation of the STX17-SNAP29-VAMP8 SNARE complex.
Deacetylation of STX17 facilitates the recruitment of the HOPS complex by STX17
In addition to its function as a SNARE protein, autophagosomal STX17 also contributes to the recruitment of the HOPS complex to the autophagosomal membranes, a process which requires its SNARE domain [7,8]. We, therefore, tested whether acetylation of STX17 at K219 and K223 affects its association with HOPS. When HA-STX17-WT was immunoprecipitated from the cells, multiple components of the HOPS complex were also co-precipitated, including endogenous VPS11, VPS16, VPS18, VPS33A, and VPS41 (Figure 6(A)). Also, STX17-Flag-WT was co-precipitated by exogenously expressed VPS39-HA from HEK293 cells (Figure 6(B)). This result confirmed an interaction between STX17 and HOPS. In comparison to STX17-WT, STX17-2KR co-precipitated higher levels of these HOPS components, whereas STX17-2KQ co-precipitated much lower levels (Figure 6(A,B)). In addition, when cell starvation or torin1 treatment increased the association of HA-STX17-WT with the HOPS components, the interactions between these HOPS components and HA-STX17-2KR or HA-STX17-2KQ were not influenced (Figure 6(A,B)). Consistent with these results, the in vitro pull-down assays using purified WT and mutant STX17 proteins demonstrated that less VPS16 and VPS33A were pulled down by GST-STX17-2KQ than GST-STX17-WT or GST-STX17-2KR (Figure 6(C)). This result suggests that acetylation directly influences the affinity of STX17 for HOPS. We then examined the colocalization of HOPS with STX17 in cells. Upon torin1 treatment, the HOPS complex, indicated by two of its components, VPS16 and VPS33A, was located in punctate structures that showed perfect colocalization with RFP-STX17-WT and RFP-STX17-2KR (Figure 6(D-I)). However, in cells expressing RFP-STX17-2KQ, VPS16-HA, and VPS33A-HA were mainly diffused in the cytoplasm, and their localization on RFP-STX17-2KQ puncta was significantly decreased (Figure 6(D-I)).
Figure 6.

Acetylation of STX17 inhibits its interaction with the HOPS complex. (A) Co-precipitation of the endogenous HOPS subunits with STX17. HA-STX17 (WT or mutants) was immunoprecipitated from HEK293T cells with anti-HA, and the precipitates were analyzed using antibodies against VPS11, VPS16, VPS18, VPS33A, and VPS41. (B) Co-precipitation of the exogenously expressed HOPS subunit VPS39 with STX17. VPS39-HA was immunoprecipitated from transfected HEK293T cells expressing STX17-Flag (WT or mutants), and the precipitates were then analyzed using anti-Flag. (C) Purified recombinant GST-STX17 (WT or mutants) was incubated with HEK293 cell lysates, and the bound VPS16 and VPS33A were detected by western blot using anti-VPS16 and anti-VPS33A, respectively. (D) Colocalization of RFP-STX17 (WT or mutants) with VPS16-HA in torin1-treated HeLa cells. (E) Colocalization of RFP-STX17 (WT or mutants) with VPS33A-HA in torin1-treated HeLa cells. (F and G) Quantification of the number of VPS16-HA- or VPS33A-HA-positive puncta in (D) or (E). (H and I) Quantification of the ratio of RFP-STX17 puncta positive for VPS16-HA or VPS33A-HA. Scale bars: 10 µm. The quantifications are shown as mean ± SEM; n = 30. ***P < 0.001
Together, these results suggest that the deacetylation of STX17 at K219 and K223, within the SNARE domain, is required for the HOPS complex recruitment.
Discussion
Here, we provide evidence for a previously unrecognized acetylation/deacetylation mechanism that regulates STX17 activity and autophagosome maturation. Our data suggest that the deacetylation of STX17 is required for STX17 to assemble the STX17-SNAP29-VAMP8 SNARE complex and to recruit the HOPS complex, both essential for autophagosome-lysosome fusion.
