

ABSTRACT
Mitophagy is a conserved intracellular catabolic process responsible for the selective removal of dysfunctional or superfluous mitochondria to maintain mitochondrial quality and need in cells. Here, we examine the mechanisms of receptor-mediated mitophagy activation, with the focus on BNIP3L/NIX mitophagy receptor, proven to be indispensable for selective removal of mitochondria during the terminal differentiation of reticulocytes. The molecular mechanisms of selecting damaged mitochondria from healthy ones are still very obscure. We investigated BNIP3L dimerization as a potentially novel molecular mechanism underlying BNIP3L-dependent mitophagy. Forming stable homodimers, BNIP3L recruits autophagosomes more robustly than its monomeric form. Amino acid substitutions of key transmembrane residues of BNIP3L, BNIP3LG204A or BNIP3LG208V, led to the abolishment of dimer formation, resulting in the lower LC3A-BNIP3L recognition and subsequently lower mitophagy induction. Moreover, we identified the serine 212 as the main amino acid residue at the C-terminal of BNIP3L, which extends to the intermembrane space, responsible for dimerization. In accordance, the phosphomimetic mutation BNIP3LS212E leads to a complete loss of BNIP3L dimerization. Thus, the interplay between BNIP3L phosphorylation and dimerization indicates that the combined mechanism of LIR phosphorylation and receptor dimerization is needed for proper BNIP3L-dependent mitophagy initiation and progression.
Abbreviations: AMBRA1: autophagy and beclin 1 regulator 1; Baf A1: bafilomycin A1; BH3: BCL2 homology 3; BNIP3: BCL2 interacting protein 3; BNIP3L/NIX: BCL2 interacting protein 3 like; CCCP: carbonyl cyanide 3-chlorophenylhydrazone; CoCl2: cobalt (II) chloride; FKBP8: FKBP prolyl isomerase 8; FUNDC1: FUN14 domain containing 1; GABARAP: GABA type A receptor-associated protein; GST: glutathione S-transferase; IMM: inner mitochondrial membrane; LIR: LC3-interacting region; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; OMM: outer mitochondrial membrane; PHB2: prohibitin 2; PI: propidium iodide; PINK1: PTEN induced kinase 1; TM: transmembrane domain; TOMM20: translocase of outer mitochondrial membrane 20
KEYWORDS: Autophagy, dimerization, mitophagy, BNIP3L/NIX, selective autophagy
Introduction
Eukaryotic cells developed two intracellular degradation mechanisms for removing unnecessary and harmful parts to maintain homeostasis: the ubiquitin-proteasome system (UPS) and the lysosome-mediated degradation pathway (autophagy). In contrast to UPS, autophagy is not restricted to protein degradation. The majority of cellular macromolecules and their complexes, even whole organelles and intracellular pathogens, are degraded and recycled by autophagy. Numerous studies have revealed a set of specific selective autophagy receptors that interact simultaneously with the cytoplasmic cargo and components of the autophagy machinery, thereby physically linking the cargo with autophagosomes [1–5]. However, the exact molecular mechanisms of selective cargo recognition are still under intense investigation (reviewed in [6]).
Here, we focus on the selective elimination of mitochondria by mitophagy, an evolutionarily conserved process essential for cellular homeostasis maintenance, metabolism, and physiology. Defective mitophagy is a characteristic phenomenon of a broad spectrum of pathologies, including cardiovascular disorders, cancer, and different neurodegenerative diseases: Parkinson, Alzheimer, and Huntington disease or amyotrophic lateral sclerosis [7,8]. Even though mitophagy primarily eliminates damaged or dysfunctional mitochondria [9], this pathway is also responsible for the degradation of normal, healthy mitochondria during the development of particular cell types by so-called programmed mitophagy [10]. Developmentally regulated mitochondrial elimination is observed during the development of the eye lens cells, sperm cells maturation [11,12], and sperm-derived mitochondria elimination during early embryogenesis [13]. Currently, the best described programmed mitophagy is the removal of mitochondria during terminal mammalian erythropoiesis [14,15], where nascent reticulocytes (erythrocytes precursors) are obliged to remove the entire mitochondrial population to become functional erythrocytes. Schweers and Sandoval independently found that the outer mitochondrial membrane protein BNIP3L/NIX is indispensable for the programmed mitochondrial elimination during terminal reticulocytes differentiation [14,15]. Furthermore, Novak et al. described BNIP3L as a selective autophagy receptor that binds to Atg8 homologs, LC3/GABARAP proteins, through a conserved LC3-interacting region (LIR) motif at the amino terminus of the BNIP3L [5,16]. Apart from BNIP3L, there are additional mitophagy receptors directing dysfunctional mitochondria to the autophagosomes, such as BNIP3 (BCL2 interacting protein 3) [17], BCL2L13 (BCL2 like 13) [18], FUNDC1 (FUN14 domain containing 1) [19], AMBRA1 (autophagy and beclin 1 regulator 1) [20], and FKBP8 (FKBP prolyl isomerase 8) [21], found on the outer mitochondrial membrane (OMM), and recently described inner mitochondrial membrane (IMM) protein, PHB2 (prohibitin 2) [22]. Interestingly, two of these proteins, BNIP3 and BNIP3L, apart from their role in mitophagy, are associated with the induction of apoptosis [17,23]. Their dual functions, with respect to cellular life or death fate, converge at the mitochondria, but detailed mechanistic insights that determine which mechanism will be initiated by these receptors are still missing.
BNIP3L, a single pass OMM protein, requires its transmembrane domain (TM) for proper OMM localization [24]. Moreover, studies have shown that BNIP3L is a 24-kDa protein, which is predominantly expressed as a 48-kDa dimer, suggesting that BNIP3L dimerization might have a functional role [5,25]. As the molecular mechanisms of receptor activation are still largely unknown, we investigated BNIP3L dimerization as a potentially novel mechanism of BNIP3L-mediated mitophagy initiation. Here, we present how the phosphorylation status of BNIP3L influences its dimerization property and consequently initiates receptor activity and subsequently induces receptor-mediated mitophagy.
Results
Glycine 204 and glycine 208 in the BNIP3L transmembrane domain are important for BNIP3L dimerization
Previous studies have shown that BNIP3L TM is responsible for its localization on the OMM and speculated that BNIP3L could, similar to its homolog BNIP3, form dimers [24,25]. To that end, Sulistijo et al. [26] biophysically described the interactions responsible for BNIP3 dimer formation and defined the marginal conserved TM pentapeptide GxxxG as the critical motif for the dimerization of BNIP3 and many other membrane proteins [27]. This pentapeptide allows lateral interactions between two transmembrane alpha-helices, resulting in the BNIP3 dimer formation [26]. When analyzed by SDS-PAGE, BNIP3 migrates predominantly as a 60 kDa dimer in addition to the 30 kDa monomer [28]. Our previous study [5] has shown that BNIP3L is a 24-kDa protein but is often expressed as a 48-kDa protein, suggesting that it also homodimerizes, which is not surprising considering the high sequence homology between the TM domains of both BNIP3 and BNIP3L (Figure 1A). We also tested the stability of these homodimers and showed that they are extremely stable and resistant to the strong detergents and high-temperature denaturation (Figure 1B and S1A). This conserved feature of extremely stable dimerization, seen both in BNIP3L and BNIP3, suggests their functional significance. Thus, considering a high homology between the TM domains of BNIP3 and BNIP3L and the results presented by Sulistijo et al. [26], we designed BNIP3LH197A, BNIP3LA200L, BNIP3LG202A, BNIP3LG204A, and BNIP3LG208V mutants in the TM domain to identify the amino acids important for dimer stabilization. To this end, we concentrated on the residues that exclusively formed monomers in BNIP3. In contrast to BNIP3 TM corresponding mutants, which all showed a complete loss of dimerization (as seen in Sulistijo et al. [26]), only BNIP3LG204A and BNIP3LG208V TM mutants resulted in the complete loss of dimer formation (Figure 1C and S1B). Surprisingly, the BNIP3LG202A mutant did not influence BNIP3L dimerization status and was, therefore, used as a control next to the WT BNIP3L. Our results further confirmed that GFP-labeled BNIP3LG204A and BNIP3LG208V dimerization-deficient mutants migrated on SDS-PAGE as monomeric forms of approximately 68 kDa in contrast to WT BNIP3L and BNIP3LG202A that migrated as two distinct protein species of approximately 68 and 135 kDa, representing monomeric and dimeric forms, respectively (Figure 1C).
Figure 1.
