

Abstract
Background
Major depressive disorders have a significant impact on children and adolescents, including on educational and vocational outcomes, interpersonal relationships, and physical and mental health and well‐being. There is an association between major depressive disorder and suicidal ideation, suicide attempts, and suicide. Antidepressant medication is used in moderate to severe depression; there is now a range of newer generations of these medications.
Objectives
To investigate, via network meta‐analysis (NMA), the comparative effectiveness and safety of different newer generation antidepressants in children and adolescents with a diagnosed major depressive disorder (MDD) in terms of depression, functioning, suicide‐related outcomes and other adverse outcomes. The impact of age, treatment duration, baseline severity, and pharmaceutical industry funding was investigated on clinician‐rated depression (CDRS‐R) and suicide‐related outcomes.
Search methods
We searched the Cochrane Common Mental Disorders Specialised Register, the Cochrane Library (Central Register of Controlled Trials (CENTRAL) and Cochrane Database of Systematic Reviews (CDSR)), together with Ovid Embase, MEDLINE and PsycINFO till March 2020.
Selection criteria
Randomised trials of six to 18 year olds of either sex and any ethnicity with clinically diagnosed major depressive disorder were included. Trials that compared the effectiveness of newer generation antidepressants with each other or with a placebo were included. Newer generation antidepressants included: selective serotonin reuptake inhibitors; selective norepinephrine reuptake inhibitors (SNRIs); norepinephrine reuptake inhibitors; norepinephrine dopamine reuptake inhibitors; norepinephrine dopamine disinhibitors (NDDIs); and tetracyclic antidepressants (TeCAs).
Data collection and analysis
Two reviewers independently screened titles/abstracts and full texts, extracted data, and assessed risk of bias. We analysed dichotomous data as Odds Ratios (ORs), and continuous data as Mean Difference (MD) for the following outcomes: depression symptom severity (clinician rated), response or remission of depression symptoms, depression symptom severity (self‐rated), functioning, suicide related outcomes and overall adverse outcomes. Random‐effects network meta‐analyses were conducted in a frequentist framework using multivariate meta‐analysis. Certainty of evidence was assessed using Confidence in Network Meta‐analysis (CINeMA). We used "informative statements" to standardise the interpretation and description of the results.
Main results
Twenty‐six studies were included. There were no data for the two primary outcomes (depressive disorder established via clinical diagnostic interview and suicide), therefore, the results comprise only secondary outcomes. Most antidepressants may be associated with a "small and unimportant" reduction in depression symptoms on the CDRS‐R scale (range 17 to 113) compared with placebo (high certainty evidence: paroxetine: MD ‐1.43, 95% CI ‐3.90, 1.04; vilazodone: MD ‐0.84, 95% CI ‐3.03, 1.35; desvenlafaxine MD ‐0.07, 95% CI ‐3.51, 3.36; moderate certainty evidence: sertraline: MD ‐3.51, 95% CI ‐6.99, ‐0.04; fluoxetine: MD ‐2.84, 95% CI ‐4.12, ‐1.56; escitalopram: MD ‐2.62, 95% CI ‐5.29, 0.04; low certainty evidence: duloxetine: MD ‐2.70, 95% CI ‐5.03, ‐0.37; vortioxetine: MD 0.60, 95% CI ‐2.52, 3.72; very low certainty evidence for comparisons between other antidepressants and placebo).
There were "small and unimportant" differences between most antidepressants in reduction of depression symptoms (high‐ or moderate‐certainty evidence).
Results were similar across other outcomes of benefit.
In most studies risk of self‐harm or suicide was an exclusion criterion for the study. Proportions of suicide‐related outcomes were low for most included studies and 95% confidence intervals were wide for all comparisons. The evidence is very uncertain about the effects of mirtazapine (OR 0.50, 95% CI 0.03, 8.04), duloxetine (OR 1.15, 95% CI 0.72, 1.82), vilazodone (OR 1.01, 95% CI 0.68, 1.48), desvenlafaxine (OR 0.94, 95% CI 0.59, 1.52), citalopram (OR 1.72, 95% CI 0.76, 3.87) or vortioxetine (OR 1.58, 95% CI 0.29, 8.60) on suicide‐related outcomes compared with placebo. There is low certainty evidence that escitalopram may "at least slightly" reduce odds of suicide‐related outcomes compared with placebo (OR 0.89, 95% CI 0.43, 1.84). There is low certainty evidence that fluoxetine (OR 1.27, 95% CI 0.87, 1.86), paroxetine (OR 1.81, 95% CI 0.85, 3.86), sertraline (OR 3.03, 95% CI 0.60, 15.22), and venlafaxine (OR 13.84, 95% CI 1.79, 106.90) may "at least slightly" increase odds of suicide‐related outcomes compared with placebo.
There is moderate certainty evidence that venlafaxine probably results in an "at least slightly" increased odds of suicide‐related outcomes compared with desvenlafaxine (OR 0.07, 95% CI 0.01, 0.56) and escitalopram (OR 0.06, 95% CI 0.01, 0.56). There was very low certainty evidence regarding other comparisons between antidepressants.
Authors' conclusions
Overall, methodological shortcomings of the randomised trials make it difficult to interpret the findings with regard to the efficacy and safety of newer antidepressant medications. Findings suggest that most newer antidepressants may reduce depression symptoms in a small and unimportant way compared with placebo. Furthermore, there are likely to be small and unimportant differences in the reduction of depression symptoms between the majority of antidepressants. However, our findings reflect the average effects of the antidepressants, and given depression is a heterogeneous condition, some individuals may experience a greater response. Guideline developers and others making recommendations might therefore consider whether a recommendation for the use of newer generation antidepressants is warranted for some individuals in some circumstances. Our findings suggest sertraline, escitalopram, duloxetine, as well as fluoxetine (which is currently the only treatment recommended for first‐line prescribing) could be considered as a first option.
Children and adolescents considered at risk of suicide were frequently excluded from trials, so that we cannot be confident about the effects of these medications for these individuals. If an antidepressant is being considered for an individual, this should be done in consultation with the child/adolescent and their family/caregivers and it remains critical to ensure close monitoring of treatment effects and suicide‐related outcomes (combined suicidal ideation and suicide attempt) in those treated with newer generation antidepressants, given findings that some of these medications may be associated with greater odds of these events. Consideration of psychotherapy, particularly cognitive behavioural therapy, as per guideline recommendations, remains important.
Keywords: Adolescent; Child; Female; Humans; Male; Antidepressive Agents; Antidepressive Agents/adverse effects; Antidepressive Agents/therapeutic use; Bias; Citalopram; Citalopram/therapeutic use; Depressive Disorder, Major; Depressive Disorder, Major/drug therapy; Depressive Disorder, Major/psychology; Desvenlafaxine Succinate; Desvenlafaxine Succinate/therapeutic use; Duloxetine Hydrochloride; Duloxetine Hydrochloride/therapeutic use; Fluoxetine; Fluoxetine/therapeutic use; Mirtazapine; Mirtazapine/therapeutic use; Paroxetine; Paroxetine/therapeutic use; Selective Serotonin Reuptake Inhibitors; Selective Serotonin Reuptake Inhibitors/therapeutic use; Sertraline; Sertraline/therapeutic use; Suicidal Ideation; Venlafaxine Hydrochloride; Venlafaxine Hydrochloride/therapeutic use; Vilazodone Hydrochloride; Vilazodone Hydrochloride/therapeutic use; Vortioxetine; Vortioxetine/therapeutic use
Plain language summary
Newer generation antidepressants for depression in children and adolescents: a network meta‐analysis
How well do newer formulations of antidepressants work for children and adolescents with clinical depression?
Children and adolescents (6 to 18 years) with depression (also called ‘major depressive disorder’) experience a range of negative impacts in all areas of their lives and have an increased risk of suicide, suicidal thinking and suicide attempts. Antidepressants have been shown to reduce symptoms of depression, but can also increase the risk of suicide‐related outcomes.
Who will be interested in this research?
The research in this Cochrane Review will interest:
‐ people who decide policy, and influence decisions about the prescription of antidepressant medicines to children and adolescents;
‐ people who prescribe these medicines to children and adolescents;
‐ children and adolescents with depression; and
‐ those who support and care for them (including their parents and caregivers and clinicians who provide treatment).
What did we want to find out?
We wanted to find out how well newer formulations (called ‘new generation’) antidepressants work to improve depression in children and adolescents aged 6 to 18 years. New generation antidepressants are those that have been developed recently. They are sometimes referred to as ‘second‐‘ and ‘third‐generation’ antidepressants; they do not include older formulations (tricyclic antidepressants or monoamine oxidase inhibitors).
We wanted to know how these antidepressants affect:
‐ symptoms of depression;
‐ recovery: no longer meeting diagnostic criteria for major depressive disorder;
‐ response or remission: scores on a scale indicating an important reduction in depression or no longer experiencing depression;
‐ ability to function in daily life;
‐ suicide‐related outcomes; and
‐ whether they cause any unwanted effects in children and adolescents.
What did we do?
We searched for studies that tested new generation antidepressants on children or adolescents (or both) who had been diagnosed with a major depressive disorder. We identified 26 such studies. We then assessed the trustworthiness of those studies, and synthesized the findings across the studies.
What does the evidence from the review tell us?
Most newer antidepressants probably reduce depression symptoms better than a placebo (a 'dummy' treatment that does not contain any medicine but looks identical to the medicine being tested). However, the reduction is small and may not be experienced as important by children and adolescents, their parents and caregivers, or clinicians. When different medications are compared against each other, there may be only small and unimportant differences between most of them for the reduction of symptoms.
Our findings reflect what happens on average to individuals, but some individuals may experience a greater response. This might lead to recommendations being made for the use of antidepressants for some individuals in some circumstances. Our findings suggest that sertraline, escitalopram, duloxetine and fluoxetine can be used if medication is being considered.
The impact of medication on depression symptoms should be closely monitored by those prescribing the medication, especially as suicide‐related thinking and behaviour may be increased in those taking these medications. Close monitoring of suicide‐related behaviours is vital in those treated with new generation antidepressants.
What should happen next?
The studies that provided this evidence largely excluded children and adolescents who:
‐ were already thinking about suicide and wanting to take their own lives (i.e. had suicidal ideation);
‐ were self‐harming;
‐ had other mental health conditions; and
‐ had psychosocial difficulties.
Future research should aim to understand the impacts of these medicines in children and adolescents with these problems, who are more typical of those who request clinical services.
Summary of findings
Summary of findings 1. Summary of findings table comparing individual antidepressants on clinician‐rated depression symptoms (CDSR‐R).
| BENEFITS | ||||||
| Population: children and/or adolescents with depression | ||||||
| Interventions: new generation antidepressants including SSRIs (e.g. fluoxetine), SNRIs (e.g. duloxetine), and TeCAs (e.g. mirtazapine) | ||||||
| Comparator: other new generation antidepressant | ||||||
| Outcome: clinician‐rated depression symptoms (CDRS‐R) | ||||||
| Setting: primary care, community settings, specialist settings | ||||||
| Anticipated absolute effect (95% CI) | ||||||
|
Total studies: 22 Total participants: 5750 |
MD on CDRS‐R (95% CI) |
Without intervention | With intervention | Certainty of evidence | Ranking (P value) | Interpretation |
| fluoxetine:desvenlafaxine | ‐2.77 (‐6.20, 0.66) | ‐ | ‐ |
Moderate due to imprecision2 |
0.72 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| fluoxetine:duloxetine | ‐0.14 (‐2.46, 2.19) | ‐ | ‐ |
Moderate due to incoherence4 |
0.67 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| fluoxetine:vilazodone | ‐2.00 (‐4.40, 0.41) | ‐ | ‐ |
Moderate due to heterogeneity5 |
0.72 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| fluoxetine:vortioxetine | ‐3.44 (‐6.56, ‐0.33) |
‐ | ‐ |
Very Low due to within‐study bias3, heterogeneity5 |
0.72 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| citalopram:desvenlafaxine | ‐2.83 (‐9.16, 3.51) | ‐ | ‐ |
Low due to within‐study bias1, imprecision,2 |
0.65 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| duloxetine:citalopram | 0.20 (‐5.62, 6.01) |
‐ | ‐ |
Very Low due to within‐study bias,3 imprecision7 |
0.67 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| escitalopram:citalopram | 0.28 (‐5.68, 6.23) | ‐ | ‐ |
Very Low due to within‐study bias,3 imprecision7 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| fluoxetine:citalopram | 0.06 (‐5.42, 5.54) | ‐ | ‐ |
Very Low due to within‐study bias,3 imprecision7 |
0.72 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| mirtazapine:citalopram | 0.11 (‐6.51, 6.73) |
‐ | ‐ |
Very Low due to within‐study bias,3 imprecision7 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| citalopram:paroxetine | ‐1.47 (‐7.34, 4.40) | ‐ | ‐ |
Very Low due to within‐study bias,1 imprecision,2 heterogeneity5 |
0.65 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| sertraline:citalopram | ‐0.61 (‐6.97, 5.74) |
‐ | ‐ |
Very Low due to within‐study bias,1 imprecision7 |
0.77 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| citalopram:venlafaxine | ‐1.00 (‐7.25, 5.25) |
‐ | ‐ |
Very Low due to within‐study bias,3 imprecision7 |
0.65 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| citalopram:vilazodone | ‐2.06 (‐7.82, 3.70) |
‐ | ‐ |
Low due to within‐study bias,1 imprecision2 |
0.65 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| citalopram:vortioxetine | ‐3.50 (‐9.67, 2.67) |
‐ | ‐ |
Very Low due to within‐study bias,3 imprecision2 |
0.65 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| duloxetine:desvenlafaxine | ‐2.63 (‐6.68, 1.42) |
‐ | ‐ |
Moderate due to imprecision2 |
0.67 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| escitalopram:desvenlafaxine | ‐2.55 (‐6.89, 1.80) |
‐ | ‐ |
Moderate due to imprecision2 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| mirtazapine:desvenlafaxine | ‐2.71 (‐7.93, 2.51) |
‐ | ‐ |
Low Due to within‐study bias,1 imprecision2 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| paroxetine:desvenlafaxine | ‐1.35 (‐5.58, 2.87) |
‐ | ‐ |
Moderate due to imprecision2 |
0.45 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| sertraline:desvenlafaxine | ‐3.44 (‐8.32, 1.44) |
‐ | ‐ |
Moderate due to imprecision2 |
0.77 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| venlafaxine:desvenlafaxine | ‐1.83 (‐6.57, 2.91) |
‐ | ‐ |
Low due to within‐study bias,1 imprecision2 |
0.53 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| vilazodone:desvenlafaxine | ‐0.77 (‐4.80, 3.26) |
‐ | ‐ |
Moderate Due to heterogeneity5 |
0.34 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| desvenlafaxine:vortioxetine | ‐0.68 (‐5.22, 3.87) |
‐ | ‐ |
Low due to within‐study bias,1 imprecision2 |
0.24 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| duloxetine: escitalopram | ‐0.08 (‐3.62, 3.46) |
‐ | ‐ | High | 0.67 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| duloxetine:mirtazapine | 0.08 (‐4.49, 4.65) |
‐ | ‐ |
Very Low due to within‐study bias,1heterogeneity6 |
0.67 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| duloxetine:paroxetine | ‐1.27 (‐4.67, 2.12) |
‐ | ‐ |
Moderate due to heterogeneity5 |
0.67 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| sertraline:duloxetine | ‐0.81 (‐4.99, 3.37) |
‐ | ‐ |
Moderate due to heterogeneity5 |
0.77 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| duloxetine:venlafaxine | ‐0.80 (‐4.82, 3.21) |
‐ | ‐ |
Low due to within‐study bias,1heterogeneity5 |
0.67 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| duloxetine:vilazodone | ‐1.86 (‐5.01, 1.29) |
‐ | ‐ |
Moderate due to imprecision2 |
0.67 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| duloxetine:vortioxetine | ‐3.30 (‐7.09, 0.48) |
‐ | ‐ |
Low due to within‐study bias,1 imprecision2 |
0.67 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| fluoxetine:escitalopram | ‐0.22 (‐3.18, 2.74) |
‐ | ‐ | High | 0.72 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| escitalopram:mirtazapine | 0.17 (‐4.58, 4.92) |
‐ | ‐ |
Very Low due to within‐study bias,1heterogeneity6 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| escitalopram:paroxetine | ‐1.19 (‐4.83, 2.44) |
‐ | ‐ |
Moderate due to heterogeneity5 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| sertraline:escitalopram | ‐0.89 (‐5.27, 3.48) |
‐ | ‐ |
Moderate due to imprecision2 |
0.77 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| escitalopram:venlafaxine | ‐0.72 (‐4.94, 3.50) |
‐ | ‐ |
Low due to within‐study bias,1heterogeneity5 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| escitalopram:vilazodone | ‐1.78 (‐5.23, 1.67) |
‐ | ‐ |
Moderate due to imprecision2 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| escitalopram:vortioxetine | ‐3.22 (‐7.32, 0.88) |
‐ | ‐ |
Low due to within‐study bias,1 imprecision2 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| fluoxetine:mirtazapine | ‐0.05 (‐4.19, 4.08) |
‐ | ‐ |
Very Low due to within‐study bias,1heterogeneity6 |
0.72 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| fluoxetine:paroxetine | ‐1.41 (‐4.20, 1.37) |
‐ | ‐ |
Moderate due to heterogeneity5 |
0.72 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| sertraline:fluoxetine | ‐0.67 (‐4.37, 3.03) |
‐ | ‐ |
Moderate due to heterogeneity5 |
0.77 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| fluoxetine:venlafaxine | ‐0.94 (‐4.45, 2.57) |
‐ | ‐ |
Low due to within‐study bias,1heterogeneity5 |
0.72 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| mirtazapine:paroxetine | ‐1.36 (‐6.00, 3.28) |
‐ | ‐ |
Low due to within‐study bias,1 imprecision2 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| sertraline:mirtazapine | ‐0.73 (‐5.97, 4.52) |
‐ | ‐ |
Very Low due to within‐study bias,1 imprecision,2 heterogeneity5 |
0.77 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| mirtazapine:venlafaxine | ‐0.89 (‐6.00, 4.23) |
‐ | ‐ |
Very Low due to within‐study bias,3 imprecision,2 heterogeneity5 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| mirtazapine:vilazodone | ‐1.94 (‐6.44, 2.56) |
‐ | ‐ |
Low due to within‐study bias,1 imprecision2 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| mirtazapine:vortioxetine | ‐3.39 (‐8.41, 1.63) |
‐ | ‐ |
Very Low due to within‐study bias,3 imprecision2 |
0.66 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| sertraline:paroxetine | ‐2.09 (‐6.35, 2.17) |
‐ | ‐ |
Moderate due to imprecision2 |
0.77 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| venlafaxine:paroxetine | ‐0.47 (‐4.57, 3.62) |
‐ | ‐ |
Low due to within‐study bias,1heterogeneity5 |
0.53 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| paroxetine:vilazodone | ‐0.58 (‐3.89, 2.72) |
‐ | ‐ | High | 0.45 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| paroxetine:vortioxetine | ‐2.03 (‐6.01, 1.95) |
‐ | ‐ |
Low due to within‐study bias,1 imprecision2 |
0.45 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| sertraline:venlafaxine | ‐1.61 (‐6.38, 3.15) |
‐ | ‐ |
Low due to within‐study bias,1 imprecision2 |
0.77 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| sertraline:vilazodone | ‐2.67 (‐6.78, 1.43) |
‐ | ‐ |
Moderate due to imprecision2 |
0.77 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| sertraline:vortioxetine | ‐4.12 (‐8.78, 0.55) |
‐ | ‐ |
Low due to within‐study bias,1 imprecision2 |
0.77 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| venlafaxine:vilazodone | ‐1.06 (‐4.99, 2.88) |
‐ | ‐ |
Low due to within‐study bias,1heterogeneity5 |
0.53 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R |
| venlafaxine:vortioxetine | ‐2.50 (‐7.02, 2.02) |
Very Low due to within‐study bias,3 imprecision2 |
0.53 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R | ||
| vilazodone:vortioxetine | ‐1.44 (‐5.21, 2.32) |
Low due to within‐study bias,1 imprecision2 |
0.34 | We proposed an equivalence range of ‐5, 5 points on the CDRS‐R | ||
MD = mean difference, CI = confidence interval, PI = prediction interval
1. studies were at moderate risk of bias based on weighted average percentage contribution to effect estimate
2. 95% CI crossed equivalence range (MD ‐5, 5 points on the CDRS‐R)
3. studies were at high risk of bias based on weighted average percentage contribution to effect estimate
4. potential inconsistency between direct and indirect evidence (but inconclusive due to limited direct evidence)
5. 95% PI crossed equivalence range (MD ‐5, 5 points on the CDRS‐R)
6. 95% PI crossed equivalence range in both directions (MD ‐5, 5 points on the CDRS‐R)
7. 95% CI crossed equivalence range in both directions (MD ‐5, 5 points on the CDRS‐R)
Summary of findings 2. Summary of findings table comparing individual antidepressants on suicidal behaviour.
| HARMS | ||||||
| Population: children and/or adolescents with depression | ||||||
| Interventions: new generation antidepressants including SSRIs (e.g. fluoxetine), SNRIs (e.g. duloxetine), and TeCAs (e.g. mirtazapine ) | ||||||
| Comparator: placebo or other new generation antidepressant | ||||||
| Outcome: suicidal behaviour according to the Columbia Classification system, e.g. suicidal ideation, suicide attempt | ||||||
| Setting: primary care, community settings, specialist settings | ||||||
| Anticipated Absolute effects (95% CI) | ||||||
|
Total studies: 21 RCTs Total participants: 6413 |
Assumed comparator risk per 1000 | Corresponding intervention risk per 1000 |
Relative effect: OR (95% CI) |
Certainty of evidence | Ranking (P value) | Interpretation |
| desvenlafaxine:fluoxetine | 82.6 | 62.47 (38.11, 102.61) |
0.74 (0.44, 1.27) |
Low due to imprecision1 |
0.73 | equivalence range from OR 0.90, 1.12 |
| duloxetine:fluoxetine | 82.6 | 74.96 (48.82,114.77) |
0.9 (0.57,1.44) |
Very Low due to imprecision, 1incoherence8 |
0.57 | equivalence range from OR 0.90, 1.12 |
| vilazodone:fluoxetine | 82.6 | 67.19 (39.77, 109.81) |
0.8 (0.46,1.37) |
Very low due to imprecision,1and within‐study bias2 |
0.68 | equivalence range from OR 0.90, 1.12 |
| fluoxetine:vortioxetine | 16.2 | 13.00 (2.46,64.54) |
0.8 (0.15,4.19) |
Very low due to imprecision1, and within‐study bias3 |
0.47 | equivalence range from OR 0.90, 1.12 |
| desvenlafaxine:citalopram | 70.1 | 39.81 (15.58,96.08) |
0.55 (0.21,1.41) |
Very low due to imprecision1, and within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| duloxetine:citalopram | 70.1 | 48.08 (19.22,113.60) |
0.67 (0.26,1.7) |
Very low due to imprecision1, and within‐study bias2 |
0.57 | equivalence range from OR 0.90, 1.12 |
| escitalopram:citalopram | 70.1 | 37.72 (12.65, 104.02) |
0.52 (0.17,1.54) |
Very low due to imprecision1, and within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| fluoxetine:citalopram | 70.1 | 52.84 (22.12, 120.06) |
0.74 (0.30,1.81) |
Very low due to imprecision1, and within‐study bias2 |
0.47 | equivalence range from OR 0.90, 1.12 |
| mirtazapine:citalopram | 70.1 | 21.39 (1.51, 283.94) |
0.29 (0.02,5.26) |
Very low due to imprecision1, and within‐study bias3 |
0.76 | equivalence range from OR 0.90, 1.12 |
| citalopram:paroxetine | 26.9 | 25.59 (8.50, 73.98) |
0.95 (0.31,2.89) |
Very low due to imprecision1, and within‐study bias2 |
0.35 | equivalence range from OR 0.90, 1.12 |
| citalopram:sertraline | 31.7 | 18.32 (2.94, 101.48) |
0.57 (0.09,3.45) |
Very low due to imprecision1, and within‐study bias2 |
0.35 | equivalence range from OR 0.90, 1.12 |
| citalopram:venlafaxine | 70.7 | 9.05 (0.76, 78.52) |
0.12 (0.01,1.12) |
Very low due to imprecision1, and within‐study bias3 |
0.35 | equivalence range from OR 0.90, 1.12 |
| vilazodone:citalopram | 70.1 | 42.58 (17.77, 97.92) |
0.59 (0.24,1.44) |
Very low due to imprecision1, and within‐study bias3 |
0.68 | equivalence range from OR 0.90, 1.12 |
| vortioxetine:citalopram | 70.1 | 64.86 (10.44, 312.51) |
0.92 (0.14,6.03) |
Very low due to imprecision1, and within‐study bias2 |
0.45 | equivalence range from OR 0.90, 1.12 |
| desvenlafaxine:duloxetine | 134.5 | 113.03 (64.00, 194.12) |
0.82 (0.44,1.55) |
Low due to imprecision1 |
0.73 | equivalence range from OR 0.90, 1.12 |
| escitalopram:desvenlafaxine | 127.45 | 120.73 (55.20,246.53) |
0.94 (0.4,2.24) |
Very low due to imprecision1, and within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| mirtazapine:desvenlafaxine | 127.45 | 71.85 (4.36, 564.11) |
0.53 (0.03,8.86) |
Very low due to imprecision1, and within‐study bias2 |
0.76 | equivalence range from OR 0.90, 1.12 |
| desvenlafaxine:paroxetine | 26.9 | 14.17 (5.77,34.17) |
0.52 (0.21,1.28) |
Very low due to imprecision1, and within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| desvenlafaxine:sertraline | 31.7 | 10.05 (1.96,51.84) |
0.31 (0.06,1.67) |
Very low due to imprecision1, and within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| desvenlafaxine:venlafaxine | 70.7 | 5.30 (0.76, 40.86) |
0.07 (0.01,0.56) |
Moderate due to within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| desvenlafaxine:vilazodone | 167.7 | 159.24 (93.18, 257.37) |
0.94 (0.51,1.72) |
Very low due to imprecision1, and within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| desvenlafaxine:vortioxetine | 16.2 | 9.78 (1.81, 52.87) |
0.6 (0.11,3.39) |
Very low due to imprecision1, and within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| escitalopram:duloxetine | 134.5 | 108.11 (48.78, 222.36) |
0.78 (0.33,1.84) |
Low due to imprecision1 |
0.73 | equivalence range from OR 0.90, 1.12 |
| mirtazapine:duloxetine | 134.5 | 62.64 (4.64,531.49) |
0.43 (0.03,7.30) |
Very low due to imprecision1, and within‐study bias2 |
0.76 | equivalence range from OR 0.90, 1.12 |
| duloxetine:paroxetine | 26.9 | 17.12 (7.14, 40.83) |
0.63 (0.26,1.54) |
Low due to imprecision1 |
0.57 | equivalence range from OR 0.90, 1.12 |
| duloxetine:sertraline | 31.7 | 12.29 (2.29, 62.03) |
0.38 (0.07,2.02) |
Low due to imprecision1 |
0.57 | equivalence range from OR 0.90, 1.12 |
| duloxetine:venlafaxine | 70.7 | 6.05 (0.76, 48.50) |
0.08 (0.01,0.67) |
Low due to within‐study bias2 and heterogeneity4 |
0.57 | equivalence range from OR 0.90, 1.12 |
| vilazodone:duloxetine | 134.5 | 120.30 (69.41,200.13) |
0.88 (0.48,1.61) |
Very low due to imprecision1, and within‐study bias2 |
0.68 | equivalence range from OR 0.90, 1.12 |
| duloxetine:vortioxetine | 16.2 | 11.72 (2.14,62.23) |
0.72 (0.13,4.03) |
Very low due to imprecision1, and within‐study bias2 |
0.57 | equivalence range from OR 0.90, 1.12 |
| escitalopram:fluoxetine | 82.6 | 59.29 (27.15, 125.23) |
0.7 (0.31,1.59) |
Low due to imprecision1 |
0.73 | equivalence range from OR 0.90, 1.12 |
| mirtazapine:escitalopram | 49.25 | 28.19 (1.55, 339.44) |
0.56 (0.03,9.92) |
Very low due to imprecision,1 and within‐study bias2 |
0.76 | equivalence range from OR 0.90, 1.12 |
| escitalopram:paroxetine | 26.9 | 13.36 (4.68,37.52) |
0.49 (0.17,1.41) |
Low due to imprecision1 |
0.73 | equivalence range from OR 0.90, 1.12 |
| escitalopram:sertraline | 31.7 | 9.40 (1.63, 53.31) |
0.29 (0.05,1.72) |
Low due to imprecision1 |
0.73 | equivalence range from OR 0.90, 1.12 |
| escitalopram:venlafaxine | 70.7 | 4.54 (0.76,40.86) |
0.06 (0.01,0.56) |
Moderate due to within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| escitalopram:vilazodone | 167.7 | 150.61 (72.86, 288.25) |
0.88 (0.39,2.01) |
Very low due to imprecision1, and within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| escitalopram:vortioxetine | 16.2 | 9.14 (1.48, 55.38) |
0.56 (0.09,3.56) |
Very low due to imprecision1, and within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| mirtazapine:fluoxetine | 82.6 | 33.92 (1.80, 369.54) |
0.39 (0.02,6.51) |
Very low due to imprecision1, and within‐study bias2 |
0.76 | equivalence range from OR 0.90, 1.12 |
| fluoxetine:paroxetine | 26.9 | 18.98 (8.22, 43.37) |
0.7 (0.3,1.64) |
Low due to imprecision1 |
0.47 | equivalence range from OR 0.90, 1.12 |
| fluoxetine:sertraline | 31.7 | 13.56 (2.61,66.90) |
0.42 (0.08,2.19) |
Low due to imprecision1 |
0.47 | equivalence range from OR 0.90, 1.12 |
| fluoxetine:venlafaxine | 70.7 | 6.80 (0.76, 52.62) |
0.09 (0.01,0.73) |
Very Low due to within‐study bias2 and heterogeneity5 |
0.47 | equivalence range from OR 0.90, 1.12 |
| mirtazapine:paroxetine | 26.9 | 7.68 (0.55, 119.94) |
0.28 (0.02,4.93) |
Very low due to imprecision1, and within‐study bias2 |
0.76 | equivalence range from OR 0.90, 1.12 |
| mirtazapine:sertraline | 31.7 | 5.21 (0.33, 118.09) |
0.16 (0.01,4.09) |
Very low due to imprecision1, and within‐study bias2 |
0.76 | equivalence range from OR 0.90, 1.12 |
| mirtazapine:venlafaxine | 70.7 | 3.03 (0.76, 79.81) |
0.04 (0.01,1.14) |
Very low due to imprecision1, and within‐study bias3 |
0.76 | equivalence range from OR 0.90, 1.12 |
| mirtazapine:vilazodone | 167.7 | 89.86 (6.01, 622.67) |
0.49 (0.03,8.19) |
Very low due to imprecision1, and within‐study bias3 |
0.76 | equivalence range from OR 0.90, 1.12 |
| mirtazapine:vortioxetine | 16.2 | 5.08 (0.16,118.84) |
0.31 (0.01,8.19) |
Very Low due to imprecision1, and within‐study bias2 |
0.76 | equivalence range from OR 0.90, 1.12 |
| paroxetine:sertraline | 31.7 | 19.26 (3.26, 103.86) |
0.6 (0.1,3.54) |
Low due to imprecision1 |
0.32 | equivalence range from OR 0.90, 1.12 |
| paroxetine:venlafaxine | 70.7 | 9.79 (0.76,81.09) |
0.13 (0.01,1.16) |
Very low due to imprecision1, and within‐study bias2 |
0.32 | equivalence range from OR 0.90, 1.12 |
| vilazodone:paroxetine | 26.9 | 15.24 (6.59, 34.95) |
0.56 (0.24,1.31) |
Very low due to imprecision1, and within‐study bias2 |
0.68 | equivalence range from OR 0.90, 1.12 |
| vortioxetine:paroxetine | 26.9 | 23.49 (3.86, 134.05) |
0.87 (0.14,5.6) |
Very low due to imprecision1, and within‐study bias2 |
0.45 | equivalence range from OR 0.90, 1.12 |
| sertraline:venlafaxine | 70.7 | 16.46 (1.52,183.80) |
0.22 (0.02,2.96) |
Very low due to imprecision1, and within‐study bias2 |
0.23 | equivalence range from OR 0.90, 1.12 |
| vilazodone:sertraline | 31.7 | 10.69 (1.96, 54.19) |
0.33 (0.06,1.75) |
Very low due to imprecision1, and within‐study bias2 |
0.68 | equivalence range from OR 0.90, 1.12 |
| vortioxetine:sertraline | 31.7 | 16.74 (1.63,150.46) |
0.52 (0.05,5.41) |
Very low due to imprecision1, and within‐study bias2 |
0.45 | equivalence range from OR 0.90, 1.12 |
| vilazodone:venlafaxine | 70.7 | 5.30 (0.76, 42.26) |
0.07 (0.01,0.58) |
Low due to within‐study bias3 |
0.68 | equivalence range from OR 0.90, 1.12 |
| vortioxetine:venlafaxine | 70.7 | 8.30 (0.76,110.33) |
0.11 (0.01,1.63) |
Very low due to imprecision1, and within‐study bias2 |
0.45 | equivalence range from OR 0.90, 1.12 |
| vilazodone:vortioxetine | 16.2 | 10.43 (1.81, 56.40) |
0.64 (0.11,3.63) |
Very low due to imprecision1, and within‐study bias2 |
0.68 | equivalence range from OR 0.90, 1.12 |
| citalopram:placebo | 37.3 | 62.48 (28.60, 130.39) |
1.72 (0.76, 3.87) |
Very low due to imprecision1, and within‐study bias3 |
0.35 | equivalence range from OR 0.90, 1.12 |
| desvenlafaxine:placebo | 37.3 | 35.14 (22.35, 55.62) |
0.94 (0.59, 1.52) |
Very low due to imprecision1, and within‐study bias2 |
0.73 | equivalence range from OR 0.90, 1.12 |
| duloxetine:placebo | 37.3 | 42.66 (27.14, 65.87) |
1.15 (0.72, 1.82) |
Very low due to imprecision,1 and incoherence6 |
0.57 | equivalence range from OR 0.90, 1.12 |
| escitalopram:placebo | 37.3 | 33.33 (16.39, 66.55) |
0.89 (0.43, 1.84) |
Low due to imprecision1 |
0.73 | equivalence range from OR 0.90, 1.12 |
| fluoxetine:placebo | 37.3 | 46.90 (32.61, 67.22) |
1.27 (0.87, 1.86) |
Low due to heterogeneity,4 imprecision7 |
0.47 | equivalence range from OR 0.90, 1.12 |
| mirtazapine:placebo | 37.3 | 19.00 (1.16, 237.52) |
0.50 (0.03, 8.04) |
Very low due to imprecision,1and within‐study bias3 |
0.76 | equivalence range from OR 0.90, 1.12 |
| paroxetine:placebo | 37.3 | 65.53 (31.88, 130.10) |
1.81 (0.85, 3.86) |
Low due to imprecision1 |
0.32 | equivalence range from OR 0.90, 1.12 |
| sertraline:placebo | 37.3 | 105.06 (22.72, 370.95) |
3.03 (0.60, 15.22) |
Low due to imprecision1 |
0.23 | equivalence range from OR 0.90, 1.12 |
| venlafaxine:placebo | 37.3 | 349.06 (64.86, 805.52) |
13.84 (1.79, 106.90) |
Low due to within‐study bias3 |
0.03 | equivalence range from OR 0.90, 1.12 |
| vilazodone:placebo | 37.3 | 37.66 (25.67, 54.23) |
1.01 (0.68, 1.48) |
Very low due to imprecision1, and within‐study bias3 |
0.68 | equivalence range from OR 0.90, 1.12 |
| vortioxetine:placebo | 37.3 | 57.69 (11.11, 249.93) |
1.58 (0.29, 8.60) |
Very low due to imprecision1, and within‐study bias2 |
0.45 | equivalence range from OR 0.90, 1.12 |
CI = confidence interval, PI = prediction interval, OR = odds ratio
1. 95% CI crossed equivalence range (OR = 0.90, 1.12) in both directions
2. studies were at moderate risk of bias based on weighted average percentage contribution to effect estimate
3. studies were at high risk of bias based on weighted average percentage contribution to effect estimate
4. 95% PI crossed equivalence range (OR = 0.90, 1.12)
5. 95% PI crossed equivalence range (OR = 0.90, 1.12) in both directions
6. minor differences between direct and indirect evidence
7. 95% CI crossed equivalence range (OR = 0.90, 1,12)
8. minor differences between direct and indirect evidence
Background
Description of the condition
'Major depression' is a category of mental health disorder within both of the two major international classification systems: the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM‐5) of the American Psychological Association (APA 2013); and the International Classification of Diseases from the World Health Organisation (WHO 1992; WHO 2019). According to the DSM‐5, the core features of major depression are persistent low mood and loss of enjoyment in once‐pleasurable activities, which are accompanied by a range of other symptoms including weight or appetite changes, inability to sleep or sleeping too much, psychomotor agitation or retardation (feeling restless, sluggish, loss of energy), inappropriate guilt or feelings of worthlessness, poor concentration and thoughts of death or suicide (APA 2000; APA 2013). Criteria differences for children and adolescents include the presence of irritability as an alternative to a depressed mood, in acknowledgement that depression in this age group often features irritability and can be characterised by mood fluctuations that are highly dependent on — or reactive to — circumstances (Thapar 2012). It has also been noted that depression in this age group can be characterised by comorbid anxiety, school refusal, social withdrawal, unexplained physical symptoms, decline in academic performance, substance misuse and behaviour problems (Thapar 2012; Maughan 2013). The presence of other psychiatric disorders is also common (Angold 1999; Maughan 2013).
Meta‐analytic estimates of prevalence of depression suggest rates of 2.8% (95% confidence interval (CI) 1.8 to 3.8) in children, and 5.7% (95% CI 5.1% to 6.3%) in adolescents (Costello 2006). By the age of 19 years, around 25% of adolescents are estimated to have experienced a depressive episode (Lewinsohn 1998; Kessler 2001), and one longitudinal study of a general population sample showed that by the age of 30, 53% of people in the cohort had experienced a major depressive episode at some point over their lifetime since the age of five years (Rhode 2013). Overall, these data indicate that many people across the lifespan are affected by depression. Approximately 25% of cases of the onset of major depression has occurred by the age of 19 years (Kessler 2005). During adolescence, twice as many girls as boys experience depression (Hyde 2008). Previous studies, including both community‐ and clinic‐based longitudinal cohort and case‐control studies have shown that in those experiencing adolescent‐onset depression, there is a high risk of a recurrence of depression in adulthood (Harrington 1990; Lewinsohn 1998; Weissman 1999; Dunn 2006; Fergusson 2007).
The impact of depression can be significant. In a large study of adults with depression who were being treated in a clinic setting, the earlier the age of onset, the more likely were there to be social and occupational difficulties, poor quality of life, and greater physical and mental health problems, more episodes of depression over their lifetime and more attempts of suicide (Zisook 2007). These types of impacts have again been shown in a prospective population‐based cohort study showing that children and adolescents who experienced depression had significantly increased odds of poor educational outcome, mental health and substance use problems, suicidal ideation and suicide attempts, criminal convictions, teenage parenthood, physical health problems, untimely death, and social isolation, even when childhood adversity such as the experience of abuse and the presence of psychiatric disorder as a young adult were controlled for (Copeland 2015). A review by Cash 2009 summarised various studies examining the association between depression and suicide and suicide attempt (for example, psychological autopsy studies summarised in this review) showed that approximately 60% of young people who die by suicide had a diagnosis of depression at the time of their death, and that 40% to 80% of adolescents who attempt suicide have depression (Cash 2009). Early longitudinal studies, summarised in this same review (Cash 2009), showed that up to 32% of children and adolescents with depression who were followed through to late adolescence and up to the age of 31 years attempted suicide, and between 2.5% and 3.3% had died by suicide (Cash 2009). A longitudinal study of a large birth cohort has shown that the more depressive episodes experienced in adolescence and young adulthood, the worse the outcomes in adulthood in terms of suicidal ideation and attempts, depression, anxiety, welfare dependence and unemployment (Fergusson 2007). Overall, it has been estimated that depression causes more disability for young people (aged 10 to 24 years in the 2004 WHO Global Burden of Disease study) than any other illness (Gore 2011).
Description of the intervention
Antidepressant medication is recommended for those children and adolescents with moderate to severe depression when there has been an inadequate response to psychotherapy (NICE 2019). While it is recommended that antidepressant treatment should happen alongside concurrent psychotherapy, provision is also made for antidepressant monotherapy (NICE 2019). Tricyclic antidepressants (TCAs), the mainstay of treatment in the past, have not been shown to be an effective pharmacological treatment for depression in young people (Weller 2000; Hazell 2002). This has meant that newer generation antidepressants have been increasingly used over the last 20 years (Vitiello 2006; John 2016), with initial studies suggesting they were well tolerated (Cooper 1988). Reviews of efficacy have shown modest effects of these antidepressants over the last two decades (e.g. Hetrick 2007; Hetrick 2012; Locher 2017) and have also raised concerns about the increased risk of suicide attempt and suicidal ideation (collectively referred to as suicide‐related behaviour; Dubicka 2006; Hammad 2006; Hetrick 2007; Hetrick 2012).
This review has included second and third generation antidepressants, which together are referred to as 'newer generation' antidepressants. Selective serotonin reuptake inhibitors (SSRIs) are sometimes referred to as 'second generation' antidepressants. In addition to SSRIs, several other classes of antidepressants are now being used, including selective norepinephrine reuptake inhibitors (SNRIs), norepinephrine reuptake inhibitors (NRIs), norepinephrine‐dopamine reuptake inhibitors (NDRIs), norepinephrine‐dopamine disinhibitors (NDDIs) and tetracyclic antidepressants (TeCAs). These newer additional classes are sometimes referred to as 'third generation' antidepressants. Rather than being a homogenous group based on mechanisms of action, however, third generation antidepressants are classed together because they are modified versions of first and second generation antidepressants (Olver 2001).
How the intervention might work
Depressive symptoms were first linked to an underlying depletion in monoamines, notably serotonin, noradrenaline and possibly dopamine in the central nervous system over 50 years ago, with evidence that monoamine‐depleting medications could precipitate depressive symptoms, while agents that increase their levels in the brain have been shown to alleviate depressive symptoms (Delgado 2000). In line with the recommendations for adults, SSRIs are considered first‐line treatment for adolescents, partly because the noradrenergic system matures later than the serotonergic system, potentially explaining observed differences in response to antidepressants by children and adolescents compared with adults (Cousins 2015).
Most antidepressant medications target monoamine transmitter function (Harmer 2017), with increases in synaptic concentrations of serotonin and noradrenaline, although the onset of the chemical effect, which is within hours, is much faster than the clinical effect, which can take days or weeks. The delayed onset of action of the medications has led to research in the following three main areas (Harmer 2017).