Our study suggests that acetylation/deacetylation is involved in connecting autophagic signals both to the early and the late steps of the autophagy process. By the coordinate facilitation of the different stages of autophagy, these acetylation/deacetylation events enable cells to achieve autophagic degradation and maintain autophagy flux to cope with the poor conditions rapidly and efficiently. Recently, it has been reported that STX17 can be phosphorylated by the protein kinase TBK1 [41]. During autophagy induction, phosphorylated STX17 is transferred from the Golgi apparatus to the pre-autophagosome structure to assemble the ULK1 complex for autophagosome formation [41]. Here, we demonstrated that the acetylation of STX17 significantly inhibits autophagosome maturation without affecting its autophagosomal localization. Our data suggest that the deacetylation of STX17 is an indispensable step for its function in autophagosome-lysosome fusion after it is recruited to autophagosomal membranes. The modification of residues facing the inside of the alpha-helical bundle may result in the incomplete zippering of the SNARE complexes and reduce SNARE-mediated membrane fusion by steric hindrance [45]. However, K219 and K223 of STX17 are facing the outside of the alpha-helical bundle of the STX17-SNAP29-VAMP8 complex [12]. We propose that STX17 acetylation originally affects the interaction between STX17 and SNAP29, and subsequently, the membrane recruitment of SNAP29 and the SNARE complex formation. This proposal is supported by previous studies showing that the phosphorylation of SNAP25 at S187 site, which is also facing the outside of the alpha-helical bundle of the STX1A-SNAP25-VAMP2/Synaptobrevin 2 SNARE complex, increases the binding of SNAP25 to STX1A and promotes the SNARE complex formation [46,47]. Possibly, acetylation at K219 and K223 disturbs the ionic interaction between STX17 and SNAP29 by neutralizing the positive charge of the lysine residues. Recently, it has been shown that multi-subunit tethering complexes, such as HOPS, promote membrane fusion not only through mediating membrane contact and stabilizing trans-SNARE complex but also by promoting fusion pore formation during the terminal stage of membrane fusion after the SNARE complex has formed [48]. Although it is currently unclear whether the same mechanism is employed in autophagosome-lysosome fusion, our results suggest that the acetylation of STX17 affects its interaction with both SNAP29 and HOPS, and the effect on the STX17-SNAP29 interaction seems to be independent of the binding of STX17 and HOPS. Considering that the SNARE domain of STX17 is required for its binding to SNAP29 and HOPS [8], we propose that there is a sequential interaction between STX17 and HOPS, and STX17 and SNAP29 during the fusion of autophagosomes and lysosomes.
The acetyltransferases CREBBP and EP300 are functional paralogues that acetylate histones, as well as non-histone proteins, and play roles in the regulation of an array of cellular processes. Accumulating pieces of evidence have indicated the functional differences between CREBBP and EP300 due to their differential associations with other proteins and substrate selectivity [49,50]. Recently, we have shown that MTORC1 can phosphorylate and activate EP300 and CREBBP [27]. During cell starvation- and torin1 treatment-induced autophagy, both EP300 and CREBBP are inactivated due to MTORC1 inhibition. Interestingly, compared to EP300, which acetylates LC3, the inactivation of CREBBP exerts relatively little effect on LC3 acetylation and autophagosome formation [27]. In this study, we have demonstrated that STX17 is a selective substrate of CREBBP but not EP300, and CREBBP-mediated STX17 acetylation plays an important role in the regulation of autophagosome maturation. Our data, therefore, suggest that EP300 and CREBBP, controlled by MTORC1, coordinately regulate the autophagy process by spatiotemporally targeting the early and late stages of autophagy in response to cell metabolic stress.
HDAC2 promotes autophagy flux in the skeletal muscle by mediating the induction of autophagic gene expression and autophagosome formation [51]. However, the inhibitory effect of HDAC2 on autophagy through deacetylation of the central transcription factor TFEB was reported recently [52]. The function of HDAC2 in autophagy regulation remains rather controversial. Here, we show that STX17 is a selective substrate of HDAC2, but not its close homolog HDAC1, by demonstrating that STX17 strongly associates with HDAC2 and is directly deacetylated by it. Nevertheless, it is notable that the association of STX17 with HDAC2 and the deacetylation activity of HDAC2 remain unchanged under cell starvation and torin1 treatment. HDAC2 likely has different actions in different cell types and responses to different stress cues.
STX17 is a well-conserved SNARE protein in different species and is involved in multiple intracellular membrane fusion processes, such as the fusion of mitochondrial-derived vesicles to late endosomes/lysosomes [53], and the fusion of ER-derived vesicles with lysosomes [54]. Therefore, we speculate that the acetylation of STX17 at K219 and K223 within the SNARE domain may play a role in the regulation of these events.