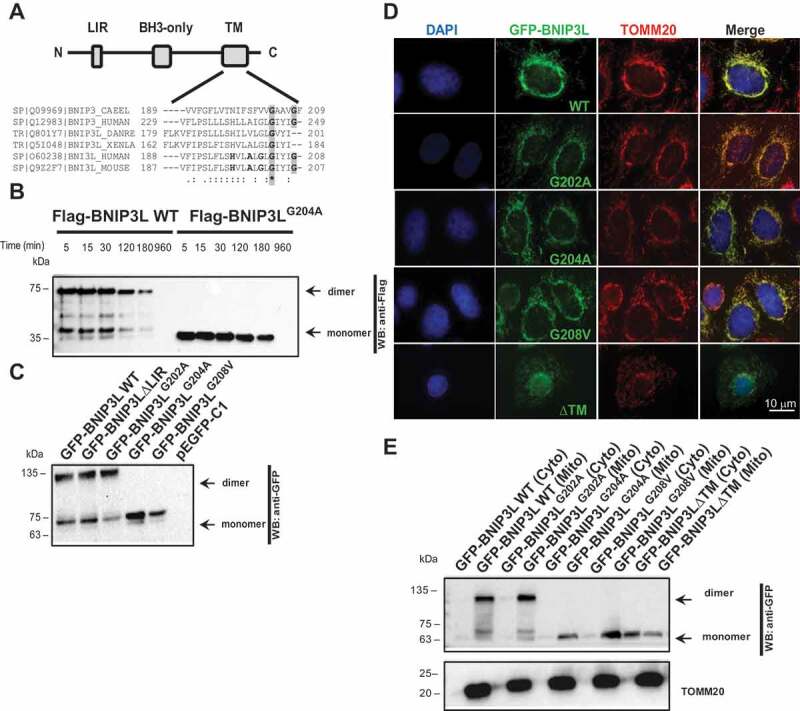
Glycine 204 and 208 in the BNIP3L transmembrane domain are important for BNIP3L dimerization. (A) A scheme of BNIP3L domain organization: LC3-interacting region (LIR), BCL2 homology domain 3 (BH3), and transmembrane domain (TM). The alignment shows amino acid sequences of BNIP3L and related BNIP3 TM domains from the indicated species. The shaded regions indicate highly conserved glycine residues in the TM domains. Consensus symbols “*” (identical residues), “:” (conserved substitution) and “.” (semi-conserved substitution). (B) Western blot analysis of BNIP3L dimer temperature stability. HEK293 cells overexpressing Flag-BNIP3L WT or Flag-BNIP3LG204A mutant were lysed in RIPA buffer containing 0.5% SDS and boiled for the indicated period of time (C) Western blot of HEK293 lysates overexpressing different GFP-BNIP3L mutants with or without dimerization ability. (D) Representative immunofluorescence images of BNIP3L dimerization mutants co-localizing to the mitochondria. Nuclei were stained with DAPI (blue), green signals are GFP-BNIP3L proteins, and red represents mitochondrial marker TOMM20. BNIP3L and TOMM20 colocalization is reflected as a yellow color. (E) Western blot of HEK293 subcellular fractionation for detecting the localization of GFP-BNIP3L WT or the indicated mutants. TOMM20 was used as a marker for the outer mitochondrial membrane or mitochondrial fraction (mito.). Abbreviations: BH3: BCL2 homology 3; BNIP3: BCL2 interacting protein 3; BNIP3L: BCL2 interacting protein 3 like; CAEEL: Caenorhabditis elegans; cyto: cytosolic fraction; DANRE: Danio reio; LIR: LC3-interacting region; BNIP3L ΔLIR: recombinant BNIP3L lacking LC3-interacting region; BNIP3LG202A: recombinant BNIP3L with substituted glycine 202 with alanine; BNIP3LG204A: recombinant BNIP3L with substituted glycine 204 with alanine; BNIP3LG208V: recombinant BNIP3L with substituted glycine 208 with valine; mito: mitochondrial fraction; TM: transmembrane; XENLA: Xenopus laevis; WT: wild type
To verify if the generated mutants still localize to the mitochondria, we performed an immunofluorescence microscopy colocalization experiment. As expected, BNIP3LG204A and BNIP3LG208V TM mutants colocalized to the TOMM20-immunolabeled mitochondria confirming their normal OMM localization (Figure 1D). In addition, the proper localization of the mutants to the mitochondria was confirmed by subcellular localization, where all mutants exclusively separate in the mitochondrial fraction (Figure 1E). Therefore, we considered that the mutations are not altering the proper receptor localization to the mitochondria and could be used for subsequent functional analysis (Figure 1D). Moreover, we found Gly to Ala monomeric mutants were also extremely stable and resistant to SDS and temperature denaturation similar to WT (Figure 1B). Together, these results confirmed the importance of the marginal GIYIG transmembrane BNIP3L sequence, particularly the conserved Gly204 and Gly208, in the stabilization of the BNIP3L dimer.
Dimerization increases BNIP3L activity as a mitophagy receptor
The interactions between autophagy receptors and autophagosomal membrane-anchored LC3 and GABARAP are essential for proper activation of selective autophagy, including BNIP3L-mediated mitophagy [5,29]. To determine whether BNIP3L dimerization is associated with receptor activation and if it is responsible for efficient mitophagy, we tested the ability of BNIP3LG204A and BNIP3LG208V to interact with LC3 and GABARAP proteins. We performed affinity-isolation assays using GST-bound LC3/GABARAP and HEK293 cell extracts overexpressing GFP-BNIP3L WT, BNIP3LG204A, BNIP3LG208V, and BNIP3LG202A mutants and compared their binding properties to BNIP3L∆LIR mutant, which is unable to bind LC3/GABARAP. As shown in the biochemical assay, monomeric BNIP3L, seen in the BNIP3LG204A and BNIP3LG208V mutants, did not affect the ability of BNIP3L to interact with LC3A (Figure 2A). This interacting pattern was observed when any of the Atg8 homologs were tested: LC3A, LC3B, GABARAPL1, or GABARAPL2 (Fig. S2). Together, this indicated that the dimerization affects the affinity of BNIP3L to interact with LC3/GABARAP via its LIR domain, and suggests that it could increase its binding avidity, and thus creating larger and longer-lasting protein complexes in vivo. Moreover, our quantification analysis of LC3A binding to BNIP3L dimer and monomer showed that the percentage of BNIP3L dimer binding to LC3A is significantly higher compared to the percentage of BNIP3L monomer binding. This effect was observed in all BNIP3L mutants with normal dimerization phenotype (Figure 2A). Next, we examined the effect of BNIP3L dimerization on mitophagy activation by analyzing the recruitment of LC3A to the mitochondria in HeLa cells overexpressing WT BNIP3L, BNIP3L∆LIR, BNIP3LG202A, and BNIP3LG204A. Transfected HeLa cells were treated with 10 µM carbonyl cyanide m-chlorophenyl hydrazine (CCCP) for 2 h to induce mitochondrial depolarization and recruit the autophagic machinery to the depolarized mitochondria (as previously described in Rogov et al. [30]). Using immunofluorescence microscopy, we analyzed the number of LC3A-positive puncta. The quantification of LC3A was performed by counting the number of LC3A-positive dots (signals) in 100 cells per each BNIP3L construct presented as fold-change. Only the clear and well-defined LC3A signals were taken into the analysis, while weak and oversize signals were excluded. Expectedly, BNIP3LG204A dimerization mutant showed decreased recruitment of LC3A-positive vesicles compared to the WT BNIP3L and BNIP3LG202A mutant (Figure 2B). The observed LC3A recruitment decrease by the BNIP3LG204A mutant was similar to the decrease previously seen with the LIR mutant, suggesting that receptor dimerization is equivalently important for LC3 recruitment, as is the intact LIR.
Figure 2.