1. Neurochemical theories
Initial research focused on the down‐regulation of post‐synaptic β‐adrenoceptors by first generation tricyclic and monoamine oxidase inhibitor antidepressants. With the advent of SSRIs, researchers focused on the initial inhibition of the reuptake of serotonin (5‐hydroxytryptamine, or 5‐HT) (Lenox 2008) which was shown, over time, to reduce sensitivity of 5‐HT auto‐receptors and was postulated to be linked to the delayed clinical effect (Castrén 2005).
2. Neuroplasticity theories
A greater understanding of pathways that regulate neuronal function has led to research that moves beyond the impact on receptor function to a greater focus on intracellular mechanisms, gene expression and protein translation as possible mediators of antidepressant action. Neuroplasticity, or the ability of the nervous system to react and adapt to environmental stimuli, appears to underpin both depression and the action of antidepressants. Synaptic plasticity is reduced by chronic stress, with a reduction in the number and function of synapses particularly in the prefrontal cortex and hippocampus. Stress also decreases the formation of neurons in the hippocampus. Depressive disorder is associated with a decrease in volume in key areas in the prefrontal cortex and hippocampus (Price 2010; MacQueen 2011). Brain‐derived neurotrophic factor (BDNF) is postulated to be a transducer of some of these effects (Björkholm 2016). BDNF has been shown to play a key role in the formation and survival of neurons and to increase synaptic plasticity. It has been shown to be decreased in 'stress in animal' studies and in post‐mortem studies of humans with depression. Longer‐term use of SSRIs has been shown to increase BDNF expression, to increase synaptic plasticity and to block stress‐related synaptic deficits (Castrén 2014; Castrén 2017).
3. Cognitive neuropsychological approaches
A negative affective bias, with differential attention to negative rather than positive stimuli, has been shown to be related to depressive symptoms. Antidepressant medications have been shown to decrease the negative attentional bias; for example, SSRIs reduce the response to negative facial expressions in the amygdala, both in the short and long term (Murphy 2009). It is postulated that the later impact on depressive symptoms may be dependent on an interaction with the environment, so that habitual negative responses to cues in the environment are re‐learnt within a more positive cognitive frame. Links between these processes and synaptic plasticity remain unclear (Harmer 2017).
Recent research on rapidly acting antidepressants such as ketamine has lent weight to some of the neurochemical and neuroplasticity theories (Harmer 2017). However, it is important to recognise that while there is progress in understanding the underlying mechanisms of antidepressant medications, further elucidation is needed. Integrating the work from different schools of thought will be needed to develop new and more effective treatments.
Why it is important to do this review
Evidence‐based guideline‐recommended treatments for depression in young people include psychotherapy (cognitive behaviour therapy (CBT) and interpersonal psychotherapy (IPT)) as well as SSRIs (fluoxetine, in the first instance) (e.g. AACAP 2007; McDermott 2011; Cheung 2018; NICE 2019). The modest effects of all guideline‐recommended treatments has been the focus of many reviews over the last two decades (e.g. Weersing 2006; Weisz 2006; Locher 2017), including the Cochrane Review of antidepressants for children and adolescents (Hetrick 2007; Hetrick 2012). However, concerns about the increased risk of suicide, suicide attempt and suicidal ideation (collectively referred to as suicide‐related behaviour) for those administered SSRIs were first raised in 2003 (Healy 2003). Meta‐analyses examining the risks of suicide‐related behaviour have shown a consistent and modest increased risk for those taking SSRIs compared with placebo (Dubicka 2006; Hammad 2006). The evidence about these risks has led to action by regulatory bodies: the Committee on Safety of Medicines/Medicines and Healthcare products Regulatory Agency (CSM/MHRA) in the UK (CSM 2004; MHRA 2014), the European Medicines Agency (EMA 2005), and the US Food and Drug Administration (FDA 2018) have all cautioned practitioners on the use of SSRIs in children and adolescents, including an FDA 'black box' warning label issued on 14 September 2004, which notifies healthcare providers of this evidence of an increased risk of suicide‐related behaviour (FDA 2018). The impact of these actions by regulatory bodies and reactions to it in the media is unclear, with some early evidence of reduced prescriptions (Gibbons 2007; Lu 2014) and more recent evidence suggesting ongoing increases in antidepressant prescribing (Plöderl 2019; Whitely 2020).
The use of medications, and in particular fluoxetine, is still recommended in guidelines (AACAP 2007; NICE 2019); however, continuing concerns about the efficacy and safety of these treatments warrant an update to the Hetrick 2007 and Hetrick 2012 Cochrane Reviews of antidepressants for children and adolescents, ensuring the inclusion of trials of all recently available newer generation antidepressants, and investigating, using network meta‐analysis (NMA), comparative effectiveness and safety outcomes. NMA combines all available direct and indirect evidence on relative intervention effects to allow effect estimates for all comparisons, even when head‐to‐head trials are not available, as is the case for this class of medications for child and adolescent depression.
Objectives
To investigate, via network meta‐analysis (NMA), the comparative effectiveness and safety of different newer generation antidepressants in children and adolescents with a diagnosed major depressive disorder (MDD). Specific objectives, in order of priority, are to:
estimate the relative effects of newer generation antidepressants compared with placebo and with each other, on depression, functioning, suicide‐related outcomes and other adverse outcomes;
estimate the relative ranking of the included newer generation antidepressants for the outcomes of depression, functioning, suicide‐related outcomes and other adverse outcomes; and,
examine whether the relative effects on clinician‐rated depression symptoms and suicide‐related outcomes estimated in objectives 1 and 2 are modified by age, treatment duration, baseline severity and pharmaceutical industry funding of the antidepressant under evaluation.
Methods
Criteria for considering studies for this review
Types of studies
All variants of randomised trials (RTs) were eligible for inclusion in the review (e.g. individually randomised, cross‐over, cluster trials). However, we only included the first period data (if possible) in cross‐over trials in which there was less than one week's washout because, in this circumstance, there is a serious risk of carry‐over effects arising from the effects of the first‐period antidepressant persisting into subsequent period(s) (Hosenbocus 2011). We did not include trials in which the treatment assignment was decided through a deterministic method, such as alternate days of the week. Similarly, non‐randomised designs to examine the effects of antidepressants on adverse effects were not included. No language restrictions were used. We included both published and unpublished trial data and reports.
Types of participants
Participant characteristics
Trials involving children and adolescents aged six to 18 years old of either sex and any ethnicity were included. Trials where both adults and children/adolescents were treated were eligible for inclusion, as long as data on the children/adolescents were available separately or by obtaining data from the trial authors where randomisation was maintained; however, no such trials were included.
Diagnosis
Trials that focused on the acute phase treatment of clinically diagnosed major depressive disorder (MDD) were included.
Trials that adopted any standardised diagnostic criteria to define participants suffering from an acute phase unipolar depressive disorder were included. Accepted diagnostic criteria included DSM‐III (APA 1980), DSM‐III‐R (APA 1987), DSM‐IV (APA 1994), DSM‐IV‐TR (APA 2000), DSM‐5 (APA 2013) or ICD‐9 or ICD‐10 (WHO 1992; WHO 2019).
Trials that focused on treatment‐resistant depression, including those where participants were receiving treatment to prevent relapse following a depressive episode (that is, where participants were not depressed at study entry), were excluded.
Trials involving people described as 'at risk of suicide' or with dysthymia or other affective disorders such as panic disorder have been included in this review as long as participants met criteria for major depression as stated above.
Comorbidities
Trials that included participants with comorbid conditions secondary to a depressive disorder were included in this review.
Setting
In this review, studies conducted in primary care and community‐based settings, or in secondary or specialist settings, including inpatient settings, and including referrals as well as volunteers, have been included. Similarly, studies focused on specific populations, for example, school refusal or suicide risk, were also eligible for inclusion if the participants all met the criteria for major depression.
Types of interventions
We developed criteria for inclusion in consideration of the transitivity assumption, such that there was sufficient clinical and methodological comparability across the planned comparisons in this NMA. All included interventions are part of the same broad class and therefore have been considered to be legitimate alternatives (i.e. they are equally likely and able to be randomised).
Trials that compared the effectiveness of newer generation antidepressants with each other or with a placebo have been included. The antidepressants formed the 'decision set' of treatments — that is, the treatments that are of direct interest for clinical decision‐making. The 'supplementary set' of treatments included placebo and no treatment. Although understanding the effectiveness of these treatments is not directly relevant to clinical practice, since many of the trials compare antidepressants to placebo, their inclusion in the network provides important indirect evidence that helps evaluate clinically relevant medications (i.e. the 'decision set') with greater precision (Hetrick 2012).
The antidepressants included in this review are those consistent with the medications included in the equivalent Cochrane Common Mental Disorders (CCMD) Group Meta‐Analysis of New Generation Antidepressants (MANGA) reviews for adult depressive disorders (Cipriani 2005; Cipriani 2007; Imperadore 2007; Nosè 2007; Nakagawa 2009; Cipriani 2009a; Cipriani 2009b; Churchill 2010; Cipriani 2010; Guaiana 2010; Omori 2010; Watanabe 2011). The set of antidepressants, grouped according to class, included:
selective serotonin reuptake inhibitors (SSRIs): fluoxetine, fluvoxamine, sertraline, paroxetine, escitalopram, citalopram, alaproclate, vilazodone, vortioxetine;
selective norepinephrine reuptake inhibitors (SNRIs): venlafaxine, duloxetine, desvenlafaxine, milnacipran, levomilnacipran, edivoxetine;
norepinephrine reuptake inhibitors (NRIs): reboxetine;
norepinephrine dopamine reuptake inhibitors (NDRIs): bupropion;
norepinephrine dopamine disinhibitors (NDDIs): agomelatine;
tetracyclic antidepressants (TeCAs): mirtazapine.
We applied no restrictions on the dose or pattern of administration of included antidepressants. The antidepressants have been grouped for the syntheses to ensure a high degree of similarity within a group (node), in terms of the class, specific antidepressant and dose.
Trials where newer‐generation antidepressants were used in combination with a co‐intervention (e.g. the YoDA‐C trial where fluoxetine and CBT were compared with placebo and CBT; Davey 2019) were not eligible for inclusion. Trials with multiple comparison arms have been included but only data from relevant treatment arms has been extracted (e.g. the Treatment for Adolescent Depression‐TADS trial; TADS 2004).
Types of outcome measures
Primary outcomes
1. Depressive disorder according to DSM or ICD criteria and established by a clinician conducting a structured or semi‐structured diagnostic interview such as the Schedule for Affective Disorders and Schizophrenia for School Aged Children, Present Episode Version (K‐SADS‐P) (Chambers 1985). We chose this as the most robust approach to establishing the resolution of a depressive episode.
2. Death by suicide established via recording of this adverse outcome within the trial period or by medical record or direct inquiry with appropriate contact person at follow‐up.
Secondary outcomes
1. Efficacy outcomes
1.1 Depression symptom severity (clinician‐rated) using the Children's Depression Rating Scale (CDRS‐R)
The CDRS‐R was adapted for children and adolescents from the Hamilton Depression Rating Scale (HAM‐D), a tool validated and commonly used in adult populations (Brooks 2001). Early reviews indicated that both the CDRS‐R and HAM‐D have good reliability and validity (Brooks 2001). The Montgomery‐Åsberg Depression Rating Scale (MADRS) was also based on the HAM‐D but designed to better assess sensitivity to change. It was not, however, designed specifically for children and adolescents (Brooks 2001). More recent evidence suggests that the psychometric properties of the CDRS mean its ability to meaningfully measure depression severity may be limited (Stallwood 2021). Nevertheless, this outcome was chosen due to its consistency of use across trials (the most commonly used tool in the previous version of this review (Appendix 1)).
1.2. Remission or response as defined by trialists
Response and remission are separate constructs for which distinct consensus definitions (related to a severity‐based and temporal criterion) have been agreed: response is an initial improvement of symptoms, usually after treatment initiation and usually attributable to the treatment. After three weeks of minimal symptoms, a patient can be said to have entered remission (Frank 1991; Rush 2006), As published specifically in relation to children and adolescents, response has been defined as there being no symptoms or a significant reduction in depressive symptoms for at least two weeks and remission as a period of at least two weeks and more than two months with no or few depressive symptoms (Birmaher 2007).
While these constructs are distinct, the way they are defined and operationalised is inconsistent across trials in this field. Typically, 'remission' and 'response' are defined by dichotomising a continuous measure of clinician‐rated depression symptoms. The labelling of remission and response varied across trials in the previous versions of this review (Hetrick 2007; Hetrick 2012), with the labelling being different even though the cut‐points were the same. In the previous versions of this review, for consistency across trials, we chose the most commonly reported cut‐point: CDRS‐R less than or equal to 28, which was generally referred to as 'remission'. When 'remission' was not reported, we used 'response', if available. An exception to this rule was: if remission was only available from observed case (OC) data but response data were available from 'last‐observation‐carried‐forward' (LOCF) data, we used response data (see 'Dealing with missing data'). We have taken the same approach in this review. We have chosen to include both continuous and dichotomised measures of clinician‐rated depression symptoms, since there are advantages and disadvantages to each. Responder analyses (based on the dichotomised continuous outcomes) are well known to be problematic (Kieser 2004), with arbitrariness in the choice of cut‐point, loss of power resulting from the dichotomisation (Altman 2006), and difficulties in interpretation (as outlined above). However, synthesising continuous outcomes is not without its difficulties. The scales used to measure depression symptoms vary across trials: there is inconsistency in the analytical methods employed (e.g. analyses of change scores, regression models), which can preclude the use of the standardised mean difference; and there are also interpretational difficulties.
1.3 Depression symptom severity ‒ self‐rated (on standardised, validated, reliable depression rating scales)
The Beck Depression Inventory (BDI)/Children's Depression Inventory (CDI) were the most commonly used across trials in Hetrick 2012, the previous version of this review, and ranked the highest in the hierarchy (see Appendix 1); therefore, we have undertaken meta‐analyses of this outcome measured by either scale (BDI for preference if both are used), with results based on other scales reported in tables.
1.4 Functioning (on standardised, validated, reliable global functioning rating scales)
The Children's Global Assessment Scale (CGAS) was the most commonly used in the previous version of this review and, therefore, has been used for meta‐analyses of this outcome, with results based on other scales reported in tables.
2. Suicide‐related outcomes
Where possible, we have chosen data based on the definitions used in the FDA review using the Columbia Classification system, e.g. suicidal ideation, suicide attempt (Hammad 2004), based on the previous version of this review.
3. Overall adverse outcomes
Experience of any adverse event.
Timing of outcome assessment
Our primary outcome was measured post‐intervention (i.e. at completion of the treatment). We have chosen this follow‐up time (post‐intervention) based on our previous review (Hetrick 2012), where all trials primarily measured the post‐intervention outcomes. However, recognising that longer‐term time frames are clinically more meaningful for antidepressant treatments, we have also collected all available outcomes and classified them into short‐term (one to six months) and long‐term (> six months) follow‐up categories. Where multiple outcomes per category were available, we selected the outcome with the longest follow‐up (e.g. in the short‐term category, outcomes measured at six months were selected in preference to outcomes at three months).
Search methods for identification of studies
We identified eligible studies (RCTs) of antidepressants for depression in children and adolescents from the Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR; all years to 2016) (Appendix 2).
Electronic searches
We conducted supplementary searches on the following bibliographic databases using relevant subject headings (controlled vocabularies) and search syntax, appropriate for each resource (Appendix 3):
Cochrane Central Register of Controlled Trials (CENTRAL; Issue 3 of 12, 2020) in the Cochrane Library;
MEDLINE Ovid (2016 to March 30, 2020);
Embase Ovid (2016 to 2020 Week 13);
PsycINFO Ovid (2016 to March Week 4 2020).
No restrictions on language or publication status were applied to the searches.
3. International registries
International trial registries via the World Health Organization's trials portal (ICTRP) and ClinicalTrials.gov were searched to identify unpublished or ongoing studies.
4. Searches already completed
We had already conducted searches up to October 2011 for other direct comparison reviews (Hetrick 2007; Hetrick 2012) (Appendix 4).
Searching other resources
Grey literature
We searched sources of grey literature including theses, clinical guidelines and reports from regulatory agencies.
Handsearching
Conference abstracts for the American Academy of Child and Adolescent Psychiatry were searched (2003 to 2005) for the original review. For this update, we now searched for these via Embase.
Reference lists
We checked the reference lists of all included studies and relevant systematic reviews to identify additional studies missed from the original electronic searches (for example unpublished or in‐press citations).
Correspondence
We contacted trialists and subject experts for information on unpublished or ongoing studies or to request additional trial data.
Data collection and analysis
Selection of studies
We considered trials from the previous reviews to already be included (Hetrick 2007; Hetrick 2012); they had been independently screened for inclusion by two review authors (MS and GC). For trials identified from the updated search, two of five review authors (PB, AB, SH, CM, GC) independently assessed the titles and abstracts against the inclusion criteria.
Where a title or abstract appeared to describe a trial eligible for inclusion, we obtained the full article to assess whether it met the inclusion criteria. Where peer‐reviewed academic publications of trials were not available, the process was to obtain the trial registry report and then identify further reports that were available on this basis. Pairs of review authors resolved disagreements in screening decisions at each stage of screening through discussion, or if necessary by consultation with a third review author. We have reported the reasons for exclusion of trials in the 'Characteristics of excluded studies' section.
Data extraction and management
For the 2012 update of the review, two review authors (SH and GC) independently extracted information on each trial, including 'Risk of bias' criteria, details about the trial and outcome data.
For this version of the review, two of five review authors (AB, PB, CM, GC, VS) independently extracted information on newly included trials, including 'Risk of bias' criteria and details of participants, interventions, comparisons, potential effect modifiers (see Subgroup analysis and investigation of heterogeneity), outcomes and results. A third review author (SH or NM) resolved disagreements. Where there were multiple reports on one trial and discrepancies between these, we typically relied on the clinical trial reports but would resolve these by discussion between those authors extracting the data. We have reported trial characteristics in the 'Characteristics of included studies' tables. These data formed the basis for discussing the internal and external validity (directness) of results.
When estimates of treatment effect or standard errors were not directly reported, we calculated these, where possible, through algebraic manipulation of available statistics (e.g. means, confidence interval limits, exact P values).
For the previous versions of the review we decided post hoc to extract suicide‐related outcomes from the Medicines and Healthcare Products Regulatory Agency (MHRA) rather than from the individual trial reports retrieved in the search. The MHRA has produced a web‐based report (www.mhra.gov.uk) that summarises the results of the majority of the trials included in the original review (Hetrick 2007). We used two additional reports: one on suicide‐related outcomes (Hammad 2004); and one on trial characteristics (Dubitsky 2004). These gave details of outcomes for 25 SSRI trials, both based on data submitted to the FDA. For this review, we again used data from these reports of suicide‐related outcomes, where it was available; and where it was not, we extracted data from the trial reports using, where possible, outcomes with a similar definition of 'suicide‐related', as defined in the above stated reports.
Assessment of risk of bias in included studies
In the original review (Hetrick 2007), we assessed the risk of bias of the included randomised trials using the quality of trials ratings devised by Moncrieff and colleagues (Moncrieff 2001). For the 2012 update, we used the first version of Cochrane's 'Risk‐of‐bias' tool and have again used this version for this update (Higgins 2009).
We assessed the following domains:
Random sequence generation
Allocation concealment
Blinding of participants and personnel
Blinding of outcome assessment
Incomplete outcome data
Selective outcome reporting
Other bias
We judged each domain as having a 'high', 'low' or 'unclear risk of bias' and have provided a supporting quotation from the study report together with a justification for our judgement in the 'Risk of bias' table. We considered blinding separately for different outcomes, where necessary (e.g. the risk of bias resulting from unblinded outcome assessment for an objective outcome, such as 'death by suicide', may differ from a subjective outcome, such as self‐reported depression). Where information on risk of bias related to unpublished data or correspondence with a trialist, we noted this in the 'Risk of bias' table. We classified the overall risk of bias for each trial into three categories based on all domains except 'Other'. These categories are: 'low risk of bias', where all domains are judged to be at a low risk of bias; 'some concerns', where at least one domain is rated at an unclear risk of bias and all other domains are rated at a low risk of bias; and 'high risk of bias', where at least one domain is rated at a high risk of bias.
Two of five review authors (AB, CM, VS, GC, SH) independently assessed the risk of bias for each trial newly included in this update. We resolved any uncertainties or disagreements by discussion or by involving another author (JM or NM).
Measures of treatment effect
Relative treatment effects
Dichotomous data
For dichotomous outcomes, we measured treatment effects using odds ratios (ORs) (e.g. remission rates and adverse effects, measured as a count of any adverse event).
Continuous data
For continuous outcomes (such as clinician‐rated depression symptom severity), we use the mean difference (MD). In the majority of trials, multiple linear regression models were fitted, and ‘covariate‐adjusted’ estimates of treatment effects from these models were reported (often as least square means or least square mean differences) (Hetrick 2007; Hetrick 2012). These models adjusted for varying factors such as age, sex, investigator site and baseline of the outcome. We had not planned and did not use the standardised mean difference (SMD) (often used where the same outcome domain is measured across trials, but using different measurement scales) because of the inconsistency in the analytical methods employed across trials (e.g. analyses of final values, change scores and regression models).
Relative treatment rankings
We estimated P‐scores (Rucker 2015), which measure the certainty that a particular treatment is more effective than competing treatments.
Unit of analysis issues
Cluster‐randomised trials
No cluster‐RCTs were included. If they had been, but had not appropriately adjusted for the correlation between participant outcomes within clusters, we would have contacted trial authors to obtain an estimate of the intra‐cluster correlation (ICC), or imputed the ICC using estimates from the other included trials or from similar external trials. We would have inflated the reported standard errors by the square root of the design effect, using the estimated/imputed ICC (Higgins 2019), and undertaken sensitivity analyses to assess the robustness of the combined intervention effects to assumptions regarding the ICCs.
Cross‐over trials
No cross‐over trials were included. If we had included cross‐over trials, and the appropriate data from a paired analysis were not available and we could not obtain them from trial authors, we would have imputed missing statistics (e.g. missing standard deviation, correlation) using data available from other trials included within the meta‐analysis, or trials outside the meta‐analysis (Elbourne 2002; Higgins 2019). We would have used sensitivity analyses to assess the robustness of the pooled treatment effect to assumptions made regarding missing statistics.
Studies with multiple treatment groups
We undertook adjustment for multi‐arm trials in the network meta‐analyses using standard methods that account for between‐arm correlations using multivariate meta‐regression models (White 2012).
Dealing with missing data
In the original and 2012 review (Hetrick 2007; Hetrick 2012), we sought additional data from the principal authors and pharmaceutical companies of trials (the latter approached by the Cochrane Common Mental Disorders group on our behalf, who also approached the National Institutes for Mental Health (NIMH) in the case of the Treatment for Adolescent Depression Study; March 2004) where the data were missing, or were in a form unsuitable for meta‐analysis. We also searched the pharmaceutical company websites for additional data on included trials.
In the original and 2012 review (Hetrick 2007; Hetrick 2012), most trials used the 'last‐observation‐carried‐forward' (LOCF) method of data imputation for the majority of outcomes: that is, the last observed value for a participant lost to follow‐up was assigned as the follow‐up value. We chose to pool LOCF data and other data derived via newer imputation methods like Mixed Effect Model Repeated Measure (rather than mix LOCF and OC data) but also undertook sensitivity analysis using OC data, where available. We have used the same approach in this review because estimates of treatment effect based on either LOCF or OC data can result in serious bias (Sterne 2009).
We contacted investigators or study sponsors in order to verify key study characteristics and obtain missing numerical outcome data, where necessary and possible (e.g. when a study was identified as abstract only). We documented all correspondence with trialists and reported which trialists responded.
As was our approach in the previous reviews (Hetrick 2007; Hetrick 2012), where least squares means and their standard errors were reported from regression models by treatment group, but no contrast between groups was reported, we estimated the variance of the treatment effect by summing the square of the standard errors in each treatment group. There may be some inaccuracy in this approach when there is imbalance in the covariates being adjusted for.
Assessment of heterogeneity
We assessed inconsistency between direct and indirect sources of evidence in several steps. First, we assessed the distribution of potential effect modifiers across treatment comparisons based on an examination of participant, intervention and methodological characteristics. Second, we conducted a global test of inconsistency using the design‐by‐treatment interaction model (Higgins 2012). Third, where there was evidence of potential inconsistency, we investigated this using side splitting in netmeta in R (Rucker 2012).
For each network, we assumed a common between‐trial heterogeneity variance of the relative treatment effects for every treatment comparison. We reported these variance estimates.
Assessment of reporting biases
There is currently no tool available to assess the risk of bias due to missing results in a synthesis. However, a framework has been proposed in which an assessment is made for each comparison regarding i) the risk and potential impact of missing results from studies (termed 'known‐unknowns'), and ii) the risk of missing studies (termed 'unknown‐unknowns') (Page 2019). We have used this framework to guide our assessments of whether there was 'undetected' or 'suspected' reporting bias for each of the comparisons in our GRADE assessment ('Summary of findings' tables).
In assessing i), we have considered our 'Risk of bias' judgement for the 'selective outcome reporting' domain since in the first version of the 'Risk of bias' tool (Higgins 2009), this captures not only selective reporting of results, but also missing results (i.e. arising from non‐reporting of outcomes or incomplete reporting of results for inclusion in a meta‐analysis). For ii), we have considered qualitative signals; and used statistical methods to visualise and model the impact of small‐study effects. As a qualitative signal, we considered the potential for reporting bias (specifically lag‐time bias) for any newly developed antidepressants that have only been evaluated in a small number of trials (Page 2019).
For 'Depression symptom severity (clinician‐rated)' and 'Suicide‐related outcomes', we planned to use comparison‐adjusted funnel plots, which are an extension of funnel plots for network meta‐analysis, to visually examine if there was evidence of small‐study effects (Chaimani 2013). Following the approach outlined in Mavridis 2016, we assumed that small‐study effects, where they exist, favour the active treatment group in placebo‐controlled comparisons, and the 'treatment group' in head‐to‐head trials. We used the following hierarchy to define the 'treatment group' in head‐to‐head trials: i) the sponsored antidepressant treatment; ii) the antidepressant treatment identified as such by the trial authors; iii) the newest antidepressant treatment.
Where the funnel plots were suggestive of small‐study effects, we modelled the impact of small studies using two network meta‐analysis regression models. In the first model, we assumed that small‐study effects only arose in placebo‐controlled trials. In the second model, we assumed that the small‐study effects arose in placebo‐controlled and head‐to‐head trials, using the assumptions noted above. Further details of the models are available in Mavridis 2016 and Chaimani 2012.
Data synthesis
Where the distribution of potential effect modifiers across the different pairwise comparisons was considered sufficiently similar, we undertook a network meta‐analysis that synthesises direct as well as indirect comparisons, enabling an analysis of comparative effects of interventions for each outcome specified above. Indirect comparisons are those made between competing interventions that have not been compared directly with each other.
For our primary analyses, we grouped antidepressants by the specific antidepressant (e.g. fluoxetine, sertraline etc.). We also undertook post hoc analyses where we grouped at a higher level ‐ the class level of antidepressant (SSRI, SNRI, TeCAs). These latter analyses were undertaken for the purpose of being able to provide general guidance about the class of antidepressant drugs, which was particularly important when considering the suicide‐related outcome. This was largely driven by observation that effects appeared to be more consistent within class so that decision‐making might be made on the basis of class.
Random‐effects network meta‐analyses were conducted for most outcomes (except suicide related outcomes, where we used a fixed effect Mantel Haenszel model as performs better for rare outcomes) in a frequentist framework using multivariate meta‐analysis. The main analyses were undertaken using the netmeta package in R (Rucker 2012). Meta‐regression analyses were conducted using the suite of network commands in Stata (Chaimani 2015; White 2015; Stata 2019). We have presented network plots, forest plots ordered by P‐scores, and displayed results for each outcome in league table format with treatments ordered in terms of their P‐scores. P‐scores are the frequentist analogue to the Surface Under the Cumulative RAnking (SUCRA) values which are commonly reported in the Bayesian framework.
We have reported effect estimates of outcomes (and their 95% confidence intervals) that are not included in the network meta‐analysis in tables structured by outcome and treatment comparison.
Subgroup analysis and investigation of heterogeneity
We conducted meta‐regression analyses for 'depression symptom severity (clinician‐rated)' and 'suicide‐related' outcomes'. There is evidence that children and adolescents may respond differently to pharmacological intervention: for example, oral tricyclic antidepressants versus placebo significantly reduce symptoms in adolescents but not in children (Hazell 2002). Children and adolescents were defined as those aged approximately 6 to 12 and 13 to 18 years, respectively. When estimates of treatment effect were not presented for children and adolescents separately, we created another subgroup that contained both children and adolescents.
There is some evidence from research on adults that suggests that baseline severity impacts the magnitude of treatment effects (Kirsch 2008); even without this evidence, those with lower scores on depression have less improvement that can be made than those who begin with higher scores. Treatment duration also potentially impacts the magnitude of treatment effects, with longer periods of intervention potentially resulting in greater improvements. We considered each of these factors as continuous covariates in the model. The impact of industry funding on treatment effects has been an important consideration across all fields of medicine, including in psychiatry (Lundh 2017). We classified industry‐sponsored trials as those that declare any pharmaceutical industry sponsorship. We classified studies that did not report a funding disclosure statement as 'non‐industry'.
Therefore, the meta‐regression model included the following covariates: treatment duration; age (children versus adolescents); baseline depression severity (baseline clinician‐rated severity of depression measure); and pharmaceutical funding (any industry sponsorship versus no industry sponsorship). We estimated regression coefficients for each comparison separately along with their 95% CIs. Continuous covariates (treatment duration and baseline severity) were centred using the mean (i.e. the difference between each trial's mean and the mean across trials).
Sensitivity analysis
We undertook sensitivity analyses to examine how the estimates of treatment effects were affected by the:
inclusion of trials judged to be at a high risk of bias ‒ we restricted sensitivity analyses of the outcomes 'Depression symptom severity (clinician‐rated)' and 'Suicide‐related outcomes' to trials we judged to be at a 'low risk of bias' or those with 'some concerns';
assumptions for missing statistics ‒ we undertook sensitivity analyses of the outcomes 'depression symptom severity (clinician‐rated)', and 'Suicide‐related outcomes' to examine the impact of any assumptions we made regarding imputation of statistics (e.g. ICCs, missing standard deviations).
Summary of findings and assessment of the certainty of the evidence
On the basis of the previous reviews (Hetrick 2007; Hetrick 2012), we knew that trialists did not include our primary outcomes of depressive diagnosis by DSM or ICD criteria and suicide completion outcomes. Therefore, we assessed our certainty in the evidence from the network meta‐analyses for the outcomes 'Depression symptom severity (clinician‐rated)' and 'Suicide‐related outcomes'. The decision to use the clinician rated measure of depression symptom severity was based on the previous review, in which we noted that few trials included a measure or reported outcomes for self‐rated depression symptom severity.
We used the CINeMA approach (Confidence In Network Meta‐Analysis) to assess our certainty in the results from the network meta‐analysis (Salanti 2014; CINeMA 2017; Nikolakopoulou 2020). CINeMA is based on the GRADE framework for assessing the certainty of the body of evidence and involves assessing the following six domains: within‐study bias (i.e. the impact of risk of bias of the included trials); reporting bias (i.e. the impact of missing studies, outcomes and results); indirectness; imprecision; heterogeneity; and incoherence. Each domain (except 'reporting bias') is judged to have no concerns, some concerns, or major concerns. 'Reporting bias' is judged as 'suspected' or 'undetected'. Judgements across the six domains are summarised to obtain four levels of confidence for each relative treatment effect: very low, low, moderate or high. We assessed our certainty in all comparisons formed by treatment groups in the decision and supplementary sets. Below are further details of the criteria we used to inform our judgements for the domains.
Within‐study bias
In judging the certainty of the evidence for each relative effect estimate in this domain, we considered the percentage contribution from studies judged to be at a low risk of bias, with some concerns, or a high risk of bias. Specifically, we calculated a weighted average of the risk of bias, where the risk of bias judgements were assigned scores of −1 (low), 0 (some concerns) and 1 (high) and these scores were weighted by the proportion contributed from studies at each level. We used these weighted averages to classify the certainty of evidence for each estimate.
Reporting bias
We used the results from our investigation of reporting bias (see 'Assessment of reporting biases' above) in judging the certainty of evidence for each relative effect estimate. This involved considering the contribution that each direct comparison made to each relative effect estimate (i.e. network estimate).
Indirectness
In judging the certainty of the evidence for each relative effect estimate in this domain, we considered for each study how directly it addressed the research question in combination with the percentage contribution the study made to the estimate. In considering directness, we considered population, intervention and outcome characteristics that are potential effect modifiers (e.g. age).
Imprecision
In judging the certainty of the evidence for each relative effect estimate in this domain, we set an 'equivalence range' for each outcome. The 'equivalence range' corresponded to clinically unimportant differences between groups. Our 'equivalence range' for 'Suicide‐related outcomes' ranged from an OR of 0.90 to 1.12. We derived this by deciding upon an important absolute risk difference (2%) and considering a range of placebo group risks for this outcome, informed from our previous review and recent research (Davey 2019). The selected 'equivalence range' for the OR is based on assuming a risk of 'suicide‐related outcome' in the placebo group of 75%. This 'equivalence range' is conservative (i.e. the range is smallest) for comparison group risks that are more common than 75% (or less common than 25%).
Our 'equivalence range' for the 'Depression symptom severity (clinician‐rated)' outcome ranged from MD −5.00 to 5.00 on the CDRS‐R. This corresponds to an 'equivalence range' of approximately SMD −0.35 to 0.35, assuming an SD of 14.471 based on the published Cochrane Review of antidepressants in children and adolescents (Hetrick 2012) and on more recent trials that have determined sample size using target differences of around 0.4 standard deviation units on the CDRS‐R (e.g. Atkinson 2014; Emslie 2014; Atkinson 2018). The context for this is that the CDRS‐R scale ranges from 17 to 113 with categories that are separated by between 10 and 15 points (categories: depressive disorder unlikely to be confirmed; it is possible a depressive disorder might be confirmed; a depressive disorder is likely to be confirmed; a depressive disorder is very likely to be confirmed; a depressive disorder is almost certain to be confirmed). This implies that any significant change would need to be at least half of 10‐15 points.
Heterogeneity
We used the clinical equivalence ranges specified for the 'imprecision' domain in combination with prediction intervals to determine whether heterogeneity in the results affected our certainty in the evidence. Decisions on whether to downgrade by one or two levels were based on agreement between the 95% confidence interval (CI) and 95% prediction interval (PI). For example, where the 95% CI was within the equivalence range, if the 95% PI crossed the equivalence range in one direction the comparison was downgraded one level for inconsistency. If the 95% PI crossed the equivalence range in both directions the comparison was downgraded by two levels. Decisions were automated using CINeMA software.
Incoherence
We used the clinical equivalence ranges specified for the 'imprecision' domain in combination with the 95% confidence intervals of direct and indirect relative treatment effects to determine if incoherence between these estimates affected our certainty in the evidence.
We have presented 'Summary of findings' tables according to the template provided in Table 2 (pg.7) of Yepes‐Nunez 2019. For 'Depression symptom severity (clinician‐rated)', we limited our 'Summary of findings' tables to include only the decision set of interventions (i.e. all interventions except placebo or no treatment). For 'Suicide‐related outcomes', we included all interventions in our 'Summary of findings' tables.
Informative statements
In describing the results, we used informative statements that incorporated the certainty of the evidence and the size of the effect estimate (Santesso 2020). For high‐certainly evidence, we used definitive terms such as 'reduces/increases' or 'results in'; for moderate‐certainty evidence, we used terms such as 'likely' or 'probably'; for low‐certainty evidence, we used terms such as 'may'; and for very low‐certainty evidence, we noted that the 'evidence is very uncertain about the effect of the drug on the outcome'. In combination with these terms, we described the size of effect as 'small and unimportant difference' or 'at least a slight difference'. The choice between these two terms is determined by whether the effect estimate falls inside (in which case the first term) or outside (in which case the second term) the equivalence range. An exception to using these terms is when there is very low certainty evidence, in this case we do not describe the size of the effect.
For outcomes where we did not assess the certainty of the evidence, we described the results using the same categories of 'small and unimportant difference' or 'at least a slight difference'. The choice between these two terms was based on whether or not the confidence interval sat completely within the equivalence range (in which case the first term was used), or at least one bound sat outside the equivalence range (in which case the second term was used). We had not specified equivalence ranges for all outcomes a‐priori. Therefore, for the continuous outcomes (Children's Depression Inventory, Children's Global Assessment Scale), we used the same equivalence range of an SMD ‐0.35 to 0.35 as we had set for 'Depression symptom severity (clinician‐rated)'. Converting this equivalence range to the original scales assuming the median SD reported by the included studies led to ranges of ‐7.99 to 7.99 for 'Children's Depression Inventory' and ‐9.34 to 9.34 for 'Children's Global Assessment Scale'. For the binary outcomes (overall adverse outcomes and response/remission) we used an equivalence range from 0.80 to 1.25 on the odds ratio scale based on agreement between authors
Results
Description of studies
Results of the search
This updated review has been based on earlier work with a similar clinical question but represents a new methodological approach to synthesis (NMA) and therefore has a newly developed protocol. Trials from the previous versions of this review remain relevant and are included.
Twelve trials were included in the original review that only included SSRIs (Hetrick 2007). The inclusion criteria for the first update of the review (Hetrick 2012) were expanded to include newer classes of antidepressants and four trials excluded from the original review were then included in this update (Emslie 2007 Trial 1; Emslie 2007 Trial 2; Mirtazapine Trial 1; Mirtazapine Trial 2), along with seven new trials, four of which were ongoing trials (Glod 2004; NCT00353028, for which no further information has been obtained in this update and two trials that are now published and included in this update, Atkinson 2014; Emslie 2014). In all, a total of 19 trials were included in the 2012 update.
Searches for this update were up to 30 March 2020. In total, 1925 records were retrieved in the search, of which 1836 were excluded on the basis of title and abstract. We attempted to retrieve 89 full‐text articles for full inspection. One publication could not be located and is awaiting assessment (see Studies awaiting classification). Of the 88 that were obtained, 45 were either already included trials or were secondary publications from already included trials (27 of these were for TADS 2004 and are not listed under the main reference for this trial; in the 2012 update we had already located 35 publications that we had not listed). Seven new trials have been included in this update, two of which were listed as ongoing trials in the 2012 version (Atkinson 2014; Emslie 2014), resulting in a total of 26 trials included in this updated version of the review (see Figure 1). In addition, 10 studies were newly identified as ongoing (see Ongoing studies) and 14 studies were newly excluded (see Excluded studies).
1.

For the included trials, we had retrieved additional reports during preparation of the original review (Hetrick 2007), including the web‐based report of the Medicines and Healthcare Product Regulatory Agency (MHRA), summarising the majority of clinical trials on SSRIs for major depressive disorder in children and adolescents at the time. When we wrote to trial authors for additional data during preparation of the original review, in many cases trial authors did not have access to any additional data. We accessed the trial reports published online by SmithKline Beecham on paroxetine (Keller 2001; Emslie 2006; Berard 2006) (http://www.gsk.com/media/paroxetine.htm). For one paroxetine trial (Paroxetine Trial 1) we only had access to a brief trial report from this website. We had also accessed trial reports published online by Forest Laboratories for escitalopram and citalopram and for this update located the report for a newly located trial of escitalopram. Eli Lilly provided additional data for a trial on fluoxetine (Emslie 2002) during preparation of the original review. For this update, the trial authors for this trial and for Emslie 1997 were able to provide additional data in response to our requests. For this update, we accessed trial reports of venlafaxine from CCDAN who had accessed them from Pfizer.
Two published trial reports include the results of two trials, Wagner Trial 1 & 2 (2003) and Emslie 2007. Data for the individual trials for Wagner Trial 1 & 2 (2003) were only available from the MHRA report. Emslie 2007 provided some data separately for each of the two trials included in the publication.
There were no published reports for the mirtazapine trials; data were available from the MHRA report and from two reports to regulatory agencies.
The trial by Simeon was discontinued early due to slow enrolment, with some information about the trial from the written report and some from the MHRA report (Dubitsky 2004).
Included studies
Design
The trials were all individual patient parallel‐group randomised trials. The number of sites ranged from 1 to 124, however, this information has not been specified for Emslie 2002 and NCT02709746. The trials included in the review compared: 1) newer generation antidepressant with a placebo, 2) different drug classes of the newer generation of antidepressants ‐ SSRI and SNRI, 3) different doses of a newer generation of antidepressants, and 4) different drugs of the same class of newer generation antidepressants. For trials where one or more of the treatment arms was not a newer generation antidepressant, data were extracted only from the newer generation antidepressant and the placebo groups (Keller 2001 and TADS 2004). For more details, see Table 3.