Materials and methods
Antibodies
Antibodies were from the following companies, anti-EP300 (sc-585, 1:1000), anti-SNAP29 (sc-390602, 1:1000), anti-VAMP8 (sc-166820, 1:1000), anti-HDAC1 (sc-81598, 1:1000), anti-VPS11 (sc-515094, 1:1000), and anti-VPS41 (sc-377046, 1:1000) for western blot were from Santa Cruz Biotechnology; anti-STX17 (HPA001204, 1:300), anti-LC3 (L7543, 1:1000), and anti-ACTB (A5316, 1:5000) were from Sigma-Aldrich; anti-CREBBP (7389, 1:1000), anti-histone H3 (9715, 1:1000), anti-acetyl-histone H3 (Lys56) (4243, 1:1000), and anti-acetyl-lysine (9441, 1:1000) were from Cell Signaling Technology; anti-HDAC2 (12922-3-AP, 1:1000), anti-VPS16 (17776-1-AP, 1:1000), anti-VPS18 (10901-1-AP, 1:1000), anti-VPS33A (16896-1-AP, 1:1000), and anti-SQSTM1 (18420-1-AP, 1:1000) were from Proteintech; anti-HA (M180-3, 1:5000) and anti-Flag (PM020, 1:5000) were from MBL; and anti-LC3 (CAC-CTB-LC3-2-IC, 1:100) for immunostaining was from Cosmo Bio. The secondary donkey anti-mouse IRDye680 (926–32222) and anti-rabbit IRDye800CW (926–32213) antibodies for western blot were from LI-COR Biosciences. Glutathione-sepharose 4B beads were from GE Healthcare Life Sciences. Protein G agarose (sc-2002) and protein A agarose (sc-2001) were from Santa Cruz Biotechnology. Anti-HA affinity beads (B23302) and anti-Flag affinity beads (B23101) were from Biotool.
Reagents and treatment
The chemicals were used as follows unless indicated otherwise: torin1 (Selleck, S2827; 500 nM) was added to the medium for 3 h. Trichostatin A (Selleck, S1054; 400 nM) were added to the culture medium for 16 h. Nicotinamide (Sigma-Aldrich, 72340; 5 mM) was added to the medium for 8 h. CTB (Sigma-Aldrich, C6499; 50 μM) was added to the medium for 6 h. C646 (Selleck, S7152; 10 μM) were added to the medium for 4 h. Starvation medium (1% BSA [Sigma-Aldrich, A1933], 140 mM NaCl [Sangon Biotech, A610476], 1 mM CaCl2 [Sangon Biotech, A501330], 1 mM MgCl2 [Sangon Biotech, B601193], 5 mM glucose [Sangon Biotech, A600219], 20 mM HEPES [Beyotime, ST092], pH 7.4) was added for 2 h.
Plasmid constructs
HA-STX17, STX17-Flag, GFP-STX17, and RFP-STX17 plasmid were made by cloning human STX17 cDNA into a PXF4H, pcDNA5-FRT-TO-3×FLAG (Invitrogen, V6010-20), pEGFP-C1 (Clontech, PT3028-5), and pDsRed2-C1 (Clontech, PT3603-5) vector separately. PXF4H vector was a gift from Xinhua Feng (Life Sciences Institute Zhejiang University, Hangzhou, China). GST-STX17 plasmid was made by cloning human STX17 DNA fragments into a pGEX-5X-1 vector (GE Healthcare, 27-4584-01). Site-directed mutagenesis was performed using QuikChange II XL (Stratagene, 210522) according to the manufacturer’s instructions. pEP-STX17-KO and pEP-CREBBP-KO were made by cloning the target DNA sequence of human STX17 (CATGAAGAGCATATCAATGC) and human CREBBP (TCGACAATGCGGGAGCGAGC) into a pEP-KO Z1779 vector, respectively. pEP-KO Z1779 vector was provided by Zongping Xia (The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China). CREBBP-Flag was provided by Qunying Lei (Fudan University School of Basic Medical Sciences, Shanghai, China). HA-SNAP29, HA-VAMP8, VPS16-HA, VPS33A-HA, VPS39-HA were gifts from Qiming Sun (Zhejiang University School of Medicine, Hangzhou, China).