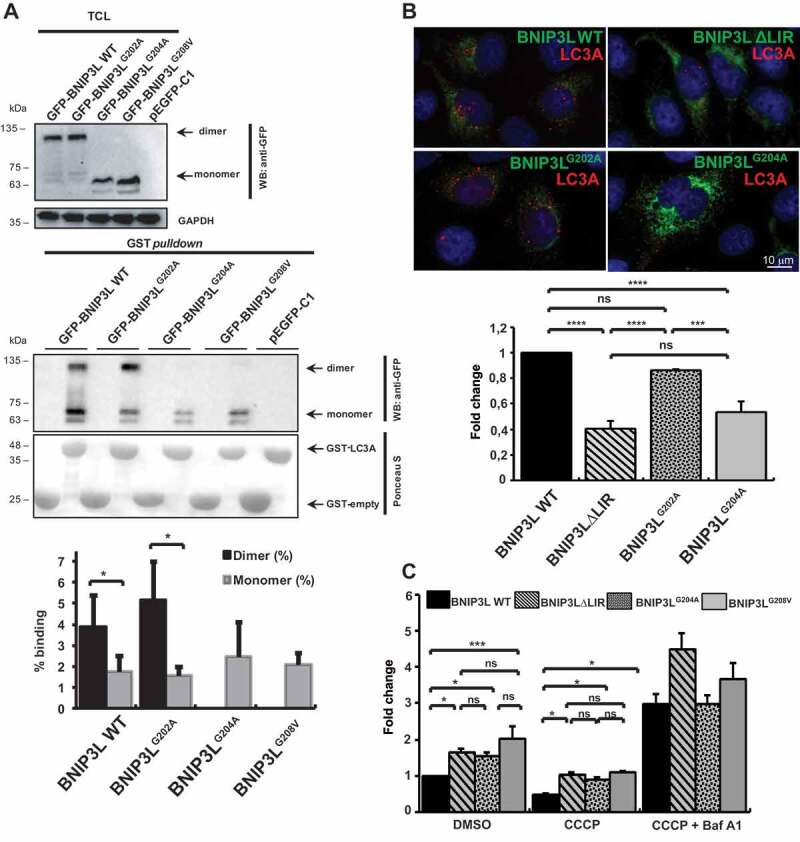
Dimerization increases BNIP3L activity as a mitophagy receptor. (A) GST affinity isolation showing BNIP3L ability for LC3A binding. Affinity isolation with purified GST-LC3A was performed against GFP-BNIP3L WT or indicated TM mutants: BNIP3LG202A, BNIP3LG204A, and BNIP3LG208V (lower blot). Upper blot shows a western blot of 10% of TCL input used for the affinity-isolation reaction. GAPDH was used as a loading control. The graph represents a quantitative analysis of GST affinity-isolation binding efficiency between BNIP3L dimer and monomer to GST-fused LC3A. Binding efficiency was shown as a percentage of GST affinity isolation from a total of 10% TCL input in an affinity-isolation assay. Densitometric scans of immunoblots were obtained from three independent experiments and analyzed in Image Lab. T-test statistical analysis was used to compare differences between GFP-WT BNIP3L dimer and monomer LC3A binding, as well as between GFP-BNIP3LG202A dimer and monomer. (B) Recruitment of autophagosomes on mitochondria overexpressing different GFP-BNIP3L mutants in HeLa cells upon mitophagy induction. HeLa cells were transfected with different GFP-BNIP3L proteins and treated with CCCP for 2 h to induce mitophagy. GFP-BNIP3L proteins are represented as green and autophagosomes labeled by LC3A as red. The quantification was based on the analysis of the total number of LC3A puncta in 100 cells per each WT BNIP3L or indicated mutants. The data are represented as fold-change relative to the WT BNIP3L (mean ± SD) from three independent experiments (significance assessed by one-way ANOVA with Tukey’s multiple comparisons test). (C) Mitochondrial removal upon CCCP treatment in cells overexpressing GFP-BNIP3L WT, BNIP3L∆LIR, BNIP3LG204A, or BNIP3LG208V proteins monitored by flow cytometry. Transfected cells were treated for 24 h with CCCP or in combination with Baf A1 to induce mitophagy. Two-way ANOVA with Tukey’s multiple comparisons test analysis was used to compare differences between mitochondrial removal in GFP-BNIP3L (WT BNIP3L, BNIP3L∆LIR, BNIP3LG204A, and BNIP3LG208V) transfected cells. All data were analyzed in GraphPad Prism 8. Statistical significance: *P = < 0.05, **P = < 0.01; ***P = < 0.001; ns, not significant; error bars indicate standard deviation, n = 3
Additionally, we performed flow cytometry analysis to further evaluate the BNIP3L dimerization effect on the mitochondrial loss during mitophagy. HEK293 cells transfected with GFP-BNIP3L WT, BNIP3LG204A, and BNIP3LG208V were treated for 24 h with 10 μM CCCP or with 10 μM CCCP/100 nM bafilomycin A1 (Baf A1) to block autophagosome-lysosome fusion. We analyzed the GFP-BNIP3L-labeled mitochondria and showed that the mitochondrial loss is more pronounced in cells transfected with WT BNIP3L compared to BNIP3LG204A- or BNIP3LG208V-transfected cells (Figure 2C). Further, mitochondrial removal was not significantly different in cells transfected with LIR-deficient BNIP3L, which implied that the dimerization mechanism could serve as an additional LIR-independent mechanism for receptor-mediated mitophagy regulation. In the cells treated with 10 μM CCCP/100 nM Baf A1, we observed an accumulation of the mitochondria in all mutants suggesting that dimerization affects the initial steps of mitophagy and not its final execution (Figure 2C).
Phosphorylation at the C-terminal BNIP3L disrupts its dimerization
Since BNIP3LG204A or BNIP3LG208V mutations are not expected biological events in cells, we further evaluated the physiological and functional significance of BNIP3L dimerization by analyzing the C-terminal of BNIP3L. The 11-amino-acids-long C-terminal that localizes to the intermembrane mitochondrial space is composed of more than 50% of the amino acids that may undergo phosphorylation (Figure 3A). As reported previously, besides the role of BNIP3L in autophagy, BNIP3L is also recognized as a pro-death protein [31]. Thus, the presence of a large number of cell death regulators in the intermembrane mitochondrial space suggests that C-terminal BNIP3L could be a common regulator of both mitophagy and apoptosis. Therefore, we investigated if C-terminal BNIP3L phosphorylation would influence the BNIP3L dimerization status and, if so, what effect it would have on cellular destiny. We first generated a series of C-terminal BNIP3L phosphomimetic mutants, including BNIP3LS212, BNIP3LT213,BNIP3LS215, BNIP3LS217, BNIP3LT218, and BNIP3LY219. Selected amino acids were changed either to alanine to generate phosphorylation-defective mutants or to glutamic acid residues for the positive phosphomimetic mutants (Figure 3A). Surprisingly, analyses of cell lysates overexpressing all C-terminal mutants revealed BNIP3LS212E phosphomimetic mutant as the only mutant unable to dimerize, which is similar to the BNIP3LG204A and BNIP3LG208V mutants or a mutant completely lacking the C-terminal intermembrane domain (Figure 1C, Figure 3B, and S3). All phosphorylation-defective C-terminal BNIP3L mutants (Thr, Ser, or Tyr to Ala) exhibited roughly equal amounts of dimeric and monomeric forms. Conversely, other C-terminal BNIP3L phosphomimetic mutants did not show a clear loss of dimerization, but some phenotypic differences could be observed for the BNIP3LT213E, BNIP3LS215E, and BNIP3LS217E mutants, where dimeric species are less pronounced when compared to WT BNIP3L (Fig. S3). Although all C-terminal mutants can regularly interact with LC3A and colocalize to positively labeled TOMM20 mitochondria (data not shown), for further analyses, we focused on BNIP3LS212E mutant to investigate the role of C-terminal BNIP3L phosphorylation in receptor dimerization and its effect on mitophagy initiation. As expected, in GST affinity-isolation assay with GST-LC3A and cell lysates overexpressing BNIP3LS212E or BNIP3LS212A mutants, both mutants interacted with LC3A, indicating that the C-terminal BNIP3L phosphorylation did not abrogate the recruitment of mitochondria into autophagosomal vesicles. The quantification of the GST affinity isolation also showed a higher percentage binding capacity of the dimerized form of BNIP3LS212A to LC3A compared to the BNIP3LS212A monomeric form, again confirming the importance of the BNIP3L dimerization in LC3A binding (Figure 3C). Further, immunofluorescence microscopy and mitochondrial fractionation confirmed that changes in phosphorylation did not disrupt the normal OMM BNIP3L localization (Figure 3D and Figure 3E), suggesting that the BNIP3LS212E and BNIP3LS212A C-terminal mutants could be used in subsequent functional analyses of the receptor.
Figure 3.