1. Comparison of study characteristics.
| Study ID | Location | Funding | Setting | Age (Mean (SD)) | Gender (ratio) Female:Male (F:M) | Method of diagnosis | Baseline depression severity | Length of current episode | Comorbidity |
| Almeida‐Montes 2005 | Mexico | Eli Lily provided fluoxetine and placebo | Setting: Outpatient; # sites: single; Total # participants = 23 |
Intervention = 13.3 (3.16); Control = 11.5 (1.58); Range: 8‐14 years |
Not stated | DSM‐IV[1] using semi‐structured interview; MINI‐KID[2] | Mean (measure): Not reported Category: Not reported by the trialists |
Not stated | Not stated |
| Atkinson 2014 | USA Finland France Germany Slovakia Estonia Russia Ukraine S. Africa |
Eli Lilly and Company | Setting: Outpatients; # sites: 65; Total #participants enrolled = 337 |
Intervention 1 (duloxetine): 13.1 (3.0); Intervention 2 (fluoxetine): 13.1 (3.3); placebo: 13.3 (3.1); Range: 7‐17 years |
Intervention 1 (duloxetine): 64:53; Intervention 2 (fluoxetine): 61:56; placebo: 51:52 |
DSM‐IV‐TR[3] criteria for MDD without psychotic features, had a CDRS‐R
total score ≥ 40 inventory at three screening visits; MDD diagnosis supported MINI‐KID conducted by 2 independent evaluators (including 1 psychiatrist) |
CDRS‐R mean (SD) score: Intervention 1 (duloxetine) = 59.2 (10.5) Intervention 2 (fluoxetine) = 58.8 (10.6) placebo = 60.2 (11.7) CGI‐S score (SD): Intervention 1 (duloxetine) = 4.5 (0.6) Intervention 2 (fluoxetine) = 4.5 (0.6) placebo = 4.6 (0.7) Category: Trialists reported moderately severe depression |
Not reported | Not stated separately for each group (Total patient population): ADHD (2.4%), ODD/CD (2.4%) |
| Atkinson 2018 | USA Chile |
Pfizer Inc. | Setting: Outpatients; # sites: 33; Total #participants enrolled = 363 |
Intervention 1 (desvenlafaxine high exposure) = 12.87(3.01); Intervention 2 (desvenlafaxine low exposure) = 13.07(2.8); placebo = 13.15 (2.68); Range: 7‐18 years |
Intervention 1 (desvenlafaxine high exposure) = 76:45; Intervention 2 (desvenlafaxine low exposure) = 69:53; placebo = 50:50 |
DSM‐IV‐TR, K‐SADS‐PL and clinical interview. A comprehensive diagnostic psychiatric evaluation, including collection of psychiatric history and treatments and confirmation of the MDD diagnosis, was performed by a psychiatrist at screening; had at least moderately severe depressive symptoms for ≥ 30 days before screening, and a CDRS‐R score >40 and CGI‐S score ≥ 4 at screening and baseline | CDRS‐R total score, M (SD) Intervention 1 (desvenlafaxine high exposure) = 58.45 (9.45); Intervention 2 (desvenlafaxine low exposure) = 58.52 (9.18); placebo = 57.28 (8.94) CGI‐S mean (SD) score, Intervention 1 (desvenlafaxine high exposure) = 4.61 (0.58); Intervention 2 (desvenlafaxine low exposure) = 4.61 (0.61); placebo = 4.55 (0.58) Category: Trialists reported moderately severe depression |
Mean (SD) months: Intervention 1 (desvenlafaxine high exposure) = 12.42
(13.24); Intervention 2 (desvenlafaxine low exposure) = 11.23 (11.21); placebo = 12.85 (12.10) |
Intervention 1: (desvenlafaxine high exposure): ADHD, 9.9%; Intervention 2: (desvenlafaxine low exposure): ADHD 9.8%; Control: ADHD 7.5% |
| Berard 2006 | Argentina Belgium Canada Holland Italy Mexico S. Africa Spain UAE UK |
SmithKline Beecham | Setting: Not stated; # sites: 33; Total # participants enrolled = 286 |
Intervention= 15.5 (1.6); Control = 15.8 (1.6); Range: 13‐19 years |
Intervention = 122:65; Control = 61:38 |
DSM‐IV (no other detail stated); GAS < 69; MADRS[4] ≥ 16; after screening 14‐day single‐blind run‐in period | Mean (MADRS): MADRS mean (SE) score: intervention = 25. 9 (0.5); control:
25.9 (0.6); CGI Intervention 4.2 (0.1); CGI placebo 4.2 (0.1) Category: Trialists reported moderately to severely ill |
Not stated | Intervention: Anxiety disorder 17.0%; ADHD[5] 1.6%;ODD/CD[6] 0.5%; Control: Anxiety disorder 18.3% ADHD 0.0%; ODD/CD 1.1% |
| Emslie 1997 | USA | National Institute of Mental Health | Setting: Outpatients; # sites: Single; Total #participants enrolled = 96 |
Intervention = 12.2 (2.7); Control = 12.5 (2.6); Range: 7‐17 years |
Intervention = 22:26; Control = 22:26 |
DSM‐III‐R[7]; K‐SADS[8] depressive items; CDSR‐R[9] ≥ 40; 3
independent diagnostic interviews and a 1‐week placebo lead‐in |
Mean (CDRS‐R): Intervention = 58.5 (10.5); Control = 57.6 (10.4); CGI not reported Category: Not reported by trialists |
Mean (SD) weeks: Intervention = 14.6 (9.7); placebo = 13.7 (7.5) | Intervention: Anxiety disorder 66.7%; ADHD 33.3%; ODD/CD[10] 27.1%;
Dysthymia 41.7% Control: Anxiety disorder 45.8%; ADHD 27.1% ; ODD/CD 33.3%; Dysthymia 29.2% |
| Emslie 2002 | USA | Eli Lilly and Company | Setting: Outpatients; #sites: Not specified (study was conducted by 15 investigators throughout the United states); Total #participants enrolled = 122 children and 97 adolescents |
Intervention = 12.70 (2.46); Control = 12.69 (2.67); Range: 8 ‐18 years | Intervention = 54:55; Control = 54:56 |
DSM‐IV Diagnostic Interview for Children and Adolescents (DICA) interview, CDRS‐R ≧ 40 and CGI[11] = 4; 3 independent diagnostic interviews and a 1‐week placebo lead‐in | CDRS‐R mean (SD) score: Intervention = 57.1 (9.9); Control = 55.1 (11.8); CGI score: Intervention 4.5 (0.6); placebo 4.4 (0.6) Category: trialists reported moderate to marked severity of illness (based on CGI‐S score) |
Mean weeks: Intervention = 60.44; placebo = 61.29 | Intervention: ADHD 14.7%; ODD/CD 20.2%; Control: ADHD 13.6%; ODD/CD 16.4% |
| Emslie 2006 | USA Canada |
GlaxoSmithkline | Setting: Outpatients; #sites: 40 centres in US, 1 in Canada; Total #participants enrolled = 206 |
Intervention = 11.9 (3.00); Control = 12.1 (2.95); Range: 7 to 17 years |
Intervention = 48:53; Control = 47:55 |
DSM‐IV, K‐SADS‐PL[12] using 1‐week screening phase | CDRS‐R mean (SD) score: Intervention = 60.7 (9.37); Control = 62.6
(8.96); CGI score: Intervention = 4; placebo = 4 Category: Trialists reported moderate to severe MDD symptomatology |
Mean (SD) months: Intervention = 26.9 (28.62); placebo = 24.9 (27.08) | Intervention: Any disorder 27.7%; Anxiety disorders 10.9%; ADHD 3.0%;
ODD/CD 4.9%; Dysthymia 2%; Control: Any disorder 17.6%; Anxiety disorder 2%; ADHD 1.0%; ODD/CD 3.9%; Dysthymia 0.0% |
| Emslie 2007 (trial 1 &2) | USA | Wyeth Research | Setting: Outpatients (academic and clinical sites); #sites: 50; Total #participants enrolled = 367 |
Intervention = 12.2 (2.6); Control = 12.3 (2.6); Range: 7 to 17 years |
Intervention = 78:101; Control: 83:92 |
DSM‐IV K_ SADS‐PL, at pre‐trial and baseline a CDRS‐R score of ≥ 40, and CGI‐S score of ≥ 4 and depressive symptoms for at least 1 month before trial entry. Single‐blind placebo run‐in period of 14 days (+/‐ 3) for trial 1 and 7 days (+/‐3) for trial 2 | CDRS‐R mean (SD) score: Intervention = 56.4 (9.2); Control = 55.8
(8.4); CGI score: Intervention = 4.5 (0.6); placebo 4.5 (0.7) Category: Not reported by trialists |
Mean (SD) weeks: Intervention = 91.1(88.2); placebo = 92.5 (91.3) | Not stated |
| Emslie 2009 | USA | Forest laboratories | Setting: Outpatients; #sites: 40; Total #participants enrolled = 312 |
Intervention = 14.7 (1.6); Control = 14.5 (1.5); Range: 12 to 17 years |
Intervention = 92:63; Control = 92:65 |
DSM‐IV with duration of current episode at least 12 weeks at screening confirmed by K‐SADS. At screening and baseline, a CDRS‐R score of ≥ 45 and a CGI‐S score of ≥ 4. Screening period of 2 weeks, and a single‐blind placebo run‐in of 1 week during 2nd week of screening | CDRS‐R mean (SD) score: Intervention = 56.0 (0.66); Control = 57.6
(0.66); CGI score: Intervention 4.6 (0.05); placebo 4.4 (0.04) Category: Not reported by trialists |
Mean months Intervention = 15.7 (17.4); placebo = 16.5 (15.4) | Intervention: Previous and/or ongoing secondary psychiatric disorder 12.9%; Control: Previous and/or ongoing secondary psychiatric disorder 16.6% |
| Emslie 2014 | USA Canada Mexico Argentina | Eli Lilly and Company | Setting: Psychiatric clinical sites; # sites: 60; Total #participants enrolled = 463 |
Intervention 1 (duloxetine 60 mg) = 12.9 (2.9), Intervention 2 duloxetine
30 mg = 12.9 (2.9), Intervention 3 (fluoxetine) = 13.0 (3.2); placebo =
13.1(2.9); Range: 7‐11 years (children);12‐17 years (adolescents) |
Intervention 1 (duloxetine 60 mg) = 60:48, Intervention 2 (duloxetine 30
mg) = 47:69; Intervention 3 (fluoxetine): 61:56; placebo = 69:53 |
DSM‐IV‐TR, The MINI‐KID for MDD, CDSR‐R and CGI‐S. MDD without psychotic features, had a CDRS‐R total score >= 40 and a CGI‐S score >= 4 at the three screening visits | CDRS‐S mean (SD) score: Intervention 1 (duloxetine 60 mg) = 59.3 (10.9),
Intervention 2 (duloxetine 30 mg) = 59.8 (11.0), Intervention 3
(fluoxetine) = 57.9 (10.1); placebo = 58.2 (9.4) CGI‐S scores: Intervention 1 (duloxetine 60 mg) = 4.6 (0.7), Intervention 2 (duloxetine 30 mg) = 4.6 (0.7), Intervention 3 (fluoxetine) = 4.6 (0.6) placebo = 4.5 (0.6) Category: Trialists reported moderately severe depression |
Not reported | Not stated separately for each group (Reported by >= 10% of the
patient population: ADHD = 10.8% Reported by >= 2% and =< 10% of the patient population: Anxiety disorder = 2.2%; ODD/CD = 4.1%) |
| Keller 2001 | USA Canada |
GlaxoSmithKline | Setting: Outpatients; #sites: 12 Total #participants enrolled = 275 |
Intervention = 14.8 (1.6); control = 15.1 (1.6); Range: 12‐18 years |
Intervention = 58:35; Control = 57:30 |
DSM‐IV diagnosis confirmed by K‐SADS‐L and current duration of episode at least 8 weeks, a score of ≥ 12 on the HAM‐D[13], a C‐GAS score of ≥ 60; screening period of 7 to 14 days, no placebo run‐in phase | K‐SADS 9‐item depression score; Intervention = 28.25; Control = 28.84;
C‐GAS[14] mean score: Intervention = 42.7; Control = 42.8; CGI score not reported Category: Not reported by trialists |
Mean (SD) months: Intervention = 14 (18); placebo = 13 (17) | Intervention: Any diagnosis 44.1%; Anxiety disorder 20.4%; ODD/CD
26.9% Control: Any diagnosis 51.7%; Anxiety disorder 32.2%; ODD/CD 23.0% |
| Mirtazapine Trial 1 | USA | Organon International | Setting: Outpatients; #sites: 17; Total #participants enrolled = 126 |
Intervention = 12.3; Control = 12.4; Range: 8‐18 years |
Intervention = 39:43; Control = 25:19 | DSM‐IV diagnosis was confirmed by K‐SADS‐L and baseline score of ≥ 15 on 1st 17 items of HAM‐D (21‐item), a C‐GAS score of < 70; CDRS‐R ≥ 40; screening period not stated | CDRS‐R mean score: Intervention = 50.93; Control = 51.93 Category: Not reported by trialists |
Not stated | Not stated |
| Mirtazapine Trial 2 | USA | Organon International | Setting: Outpatients; #sites: 15; Total #participants enrolled = 133 |
Intervention = 11.9; Control = 12.3; Range: 8‐18 years |
Intervention = 46:42; Control: 24:21 | DSM‐IV diagnosis was confirmed by K‐SADS‐L and baseline score of ≥ 15 on 1st 17 items of HAM‐D (21 item), a C‐GAS score of < 70; CDRS‐R ≥ 40; screening period not stated | CDRS‐R mean score: Intervention = 48.87; Control = 47.57 Category: Not reported by trialists |
Not stated | Not stated |
| Paroxetine Trial 1 | Japan | GlaxoSmithkline | Setting: Unclear; # sites: 19; Total #participants enrolled = 56 |
Intervention = 14.4 (1.99); placebo = 14.8 (2.62) Range: 7‐17 years |
Intervention = 18:9; placebo = 16:13 |
DSM‐IV (no other detail available); CDRS‐R score of ≥ 45 | CDRS‐R mean (SD) Intervention = 55.4 (7.3); placebo = 56.8 (8.46) Category: Not reported by trialists |
Not stated | Not stated |
| Simeon 1990 | Canada | Not stated | Setting: Outpatients; # sites: single; Total #participants enrolled = 40 |
Mean age = 16 (group ages not stated) Range: not stated | Gender (total): 22:18 (group gender not stated) |
DSM‐III criteria with HAM‐D score of ≥ 20, 1‐week placebo run‐in period | Not stated | Not stated | Not stated |
| TADS 2004 | USA | National Institute of Mental Health | Setting: Outpatients; # sites:13; Total #participants enrolled = 439 |
Total participants: 14.6 (1.5); Range: 12‐18 years |
Gender (total): 239:200 (group gender not stated) | DSM‐IV confirmed using K‐SADS‐PL and a CDRS‐R score of ≥ 45; assessment (not interview) at consent and baseline | CDRS‐R raw mean (SD) score: Intervention: 58.96 (10.16) (T‐score 74.73
(6.74)); Control: 61.11 (10.50) (T‐score 76.14 (6.11)): CGI score: Intervention 4.66; placebo 4.84 Category: Trialists reported moderate to moderately severe MDD |
Median weeks Intervention = 38; placebo = 35.5 | Intervention: Any psychiatric disorder 56.9%; Anxiety 25.7%; ADHD 11.9%;
ODD/CD 22.9%; Dysthymia 5.5% Control: Any psychiatric disorder 48.7%; Anxiety 28.6%; ADHD 16.7%; ODD/CD 25.0%; Dysthymia 10.7% |
| VLZ‐MD‐22 | USA Canada |
Forest Laboratories | Setting: Outpatients; # sites:55; Total #participants enrolled = 470 |
Intervention 1 (vilazodone) = 13 (2.9); Intervention 2 (fluoxetine) =
13.2 (2.8); placebo = 13 (2.9); Range: 7 to 17 years |
Vilazodone = 126:61; Fluoxetine = 51:46; placebo = 106:80 |
DSM‐IV‐TR criteria for MDD using semi‐structured interview; K‐SADS‐PL; CDRS‐R score ≥ 40, and CGI‐S score ≥ 4 | CDRS‐R mean (SD) score: Intervention 1 (vilazodone) = 58.3 (9.2); Intervention 2 (fluoxetine) = 58 (8.8); placebo = 57.3 (9.2) CGI‐S scores: Intervention 1 (vilazodone) = 4.7 (0.6); Intervention 2 (fluoxetine)= 4.6 (0.6); placebo = 4.6 (0.6) Category: Trialists reported moderate‐to‐marked illness severity (based on CGI‐S) |
Mean (SD) months: Intervention 1 (vilazodone) = 11.2 (16.2); Intervention 2 (fluoxetine) = 11.9 (14.0); placebo = 11.1 (12.8) |
Not stated |
| Von Knorring 2006 | Denmark Estonia Finland Germany Norway Sweden Switzerland |
H. Lundbeck A/S | Setting: Inpatients and outpatients; # sites: 31; Total #participants enrolled = 244 |
Intervention = 16 (1); Control = 16 (1); Range: 13‐18 years |
Not stated | DSM‐IV including 5‐minute interview with parents. GAF< 60 on either symptoms, activities, relationships or personal care; BDI < 21 for girls and < 16 for boys | K‐SADS‐P score: Intervention = 32.5; Control = 32.3; Totals only for MADRS 30 (SD = 5/6), GAF 55 (SD = 7); CGI not reported Category: Not reported by trialists |
Not reported | Not stated |
| NCT02709746 | USA
Russian Federation Mexico Colombia Serbia Ukraine Korea South Africa Canada Poland Spain UK Bulgaria Estonia France Germany Hungary Italy Latvia |
H. Lundbeck A/S | Setting: Not reported; # sites: 124; Total #participants enrolled = 784 |
Intervention 1 (vortioxetine 10 mg): 14.8 (1.66);
Intervention 2
(vortioxetine 20 mg): 14.5 (1.63);
Intervention 3 (fluoxetine 20
mg): 14.8 (1.6);
placebo: 14.6 (1.6); Range: 12‐17 years |
Intervention 1 (vortioxetine 10 mg) = 93:54; Intervention 2 (vortioxetine 20 mg) = 97:65; Intervention 3 (fluoxetine 20 mg) = 103:50; placebo: 105:49 | DSM‐5 ™ (no other details available) | CDRS‐R mean (SD) score (measure): Intervention 1 (vortioxetine 10 mg): 64.82 (9.38) Intervention 2 (vortioxetine 20 mg): 65.29 (9.73) Intervention 3 (fluoxetine 20 mg): 64.06 (8.65) placebo: 64.02 (8.96) CGI‐S mean (SD): Intervention 1 (vortioxetine 10 mg): 4.99 (0.77) Intervention 2 (vortioxetine 20 mg): 5.00 (0.71) Intervention 3 (fluoxetine 20 mg): 4.97 (0.68) Category: Not reported by trialists |
Not reported | Not stated |
| Wagner 2004 | USA | Forest Pharmaceuticals | Setting: Outpatients; # sites: 21; Total #participants enrolled = 178 |
Intervention = 12.1 (2.8); Control = 12.1 (3.1); Range: 7‐17 years |
Intervention = 54:39; Control = 43:42 |
DSM‐IV confirmed using K‐SADS‐P and L and a CDRS‐R score of ≥ 40 | CDRS‐R mean (SD) score: Intervention = 58.8 (10.9); Control = 57.8
(11.1); CGI not reported Category: Trialists reported moderately severe illness |
Mean (SD) months: Intervention = 20.8 (21.4); placebo =18.6 (16.4) | Intervention: ADHD 4.5%; dysthymia 5.6% Control: ADHD 1.2%; dysthymia 1.2% |
| Wagner 2006 | USA | Forest Laboratories, Inc | Setting: Outpatients; # sites: 25 Total #participants enrolled = 268 |
Intervention = 12.2 (2.9); Control = 12.4 (3.0) Range: 6‐17 years |
Intervention = 68:63; Control = 69:64 | DSM‐IV confirmed using K‐SADS‐PL and a CDRS‐R score of ≥ 40; 1‐week placebo run‐in period | CDRS‐R mean score: Intervention = 54.5; Control = 56.6; CGI score: Intervention 4.4; placebo 4.2 Category: Not reported by trialists |
Mean (SD) months: Intervention = 16.7 (15.3); placebo = 15.6 (13.6) | Intervention: Anxiety disorder 4.5%; Control: Anxiety disorder 7.5% |
| Wagner Trial 1 & 2 (2003) | USA India Canada Costa Rica Mexico |
Pfizer | Setting: Outpatients; # sites: 53; Total #participants enrolled = 376 |
Not stated for either group Range: 6 ‐17 years | Intervention = 108:81; Control = 84:103 | DSM‐IV confirmed using K‐SADS‐PL, a CDRS‐R score of ≥ 45 and a CGI‐S score of ≥ 4 | CDRS‐R mean (SD) score: Intervention = 64.3 (11.0); Control = 64.6
(11.0); CGI score (SD): Intervention 4.6 (0.6); placebo 4.5 (0.7) Category: Not reported by trialists |
Not reported | Not stated separately for each group (Total patient population with at least 1 comorbid condition 40%; Conditions that occurred in at least 5% of patient population included anxiety; phobic disorder; adjustment reaction; ODD) |
| Weihs 2018 | USA Mexico |
Pfizer Inc | Setting: Outpatients; # sites: 37; Total #participants enrolled = 339 |
CHILDREN
Intervention 1 (desvenlafaxine) = 9.3
(1.4);
Intervention 2 (fluoxetine) = 9.6 (1.3);
Control = 9.4
(1.3);
Range: 7–11 years ADOLESCENTS Intervention 1 (desvenlafaxine) = 15.0 (1.5); Intervention 2 (fluoxetine) = 14.7 (1.6); Control = 14.6 (1.5); Range: 12–17 years |
Not reported according to treatment groups Children = 57:73; Adolescents = 127:82 |
DSM‐IV‐TR criteria for MDD as the primary diagnosis, supported by the K‐SADS‐PL | CDRS‐R total score, M (SD):Children
Intervention 1 (desvenlafaxine)
= 56.4 (10.9)
Intervention 2 (fluoxetine) = 55.0 (8.7)
Control
= 57.0 (8.6) Adolescents Intervention 1 (desvenlafaxine) = 56.3 (8.8) Intervention 2 (fluoxetine) = 57.0 (8.1) Control = 57.1 (9.1) Category: Not reported by trialists |
Median (range) months: Children Intervention 1 (desvenlafaxine) = 8 (1–71) Intervention 2 (fluoxetine) = 6 (1–42) Control = 11 (1–57) Adolescents Intervention 1 (desvenlafaxine) = 7 (1–61) Intervention 2 (fluoxetine) = 7 (1–96) Control = 8 (1–69) |
None (exclusion criteria included comorbid primary psychiatric condition other than MDD) |
ADHD: attention deficit hyperactivity disorder BDI: Beck Depression Inventory CD: conduct disorder CDSR: Children's Depression Rating Scale C‐GAS: Children's Global Assessment Scale CGI: Clinical Global Impressions Scale CGI‐S: Clinical Global Impressions Scale ‐ Severity DICA: Diagnostic Interview for Children and Adolescents DSM‐III(‐R): Diagnostic and Statistical Manual of Mental Disorders III ‐ Revised DSM‐IV(‐TR): Diagnostic and Statistical Manual of Mental Disorders IV ‐ Text Revision GAF: Global assessment of functioning HAM‐D: Hamilton Rating Scale for Depression K‐SADS(‐PL): Kiddie Schedule for Affective Disorders and Schizophrenia for School Aged Children (Present and Lifetime version) MADRS: Montgomery‐Asberg Depression Rating Scale MDD: major depressive disorder MINI‐KID: Mini International Neuropsychiatric Interview for children and adolescents ODD: oppositional defiant disorder SD:standard deviation SE: standard error
Sample sizes
The number of participants randomised to the relevant arms in these trials ranged from 23 to 784 (median 275).
Study Setting
The included trials had study sites in a number of countries (Denmark, Estonia, Germany, Norway, Sweden, Switzerland, Argentina, Belgium, Holland, Italy, Japan, Mexico, South Africa, Spain, United Arab Emirates, UK, India, Costa Rica, USA, Canada, Russian Federation, Colombia, Serbia, Ukraine, Korea, Poland, Bulgaria, France, Hungary, Latvia, Chile, Finland, Slovakia). The majority of the trials included outpatients only except for two trials (Simeon 1990; Von Knorring 2006) where inpatients were also included. Information about the study settings was not available for Simeon 1990 or Berard 2006 (although the MHRA report (Dubitsky 2004) stated that Simeon 1990 only included outpatients; however, we did not receive confirmation from the author) or NCT02709746 and the study setting was unclear for Paroxetine Trial 1.
Participants
Eight trials included only adolescents, with an age range of 12 or 13 years to 17 or 18 years. Sixteen trials of children and adolescents had a lower age limit of between six to eight years. The mean age ranged from 14.4 to 16.0 years in the adolescent trials and 11.5 to 13.3 years in the trials of children and adolescents; one study reported a mean age for children and adolescents separately: 9.4 and 14.8 years, respectively (Weihs 2018).
Most trials reported that there were more female than male participants, but the ratio of females to males varied enormously both within trials (across arms) and across trials, from very small differences to there being twice the number of females to males. One trial had a similar proportion of females and males and five trials had more males than females. Three trials did not provide information on gender by treatment arm and two trials provided no information on gender of participants.
All trials included participants with major depressive disorder with most of them basing the diagnosis at entry on DSM‐IV or DSM‐IV‐TR criteria; with three trials basing diagnoses on DSM‐III or DSM‐III‐R criteria and one more recent trial on DSM‐5 criteria. The majority used a semi‐structured clinical interview (K‐SADS/K‐SADS‐PL) and three trials used the MINI‐KID; this information was not available for two trials. Only one trial (Atkinson 2018) used both K‐SADS and a clinical interview, while Von Knorring 2006, in contrast to all the other trials, used only a five‐minute clinical interview with parents. In addition to a diagnostic interview, the majority of trials (except Emslie 2006 and Durgam 2018) used a cut‐off score on a measure of depression symptom severity to establish eligibility. Most studies used a cut‐off greater than 40 points on the CDRS‐R; while four trials used a cut‐off of 45. In addition to CDRS‐R, eight studies used the Clinical Global Impressions‐Severity (CGI‐S) score (>= 4). Few other scales were used, including the HAM‐D scale (four studies), and (in one study each) the MADRS scale and CDI. Some trials also used a measure of functioning to confirm diagnosis.
Some trials included a screening process that was undertaken over a period of several weeks. This comprised up to two or three independent diagnostic interviews, taking place over a period of up to three weeks. In 13 trials, all participants were treated with placebo for a lead‐in period and those whose depressive disorder improved during this time were excluded.
In all trial reports, except two (Simeon 1990; Almeida‐Montes 2005), there was a description of depression symptom severity at baseline for the treatment and placebo groups. Mean severity scores at baseline across the included trials ranged from 47.6 to 65.5 on the CDRS‐R (the total range on this measure is 17 to 113). Nine trialists deemed these baseline scores as indicative of moderate to severe depression. Two trials (Emslie 2002; VLZ‐MD‐22) indicated participants had moderate to severe depression based on CGI‐S scores; no categorisation was available for the other thirteen trials. There was no clinically important imbalance between treatment groups in depression symptom severity at baseline in any trial. Clinical Global Impression (CGI) scores were reported in 13 trials and ranged from a mean of 3.9 to 5.0 (with a median of four being reported in Emslie 2006), which is in the moderately ill range. The length of the current episode was reported in 12 trials and the mean duration ranged from approximately 15 weeks to 108 weeks in the intervention group and 14 to 100 weeks in the placebo group, with most of the trials reporting episode lengths of over 40 weeks. In the TADS trial, the median duration of episode length was reported as 38 weeks in the intervention and 35.5 weeks in the placebo groups, and in Weihs 2018 the median duration ranged from six to 11 weeks across children and adolescent intervention and control groups.
It is clear from research that comorbidity may affect the clinical outcome (Birmaher 1996; Kovacs 1989); however, it is difficult to examine this, given the non‐standard way in which comorbidity is reported and because some comorbid disorders form part of the exclusion criteria in some trials (refer to Characteristics of included studies table ‐ inclusion and exclusion criteria). Nine trials provided no detail about the comorbid mental health conditions of participants, three studies provided combined information for all the participants, while one study excluded participants with any primary psychiatric condition other than MDD (NCT02709746). Trials included young people with a range of mental health comorbidity. The percentages of some of the commonly reported comorbid conditions varied markedly ‐ anxiety disorders (ranging from 4.5% to 66.7% in the intervention arm and 2% to 45.8% in the control arm), dysthymia (ranging from 5.5% to 41.7% in the intervention arm and 1.2% to 29.2% in the control arm), Attention Deficit Hyperactivity Disorder (ADHD) (ranging from 1.6% to 33.3% in the intervention arm and 0.0% to 27.1% in the control arm), and Oppositional Defiant Disorder/Conduct Disorder (ODD/CD) (ranging from 0.5% to 27.1% in the intervention arm and 1.1% to 3.3% in the control arm) (see Table 3).
Twelve trials explicitly excluded those who had previously not responded to antidepressant treatment. Further, participants who were considered at risk for suicide at baseline were specifically excluded in all but four trials (Emslie 1997; Von Knorring 2006; Paroxetine Trial 1; NCT02709746). The method to define risk varied across trials; some gave no definition of 'serious suicidality risk' or acute suicidality or defined it as "in the opinion of the investigator". Most studies used more than one parameter to determine suicidal risk which variously included previous history of suicide attempts, previous history or active suicidal ideation or plan, hospitalisation for suicidal attempt, or having a first degree relative who died by suicide. Only three studies used the Columbia Suicide Severity Rating Scale (C‐SSRS) responses to assess suicide risk. The FDA carried out a stratified analysis of those trials available at the time based on history of suicide attempt or ideation to investigate if risk of suicide attempt or ideation for those receiving SSRIs varied by stratum. They concluded that there was no evidence of this (Hammad 2004).
Interventions
See Table 4 for full description of interventions. In the SSRI class, there were four trials of paroxetine, five trials of fluoxetine, two trials of citalopram, two trials of escitalopram oxalate (the therapeutically active component of citalopram), two trials of sertraline, one trial of vilazodone and one trial of vortioxetine. Only one trial included intervention arms of two different drugs within the SSRI class. In the SNRI class, there were two trials of venlafaxine, desvenlafaxine and in the TeCA class there were two trials of mirtazapine. Three trials had an intervention arm each of a SSRI and a SNRI. The treatment period of the included trials was between six and 12 weeks.
2. Comparison of interventions.
| Study ID | Intervention Drugs | Intervention Regimen | Comparison groups | Exclusion based on improvement in depressive symptoms (lead‐in period) | Exclusion based on risk for suicide, H/O suicide attempts, suicidal ideation[1] | Exclusion based on H/O of non‐response to anti‐depressant treatment | Frequency of data collection for efficacy measures |
| Almeida‐Montes 2005 | Fluoxetine | Dosage: 20 mg/day; Length of treatment: 6 weeks |
Placebo | Yes. Placebo lead‐in:1 week | Suicide attempt in the preceding 4 weeks | Not excluded | Weekly |
| Atkinson 2014 | Duloxetine | Duloxetine: Dosage: 60–120 mg/day; Length of treatment: 10 weeks |
Placebo | No information | Serious suicide risk | Not excluded | Nearly weekly during screening and acute treatment period. Nearly fortnightly during the long term treatment period |
| Fluoxetine | Dosage: 20–40 mg/day; Length of treatment: 10 weeks |
||||||
| Atkinson 2018 | Desvenlafaxine | High exposure dosage: 10–50 mg/day; Low exposure dosage: 10–35 mg/day; Length of treatment: 8 weeks |
Placebo | No placebo lead‐in | High risk of suicide (including first‐degree relative who committed suicide) | Not excluded | Weekly till week 4, thereafter fortnightly until week 8 |
| Berard 2006 | Paroxetine | Dosage: 20‐40 mg/day; Length of treatment: 12 weeks |
Placebo | Yes. Placebo lead‐in: 2‐week single‐blind | Patients with current serious suicidal ideation |
Not excluded | Near weekly |
| Durgam 2018 | Vilazodone | Low exposure dosage: initial dosage of 5 mg/day gradually increased to 15 mg/day; High exposure dosage: Initial dosage of 5 mg/day gradually increased to 30 mg/day; Length of treatment: 10 weeks | Placebo | No placebo lead‐in | Significant suicide risk judged by the investigator based on the psychiatric interview or information collected from the Columbia‐Suicide Severity Rating Scale (C‐SSRS), or suicide attempt within the past year |
Yes, excluded | Weekly till week 4, thereafter fortnightly until week 8 |
| Emslie 1997 | Fluoxetine | Dosage: 20 mg/day Length of treatment: 8 weeks |
Placebo | Yes. Placebo lead‐in:1‐week single‐blind (Those patients whose conditions improved during the 1‐week placebo run‐in period continued to receive placebo for an additional week to determine if the symptoms returned. If the patients’ conditions still improved, they were withdrawn from the study) | Not specifically stated | Not excluded | Weekly |
| Emslie 2002 | Fluoxetine | Dosage: 20 mg/day (except first 1 week ‐ 10 mg); Length of treatment: 9 weeks |
Placebo | Yes. Placebo lead‐in: 1‐week single‐blind | Serious suicide risk (no further definition) | Yes, excluded | Weekly |
| Emslie 2006 | Paroxetine | Dosage: 10 to 50 mg; Length of treatment: 8 weeks |
Placebo | No placebo lead‐in | Suicidal or homicidal risk (no further definition) | Yes, excluded | Near weekly |
| Emslie 2007 | Venlafaxine extended release | Dosage: 37.5 to 225 mg/day; Length of treatment: 8 weeks |
Placebo | Yes. Placebo lead‐in: trial 1: single‐blind for 14 days (+/‐3 days);
trial 2: single‐blind for 7 days (+/‐3 days) |
Acute suicidality (no further definition) | Not excluded | Near weekly |
| Emslie 2009 | Escitalopram | Dosage: 10 to 20 mg/day; Length of treatment: 8 weeks |
Placebo | Yes. Placebo lead‐in: 1‐week single‐blind | Patients considered a suicide risk by the investigator, including those
who had active suicidal ideation, had made a suicide attempt, or had ever been hospitalised because of a suicide attempt |
Yes, excluded | Weekly till week 4, thereafter fortnightly until week 8 |
| Emslie 2014 | Duloxetine | Acute treatment phase: Dosage: 30 or 60 mg QD; Length of treatment: 10 weeks; Long‐term treatment/ extension phase: Dosage: 60‐120 mg QD Length of treatment: 26 weeks; Taper phase: Dosage: Dosage decreases; Length of treatment: 2 weeks |
Placebo | No placebo lead‐in | Serious suicide risk (no further definition) | Not excluded | Weekly |
| Fluoxetine | Acute treatment phase: Dosage: 20 mg QD Length of treatment: 10 weeks; Long‐term treatment/extension phase: Dosage: 20‐40 mg QD; Length of treatment: 26 weeks; Taper phase: Dosage: Dosage decreases; 20 mg QD Length of treatment: 2 weeks |
||||||
| Keller 2001 | Paroxetine | Dosage: 20 to 40 mg/day; Length of treatment: 8 weeks; |
Placebo | No placebo lead‐in | Current suicidal ideation with intent or specific plan; history of suicide attempt by drug overdose | Not excluded | Weekly |
| Mirtazapine Trial 1 | Mirtazapine | Dosage: 15 to 45 mg/day; Length of treatment: 8 weeks; |
Placebo | No placebo lead‐in | Serious suicide attempt during the current major depressive episode, or any previous suicide attempt resulting in hospitalisation | Yes, excluded | Near weekly |
| Mirtazapine Trial 2 | Mirtazapine | Dosage: 15 to 45 mg/day; Length of treatment: 8 weeks |
Placebo | No placebo lead‐in | Serious suicide attempt during the current major depressive episode, or
any previous suicide attempt resulting in hospitalisation |
Yes, excluded | Near weekly |
| Paroxetine Trial 1 | Paroxetine | Dosage: 10 to 40 mg/day; Length of treatment: 8 weeks |
Placebo | Yes, Placebo lead‐in: 2‐week period | Not stated | Not excluded | Near weekly |
| Simeon 1990 | Fluoxetine | Dosage: 20 to 60 mg/day; Length of treatment: 7 weeks; |
Placebo | Yes. Placebo lead‐in: 1‐week single‐blind | Serious suicidal risk (no further definition) | Not excluded | No information available |
| TADS 2004 | Fluoxetine | Dosage: 20 to 40 mg/day; Length of treatment: 12 weeks |
Placebo (We are just using data from the two relevant arms but there were comparison groups: CBT alone and CBT with fluoxetine) |
No placebo lead‐in | Suicidality or homicidality (patients were excluded for dangerousness to
self or others if they had been hospitalised for dangerousness within 3 months of consent or were deemed by a cross‐site panel to be “high risk” because of a suicide attempt requiring medical attention within 6 months, clear intent or an active plan to commit suicide, or suicidal ideation with a disorganised family unable to guarantee adequate safety monitoring) |
Yes, excluded | At baseline, 6, 12, 18, 24, 30 and 36 weeks |
| VLZ‐MD‐22 | Vilazodone | Dosage: 15‐30 mg Length of treatment: 9 weeks (8 weeks acute treatment, 1‐week taper) | Placebo | Yes. Placebo lead‐in: All participants received placebo (3 tablets, 1 capsule) at screening visit (1 week prior to baseline) to “confirm their ability to swallow investigational product”. Those unable were ineligible for participation | History of a suicide attempt within the past year or a current significant suicide risk as judged by the investigator based on interview or information collected in the C‐SSRS |
Not excluded | Weekly |
| Fluoxetine | Dosage: 20 mg; Length of treatment: 9 weeks (8 weeks acute treatment, 1‐week taper) | ||||||
| Von Knorring 2006 | Citalopram | Dosage: 10 to 40 mg/day; Length of treatment: 12 weeks |
Placebo | No placebo lead‐in | Not explicitly stated | Not excluded | Weekly for the first 2 weeks, and then every 3‐4 weeks |
| NCT02709746 | Vortioxetine | Intervention group 1 and 2 (vortioxetine) Dosage: 10 mg or 20 mg; Length of treatment: 8 weeks | Placebo | Yes. Placebo lead‐in: nonrandomised single‐blind treatment period comprising placebo and brief psychosocial intervention for 4 weeks | Very limited information available about the exclusion criterion | Yes, excluded | Unclear |
| Fluoxetine | Dosage: 20 mg; Length of treatment: 8 weeks | ||||||
| Wagner 2004 | Citalopram | Dosage: 20 mg to 40 mg; Length of treatment: 8 weeks |
Placebo | Yes. Placebo lead‐in: 1‐week single‐blind | Patients who were considered a suicide risk, who had made an active suicide attempt within the past year, or had been hospitalised because of an attempt | Yes, excluded | Weekly for 2 weeks, fortnightly thereafter until week 8 |
| Wagner 2006 | Escitalopram oxalate | Dosage: 10 mg for the first 4 weeks; thereafter flexible dosage 10 to 20
mg/day Length of treatment: 8 weeks |
Placebo | Yes. Placebo lead‐in: 1‐week | Suicide risk based on clinical judgement of investigator or ever hospitalised for suicide attempt or had made a suicide attempt within the past year | Yes, excluded | Weekly for 2 weeks, fortnightly thereafter until week 8 |
| Wagner Trial 1 & 2 (2003) | Sertraline | Dosage: 25 to 200 mg/day; Length of treatment: 10 weeks | Placebo | No placebo lead‐in | Previous suicide attempt or current significant suicidal or homicidal risk | Yes, excluded | Frequent follow‐up visits but no further details available |
| Weihs 2018 | Desvenlafaxine | Dosage: 25–50 mg/day; Length of treatment: 8 weeks | Placebo | No placebo lead‐in | History or current evidence of suicidal behaviour or suicidal ideation associated with actual intent and/or plan at any time in their lifetime based on clinical judgement or C‐SSRS responses at the screening or baseline visit, or first‐degree relative who had committed suicide |
Yes, excluded | Weekly till week 4, fortnightly thereafter until week 8 |
| Fluoxetine | Dosage: 20 mg/d; Length of treatment: 8 weeks. |
C‐SSRS: Columbia suicide severity rating scale QD: quaque die: once a day
Efficacy measures were collected throughout the treatment period and at completion of the trial. For six trials, this was described as weekly and for ten trials, this was nearly weekly. For five trials, this was described as weekly until weeks two to four followed by fortnightly data collection until the end of the data collection period. Three trials did not provide specific details about follow‐up visits. TADS 2004 described assessments at baseline, six, 12, 18, 24, 30 and 36 weeks. Emslie 2009 specifically stated that the high placebo response rate may be due to "extensive contact" (pg.728), which refers to this regular assessment.
With the exception of five trials (Emslie 1997; Emslie 2002; Almeida‐Montes 2005; Emslie 2009; NCT02709746), a flexible dosing scheme was used. Three studies did not use flexible dosing for the entire treatment period: Wagner 2006 offered flexible dosing only after the first four weeks of treatment; Emslie 2014 offered flexible dosing only in the long‐term treatment/extension phase; VLZ‐MD‐22 offered flexible dosing only in one intervention arm.
Outcomes
Table 5 describes the outcomes measures in each trial. Our primary outcomes (depressive disorder established via a clinician administered diagnostic interview and death by suicide) were not measured in any trial. The assessor‐rated CDRS‐R was used to measure depression symptom severity in the majority of trials (N = 18). A mix of remission and response were used as outcomes across the studies; at times. the definitions of response were the same as the definition of remission. Typically, these definitions were based on a cut‐point on the assessor‐rated continuous measure of depression symptom severity. Where both response and remission were measured and reported, by preference we used remission data in the analysis. Self‐rated depression was seldom measured. Fewer than half of the studies (N = 10) used the Children's Global Assessment Scale to measure functioning, though other measures were also used.