Cell culture and transfection
HEK293 (ATCC, CRL-1573), HEK293T (ATCC, CRL-3216), HeLa cells (ATCC, CCL-2), and MEFs were cultured in Dulbecco Modified Eagle Medium (Gibco, 11965092) supplemented with 10% fetal bovine serum (Gibco, 10091148) in a 37°C incubator with a humidified, 5% CO2 atmosphere. WT, tsc1−/- and tsc2−/- MEFs were gifts from Han-Ming Shen (National University of Singapore Yong Loo Lin School of Medicine, Singapore). HEK293 cells stably expressing GFP-LC3 were generated as described previously [21]. HEK293T cell lines stably expressing HA-STX17 were generated by transient transfection and selected with G418 (Sigma-Aldrich, A1720). STX17-KO and CREBBP-KO HEK293 cells were created by transient transfection of pEP-STX17-KO and pEP-CREBBP-KO plasmid, respectively, followed by selection with puromycin (Selleck, S7417). Lipofectamine 2000 (Invitrogen, 11668019) was used for plasmid transient transfection according to the manufacturer’s instructions. Cells were analyzed 16–24 h after transfection.
For RNA interference, siRNA duplexes were transfected for 48 h, as specified by the manufacturer. The following siRNA duplexes (GenePharma) were used: EP300 siRNA: CUAGAGACACCUUGUAGUATT; CREBBP siRNA: AAUCCACAGUACCGAGAAAUGTT; HDAC1 siRNA: CGUUCUUAACUUUGAACCAUA; HDAC2 siRNA: CAGUCUCACCAAUUUCAGAAA; non-targeting siRNA: UUCUCCGAACGUGUCACGUTT.
Confocal microscopy
Cells were fixed in 4% formaldehyde [Sangon Biotech, A501912] for 10 min at room temperature. After washing twice with PBS [Sangon Biotech, E607008], cells were incubated in PBS containing 10% fetal calf serum (Sigma-Aldrich, F0685) to block nonspecific sites of antibody adsorption. The cells were then incubated with the appropriate primary antibodies and secondary antibodies in PBS containing 0.1% saponin (Sigma-Aldrich, S7900) and 10% fetal calf serum as indicated. Confocal images were captured in multitracking mode on a Meta laser-scanning confocal microscope 880 (Carl Zeiss) with a 63× Plan Apochromat 1.4 NA objective (Carl Zeiss) and analyzed with the LSM 880 software (Carl Zeiss).
Western blot and immunoprecipitation
For western blot, cells were harvested and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl [Sangon Biotech, A610476], 1% TritonX-100 [Sangon Biotech, A600198], 1% sodium deoxycholate [Sangon Biotech, A600150], 0.1% SDS [Sangon Biotech, A100227], 1 mM EDTA [Sangon Biotech, B540625]) supplemented with complete protease inhibitor cocktail (Roche, 4693159001). Proteins were resolved on SDS polyacrylamide gels and then transferred to a polyvinylidene difluoride membrane (Millipore, ISEQ00010). After blocking with 3% (w/v) bovine serum albumin, the membrane was stained with the corresponding primary antibodies. After incubation with the corresponding secondary antibodies, specific bands were analyzed using an Odyssey infrared imaging system (LI-COR Biosciences).
For immunoprecipitation, cells were lysed in NP-40 lysis buffer (50 mM Tris-HCl, 1% NP-40 [Sangon Biotech, A100109], 150 mM NaCl [Sangon Biotech, A610476], 1 mM MgCl2 [Sangon Biotech, B601193], 10% glycerol [Sangon Biotech, A600232], 1 mM EGTA [Sangon Biotech, A600077], pH 7.4) supplemented with complete protease inhibitor cocktail (Roche, 4693159001). Then the cell lysate was mixed with antibodies at 4°C overnight followed by the addition of protein A (Santa Cruz Biotechnology, sc-2001) or G sepharose beads (Santa Cruz Biotechnology, sc-2002), incubation was continued for 2 h, and then immunocomplexes were washed four times using lysis buffer, resolved by SDS-PAGE, and analyzed by western blot.
Protein purification
GST-STX17 (WT, 2KR and 2KQ), GST-HDAC1 and GST-HDAC2 were expressed in Escherichia coli BL21 (Tsingke, TSV-A09). Bacteria by induction with 0.1 mM IPTG (Sangon Biotech, A600168-0025) for 16 h at 18°C to induce protein expression. The recombinant proteins were purified using glutathione-sepharose 4B beads (GE Healthcare Life Sciences,17-0756-01), eluted with glutathione (Beyotime, S0073) at 4°C for 4 h to release the proteins.