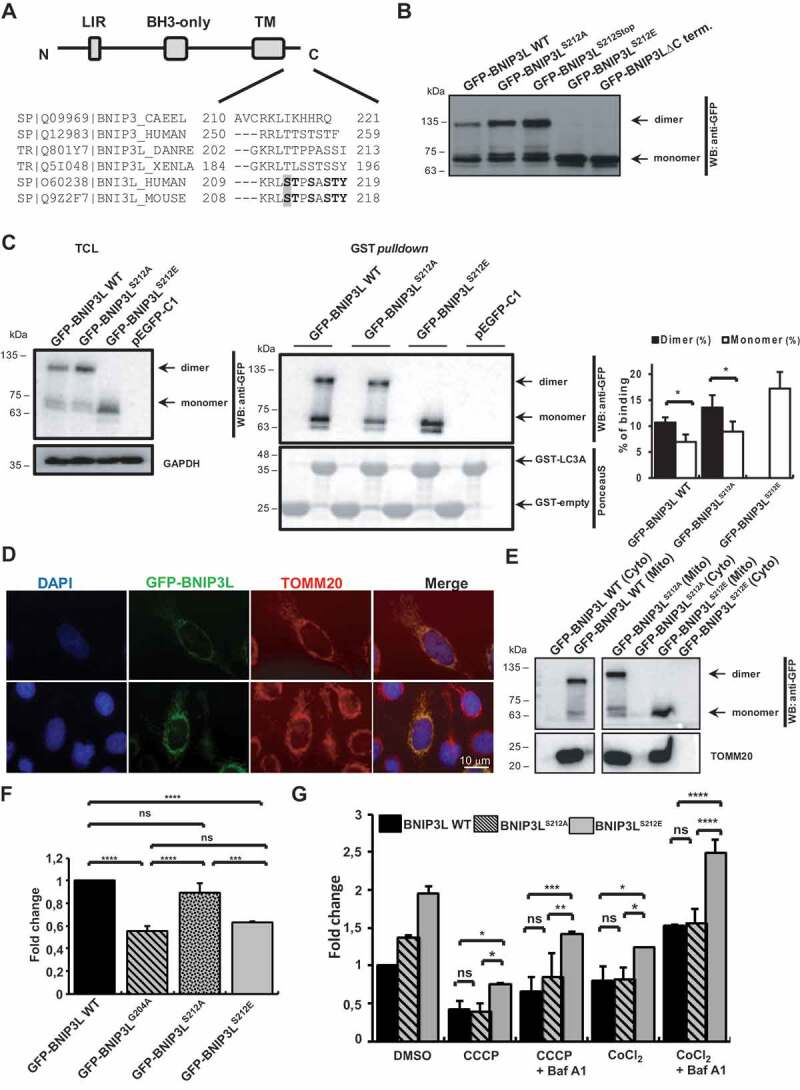
Phosphorylation at the C-terminal BNIP3L disrupts its dimerization. (A) Domain organization of the BNIP3L protein and amino acid alignment of the C-terminal of BNIP3L and BNIP3 proteins from the indicated species. The shaded region indicates the Ser212 residue that is important for dimerization. (B) Western blot analysis of HEK293 overexpressing GFP-BNIP3L WT, GFP-BNIP3LS212A, GFP-BNIP3L212Stop, GFP-BNIP3LS212E, and GFP-BNIP3L∆C-terminal mutants. (C) GST affinity isolation of GST-LC3A against GFP-BNIP3L WT, GFP-BNIP3LS212A, and GFP-BNIP3LS212E mutants (right blot). Left blot shows BNIP3L expression in 10% of cell lysates used in the affinity-isolation reaction with GAPDH as a loading control. Quantification of the GST affinity-isolation binding efficiency between BNIP3L dimer and monomer to GST-fused LC3A was showed as a percentage of GST binding from a total of 10% TCL input in the affinity-isolation assay. Densitometric scans of immunoblots were obtained from three independent experiments and analyzed in Image Lab. T-test statistical analysis was used to compare differences in LC3A binding between GFP-BNIP3L WT or BNIP3LS212A dimer and monomer. (D) Immunofluorescence microscopy of the BNIP3L C-terminal mutant and its localization to the mitochondria. Nuclei were stained with DAPI (blue), green signals are GFP-BNIP3L proteins, and red represents mitochondrial marker TOMM20. BNIP3L and TOMM20 colocalization is reflected by a yellow color. (E) Western blot analysis of HEK293 subcellular fractionation for detecting the localization of GFP-BNIP3L WT or GFP-BNIP3LS212A/E mutants. TOMM20 was used as a marker for the outer mitochondrial membrane. (F) Recruitment of autophagosomes on damaged mitochondria overexpressing GFP-BNIP3L WT, BNIP3LG204A, BNIP3LS212A, or BNIP3LS212E. HeLa cells were transfected with indicated BNIP3L C-terminal dimerization mutants and treated with CCCP for 2 h to induce mitophagy. Quantification of the LC3A puncta was performed by analyzing the number of LC3A dots for 100 cells per each BNIP3L plasmid in three independent experiments (the data are represented as mean ± SD of fold-change against GFP-WT BNIP3L). One-way ANOVA with Tukey’s multiple comparisons test was used to compare the difference in autophagosomal recruitment between BNIP3L mutants. (G) Quantification of mitochondrial removal using GFP-BNIP3L WT, BNIP3LS212A, or BNIP3LS212E by flow cytometry. Transfected HEK293 cells were treated with CCCP or CoCl2 and in combination with Baf A1 for 24 h. Two-way ANOVA with Tukey’s multiple comparisons test was used to compare differences between mitochondrial removal in GFP-BNIP3L WT, BNIP3LS212A, and BNIP3LS212E. Data were analyzed in GraphPad Prism 8. Statistical significance: *P = < 0.05, **P = < 0.01, ***P = < 0.001; ns, not significant; error bars indicate standard deviation, n = 3 (F), n = 2 (G)
To confirm our biochemical data and test the effect of C-terminal phosphorylation on dimerization and mitophagy initiation at a cellular level, we analyzed the recruitment of LC3A to the mitochondria in HeLa cells overexpressing WT BNIP3L, BNIP3LS212E, and BNIP3LS212A mutants. Transfected HeLa cells were treated with 10 µM CCCP for 2 h, and the number of LC3A-positive vesicles was evaluated, as previously described. The phosphomimetic BNIP3LS212E mutant showed decreased recruitment of LC3A-positive vesicles compared to the WT BNIP3L and BNIP3LS212A mutant, both able to dimerize, suggesting once again that BNIP3L dimerization could be a probable mechanism of BNIP3L-mediated mitophagy initiation (Figure 3F).
Furthermore, we performed flow cytometry analysis to evaluate the effect of the phosphomimetic BNIP3LS212E mutant on the receptor’s activity by monitoring mitochondrial clearance during mitophagy progression. GFP-BNIP3L WT and BNIP3LS212E or BNIP3LS212A mutants were transfected into HEK293 cells and treated for 24 h with 10 μM CCCP. In this experiment, we also treated the cells with the hypoxia mimetic CoCl2 to chemically simulate hypoxia-induced mitophagy since BNIP3L expression is shown to be HIF1A-dependent [32]. We analyzed the fluorescently labeled mitochondria and showed that mitochondrial loss is more pronounced in cells transfected with BNIP3LS212A mutant that could undergo dimerization compared to the BNIP3LS212E phosphomimetic mutant that is unable to dimerize (Figure 3G). Additionally, we analyzed the mitochondrial retention in the cells treated with CCCP or CoCl2, in combination with Baf A1, and the accumulation of the mitochondria was detected in all mutants (Figure 3G). Together, our results suggest that C-terminal BNIP3L phosphorylation and its consequent dimerization loss could decrease the induction of BNIP3L-mediated mitophagy.
LIR and receptor dimerization jointly enhance BNIP3L-mediated mitophagy
Previous studies suggested that, in addition to the LIR domain, other properties of BNIP3L are also important for the BNIP3L-mediated mitochondrial clearance [5]. Since our results indicated that dimerization, for itself or in combination with the dephosphorylation of C-terminal BNIP3L, is a possible new mechanism for BNIP3L-mediated mitophagy activation, we explored the combined effect of dimerization loss with previously described LIR-dependent recruitment of LC3/GABARAP proteins on mitophagy [5,30]. Therefore, we designed double ∆LIR and TM/C-terminal BNIP3L mutants: BNIP3L∆LIRG202A, BNIP3L∆LIRG204A, BNIP3L∆LIRS212A, and BNIP3L∆LIRS212E. Before testing their effect on mitophagy, we first examined their expressions and LC3A recruitment. As expected, all BNIP3L mutants with the lack of dimerization ability appeared on the western blot solely in monomeric forms (Figure 4A). Additionally, the GST affinity isolation with GST-LC3A showed that the double mutants could not interact with LC3A due to the lack of LIR motif crucial for the LC3/GABARAP binding independent of its dimerization property (Figure 4B).
Figure 4.