3. Outcomes measured in each trial.
| Study ID | Disorder diagnosis (ICD, DSM) | Death by suicide | Clinician‐rated CDRS‐R depression symptom severity | Remission/response definition |
Self‐rated depression symptom severity Using BDI/CDI |
Functioning (C‐GAS) |
Suicide related outcomes (FDA) |
Suicidal ideation (SIQ‐Jr) |
Overall adverse outcomes | Other measures they included |
| Atkinson 2014 | No | No | Yes | Remission CDRS‐R total score < 28 (response 50% improvement on the CDRS‐R total score after subtracting the 17‐item base score) | No | No | No. Columbia‐Suicide Severity Rating Scale (C‐SSRS) | No | Yes | CGI‐S |
| Atkinson 2018 | No | No | Yes | Response CGI improvement of 1 or 2 | No | No | No. C‐SSRS | No | Yes | CGI‐S; CGI‐I |
| Almeida‐Montes 2005 | No | No | No | Remission 50% reduction in Hamilton Depression Rating Scale (HAM‐D) scores (also used CGI‐I score of 1 or 2; 50% reduction in CDSR‐S and HAM‐D scores) |
No | Yes. Foreign language and data not reported in a form that could be used | No | No | No | Hamilton Anxiety Rating Scale; Birleson Depression Self‐Rated Scale |
| Berard 2006 | No | No | No | Response 50% reduction on MADRS. Post hoc analysis response CGI‐I score of 1 or 2 |
No | Yes | FDA | No | Yes | Montgomery‐Asberg Depression Rating Scale (MADRS); Clinical Global Impressions Scale Improvement (CGI‐I); The Schedule for Affective Disorders and Schizophrenia for School‐Age Children—Lifetime Version K‐SADS‐L (Present version = P) depression subscale score; Mood and Feeling Questionnaire |
| Durgam 2018 | No | No | Yes | Remission CDRS‐R ≤ 28 (response CDRS‐R ≥ 40% total score improvement from baseline at week 8; CGI response score 1 or 2) | No | No | No. C‐SSRS | No | Yes | CGI‐S; CGI‐I; K‐SADS‐PL |
| Emslie 1997 | No | No | Yes | Remission CDRS‐R ≤ 28 (response CGI score of 1 or 2) | Yes | Yes | FDA | No | No | Weinberg Screening Affective Scale (WSAS); Brief Psychiatry Rating Scale ‐ Children’s (BPRS‐C) |
| Emslie 2002 | No | No | Yes | Remission CDRS‐R ≤ 28 (response 30% decrease CDRS‐R & CGI of 1 or 2) |
Yes | No | FDA | No | Yes | Clinical Global Impressions Scale Severity (CGI‐S); CGI‐I; Hamilton Anxiety Rating Scale;MADRS; Global Assessment of Functioning Scale (GAF) |
| Emslie 2006 | No | No | Yes | Remission CDRS‐R ≤ 28 (response CGI improvement of 1 or 2) | No | No | Report of events based on Columbia classification; no report of continuous measure | No | Yes | CGI‐S; CGI‐I); Kutcher Adolescent Depression Rating Scale (KADS); GAF |
| Emslie 2007 | No | No | Yes | Response > 35% decrease in CDRS‐ R (also used ≥ 50% decrease in HAM‐D or MADRS or CGI) | No | No | FDA | No | Data not available in most cases | MADRS; CGI‐S; HAM‐D; GAF |
| Emslie 2009 | No | No | Yes | Remission CDRS‐R ≤ 28 (also used: CGI improvement of 1 or 2 or CDRS‐R reduction of ≥ 40%) | No | Yes | Modified Columbia Suicide Severity Rating Scale (MC‐SSRS) | Suicidal Ideation Questionnaire‐Junior High School Version | Yes | |
| Emslie 2014 | No | No | Yes | Remission CDRS‐R ≤ 28 (response 50% improvement on the CDRS‐R total score after subtracting the 17‐item base score) |
No | No | No. C‐SSRS | No | Yes | CGI‐I |
| Keller 2001 | No | No | No | Response HAM‐D ≦ 8 or ≥ 50% reduction in baseline HAM‐D | No | No | FDA data | No | Yes | K‐SADS‐L and HAM‐D; CGI‐I; Self Perception Profile; Sickness Impact Scale; Autonomous Function Checklist |
| Mirtazapine Trial 1 | No | No | Yes | Not stated | Yes but HAM‐D self‐rating | C‐GAS used but not reported | Events reported as adverse events | No | No | CGI‐I; Self‐Report Childhood Anxiety Related Disorder (SCARED), Connors' Global Index (Parent and Teacher Versions) |
| Paroxetine Trial 1 | No | No | Yes | Response CGI improvement of 1 or 2 | No | No report of measure used | Events reported as adverse events; no report of continuous data | No | Yes | Not stated |
| Simeon 1990 | No | No | No | Not stated | No | No or not reported | No report | No | No | CG‐I; HAM‐D; Raskin Depression Scale; Covi Anxiety Scale; Hopkins Symptom Checklist Follow‐up assessment, current activities and functioning with family and peers |
| TADS 2004 | No | No | Yes | Remission CDRS‐R ≤ 28 (response CGI‐I improvement score of 1 or 2) |
No | Yes | No FDA. Report of events based on Columbia classification | Suicidal Ideation Questionnaire‐Junior High School Version | Yes | CGI‐I; Reynolds Adolescent Depression Scale (RADS) |
| Wagner 2004 | No | No | Yes | Response CDRS‐R ≤ 28 (called remission in other trials) | No | Yes | FDA data | No | Yes | K‐SADS‐PL |
| Wagner 2006 | No | No | Yes | Response CDRS‐R ≤ 28 (called remission in other trials) (also analysed CGI‐I improvement of 1 or 2) |
No | Yes | Events reported as adverse events | No | Yes | CGI‐S; CGI‐I |
| Wagner Trial 1 & 2 (2003) | No | No | Yes | Response ≥ 40% decrease on CDRS‐R | No | Yes | Events reported as adverse events | No | No | CGI‐S; CGI‐I; Multidimensional Anxiety Scale for Children (MASC); Pediatric Quality of Life Enjoyment and Satisfaction Questionnaire (PQ‐LES‐Q); adverse events |
| VLZ‐MD‐22 | No | No | Yes | Not stated. | No | No | No. C‐SSRS | No | Yes | K‐SADS‐PL, CGI‐S, CGI‐I |
| Von Knorring 2006 | No | No | No | Remission MADRS < 12 (response ≤ K‐SADS‐P depression and anhedonia items or with a reduction of at least 50% from baseline of the MADRS total score) | Yes | No | FDA data | No | Yes | K‐SADS‐P; MADRS; GAF |
| NCT02709746 | No | No | Yes | Remission CDRS‐R ≤ 28 (response ≥ 50% decrease in CDRS‐R total score) | No | Yes | Not reported | No | Yes | General Behaviour Inventory (GBI) depression subscale; Parent Global Assessment–Global Improvement (PGA); Symbol Digit Modalities Test (SDMT); CGI‐S; (CGI‐I); PQ‐LES‐Q |
| Weihs 2018 | No | No | Yes | Response CGI improvement of 1 or 2 | No | No | No. C‐SSRS | No | Yes | CGI‐S; CGI‐I |
BDI: Beck Depression Inventory BPRS‐C: Brief Psychiatry Rating Scale ‐ Children's CDI:Children's Depression Inventory CDRS‐R:Children's Depression Rating Scale ‐ Revised C‐GAS:Children's Global Assessment Scale CGI(‐S)(‐I): Clinical Global Impressions Scale ‐ Improvement and Severity C‐SSRS:Columbia SuicideSeverity Rating Scale DSM: Diagnostic and Statistical Manual of Mental Disorder FDA: US Food and Drug Administration GAF:Global Assessment of Functioning GBI: General Behaviour Inventory HAM‐D: Hamilton Rating Scale for Depression ICD: International Classification of Diseases KADS: Kutcher Adolescent Depression Scale K‐SADS(‐PL):Kiddie Schedule for Affective Disorders and Schizophrenia for School Aged Children (Present and Lifetime version) MADRS: Montgomery‐Asberg Depression Rating Scale MASC: Multidimensional Anxiety Scale for Children MC‐SSRS: Modified Columbia Suicide Severity Rating Scale PGA: Physician Global Assessment PQ‐LES‐Q: Pediatric Quality of Life Enjoyment and Satisfaction Scale RADS: Reynolds adolescent depression scale SCARED: Screen for Child Anxiety Related Disorders SDMT:Symbol Digit Modalities Test WSAS: Weinberg Screening Affective Scale
Suicide‐related outcomes were classified and reported in various ways in each of the trials. As described in the Methods section in the original version of this review, a post hoc decision was made to use the data provided in an FDA report (Hammad 2004) in order to overcome inconsistent reporting of these outcomes across trial reports. The process of the FDA in establishing the rate of suicide‐related outcomes for each trial was based on the following process. A group of 10 suicidology experts were assembled by Columbia University (led by Dr Kelly Posner). Suicide‐related outcomes were defined after careful deliberation by this expert panel as including 'definitive suicidal behaviour/ideation' (pg.8, Hammad 2004) and where more than one event was recorded for an individual, the most severe event was used. The group of experts reviewed all of the suicide‐related adverse events, all serious adverse events and all accidental injuries identified by the sponsors of SSRI trials. There was some discrepancy between the sponsors' classifications and the expert panel classification (with 22 new events added, and 26 old events removed). Overall, there were no completed suicides in any of the trials. The report highlighted the important point that none of these trials had adequate power for safety analysis. We included data from the trial reports as follows: for TADS 2004, data were extracted from the Emslie 2006 report where it was stated that rates were based on a reanalysis by the Columbia Group using the Columbia‐Classification Algorithm for Suicidal Assessment); Emslie 2009 stated that their data were based on an increase in suicidal ideation and behaviour on the Modified Columbia Suicide Severity Rating Scale (MC‐SSRS), a clinician‐rated instrument; Wagner 2006 stated that "potential suicide‐related events were identified" and described these in the results as adverse events, which was not equivalent to the data based on the Columbia Classification; for the mirtazapine trials (Mirtazapine Trial 1 & 2), the MHRA report gave a description of events stating there was one case of suicidal ideation in the mirtazapine group (both trials combined) and one case of self‐mutilation in the placebo group (both trials combined). The data for Paroxetine Trial 1 were suicidal ideation reported as an adverse event. The trial by Almeida‐Montes 2005 did not provide data for this outcome. The newly included trials largely used the Columbia Suicide Severity Rating Scale (C‐SSRS), a clinician‐rated instrument (6/7). Suicidal ideation as an continuous outcome was very seldom measured (N = 2). Finally, data on overall adverse outcomes were available in 17 trials.
Ongoing studies
Two studies were previously identified as ongoing for CD004851 (Glod 2004; NCT00353028), for which no new information has been identified for this update.
Ten studies were newly identified as ongoing for this update (see Ongoing studies for full details). One was a multicentre, individually randomised, parallel group trial comparing bupropion and placebo and was classified as “active, not yet recruiting” according to its trial registry entry (NCT02129751‐bupropion). Two further studies were classified as “active, recruiting” according to their trial registry entries (JPRN‐JapicCTI‐194585‐escitalopram; NCT02709655‐Vortioxetine). Both were multicentre, individually randomised, parallel group trials, one comparing escitalopram and placebo, and the second a four‐arm trial comparing vortioxetine 10mg, vortioxetine 20mg, fluoxetine and placebo. Three further studies were classified as “complete, no results posted” according to their trial registry entries, suggesting recruitment to these studies was complete but results were not publicly available (NCT01185977‐fluoxetine; IRCT138901093607N1‐fluoxetine; NCT03569475‐Levomilnacipran). All were individually randomised, parallel group trials, one comparing fluoxetine and placebo, the second comparing fluvoxamine and fluoxetine and the third a three‐arm multicentre trial comparing levomilnacipran, fluoxetine and placebo. Three additional studies were initially classified as “complete, no results posted” at the time of data extraction (EUCTR2015‐002181‐23‐Agomelatine; NCT03315793‐Duloxetine; NCT02431806‐Levomilnacipran). All were multicentre, individually randomised, parallel group trials, one comparing duloxetine and placebo, the second a four‐arm trial comparing agomelatine 10mg, agomelatine 25mg, fluoxetine and placebo, and the third a four‐arm trial comparing levomilnacipran 40mg, levomilnacipran 80mg, fluoxetine and placebo. Pharmaceutical company sponsors have subsequently made results for these three studies publicly available via trial registry entries, and we’ve reclassified them as “complete, results posted”. Given this occurred after our data inclusion cut‐off date of 6 May 2020, these results have not been included in the current analysis but will be included in the next update of this review. The final study compared reboxetine and fluoxetine, and was classified as “unknown status” according to its trial registry entry (NCT00426946‐Reboxetine). In attempting to ascertain trial status, a recent publication appearing to report partial trial results was located after our data inclusion cut‐off date of 6 May 2020 (Toren 2019, not retrieved in search). Data from these four studies will be assessed for inclusion in a subsequent review update.
Excluded studies
In the original review, there were eight excluded studies. These were excluded due to the intervention not being an SSRI; one trial was a head‐to‐head trial of antidepressants that did not include a placebo; one trial was a case‐control trial and one trial included participants with bipolar disorder, not depressive disorder. Of these, the two trials on venlafaxine and the two trials on mirtazapine were then included in the 2012 update (Hetrick 2012) due to the inclusion criteria changing to include SSRIs as well as newer generation antidepressants.
In the 2012 update review, seven new studies were excluded. The primary reasons for exclusion included the following: two did not have a pure newer generation antidepressant or placebo treatment arm; two focused on comorbid substance use; one used an antidepressant that did not meet the inclusion criteria for antidepressants considered in this review; and two were not randomised trials.
In this version of the review, 14 studies were excluded. The primary reasons for exclusion included the following: one study compared a single dose of fluoxetine to peppermint syrup with no depression outcomes planned; one recruited a mixed sample of major depressive disorder and/or anxiety disorder, one study recruited those with a primary diagnosis of bipolar disorder; one did not have a pure antidepressant or placebo treatment arm; three recruited young adults or adults; three were not randomised trials during their acute phases; and four were not acute phase trials (e.g. maintenance or discontinuation trials).
Risk of bias in included studies
See Figure 2 for the 'Risk of bias' graph that shows the proportion of studies with each of the judgements and Figure 3 for the 'Risk of bias' summary showing all the judgements in a cross‐tabulation by trial.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Thirteen trials were rated as having described adequate sequence generation. There were no full reports of allocation concealment in any of the included trials. In three of the newly included trials an Interactive Voice Response System was used that is presumed to ensure allocation concealment (Atkinson 2014; Emslie 2014; Durgam 2018).
Blinding
All trials were described as being 'double‐blind', of having the relevant treatment arms double‐blind, as quadruple‐blind (Atkinson 2014; VLZ‐MD‐22) or as having 'patients, investigators and study site personnel' blinded (Durgam 2018). There was little description of the blinding in 10 trials, so that it was unclear what 'double‐blind' referred to (Simeon 1990; Wagner 2004; Von Knorring 2006; Wagner 2006; Emslie 2007 Trial 1; Emslie 2007 Trial 2; Emslie 2009; Mirtazapine Trial 1 & 2; Paroxetine Trial 1); in two trials described as doubled‐blinded, it was stated that the 'subject and investigator' were blinded. Emslie 1997 mentioned that the pharmacy staff were blind. Almeida‐Montes 2005 and TADS 2004 stated that there were independent evaluators who were also 'blind'. In two trials (Emslie 1997; Emslie 2002), the description of blinding indicated that the antidepressant and placebo medications were identical; in three trials, there was mention of the placebo being matched (Durgam 2018; VLZ‐MD‐22; Weihs 2018); and one trial described the placebo being identical in appearance, colour, taste and smell (Emslie 2014). There were no reports on the success of blinding in any of the trials, and the possibility of clinicians or patients guessing the nature of the intervention from side effects was not discussed. Given outcomes were based on ratings by participants and clinicians, this could be an important omission, although the updated CONSORT guidelines highlight that asking participants or healthcare providers what intervention they received as a test of blinding at the end of the trial is confounded because usually by this stage they know what intervention they received (Moher 2010).
Incomplete outcome data
One trial was discontinued early (Simeon 1990) and it was unclear whether this was also the case for Glod 2004. One trial of paroxetine was aiming to recruit 65 participants in each of the treatment and placebo arms; however, it appeared to stop recruitment with fewer than half of this number recruited to each group. The attrition rate for the 27 trials varied between 10% and 82% in the control groups and 10% and 58% in the intervention groups (see Figure 4). The disparity in attrition between treatment arms was of particular concern in the trials of fluoxetine and in the Atkinson 2014 trial of duloxetine, fluoxetine and placebo.
4.

Dropouts from each trial (by treatment arm where available)
All authors stated that intention‐to‐treat analyses (ITT) had been undertaken except for NCT02709746. However, a full application of the intention‐to‐treat principle is only possible when complete outcome data are available for all randomised participants (Hollis 1999). Only three trials (Emslie 1997; TADS 2004; Emslie 2014) appear to have included all patients randomised in their analyses (the Paroxetine Trial 1 appears to have included all randomised patients only in their primary analysis). In the other trials, analyses were carried out on fewer patients than the number randomised. For the majority of trials, only those who received at least one dose of medication or placebo, or had at least one post‐baseline efficacy or safety evaluation were included in the analyses.
Selective reporting
There is some evidence of reporting bias in some of the trials, though this is difficult to assess in most trials, since it was not possible to obtain the trial protocol for studies previously included; in trials included in this update, while clinical trial registry records have been obtained, these often lack necessary detail and there are seldom full protocols available (except in VLZ‐MD‐22). Table 5 documents the outcomes measured (both those relevant to this review and additional outcomes measured) for each trial.
Some of the notable concerns with regard to possible selective reporting of results and missing results follow. We denote concerns arising from missing results with an asterisk.
*The trial report by Emslie 2002 emphasised CDRS‐R scores and remission rates rather than response rate, although response rate was specified as the primary outcome in the Methods section. Additionally, the cut‐off used for remission rate differed from that stated in the Methods section.
Emslie 1997 reported outcomes at five weeks rather than at the completion of the trial; however, data for the end of the trial was provided and used in the review.
In a letter to the editor, Keller 2001 was criticised for changing the definition of response post‐data analysis to a cut‐off that showed treatment effectiveness (Jureidini 2003). In response, Keller 2001 changed their claim of finding a significant effect to stating that the findings showed a strong signal for efficacy (Keller 2003; Jureidini 2004). Subsequently, this study has been reanalysed under the restoring invisible and abandoned trials (RIAT) initiative, where there was full access to and reanalysis of the original full dataset, again highlighting that the original published conclusions were not supported by the data or analysis according to the originally defined primary outcomes (Le Noury 2015). Results of the reanalysis showed that neither paroxetine nor imipramine were statistically or clinically different from placebo on any of the prespecified primary or secondary outcomes and the authors concluded that neither of these antidepressants showed efficacy for major depression in adolescents.
In many trials, response and remission are defined, measured, and reported in many different ways within the trial, without it being clear what the primary outcome is, e.g. Emslie 2009 reported two different results for response using two different definitions. The report by Wagner Trial 1 & 2 (2003) and Emslie 2007 combined the results of two trials and, in most cases, reported the overall outcomes while being vague about the actual definitions.
The outcomes for TADS 2004 have been reported in multiple publications, with the reporting of outcome results that are not consistent across papers.
For trials where results were only reported in the MHRA report, there were few data reported for the outcomes of functioning, adverse outcomes (e.g. Mirtazapine Trial 1 & 2 data were only available for CDRS‐R and suicidal behaviour but not for any other outcome).
For one trial of paroxetine, there was no publication except a brief pharmaceutical company trial report (Paroxetine Trial 1).
It appears that one of the included trials and one trial still included as ongoing have been stopped early (Simeon 1990; Glod 2004). There were no data reported from Simeon on the 40 participants who were included. Glod 2004 reported data on depression symptom severity (but not by group) on the first 18 participants and we have been unable to find publication of the full trial, despite our efforts to contact the author and pharmaceutical company.
In the trial of vortioxetine (NCT02709746), there was very little information provided about planned analyses such that it was unclear whether the reported outcomes were according to a prespecified plan. This included but was not limited to there being no clear indication of for which weeks CDRS‐R scores would be reported, and which subscale scores for the CDRS‐R would be reported.
In many cases, trials appear not to have measured or reported the specified outcomes of this review, or have reported data in a way that they cannot be used in meta‐analysis, so that there were data missing from the meta‐analyses.
In summary, there are a number of concerns relating to these trials, with some clear inconsistencies between aims, methods and results, and unclear and, at times, inconsistent reports of results.
Reporting bias
As a qualitative signal, we considered there to be the potential for reporting bias (specifically lag‐time bias) for the newly developed antidepressants given they have only been evaluated in a small number of trials.
The comparison‐adjusted funnel plots for individual antidepressants (Figure 5) and antidepressant class (Figure 6) for the outcomes clinician‐rated depression (CDRS‐R), and for suicide‐related outcomes (individual antidepressants Figure 7; antidepressant class) were not suggestive of small‐study effects.
5.
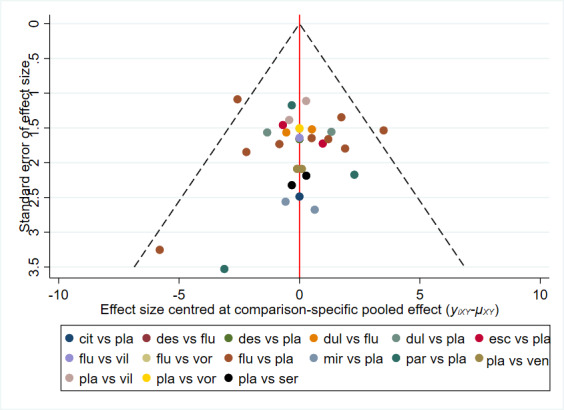
Comparison‐adjusted funnel plot individual antidepressants for clinician‐rated depression symptoms (CDRS‐R)
6.
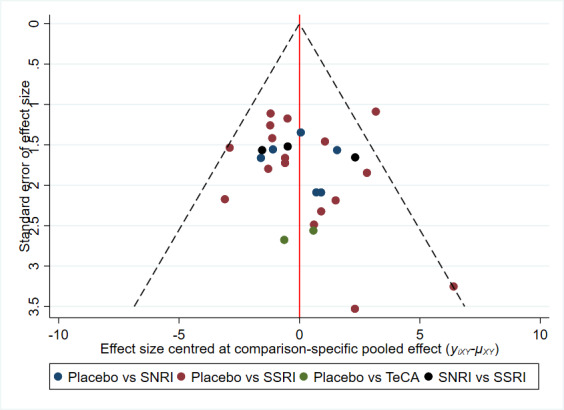
Comparison‐adjusted funnel plot antidepressant classes for clinician‐rated depression symptoms (CDRS‐R)
7.
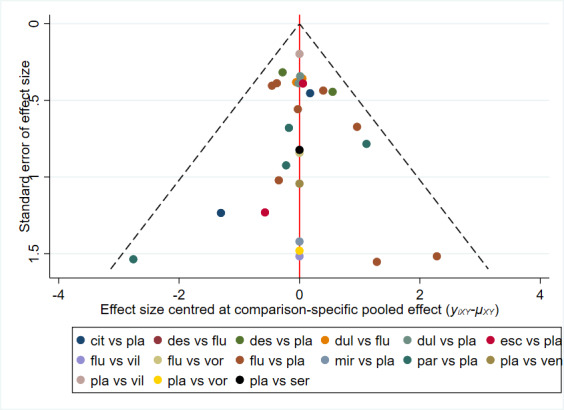
Comparison‐adjusted funnel plot individual antidepressants for suicide‐related outcomes
Other potential sources of bias
Funding
Most trials, with the exception of Emslie 1997, were pharmaceutically funded. The TADS 2004 trial was funded by an NIMH contract but had an "unrestricted educational grant from Eli Lily" (pg.531 of the 2003 publication).
Compliance
Eleven trial reports did not describe any method for assessing compliance with the intervention. Three trials (all of paroxetine) attempted to assess compliance by pill count (Keller 2001; Berard 2006; Emslie 2006) and six trials assessed plasma blood levels of the investigative trial medication (Simeon 1990; Emslie 1997; Von Knorring 2006; Mirtazapine Trial 1; Mirtazapine Trial 2; Paroxetine Trial 1).
Additional therapy
Some trials gave details about additional support or psychotherapy provided to participants in the medication and placebo arms of trials. Psychotherapy was not permitted in Wagner 2004, Wagner 2006 and the mirtazapine trials (Mirtazapine Trial 1 & 2), although in the mirtazapine trials 'supportive care' was permitted, with no detail about how many received this. Non‐directive supportive therapy was permitted in Berard 2006 but again no details were provided about how many young people received this. Supportive case management (including CBT and interpersonal therapy interventions) was provided to all participants in Keller 2001. Therapy was permitted in the sertraline trials (Wagner Trial 1 & 2 (2003)) and it is unclear how many received this; Von Knorring 2006 reported that psychotherapy was permitted and three‐quarters of participants received it. In TADS 2004, each participant received six 20 to 30‐minute medication visits spread across 12 weeks of treatment (pg.809) during which their pharmacotherapist monitored their clinical status and medication effects, and offered general encouragement about the effectiveness of pharmacotherapy for MDD. In Atkinson 2018, supportive non‐behavioural psychotherapy, family therapy, counselling, or play therapy with a focus other than on depressive symptoms was permitted, provided that no changes in intensity or frequency were made within 90 days before study baseline and no change was anticipated for the duration of the study. In Durgam 2018, psychotherapy or behaviour therapy were allowed if either was initiated at least three months prior to screening and there was no plan to change such therapies during the study. In Weihs 2018, participants who required concomitant psychotherapy were excluded. For the remainder of the trials, there was no detail given about the provision of support or therapy (Simeon 1990; Emslie 1997; Emslie 2002; Almeida‐Montes 2005; Emslie 2006; Emslie 2007 Trial 1; Emslie 2007 Trial 2; Emslie 2009; Atkinson 2014; Emslie 2014; NCT02709746; VLZ‐MD‐22).
Effects of interventions
A. Network meta‐analyses (NMA) of individual new generation antidepressants
Primary outcomes
1. Depressive disorder according to DSM or ICD criteria and established by a clinician conducting a structured or semi‐structured diagnostic interview
No data were provided for this outcome.
2. Suicide completion
No data were provided for this outcome.
Secondary outcomes: efficacy outcomes
1. Depression symptom severity (clinician‐rated) using the Children's Depression Rating Scale (CDRS‐R)
Figure 8 presents a network plot for the depression symptom severity outcome (scale range 17 to 113). Nodes were weighted by the number of studies and width of the edges was weighted by the inverse of the variance. Most interventions were compared with placebo and fluoxetine was the most common active comparator in trials. Twenty‐two RCTs including 5750 participants were included in the NMA.
8.
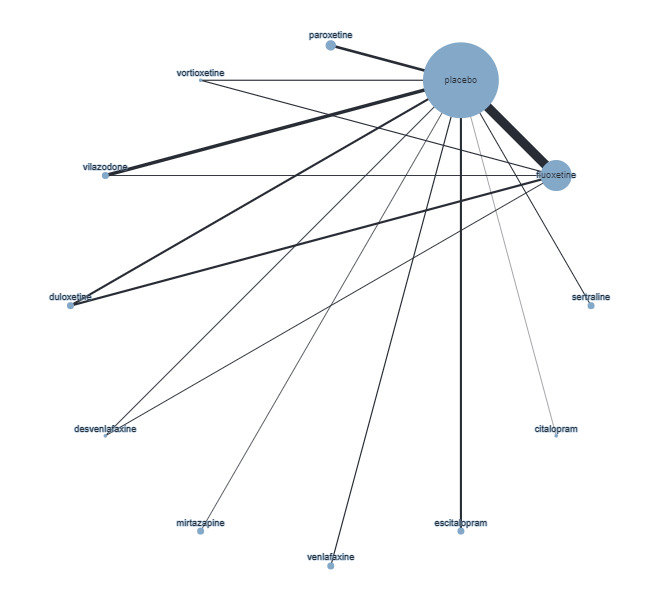
Network plot comparing individual antidepressants on depression symptom severity (clinician‐rated) using the Children's Depression Rating Scale (CDRS‐R). Nodes are weighted by number of studies and the width of the edges are weighted by the inverse of the variance.
NMA findings for all comparisons of antidepressants are in Table 6 and Figure 9 plots the effectiveness of all included antidepressants versus placebo.
4. League table comparing individual antidepressants with placebo and one another for clinician‐rated depression symptoms (CDRS‐R) ordered by the P value: MD (95% CI).
| sertraline | |||||||||||
| ‐0.67 (‐4.37 to 3.03) | fluoxetine | ||||||||||
| ‐0.81 (‐4.99 to 3.37) | ‐0.14 (‐2.46 to 2.19) | duloxetine | |||||||||
| ‐0.89 (‐5.27 to 3.48) | ‐0.22 (‐3.18 to 2.74) | ‐0.08 (‐3.62 to 3.46) | escitalopram | ||||||||
| ‐0.73 (‐5.97 to 4.52) | ‐0.05 (‐4.19 to 4.08) | 0.08 (‐4.49 to 4.65) | 0.17 (‐4.58 to 4.92) | mirtazapine | |||||||
| ‐0.61 (‐6.97 to 5.74) | 0.06 (‐5.42 to 5.54) | 0.20 (‐5.62 to 6.01) | 0.28 (‐5.68 to 6.23) | 0.11 (‐6.51 to 6.73) | citalopram | ||||||
| ‐1.61 (‐6.38 to 3.15) | ‐0.94 (‐4.45 to 2.57) | ‐0.80 (‐4.82 to 3.21) | ‐0.72 (‐4.94 to 3.50) | ‐0.89 (‐6.00 to 4.23) | ‐1.00 (‐7.25 to 5.25) | venlafaxine | |||||
| ‐2.09 (‐6.35 to 2.17) | ‐1.41 (‐4.20 to 1.37) | ‐1.27 (‐4.67 to 2.12) | ‐1.19 (‐4.83 to 2.44) | ‐1.36 (‐6.00 to 3.28) | ‐1.47 (‐7.34 to 4.40) | ‐0.47 (‐4.57 to 3.62) | paroxetine | ||||
| ‐2.67 (‐6.78 to 1.43) | ‐2.00 (‐4.40 to 0.41) | ‐1.86 (‐5.01 to 1.29) | ‐1.78 (‐5.23 to 1.67) | ‐1.94 (‐6.44 to 2.56) | ‐2.06 (‐7.82 to 3.70) | ‐1.06 (‐4.99 to 2.88) | ‐0.58 (‐3.89 to 2.72) | vilazodone | |||
| ‐3.44 (‐8.32 to 1.44) | ‐2.77 (‐6.20 to 0.66) | ‐2.63 (‐6.68 to 1.42) | ‐2.55 (‐6.89 to 1.80) | ‐2.71 (‐7.93 to 2.51) | ‐2.83 (‐9.16 to 3.51) | ‐1.83 (‐6.57 to 2.91) | ‐1.35 (‐5.58 to 2.87) | ‐0.77 (‐4.80 to 3.26) | desvenlafaxine | ||
| ‐3.51 (‐6.99 to ‐0.04) | ‐2.84 (‐4.12 to ‐1.56) | ‐2.70 (‐5.03 to ‐0.37) | ‐2.62 (‐5.29 to 0.04) | ‐2.79 (‐6.72 to 1.14) | ‐2.90 (‐8.23 to 2.43) | ‐1.90 (‐5.17 to 1.37) | ‐1.43 (‐3.90 to 1.04) | ‐0.84 (‐3.03 to 1.35) | ‐0.07 (‐3.51 to 3.36) | placebo | |
| ‐4.12 (‐8.78 to 0.55) | ‐3.44 (‐6.56 to ‐0.33) | ‐3.30 (‐7.09 to 0.48) | ‐3.22 (‐7.32 to 0.88) | ‐3.39 (‐8.41 to 1.63) | ‐3.50 (‐9.67 to 2.67) | ‐2.50 (‐7.02 to 2.02) | ‐2.03 (‐6.01 to 1.95) | ‐1.44 (‐5.21 to 2.32) | ‐0.68 (‐5.22 to 3.87) | ‐0.60 (‐3.72 to 2.52) | vortioxetine |
The effect in each cell represents the difference in mean CDRS‐R scores between the column treatment and the row treatment
CI: confidence interval MD: mean difference
9.
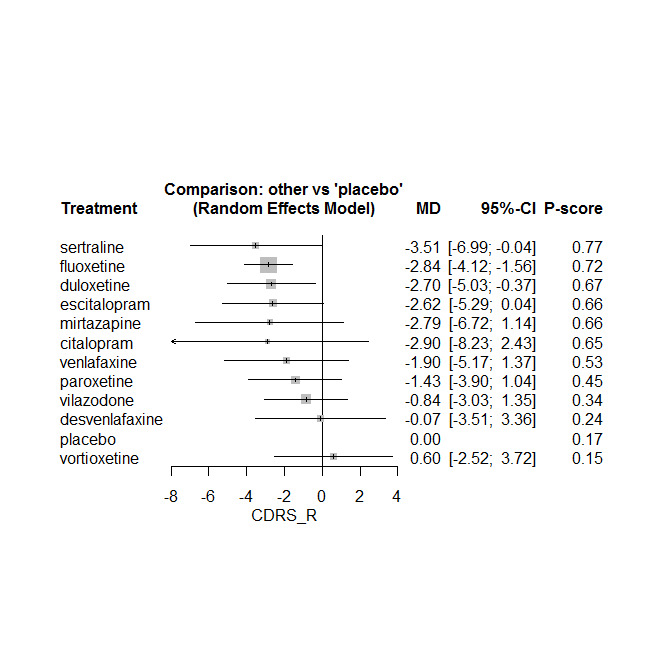
Forest plot effectiveness of individual antidepressants versus placebo CDRS‐R depression scale (ordered by ranking)
Comparisons with placebo
Below we structure the reporting of our results on the basis of our certainty judgements (i.e. CINeMA judgements). Note that none of the effect estimates comparing newer generation antidepressants (NGAs) exceeded our equivalence range (CDRS‐R from ‐5 to 5 points) and are therefore considered small and unimportant. The size of an effect, in combination with the certainty, determines how we describe the result, as outlined in section 'informative statements' in the methods. In addition, we report P‐scores (higher scores reflected a higher probability of being the most effective treatment).
High certainty evidence: there was a small unimportant difference between the following NGAs and placebo:
paroxetine: (MD ‐1.43, 95% CI ‐3.90, 1.04), P‐score=0.45
vilazodone: (MD ‐0.84, 95% CI ‐3.03, 1.35), P‐score=0.34
desvenlafaxine (MD ‐0.07, 95% CI ‐3.51, 3.36), P‐score=0.24
Moderate certainty evidence: there was probably a small unimportant difference between the following NGAs and placebo:
sertraline: (MD ‐3.51, 95% CI ‐6.99, ‐0.04), P‐score=0.77
fluoxetine: (MD ‐2.84, 95% CI ‐4.12, ‐1.56), P‐score=0.72
escitalopram: (MD ‐2.62, 95% CI ‐5.29, 0.04), P‐score=0.66
Low certainty evidence: there may be a small unimportant difference between the following NGAs and placebo:
duloxetine: (MD ‐2.70, 95% CI ‐5.03, ‐0.37), P‐score=0.67
vortioxetine: (MD 0.60, 95% CI ‐2.52, 3.72), P‐score=0.15
There was very low certainty for all other comparisons between NGAs and placebo.
Comparisons between SSRIs
Below we summarise comparisons on the basis of CINeMA ratings (Table 1).
High certainty evidence: there was a small unimportant difference between the following SSRIs:
escitalopram and fluoxetine: (MD 0.22, 95% CI ‐2.74, 3.18)
paroxetine and vilazodone: (MD ‐0.58, 95% CI ‐3.89, 2.72)
Moderate certainty evidence: there was probably a small unimportant difference between the following SSRIs:
escitalopram and sertraline: (MD 0.89, 95% CI ‐3.48, 5.27)
fluoxetine and paroxetine: (MD ‐1.41, 95% CI ‐4.20, 1.37)
fluoxetine and sertraline: (MD 0.67, 95% CI ‐3.03, 4.37)
paroxetine and escitalopram: (MD 1.19, 95% CI ‐2.44, 4.83)
fluoxetine and vilazodone: (MD ‐2.00, 95% CI ‐4.40, 0.41)
sertraline and paroxetine: (MD ‐2.09, 95% CI ‐6.35, 2.17)
sertraline and vilazodone: (MD ‐2.67, 95% CI ‐6.78, 1.43)
escitalopram and vilazodone: (MD ‐1.78, 95% CI ‐5.23, 1.67)
There was low or very low certainty for all other SSRI comparisons.
Comparisons with SNRIs
High certainty evidence: there was a small unimportant difference between duloxetine (an SNRI) and escitalopram (an SSRI) (MD ‐0.08, 95% CI ‐3.62, 3.46).
Moderate certainty evidence: there was probably a small unimportant difference between duloxetine and the following SSRIs:
fluoxetine: (MD 0.14, 95% CI ‐2.19, 2.46)
sertraline: (MD 0.81, 95% CI ‐3.37, 4.99)
paroxetine: (MD ‐1.27, 95% CI ‐4.67, 2.12)
vilazodone: (MD ‐1.86, 95% CI ‐5.01, 1.29)
Moderate certainty evidence: there was probably a small unimportant difference between the SNRIs: desvenlafaxine and duloxetine (MD 2.63, 95% CI ‐1.42, 6.68).
Moderate certainty evidence: there was probably a small unimportant difference between desvenlafaxine and these SSRIs:
escitalopram: (MD 2.55, 95% CI ‐1.80, 6.89)
sertraline: (MD 3.44, 95% CI ‐1.44, 8.32)
paroxetine: (MD 1.35, 95% CI ‐2.87, 5.58)
vilazodone: (MD 0.77, 95% CI ‐3.26, 4.8)
fluoxetine: (MD 2.77, 95% CI ‐0.66, 6.20)
For the certainty of evidence for all comparisons, see Table 1.
There was moderate heterogeneity (tau2 = 1.199, I2 = 29.5% (0%, 60.7%)) but no evidence of inconsistency (design x treatment interaction: Х2= 8.10, df = 5, P = 0.15), although all direct comparisons between antidepressants were based on only one or two trials, therefore it was not possible to rule out the potential for inconsistency.
We did not find evidence of reporting biases in the comparison‐adjusted funnel plot (see Figure 5).
Overall, improvements in reduction of depression symptoms for antidepressants compared with placebo were small and unimportant.
Sensitivity analyses
Removing trials judged to be at high risk of bias had limited impact on estimates (see Figure 10 for comparisons versus placebo). However, we were no longer able to estimate effectiveness of mirtazapine, citalopram, venlafaxine or vortioxetine. Surprisingly, heterogeneity (tau2= 2.01, I2 = 44.1% (0%, 70.8%)) was higher in the sensitivity analyses and there was now potential evidence of inconsistency (Χ2 = 9.72, df = 4, P = 0.05). However, this may indicate that estimates are relatively unstable due to sparse data for most comparisons.
10.
Forest plot on the effectiveness of individual antidepressants on CDRS‐R (sensitivity analysis removing studies at high risk of bias)
Intention‐to‐treat (ITT) data were available for all included studies, therefore, we did not conduct sensitivity analyses that included both observed cases and ITT data.
2. Remission or response as defined by trialists
Figure 11 presents a network plot for remission or response as defined by trialists. Nodes were weighted by number of studies and width of edges was weighted by sample size. Most interventions were compared with placebo and fluoxetine was the most common active comparator in trials. Nineteen RCTs including 4627 participants were included in the NMA.
11.
Network plot comparing individual antidepressants on remission or response as defined by trialists. Nodes are weighted by number of studies and the width of the edges are weighted by the inverse of the variance.
NMA findings for all comparisons of antidepressants are in Table 7 and Figure 12 plots the effectiveness of all included antidepressants versus placebo.
5. League table comparing individual antidepressants with placebo and one another for remission (sorted by the P value): OR (95% CI).
| duloxetine | |||||||||
| 1.01 (0.49 to 2.10) | vilazodone | ||||||||
| 1.05 (0.53 to 2.07) | 1.03 (0.46 to 2.32) | venlafaxine | |||||||
| 1.09 (0.53 to 2.25) | 1.08 (0.46 to 2.50) | 1.04 (0.47 to 2.32) | sertraline | ||||||
| 1.22 (0.81 to 1.86) | 1.21 (0.62 to 2.37) | 1.17 (0.63 to 2.19) | 1.13 (0.58 to 2.19) | fluoxetine | |||||
| 1.27 (0.69 to 2.33) | 1.25 (0.59 to 2.63) | 1.21 (0.60 to 2.44) | 1.16 (0.56 to 2.44) | 1.03 (0.60 to 1.77) | escitalopram | ||||
| 1.39 (0.72 to 2.68) | 1.37 (0.63 to 3.01) | 1.33 (0.63 to 2.80) | 1.28 (0.58 to 2.79) | 1.13 (0.63 to 2.05) | 1.10 (0.56 to 2.15) | citalopram | |||
| 1.63 (0.82 to 3.24) | 1.61 (0.69 to 3.74) | 1.56 (0.70 to 3.48) | 1.50 (0.65 to 3.46) | 1.33 (0.74 to 2.38) | 1.29 (0.62 to 2.69) | 1.17 (0.54 to 2.56) | vortioxetine | ||
| 1.61 (0.91 to 2.85) | 1.59 (0.78 to 3.25) | 1.54 (0.79 to 3.00) | 1.48 (0.73 to 3.00) | 1.31 (0.80 to 2.15) | 1.27 (0.70 to 2.29) | 1.16 (0.61 to 2.20) | 0.99 (0.49 to 2.00) | paroxetine | |
| 1.68 (1.11 to 2.56) | 1.66 (0.91 to 3.03) | 1.61 (0.93 to 2.77) | 1.55 (0.86 to 2.80) | 1.37 (1.01 to 1.86) | 1.33 (0.85 to 2.07) | 1.21 (0.73 to 2.02) | 1.03 (0.57 to 1.87) | 1.05 (0.71 to 1.55) | placebo |
The effect in each cell represents the difference in remission/response rates between the column treatment and the row treatment
CI: confidence interval OR: odds ratio
12.
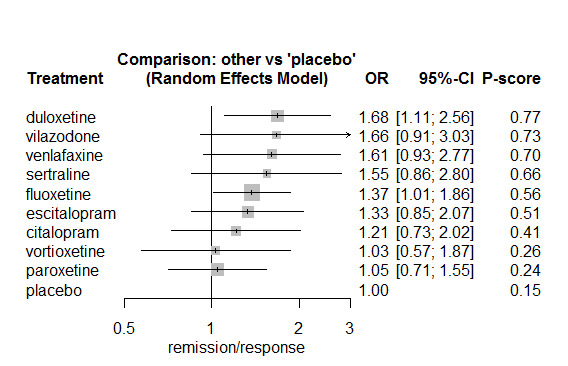
Forest plot comparing individual antidepressants with placebo on remission/response (ordered by ranking)
We did not conduct CINeMA ratings for remission or response. Therefore, we summarized comparisons according to the equivalence range (OR 0.80 to 1.25) (see the 'Informative statements' section for further details).
Most antidepressants at least slightly increase the odds of remission/response compared with placebo. However, the 95% CIs included values within the equivalence range. For example:
duloxetine: (OR 1.68, 95% CI 1.11, 2.56), P‐score=0.77
vilazodone: (OR 1.66, 95% CI 0.91, 3.03), P‐score=0.73
venlafaxine: (OR 1.61, 95% CI 0.93, 2.77), P‐score=0.70
sertraline: (OR 1.55, 95% CI 0.86, 2.80), P‐score=0.66
fluoxetine: (OR 1.37, 95% CI 1.01, 1.86), P‐score=0.56
escitalopram: (OR 1.33, 95% CI 0.85, 2.07), P‐score=0.51
Similarly, for all comparisons between NGAs, it was uncertain whether there were any differences between interventions due to wide 95% CIs and potential incoherence (see below).
Magnitude of heterogeneity was unlikely to be important but the 95% CI for I2 was wide (tau2 = 0.04, I2 = 29.5% (0%, 62.8%)). There was statistically significant evidence for inconsistency from the global assessment (design x treatment interaction: Χ2 = 7.86, df = 2, P = 0.02). However, local assessment of inconsistency is extremely limited as only three trials included direct comparisons between fluoxetine and duloxetine (Atkinson 2014; Emslie 2014), or vortioxetine and fluoxetine (NCT02709746). Of these three multi‐arm trials, two are the only source of data for duloxetine versus placebo and the other trial is the only source of data for vortioxetine versus placebo. Therefore, it is not possible to draw conclusions on the validity of the transitivity assumption.
Overall, most antidepressants were associated with a greater odds of responding or remitting. It was unclear whether any antidepressants were more effective than others.
3. Depression symptom severity ‒ self‐rated (on standardised, validated, reliable depression rating scales)
Figure 13 presents a network plot for self‐rated depression symptom severity (scale range 0 to 54‐CDI or 63‐BDI). Nodes were weighted by number of studies and width of edges was weighted by sample size. Three RCTs were included in the NMA including 543 participants.
13.
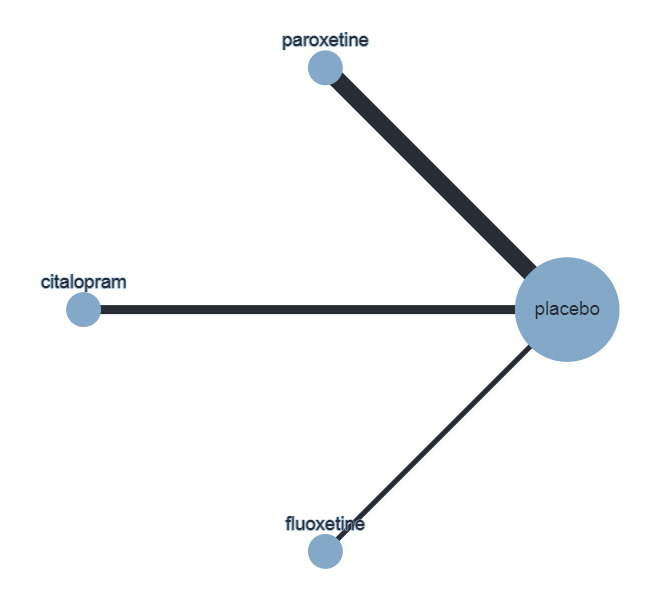
Network plot of individual antidepressants for self‐rated depression. Nodes are weighted by number of studies and the width of the edges are weighted by the inverse of the variance.