In vitro pull-down assay
For pull-down assays, purified GST-STX17 (WT, 2KR, and 2KQ), GST-HDAC1 and GST-HDAC2 proteins were incubated with whole-cell extract overnight at 4°C, GST agarose beads (GE Healthcare Life Sciences,17-0756-01) were then added to the mixture, followed by further incubation for 2 h at 4°C. Immunocomplexes were washed and used for western blot.
In vitro acetylation assay
GST-STX17-WT protein (10 μg) was incubated with CREBBP-Flag or CREBBP-mut-Flag immunoprecipitated from cell lysates, in the presence of acetyl-coenzyme A (4 μg; Sigma-Aldrich, 10101893001) and HAT assay buffer (250 mM Tris-HCl, pH 8.0, 50% glycerol [Sangon Biotech, A600232], 0.5 mM EDTA [Sangon Biotech, B540625], 5 mM dithiothreitol [Sangon Biotech, A620058]) in a total volume of 50 μl. The samples were then incubated at 37°C by gently shaking for 4 h. Then the reaction was stopped by the addition of SDS sample buffer, and the mixture was boiled for 5 min. The reaction products were separated by SDS-PAGE and immunoblotted with anti-acetyl-lysine.
Fluorometric HDAC2 activity assay
HDAC2 activity was measured using an HDAC Activity Fluorometric Assay Kit (BioVision Technologies, K330-100) with the HeLa nuclear extract provided by the kit as the positive control. Endogenous HDAC2 was immunoprecipitated from HEK293 cells treated as indicated. The immunoprecipitated proteins were incubated with fluorogenic HDAC substrate Boc-Lys (Ac)-AMC and HDAC assay buffer in a total volume of 100 μl at 37°C for 2 h by gently shaking according to the manufacturer’s instructions. The reaction was stopped by adding Lysine Developer with further incubation at 37°C for 30 min. The endogenous activity of HDAC2 was assessed by measuring the fluorescent emission at 450 nm following excitation at 360 nm.
HPLC-MS/MS
HEK293T cells were transfected with HA-STX17 and then treated with TSA. Immunoprecipitation was performed 24 h after transfection with anti-HA affinity beads (Biotool, B23302). The agarose beads were washed using Tris-HCl (100 mM, pH 8.5), and then dissolved with urea (Sangon Biotech, A610148; 8 M)/dithiothreitol (Sangon Biotech, A620058; 10 mM) buffer. The mixture was sonicated for 30 min at room temperature, and then treated with IAA (Sangon Biotech, A600723; 10 mM) sequentially and trypsin (Sangon Biotech, A003736) to alkylate the resulting thiol group and digest the proteins overnight at 37°C. The digested peptides were desalted and loaded on a capillary reverse-phase C18 column packed in-house (15 cm in length, 100 µm ID × 360 µm OD, 3 µm particle size, 100 Å pore diameter) connected to an Easy LC 1000 system. The samples were analyzed with a 180 min HPLC gradient from 0% to 100% buffer B (buffer A: 0.1% formic acid in water; buffer B: 0.1% formic acid in acetonitrile) at 300 nL/min. The eluted peptides were ionized and directly introduced into a Q-Exactive or Fusion mass spectrometer (Thermo Fisher Scientific) using a nano-spray source. Survey full-scan MS spectra (m/z 300–1800) were acquired in the Orbitrap analyzer with resolution r = 70,000 at m/z 400.
In vitro autophagosome-lysosome fusion assay
GFP-STX17 (WT, 2KR, and 2KQ) and LAMP1-RFP were overexpressed separately in two groups of STX17-KO HEK293 cells with torin1 treatment. Cells are cracked 24 h after transfection using 1 ml disposable syringes in homogenization buffer (250 mM sucrose [Sangon Biotech, A502792], 20 mM HEPES-KOH, pH 7.4) followed by centrifugation at 1,200 g for 10 min and postnuclear supernatant (PNS) are prepared. The PNS from GFP-STX17 expressing cells containing GFP-STX17-positive autophagosomes was mixed with the PNS from LAMP1-RFP expressing cells containing LAMP1-RFP-positive lysosomes in the presence of the ATP-regeneration system (10 mM creatine phosphate [Sangon Biotech, A610821], 1 mM ATP [Selleck, S1985], 20 μg/ml creatine kinase [BioVision Technologies, P1301-1]). The samples were then incubated at 37°C by gently shaking for 2 h. After incubation, all the samples were centrifuged by centrifugation at 100,000 g for 15 min and immobilized on glass coverslips for confocal microscopy analysis.