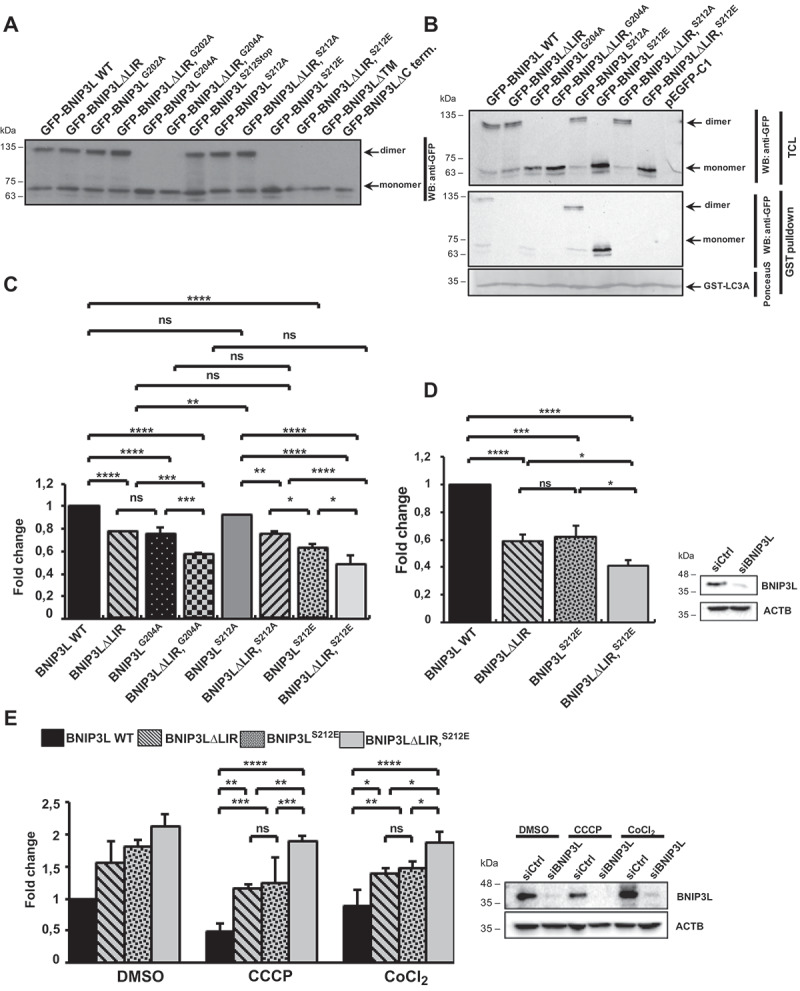
LIR and receptor dimerization jointly enhanced BNIP3L-mediated mitophagy. (A) Western blot analysis of the double ∆LIR and TM/C-terminal BNIP3L mutants. (B) GST affinity isolation of GST-LC3A and GFP-BNIP3L WT, BNIP3L∆LIR, BNIP3LG204A, BNIP3L∆LIRG204A, BNIP3LS212A, BNIP3LS212E, BNIP3L∆LIRS212A, or BNIP3L∆LIRS212E. (C) Recruitment of autophagosomes on damaged mitochondria overexpressing GFP-BNIP3L WT, BNIP3L∆LIR, BNIP3LG204A, BNIP3L∆LIRG204A, BNIP3LS212A, BNIP3LS212E, BNIP3L∆LIRS212A, or BNIP3L∆LIRS212E. Quantification of LC3A puncta was performed by analyzing the number of LC3A dots for 100 cells per each BNIP3L plasmid in three independent experiments (the data are represented as mean ± SD). One-way ANOVA with Tukey’s multiple comparisons test was used to compare the difference in autophagosome recruitment between GFP-BNIP3L WT, BNIP3L∆LIR, BNIP3LG204A, BNIP3L∆LIRG204A, BNIP3LS212A, BNIP3LS212E, BNIP3L∆LIRS212A, and BNIP3L∆LIRS212E. *P = < 0.05, ** = P < 0.01, *** = P < 0.001. (D) Western blot confirmation of BNIP3L silencing in Hela cells transfected with control siRNA or siRNA against BNIP3L for 48 h that were used for the quantification of autophagosome recruitment on CCCP-damaged mitochondria overexpressing the indicated GFP-BNIP3L proteins. (E) Western blot analysis of HEK293 with silenced endogenous BNIP3L and transfected with different GFP-BNIP3L dimerization mutants upon CCCP- or CoCl2-induced mitophagy (right). Mitochondrial removal in CCCP- or CoCl2-treated HEK293 cells lacking endogenous BNIP3L overexpressing GFP-BNIP3L dimerization mutants were analyzed by flow cytometry (left). Two-way ANOVA with Tukey’s multiple comparisons test was used to compare the differences in mitochondrial removal in cells with GFP- BNIP3L WT, BNIP3LS212E, BNIP3L∆LIR, or BNIP3L∆LIRS212E proteins. *P = < 0.05, **P = < 0.01; ns, not significant; error bars indicate standard deviation, n = 3
Next, we tested the combined effect of BNIP3L dimerization loss and LIR-dependent recruitment of autophagosomes on damaged mitochondria at a cellular level using a previously described immunofluorescence method. Double mutants, BNIP3L∆LIRG204A and BNIP3L∆LIRS212E, both without dimerization ability, recruited significantly fewer autophagosomes on the damaged mitochondria compared to the single BNIP3L mutants, BNIP3L∆LIR or BNIP3LG204A and BNIP3LS212E, respectively (Figure 4C) suggesting the importance of both mechanisms for the initiation of BNIP3L-dependent mitophagy.
Finally, we performed a series of experiments using WT BNIP3L, BNIP3L∆LIRS212E, and BNIP3L∆LIRS212E mutants in BNIP3L knockdown background using RNA interference. We tested the recruitment of autophagosomes to the damaged mitochondria by an immunofluorescence method (Figure 4D), as well as mitochondrial retention by flow cytometry (Figure 4E). As expected, the analyzed mutants showed the same phenotype in the siCtrl and siBNIP3L background, where monomeric BNIP3LS212E mutant, similar to the ∆LIR mutant, recruited less LC3A to the mitochondria and was less efficient in mitochondrial removal through mitophagy. BNIP3L∆LIRS212E double mutant showed accumulating defects in LC3A recruitment and mitophagy progression. Lastly, this confirms our hypothesis that both functional LIR and dimerized receptors are needed for strong mitophagy induction in BNIP3L-dependent mitophagy.
Discussion
Mitophagy is known as a fundamental cellular process critical for maintaining normal mitochondrial function [33], and growing evidence suggests that mitophagy dysregulation is one of the fundamental processes involved in numerous pathologies, including neurodegenerations and tumors [7,8]. In mammals, mitophagy is essential during basic physiological processes, such as eye development [11,12] or reticulocyte maturation [14,15]. This development-induced mitophagy is receptor-mediated, and the most investigated mitophagy receptor, BNIP3L, was shown to govern mitochondrial clearance in an LIR-dependent manner [5,30]. However, there are still missing knowledge on the exact molecular mechanism of cargo selection and, even more interesting, receptor activation. Here, we investigated the new molecular mechanism of receptor activation required for the BNIP3L-mediated mitophagy. It is known that the binding of BNIP3L to LC3s and GABARAPs, achieved through the LIR motif, is required for proper receptor-mediated mitophagy [5,29], and the phosphorylation of amino acids juxtaposed to LIR is highly important for receptor activation [30]. This mechanism is analogous to the interaction of other autophagy receptors with LC3/GABARAP [3,34], including mitophagy receptors, OPTN, FUNDC1, and BNIP3 [17,19,35]. In our recent study [30], we demonstrate how the effect of BNIP3L LIR phosphorylation on mitophagy initiation and progression is not sufficient to fully activate the receptor since the abolishment of the LIR phosphorylation resulted only in partially deficient mitophagy, seen by the increased mitochondrial retention. Therefore, additional mechanisms required for the receptor activation were examined. The biochemical analysis of BNIP3L, similar to BNIP3, showed two distinct species of BNIP3L in the cells: monomeric and dimeric forms (Figure 1B). Furthermore, both dimeric and monomeric forms of the protein were extremely stable under stringent denaturing conditions (Figure 1B). Analyzing BNIP3, Sulistijo et al. found that polar substitutions in the transmembrane (TM) domain of BNIP3 decreased the fraction of dimeric forms. This dimerization loss is particularly evident in substitutions of the amino acids (Ser172, His173, Ala176, Gly180, Ile183, and Gly184) that were also conserved in the BNIP3L TM domain ([26]; Figure 1A). Accordingly, the mutation of a single glycine to alanine at 204 or valine at 208 positions in the BNIP3L TM domain was sufficient to achieve dimerization loss (Figure 1C and S1B). Both glycine residues are conserved between BNIP3 and BNIP3L, indicating the importance of the GxxxG motif in establishing interactions needed for the stable dimer formation. Interestingly, BNIP3LG204A and BNIP3LG208V mutations did not influence BNIP3L localization to the mitochondria. Furthermore, since the analysis of the LC3A recruitment to the mitochondria in cells overexpressing BNIP3L TM mutants was lower compared to the WT and very similar to the behavior of BNIP3L∆LIR mutant and that monomeric forms of BNIP3L interacted with LC3A with lower affinity, these results strongly suggest that dimerization is a probable mechanism of BNIP3L-mediated mitophagy initiation in vivo (Figure 2B) [30]. Although other effects could also be possible. Additionally, the dimerization defect influenced the total mitochondrial removal indicating that the lower mitophagy initiation ability of the BNIP3L receptor directly affects mitophagy progression (Figure 2B and 2C).
The short amino acid stretch at the C-terminal end of the BNIP3L that localizes to the intermembrane mitochondrial space contains several potential phosphorylation sites. The phosphorylation of serine on position 212 revealed the highest possible regulatory mechanism of BNIP3L receptor activation since the expression of phosphomimetic BNIP3LS212E in cells significantly decreased both the initiation and the progression of the receptor-mediated mitophagy (Figure 3A, Figure 3E, and Figure 3F). This result was due to the complete loss of the dimerization ability of the phosphomimetic BNIP3LS212E that still localized to the mitochondria and retained its LC3A-interacting function (Figure 3B-G). Finally, this study demonstrated how both mechanisms, LIR:LC3 interaction and receptor dimerization, contribute together to mitochondrial recruitment and removal by receptor-mediated mitophagy (Figure 4).