NMA findings for all comparisons of antidepressants are in Table 8 and Figure 14 plots the effectiveness of individual antidepressants compared with placebo.
6. League table comparing individual antidepressants with placebo and one another on self‐rated depression: MD (95% CI).
| fluoxetine | |||
| ‐0.87 (‐6.07 to 4.33) | paroxetine | ||
| ‐1.02 (‐6.74 to 4.70) | ‐0.15 (‐4.39 to 4.09) | citalopram | |
| ‐1.30 (‐5.87 to 3.27) | ‐0.43 (‐2.91 to 2.05) | ‐0.28 (‐3.72 to 3.16) | placebo |
The effect in each cell represents the difference in mean CDI scores between the column treatment and the row treatment
CI: confidence interval MD: mean difference
14.
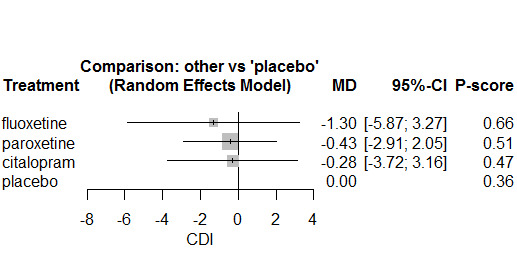
Forest plot comparing individual antidepressants with placebo for self‐rated depression (CDI)
We did not conduct CINeMA ratings for self‐rated depression symptom severity. Therefore, we summarized comparisons according to the equivalence range (MD ‐7.99 to 7.99) (see the 'Informative statements' section for further details).
Differences between NGAs and placebo are small and unimportant, the 95% CI is within the equivalence range:
fluoxetine: (MD ‐1.30, 95% CI ‐5.87, 3.27), P‐score=0.66
paroxetine: (MD ‐0.43, 95% CI ‐2.91, 2.05), P‐score=0.51
citalopram: (MD ‐0.28, 95% CI ‐3.72, 3.16), P‐score=0.47
It was also uncertain whether there were differences in effectiveness between these individual antidepressants.
There were insufficient data to accurately estimate heterogeneity as only three trials were included in the NMA, and for this reason, we do not report an estimate. We were also unable to investigate the validity of the transitivity assumption as all three trials compared NGAs with placebo and none compared NGAs head‐to‐head.
4. Functioning (on standardised, validated, reliable global functioning rating scales)
Figure 15 presents a network plot for the CGAS (scale range from 1‐100). Nodes and width of edges were weighted by number of studies. Ten RCTs were included in the NMA including 2134 participants.
15.
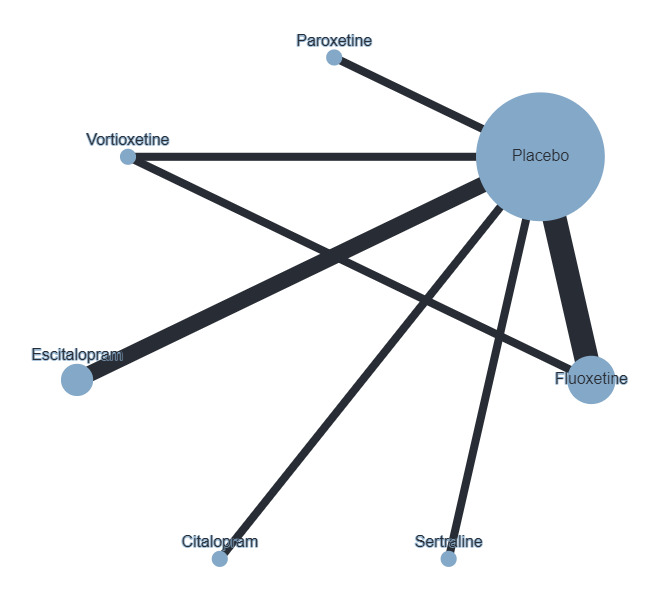
Network plot of individual antidepressants on functioning (C‐GAS). Nodes are weighted by number of studies and the width of the edges are weighted by the inverse of the variance.
NMA findings for all comparisons of antidepressants are in Table 9 and Figure 16 plots the effectiveness of individual antidepressants compared with placebo.
7. League table comparing individual antidepressants with placebo and one another on functioning (ordered by the P value): MD (95% CI).
| escitalopram | ||||||
| ‐0.22 (‐4.73 to 4.29) | citalopram | |||||
| 0.36 (‐1.71 to 2.42) | 0.58 (‐3.45 to 4.61) | fluoxetine | ||||
| 0.68 (‐3.89 to 5.24) | 0.90 (‐4.83 to 6.63) | 0.32 (‐3.77 to 4.41) | paroxetine | |||
| 0.97 (‐2.60 to 4.53) | 1.19 (‐3.78 to 6.16) | 0.61 (‐2.32 to 3.54) | 0.29 (‐4.73 to 5.31) | sertraline | ||
| 2.28 ( 0.23 to 4.32) | 2.50 (‐1.52 to 6.52) | 1.92 ( 1.64 to 2.20) | 1.60 (‐2.48 to 5.68) | 1.31 (‐1.61 to 4.23) | placebo | |
| 3.68 ( 1.62 to 5.74) | 3.91 (‐0.12 to 7.94) | 3.33 ( 3.06 to 3.59) | 3.01 (‐1.08 to 7.09) | 2.72 (‐0.22 to 5.65) | 1.41 ( 1.14 to 1.67) | vortioxetine |
The effect in each cell represents the difference in mean functioning scores between the column treatment and the row treatment
CI: confidence interval MD: mean difference
16.
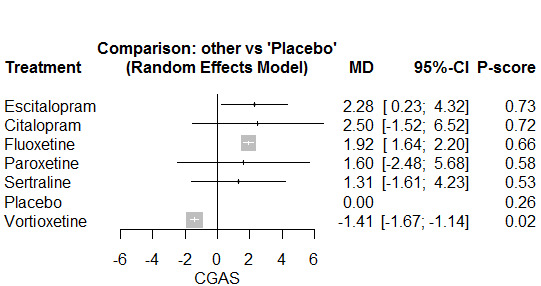
Forest plot comparing individual antidepressants with placebo on the CGAS (sorted by P value)
We did not conduct CINeMA ratings on functioning. Therefore, we summarized comparisons according to the equivalence range (MD ‐9.34 to 9.34) (see the 'Informative statements' section for further details).
NGAs s are associated with small and unimportant differences in functioning compared with placebo, the 95% CI is also within the equivalence range:
escitalopram: (MD 2.28, 95% CI 0.23, 4.32), P‐score=0.73
citalopram: (MD 2.50, 95% CI ‐1.52, 6.52), P‐score=0.72
fluoxetine: (MD 1.92, 95% CI 1.64, 2.20), P‐score=0.66
paroxetine: (MD 1.60, 95% CI ‐2.48, 5.68), P‐score=0.58
sertraline: (MD 1.31, 95% CI ‐1.61, 4.23), P‐score=0.53
vortioxetine: (MD ‐1.41, 95% CI ‐1.67, ‐1.14), P‐score=0.02
Magnitude of heterogeneity was unlikely to be important (tau2 = 0.01, I2 = 0% (0%, 34.5%)). There was no statistically significant evidence for inconsistency from the global assessment (design x treatment interaction: Χ2 = 0.6, df = 1, P = 0.44).
Overall, most antidepressants were associated with a small improvement in functioning. However, vortioxetine was associated with a small decline in functioning.
Secondary outcomes: suicide‐related outcomes
Figure 17 presents a network plot for suicide‐related outcomes. Nodes were weighted by number of studies and width of edges was weighted by sample size. As above, most interventions were compared with placebo and fluoxetine was the most common active comparator in trials. Twenty‐one RCTs including 6318 participants were included in the NMA.
17.
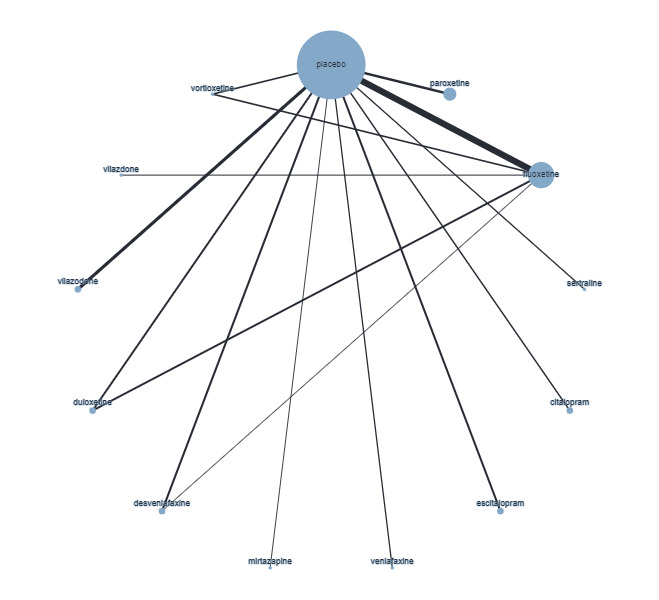
Network plot comparing individual antidepressants on suicide‐related outcomes. Nodes are weighted by number of studies and the width of the edges are weighted by the inverse of the variance.
Below we structure the reporting of our results on the basis of our certainty judgements (i.e. CINeMA judgements) and equivalence range (OR 0.90 to 1.11). The size of an effect, in combination with the certainty, determines how we describe the result, as outlined in the 'informative statements' section of the methods.
Comparisons with placebo
NMA findings for all comparisons of antidepressants are in Table 10 and Figure 18 plots the effectiveness of individual antidepressants compared with placebo.
8. League table comparing individual antidepressants and placebo for suicide related outcomes (sorted by the P value): OR (95% Cl).
| mirtazapine | |||||||||||
| 0.56 (0.03 to 9.92) | escitalopram | ||||||||||
| 0.53 (0.03 to 8.86) | 0.94 (0.40 to 2.24) | desvenlafaxine | |||||||||
| 0.50 (0.03 to 8.04) | 0.89 (0.43 to 1.84) |
0.94 (0.59 to 1.52) | placebo | ||||||||
| 0.49 (0.03 to 8.19) | 0.88 (0.39 to 2.01) | 0.94 (0.51 to 1.72) | 0.99 (0.67 to 1.46) | vilazodone | |||||||
| 0.43 (0.03 to 7.30) | 0.78 (0.33 to 1.84) | 0.82 (0.44 to 1.55) | 0.87 (0.55 to 1.39) | 0.88 (0.48 to 1.61) | duloxetine | ||||||
| 0.39 (0.02 to 6.51) | 0.70 (0.31 to 1.59) | 0.74 (0.44 to 1.27) | 0.79 (0.54 to 1.15) | 0.80 (0.46 to 1.37) | 0.90 (0.57 to 1.44) | fluoxetine | |||||
| 0.31 (0.01 to 8.19) | 0.56 (0.09 to 3.56) | 0.60 (0.11 to 3.39) | 0.63 (0.12 to 3.45) | 0.64 (0.11 to 3.63) | 0.72 (0.13 to 4.03) | 0.80 (0.15 to 4.19) | vortioxetine | ||||
| 0.29 (0.02 to 5.26) | 0.52 (0.17 to 1.54) | 0.55 (0.21 to 1.41) | 0.58 (0.26 to 1.31) | 0.59 (0.24 to 1.44) | 0.67 (0.26 to 1.70) | 0.74 (0.30 to 1.81) | 0.92 (0.14 to 6.03) | citalopram | |||
| 0.28 (0.02 to 4.93) | 0.49 (0.17 to 1.41) | 0.52 (0.21 to 1.28) | 0.55 (0.26 to 1.18) | 0.56 (0.24 to 1.31) | 0.63 (0.26 to 1.54) | 0.70 (0.30 to 1.64) | 0.87 (0.14 to 5.60) | 0.95 (0.31 to 2.89) | paroxetine | ||
| 0.16 (0.01 to 4.09) | 0.29 (0.05 to 1.72) | 0.31 (0.06 to 1.67) | 0.33 (0.07 to 1.66) | 0.33 (0.06 to 1.75) | 0.38 (0.07 to 2.02) | 0.42 (0.08 to 2.19) | 0.52 (0.05 to 5.41) | 0.57 (0.09 to 3.45) | 0.60 (0.10 to 3.54) | sertraline | |
| 0.04 (0.00 to 1.14) | 0.06 (0.01 to 0.56) | 0.07 (0.01 to 0.56) | 0.07 (0.01 to 0.56) | 0.07 (0.01 to 0.58) | 0.08 (0.01 to 0.67) | 0.09 (0.01 to 0.73) | 0.11 (0.01 to 1.63) | 0.12 (0.01 to 1.12) | 0.13 (0.01 to 1.16) | 0.22 (0.02 to 2.96) | venlafaxine |
The effect in each cell represents the difference in suicide‐related outcomes between the column treatment and the row treatment
CI: confidence interval OR: odds ratio
18.
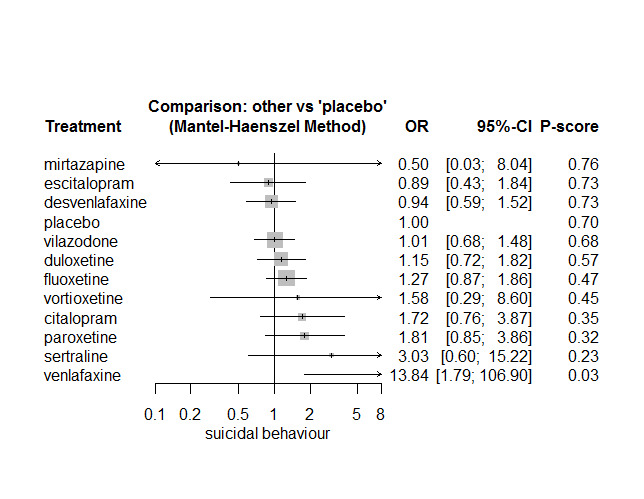
Forest plot comparing individual antidepressants with placebo for suicide related outcomes (ordered by ranking)
Proportions of suicide‐related outcomes were low for most included studies and 95% confidence intervals were wide for all comparisons.
Low certainty evidence: The following may at least slightlyincrease odds of suicide‐related events compared with placebo:
fluoxetine (OR 1.27, 95% CI 0.87, 1.86), P‐score=0.47
paroxetine (OR 1.81, 95% CI 0.85, 3.86), P‐score=0.32
sertraline (OR 3.03, 95% CI 0.60, 15.22), P‐score=0.23
venlafaxine (OR 13.84, 95% CI 1.79, 106.90), P‐score=0.03
Low certainty evidence: Escitalopram may at least slightly reduce odds of suicide‐related outcomes compared with placebo (OR 0.89, 95% CI 0.43, 1.84; P‐score=0.73)
Very low certainty evidence: Differences between the following NGAs and placebo are uncertain:
mirtazapine (OR 0.50, 95% CI 0.03, 8.04), P‐score=0.76
duloxetine (OR 1.15, 95% CI 0.72, 1.82), P‐score=0.57
vilazodone (OR 1.01, 95% CI 0.68, 1.48), P‐score=0.68
desvenlafaxine (OR 0.94, 95% CI 0.59, 1.52), P‐score=0.73
citalopram (OR 1.72, 95% CI 0.76, 3.87), P‐score=0.35
vortioxetine (OR 1.58, 95% CI 0.29, 8.60), P‐score=0.45
Comparison between NGAs
Moderate certainty evidence: Venlafaxine probably at least slightly increases odds of suicide‐related outcomes compared with the following antidepressants:
desvenlafaxine (OR 0.07, 95% CI 0.01, 0.56)
escitalopram (OR 0.06, 95% CI 0.01, 0.56)
For comparisons of NGAs with low‐ and very low‐certainty evidence, see Table 2.
The test for heterogeneity was not statistically significant (Q = 4.84, df = 3, P = 0.18). For this outcome we fitted a fixed‐effect model using the Mantel‐Haenszel method. Side‐splitting analyses did not identify evidence of inconsistency between direct (OR 0.82) and indirect estimates (OR 0.63) comparing desvenlafaxine and fluoxetine (ratio of direct and indirect ratios: 1.31, z = 0.50, P = 0.62). As above, conclusions on inconsistency are limited by direct comparisons between antidepressants.
We did not find evidence of reporting biases in the comparison‐adjusted funnel plot (see Figure 7).
Overall, it is hard to make any clear conclusions about suicide‐related outcomes. Results are contradictory with some data showing a reduced risk, some showing no change, and some showing an increased risk. Overall, the risk appears increased in a number of comparisons (venlafaxine, in particular) but the wide confidence intervals make it hard to be certain.
Sensitivity analyses
For most antidepressants, removing studies at high risk of bias had minimal impact on effect estimates (see Figure 19). However, odds ratios for desvenlafaxine (OR 1.18, 95% CI 0.74, 1.88) and paroxetine (OR 2.55, 95% CI 1.08, 6.02) compared to placebo were higher than in the main analyses (i.e. a higher odds of suicidal behaviour) and led to a lower ranking in effectiveness for these antidepressants. However, it should also be noted that this may just reflect the sparse nature of the data and instability of estimates, as the sensitivity analyses led to much wider 95% CIs for these interventions. The test for heterogeneity was not statistically significant (Χ2 = 3.57, df = 2 P = 0.17). There were insufficient data to compare the consistency of direct and indirect evidence.
19.
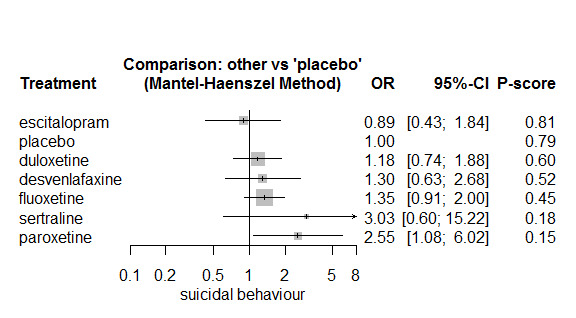
Forest plot comparing effectiveness of individual antidepressants with placebo on suicide‐related outcomes (studies at high risk of bias removed)
Intention‐to‐treat (ITT) data were available for all included studies, therefore, we did not conduct sensitivity analyses that included both observed cases and ITT data.
Secondary outcomes: overall adverse outcomes
Figure 20 presents a network plot for overall adverse outcomes. Nodes were weighted by number of studies and width of edges was weighted by sample size. As above, most interventions were compared with placebo and fluoxetine was the most common active comparator in trials. Seventeen RCTs including 5249 participants were included in the NMA. No data were available for venlafaxine, sertraline or mirtazapine.
20.
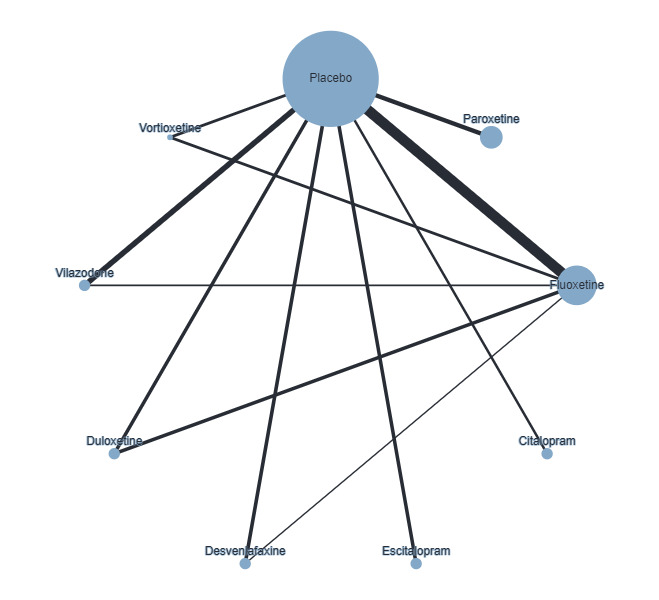
Network plot of individual antidepressants for overall adverse outcomes. Nodes are weighted by number of studies and the width of the edges are weighted by the inverse of the variance.
NMA findings for all comparisons of antidepressants are in Table 11 and Figure 21 plots the effectiveness of individual antidepressants compared with placebo.
9. League table comparing individual antidepressants with placebo and one another for overall adverse outcomes (ordered by the P value): OR (95% CI).
| placebo | ||||||||
| 1.02 (0.54 to 1.94) | desvenlafaxine | |||||||
| 0.90 (0.48 to 1.67) | 0.88 (0.37 to 2.09) | duloxetine | ||||||
| 0.89 (0.45 to 1.77) | 0.87 (0.34 to 2.23) | 0.99 (0.39 to 2.49) | escitalopram | |||||
| 0.86 (0.58 to 1.28) | 0.84 (0.42 to 1.68) | 0.96 (0.52 to 1.78) | 0.97 (0.44 to 2.15) | fluoxetine | ||||
| 0.59 (0.28 to 1.25) | 0.58 (0.22 to 1.55) | 0.66 (0.25 to 1.74) | 0.67 (0.24 to 1.85) | 0.69 (0.30 to 1.60) | citalopram | |||
| 0.55 (0.32 to 0.94) | 0.54 (0.23 to 1.24) | 0.61 (0.27 to 1.39) | 0.62 (0.26 to 1.49) | 0.64 (0.33 to 1.25) | 0.93 (0.37 to 2.32) | paroxetine | ||
| 0.35 (0.09 to 1.44) | 0.35 (0.08 to 1.59) | 0.39 (0.09 to 1.76) | 0.40 (0.08 to 1.90) | 0.41 (0.10 to 1.63) | 0.60 (0.12 to 2.91) | 0.64 (0.14 to 2.89) | vortioxetine | |
| 0.44 (0.24 to 0.82) | 0.43 (0.18 to 1.04) | 0.49 (0.21 to 1.15) | 0.50 (0.20 to 1.26) | 0.52 (0.26 to 1.01) | 0.75 (0.28 to 1.96) | 0.81 (0.36 to 1.83) | 1.25 (0.28 to 5.69) | vilazodone |
The effect in each cell represents the differences in overall adverse outcomes between the column treatment and the row treatment
CI: confidence interval OR: odds ratio
21.
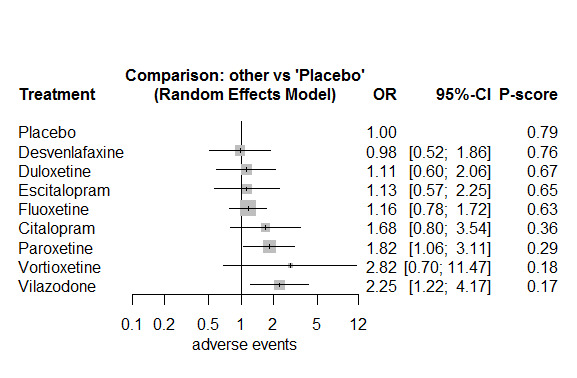
Forest plot comparing individual antidepressants versus placebo on overall adverse outcomes
We did not conduct CINeMA ratings for overall adverse outcomes. Therefore, we summarized comparisons according to the equivalence range (OR 0.80 to 1.25) (see the 'Informative statements' section for further details).
There is a small and unimportant difference in overall adverse outcome compared with placebo for the following interventions, 95% CI crosses the equivalence range in both directions:
desvenlafaxine: (OR 0.98, 95% CI 0.52, 1.86), P‐score=0.76
duloxetine: (OR 1.11, 95% CI 0.60, 2.06), P‐score=0.67
escitalopram: (OR 1.13, 95% CI 0.57, 2.25), P‐score=0.65
fluoxetine: (OR 1.16, 95% CI 0.78, 1.72), P‐score=0.63
There is at least a slight increase in odds of an adverse outcome compared with placebo for the following interventions, 95% CI crosses the equivalence range in both directions:
paroxetine: (OR 1.82, 95% CI 1.06, 3.11), P‐score=0.29
vilazodone: (OR 2.25, 95% CI 1.22, 4.17), P‐score=0.17
vortioxetine: (OR 2.82, 95% CI 0.70, 11.47), P‐score=0.18
For citalopram, there was at least a slight increase in odds of an adverse outcome compared with placebo, the 95% CI crosses the equivalence range in one direction (OR 1.68, 95% CI 0.80, 3.54).
For all comparisons between NGAs, it was uncertain whether there were differences between interventions due to wide 95% CIs and substantial heterogeneity (see below).
There was substantial heterogeneity in the effect estimates of studies included in the NMA (tau2 = 0.18, I2 = 67.6% (95% CI 44.6%, 81.1%)). The global test found potential evidence of inconsistency (design x treatment interaction: χ2 = 13.23, df = 6, P = 0.04). However, side‐splitting analyses did not identify evidence of inconsistency between direct (OR 0.83), and indirect estimates (OR 1.05), comparing desvenlafaxine and fluoxetine (ratio of direct and indirect ratios (0.79, z = 0.31, P = 0.76). Similarly, there was no evidence of inconsistency between direct (OR = 0.36) and indirect estimates (OR = 0.76), comparing fluoxetine and vilazodone (ratio of direct and indirect ratios (0.47, z = 1.02, P = 0.31).
Overall, it was difficult to draw conclusions on overall adverse outcomes. There was increased risks for most antidepressants. Although for some interventions (e.g. desvenlafaxine, duloxetine) there may be no increased risk but 95% CIs were wide so it is difficult to be certain of this.
B. Network meta‐analyses (NMA) of new generation antidepressant classes
Primary outcomes
1. Depressive disorder according to DSM or ICD criteria and established by a clinician conducting a structured or semistructured diagnostic interview
No data were provided for this outcome.
2. Suicide completion
No data were provided for this outcome.
Secondary outcomes: efficacy outcomes
1. Depression symptom severity (clinician‐rated) using the Children's Depression Rating Scale (CDRS‐R)
Figure 22 presents a network plot for the depression symptom severity outcome (scale range 17 to 113). Nodes were weighted by number of studies and width of edges was weighted by the inverse of the variance. Most studies compared an SSRI with placebo and SSRIs were also the most common active comparator in trials. Twenty‐two RCTs including 5750 participants were included in the NMA.
22.
Network plot comparing antidepressants classes on depression severity (CDRS‐R). Nodes are weighted by number of studies and the width of the edges are weighted by the inverse of the variance.
NMA findings for all comparisons of classes of antidepressants are in Table 12. Figure 23 plots the effectiveness of all included antidepressants versus placebo.
10. League table comparing antidepressants classes with placebo and one another for clinician‐rated depression (CDRS‐R) (ordered by the P value): MD (95% CI) .
| SSRI | |||
| 0.49 (‐3.47 to 4.45) | TeCA | ||
| ‐0.70 (‐2.23 to 0.83) | ‐1.19 (‐5.31 to 2.92) | SNRI | |
| ‐2.30 (‐3.20 to ‐1.39) | ‐2.79 (‐6.64 to 1.07) | ‐1.59 (‐3.02 to ‐0.17) | placebo |
The effect in each cell represents the difference in mean CDRS‐R scores between the column treatment class and the row treatment class
CI: confidence interval MD: mean difference SNRI:serotonin norepinephrine reuptake inhibitor SSRI: selective serotonin reuptake inhibitor TeCA: tetracyclic antidepressants
23.
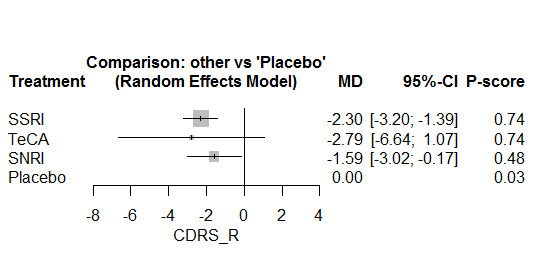
Forest plot effectiveness of antidepressant classes versus placebo CDRS‐R depression scale
Below we structure the reporting of our results on the basis of our certainty judgements (i.e. CINeMA judgements). Note that none of the effect estimates comparing NGAs exceeded our equivalence range (CDRS‐R from ‐5 to 5 points) and are therefore considered small and unimportant. The size of an effect, in combination with the certainty, determines how we describe the result, as outlined in section 'informative statements' in the methods.
Comparisons with placebo
High certainty evidence: there was a small unimportant difference between SSRIs and placebo (MD ‐2.30, 95% CI ‐3.20, ‐1.39; P‐score=0.74).
Moderate certainty evidence: there was probably a small unimportant difference betweenSNRIs and placebo (MD ‐1.59, 95% CI ‐3.02, ‐0.17; P‐score=0.48).
Very low certainty evidence: Differences between TeCAs and placebo are uncertain (MD ‐2.79, 95% CI ‐6.64, 1.07; P‐score=0.74).
Comparisons between NGA classes
None of the differences between NGA classes exceeded the equivalence range.
High certainty evidence: There was a small unimportant difference between SSRIs and SNRIs (MD ‐0.70, 95% CI ‐2.23, 0.83).
Low certainty evidence: There may be asmall unimportantdifference between the following comparisons:
SSRIs and TeCAs: (MD 0.49, 95% CI ‐3.47, 4.45)
SNRIs and TeCAs: (MD ‐1.19, 95% CI ‐2.92, 5.31)
Analysing by antidepressant classes enabled us to estimate random effects more precisely. Heterogeneity in the NMA may not be important (tau2 = 0.89, I2 = 24.7% (0%, 54.9%)), although the confidence interval for I2 was compatible with moderate heterogeneity. There was no evidence of inconsistency (design x treatment interaction: X2 = 2.89, df = 2, P = 0.24). All direct comparisons between antidepressants were based on only one or two trials, therefore, it was not possible to rule out the potential for inconsistency.
We did not find evidence of reporting biases in the comparison‐adjusted funnel plot (see Figure 6).
Overall, there were small benefits for all antidepressant classes. Most probably, there were no differences between antidepressant classes, but data for TeCAs was very limited, so it is difficult to draw conclusions on their comparative effectiveness.
Sensitivity analyses
Removing studies at high risk of bias from analyses had minimal impact on effectiveness estimates (see Figure 24). However, there were no longer data on TeCAs included in the analyses. Similar to the analyses of individual antidepressants, heterogeneity was higher (tau2 = 1.86, I2 = 43.9% (0.7%, 68.3%)) and some evidence of inconsistency (Χ2 = 3.50, df = 1, P = 0.06).
24.
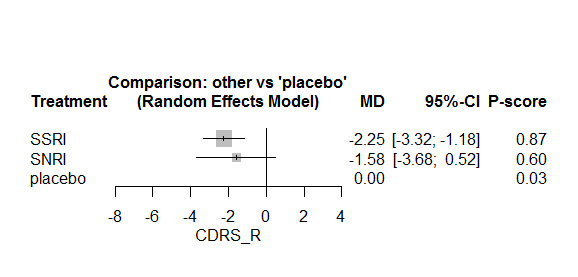
Forest plot comparing effectiveness of antidepressant classes with placebo on CDRS‐R (studies at high risk of bias removed)
Intention‐to‐treat (ITT) data were available for all included studies, therefore, we did not conduct sensitivity analyses that included both observed cases and ITT data.
2. Remission or response as defined by trialists
Figure 25 presents a network plot for remission/response. Nodes were weighted by number of studies and width of edges was weighted by sample size. Data were only available for SSRIs, SNRIs, and placebo. Nineteen RCTs including 4627 participants were included in the NMA.
25.
Network plot comparing antidepressants classes for remission/response. Nodes are weighted by number of studies and the width of the edges are weighted by the inverse of the variance.
Figure 26 plots the effectiveness of different classes of antidepressants versus placebo.
26.
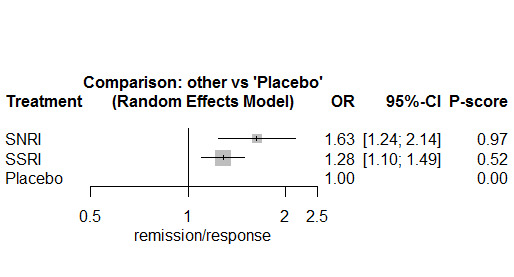
Forest plot effectiveness of antidepressant classes versus placebo for remission/response (ordered by ranking)
We did not conduct CINeMA ratings for remission or response. Therefore, we summarized comparisons according to the equivalence range (OR 0.80 to 1.25) (see the 'Informative statements' section for further details).
SNRIs (OR 1.63, 95% CI 1.24, 2.14; P‐score=0.97) and SSRIs (OR 1.28, 95% CI 1.10, 1.49; P‐score=0.52) at least slightly increased odds of remission or response compared with placebo, 95% CI crossed the equivalence range.
SNRIs at least slightly increased odds compared with SSRIs (OR 1.27, 95% CI 0.95, 1.70), but the 95% CI crossed the equivalence range.
Heterogeneity may not be important (tau2 = 0.01, I2 = 11.4% (0%, 46.8%)) although the 95% CI for the I2 statistic was compatible with moderate heterogeneity. There was no evidence of inconsistency (X2= 3.93, df = 2, P = 0.14). All direct comparisons between antidepressants were based on only one or two trials, therefore, it was not possible to rule out the potential for inconsistency.
Overall, SSRIs and SNRIs improved the odds of remission or response, but it was unclear whether there were any differences between these antidepressant classes.
3. Depression symptom severity ‒ self‐rated (on standardised, validated, reliable depression rating scales)
No comparisons between antidepressant classes were possible as all three trials compared an SSRI with placebo. Therefore we did not conduct an NMA for this outcome.
4. Functioning (on standardised, validated, reliable global functioning rating scales)
No comparison between antidepressant classes were possible as all 10 trials compared an SSRI with placebo. Therefore we did not conduct an NMA for this outcome.
Secondary outcomes: suicide‐related outcomes
Figure 27 presents a network plot for suicide‐related outcomes. Nodes were weighted by number of studies and width of edges was weighted by sample size. Most evidence was available for SSRIs and SNRIs compared with placebo. Twenty‐one RCTs including 6318 participants were included in the NMA.
27.
Network plot comparing antidepressant classes for suicide‐related outcomes. Nodes are weighted by number of studies and the width of the edges are weighted by the inverse of the variance.
NMA findings for all comparisons of antidepressants are in Table 13. Figure 28 plots the effectiveness of classes of antidepressants compared with placebo.
11. League table comparing antidepressant classes with placebo and one another on suicide related outcomes (ordered by the P value): OR (95% CI).
| TeCA | |||
| 0.50 (0.03 to 8.04) | placebo | ||
| 2.44 (0.15 to 50.00) | 1.22 (0.90 to 1.67) | SNRI | |
| 2.63 (0.16 to 50.00) | 1.30 (1.04 to 1.63) | 1.06 (0.76 to 1.49) | SSRI |
The effect in each cell represents the difference in suicide related outcomes between the column treatment class and the row treatment class
CI: confidence interval MD: mean difference SNRI: serotonin–norepinephrine reuptake inhibitor SSRI: selective serotonin reuptake inhibitor
28.
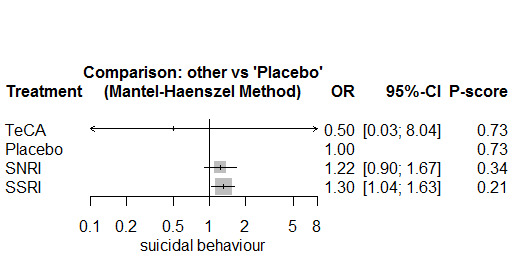
Forest plot effectiveness of antidepressant classes versus placebo for suicide related outcomes (ordered by ranking)
Below we structure the reporting of our results on the basis of our certainty judgements (i.e. CINeMA judgements) and equivalence range (OR 0.90 to 1.11). The size of an effect, in combination with the certainty, determines how we describe the result, as outlined in the 'informative statements' section of the methods.
Comparisons with placebo
Low certainty evidence: There may be at least a slight increase in odds of suicide‐related outcomes compared with placebo for the following interventions:
SNRIs: (OR 1.22, 95% CI 0.90, 1.67; P‐score=0.34)
SSRIs: (OR 1.30, 95% CI 1.04, 1.63; P‐score=0.21)
Very low certainty evidence: It is uncertain whether TeCAs reduce the odds of suicide‐related outcomes compared with placebo (OR 0.50, 95% CI 0.03, 8.04; P‐score=0.73).
Comparisons of NGA classes
Low certainty evidence: SNRIs may at least slightly increase the odds of suicide‐related outcomes compared with SSRIs (OR 1.06, 95% CI 0.76, 1.49 ).
Very low certainty evidence: It is uncertain whether TeCAs reduce the odds of suicide‐related outcomes compared with other NGAs:
SSRIs vs TeCAs: (OR 2.63, 95% CI 0.16, 50.00 )
SNRIs vs TeCAs: (OR 2.44, 95% CI 0.15, 50.00 )
There was no evidence of heterogeneity (Q = 0.2, df = 2, P = 0.90). Side‐splitting analyses did not find evidence of inconsistency between direct and indirect evidence for the SSRI vs SNRI comparison (direct: OR = 0.98, indirect: OR = 0.96; ratio of ratios: 1.05, z = 0.06, P = 0.95).
We did not find evidence of reporting biases in the comparison‐adjusted funnel plot (see Figure 29).
29.
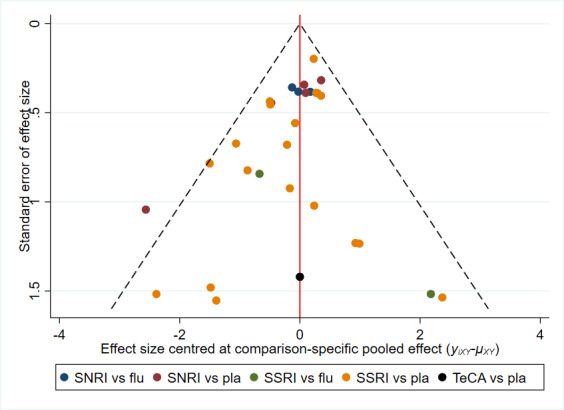
Comparison‐adjusted funnel plot antidepressant classes for suicide‐related outcomes
Overall, antidepressants may be associated with increased odds of suicide‐related outcomes. But it is uncertain whether there are any differences in suicide‐related outcomes between antidepressant classes.
Sensitivity analyses
Similar to the analyses of individual antidepressants, removing studies at higher risk of bias led to slightly higher odds ratios for suicide‐related outcomes compared with placebo (see Figure 30). The test for heterogeneity was not statistically significant (Χ2 = 1.38, df = 1, P = 0.24). There was insufficient evidence to compare the consistency between direct and indirect evidence.
30.
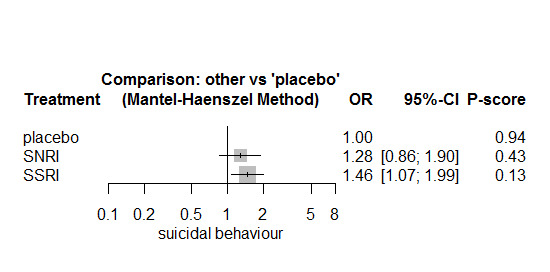
Forest plot comparing effectiveness of antidepressant classes on suicide‐related behaviour (removing studies at high risk of bias)
Intention‐to‐treat (ITT) data were available for all included studies, therefore, we did not conduct sensitivity analyses that included both observed cases and ITT data.
Secondary outcomes: overall adverse outcomes
Figure 31 presents a network plot for overall adverse outcomes. Nodes were weighted by number of studies and width of edges was weighted by sample size. As above, most data were available for SSRIs and SNRIs compared with placebo. Seventeen RCTs including 5249 participants were included in the NMA.
31.
Network plot comparing antidepressants classes on overall adverse outcomes. Nodes are weighted by number of studies and the width of the edges are weighted by the inverse of the variance.
We did not conduct CINeMA ratings for overall adverse outcomes. Therefore, we summarized comparisons according to the equivalence range (OR 0.80 to 1.25) (see the 'Informative statements' section for further details).
Figure 32 compares NGA classes with placebo. There is at least a slight increased odds of overall adverse outcomes for SSRIs compared with placebo (OR 1.47, 95% CI 1.16, 1.87; P‐score=0.06), but the 95% CI crosses the equivalence range. The difference between SNRI and placebo (OR 1.16, 95% CI 0.77, 1.73; P‐score=0.56) is small and unimportant but the 95% CI crosses the equivalence range in both directions.
32.
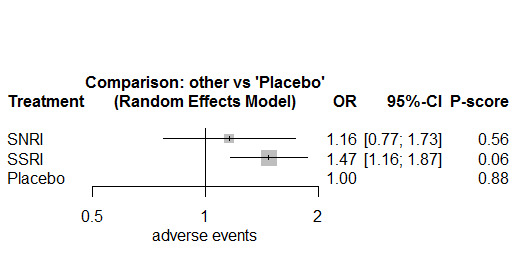
Forest plot comparing effectiveness of antidepressant classes on overall adverse outcomes
There is at least a slight reduction in odds of overall adverse outcomes for SNRIs compared with SSRIs (OR 0.79, 95% CI 0.52, 1.19), however, the 95% CI crosses the equivalence range.
There was moderate heterogeneity (tau2 = 0.14, I2 = 63.6% (41.2%, 77.4%)). The global test identified inconsistency between direct and indirect evidence (χ2 = 10.50, df = 2, P = 0.005). However, side‐splitting analyses did not find evidence of inconsistency between SSRIs and SNRIs (direct: OR 0.96, indirect: OR 0.77; ratio of ratios: 1.24, z = 0.40, P = 0.69).
C. Meta‐regression analyses (age, baseline severity, treatment duration, industry funding)
We conducted meta‐regression analyses for clinician‐rated depression (CDRS‐R) and suicide‐related outcomes. There were insufficient data to conduct analyses for individual antidepressants, therefore, we limited analyses to classes of antidepressants (SSRIs and SNRIs).
We did not assess the impact of funding as a covariate in the meta‐regression analyses since there were insufficient data (only two included studies not funded by industry). The impact of age was assessed in a separate meta‐regression model because for some included studies, we had separate data for child and adolescent participants but age‐specific data were not available for other covariates. In the following, we present regression coefficients (beta) and their 95%CIs. For the CDRS‐R outcome, the beta coefficients yield an estimate of the difference in MDs between levels of categorical covariates (i.e. categories of age) or for a 1‐unit increase in continuous covariates (i.e. baseline depression and treatment duration). For example, in the comparison of SSRIs versus placebo, one of the beta coefficients yields an estimate of the MD in children minus the MD in adolescents; while for baseline depression and treatment duration, the beta coefficient yields an estimate of the change in the MD for a 1‐unit increase in baseline depression and treatment duration (measured in weeks). For the suicide‐related outcome, the beta coefficients yield an estimate of the difference in ln(ORs) between the covariate levels as described above.
Depression symptom severity (clinician‐rated) using the CDRS‐R
We found no evidence that age impacted on depression symptom severity (SSRIs versus placebo: child versus adolescent, beta = ‐0.53, 95% CI ‐3.82, 2.76; child versus child + adolescent, beta = ‐2.52, 95% CI ‐5.58, 0.54; SNRIs versus placebo: child versus adolescent, beta = 4.24, 95% CI ‐0.82, 9.29; child versus child + adolescent, beta = 2.58, 95% CI ‐2.18, 7.34).