Statistical analysis
Statistical analyses were conducted with two-tailed Student’s t-test, *P < 0.05 was considered to be statistically significant. All the statistical data are presented as mean ± SEM.
Supplementary Material
Acknowledgments
We are grateful to the Imaging Center of Zhejiang University School of Medicine for their assistance in confocal microscopy. We thank Qunying Lei for sharing CREBBP-Flag plasmid, Zongping Xia for sharing pEP-KO Z1779 vector and Han-Ming Shen for sharing tsc1−/- and tsc2−/- MEFs.
Funding Statement
This study was supported by the National Basic Research Program of China [2017YFA0503402] and the National Natural Science Foundation of China [31790402, 31530040, and 31671434].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007. November;8(11):931–937. PubMed PMID: 17712358. [DOI] [PubMed] [Google Scholar]
- [2].Mizushima N. Autophagy: process and function. Genes Dev. 2007. November 15;21(22):2861–2873. PubMed PMID: 18006683. [DOI] [PubMed] [Google Scholar]
- [3].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014. January;24(1):24–41. PubMed PMID: 24366339; PubMed Central PMCID: PMC3879710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lamb CA, Dooley HC, Tooze SA. Endocytosis and autophagy: shared machinery for degradation. Bioessays. 2013. January;35(1):34–45. PubMed PMID: 23147242. [DOI] [PubMed] [Google Scholar]
- [5].Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008. September;9(9):859–864. PubMed PMID: 18704115; PubMed Central PMCID: PMC2529362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. PubMed PMID: 21801009. . [DOI] [PubMed] [Google Scholar]
- [7].Jiang P, Nishimura T, Sakamaki Y, et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell. 2014. April;25(8):1327–1337. PubMed PMID: 24554770; PubMed Central PMCID: PMC3982997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Takáts S, Pircs K, Nagy P, et al. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol Biol Cell. 2014. April;25(8):1338–1354. PubMed PMID: 24554766; PubMed Central PMCID: PMC3982998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang X, Wang L, Lak B, et al. GRASP55 senses glucose deprivation through O-GlcNAcylation to promote autophagosome-lysosome fusion. Dev Cell. 2018. April 23;45(2):245–261. PubMed PMID: 29689198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012. December 7;151(6):1256–1269. PubMed PMID: 23217709. [DOI] [PubMed] [Google Scholar]
- [11].Takáts S, Nagy P, Varga Á, et al. Autophagosomal Syntaxin 17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol. 2013. May 13;201(4):531–539. PubMed PMID: 23671310; PubMed Central PMCID: PMC3653357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015. April 23;520(7548):563–566. PubMed PMID: 25686604; PubMed Central PMCID: PMC4442024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cheng X, Ma X, Ding X, et al. Pacer mediates the function of class III PI3K and HOPS complexes in autophagosome maturation by engaging Stx17. Mol Cell. 2017. March 16;65(6):1029–1043. PubMed PMID: 28306502. [DOI] [PubMed] [Google Scholar]
- [14].Ding X, Jiang X, Tian R, et al. RAB2 regulates the formation of autophagosome and autolysosome in mammalian cells. Autophagy. 2019. October;15(10):1774–1786. PubMed PMID: 30957628; PubMed Central PMCID: PMC6735470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fujita N, Huang W, Lin TH, et al. Genetic screen in Drosophila muscle identifies autophagy-mediated T-tubule remodeling and a Rab2 role in autophagy. Elife. 2017. January 7;6:pii: e23367. PubMed PMID: 28063257; PubMed Central PMCID: PMC5249261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lőrincz P, Tóth S, Benkő P, et al. Rab2 promotes autophagic and endocytic lysosomal degradation. J Cell Biol. 2017. July 3;216(7):1937–1947. PubMed PMID: 28483915; PubMed Central PMCID: PMC5496615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009. August 14;325(5942):834–840. PubMed PMID: 19608861. [DOI] [PubMed] [Google Scholar]