Together, we suggest the dimerization of mitophagy receptor BNIP3L as a novel molecular mechanism of its activation. The tendency of BNIP3L to form higher-order structures is in line with the aggregation of the receptors, such as SQSTM1 and NBR1, that is required for stronger autophagy recruitment and better autophagic cargo sequestration [36]. Oligomerization of BNIP3L and consequent LIR accumulation, hence, would be a requisite for sufficient avidity of the receptor to activate programmed mitochondrial clearance.
With this, our study now opens a new set of questions regarding the regulation of BNIP3L-dependent mitophagy. The recently published data by Rogov et al. highlights how phosphorylation of the BNIP3L LIR enhances mitophagy receptor engagement [30]. Consequently, a question arises on what would be the upstream signals activating LIR phosphorylation and dimerization mechanisms and whether one precedes the other? Also, the next obvious matter to investigate would be what the phosphatase that regulates BNIP3L dimerization is and logically, which kinase is responsible for maintaining BNIP3L in an inactive monomeric stage? Legitimate candidates for both phosphatases and kinases would have to reach the C-terminal part of BNIP3L exposed to intermembrane space, and enzymes localizing to mitochondria should be first explored. One possible candidate is PGAM5 (PGAM family member 5, mitochondrial serine/threonine protein phosphatase) already associated with PINK1-PRKN-dependent mitophagy [37–39]. However, since PINK1-PRKN-dependent and receptor-dependent mitophagy differ in many aspects, other phosphatases should not be overlooked. Finally, our findings would be particularly interesting to assess in the context of reticulocyte differentiation and their imperative in mitochondrial removal by BNIP3L-mediated mitophagy.
Material and methods
Plasmids
Plasmids used in the study were generated by site-directed mutagenesis PCR to introduce desired mutations in the BNIP3L constructs. The correctness of the DNA sequence was verified by sequencing. Plasmids are described in Table 1.
Table 1.
Plasmids used in the study
| Vector | Descripton | Source |
|---|---|---|
| pEGFP-C1/BNIP3L | GFP-tagged human BNIP3L | ref [30] |
| pEGFP-C1/BNIP3L∆LIR | GFP-tagged human BNIP3L, ∆LIR (∆WVEL) | ref [5] |
| pEGFP-C1/BNIP3LH197A | GFP-tagged human BNIP3L, His 197 mutated to Ala | this study |
| pEGFP-C1/BNIP3LA200L | GFP-tagged human BNIP3L, Ala 200 mutated to Leu | this study |
| pEGFP-C1/BNIP3LG202A | GFP-tagged human BNIP3L, Gly 202 mutated to Ala | this study |
| pEGFP-C1/BNIP3LG204A | GFP-tagged human BNIP3L, Gly 204 mutated to Ala | this study |
| pEGFP-C1/BNIP3LG208V | GFP-tagged human BNIP3L, Gly 208 mutated to Val | this study |
| pEGFP-C1/BNIP3L∆TM | GFP-tagged human BNIP3L, ∆TM (188–208) | this study |
| pEGFP-C1/BNIP3LS212A | GFP-tagged human BNIP3L, Ser 212 mutated to Ala | this study |
| pEGFP-C1/BNIP3LS212E | GFP-tagged human BNIP3L, Ser 212 mutated to Glu | this study |
| pEGFP-C1/BNIP3LS212 Stop | GFP-tagged human BNIP3L, Ser 212 mutated to Stop | this study |
| pEGFP-C1/BNIP3LT213A | GFP-tagged human BNIP3L, Thr 213 mutated to Ala | this study |
| pEGFP-C1/BNIP3LT213E | GFP-tagged human BNIP3L, Thr 213 mutated to Glu | this study |
| pEGFP-C1/BNIP3LS215A | GFP-tagged human BNIP3L, Ser215 mutated to Ala | this study |
| pEGFP-C1/BNIP3LS215E | GFP-tagged human BNIP3L, Ser 215 mutated to Glu | this study |
| pEGFP-C1/BNIP3LS217A | GFP-tagged human BNIP3L, Ser 217 mutated to Ala | this study |
| pEGFP-C1/BNIP3LS217E | GFP-tagged human BNIP3L, Ser 217 mutated to Glu | this study |
| pEGFP-C1/BNIP3LT218A | GFP-tagged human BNIP3L, Thr 218 mutated to Ala | this study |
| pEGFP-C1/BNIP3LT218E | GFP-tagged human BNIP3L, Thr 218 mutated to Glu | this study |
| pEGFP-C1/BNIP3LY219A | GFP-tagged human BNIP3L, Tyr 219 mutated to Ala | this study |
| pEGFP-C1/BNIP3LY219E | GFP-tagged human BNIP3L, Tyr 219 mutated to Glu | this study |
| pEGFP-C1/BNIP3L∆LIR,G202A | GFP-tagged human BNIP3L, double ∆LIR (∆WVEL), Gly 202 mutated to Ala | this study |
| pEGFP-C1/BNIP3L∆LIR,G204A | GFP-tagged human BNIP3L, double ∆LIR (∆WVEL), Gly 204 mutated to Ala | this study |
| pEGFP-C1/BNIP3L∆LIR,S212A | GFP-tagged human BNIP3L, double ∆LIR (∆WVEL), Ser 212 mutated to Ala | this study |
| pEGFP-C1/BNIP3L∆LIR,S212E | GFP-tagged human BNIP3L, double ∆LIR (∆WVEL), Ser 212 mutated to Glu | this study |
| pEGFP-C1/BNIP3L∆C terminus | GFP-tagged human BNIP3L, ∆C terminus (209–219) | this study |
| pcDNA3.1/Flag-BNIP3L | Flag-tagged human BNIP3L | ref [5] |
| pcDNA3.1/Flag-BNIP3L∆LIR | Flag-tagged human BNIP3L, ∆LIR (∆WVEL) | this study |
| pcDNA3.1/Flag-BNIP3LG202A | Flag-tagged human BNIP3L, Gly 202 mutated to Ala | this study |
| pcDNA3.1/Flag-BNIP3LG204A | Flag-tagged human BNIP3L, Gly 204 mutated to Ala | this study |
| pcDNA3.1/Flag-BNIP3LS212A | Flag-tagged human BNIP3L, Ser 212 mutated to Ala | this study |
| pcDNA3.1/Flag-BNIP3LS212E | Flag-tagged human BNIP3L, Ser 212 mutated to Glu | this study |
| pcDNA3.1/Flag-LC3 | Flag-tagged human LC3 | ref [2] |
| pGEX-4 T-1/hLC3-A | GST-tagged human LC3-A | ref [2] |
| pGEX-4 T-1/hLC3-B | GST-tagged human LC3-B | ref [2] |
| pGEX-4 T-1/hGABARAP | GST-tagged human GABARAP | ref [2] |
| pGEX-4 T-1/hGABARAP-L1 | GST-tagged human GABARAP-L1 | ref [2] |
| pGEX-4 T-1/hGABARAP-L2 | GST-tagged human GABARAP-L2 | ref [2] |
| pEGFP-C1/BNIP3 | GFP-tagged human BNIP3 | this study |
| pEGFP-C1/BNIP3H173A | GFP-tagged human BNIP3, His 173 mutated to Ala | this study |
| pEGFP-C1/BNIP3H176L | GFP-tagged human BNIP3, His 176 mutated to Leu | this study |
| pEGFP-C1/BNIP3G180A | GFP-tagged human BNIP3, Gly 180 mutated to Ala | this study |
Antibodies and reagents
In this study, the following antibodies were used: mouse monoclonal anti-Flag (Sigma, F1804; 1:1000), mouse monoclonal anti-GFP (Roche, 11 814 460 001; 1:1000), rabbit polyclonal anti-GFP (Clontech, 632592; 1:1000), rabbit polyclonal anti-TOMM20 (Santa Cruz Biotechnology, sc-17764; 1:1000), mouse monoclonal anti-ACTB/β-ACTIN (Sigma Aldrich, A2228; 1:5000), rabbit monoclonal anti-GAPDH (Sigma Aldrich, G9545; 1:1000), rabbit monoclonal (clone D4R4B) anti-BNIP3L/NIX (Cell Signaling Technology, 12396; 1:1000). Secondary HRP-conjugated antibodies, goat anti-mouse (Bio-Rad, 1706516; 1:5000) and goat anti-rabbit (Dako, P0448; 1:5000) IgGs were used for immunoblotting. Donkey anti-mouse Alexa Fluor 488 (Thermo Fisher Scientific, R37114, 1:1000), donkey anti-rabbit Alexa Fluor 488 (Thermo Fisher Scientific, A-21206; 1:10009), goat-anti-mouse Alexa Fluor 568 (Thermo Fisher Scientific, A-11004; 1:1000) and goat-anti-rabbit Alexa Fluor 568 secondary antibody (Thermo Fisher Scientific, A-11011; 1:1000) were used for immunofluorescence studies. CoCl2 (Sigma Aldrich, 60818) was applied at 100 µM for 24 h. CCCP (Sigma Aldrich, C2759) was applied to cells at a final concentration of 10 μM for 2 or 24 h. 100 nM Baf A1 (Sigma Aldrich, 19–148) was used in combination with CCCP and CoCl2 for 24 h.