The meta‐regression analysis including baseline depression severity (SSRIs versus placebo, beta = ‐0.08, 95% CI ‐0.42 to 0.26; SNRIs versus placebo, beta = 0.17, 95% CI ‐1.31, 1.66) and treatment duration (SSRIs versus placebo, beta = 0.60, 95% CI ‐0.16, 1.36; SNRIs versus placebo, beta = ‐0.55, 95% CI ‐2.78 to 1.67) found no evidence that these covariates were associated with effectiveness of interventions for reducing symptom severity.
Suicide‐related outcomes
We found no evidence that age impacted on effects of the interventions on suicide‐related outcomes (SSRIs versus placebo: child versus adolescent, beta = 0.01, 95% CI ‐0.99, 1.02; child versus child + adolescent, beta = 0.84, 95% CI ‐0.81, 2.48; SNRIs versus placebo: child versus adolescent, beta = ‐0.22, 95% ‐1.76, 1.32).
The meta‐regression analysis including baseline depression severity (SSRIs versus placebo, beta = 0.10, 95% CI ‐0.07, 0.26; SNRIs versus placebo, beta = ‐0.14, 95% CI ‐0.48, 0.20) and treatment duration (SSRIs versus placebo, beta = 0.09, 95% CI ‐0.23, 0.41; SNRIs versus placebo, beta = 0.19, 95% CI ‐0.30, 0.68) found no evidence that these covariates were associated with effects of the interventions for suicide‐related outcomes.
Discussion
Summary of main results
Twenty‐six studies have been included in this updated review, with seven newly included. These new trials investigate the efficacy of antidepressants that were not tested in trials in the previous version of the review, including duloxetine, desvenlafaxine, vilazodone and vortioxetine. Of the seven new trials, five also included a comparator arm comprising fluoxetine.
There was some evidence from 22 trials (N = 5750) that all newer antidepressant classes (noting that TeCAs have been less investigated) and most newer antidepressants may be associated with small unimportant reductions in depression symptoms (measured by the clinician‐administered CDRS‐R) compared with placebo. The largest difference on the CDRS‐R scale was 3.5 points for sertraline compared with placebo, while for fluoxetine (the only treatment recommended for first‐line prescribing), this difference was 2.8 points. This is in the context of each of the 'severity' categories on the CDRS‐R scale (total 17‐113 points) being approximately 10 points (each category represents the likelihood of depressive disorder being diagnosed: coded as "unlikely", "possible", "likely", "very likely " and "almost certain"). These findings reflect the average effects of treatments, and given depression is a heterogeneous condition, some individuals may experience a greater response.
The P‐scores for depression symptoms (CDR‐R) suggested a possible ranking of medication class (SSRIs, followed by TeCAs, followed by SNRIs), and specific medications (sertraline, fluoxetine, escitalopram and duloxetine ranked above others). However, given all comparisons yielded small and unimportant differences in depression, this ranking has little meaning.
Results from 20 trials (N = 4692) suggest most antidepressants may at least slightly increase the odds of remission/response. Confidence intervals were wide and overlapped for all comparisons making conclusions with regard to comparative efficacy limited, consistent with clinician‐rated depression symptoms. A global test found evidence of potential inconsistency but there were insufficient data to conduct more detailed local assessments to assess evidence of inconsistency for specific comparisons. In terms of antidepressant class, there may be some evidence that both SSRIs and SNRIs at least slightly increase remission/response, and SNRIs may increase at least slightly the odds of remission/response compared with SSRIs. There were few trials and uncertain evidence for self‐rated depression (K = 3; N = 543). Most antidepressants may be associated with small and unimportant improvements in functioning (K = 10; N = 2134) but vortioxetine may be associated with a small decline in functioning. Again, confidence intervals were mostly wide and overlapped for all comparisons, although fluoxetine was highly ranked and had the most precise estimate of benefit.
The efficacy results need to be balanced with evidence about adverse outcomes. Twenty‐two trials (N = 6590) provided data on suicide‐related outcomes; numbers of events were small in most trials and confidence intervals were wide for all comparisons. Results suggest that as a class, SSRIs and SNRIs may at least slightly increase the odds of suicide‐related outcomes compared with placebo and SNRIs may increase, at least slightly, odds of suicide‐related outcomes compared with SSRIs. The evidence for TeCAs is uncertain for all comparisons. In terms of individual antidepressants, results were uncertain for mirtazapine, duloxetine, desvenlafaxine, citalopram, vortioxetine or vilazodone compared with placebo. Low certainty evidence suggests escitalopram may at least slightly reduce the odds of suicide‐related outcomes compared with placebo. Fluoxetine, sertraline, paroxetine and, particularly, venlafaxine may be associated with at least a slight increase in odds of suicide‐related outcomes compared with placebo. While confidence intervals were wide and overlapping, based on P‐scores, there is a higher probability of mirtazapine, escitalopram, desvenlafaxine, vilazodone, being associated with lower odds of a suicide‐related outcome.
Eighteen trials (N = 5524) provided data on overall adverse outcomes (no data were available for venlafaxine, sertraline or mirtazapine). Desvenlafaxine, duloxetine, escitalopram, and fluoxetine may be associated with small and unimportant differences in overall adverse outcomes compared with placebo; however, citalopram, paroxetine, vilazodone and vortioxetine were associated with at least a slight increase in odds of an adverse outcome.
Overall completeness and applicability of evidence
In this review, to our knowledge, we have presented data on efficacy and adverse outcomes, including suicide‐related outcomes, from all published and unpublished trials examining the use of newer antidepressants for child and adolescent depressive disorder. Despite attempts to contact trial authors, as well as pharmaceutical companies responsible for funding the included trials, there were many instances of missing data in terms of effect estimates. In seven cases, there was very limited reporting of trials by the Medicines and Healthcare Products Regulatory Agency (MHRA) (Mirtazapine Trial 1 & 2), ClinicalTrials.gov (VLZ‐MD‐22; NCT02709746), and the GlaxoSmithKleine website (Paroxetine Trial 1) with publication in peer‐reviewed journals not yet available. Likewise, the trial by Simeon 1990, which was stopped early, has never been published. We were unable to obtain any further report of the trial Glod 2004, the preliminary findings of which were published in a conference abstract.
The care given to the comparator group may impact the size of the effects and the large improvement from baseline observed in the placebo groups in these trials has been previously commented upon (e.g. Jureidini 2004), including by authors of the included trial reports, who have suggested that this may be due to the large amount of contact trial participants received. Trials consistently include regular (often weekly) assessments and, in some trials, some supportive contact or therapy was allowed. The interaction between participants and trial investigators was seldom standardised, as shown in a trial specifically investigating this issue, and while this interaction is typical of what takes place in real world clinical encounters, it impacts in an unknown way on the detection of differences between the active drug and placebo (Dunlop 2010).
The characteristics of the participants also may impact on the size of the intervention effects. For example, the exclusion of placebo responders in the lead‐in time to the start of the trial may have an impact, and is not representative of clinical populations typically seen in child and adolescent mental health services. This exclusion of placebo responders occurred variably across trials (Table 4), but was a characteristic of the majority of trials of fluoxetine and escitalopram (but not sertraline and duloxetine). The placebo remission rate was often lower in fluoxetine, for example, than other antidepressants (Table 14).
12. Remissions rates.
| Study ID | Intervention Drugs | Data used | Remission rate intervention | Remission rate placebo |
| Atkinson 2014 | Duloxetine | CDRS‐R Remission LOCF | 46/113; 41% | 42/103; 41% |
| Fluoxetine | 37/113; 33% | |||
| Atkinson 2018 | Desvenlafaxine | CGI response OC | 66/106; 62% | 57/102; 56% |
| Almeida‐Montes 2005 | Fluoxetine | LOCF HAM‐D | 4/7; 57% | 6/9; 66% |
| Berard 2006 | Paroxetine | LOCF MADRS | 107/177; 60% | 53/91; 58% |
| Durgam 2018 | Vilazodone | CDRS‐R Response OC | Low: 62/148; 42% High: 72/163; 44% |
63/143; 44% |
| Emslie 1997 | Fluoxetine | CDRS‐R Remission LOCF | 15/48; 31% | 11/48; 23% |
| Emslie 2002 | Fluoxetine | CDRS‐R Remission LOCF | 45/109; 41% | 20/101; 20% |
| Emslie 2006 | Paroxetine | CDRS‐R Remission LOCF | 23/101; 23% | 29/102; 28% |
| Emslie 2007 | Venlafaxine extended release | CDRS‐R Response LOCF | Study 1: 43/68; 63% Study 2: 77/101; 76% |
Study 1: 37/73; 51% Study 2: 62/92; 67% |
| Emslie 2009 | Escitalopram | CDRS‐R Remission LOCF | 64/154; 42% | 56/157; 36% |
| Emslie 2014 | Duloxetine | CDRS‐R remission LOCF | High: 42/105; 40% Low: 52/114; 46% |
35/117; 30% |
| Fluoxetine | 36/112; 32% | |||
| Keller 2001 | Paroxetine | From Le Noury publication: HAM‐D Response LOCF | 60/90; 67% | 48/87; 46% |
| Mirtazapine Trial 1 | Mirtazapine | No data reported | ||
| Mirtazapine Trial 2 | Mirtazapine | No data reported | ||
| Paroxetine Trial 1 | Paroxetine | CGI response LOCF | 15/29; 52% | 11/27; 41% |
| Simeon 1990 | Fluoxetine | No data reported | ||
| TADS 2004 | Fluoxetine | CDRS‐R Remission LOCF | 25/109; 23% | 19/112; 17% |
| Von Knorring 2006 | Citalopram | Remission MADRS LOCF | 40/121; 33% | 40/112; 36% |
| Wagner 2004 | Citalopram | CDRS‐R Response LOCF | 32/89; 36% | 20/85; 24% |
| Wagner 2006 | Escitalopram | CDRS‐R Response LOCF | 59/129; 46% | 50/132; 38% |
| Wagner Trial 1 & 2 (2003) | Sertraline | CDRS‐R Response LOCF | 128/185; 69% | 106/179; 59% |
| VLZ‐MD‐22 | Vilazodone | No data reported | ||
| Fluoxetine | ||||
| NCT02709746 | Vortioxetine | CDRS‐R Remission LOCF | Low: 21/126; 17% High: 24/139; 17% |
20/137; 15% |
| Fluoxetine | 32/137; 23% | |||
| Weihs 2018 | Desvenlafaxine | CGI Response OC | 68/99; 69% | 62/99; 63% |
| Fluoxetine | 79/101; 78% |
CDRS‐R: Children's Depression Rating Scale ‐ Revised CGI: Clinical Global Impressions Scale HAM‐D: Hamilton Rating Scale for Depression LOCF:last‐observation‐carried‐forward MADRS: Montgomery‐Asberg Depression Rating Scale OC:observed case
The trial populations were also uncharacteristic of public child and adolescent mental health services in terms of the exclusion of those with comorbid disorders, and the exclusion of those at risk of suicide. The presence of suicidal ideation and suicidal behaviour in young people presenting to services with depression is common (Birmaher 1996; Lewinsohn 1998; Davey 2019). The baseline severity of depression of young people included was in the moderately severely ill range (Table 3). The effectiveness of newer generation antidepressants in young people and children with more severe disorders and complex presentations, including comorbid conditions and suicide risk, is therefore unknown.
Finally, several studies included a large range of treatment doses and one study each of desvenlafaxine and vilazodone had separate arms for high and low doses of these medications that we combined in the analysis. This means that consideration of optimal dosing has not been considered in this review and we point readers to Table 4 to examine dosage of the various medication across trials’
Quality of the evidence
There was limited information on the conduct of trials in relation to allocation concealment, blinding and compliance. Blinding is an issue when clinician‐rated scales are the main outcome, particularly in the context of an inactive placebo where it may be possible to guess the assigned treatment group given side and other physiological effects likely in this group (Moncrieff 2004). In most cases, there was not an explicit description of who in the trial was blinded and there were very few trials with details about the placebo capsules in terms of ensuring blinding against the medication capsules.
The issue of reporting bias is important. Kirsch 2008 highlight, in their meta‐analysis of all trials of antidepressants submitted to the Food and Drug Administration (FDA), that effect sizes are smaller when unpublished studies are included and Turner 2008 showed that whether and how trials of antidepressants were published depended on the outcome of the trial. For the trials included in this review, the possibility of reporting bias (across trials) was initially highlighted in a letter to the editor regarding post hoc alterations of response definitions in the trial by Keller 2001 (Jureidini 2003). Generally though, reporting bias within the published reports of included trials is difficult to assess, given the conduct of a trial can be obscured by the write‐up for publication. Even though clinical trial registration and publication of protocols is more common in recent times, our observation is that details in these documents can be sparse. Within our included studies, full and explicit reporting of changes in outcome definition was only undertaken by one investigator, however, the primary outcome was reported and findings discussed (Emslie 1997; Jureidini 2004). It is notable that we located several trials that had not been fully reported or published at all. It is also worth noting that, in many cases, we were unable to obtain the required data from the published paper but had to contact authors or the pharmaceutical company.
What level of improvement constitutes a meaningful clinical outcome is uncertain, given response and remission were defined and reported variously both within and across trials, with the noted possibility of alteration of this definition, and the possibility of reporting bias as a result. A standard definition of remission or response would have been ideal; however, to calculate this, individual patient data would have been necessary. Further, given a diagnosis of major depressive disorder on DSM criteria was an entry criterion for most of the trials, this may be considered the most desirable outcome measure. The appropriateness of outcome measurement remains an issue, as highlighted in the previous version of this review (Hetrick 2012); symptom improvement does not necessarily equate to outcomes that young people with depression may define as important or meaningful. In a meta‐analysis of antidepressants for all age groups, Ioannidis 2008 questioned the appropriateness of outcome measures used and highlighted issues related to selective and distorted reporting and interpretation, calling into question the effectiveness of antidepressants, even in adult populations.
Since the protocol for this review was published, a core outcome set for common mental disorders in children and young people has been published (Krause 2021), which represents an important development in the field. Our review outcomes are largely consistent with the core outcome set recommended by Krause 2021. Of note, the core outcome set recommendations include measurement of depression symptoms using a self‐report scale (the ‘Revised Children’s Anxiety and Depression Scale’), and not a clinician‐rated scale. In our review, only 3 of the 26 included trials measured and reported depression symptoms using a self‐report scale (with none using the recommended scale), while 22 reported clinician‐rated symptom severity (using the CDRS‐R scale). It is clear therefore, that at this point in time, there exists a disconnect between recommended depression symptom measures, and those which have been used.
There was evidence of inappropriate methods of imputation with trialists in older trials often using last‐observation‐carried‐forward data (Sterne 2009). It was often the case that some randomised patients were not included in the final analysis.
The majority of trials were pharmaceutically funded. Two of the four fluoxetine trials were not pharmaceutically funded (Emslie 1997; TADS 2004) (the TADS trial had an unrestricted education grant from Eli Lily). Research has shown that, across different health fields, pharmaceutically funded studies are more likely to have results favouring the pharmaceutical company's product (Lexchin 2003; Sismondo 2008).
Finally, the trials were designed only to examine the short‐term effects of antidepressant medication, however, this does not preclude the possibility that the effectiveness of treatment is only apparent over a longer period of time. Long‐term follow‐up would be required to assess this.
Potential biases in the review process
It should be noted that the review process included collection of data from various sources. Information and data for included trials were taken variously from scientific journal publications, from the MHRA data and, in some cases, obtained directly from trial authors and pharmaceutical companies. For some trials, there were very limited data and, even in the case where there was more complete reporting and across several sources, as noted above, we cannot rule out the possibility that some relevant outcome data may be missing from this review. As noted above, there are now consensus based core outcome sets that are relevant to this review, but that were not available to inform the development of this review.
Comparison adjusted funnel plots for the outcomes clinician‐rated depression and suicide‐related outcomes were not suggestive of small‐study effects.
Agreements and disagreements with other studies or reviews
A number of reviews have been undertaken investigating the efficacy and safety of antidepressants in children and adolescents, particularly since the Food and Drug Administration issued a 'black box' warning label in 2004 (FDA 2004). As the field has evolved, there has been convergence on conclusions that support the modest efficacy of these medications in the context of concerns about the quality of the evidence base (Cohen 2004; Jureidini 2004; Whittington 2004; Hetrick 2007; Tsapakis 2008; Hetrick 2012; Locher 2017). In particular, fluoxetine has been shown to be supported by the most evidence and guidelines consistently recommend the use of fluoxetine as first‐line medication (NICE 2019). Also consistent is evidence of increased risks of suicide‐related outcomes for those taking these medications and recommendations of the need for vigilant monitoring of those prescribed antidepressants (Cohen 2004; Jureidini 2004; Whittington 2004; Hetrick 2007; Tsapakis 2008; Hetrick 2012; Locher 2017; NICE 2019).
Conclusions about comparative efficacy have been more circumspect, given few head‐to‐head trials and the lack of methodology to support examination of comparative efficacy within a meta‐analysis (Hetrick 2012). In our previous review, using subgroup analysis, we concluded that there were no meaningful differences between treatment effects. The emergence of network meta‐analysis methodology has allowed more complex analyses. In 2016, Cipriani and colleagues published a network meta‐analysis that was able to examine comparative efficacy of all antidepressants (Cipriani 2016). Again, the quality of the evidence was rated as very low for the majority of comparisons, consistent with our findings. The authors showed that only fluoxetine was more effective than placebo on the basis of depression severity and that fluoxetine, desipramine and duloxetine had the highest probabilities of being ranked as most effective of all the treatments. Venlafaxine was associated with greater suicide‐related outcomes than placebo, escitalopram, imipramine, duloxetine, fluoxetine, and paroxetine.
Consistent with Cipriani 2016, and in the context that the effects for all treatments were smaller than our threshold for an important different, fluoxetine, may have a greater probability of being the most effective. However, we found some data to support sertraline, escitalopram and duloxetine, being considered for first‐line treatment along with fluoxetine, which is a new finding compared with Cipriani 2016. Consistent with Cipriani 2016, we found venlafaxine may at least slightly increase odds of suicide‐related outcomes, but conclude that fluoxetine, paroxetine and sertraline should be similarly considered as at least slightly increasing the odds of these outcomes.
Overall, the results of our review are consistent with Cipriani 2016, the previous version of this review, and other reviews, in that it highlights the lack of robust evidence on which clinicians can base their treatment of young people with depressive disorders. We rated most comparisons of antidepressants to be of very low‐certainty evidence. The results of this review differ somewhat from previous reviews with regard to fluoxetine, which is likely due to the inclusion of more recent trials where the effect sizes for fluoxetine have decreased; further trials of other antidepressant treatments may well see a similar phenomenon. We are also aware of findings from other previous reviews of medication (Locher 2017) and psychotherapy (Weisz 2017) that highlight similar treatment effects for both of these interventions for depression (Merry 2017).
There remain important questions about the clinical effectiveness of these treatments and, even though they may reduce depression symptoms in comparison to placebo, the effects are small and unimportant. Confidence intervals are mostly overlapping so that, although there is some direction in terms of comparative efficacy, this is certainly not definitive. The trials are of young people who are not representative of those typically presenting for treatment in clinics. Furthermore, the trials had some significant methodological shortcomings, making it difficult to draw firm conclusions. Potential benefit must be balanced with the finding that newer generation antidepressants are associated with an increased risk of suicide‐related outcomes (a combination of suicidal ideation and suicide attempt). It is unknown how children and adolescents with a depressive disorder and comorbid conditions, who are at risk of suicide (i.e. those more typical of the young people who present at health services), would respond to newer generation antidepressants because these young people are largely excluded from the trials. Clinically, depressive disorder has an increased risk of suicide completion, as well as impacts on academic and social functioning, which is often used as a justification of using medications. However, it is important that our concerns about young people do not lead to prescription of ineffective treatments that have the potential to do harm.
It is of concern that after 26 trials involving children and adolescents, we are still at a point where there are no trials that report convincing evidence of remission of a diagnosed major depressive disorder, or even of a substantial reduction in symptoms and that the quality of evidence remains low. The dilemma for clinicians working with children and adolescents with depression remains. Ensuring clear information about the risks and benefits of newer generation antidepressants with children, adolescents, and their parents/carers/families, providing information about possible alternative treatments and engaging in shared decision‐making, with ongoing monitoring to determine effect in an individual is ever more important (Simmons 2017).
Authors' conclusions
Implications for practice.
While this review has provided an update to the evidence, including seven new trials, overall, the methodological shortcomings of the trials make it difficult to interpret the outcome data with regard to the efficacy and safety of newer antidepressant medications. It is unclear whether the reduction in depression symptom severity is of clinical importance to children, adolescents and their parents/carers/families. There were no data to inform the comparison of greatest interest: what effect do antidepressants have on resolution of a diagnosis of major depressive disorder. On our secondary outcomes, we have used an approach based on equivalence ranges (where differences between interventions within this range of values are not expected to be of clinical importance); however, it is unclear if this definition and child and adolescent definitions of meaningful change differ. Our findings based on these secondary outcomes suggest that most newer antidepressants may be associated with small and unimportant reductions in depression symptoms compared with placebo, which raises the question of whether they should be used at all. The inclusion of new trials has allowed a greater number of comparisons to be made, although overall the differences between the majority of treatments appear small and unimportant. Findings from the NMA suggest that if medications are to be used, there is evidence to support a greater range of options for first‐line prescribing of antidepressants including sertraline, escitalopram, duloxetine as well as fluoxetine. This is in contrast to guideline recommendations that recommend fluoxetine alone (NICE 2019).
It is vital to discuss the options with the child or young person and family and it remains critical to ensure they understand the data and that there is close monitoring of suicide‐related outcomes (combined suicidal ideation and suicide attempt) in those treated with newer generation antidepressants, given findings many are associated with at least slightly greater odds of these outcomes. The evidence is very uncertain, but those treatments associated with lower odds may be escitalopram, desvenlafaxine, vilazodone, duloxetine and fluoxetine.
There remains uncertainty, on the basis of these findings, about how children and adolescents with comorbid conditions and/or who are already experiencing suicidal ideation or have attempted suicide (i.e. those more typically seen in mental health services) would respond to these medications, given that trials have largely excluded these children and adolescents. Again, close monitoring is required.
In the context of following guideline recommendations about the use of psychotherapy for depression in children and adolescents, if indicated, clinicians should make every effort to present the information on the potential benefits and risks of newer generation antidepressants, including the risks associated with depression, and together with the child or adolescent and their family, consider the various options for treatment. If a newer generation antidepressant is used, both the response to treatment in terms of depression symptom severity should be monitored, and there should be close monitoring of suicide‐related outcomes in line with guideline recommendations (NICE 2019). Depressive disorder is heterogeneous so that the effects of newer generation antidepressants in young may be variable. Routinely collected monitoring data may be important to examine over time, particularly with regard to specific populations who are often excluded from trials.
Implications for research.
It is clear from the results that we need more effective treatments for depressive disorders in children and young people and there is no one clearly effective treatment to date. Children and adolescents with a depressive disorder who present for treatment are likely to be different to those in the included trials in this review; moreover, those presenting for treatment in clinical services are heterogeneous. Trials should include children and adolescents more typical of those presenting to clinical services, such as those with comorbid mood disorders, as well as those with comorbid neurodevelopmental disorders such as Autism Spectrum Disorder (ASD) and Attention Deficit Hyperactivity Disorder (ADHD), and allow analysis of different subgroups of these children and adolescents. Identifying minimally important different metrics for key outcomes that are anchored in child and adolescent (and their parents and caregivers) perceptions of meaningful change, and ensuring these are used routinely in trials would help the field. For example, remission or response as per diagnostic assessment maybe a better metric on which to base an understanding the effects of treatment more rigorously.
Trials that are undertaken should address the methodological shortcomings identified in this review, including adequate blinding, particularly of outcome assessors, consistent definition and use of clinically important outcomes and longer‐term follow‐up. In terms of medication responsiveness and adherence, assessing family factors such as parental mental health and family functioning may also highlight important influential factors. Non‐industry funded large studies that include comparisons with the current first‐line recommended treatment (fluoxetine) (NICE 2019), studies that investigate differences in responsiveness to medication, optimal dosing, and cost‐effectiveness analyses would add to the field. Well designed syntheses of data on adverse outcomes and further investigation of these from the perspective of young people would be useful.
Within the context of the currently available trial data, individual patient data meta‐analyses may be useful in examining whether the effect of treatment differs in particular subgroups.
History
Protocol first published: Issue 7, 2020
Acknowledgements
We would like to acknowledge the support of the editorial team of the Cochrane Common Mental Disorders Group; and, in particular, Sarah Dawson, the Information Specialist for the Group, who helped develop the search strategies and Jess Hendon who provides consistent and high‐quality editorial support.
The authors and the CCMD Editorial Team are grateful to the peer reviewers for their time and comments including: Karolin Krause, Stella Le, Aileen Parke and Peter Szatmari. They would also like to thank Cochrane Copy Edit Support for the team's help.
CRG funding acknowledgement: the National Institute for Health Research (NIHR) is the largest single funder of the CCMD Group.
Disclaimer: the views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the NIHR, the National Health Service or the Department of Health and Social Care.
Appendices
Appendix 1. Hierarchy of depression symptom severity measurement scales
Where different depression symptom severity rating scales were used, for the purpose of pooling results we chose the single best available outcome measure according to a hierarchy based on psychometric properties and appropriateness for use with children and adolescents. The hierarchy has been updated since the first publication of the review and is based on the reviews of Hazell and colleagues (Hazell 2002), Petti (Petti 1985) and Brooks and Kutchers (Brooks 2001). We also took into consideration the most commonly used tools in the trials included in the original Cochrane Review by Hetrick and colleagues (Hetrick 2007). Finally, in this version of the review, we have also included self‐rated depression symptom severity tools and separated the hierarchy according to whether the tool was clinician‐ or self‐rated. The hierarchy is as follows.
Clinician‐rated instruments
Children's Depression Rating Scale (CDRS)
Hamilton Depression Rating Scale (HAM‐D)
Montgomery–Åsberg Depression Rating Scale (MADRS)
Schedule for Affective Disorders and Schizophrenia for School Aged Children (K‐SADS)
Bellevue Index of Depression (BID)
(Note: CDRS‐R was adapted for children and adolescents from the Hamilton Depression Rating Scale (HAM‐D), a tool validated and commonly used in adult populations (Brooks 2001). Both the CDRS‐R and HAM‐D have good reliability and validity. The MADRS was also based on the HAM‐D but designed to better assess sensitivity to change. It was not designed specifically for children and adolescents (Brooks 2001).
Self‐report measures
Beck Depression Inventory (BDI)
Childrens Depression Inventory (CDI)
Mood and Feeling Questionnaire (MFQ)
Reynolds Adolescent Depression Scale (RADS)
Kutcher Adolescent Depression Scale (KADS)
Depressive Adjective Checklist (DACL)
Child Depression Scale (CDS)
Centre for Epidemiologic Studies Depression Scale (CES‐D)
Appendix 2. Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR)
Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR)
Cochrane Common Mental Disorders (CCMD) maintains two archived clinical trials registers at its editorial base in York, UK: a references register and a studies‐based register. The CCMDCTR‐References Register contains over 40,000 reports of RCTs in depression, anxiety and neurosis. Approximately 50% of these references have been tagged to individual coded trials. The coded trials are held in the CCMDCTR‐Studies Register and records are linked between the two registers through the use of unique Study ID tags. Coding of trials is based on the EU‐Psi coding manual, using a controlled vocabulary; (please contact the CCMD Information Specialists for further details). Reports of trials for inclusion in the Group's registers are collated from routine (weekly), generic searches of MEDLINE (1950 to 2016), Embase (1974 to 2016) and PsycINFO (1967 to 2016); quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review‐specific searches of additional databases. Reports of trials are also sourced from international trial registers via the World Health Organization's trials portal (the International Clinical Trials Registry Platform (ICTRP)), pharmaceutical companies, the handsearching of key journals, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses.
Details of CCMD's generic search strategies (used to identify RCTs) can be found on the Group's website, (cmd.cochrane.org/specialised-register), with an example of the core MEDLINE search (used to inform the register) listed below. The Group’s Specialised Register has fallen out‐of‐date with the Editorial Group’s move from Bristol to York in the summer of 2016.
Core search strategy used to inform the Cochrane Common Mental Disorders Group's Specialised Register: OVID MEDLINE (to June 2016)
A weekly search alert based on condition + RCT filter only 1. [MeSH Headings]: eating disorders/ or anorexia nervosa/ or binge‐eating disorder/ or bulimia nervosa/ or female athlete triad syndrome/ or pica/ or hyperphagia/ or bulimia/ or self‐injurious behavior/ or self mutilation/ or suicide/ or suicidal ideation/ or suicide, attempted/ or mood disorders/ or affective disorders, psychotic/ or bipolar disorder/ or cyclothymic disorder/ or depressive disorder/ or depression, postpartum/ or depressive disorder, major/ or depressive disorder, treatment‐resistant/ or dysthymic disorder/ or seasonal affective disorder/ or neurotic disorders/ or depression/ or adjustment disorders/ or exp antidepressive agents/ or anxiety disorders/ or agoraphobia/ or neurocirculatory asthenia/ or obsessive‐compulsive disorder/ or obsessive hoarding/ or panic disorder/ or phobic disorders/ or stress disorders, traumatic/ or combat disorders/ or stress disorders, post‐traumatic/ or stress disorders, traumatic, acute/ or anxiety/ or anxiety, castration/ or koro/ or anxiety, separation/ or panic/ or exp anti‐anxiety agents/ or somatoform disorders/ or body dysmorphic disorders/ or conversion disorder/ or hypochondriasis/ or neurasthenia/ or hysteria/ or munchausen syndrome by proxy/ or munchausen syndrome/ or fatigue syndrome, chronic/ or obsessive behavior/ or compulsive behavior/ or behavior, addictive/ or impulse control disorders/ or firesetting behavior/ or gambling/ or trichotillomania/ or stress, psychological/ or burnout, professional/ or sexual dysfunctions, psychological/ or vaginismus/ or Anhedonia/ or Affective Symptoms/ or *Mental Disorders/ 2. [Title/ Author Keywords]: (eating disorder* or anorexia nervosa or bulimi* or binge eat* or (self adj (injur* or mutilat*)) or suicide* or suicidal or parasuicid* or mood disorder* or affective disorder* or bipolar i or bipolar ii or (bipolar and (affective or disorder*)) or mania or manic or cyclothymic* or depression or depressive or dysthymi* or neurotic or neurosis or adjustment disorder* or antidepress* or anxiety disorder* or agoraphobia or obsess* or compulsi* or panic or phobi* or ptsd or posttrauma* or post trauma* or combat or somatoform or somati#ation or medical* unexplained or body dysmorphi* or conversion disorder or hypochondria* or neurastheni* or hysteria or munchausen or chronic fatigue* or gambling or trichotillomania or vaginismus or anhedoni* or affective symptoms or mental disorder* or mental health).ti,kf. 3. [RCT filter]: (controlled clinical trial.pt. or randomized controlled trial.pt. or (randomi#ed or randomi#ation).ab,ti. or randomly.ab. or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or distribut* or expose* or fashion or number* or place* or recruit* or subsitut* or treat*)).ab. or placebo*.ab,ti. or drug therapy.fs. or trial.ab,ti. or groups.ab. or (control* adj3 (trial* or study or studies)).ab,ti. or ((singl* or doubl* or tripl* or trebl*) adj3 (blind* or mask* or dummy*)).mp. or clinical trial, phase ii/ or clinical trial, phase iii/ or clinical trial, phase iv/ or randomized controlled trial/ or pragmatic clinical trial/ or (quasi adj (experimental or random*)).ti,ab. or ((waitlist* or wait* list* or treatment as usual or TAU) adj3 (control or group)).ab.) 4. (1 and 2 and 3) Records are screened for reports of RCTs within the scope of the Cochrane Common Mental Disorders Group. Secondary reports of RCTs are tagged to the appropriate study record. Similar weekly search alerts are also conducted on OVID Embase and PsycINFO, using relevant subject headings (controlled vocabularies) and search syntax, appropriate to each resource.
Search strategy for this review
The CCMDCTR was searched for this review using the following terms:
CCMDCTR‐Studies Register
Condition = (depress* or dysthymi*) AND Intervention = ("Selective Serotonin Reuptake Inhibitors" or Agomelatine or Alaproclate or Bupropion or Citalopram or Desvenlafaxine or Duloxetine or Escitalopram or Fluoxetine or Fluvoxamine or Levomilnacipran or Milnacipran or Mirtazapine or Paroxetine or Reboxetine or Sertraline or Venlafaxine or Vortioxetine) AND Age Group = (child* or adolescent* or "not stated" or unclear).
CCMDCTR‐References Register
The References register was searched using a more sensitive set of terms to identify additional untagged/uncoded reports of RCTs.
Free‐text = (depress* or dysthymi*) AND Free‐Text = (Agomelatin* or Alaprocat* or Bupropion or Citalopram or (Desvenlafaxin* or DVS‐233 or B2061014) or Duloxetin* or Escitalopram or Fluoxetin* or Fluvoxamin* or Milnacipran or Mirtazapin* or Paroxetin* or Reboxetin* or Sertralin* or Venlafaxin* or Levomilnacipran or (Vortioxetin* or Lu AA21004) or Lu AA24530 or (LY2216684 or Edivoxetin*) or (CX157 or Tyrima)) or (serotonin and (uptake or reuptake or re‐uptake)) or SSRI*) AND Free‐Text = (adolesc* or child* or boys or girls or juvenil* or minors or paediatric* or pediatric* or pubescen* or school* or students or teen* or (young not mania) or youth*)
Note: the Cochrane Common Mental Disorders Group (CCMD) was previously called the Cochrane Collaboration Depression, Anxiety and Neurosis (CCDAN) review group. It changed name in 2015 and the re‐naming of the specialised register from CCDANCTR to CCMDCTR reflects this change. In 2016, the specialised register fell out‐of‐date with the Editorial Group’s move from Bristol to York.
Appendix 3. Other database searches (NMA)
A number of update searches have been conducted for this network meta‐analysis, since the publication of two earlier, direct comparison reviews (Hetrick 2007; Hetrick 2012).
Reports of RCTs from MEDLINE, Embase and PsycINFO were captured via the Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR) to June 2016. Searches after this date were conducted directly on MEDLINE, Embase and PsycINFO via the Ovid platform.
Update Search‐1 (2010 to 13 May 2017): Cochrane Specialised Register = 309; CENTRAL = 455; MEDLINE (2016/17) = 185; Embase (2016/17) = 72; PsycINFO (2016/17) = 75; ClinicalTrials.gov = 71; WHO Trials Portal = 90 Total = 1257 (after de‐duplication = 979) Update Search‐2 (2017 to 28‐Nov‐2018): MEDLINE = 246; Embase = 117; PsycINFOn = 167; CENTRAL = 252 Total = 782 (after de‐duplication = 443)
Update Search‐3 (2018 to 30 March 2020): MEDLINE = 206; Embase = 132; PsycINFO = 103; CENTRAL = 447 Total = 888 (after de‐duplication = 503)
Search strategies
Ovid MEDLINE(R) and Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations and Daily <2016 to March 30, 2020>
Search Strategy:
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
1 antidepressive agents/ or antidepressive agents, second‐generation/
2 "serotonin and noradrenaline reuptake inhibitors"/ or serotonin uptake inhibitors/
3 neurotransmitter uptake inhibitors/
4 (antidepressant* or antidepressi*).ti,kf.
5 ((serotonin adj2 (uptake or reuptake or re‐uptake)) or SSRI* or SNRI*).ti,ab,kf.
6 (Agomelatin* or Alaprocat* or Bupropion or Citalopram or (Desvenlafaxin* or DVS‐233 or B2061014) or Duloxetin* or Escitalopram or Fluoxetin* or Fluvoxamin* or Milnacipran or Mirtazapin* or Paroxetin* or Reboxetin* or Sertralin* or Venlafaxin* or Vilazodon* or Levomilnacipran or (Vortioxetin* or Lu AA21004) or Lu AA24530 or (LY2216684 or Edivoxetin*) or (CX157 or Tyrima)).ti,ab,kf,hw,rn.
7 or/1‐6
8 Depression/
9 depressive disorder/ or depressive disorder, major/
10 *Mood Disorders/dt
11 depress*.ti,ab,kf.
12 or/8‐11
13 adolescent/ or young adult/ or child/
14 (adolesc* or child* or boys or girls or juvenil* or minors or paediatric* or pediatric* or pubescen* or school* or students or teen* or (young not mania) or youth*).ti,ab,kf,jw.
15 (13 or 14)
16 (7 and 12 and 15)
17 randomi#ed.ab,ti.
18 randomized controlled trial.pt.
19 controlled clinical trial.pt.
20 placebo.ab.
21 double blind method.sh.
22 (RCT or at random or (random* adj (assign* or allocat* or divid* or division or number))).ti,ab,kf.
23 randomly.ab.
24 ((single or double or triple) adj3 (blind* or mask* or dummy)).ti,ab,kf.
25 trial.ti.
26 (animals not (humans and animals)).sh.
27 or/17‐25
28 (27 not 26)
29 (16 and 28)
30 (in‐data‐review or in‐process or publisher).st.
31 (2016* or 2017* or 2018* or 2019* or 2020*).yr,dp,dt,ep,ez.
32 29 and (30 or 31)
***************************
Ovid Embase <2016 to 2020 Week 13>
Search Strategy:
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
1 *antidepressant agent/
2 Serotonin Receptor Affecting Agent/ or Serotonin Uptake Inhibitor/ or Serotonin Noradrenalin Reuptake Inhibitor/ or Triple Reuptake inhibitor/
3 (antidepressant* or antidepressi*).ti,kw.
4 ((serotonin adj2 (uptake or reuptake or re‐uptake)) or SSRI* or SNRI*).ti,ab,kw.
5 (Agomelatin* or Alaprocat* or Bupropion or Citalopram or (Desvenlafaxin* or DVS‐233 or B2061014) or Duloxetin* or Escitalopram or Fluoxetin* or Fluvoxamin* or Milnacipran or Mirtazapin* or Paroxetin* or Reboxetin* or Sertralin* or Venlafaxin* or Vilazodon* or Levomilnacipran or (Vortioxetin* or Lu AA21004) or Lu AA24530 or (LY2216684 or Edivoxetin*) or (CX157 or Tyrima)).ti,ab,kw,hw,rn.
6 or/1‐5
7 major affective disorder/dt [Drug Therapy]
8 *mood disorder/dt [Drug Therapy]
9 *Depression/
10 Depression/dt [Drug Therapy]
11 major depression/
12 depress*.ti,kw.
13 ((depress* adj2 (disorder* or major)) or MDD or with depress*).ti,ab,kw.
14 or/7‐13
15 child/
16 juvenile/ or exp adolescent/ or exp child/
17 (adolesc* or child* or boys or girls or juvenil* or minors or paediatric* or pediatric* or puberty or pubescen* or school* or students or teen* or young or youth*).ti,kw,jw.
18 (depress* adj (adolesc* or child* or boys or girls or juvenil* or minors or paediatric* or pediatric* or puberty or pubescen* or school* or students or teen* or young or youth*)).ti,ab,kw.
19 or/15‐17
20 (6 and 14 and 19) or (6 and 18)
21 randomi#ed.ab,ti,kw.
22 randomized controlled trial/
23 controlled clinical trial/
24 placebo/
25 placebo.ab.
26 double blind procedure/
27 randomization/
28 (RCT or at random or (random* adj (assign* or allocat* or divid* or division or number))).ti,ab,kw.
29 or/21‐28
30 (20 and 29)
31 limit 30 to (article‐in‐press status or in‐process status)
32 (2016* or 2017* or 2018* or 2019* or 2020*).yr,dp,dc.
33 30 and 32
34 31 or 33
***************************
Ovid APA PsycInfo <2016 to March Week 4 2020>
Search Strategy:
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
1 Neurotransmitter Uptake Inhibitors/ or exp Serotonin Norepinepherine Reuptake Inhibitors/ or exp Serotonin Reuptake Inhibitors/
2 ((serotonin adj2 (uptake or reuptake or re‐uptake)) or SSRI* or SNRI*).ti,ab,id.
3 (Agomelatin* or Alaproclat* or Bupropion or Citalopram or (Desvenlafaxin* or DVS‐233 or B2061014) or Duloxetin* or Escitalopram or Fluoxetin* or Fluvoxamin* or Milnacipran or Mirtazapin* or Paroxetin* or Reboxetin* or Sertralin* or Venlafaxin* or Vilazodon* or Levomilnacipran or (Vortioxetin* or Lu AA21004) or Lu AA24530 or (LY2216684 or Edivoxetin*) or (CX157 or Tyrima)).ti,ab,id.
4 *Antidepressant Drugs/
5 or/1‐4
6 (depress* adj3 (adolesc* or child* or boys or girls or juvenil* or minors or paediatric* or pediatric* or puberty or pubescen* or school* or students or teen* or young or youth*)).ti,ab,id.
7 (5 and 6)
8 major depression/
9 depress*.ti,id.
10 ((depress* adj2 (disorder* or major)) or MDD or with depress*).ti,ab,id.
11 or/8‐10
12 treatment effectiveness evaluation.sh.
13 clinical trials/
14 drug therapy/ or placebo/
15 randomi#ed.ti,ab,id.
16 (RCT or at random or (random* adj (assign* or allocat* or divid* or division or number))).ti,ab,id.
17 ((single or double or triple) adj3 (blind* or mask* or dummy)).ti,ab,id.
18 trial.ti.
19 (empirical study and quantitative study).md.
20 or/12‐19
21 (adolesc* or child* or boys or girls or juvenil* or minors or paediatric* or pediatric* or pubescen* or school* or students or teen* or (young not mania) or youth*).ti,ab,id,jw.
22 ((7 or (5 and 11 and 21)) and 20)
23 (2016* or 2017* or 2018* or 2019* or 2020*).yr,an.