- [18].Zhao S, Xu W, Jiang W, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010. February 19;327(5968):1000–1004. PubMed PMID: 20167786; PubMed Central PMCID: PMC3232675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. PubMed PMID: 19653858; PubMed Central PMCID: PMC2831538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008. January 11;132(1):27–42. PubMed PMID: 18191218; PubMed Central PMCID: PMC2696814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Huang R, Xu Y, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015. February 5;57(3):456–466. PubMed PMID: 25601754. [DOI] [PubMed] [Google Scholar]
- [22].Lee IH, Cao L, Mostoslavsky R, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008. March 4;105(9):3374–3379. PubMed PMID: 18296641; PubMed Central PMCID: PMC2265142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. J Biol Chem. 2009. March 6;284(10):6322–6328. PubMed PMID: 19124466; PubMed Central PMCID: PMC5405322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Su H, Yang F, Wang Q, et al. VPS34 acetylation controls its lipid kinase activity and the initiation of canonical and non-canonical autophagy. Mol Cell. 2017. September 21;67(6):907–921. PubMed PMID: 28844862. [DOI] [PubMed] [Google Scholar]
- [25].Chang C, Su H, Zhang D, et al. AMPK-dependent phosphorylation of GAPDH triggers Sirt1 activation and is necessary for autophagy upon glucose starvation. Mol Cell. 2015. December 17;60(6):930–940. PubMed PMID: 26626483. [DOI] [PubMed] [Google Scholar]
- [26].Park S, Stanfield RL, Martinez-Yamout MA, et al. Role of the CBP catalytic core in intramolecular SUMOylation and control of histone H3 acetylation. Proc Natl Acad Sci U S A. 2017. July 3;114(27):E5335–E5342. PubMed PMID: 28630323; PubMed Central PMCID: PMC5502626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wan W, You Z, Xu Y, et al. mTORC1 phosphorylates acetyltransferase EP300 to regulate autophagy and lipogenesis. Mol Cell. 2017. October 19;68(2):323–335. PubMed PMID: 29033323. [DOI] [PubMed] [Google Scholar]
- [28].Mariño G, Pietrocola F, Eisenberg T, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014. March 6;53(5):710–725. PubMed PMID: 24560926. [DOI] [PubMed] [Google Scholar]
- [29].Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004. December 3;279(49):50754–50763. PubMed PMID: 15381699. [DOI] [PubMed] [Google Scholar]
- [30].Lee JY, Koga H, Kawaguchi Y, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010. March 3;29(5):969–980. PubMed PMID: 20075865; PubMed Central PMCID: PMC2837169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang R, Tan J, Chen T, et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J Cell Biol. 2019. January 7;218(1):267–284. PubMed PMID: 30538141; PubMed Central PMCID: PMC6314552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sun T, Li X, Zhang P, et al. Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat Commun. 2015. May 26;6:7215. PubMed PMID: 26008601; PubMed Central PMCID: PMC4455096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cheng X, Ma X, Zhu Q, et al. Pacer is a mediator of mTORC1 and GSK3-TIP60 signaling in regulation of autophagosome maturation and lipid metabolism. Mol Cell. 2019. February 21;73(4):788–802. PubMed PMID: 30704899. [DOI] [PubMed] [Google Scholar]
- [34].Bordoli L, Hüsser S, Lüthi U, et al. Functional analysis of the p300 acetyltransferase domain: the PHD finger of p300 but not of CBP is dispensable for enzymatic activity. Nucleic Acids Res. 2001. November 1;29(21):4462–4471. PubMed PMID: 11691934; PubMed Central PMCID: PMC60180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lin CC, Kitagawa M, Tang X, et al. CoA synthase regulates mitotic fidelity via CBP-mediated acetylation. Nat Commun. 2018. March 12;9(1):1039. PubMed PMID: 29531224; PubMed Central PMCID: PMC5847545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ma L, Gao JS, Guan Y, et al. Acetylation modulates prolactin receptor dimerization. Proc Natl Acad Sci U S A. 2010. November 9;107(45):19314–19319. PubMed PMID: 20962278; PubMed Central PMCID: PMC2984224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bánréti A, Sass M, Graba Y. The emerging role of acetylation in the regulation of autophagy. Autophagy. 2013. June 1;9(6):819–829. PubMed PMID: 23466676; PubMed Central PMCID: PMC3672293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Oehme I, Linke JP, Böck BC, et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci U S A. 2013. July 9;110(28):E2592–E2601. PubMed PMID: 23801752; PubMed Central PMCID: PMC3710791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tsai SC, Seto E. Regulation of histone deacetylase 2 by protein kinase CK2. J Biol Chem. 2002. August 30;277(35):31826–31833. PubMed PMID: 12082111. [DOI] [PubMed] [Google Scholar]