SDS PAGE of BNIP3L proteins
Indicated GFP or Flag-tagged BNIP3L proteins (LIR, TM, C-terminal, or combined LIR dimerization mutants) were overexpressed in HEK293 cells (ADCC, CRL-1573) using jetPRIME transfection kit (Polyplus, 114–07). 24 h post-transfection cells were lysed in 50 mM HEPES (Sigma Aldrich, H3375), pH 7.5, 150 mM NaCl (Kemika, 1417506), 1 mM EDTA (Fluka, 03610), 1 mM EGTA (Fluka, 03779), 10% glycerol (Kemika, 0711901), 1% Triton X-100 (Sigma Aldrich, 11332481001), 25 mM NaF (Kemika, 1407908), 10 mM ZnCl2 (Kemika, 0314708) with proteases inhibitors (Roche, 4693159001). Lysates were boiled in 6x SDS-PAGE loading buffer and loaded onto 10% or 12% SDS-PAGE gels.
For boiling experiments HEK293 with overexpressed BNIP3L mutants were lysed in RIPA buffer (150 mM NaCl [Kemika, 1417506], 1% NP-40 [Sigma Aldrich, 492018], 0.5% Na-deoxycholate [Sigma Aldrich, D6750], 0.2% SDS [Carl Roth 2326.2], 25 mM Tris [Carl Roth, 5429.2], pH 7.4) and boiled for different time points in 6x SDS-PAGE loading buffer.
Preparation of GST fusion proteins
GST fusions proteins (LC3A, LC3B, GABARAPL1 and GABARAPL2) were expressed in BL21 DE3 E. coli (New England Biolabs, C2527). Protein expression was induced with 0.5 mM IPTG (Carl Roth, 2316.5) for 4 h. Bacteria were lysed in 20 mM Tris-HCl (Carl Roth, 5429.2), pH 7.5, 10 mM EDTA (Fluka, 03610), pH 8.0, 5 mM EGTA (Fluka, 03779), pH 8.5, 150 mM NaCl (Kemika, 1417506). GST fusion proteins were subsequently bound to pre-washed Glutathione-Sepharose 4B beads (GE Healthcare, 17-0756-01). After several washes, fusion protein-bound beads were loaded on a polyacrylamide gel to determine the appropriate amount of GST-fused beads that would be used directly in GST affinity-isolation assays.
GST affinity-isolation assay
HEK293 cells were transfected with GFP-BNIP3L or Flag-BNIP3L constructs encoding the protein of interest using jetPRIME transfection kit (Polyplus, 114–07). 24 h post-transfection cells were lysed in 50 mM HEPES (Sigma Aldrich, H3375), pH 7.5, 150 mM NaCl (Kemika, 1417506), 1 mM EDTA (Fluka, 03610), 1 mM EGTA (Fluka, 03779), 10% glycerol (Kemika, 0711901), 1% Triton X-100 (Sigma Aldrich, 11332481001), 25 mM NaF (Kemika, 1407908), 10 mM ZnCl2 (Kemika, 0314708) with proteases inhibitors (Roche, 4693159001) and lysates were incubated overnight with immobilized GST fusion proteins. Following 5 washes, beads with co-precipitated proteins were resuspended in 2x SDS-PAGE loading buffer, boiled and loaded onto 10% or 12% SDS-PAGE gels for analysis.
Immunofluorescence microscopy and colocalization study
HeLa cells were seeded on 12-mm coverslips and transfected with GFP-BNIP3L WT, BNIP3LΔLIR, BNIP3LG202A, BNIP3LG204A, BNIP3LG208V, BNIP3L∆TM, BNIP3LS212A, or BNIP3LS212E constructs using jetPRIME transfection kit (Polyplus, 114–07). 24 h post-transfection, CCCP (Sigma Aldrich, C2759) was applied to cells at a final concentration of 10 μM for 2 h. Cells were washed once with PBS (Sigma Aldrich, D8537), fixed in 1.5% paraformaldehyde (Sigma Aldrich, 158127) and permeabilized with a 0.15% Triton X-100 (Sigma Aldrich, 11332481001) solution in PBS at room temperature for 20 min and finally blocked in PBS containing 3% BSA (Carl Roth, 8076.1) at 4°C overnight. Primary antibodies (rabbit polyclonal anti-TOMM20 (Santa Cruz Biotechnology, sc-17764 1:1000) and mouse monoclonal anti-GFP (Roche, 11 814 460 001, 1:1000) were diluted in the blocking solution and washes were performed in PBS. Secondary antibodies (Goat-anti-rabbit Alexa Fluor 568 and Donkey anti-mouse Alexa Fluor 488) were prepared the same way. DAPI (Sigma Aldrich, D9542) was used for nuclei staining. Coverslips were mounted with ProLong Antifade Kit (Thermo Fisher Scientific, P36930) on a glass slide. Microscopy was performed using Axioimager D1, Carl 165 Zeiss, Inc. (software: AxioVision software version 4.4; Carl Zeiss, Inc.).
Isolation of the mitochondrial protein fractions
Isolation of mitochondrial protein fractions has been performed using slightly modified Frezza et al. 2007 protocol for organelle isolation [40]. Briefly, to obtain a sufficient amount of mitochondria, HEK293 cells were transfected in 10-cm dishes with GFP-BNIP3L WT or indicated mutants using jetPRIME transfection kit (Polyplus, 114–07). 24 h post-transfection cells were washed and detached from the dish with PBS (Sigma Aldrich, D8537) and transferred in Falcon tube and centrifuged at 600x g for 20 min. All procedures were carried out at 4°C to minimize protease activity. Pellet was homogenized in 2 ml of IBc buffer (0.1 M MOPS/Tris [Carl Roth, 6979.2], 0.1 M EGTA/Tris [Fluka, 03779], 1 M sucrose [Kemika, 1800408], pH 7.4) using a glass homogenizer. Up and down movements were carried out until getting a homogeneous suspension with preserved mitochondrial integrity (~50 times). Homogenate was centrifuged at 600x g for 20 min to sediment nuclei, cell debris, and unbroken cells, and the supernatant was additionally centrifuged at higher speed (7000x g, 20 min) to collect crude mitochondrial fraction (the supernatant from this step has been used as a cytoplasmic fraction). To obtain a purified fraction of mitochondria, a sample of the crude mitochondrial pellet was resuspended in 200 µl of IBc buffer followed by high-speed centrifugation. Finally, purified mitochondria were lysed in modified RIPA buffer (50 mM Tris-HCl [Carl Roth, 5429.2], 150 mM NaCl [Kemika, 1417506], 1 mM EDTA [Fluka, 03610], 1% NP-40 [Sigma Aldrich, 492018], 0.1% Na-deoxycholate [Sigma Aldrich, D6750], pH 7.5) supplemented with protease inhibitor cocktail (Roche, 4693159001). Lysed mitochondria were centrifuged at 16000x g for 20 min, and supernatant from this step was used as the mitochondrial fraction.
RNA interference
To silence endogenous BNIP3L ON-TARGETplus human BNIP3L (665) siRNA SMARTpool (Dharmacon, L-011815-00-0005) or ON-TARGETplus Non-targeting (Ctrl) pool was used. Cells were reverse transfected with 40 nM siBNIP3L or siCtrl pool using Lipofectamine RNAiMax Transfection reagent (Thermo Fisher Scientific,13778150) following the protocol recommended by the manufacturer. 48 h post-transfection, cells were transfected with appropriate plasmid using jetPRIME (Polyplus, 114–07) reagent as described previously. 24 h post-transfection, cells were treated for the indicated amount of time with DMSO (Sigma Aldrich, D8418), CCCP (Sigma Aldrich, C2759), or CoCl2 (Sigma Aldrich, 60818). Following the treatment, cells were analyzed by immunofluorescent microscopy or flow cytometry, as previously described. Small amounts of cells were also used for the western blot analysis to confirm both RNAi knockdown as well as plasmid transfection efficiency.
Mitophagy monitoring
Mitophagy monitoring experiment was made in triplicate. Hela cells were seeded on coverslips, and co-transfected with GFP-WT BNIP3L or GFP-BNIP3L∆LIR, BNIP3LG202A, BNIP3LG204A, BNIP3LS212A, BNIP3LS212E and Flag-LC3A (0.5 μg plasmid per well altogether) constructs using jetPRIME (Polyplus, 114–07). 24 h post-transfection, CCCP (Sigma Aldrich, C2759) was applied to cells at a final concentration of 10 μM for 2 h. Fixation, permeabilization, blocking, and antibody application were performed as described before. Mouse monoclonal anti-Flag (Sigma, F1804; 1:1000), rabbit polyclonal anti-GFP (Clontech, 632592; 1:1000), goat-anti-mouse Alexa Fluor 568 (Thermo Fisher Scientific, A-11004; 1:1000) and donkey anti-rabbit Alexa Fluor 488 (Thermo Fisher Scientific, A-21206; 1:1000) were used. 100 cells, co-transfected with GFP-BNIP3L and Flag-LC3A constructs, were photographed, and the number of LC3A dots was counted for each construct. Only clear and well-defined LC3A signals are taken into consideration, while weak and oversize signals were excluded from the analysis.