24 (22 and 23)
***************************
Cochrane Central Register of Controlled Trials (CENTRAL), Issue 3 of 12, 2020
#1 (“antidepressive agents” or “antidepressive agent” or (serotonin next “noradrenaline reuptake inhibitors") or “serotonin uptake inhibitors” or “Serotonin Receptor Affecting Agent” or “Serotonin Uptake Inhibitor” or “Serotonin Reuptake Inhibitors” or “Serotonin Noradrenalin Reuptake Inhibitor” or “Serotonin Norepinepherine Reuptake Inhibitors” or “Triple Reuptake inhibitor“ or “neurotransmitter uptake inhibitors”):kw
#2 (antidepress* or (anti‐depress*):ti
#3 ((serotonin near/2 (uptake or reuptake or re‐uptake)) or SSRI* or SNRI):ti,ab
#4 (Agomelatin* or Alaprocat* or Bupropion or Citalopram or (Desvenlafaxin* or DVS‐233 or B2061014) or Duloxetin* or Escitalopram or Fluoxetin* or Fluvoxamin* or Milnacipran or Mirtazapin* or Paroxetin* or Reboxetin* or Sertralin* or Venlafaxin* or Vilazodon* or Levomilnacipran or (Vortioxetin* or “Lu AA21004”) or “Lu AA24530” or (LY2216684 or Edivoxetin*) or (CX157 or Tyrima)):ti,ab,kw
#5 (#1 or #2 or #3 or #4)
#6 (depression or “depressive disorder” or “major depression” or “major affective disorder” or “mood disorder” or “mood disorders”):kw
#7 (depress*):ti
#8 ((depress* NEAR/2 (disorder* or major)) or MDD or “with depression” or “with depressive”):ab
#9 (#6 or #7 or #8)
#10 (adolescent* or “young adult” or child* or juvenile):kw
#11 (adolesc* or child* or boys or girls or juvenil* or minors or paediatric* or pediatric* or pubescen* or school* or students or teen* or youth*):ti,ab
#12 (young not mania):ti,ab
#13 (#10 or #11 or #12)
#14 (#9 and #13)
#15 (depress* NEXT (adolesc* or child* or boys or girls or juvenil* or minors or paediatric* or pediatric* or puberty or pubescen* or school* or students or teen* or young or youth*)):ti,ab,kw
#16 (#14 or #15)
#17 (#5 and #16)
***************************
Appendix 4. Earlier searches (direct comparison reviews to 2011)
Cochrane Common Mental Disorders Group Controlled Trials Register (CCMDCTR) [Known as the Cochrane Collaboration Depression, Anxiety and NeurosisControlled Trials Register(CCDANCTR) at the time]
In 2005, the studies register was searched using the following terms: Diagnosis = (Depress* or Dysthymi*) AND Intervention = ("Selective Serotonin Reuptake Inhibitors" or Alaproclate or Citalopram or Escitalopram or Femoxetine or Fluoxetine or Fluvoxamine or Paroxetine or Sertraline) AND Age Group = (Child or Adolescent)
The update searches conducted 28 October 2011 included additional search terms for newer generation antidepressants: CCMDCTR‒Studies Register Diagnosis = (depress* or dysthymi*) AND Intervention = ("Selective Serotonin Reuptake Inhibitors" or Agomelatine or Alaproclate or Bupropion or Citalopram or Desvenlafaxine or Duloxetine or Escitalopram or Fluoxetine or Fluvoxamine or Milnacipran or Mirtazapine or Paroxetine or Reboxetine or Sertraline or Venlafaxine) AND Age Group = (child* or adolescent* or "not stated" or unclear) CCMDCTR‒References Register The references register was searched using a more sensitive set of terms to identify additional untagged/uncoded references: Title/Abstract/Keywords = (depress* or dysthymi*) AND Free‐Text = (Agomelatine or Alaproclate or Bupropion or Citalopram or Desvenlafaxine or Duloxetine or Escitalopram or Fluoxetine or Fluvoxamine or Milnacipran or Mirtazapine or Paroxetine or Reboxetine or Sertraline or Venlafaxine or (serotonin and (uptake or reuptake or re‐uptake)) or SSRI*) AND Free‐Text = (adolesc* or child* or boys or girls or juvenil* or minors or paediatric* or pediatric* or pubescen* or school* or students or teen* or young or youth*)
Other databases searched included the National Research Register (now archived), ClinicalTrials.gov and Controlled‐Trials.com and pharmaceutical industry registers to 2005.
Eli Lilly and Company
Forest Laboratories
Merck Pharmaceuticals
Lundbeck Pharmaceuticals
GlaxoSmithKline
Brystol‐Myers Squibb
Pfizer Pharmaceuticals (Wyeth, the company that was searched in the original review, has been taken over by Pfizer)
After this date, searches were conducted on pharmaceutical industry registers (and others) via the WHO International Clinical Trials Registry Platform (WHO‐ICTRP).
Australian New Zealand Clinical Trials Registry
ClinicalTrials.gov
ISRCTN (ControlledTrials.com)
Chinese Clinical Trial Registry
Clinical Trials Registry ‐ India
German Clinical Trials Register
Iranian Registry of Clinical Trials
Japan Primary Registries Network
Pan African Clinical Trial Registry
Sri Lanka Clinical Trials Registry
Netherlands National Trial Register
The original searches of MEDLINE, Embase and PsycINFO were undertaken by the author team to October 2005, and of CENTRAL (the Cochrane Central Register of Controlled Trials) to Issue 2, 2004.
Bibliographic database search strategies:
MEDLINE (all years to October 2005) 1. exp Serotonin Uptake Inhibitors/ 2. (serotonin adj (uptake or reuptake or re‐uptake)).mp 3. ssri$.mp 4. alaproclat$ or citalopram or escitalopram or femoxetin$ or fluoxetin$ or fluvoxamin$ or paroxetin$ or sertralin$ 5. or/1‐4 6. clinical trial.pt 7. (random$ or rct$).mp 8. ((singl$ or doubl$) adj5 (blind$ or mask$)).mp 9. Placebos/ 10. placebo$.mp 11. Cross‐Over Studies/ 12. (crossover$ or cross over$ or cross‐over$).mp 13.or/6‐12 14.5 and 13 15. limit 14 to all child<0‐18>
Embase (all years to October 2005) 1. exp Serotonin Uptake Inhibitors/ 2. (serotonin adj (uptake or reuptake or re‐uptake)).mp. 3. ssri$.mp. 4. alaproclat$.mp. 5. citalopram.mp. 6. escitalopram.mp. 7. femoxetin$.mp. 8. fluvoxamin$.mp. 9. paroxetin$.mp. 10. sertralin$.mp. 11. or/1‐10 12. Controlled study/ or randomized controlled trial/ 13. double blind procedure/ 14. single blind procedure/ 15. crossover procedure/ 16. drug comparison/ 17. placebo/ 18. random$.ti,ab,hw,tn,mf. 19. latin square.ti,ab,hw,tn,mf. 20. crossover.ti,ab,hw,tn,mf. 21. cross‐over.ti,ab,hw,tn,mf. 22. placebo$.ti,ab,hw,tn,mf. 23. ((doubl$ or singl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).ti,ab,hw,tn,mf. 24. (comparative adj5 trial$).ti,ab,hw,tn,mf. 25. (clinical adj5 trial$).ti,ab,hw,tn,mf. 26. or/12‐25 27. nonhuman/ 28. animal/ not (human/ and animal/) 29. or/27‐28 30. 26 not 29 31. 11 and 30 32. limit 31 to (child or preschool child <1 to 6 years> or school child <7 to 12 years> or adolescent <13 to 17 years>) PsycINFO (all years to October 2005) 1. exp serotonin reuptake inhibitors/ 2. (serotonin adj (uptake or reuptake or re‐uptake)).mp. 3. ssri$.mp. 4. (Alaproclat$ or Citalopram or Escitalopram or Femoxetin$ or Fluoxetin$ or Fluvoxamin$ or Paroxetin$ or Sertralin$).mp. [mp=title, abstract, cas registry/ec number word, MeSH subject heading] 5. or/1‐4 6. (trial$ or random$ or rct$).mp. [mp=title, abstract, cas registry/ec number word, MeSH subject heading] 7. (child$ or adolescen$ or teenage$).mp. 8. (young adj (person$ or people or adult$)).mp. 9. or/7‐8 10. and/5‐6,9
CENTRAL (all years to Issue 2, 2004) 1. exp Serotonin Uptake Inhibitors/ 2. (serotonin adj (uptake or reuptake or re‐uptake)).mp. 3. ssri$.mp. 4. alaproclat$.mp. 5. citalopram.mp. 6. escitalopram.mp. 7. femoxetin$.mp. 8. fluvoxamin$.mp. 9. paroxetin$.mp. 10. sertralin$.mp. 11. or/1‐10 12. Controlled study/ or randomized controlled trial/ 13. double blind procedure/ 14. single blind procedure/ 15. crossover procedure/ 16. drug comparison/ 17. placebo/ 18. random$.ti,ab,hw,tn,mf. 19. latin square.ti,ab,hw,tn,mf. 20. crossover.ti,ab,hw,tn,mf. 21. cross‐over.ti,ab,hw,tn,mf. 22. placebo$.ti,ab,hw,tn,mf. 23. ((doubl$ or singl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).ti,ab,hw,tn,mf. 24. (comparative adj5 trial$).ti,ab,hw,tn,mf. 25. (clinical adj5 trial$).ti,ab,hw,tn,mf. 26. or/12‐25 27. nonhuman/ 28. animal/ not (human/ and animal/) 29. or/27‐28 30. 26 not 29 31. 11 and 30 32. limit 31 to (child or preschool child <1 to 6 years> or school child <7 to 12 years> or adolescent <13 to 17 years>)
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Almeida‐Montes 2005.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; single site
Power
calculation: 0.90 power to detect a large effect size (0.80)
Use of
diagnostic criteria (or clear specification of inclusion criteria):
DSM‐IV‐TR criteria for depressive disorder plus a score of 13 in the
DSDR
Intervention integrity: not described Outcome measures described or validated measures used: yes Follow‐up assessment points: weekly for 7 weeks No. crossed: none Funded by: Eli Lilly provided fluoxetine and placebo |
|
| Participants | Setting of care: outpatient
Recruitment: no statement
Mean age
(SD): intervention = 13.3 (3.16); control = 11.5 (1.58)
Age range: 8
to 14
Gender (F:M): intervention = not stated; control = not
stated
Methods used to diagnose: DSM‐IV using semi‐structured
interview; The Mini
International Neuropsychiatric Interview for
children and adolescents (MINI‐KID)
Diagnosis: MDD
Baseline
severity of depression: not reported Length of current episode: not reported % first episode: not reported Comorbidity (intervention): not reported Comorbidity (control): not reported Location: Mexico Inclusion criteria: major depressive disorder (DSM‐IV‐TR) plus a score of 13 in the DSDR Exclusion criteria: History of chronic physical illness, intellectual disability, bipolar disorder, substance use or dependence, Attention Deficit Hyperactivity Disorder, severe anxiety, behavioural disorder, hospital admission or increased treatment intensity due to a depressive episode in the preceding 4 weeks, antidepressive treatment in the preceding 4 weeks, any lab test which was considered to be abnormal by the clinician, oppositional defiant disorder Exclusion of suicidality: suicide attempt in the preceding 4 weeks |
|
| Interventions | Intervention group
Drug: fluoxetine
Dosage: 20
mg
Regimen: daily
Length of treatment: 6 weeks Control group: placebo |
|
| Outcomes | Definition and assessment of response: we used OC response data defined as
50% reduction in HDRS scores (they stated they used CGI‐I score of 1 or 2;
50% reduction in DSRS and HAM‐D scores
Depressive symptoms: DSRS,
HAM‐D Functioning: Children's Global Assessment Scale (C‐GAS) Suicidal behaviour: no report Other measures: Hamilton Anxiety Rating Scale HARS |
|
| Notes | Type of data used for remission/response: observed case | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random numbers using SPSS, pg.34 |
| Allocation concealment (selection bias) | Low risk | An independent clinician who was not part of the trial allocated the 2 treatment conditions to either ‘0’ or ‘1’. The trial researchers remained blind to treatment allocation throughout the course of the trial, pg.34. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No statement |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | 2 clinicians who remained blind to treatment allocation assessed the participants weekly, pg.34 |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number eligible: 38 Number randomised: 23 Fluoxetine: 12 (inconsistencies in reporting noted); placebo: 11; total: 23 Number started trial: 23 Fluoxetine: 12 (inconsistencies); placebo: 11; total: 23 Number of withdrawals: Fluoxetine: 7; placebo: 9; total: 16 Number analysed post‐intervention: Fluoxetine: ITT = 7, LOCF = 10; placebo: ITT = 9, LOCF = 10; total: ITT = 16, LOCF = 20 Reasons for dropout: fluoxetine group lost to follow‐up N = 5, withdrawn due to suicide risk N = 1, did not complete N = 7; placebo lost to follow‐up N = 2, withdrawn due to suicide risk N = 0, did not complete N = 9 ITT analysis: additionally we analysed outcomes using ITT analysis in the following way: we divided the number of patients who completed the trial and were considered to be ‘responders’ by the total sample pg. 34; ITT population did not include all who were randomised. Statistical analysis: LOCF |
| Selective reporting (reporting bias) | Unclear risk | All outcomes specified in Methods were reported, however 2 outcomes (adverse and clinician‐reported depression symptoms) were reported in a graph. No access to trial protocol |
| Other bias | Unclear risk | Contact: assessment undertaken weekly Screening: unclear Placebo lead‐in: 1 week Baseline imbalance: data not reported |
Atkinson 2014.
| Study characteristics | ||
| Methods | Trial design: Double‐blind randomised placebo‐controlled trial. Three
parallel groups. 65 sites Power calculation: Based on an anticipated enrolment of 336 patients, randomised in a 1:1:1 ratio across the three groups. Assumed approximately 10% dropout, leaving approximately 100 patients per treatment arm post‐baseline. Estimated 80% power to detect an effect size of 0.40 (duloxetine efficacy relative to placebo on the CDRS‐R total score) using a two group t‐test with a 0.05 two‐sided significance level. The effect size of 0.4 was determined based on historical data for the effect size of duloxetine 60 mg QD in adult patients with MDD. Use of diagnostic criteria (or clear specification of inclusion criteria): Yes. MDD without psychotic features, single or recurrent episode, as defined by DSM‐IV‐TR. MDD diagnosis was supported by the Mini International Neurospychiatric Interview for children and adolescents (MINI‐KID). Intervention integrity: Not described Outcome measures described or validated measures used: Yes. Children’s Depression Rating Scale‐Revised (CDRS‐R). Follow‐up assessment points: Weeks 1, 2, 4, 7, and 10 during the placebo‐controlled acute treatment period, and at weeks 12, 14, 16, 20, 24, 28, 32, and 36 during the double‐blind long‐term treatment period No. crossed: None Funded by: Eli Lilly and Company |
|
| Participants | Setting of care: Outpatient Recruitment: Not described Mean age (SD): Duloxetine = 13.1 (3.0) Fluoxetine = 13.1 (3.3) Placebo = 13.3 (3.1) Age range: 12 – 17 Gender (F:M) Duloxetine = 64:53 Fluoxetine = 61:56 Placebo = 51:52 Methods used to diagnose: MDD without psychotic features, single or recurrent episode, as defined by DSM‐IV‐TR. MDD diagnosis was supported by the Mini International Neurospychiatric Interview for children and adolescents (MINI‐KID). Conducted by two independent evaluators, with at least one evaluator being a psychiatrist Diagnosis: MDD Baseline severity of depression: (Note: The SDs were incorrectly reported in the primary paper as SEs) CDRS‐R total score, Mean (SD) Duloxetine = 59.2 (10.5) Fluoxetine = 58.8 (10.6) Placebo = 60.2 (11.7) CGI‐Severity score, Mean (SD) Duloxetine = 4.5 (0.6) Fluoxetine = 4.5 (0.6) Placebo = 4.6 (0.7) Length of current episode: Not reported. % first episode: 71.5% Comorbidity (intervention): Not reported, however those with significant suicidal risk, comorbid psychiatric conditions requiring medication to manage, or other significant or unstable medical conditions, were excluded. Comorbidity (control): Not reported, however those with significant suicidal risk, comorbid psychiatric conditions requiring medication to manage, or other significant or unstable medical conditions, were excluded. Location: 65 psychiatric clinical sites in nine countries (United States, Finland, France, Germany, Slovakia, Estonia, Russia, Ukraine, and South Africa) Subjects enrolled per country: United States: 140 Finland: 5 France: 8 Germany: 4 Slovakia: 6 Ukraine: 66 Russian Federation: 40 Estonia: 1 South Africa: 67 Inclusion criteria: · Outpatient, diagnosed with major depressive disorder (MDD) as defined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM‐IV‐TR) and supported by the Mini International Neuropsychiatric Interview for children and adolescents (MINI‐KID) · Diagnosis of moderate or greater severity of MDD as determined by Children's Depression Rating Scale ‐ Revised (CDRS‐R) with a total score greater than or equal to 40 at screen, and randomisation and a Clinical Global Impression of Severity (CGI‐Severity) rating of greater than or equal to 4 at screen, and randomisation · Female patients must test negative for pregnancy during screening. · Judged to be reliable by the investigator to keep all appointments for clinical visits, tests, and procedures required by the protocol · Has a degree of understanding such that they can communicate intelligently with the investigator and study coordinator · Capable of swallowing study drug whole · Patients must have venous access sufficient to allow blood sampling and are compliant with blood draws as per the protocol. Exclusion criteria: · Children of site personnel directly affiliated with this study and/or their immediate families · Children of Lilly employees or employees of the designated clinical research organisation (CRO) assisting with the conduct of the study · Have received treatment within the last 30 days with a drug that has not received regulatory approval for any indication at the time of study entry · Have a current or previous diagnosis of bipolar disorder, psychotic depression, schizophrenia or other psychotic disorder, anorexia, bulimia, obsessive compulsive disorder, or pervasive development disorder, as judged by the investigator · Have a history of DSM‐IV‐TR‐defined substance abuse or dependence within the past year, excluding caffeine and nicotine · Have a current primary DSM‐IV‐TR Axis I disorder other than MDD or a current secondary DSM‐IV‐TR Axis I disorder that requires any pharmacologic treatment · Have 1 or more first‐degree relatives with diagnosed bipolar I disorder · Have a significant suicide attempt within 1 year of screening or are currently at risk of suicide in the opinion of the investigator · Have a weight less than 20 kilogram (kg) at screening · Have a lack of response to 2 or more adequate treatment trials of antidepressants at a clinically appropriate dose for a minimum of 4 weeks for the same MDD episode · Have initiated, stopped, or changed the type or intensity of psychotherapy within 6 weeks prior to screening · Have a history of seizure disorder (other than febrile seizures) · Have a history of electroconvulsive therapy within 1 year of screening · Have had treatment with a monoamine oxidase inhibitor (MAOI) within 14 days or fluoxetine within 30 days of randomisation; or the potential need to use an MAOI during the study or within 5 weeks of discontinuation of study drug · Have previously enrolled, completed, or withdrawn from this study or any other study investigating duloxetine or fluoxetine · Have a positive urine drug screen for any substances of abuse or excluded medication · Are taking any excluded medications that cannot be discontinued by screening · Have known hypersensitivity to duloxetine, fluoxetine, or their inactive ingredients; or have frequent or severe allergic reactions to multiple medications · Have uncontrolled narrow‐angle glaucoma · Have acute liver injury or severe cirrhosis · Have a serious or unstable medical illness, psychological condition, or clinically significant laboratory or electrocardiogram (ECG) result that, in the opinion of the investigator, would compromise participation in the study or be likely to lead to hospitalisation · Have abnormal thyroid‐stimulating hormone concentration · Have initiated or discontinued hormone therapy within the previous 3 months · Female patients who are either pregnant, nursing or have recently given birth · Need to use thioridazine during the study or within 5 weeks after discontinuation of study drug or need to use pimozide during the study Exclusion of suicidality: Have a significant suicide attempt within 1 year of screening or are currently at risk of suicide in the opinion of the investigator |
|
| Interventions | Intervention group 1: Drug: duloxetine Dosage: 60–120 mg per day Regimen: 30 mg per day for 2 weeks, increased to 60 mg per day at the 2‐week time point. Could be increased to 90 mg per day at the 4‐week time point or later, and subsequently increased to 120 mg per day at the 7‐week time point or later. Lowest dose allowed after the 2‐week time point was 60 mg per day. Length of treatment: 10 weeks Intervention group 2: Drug: fluoxetine Dosage: 20–40 mg per day Regimen: 10 mg per day for 2 weeks, increased to 20 mg per day at the 2‐week time point. Could be increased to 40 mg per day at the 4‐week time point or at any later time point. Lowest dose allowed after the 2‐week time point was 20 mg per day. Length of treatment: 10 weeks Control group: Patients randomised to placebo remained on placebo throughout the 10‐week acute treatment period. |
|
| Outcomes | Definition and assessment of response: Primary outcome was change from
baseline in Children's Depression Rating Scale‐Revised (CDRS‐R) total score
at week‐10 endpoint. Depressive symptoms: Children’s Depression Rating Scale–Revised (CDRS‐R) Functioning: Not assessed Suicidal behaviour: Columbia‐Suicide Severity Rating Scale (C‐SSRS) Other measures: Clinical Global Impressions (CGI)‐Severity |
|
| Notes | Type of data used for remission/response: Intention‐to‐treat | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | No statement. “Patients meeting entry criteria were randomly assigned 1:1:1
to either duloxetine flexible dose (60–120 mg once daily [QD]), fluoxetine
flexible dose (20–40 mg QD), or placebo, via Interactive Voice Response
System (IVRS).” pg.181. Stratified randomisation by age: children (7‐11 years) and adolescents (12‐17 years). FDA statistical review. Comment: Likely implemented adequate random sequence generation given use of IVRS, no baseline imbalance of major concern, however no statement reported |
| Allocation concealment (selection bias) | Low risk | Patients meeting entry criteria were randomly assigned 1:1:1 to either duloxetine flexible dose (60–120 mg once daily [QD]), fluoxetine flexible dose (20–40 mg QD), or placebo, via Interactive Voice Response System (IVRS). pg.181 Atkinson 2014 |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Trial registry entry (NCT00849901) stated “Masking: Quadruple (Participant,
Care Provider, Investigator, Outcomes Assessor)” “double‐blind” “…because of the need to blind multiple doses of two different drugs, all patients were required to take six capsules of study drug per day” pg.188 Atkinson 2014. Comment: likely adequate blinding of participants and personnel, although no statement on how blinding was maintained or evaluated and differential dropout due to adverse events |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Trial registry entry (NCT00849901) stated “Masking: Quadruple (Participant,
Care Provider, Investigator, Outcomes Assessor)”. Comment: likely outcome assessor was blind, however no statement in Atkinson 2014, and differential dropout due to adverse events |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: not reported, 438 patients screened Number randomised: 337; duloxetine: 117; fluoxetine: 117; placebo: 103 Number started trial: 337; duloxetine: 117; fluoxetine: 117; placebo: 103 Number of withdrawals: 72; duloxetine: 30; fluoxetine: 26; placebo: 16 Number analysed post‐intervention: 329; duloxetine: 113; fluoxetine: 113; placebo: 103 Reasons for dropout: Adverse events: duloxetine = 9; fluoxetine = 1; placebo = 3 Lack of efficacy: duloxetine = 2; fluoxetine = 3; placebo = 2 Physician decision: duloxetine = 1; fluoxetine = 1; placebo = 1 Parent/carer decision: duloxetine = 11; fluoxetine = 5; placebo = 4 Patient decision: duloxetine = 4; fluoxetine = 10; placebo = 4 Lost to follow‐up: duloxetine = 2; fluoxetine = 4; placebo = 1 Sponsor decision: duloxetine = 1; fluoxetine = 0; placebo = 0 Protocol violation: duloxetine = 0; fluoxetine = 2; placebo = 1 Comment: differential dropout rate, duloxetine = 25.6%, fluoxetine = 22.2%, placebo = 15.5%. Dropout due to adverse events and parent/carer decision higher in duloxetine, patient withdrawal higher in fluoxetine. ITT analysis: modified ITT population included all randomised patients with both a baseline and at least one post‐baseline value for CDRS‐R (corresponded to 329 patients) pg.182 Atkinson 2014. Statistical analysis: using the ITT population, the primary efficacy parameter of (LS) mean change from baseline on the CDRS‐R was estimated using the restricted maximum likelihood (REML)‐based mixed‐effects model for repeated measure (MMRM). All available observations from each post‐baseline visit were included (OC). The MMRM model included the fixed categorical effects of treatment, pooled investigative site, visit, treatment‐by‐visit interaction, age category, and age category‐by‐visit interaction, as well as the continuous, fixed covariates of the baseline CDRS‐R, and the baseline CDRS‐R‐by‐visit interaction. An unstructured covariance structure was used to model the within‐patient errors. A Kenward–Roger correction was used to estimate denominator degrees of freedom. Significance tests were based on least‐squares means (LS means) using a two‐sided ⍶ = 0.05. Additional analyses are reported using ANOVA and ANCOVA (baseline CDRS‐R, age category as covariates) models with LOCF approach. Secondary outcomes of CDRS‐R response and remission were estimated as probabilities using a categorical MMRM approach, in which a marginal model based on a pseudolikelihood method was utilised and implemented in SAS PROC GLIMMIX. Model included the fixed categorical effects of treatment, pooled investigative site, visit, treatment‐by‐visit interaction, age category, and age category‐by‐visit interaction, as well as the continuous, fixed covariates of the baseline CDRS‐R, and the baseline CDRS‐R‐by‐visit interaction. |
| Selective reporting (reporting bias) | Unclear risk | All outcomes in methods reported? Yes Data available for use in MA? Yes Note: Atkinson 2014 reported change from baseline scores for CDRS‐R but no standard errors. These are fully reported in the trial registry entry (NCT00849901). CDRS‐R “probabilities” of response and remission are reported as percentages only, without events or denominators. pg.185 Atkinson 2018. Clinical trials registry entry but no access to protocol. Atkinson 2014 pg.181 stated the protocol was filed with United States FDA prior to study initiation. Protocol and Statistical Analysis Plan have not been published. Available sources are trial registry entry (NCT00849901), F1J‐MC‐HMCK clinical study report synopsis and FDA statistical review. |
| Other bias | Unclear risk | Contact: clinic visits scheduled weekly during screening and at weeks 1, 2,
4, 7, 10 (acute phase endpoint) Screening: weekly, 2‐4 weeks prior to baseline. “Participants met DSM‐IV‐TR criteria for MDD without psychotic features, had a CDRS‐R total score >/= 40 and a CGI‐S score >/= 4 at the three screening visits” pg.181. Placebo lead‐in: no statement Baseline imbalance: “There were no significant between‐group differences in baseline demographics or psychiatric profile (Table 1).” pg.183 Atkinson, however supporting analysis not reported. Baseline CDRS, CGI‐S, age, sex, BMI, race, and ethnicity appear balanced. Other potentially important covariates (e.g. comorbidities) not reported |
Atkinson 2018.
| Study characteristics | ||
| Methods | Trial design: Double‐blind randomised placebo‐controlled trial. Three
parallel groups. 33 sites Power calculation: It was estimated that 111 participants per treatment arm (N = 333) were sufficient to demonstrate a 5‐point difference in the primary endpoint between the desvenlafaxine and placebo groups at a significance level of 5% and a power of 85%, assuming a pooled standard deviation (SD) of 12, and that no more than 5% of randomised subjects would fail to qualify for the primary analysis. Use of diagnostic criteria (or clear specification of inclusion criteria): Yes. Primary diagnosis of Major Depressive Disorder (MDD) according to Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (Text Revision) (DSM‐IV‐TR) criteria. Assessed by the Schedule for Affective Disorders and Schizophrenia for School‐Age Children–Present and Lifetime Version (K‐SADS‐PL) and clinical interview Intervention integrity: Not described Outcome measures described or validated measures used: Yes. Children’s Depression Rating Scale‐Revised (CDRS‐R). Follow‐up assessment points: Weeks 1, 2, 3, 4, 6, and 8, (and/or at early termination) in the double‐blind phase No. crossed: None Funded by: Pfizer Inc. |
|
| Participants | Setting of care: Outpatient Recruitment: Not described Mean age (SD): Desvenlafaxine high exposure = 12.87 (3.01) Desvenlafaxine low exposure = 13.07 (2.80) Placebo = 13.15 (2.68) Age range: 7–17 Gender (F:M): Desvenlafaxine high exposure = 76:45 Desvenlafaxine low exposure = 69:53 Placebo = 60:60 Methods used to diagnose: Diagnosis of MDD according to DSM‐IV‐TR criteria, as assessed by the K‐SADS‐PL and clinical interview. A comprehensive diagnostic psychiatric evaluation, including collection of psychiatric history and treatments and confirmation of the MDD diagnosis, was performed by a psychiatrist at screening. Enrolled patients were required to have at least moderately severe depressive symptoms for ≥ 30 days before screening, and a CDRS‐R score > 40 and Clinical Global Impressions‐Severity scale (CGI‐S) score ≥ 4 at screening and baseline. Eligible patients were judged, in the investigator’s opinion, to be likely to respond to antidepressant therapy without the need for concomitant psychotherapy. Diagnosis: MDD Baseline severity of depression: CDRS‐R total score, Mean (SD) Desvenlafaxine high exposure = 58.45 (9.45) Desvenlafaxine low exposure = 58.52 (9.18) Placebo = 57.28 (8.94) CGI‐Severity score, Mean (SD) Desvenlafaxine high exposure = 4.61 (0.58) Desvenlafaxine low exposure = 4.61 (0.61) Placebo = 4.55 (0.58) % first episode: Not reported Comorbidity (intervention): Not reported, although the inclusion and exclusion criteria were designed to enrol individuals without comorbid psychiatric conditions, other unstable medical illnesses, or risk for suicide. Comorbidity (control): Not reported, although the inclusion and exclusion criteria were designed to enrol individuals without comorbid psychiatric conditions, other unstable medical illnesses, or risk for suicide. Location: 33 sites in the United States and Chile. Note: Only one subject was enrolled in Chile, thus “country” was not included as a factor in statistical analyses. The original protocol stated that there were 43 sites in the United States, 2 sites in Chile, and 1 site in Mexico, however thirteen sites did not randomise any subjects. Inclusion criteria: · Being ≥ 7 and < 18 years of age and meeting the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM‐IV‐TR) criteria for MDD, as assessed by the Schedule for Affective Disorders and Schizophrenia – Present and Lifetime Version (K‐SADS‐PL) and clinical interview · Weight ≥ 20 kg, a CDRS‐R score > 40, a CGI‐S score ≥ 4, a current DSM‐IV‐TR major depressive episode of at least moderate severity with symptoms for at least 1 month before screening, depression that could, in the investigator’s opinion, respond to therapy with antidepressant(s) alone without need for concomitant psychotherapy and who could successfully pass the placebo swallow test · Subjects and their parent(s)/legal guardians had to be willing and able to comply with the scheduled visits, treatment plan, laboratory tests, and other study procedures. Subjects that were sexually active had to agree to use the required contraception as defined in the Life Styles Guidelines section of the protocol consistently and correctly for the duration of the active treatment period and for at least 28 days after the last dose of study drug. Exclusion criteria: · Pre‐randomisation blood pressure elevation · A previous lifetime history of suicidal behaviours or suicide ideation associated with actual intent and/or plan at any time in their lifetime as indicated by a positive response on items 4 or 5 of the Columbia‐Suicide Severity Rating Scale (C‐SSRS) (baseline version at screening visit) or based on the clinical judgement of the investigator, history of suicidal behaviours or suicide ideation associated with actual intent and/or plan as indicated by a positive response on items 4 or 5 of the Columbia‐Suicide Severity Rating Scale (C‐SSRS) (Since Last Visit version at baseline visit) or based on the clinical judgement of the investigator, history of suicidal behaviours indicated by a positive response to “Actual Attempt”, “Interrupted Attempt”, “Aborted Attempt”, “Preparatory Acts or behavior” or “Suicidal Behavior” on the C‐SSRS (Since Last Visit version at baseline visit) · A history or current evidence of gastrointestinal disease known to interfere with absorption or excretion of drugs, a history of surgery known to interfere with absorption or excretion of drugs, a major acute illness within 90 days before the screening visit, severe acute or chronic medical or psychiatric condition or laboratory abnormality that may increase the risk associated with study participation or may interfere with the interpretation of study results and in the judgement of the investigator would make the subject inappropriate for entry in the study · Meeting the DSM‐IV‐TR criteria for current (within 12 months before baseline) psychoactive substance or alcohol abuse or dependence, post‐traumatic stress disorder or obsessive compulsive disorder, current (within 12 months before baseline) generalised anxiety disorder, panic disorder, social anxiety disorder or attention deficit hyperactivity disorder or the criteria for borderline personality disorder, had a history of recurrent, intentional self‐injurious behaviour in the opinion of the investigator or had any clinically important personality disorder that may have interfered with the subject’s clinical evaluation · Pregnant females and female subjects with a positive serum beta human chorionic gonadotropin (β‐hCG) pregnancy test result Exclusion of suicidality: A previous lifetime history of suicidal behaviours or suicide ideation associated with actual intent and/or plan at any time in their lifetime as indicated by a positive response on items 4 or 5 of the Columbia‐Suicide Severity Rating Scale (C‐SSRS) (Baseline Version at screening visit) or based on the clinical judgement of the investigator, history of suicidal behaviours or suicide ideation associated with actual intent and/or plan as indicated by a positive response on items 4 or 5 of the Columbia‐Suicide Severity Rating Scale (C‐SSRS) (Since Last Visit version at baseline visit) or based on the clinical judgement of the investigator, history of suicidal behaviours indicated by a positive response to “Actual Attempt”, “Interrupted Attempt”, “Aborted Attempt”, “Preparatory Acts or behavior” or “Suicidal Behavior” on the C‐SSRS (Since Last Visit version at baseline visit) |
|
| Interventions | Intervention group 1: Drug: Desvenlafaxine (high exposure) Dosage: Titration (baseline/day 1–7) 20 to < 35 kg = 10 mg/day 35 to < 70 kg = 10 mg/day > 70 kg = 20 mg/day Treatment (week 1–8) 20 to < 35 kg = 25 mg/day 35 to < 70 kg = 35 mg/day > 70 kg = 50 mg/day Regimen: Once daily Length of treatment: 8 weeks. Subjects then entered either a 1‐week taper phase with 4‐week follow‐up period, or a 6‐month flexible dose extension study (reported separately). Intervention group 2: Drug: Desvenlafaxine (low exposure) Dosage: Titration (baseline/day 1–7) 20 to < 35 kg = 10 mg/day 35 to < 70 kg = 10 mg/day > 70 kg = 20 mg/day Treatment (week 1–8) 20 to < 35 kg = 20 mg/day 35 to < 70 kg = 25 mg/day > 70 kg = 35 mg/day Regimen: Once daily Length of treatment: 8 weeks. Subjects then entered either a 1‐week taper phase with 4‐week follow‐up period, or a 6‐month flexible dose extension study (reported separately). Control group: Matched placebo tablets administered once daily for 8 weeks (treatment phase), followed by placebo tablets administered once daily for 1‐week (taper phase) only for those participants not entering the extension study |
|
| Outcomes | Definition and assessment of response: Primary efficacy outcome was change
from baseline in CDRS‐R total score at week 8 (endpoint). Depressive symptoms: Children’s Depression Rating Scale‐Revised (CDRS‐R) Functioning: Not assessed Suicidal behaviour: Columbia‐Suicide Severity Rating Scale (C‐SSRS) Other measures: Clinical Global Impressions‐Severity scale (CGI‐S) and Clinical Global Impressions‐Improvement (CGI‐I) |
|
| Notes | Type of data used for remission/response: Intention‐To‐Treat (ITT) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Eligible patients were randomly assigned (1:1:1) to placebo,
desvenlafaxine low exposure (based on body weight at baseline), or
desvenlafaxine higher exposure (based on body weight at baseline) arms, and
stratified by age group (child [7–11 years] or adolescent [12–17 years]) and
country” (Pg.56, under study design). “Thirteen (13) centres did not randomize any subjects” (pg.1 of 19, Pfizer confidential public disclosure synopsis)” (however these not included in analysis). |
| Allocation concealment (selection bias) | Unclear risk | No information on attempts to conceal allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The term ‘double‐blind’ was used throughout manuscript. Pg.4: subject disposition stated both subject and investigator blinded. No information was provided on how blinding was implemented or maintained. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The term ‘double‐blind’ was used throughout, however, no explicit information on whether this included outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible (randomised): 363 (fig 1) Number randomised: 363; desvenlafaxine high exposure = 121; desvenlafaxine low exposure = 122; placebo = 120 Number started trial: 363 Number of withdrawals: desvenlafaxine high exposure = 17; desvenlafaxine low exposure = 21; placebo = 24 Number analysed post‐intervention: (ITT) desvenlafaxine high exposure = 121; desvenlafaxine low exposure = 120; placebo = 119 Reasons for dropout – desvenlafaxine high exposure Adverse events = 3 Lost to follow‐up = 1 Protocol violation = 2 No longer willing to participate = 9 Other = 2 Reasons for dropout – desvenlafaxine low exposure Adverse events = 8 Lack of efficacy = 2 Lost to follow‐up = 3 Protocol violation = 1 No longer willing to participate = 4 Other = 1 Reasons for dropout – placebo Adverse events = 8 Lack of efficacy = 2 Lost to follow‐up = 5 Protocol violation = 3 No longer willing to participate = 3 Other = 2 ITT analysis: 360 Desvenlafaxine high exposure = 121 Desvenlafaxine low exposure = 120 Placebo = 119 “TEAEs were reported by 81 patients (66.4%) in the desvenlafaxine low exposure group, 81 patients (66.9%) in the desvenlafaxine high exposure group, and 73 patients (60.8%) in the placebo group”. Statistical analysis: “Efficacy evaluations were conducted in the intent to‐treat (ITT) population, defined as all patients who were randomly assigned to treatment, received at least one dose of study drug, and had a baseline and at least one post‐baseline primary efficacy assessment. Change from baseline in CDRS‐R (primary analysis) was assessed using a mixed‐effects model for repeated measures (MMRM) with terms for treatment, week, interaction of treatment and week, age group, gender, and baseline CDRS‐R score. Change from baseline in CGI‐S score was assessed using the same approach as used with the CDRS‐R total score” (pg.57 under Efficacy). |
| Selective reporting (reporting bias) | Unclear risk | All outcomes stated in the protocol were reported. Data available for use in MA? Yes Access to trial protocol? Clinical trials registry entry but no access to protocol. Available sources are trial registry entry (NCT01371734), EUCTR2008 results synopsis and B2061032 Pfizer public disclosure synopsis and FDA statistical review. |
| Other bias | Low risk | Contact: All efficacy assessments were administered at weeks 1, 2, 3, 4, 6,
and 8. Placebo lead‐in: Different placebo lead‐in (once weekly) compared with 1 x daily for first week for desvenlafaxine (pg.6, Clinical Trials register document) Baseline imbalance: No significant differences reported between groups |
Berard 2006.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre
Power
calculation: yes
Use of diagnostic criteria (or clear specification of
inclusion criteria): yes
Intervention integrity: yes ‐ returned pill
pack. "Every effort was to be made to encourage patient compliance with the
dosage regimen as per protocol. All patients were instructed to return their
medication pack, with any unused drug, to the investigator at their next
visit. A record of the supplies dispensed, taken and returned was made in
the Case Report Form (CRF) at each visit", Section 3.6, Final report. Outcome measures described or validated measures used: yes Follow‐up assessment points: post‐intervention No. crossed over: none Funded by: SmithKline Beecham |
|
| Participants | Setting of care: not stated
Recruitment: no information
Mean
age (SD): intervention = 15.5 (SD 1.6); control = 15.8 (SD 1.6)
Age
range: 13 to 19 years
Gender (F:M): intervention = 122:65; control =
61:38
Methods used to diagnose: DSM‐IV; GAS < 69; Montgomery‐Asberg
Depression Rating Scale (MADRS) ≥ 16; after screening 14‐day single‐blind
run‐in period
Diagnosis: MDD
Baseline severity of depression:
MADRS mean (SE) score: intervention = 25. 9 (0.5); control: 25.9 (0.6) (both
groups moderately to severely ill); CGI intervention 4.2 (0.1); CGI placebo
4.2 (0.1) Length of current episode: not reported % first episode: intervention 70.9%; placebo 68.8% Comorbidity (intervention): specific phobia 6; separation anxiety 5; panic disorder 3; social phobia 3; Generalised Anxiety Disorder (GAD) 13; Post Traumatic Stress Disorder (PTSD) 1; Attention Deficit Hyperactivity Disorder (ADHD) 3; Oppositional Deficiant Disorder (ODD) 1; Anorexia Nervosa (AN) 1; Bulimia Nervosa (BN) 2; substance abuse 0 Comorbidity (control): specific phobia 3; separation anxiety; panic disorder 0; social phobia 4; GAD 4; PTSD 3; ADHD 0; ODD 1; AN 0; BN 0; substance abuse 1 Location: Argentina, Belgium, Canada, Holland, Italy, Mexico, South Africa, Spain, United Arab Emirates, United Kingdom Inclusion criteria: unipolar MDD for at least 8 weeks' duration; negative pregnancy test Exclusion criteria: prepubertal; diagnosis of conduct disorder, autism, pervasive developmental disorder, organic psychiatric disorder including schizophrenia and epilepsy; obsessive compulsive disorder, panic disorder, social phobia, PTSD that preceded MDD; medical illness that contraindicated use of paroxetine; previous response to psychotherapy; planned long‐term psychotherapy; electroconvulsive therapy (ECT) in previous 3 months or planned for trial period; drug or alcohol dependency; concomitant psychotropic medication or other drugs interfering with central nervous system (CNS) activity; use of sumatriptan, oral anticoagulants or type 1C antiarrhythmics, i.e. encainide, flecainide, lorcainide and propafenone; previous use of paroxetine or other SSRI; sensitivity to SSRI; sexually active and not using contraceptive or pregnant or lactating; use of other investigational drug Exclusion of suicidality: although a history of suicide attempt(s) was not exclusionary, patients with current serious suicidal ideation were excluded. |
|
| Interventions | Intervention group
Drug: paroxetine
Dosage: 20 to 40
mg
Regimen: daily
Length of treatment: 12 weeks Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: we used OC response defined as ≥ 50%
MADRS (they used responders defined as at least a 50% reduction on MADRS.
Post hoc analysis on responder rate based on a CGI‐I score of 1 or 2 was
also conducted).
Depressive symptoms: change from baseline in the
K‐SADS‐L depression subscale score Functioning: Children's Global Assessment Scale (C‐GAS) Suicidal behaviour: FDA data; no report of continuous measure Other measures: Montgomery‐Asberg Depression Rating Scale (MADRS); K‐SADS‐L; Clinical Global Impressions Scale Improvement (CGI‐Improvement); Mood and Feeling Questionnaire; adverse events |
|
| Notes | Additional data were sought and received from the authors.
MHRA #
377
MHRA contacted for additional data some of which were
provided
Data in MA taken from GlaxoKline Beecham web‐based report Type of data used for remission/response: last‐observation‐carried‐forward |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "a computer generated randomization list": GlaxoKline Beecham "centralised computer‐generated randomisation list" pg.61 |
| Allocation concealment (selection bias) | Low risk | "masterlist held by SB...individual sealed code breaks held by
investigators...could be broken in case of emergency": GlaxoKline
Beecham "centralised": pg.61 |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "paroxetine and placebo capsules were identical and all packaging
maintained the double blind nature of the trial": GlaxoKline Beecham
pg.32 "placebo and paroxetine capsules were centrally prepared and packaged and were identical in appearance so that all trial personnel and patients were blind": pg.61 |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No statement |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number eligible: 286 Number randomised: paroxetine: 187; placebo: 99; total: 286 Number started trial: paroxetine: 187; placebo: 99; total: 286 Number of withdrawals: paroxetine: 60; placebo: 30; total: 90 Number analysed post‐intervention: paroxetine: 182; placebo: 93; total: 275 Reasons for dropout: figure 1 in Berard 2006 publication. Higher rate of dropout in the paroxetine group including higher rate of discontinuation due to adverse events and lost to follow‐up. Higher rate of dropout due to lack of efficacy in the placebo group. ITT analysis: intention‐to‐treat (ITT) population was all patients randomised who received at least one dose of double‐blind medication and at least one treatment assessment was available. GlaxoKline Beecham and pg.63 of Berard 2006 publication Statistical methods: last‐observation‐carried‐forward (LOCF) analysis was used, but authors also did OC analysis. Used logistic regression and analysis of covariance; included treatment group, country group and covariates of age and baseline scores (pg.63) |
| Selective reporting (reporting bias) | Low risk | Authors undertook analysis of response in multiple ways. Authors undertook a post hoc analysis of participants > 16 years of age. Reported least square means and SEs for depression scores. Trial protocol contained in final report |
| Other bias | Unclear risk | Contact: assessment undertaken at weeks 1, 2, 3, 4, 6, 8 and 12 weeks.