- [40].Adenuga D, Rahman I. Protein kinase CK2-mediated phosphorylation of HDAC2 regulates co-repressor formation, deacetylase activity and acetylation of HDAC2 by cigarette smoke and aldehydes. Arch Biochem Biophys. 2010. June 1;498(1):62–73. PubMed PMID: 20388487; PubMed Central PMCID: PMC2874641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kumar S, Gu Y, Abudu YP, et al. Phosphorylation of syntaxin 17 by TBK1 controls autophagy initiation. Dev Cell. 2019. April 8;49(1):130–144. PubMed PMID: 30827897; PubMed Central PMCID: PMC6907693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tsuboyama K, Koyama-Honda I, Sakamaki Y, et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016. November 25;354(6315):1036–1041. PubMed PMID: 27885029. [DOI] [PubMed] [Google Scholar]
- [43].Barysch SV, Jahn R, Rizzoli SO. A fluorescence-based in vitro assay for investigating early endosome dynamics. Nat Protoc. 2010. June;5(6):1127–1137. PubMed PMID: 20539288. [DOI] [PubMed] [Google Scholar]
- [44].Matsui T, Jiang P, Nakano S, et al. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J Cell Biol. 2018. August 6;217(8):2633–2645. PubMed PMID: 29789439; PubMed Central PMCID: PMC6080929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Malmersjö S, Di Palma S, Diao J, et al. Phosphorylation of residues inside the SNARE complex suppresses secretory vesicle fusion. EMBO J. 2016. August 15;35(16):1810–1821. PubMed PMID: 27402227; PubMed Central PMCID: PMC5010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xu NJ, Yu YX, Zhu JM, et al. Inhibition of SNAP-25 phosphorylation at Ser187 is involved in chronic morphine-induced down-regulation of SNARE complex formation. J Biol Chem. 2004. September 24;279(39):40601–40608. PubMed PMID: 15277518. [DOI] [PubMed] [Google Scholar]
- [47].Yang Y, Craig TJ, Chen X, et al. Phosphomimetic mutation of Ser-187 of SNAP-25 increases both syntaxin binding and highly Ca2+-sensitive exocytosis. J Gen Physiol. 2007. March;129(3):233–244. PubMed PMID: 17325194; PubMed Central PMCID: PMC2151612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].D’Agostino M, Risselada HJ, Lürick A, et al. A tethering complex drives the terminal stage of SNARE-dependent membrane fusion. Nature. 2017. November 30;551(7682):634–638. PubMed PMID: 29088698. [DOI] [PubMed] [Google Scholar]
- [49].Henry RA, Kuo YM, Andrews AJ. Differences in specificity and selectivity between CBP and p300 acetylation of histone H3 and H3/H4. Biochemistry. 2013. August 27;52(34):5746–5759. PubMed PMID: 23862699; PubMed Central PMCID: PMC3756530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kalkhoven E. CBP and p300: hATs for different occasions. Biochem Pharmacol. 2004. September 15;68(6):1145–1155. PubMed PMID: 15313412. [DOI] [PubMed] [Google Scholar]
- [51].Moresi V, Carrer M, Grueter CE, et al. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc Natl Acad Sci U S A. 2012. January 31;109(5):1649–1654. PubMed PMID: 22307625; PubMed Central PMCID: PMC3277131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang J, Wang J, Zhou Z, et al. Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors. Autophagy. 2018;14(6):1043–1059. PubMed PMID: 30059277; PubMed Central PMCID: PMC6103407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].McLelland GL, Lee SA, McBride HM, et al. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol. 2016. August 1;214(3):275–291. PubMed PMID: 27458136; PubMed Central PMCID: PMC4970327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fregno I, Fasana E, Bergmann TJ, et al. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 2018. September 3;37(17):pii: e99259. PubMed PMID: 30076131; PubMed Central PMCID: PMC6120659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.