Flow cytometry
HEK293 cells were co-transfected with GFP expression constructs encoding the protein of interest using jetPRIME (Polyplus, 114–07). Twenty-four h post-transfection cells were treated with 10 µM CCCP (Sigma Aldrich, C2759), 100 µM CoCl2 (Sigma Aldrich, 60818, or in combination with 100 nM Baf A1 (Sigma Aldrich, 19–148) for 24 h. After treatment, cells were washed with PBS (Sigma Aldrich, D8537), trypsinized (Sigma Aldrich, T4049) for 5 min and resuspended by gentle pipetting. Trypsin-mediated digestion was arrested by the addition of DMEM (Sigma Aldrich, D5796) supplemented with 10% FBS (Sigma Aldrich, F2442). To remove cell clumps, cell pellets were resuspended in PBS and filtered through the 70 µm Cell Strainer (Life Sciences, 352350). Approximately 106 cells were acquired for further flow cytometry analysis. Before fixation, cells were washed twice with PBS and each time centrifuged at 500x g for 5 min. Cells were fixed with freshly prepared 4% paraformaldehyde for 10 min at room temperature, washed twice with PBS, stained with PI (Sigma Aldrich, P4170) to eliminate dead cells from further analyze and finally resuspended in PBS before the analysis. Mean fluorescence intensity values in FL1 (GFP) and FL2 (PI) channels were collected in 105 events for each BNIP3L construct in different conditions (DMSO, CCCP, CCCP + Baf A1, CoCl2, or CoCl2 + Baf A1) in three independent experiments. The acquired data were gated for singlets by generating FSC-A vs. FSC-H plot and by the exclusion of PI-positive cells. Results were analyzed using FlowLogic software.
Quantification and statistical analysis
Statistical parameters and significance are reported in the figures and the figure legends. GraphPad Prism 8 was used for data analysis. Student’s t-test was used to compare the probability of the statistical significance of binding efficiency between BNIP3L dimer and monomer to LC3A protein in GST affinity-isolation assays (n = 3). One-way ANOVA with Tukey’s multiple comparisons test was used to compare the difference between autophagosomal recruitment to the damaged mitochondria in cells overexpressing different GFP-BNIP3L mutants (n = 3). For flow cytometry, two-way ANOVA with Tukey’s multiple comparisons test was used to analyze the difference between mitochondrial removal in cells transfected with GFP-BNIP3L dimerization mutants (n = 3). All values are expressed as mean ± SD of at least three independent experiments. *P = < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001 were used as thresholds for statistical significance.
Supplementary Material
Acknowledgments
We especially thank prof. Ivan Dikic for his critical comments on the manuscript and help with the initial experiments. We thank Dr. Jelena Korać Prlić for constructive help with statistical analysis. We also thank Dr. Petra Beli and Thomas Juretschke for their help in the revision process. We thank prof. Jasna Puizina for the use of the microscope facility.
Funding Statement
This work was supported by the Hrvatska Zaklada za Znanost [UIP-11-2013-5246]; European Cooperation in Science and Technology, Transautophagy [CA15138].
Disclosure statement
The authors declare no potential conflict of interest.
Supplementary Material
Supplemental data for this article can be accessed here.
References
- [1].Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005;171(4):603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kirkin V, Lamark T, Sou Y-S, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33(4):505–516. [DOI] [PubMed] [Google Scholar]
- [3].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007. August;282(33):24131–24145. [DOI] [PubMed] [Google Scholar]
- [4].N. von M. & F. R, Thurston TLM, Ryzhakov G, Bloor S.. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10(11):1215–1222. [DOI] [PubMed] [Google Scholar]
- [5].Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Reports. 2010. January;11(1):45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. 2020;432(1):80–103. [DOI] [PubMed] [Google Scholar]
- [7].Rodolfo C, Campello S, Cecconi F. Mitophagy in neurodegenerative diseases. Neurochem Int. 2018. July 01;117 : 156–166. [DOI] [PubMed] [Google Scholar]
- [8].Palikaras K, Daskalaki I, Markaki M, et al. Mitophagy and age-related pathologies: development of new therapeutics by targeting mitochondrial turnover. Pharmacol Ther. 2017. October 01;178: 157–174. [DOI] [PubMed] [Google Scholar]
- [9].Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013. January;20(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ney PA. Mitochondrial autophagy: origins, significance, and role of BNIP3 and NIX. Biochim Biophys Acta Mol Cell Res. 2015. October 01;1853(10):2775–2783. [DOI] [PubMed] [Google Scholar]
- [11].Costello MJ, Brennan LA, Basu S, et al. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp. Eye Res. 2013. November;116:141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Esteban-Martínez L, Sierra‐Filardi E, McGreal RS, et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. The EMBO Journal. 2017;36(12):1688–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sato M, Sato K. Degradation of paternal mitochondria. Science. 2011;37(November):1141–1144. [DOI] [PubMed] [Google Scholar]
- [14].Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. 2007. December;104(49):19500–19505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008. July;454(7201):232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Noda NN, Ohsumi Y, Inagaki F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett. 2010. April;584(7):1379–1385. [DOI] [PubMed] [Google Scholar]
- [17].Zhu Y, Massen S, Terenzio M, et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013. January;288(2):1099–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Murakawa T, Yamaguchi O, Hashimoto A, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015. July;6(1):7527–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012. February;14(2):177–185. [DOI] [PubMed] [Google Scholar]
- [20].Di Rita A, Peschiaroli A, D Acunzo P, et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat Commun. 2018; December;9(1):3755–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bhujabal Z, Birgisdottir ÅB, Sjøttem E, et al. FKBP8 recruits LC3A to mediate Parkin‐independent mitophagy. EMBO Reports. 2017. June;18(6):947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wei Y, Chiang WC, Sumpter R, et al. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168(1–2):224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16(7):939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yasuda GCM, Theodorakis P, Subramanian T. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J Biol Chem. 1998;273(20):12415–12421. [DOI] [PubMed] [Google Scholar]
- [25].Chen G, Cizeau J, Vande Velde C, et al. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J. Biol. Chem. 1999. January;274(1):7–10. [DOI] [PubMed] [Google Scholar]
- [26].Sulistijo ES, MacKenzie KR. Sequence dependence of BNIP3 transmembrane domain dimerization implicates side-chain hydrogen bonding and a tandem GxxxG motif in specific helix-helix interactions. J Mol Biol. 2006. December;364(5):974–990. [DOI] [PubMed] [Google Scholar]
- [27].Russ WP, Engelman DM. The GxxxG motif: A framework for transmembrane helix-helix association. J Mol Biol. 2000. February;296(3):911–919. [DOI] [PubMed] [Google Scholar]
- [28].Chen G, Ray R, Dubik D, et al. The E1B 19K/Bcl-2–binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med. 1997. December;186(12):1975–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schwarten M, Mohrlüder J, Ma P, et al. Nix directly binds to GABARAP: A possible crosstalk between apoptosis and autophagy. Autophagy. 2009;5(5):690–698. [DOI] [PubMed] [Google Scholar]
- [30].Rogov VV, Suzuki H, Marinković M, et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017. December;7(1):1131–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Imazu T, Shimizu S, Tagami S, et al. Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with Bcl-2/Bcl-x(L) and induces apoptosis by altering mitochondrial membrane permeability. Oncogene. 1999. August;18(32):4523–4529. [DOI] [PubMed] [Google Scholar]
- [32].Vengellur A, LaPres JJ. The role of hypoxia inducible factor 1α in cobalt chloride induced cell death in mouse embryonic fibroblasts. Toxicol Sci. 2004. December;82(2):638–646. [DOI] [PubMed] [Google Scholar]
- [33].Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kirkin V, McEwan DG, Novak I, et al. A role for ubiquitin in selective autophagy. Mol Cell. 2009. May 15;34(3):259–269. [DOI] [PubMed] [Google Scholar]
- [35].Wild P, Farhan H, McEwan DG, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333(6039):228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16(6):495–501. [DOI] [PubMed] [Google Scholar]
- [37].Yan C, Gong L, Chen L, et al. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy. 2020;16(3):419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Park YS, Choi SE, Koh HC. PGAM5 regulates PINK1/Parkin-mediated mitophagy via DRP1 in CCCP-induced mitochondrial dysfunction. Toxicol Lett. 2018;284(December 2017):120–128. [DOI] [PubMed] [Google Scholar]
- [39].Chen G, Han Z, Feng D, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54(3):362–377. [DOI] [PubMed] [Google Scholar]
- [40].Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured filroblasts. Nat Protoc. 2007;2(2):287–295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.