Participants were able to have non‐directive supportive therapy during
treatment. Screening: 2 week‐screening period from screening assessment to baseline assessment Placebo lead‐in: 2‐week single‐blind Baseline imbalance: none reported ‐ authors stated baseline characteristics were similar. Table 1 reported all demographic and clinical characteristics. |
Durgam 2018.
| Study characteristics | ||
| Methods | Trial design: Double‐blind randomised placebo‐controlled trial. Three
parallel groups. 56 sites Power calculation: Sample size of 495 patients (165 per group) was planned to provide 85% power to detect an effect size of 0.36 at a two‐sided significance level of 0.5%. Use of diagnostic criteria (or clear specification of inclusion criteria): Diagnosis of MDD based on DSM‐IV‐TR criteria for a minimum of 6 weeks, confirmed by K‐SADS‐PL interview administered by a trained clinician, plus CDRS‐R total score ≥ 40 and a CGI‐S score ≥ 4. Intervention integrity: Not described Outcome measures described or validated measures used: Yes (CDRS‐R; CGI‐S; CGI‐I) Follow‐up assessment points: Weeks 1, 2, 3, 4, 6, and 8 No. crossed: None Funded by: Forest Research Institute, an Allergan affiliate |
|
| Participants | Setting of care: Outpatient Recruitment: Not described Mean age (SD): Vilazodone 15 mg = 14.9 (1.6) Vilazodone 30 mg = 14.6 (1.6) Control = 14.9 (1.7) Age range: 12–17 Gender (F:M): Vilazodone 15 mg = 103:72 Vilazodone 30 mg = 107:73 Control = 103:68 Methods used to diagnose: Diagnosis of MDD based on DSM‐IV‐TR criteria, confirmed by K‐SADS‐PL interview administered by a trained clinician Diagnosis: MDD Baseline severity of depression: CDRS‐R total score, Mean (SD) Vilazodone 15 mg = 57.8 (8.7) Vilazodone 30 mg = 56.8 (8.5) Control = 57.5 (8.6) Length of current episode: months Mean (SD) Vilazodone 15 mg = 12.0 (14.2) Vilazodone 30 mg = 12.6 (14.7) Control = 11.0 (12.0) % first episode: not reported Comorbidity (intervention): not reported, however exclusion criteria included a principal DSM‐IV‐TR Axis I diagnosis other than MDD in the past 3 months. Comorbidity (control): not reported, however exclusion criteria included a principal DSM‐IV‐TR Axis I diagnosis other than MDD in the past 3 months. Location: United States Inclusion criteria: Diagnosis of MDD for a minimum of 6 weeks based on Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM‐IV‐TR), and Children’s Depression Rating Scale–Revised (CDRS‐R) total score ≥ 40 and a Clinical Global Impressions–Severity (CGI‐S) score ≥ 4 Exclusion criteria: A principal DSM‐IV‐TR Axis I diagnosis other than MDD in the past 3 months or prior diagnosis of mental retardation or other cognitive disorders (Note: comorbid diagnoses of learning disorders, attention deficit disorder with or without hyperactivity, communication disorders, separation anxiety disorder, dysthymic disorder, oppositional defiant disorder, and anxiety disorders were not exclusionary.) Nonresponse to adequate treatment (i.e. at least 8 weeks’ duration) with two or more SSRIs or serotonin and norepinephrine reuptake inhibitors (SNRIs) or the need for concomitant psychotropic medication. History of drug or alcohol abuse or dependence within the past year. Significant suicide risk judged by the investigator based on the psychiatric interview or information collected from the Columbia‐Suicide Severity Rating Scale (C‐SSRS) or suicide attempt within the past year. Any unstable medical condition or any condition that could interfere with study conduct, confound interpretation of study results, or endanger patient well‐being Exclusion of suicidality: Significant suicide risk judged by the investigator based on the psychiatric interview or information collected from the Columbia‐Suicide Severity Rating Scale (C‐SSRS) or suicide attempt within the past year |
|
| Interventions | Intervention group 1:
Drug: viladozone
Dosage: 15 mg (5 mg/day
for days 1–3 and 10 mg/day for days 4–7, then 15 mg/day)
Regimen:
Daily
Length of treatment: 10 weeks (1‐week screening/taper‐up period,
8‐week double‐blind treatment period, 1‐week double‐blind taper‐down
period) Intervention group 2: Drug: viladozone Dosage: 30 mg Regimen: Daily (5 mg/day for days 1–3 and 10 mg/day for days 4–7, then 20 mg/day starting at week 2 and 30 mg/day starting at week 3) Length of treatment: 10 weeks (1‐week screening/taper‐up period, 8‐week double‐blind treatment period, 1‐week double‐blind taper‐down period) Control group: Placebo tablets, identical in appearance and packaging to viladozone |
|
| Outcomes | Definition and assessment of response: Primary – change from baseline to
week 8 in CDRS‐R total score. Secondary – change from baseline to week 8 in
CGI‐S score, CGI–Improvement (CGI‐I) score, CDRS‐R response (≥ 40% reduction
from baseline in CDRS‐R total score), and CDRS‐R remission (total score ≤
28) Depressive symptoms: Children’s Depression Rating Scale–Revised (CDRS‐R) Functioning: Not assessed Suicidal behaviour: Columbia‐Suicide Severity Rating Scale (C‐SSRS) Other measures: Clinical Global Impressions–Severity (CGI‐S), Clinical Global Impressions–Improvement (CGI‐I). |
|
| Notes | Type of data used for remission/response: Observed cases (OC) approach without imputation of missing values | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerised randomisation codes were generated (pg.355, section 2.2). |
| Allocation concealment (selection bias) | Low risk | “…blinding of patients, investigators, and study site personnel was implemented and maintained by interactive voice/web response systems” (pg.355, section 2.2). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | “…blinding of patients, investigators, and study site personnel was
implemented and maintained by interactive voice/web response systems….Study
medication was dispensed as vilazodone 5‐, 10‐, and 20‐mg tablets and
matching placebo tablets, identical in appearance and packaging.”. (pg.355,
section 2.2). “Breaking the blind for any reason resulted in discontinuation from the study” (pg.355, section 2.2). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | “…blinding of patients, investigators, and study site personnel was
implemented and maintained by interactive voice/web response systems….Study
medication was dispensed as vilazodone 5‐, 10‐, and 20‐mg tablets and
matching placebo tablets, identical in appearance and packaging.” (pg.355,
section 2.2). “Breaking the blind for any reason resulted in discontinuation from the study” (pg.355, section 2.2). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: 529 Number randomised: viladozone 30 mg: 180; viladozone 15 mg: 175; placebo: 174 Number started trial (safety population): viladozone 30 mg: 180; viladozone 15 mg: 175; placebo: 171 Number of withdrawals: viladozone 30 mg: 19; viladozone 15 mg: 25; placebo: 28 Number analysed post‐intervention: viladozone 30 mg: 161; viladozone 15 mg: 149; placebo: 142 Reasons for dropout: Withdrawal of consent (4.6%), AEs (4.0%); discontinuation due to AEs was higher in patients treated with vilazodone (15 mg/day = 5.1%; 30 mg/day = 4.4%) than in patients treated with placebo (2.3%) Vilazodone 30 mg: AE 8, withdrew consent 8, protocol violation 2, lost to follow‐up 1. Vilazodone 15 mg: AE 9, withdrew consent 7, protocol violation 4, insufficient response 3, lost to follow‐up 3. Placebo: AE 4, withdrew consent 9, protocol violation 3, insufficient response 5, lost to follow‐up 6, other 2. ITT analysis: ITT population comprised all patients in the safety population who had a baseline and one or more post‐baseline CDRS‐R total score assessments, ITT N = 524 Vilazodone 30 mg: 180 Vilazodone 15 mg: 174 Placebo: 170 Statistical analysis: MMRM with treatment group, study centre, visit, and treatment group‐by‐visit interaction as the fixed effects and the baseline value and baseline value‐by‐visit interaction as covariates. Analysis was based on all post‐baseline scores using the observed cases (OC) approach without imputation of missing values. Secondary efficacy parameters were analysed using an MMRM approach that was similar to the one used for the primary efficacy parameter. |
| Selective reporting (reporting bias) | Unclear risk | Data available for use in MA? yes Access to trial protocol? Yes: NCT01878292 clinicaltrials.gov/ct2/show/NCT01878292?term=Vilazodone&cond=depression&draw=2&rank=8 |
| Other bias | Low risk | Contact: Visits occurred on weeks 1, 2, 3, 4, 6, and 8. Screening: 1‐week screening period Placebo lead‐in: None Baseline imbalance: In the primary paper, baseline characteristics were reported and the statement was made that “Baseline characteristics were similar across treatment groups”, but no statistical comparisons were presented. “The sponsor was involved in conducting the analyses, interpreting the results, and the decision to submit this manuscript for publication” (pg.361, Funding) **rated low risk given the non‐significant nature of the results presented |
Emslie 1997.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; single site Power calculation: not reported Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: assessed by clinical chemistry profile Outcome measures described or validated measures used: yes Follow‐up assessment points: post‐intervention No. crossed over: none Funded by: National Institute of Mental Health | |
| Participants | Setting of care: outpatients
Recruitment: self‐referred or referred
to mood disorders programme; none were recruited by media.
Mean age
(SD): intervention = 12.2 (2.7); control: 12.5 (2.6)
Age range: 7 to
17 years
Gender (F:M): intervention = 22:26; control =
22:26
Methods used to diagnose: DSM‐II‐RK‐SADS depressive items;
CDRS‐R ≥ 40; 3 independent diagnostic interviews and a 1‐week placebo
lead‐in
Diagnosis: MDD
Baseline severity of depression: CDRS‐R
mean (SD) score: intervention = 58.5 (10.5); control = 57.6 (10.4); CGI not
reported at baseline. Length of current episode: (mean, weeks) intervention 14.6 (9.7); placebo 13.7 (7.5) % first episode: intervention 47.9%; placebo 47.9% Comorbidity (intervention): none 7; dysthymia 20; anxiety disorders 32; ADHD 16; ODD/CD 13 Comorbidity (control): none 11; dysthymia 14; anxiety disorders 22; ADHD 13; ODD/CD 16 Location: USA Inclusion criteria: non‐psychotic MDD, single or recurrent; good general medical health; normal intelligence Exclusion criteria: bipolar I and II; psychotic depression; independent sleep‐wake disorder; alcohol and other substance abuse; anorexia nervosa; bulimia nervosa; previous adequate treatment with fluoxetine; at least 1 first‐degree relative with bipolar I disorder Exclusion of suicidality: not specifically stated |
|
| Interventions | Intervention group
Drug: fluoxetine
Dosage: 20
mg
Regimen: taken daily
Length of treatment: 8 weeks (following
acute treatment, participants were given the option to continue treatment
blindly or be treated openly) Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: we used remission CDRS‐R ≤ 28 (they
used responders defined as CGI improvement rating of 1 or
2)
Depressive symptoms: Children's Depression Rating Scale ‐ Revised
(CDRS‐R) Functioning: Children's Global Assessment Scale (C‐GAS) Suicidal behaviour: FDA report; no report of continuous measure Other outcomes: Clinical Global Impressions Scale Improvement (CGI); Children's Depression Inventory (CDI); Beck Depression Inventory (BDI); Weinberg Screening Affective Scale (WSAS); Brief Psychiatry Rating Scale ‐ Children's (BPRS‐C) |
|
| Notes | Additional data were sought and supplied by the authors. Data in the MA for
child, adolescent and total populations taken from paper publication and
these additional data
Child and adolescent data from author. MHRA #
X065
MHRA contacted for additional data some of which was provided Type of data used for remission/response: last‐observation‐carried‐forward |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomisation was by a table of random numbers" pg.1032. |
| Allocation concealment (selection bias) | Unclear risk | "randomisation was conducted by the pharmacy and clinicians who remained blind to assignment until the end of the trial" pg.1032; "pharmacy provided blinded medication" pg.1033; MHRA report stated that an interactive voice response system was used to maintain blinding through follow‐up phase. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "clinicians who remained blind to assignment" pg.1032; "pharmacy provided blinded medication"; "results of blood chemistry levels not provided to clinicians" pg.1033. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "clinicians who remained blind to assignment" pg.1032 "blood chemistry levels were not provided to clinicians" pg.1032. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: 106 Number randomised: fluoxetine: 48; placebo: 48; total: 96 Number started trial: fluoxetine: 48; placebo: 48; total: 96 Number of withdrawals: fluoxetine: 14; placebo: 22; total: 36 Number analysed post‐intervention: fluoxetine: 48; placebo: 48; total: 96 Reasons for dropout: numbers of dropouts and reasons for dropout described in Table 2. There were greater numbers in the placebo group who dropped out due to lack of efficacy (19 versus 7) and greater numbers in the fluoxetine group who dropped out due to side effects (4 versus 1). ITT analysis: all those randomised completed and were included in responder outcome, pg.1033. Statistical methods: last‐observation‐carried‐forward (LOCF) used for all 96 subjects randomised for Child Depression Rating Scale‐Revised (CDRS‐R) outcome. Undertook linear regression and analysis of covariance with baseline measurement as covariate for secondary outcomes. |
| Selective reporting (reporting bias) | High risk | pg.1033 stated that a secondary analysis to explore time to remission was
intended; this was never reported nor were remission rates reported. Overall, adverse outcomes were not reported. |
| Other bias | Unclear risk | Contact: participants were seen weekly for 8 weeks and outcomes were
measured at each visit (pg.1033). No other details given Screening: phone screening followed by 3 independent full evaluations over 3 weeks Placebo lead‐in: 1‐week single‐blind Baseline imbalance: pg.1033 stated there were not differences on any clinical or demographic features except the fluoxetine group had a greater incidence of lifetime anxiety disorders. Other: small trial; Beck Depression Inventory and Childrens Depression Inventory scores combined to give a total score; while stratified for age, no outcome reporting by age was given |
Emslie 2002.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre
Power
calculation: yes
Use of diagnostic criteria (or clear specification of
inclusion criteria): yes
Intervention integrity: not
described
Outcome measures described or validated measures used:
yes
Follow‐up assessment points: post‐assessment
No. crossed
over: none Funded by: Eli Lily |
|
| Participants | Setting of care: outpatients Recruitment: academic hospitals and private research psychiatric clinics as well as newspaper and radio recruitment Mean age (SD): intervention = 12.70 (2.46); control = 12.69 (2.67) Age range: 8 to < 18 years Gender (F:M): intervention = 54:55; control = 54:56 Methods used to diagnose: DSM‐IV Diagnostic Interview for Children and Adolescents (DICA) interview, CDRS‐R ≧ 40 and CGI = 4, 3 independent diagnostic interviews and a 1‐week placebo lead‐in Diagnosis: MDD Baseline severity of depression: CDRS‐R mean (SD) score: intervention = 57.1 (9.9); control = 55.1 (11.8); CGI intervention 4.5 (0.6); placebo 4.4 (0.6) Length of current episode: (mean, weeks) intervention: 60.44; placebo: 61.29 % first episode: intervention 79.8%; placebo 78.2% Comorbidity (intervention): ADHD 16; ODD 17; CD 3 Comorbidity (control): ADHD 15; ODD 17; CD 1 Location: USA Inclusion criteria: outpatients; aged 8 to < 18; primary diagnosis of non‐psychotic major depressive disorder, single or recurrent; depressive symptoms of at least moderate severity; no clinically significant ECG abnormalities; able to keep appointments; normal intelligence as judged by investigator Exclusion criteria: serious illness that was not stabilised; abnormal thyroid function; seizure disorder; bipolar I or II; sleep‐wake disorder; psychotic depression; anorexia nervosa; bulimia nervosa; borderline personality disorder; substance abuse disorder; 1 or more first degree relatives with bipolar I disorder; organic brain diseases; previous failed response to antidepressant medication; prior adequate treatment with fluoxetine; receipt of fluoxetine within 3 months prior to trial entry; regular use of other psychotropic drugs Exclusion of suicidality: serious suicide risk (no further definition) |
|
| Interventions | Intervention group
Drug: fluoxetine
Dosage: 20
mg
Regimen: 1 week 10 mg daily, then 20 mg daily for 8
weeks
Length of treatment: 9 weeks Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: we used remission CDRS‐R ≤ 28 (they
used responders defined as CGI improvement rating of 1 or 2 or at least a
30% reduction on CDRS‐R). Remission was defined as a score of ≤ 28 on the
CDRS‐R.
Depressive symptoms: Children's Depression Rating Scale ‐
Revised (CDRS‐R)
Functioning: Global Assessment of Functioning Scale
(GAF) Suicidal behaviour: FDA report; no report of continuous measure Adverse events Other outcomes: Clinical Global Impressions Scale Severity (CGI‐Severity), Clinical Global Impressions Scale Improvement (CGI‐Improvement), Hamilton Anxiety Rating Scale (HAMA), Beck Depression Inventory (BDI) Children's Depression Inventory (CDI), Montgomery‐Asberg Depression Rating Scale (MADRS) |
|
| Notes | Additional data were sought from authors. They did not have the additional
data but gave a contact in Eli Lily. Eli Lily provided additional data. Data
in the MA from the paper and from additional data supplied by Eli
Lily.
MHRA # HCJE
MHRA contacted for additional data some of
which were provided
All data from paper (Table 3)
Assumed the P
value (that goes with the adjusted treatment effect of 7.1; effect size
0.51; CI 3.3, 10.9) was adjusted but the means presented in table 3 and
provided by the author were probably not. JM calculated SE from SDs (in
STATA file) for depression symptom outcome. Type of data used for remission/response: last‐observation‐carried‐forward |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "computer generated random sequence" pg.1206 |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "both groups took three capsules daily" pg.1209. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No complete statement "clinicians who were blinded to treatment group" pg.1209 plus patient and parent report pg.1206 |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number randomised: fluoxetine: 109; placebo: 110; total: 219 Number started trial: fluoxetine: 109; placebo: 110; total: 219 Number of withdrawals: fluoxetine: 19; placebo: 42; total: 61 Number analysed post‐intervention: fluoxetine: 109; placebo: 110; total: 219 Reasons for dropout: given in Figure 1, pg.1208. More dropouts were due to lack of efficacy in the placebo versus the fluoxetine group (12 versus 5); clinician decision (11 versus 3); lost to follow‐up (7 versus 1) and adverse events (9 versus 5). ITT analysis: "analysis of response and remission included only those patients treat(ed) at least two weeks with trial drug" pg.1208. Statistical methods: last‐observation‐carried‐forward (LCOF); ANOVA for CDRS‐R total score with baseline and each post‐baseline visit included as dependent variables pg.1208 |
| Selective reporting (reporting bias) | High risk | Remission data and overall suicide‐related event data were not reported in the paper but obtained from Eli Lily. |
| Other bias | High risk | Contact: each patient had 6 visits over the 8‐week treatment period with
outcome data collected at each visit. Screening: 3‐week screening period with 3 independent evaluations Placebo lead‐in: 1‐week single‐blind placebo lead‐in Baseline imbalance: report that there were no statistically significant differences in baseline patient characteristics (Table 2) and reasonably balanced for current comorbidities except for conduct disorder Other: none noted |
Emslie 2006.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre
Power
calculation: yes
Use of diagnostic criteria (or clear specification of
inclusion criteria): yes
Intervention integrity: yes ‐ "Every effort
was made to encourage patient compliance with the dosing regimen as per
protocol. All patients were instructed to return their medication bottles
with any unused drug to the investigator when they returned for each visit".
Section 3.6 compliance with trial medication, Final Report
Outcome
measures described or validated measures used: yes
Follow‐up
assessment points: post‐intervention
No. crossed over: none Funded by: GlaxoSmithKline |
|
| Participants | Setting of care: outpatient
Recruitment: no information
Mean
age (SD): intervention = 11.9 (3.00); control = 12.1 (2.95)
Age range:
7 to 17 years
Gender (F:M): intervention = 48:53; control =
47:55
Methods used to diagnose: DSM‐IV, K‐SADS‐PL using 1‐week
screening phase
Diagnosis: MDD
Baseline severity of depression:
CDRS‐R mean (SD) score: intervention = 60.7 (9.37); control = 62.6 (8.96);
CGI intervention 4; CGI placebo 4 Length of current episode: (mean months) intervention: 26.9 (28.62); placebo: 24.9 (27.08) % first episode: intervention 53.5%; placebo 52.9% Comorbidity (intervention): ODD 5; GAD 4; overanxious disorder 3; attention deficit disorder 3; separation anxiety disorder 2; simple phobia 1; PTSD 1; enuresis 1; adjustment disorder with depressed mood 0 Comorbidity (control): ODD 4; GAD 1; overanxious disorder 1; attention deficit disorder 1; separation anxiety disorder 0; simple phobia 0; PTSD 0; enuresis 0; adjustment disorder with depressed mood 1 Location: USA and Canada Inclusion criteria: 7 to 17 years; MDD Exclusion criteria: clinically predominant Axis I disorder other than MDD; history of psychotic episode or disorder; bipolar disorder; mental retardation or pervasive developmental disorder; substance abuse or dependence within 3 months of screening or current positive test on drug screen; epilepsy; ECT within 3 months of screening; lactating or pregnant; sexually active female and not using contraception; requirement of concurrent psychotherapy; clear history of non‐response to SSRIs Exclusion of suicidality: suicidal or homicidal risk (no further definition) |
|
| Interventions | Intervention group
Drug: paroxetine
Dosage: 10 to 50
mg
Regimen: week one 10 mg daily with option to increase up to 10 mg
weekly to a maximum of 50 mg; reduction/tapering over 4 weeks post‐8‐week
treatment
Length of treatment: 8 weeks Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: we used OC remission CDRS‐R ≤ 28 for
total; OC response for child and adolescent data CGI ≤ 2 (they used response
defined as CGI Improvement of 1 or 2)
Depressive symptoms: Children's
Depression Rating Scale ‐ Revised (CDRS‐R) Functioning: Global Assessment of Functioning (GAF) Suicidal behaviour: report of events based on Columbia classification; no report of continuous measure Adverse events: gathered by spontaneous report from patient and family Other outcomes: Clinical Global Impressions Scale Severity (CGI‐Severity); Clinical Global Impressions Scale Improvement (CGI‐Improvement); Kutcher Adolescent Depression Rating Scale (KADS) |
|
| Notes | MHRA #701
MHRA contacted for additional data some of which were
provided
Data in MA taken from GlaxoKline Beecham web‐based report Type of data used for remission/response: last‐observation‐carried‐forward |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "a computer generated randomisation list was generated...stratified by age
subgroup and performed in blocks", GlaxoKlineBeecham report "computer generated randomization list" pg.711 |
| Allocation concealment (selection bias) | Unclear risk | "individual sealed envelopes indicating treatment assigned to each patient at a particular visit were lodged with the investigators/pharmacist....the master randomisation list was held by the sponsor". The investigators were blind to the trial medication except in the instance of a serious adverse event. GlaxoKlineBeecham report |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double blind. GlaxoKlineBeecham report...paroxetine and placebo...identical in size shape and colour...blinding of trial medication was maintained by referring to daily medication dose as dose level", pg.33 |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No statement |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: 305 Number randomised: paroxetine: 104; placebo: 102; total: 206 Number started trial: paroxetine: 104; placebo: 102; total: 206 Number of withdrawals: paroxetine: 34; placebo: 23; total: 57 Number analysed post‐intervention: paroxetine: 101; placebo: 102; total: 203 Reasons for dropout were reported in Figure 1. There were more dropouts in the paroxetine group, including more adverse events, more lost to follow‐up and more who withdrew for any reason. There were more dropouts due to lack of efficacy. ITT analysis: "the Intention‐to‐Treat (ITT) population...was all patients...who received at least one dose of randomised double blind treatment, and for whom at least one valid post‐baseline evaluation was available". GlaxoKlineBeecham pg.55; "All of patients, who were randomised to the treatment phase, received at least one dose of trial medication and had at least one post baseline safety or efficacy assessment, were included in the ITT population". pg.711 Statistical methods: last‐observation‐carried‐forward (LOCF) using the ITT population and observed case (OC) data analysis undertaken. Analysis of variance techniques and logistic regression. Adjusted for age group, gender, baseline scores and presence/absence of psychiatric comorbidity pg.711‐712 |
| Selective reporting (reporting bias) | Low risk | Remission appeared to be a post hoc analysis. |
| Other bias | Unclear risk | Contact: assessments undertaken at week 1, 2, 3, 4, 6 and 8 Screening: 1‐week screening phase Placebo lead‐in: no Baseline imbalance: stated that 2 groups were similar at baseline. Reported in Table 1 |
Emslie 2007.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre Power calculation: yes Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not described Outcome measures described or validated measures used: yes Follow‐up assessment points: post‐intervention No. crossed over: none Funded by: Wyeth Research | |
| Participants | Setting of care: outpatient (consisting of academic and clinical
sites)
Recruitment: no information
Mean age (SD): intervention =
12.2 (2.6); control = 12.3 (2.6)
Age range: 7 to 17 years
Gender
(F:M): intervention = 78:101; control: 83:92
Methods used to diagnose:
DSM‐IV Schedule for Affective Disorder and Schizophrenia for School‐Age
Children‐present and Lifetime version (K_SADS‐PL), at pre‐trial and baseline
a CDRS‐R score of ≥ 40, and CGI‐S score of ≥ 4 and depressive symptoms for
at least 1 month before trial entry. Single‐blind placebo run‐in period of
14 days (+/‐ 3) for trial 1 and 7 days (+/‐3) for trial 2
Diagnosis:
MDD
Baseline severity of depression: CDRS‐R mean (SD) score:
intervention = 56.4 (9.2); control = 55.8 (8.4); CGI intervention 4.5 (0.6);
CGI placebo 4.5 (0.7) Length of current episode: (mean (weeks)) intervention 91.1 (88.2); placebo 92.5 (91.3) % first episode: intervention 84.9%; placebo 86.9% Comorbidity: not stated for either group Location: USA Inclusion criteria: DSM‐IV criteria for MDD, pre‐trial and baseline scores > 40 on the CDRS‐R with ≤ 30 decrease between pretrial and baseline, a CGI‐S score of ≥ 4 at pretrial and baseline, and depressive symptoms for at least 1 month prior to trial entry Exclusion criteria: history of any psychotic disorder or bipolar disorder; MDD with psychotic features, anorexia or bulimia, conduct disorder, panic disorder, or obsessive‐compulsive disorder; first‐degree relative with bipolar disorder; recent drug or alcohol dependence or abuse; mental disorder caused by medical condition Exclusion of suicidality: acute suicidality (no further definition) |
|
| Interventions | Intervention group
Drug: venlafaxine‐extended release
Dosage:
flexible dose based on body weight (37.5 mg/day to 225 mg/day). Mean daily
dose was 109.2 mg/day for adolescents and 80.4 mg/day for
children.
Regimen: delivered once daily for 8 weeks followed by a
taper period of up to 14 days
Length of treatment: 8 weeks Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: we used response > 35% decrease
in CDRS‐R (they used ≥ 35% decrease in CDRS‐R scores, ≥ 50% decrease in
HAM‐D or MADRS or Clinical Global Impression Severity Scale (CGI‐I; Guy 1976). A score of 1 (very much
improved) or 2 (much improved) defining response). Depressive symptoms: Childhood Depression Rating Scale (CDRS‐R; Poznanski 1996) Suicidal behaviour: FDA data; no report of continuous measure Functioning: GAF used but no report of data Adverse events |
|
| Notes | Type of data used for remission/response: last‐observation‐carried‐forward | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Two similarly designed multi‐centre, randomized...eligible subjects were randomly assigned to receive venlafaxine ER or placebo...” pg.480 (Method) |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Two similarly designed multi‐center, randomized, double‐blind...” pg.480 (trial design) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Two similarly designed multi‐center, randomized, double‐blind...” pg.480 (trial design) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: no statement (575 screened in total) Number randomised: venlafaxine ER: 184; placebo: 183; total: 367 Number started trial: venlafaxine ER: 182; placebo: 179; total: 361 Number of withdrawals: venlafaxine ER: 59; placebo: 50; total: 109 Number analysed post‐intervention: venlafaxine ER: 169; placebo: 165; total: 334 Reasons for dropout: dropouts and reasons were reported in Figure 1. There were many more dropouts due to adverse events in the venlafaxine arms; and more in the venlafaxine arm who failed to return. ITT analysis: the primary efficacy analysis population was the intent‐to‐treat population, which included all randomised subjects who had taken at least 1 dose of assigned medication and were evaluated for the primary efficacy outcome measure at baseline and at least once during therapy or within 3 days of the last full dose of treatment, pg.481. Statistical methods: last‐observation‐carried‐forward on therapy evaluation; also did observed case analysis. Parametric 2‐way analysis of covariance with treatment and investigator as main effects and baseline score as covariate |
| Selective reporting (reporting bias) | High risk | Overall adverse events were not reported; standard errors rather than standard deviations were reported; functioning and remission, apparently not trial outcomes |
| Other bias | High risk | Contact: visits on day 4, 7, 14, 21, 28, 42, 56 and at end of taper Screening: yes; described a pre‐trial and baseline visit (unclear what time points these were) Placebo lead‐in: trial 1: single‐blind for 14 days (+/‐3 days); trial 2: single‐blind for 7 days (+/‐3 days) Baseline imbalance: authors reported that there were no statistically significant differences seen in demographic or clinical characteristics. Baseline characteristics not reported by individual trial Other: there were 2 studies reported in the one paper. Trial 1 had a higher dropout. One site was excluded from trial 1. Results were inconsistently reported by trial. |
Emslie 2007 Trial 1.
| Study characteristics | ||
| Methods | See Emslie 2007 | |
| Participants | See Emslie 2007 | |
| Interventions | See Emslie 2007 | |
| Outcomes | See Emslie 2007 | |
| Notes | See Emslie 2007 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Two similarly designed multi‐center, randomized...eligible subjects were randomly assigned to receive venlafaxine ER or placebo...” pg.480 (Method) |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Two similarly designed multi‐centre, randomized, double‐blind...” pg.480 (trial design) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Two similarly designed multi‐center, randomized, double‐blind...” pg.480 (trial design) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: no statement (575 screened in total) Number randomised: venlafaxine ER: 184; placebo: 183; total: 367 Number started trial: venlafaxine ER: 182; placebo: 179; total: 361 Number of withdrawals: venlafaxine ER: 59; placebo: 50; total: 109 Number analysed post‐intervention: venlafaxine ER: 169; placebo: 165; total: 334 Reasons for dropout: dropouts and reasons were reported in Figure 1. There were many more dropouts due to adverse events in the venlafaxine arms; and more in the venlafaxine arm who failed to return. ITT analysis: the primary efficacy analysis population was the intent‐to‐treat population, which included all randomised subjects who had taken at least 1 dose of assigned medication and were evaluated for the primary efficacy outcome measure at baseline and at least once during therapy or within 3 days of the last full dose of treatment, pg.481. Statistical methods: last‐observation‐carried‐forward on therapy evaluation; also did observed case analysis. Parametric 2‐way analysis of covariance with treatment and investigator as main effects and baseline score as covariate |
| Selective reporting (reporting bias) | High risk | Overall adverse events were not reported; standard errors rather than standard deviations were reported; functioning, remission, apparently not trial outcomes |
| Other bias | High risk | Contact: visits on day 4, 7, 14, 21, 28, 42, 56 and at end of taper Screening: not reported Placebo lead‐in: trial 1: single‐blind for 14 days (+/‐3 days); trial 2: single‐blind for 7 days (+/‐3 days) Baseline imbalance: authors reported that there were no statistically significant differences seen in demographic or clinical characteristics. Baseline characteristics not reported by individual trial Other: there were 2 studies reported in the one paper. Trial 1 had a higher dropout. One site was excluded from trial 1. Results were inconsistently reported by trial. |
Emslie 2007 Trial 2.
| Study characteristics | ||
| Methods | See Emslie 2007 | |
| Participants | See Emslie 2007 | |
| Interventions | See Emslie 2007 | |
| Outcomes | See Emslie 2007 | |
| Notes | See Emslie 2007 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Two similarly designed multi‐centre, randomized...eligible subjects were randomly assigned to receive venlafaxine ER or placebo...” pg.480 (Method) |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Two similarly designed multi‐center, randomized, double‐blind...” pg.480 (trial design) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Two similarly designed multi‐centre, randomized, double‐blind...” pg.480 (trial design) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: no statement (575 screened in total) Number randomised: venlafaxine ER: 184; placebo: 183; total: 367 Number started trial: venlafaxine ER: 182; placebo: 179; total: 361 Number of withdrawals: venlafaxine ER: 59; placebo: 50; total: 109 Number analysed post‐intervention: venlafaxine ER: 169; placebo: 165; total: 334 Reasons for dropout: dropouts and reasons were reported in Figure 1. There were many more dropouts due to adverse events in the venlafaxine arms; and more in the venlafaxine arm who failed to return. ITT analysis: the primary efficacy analysis population was the intent‐to‐treat population, which included all randomised subjects who had taken at least 1 dose of assigned medication and were evaluated for the primary efficacy outcome measure at baseline and at least once during therapy or within 3 days of the last full dose of treatment, pg.481 Statistical methods: last‐observation‐carried‐forward on therapy evaluation; also did observed case analysis. Parametric 2‐way analysis of covariance with treatment and investigator as main effects and baseline score as covariate |
| Selective reporting (reporting bias) | High risk | Overall adverse events were not reported; standard errors rather than standard deviations were reported; functioning, remission, apparently not trial outcomes |
| Other bias | High risk | Contact: visits on day 4, 7, 14, 21, 28, 42, 56 and at end of taper Screening: not reported Placebo lead‐in: trial 1: single‐blind for 14 days (+/‐3 days); trial 2: single‐blind for 7 days (+/‐3 days) Baseline imbalance: authors reported that there were no statistically significant differences seen in demographic or clinical characteristics. Baseline characteristics not reported by individual trial Other: there were 2 studies reported in the 1 paper. Trial 1 had a higher dropout. One site was excluded from trial 1. Results were inconsistently reported by trial. |
Emslie 2009.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre Power calculation: yes Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not described Outcome measures described or validated measures used: yes Follow‐up assessment points: post‐intervention No. crossed over: none Funded by: Forest laboratories | |
| Participants | Setting of care: outpatient
Recruitment: no information
Mean
age (SD): intervention = 14.7 (1.6); control = 14.5 (1.5)
Age range:
12 to 17 years
Gender (F:M): intervention = 92:63; control =
92:65
Methods used to diagnose: DSM‐IV with duration of current
episode at least 12 weeks at screening confirmed by K‐SADS. At screening and
baseline a CDRS‐R score of ≥ 45 and a CGI‐S score of ≥ 4. Screening period
of 2 weeks, and a single‐blind placebo run‐in of 1 week during 2nd week of
screening
Diagnosis: MDD
Baseline severity of depression: CDRS‐R
mean (SD) score: intervention = 56.0 (0.66); control = 57.6 (0.66); CGI
intervention 4.6 (0.05); CGI placebo 4.4 (0.04) Length of current episode: (mean (months)) intervention 15.7 (17.4); placebo 16.5 (15.4) % first episode: intervention 70.3%; placebo 72% Comorbidity (intervention): previous and/or ongoing secondary psychiatric disorder 16.6% Comorbidity (control): previous and/or ongoing secondary psychiatric disorder 12.9% Location: USA Inclusion criteria: current DSM‐IV‐defined MDD episode of at least 12 weeks, CDRS‐R score ≥ 45 at screening and baseline visits, CGI‐S score of ≥ 4 and a score of ≥ 80 on the Kaufman Brief Intelligence Test Exclusion criteria: a principal diagnosis meeting DSM‐IV criteria for an Axis 1 disorder other than MDD or who currently met DSM‐IV criteria at screening for attention‐deficit/hyperactivity disorder, obsessive‐compulsive disorder, post‐traumatic stress disorder, bipolar disorder, pervasive developmental disorder, mental retardation, conduct disorder, or oppositional defiant disorder; or who had any psychotic features or a history of any psychotic disorder; or any personality disorder that, as judged by the investigator, would interfere with participation in the trial; a history of manic, or hypomanic episodes, a history of bulimia anorexia nervosa or substance abuse or dependence within the last year Exclusion of suicidality: patients considered a suicide risk by the investigator, including those who had active suicidal ideation, had made a suicide attempt, or had ever been hospitalised because of a suicide attempt |
|
| Interventions | Intervention group
Drug: escitalopram
Dosage: 10 to 20
mg/day
Regimen: daily
Length of treatment: 8 weeks Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: we used remission CDRS‐R ≤ 28 (they
used the Clinical Global Impressions‐Improvement Scale (CGI‐I). A score of 1
(very much improved) or 2 (much improved) or CDRS‐R reduction of ≥ 40%
defined response (remission CDRS‐R ≤ 28)) Depressive symptoms: the Childrens Depression Rating Scale (CDRS‐R; Poznanski 1996) Functioning: Children’s Global Assessment Scale (C‐GAS; Shaffer 1985) Suicidal behaviours: Modified Columbia Suicide Severity Rating Scale (MC‐SSRS) report of suicidal ideation, presence and type of suicidal behaviour since last visit; continuous measure using the Suicidal Ideation Questionnaire‐Junior High School Version (SIQ‐JR; Reynolds 1987) Adverse events: either spontaneously reported by patient or parent, or noted by investigator |
|
| Notes | Type of data used for remission/response: last‐observation‐carried‐forward | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “...a prospective, randomized, double‐blind placebo controlled trial...” pg.721 (Abstract) |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | “This was a randomized, double‐blind, placebo controlled trial...” pg.722 (Method). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | “Evaluations were scheduled at the end of...weeks of double‐blind treatment” pg.723 (Trial design). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: not stated Number randomised: escitalopram: 158; placebo: 158; total: 316 Number started trial: escitalopram: 155; placebo: 157; total: 312 Number of withdrawals: escitalopram: 32; placebo: 25; total: 57 Number analysed post‐intervention: escitalopram: 154; placebo: 157; total: 311 Reasons for dropout: described in Figure 1 and appeared relatively well balanced, with slightly more dropouts due to adverse events in the escitalopram group. Authors reported no statistically significant differences. ITT analysis: efficacy analyses were performed on the intent‐to‐treat (ITT) population, which included all patients in the safety population who had at least 1 post‐baseline CDRS‐R assessment pg.723. Statistical methods: reported results are LOCF unless otherwise specified, pg.724 (Statistical Methods); baseline imbalance was tested. 2‐way analysis of variance model with treatment and trial centre as factors for continuous variables and a Cochrane‐Mantel Haenzel test controlling for trial centre for categorical variables. ANCOVA with treatment group and trial centre as factors and baseline scores as covariates. Logistic regression with treatment group and baseline scores as explanatory variables. Dichotomous data. Also did mixed modelling for repeated measures for primary efficacy variable |
| Selective reporting (reporting bias) | High risk | 2 measures of response were reported (one significant and one not) |
| Other bias | Low risk | Contact: evaluation at the end of week 1, 2, 3, 4, 6 and 8. On page 728,
authors explained the placebo response as being due to the "extensive
contact". Psychotherapy was not allowed. Screening: 2‐week screening with 2 visits Placebo lead‐in: 1‐week single‐blind Baseline imbalance: authors reported no statistically significant differences, although there appeared to be a difference in the baseline CGI‐S score. Other: none noted |
Emslie 2014.
| Study characteristics | ||
| Methods | Trial design: Randomised, double‐blind, placebo‐controlled. Parallel
group. Power calculation: Sample of 111 per group was considered sufficient to demonstrate a 5‐point difference in the primary endpoint between the desvenlafaxine and placebo groups at a significance level of 5% and a power of 85%, assuming a pooled standard deviation of 12, and that no more than 5% of randomised subjects would fail to qualify for the primary analysis. Use of diagnostic criteria (or clear specification of inclusion criteria): Yes. DSM‐IV‐TR criteria for MDD as the primary diagnosis, supported by the Schedule for Affective Disorders and Schizophrenia–Present and Lifetime Version (K‐SADS‐PL) Intervention integrity: Not described Outcome measures described or validated measures used: Yes. CDRS‐R, CGI‐S, CGI‐I, C‐SSRS. Follow‐up assessment points: Weeks 1, 2, 3, 4, 6, 8, and/or at early termination in the double‐blind phase No. crossed: None Funded by: Pfizer Inc. |
|
| Participants | Setting of care: Outpatient Recruitment: Not described CHILDREN Total mean age (SD): 9.4 (1.3) Desvenlafaxine = 9.3 (1.4) Fluoxetine = 9.6 (1.3) Control = 9.4 (1.3) Age range: 7–11 years Gender (F:M): 57:73 Desvenlafaxine = 20:23 Fluoxetine = 14:31 Control = 23:19 ADOLESCENTS Total mean age (SD): 14.8 (1.5) Desvenlafaxine = 15.0 (1.5) Fluoxetine = 14.7 (1.6) Control = 14.6 (1.5) Age range: 12–17 years Gender (F:M): 127:82 Desvenlafaxine = 43:29 Fluoxetine = 43:24 Control = 41:29 Methods used to diagnose: DSM‐IV‐TR criteria for MDD as the primary diagnosis, with depressive symptoms of at least moderate severity for at least 30 days. The MDD diagnosis was confirmed by a psychiatrist at the study site and supported by the K‐SADS‐PL. Diagnosis: MDD Baseline severity of depression: CDRS‐R total score, Mean (SD) CHILDREN Total = 56.1 (9.4) Desvenlafaxine = 56.4 (10.9) Fluoxetine = 55.0 (8.7) Control = 57.0 (8.6) ADOLESCENTS Total = 56.8 (8.7) Desvenlafaxine = 56.3 (8.8) Fluoxetine = 57.0 (8.1) Control = 57.1 (9.1) Length of current (most recent) episode: median (range), months CHILDREN Total = 7 (1–71) Desvenlafaxine = 8 (1–71) Fluoxetine = 6 (1–42) Control = 11 (1–57) ADOLESCENTS Total = 7 (1–96) Desvenlafaxine = 7 (1–61) Fluoxetine = 7 (1–96) Control = 8 (1–69) % first episode: Not reported Comorbidity (intervention): Not reported, however comorbid primary psychiatric condition other than MDD was an exclusion criterion. Comorbidity (control): Not reported, however comorbid primary psychiatric condition other than MDD was an exclusion criterion. Location: United States (35 sites) and Mexico (2 sites) Inclusion criteria: · Outpatients aged 7 to < 18 years who weighed at least 20 kg at the screening and baseline visits, who met DSM‐IV‐TR criteria for MDD as the primary diagnosis, had depressive symptoms of at least moderate severity for at least 30 days (CDRS‐R total score > 40 and CGI‐S score of ≥ 4 at screening and baseline), and would not require concomitant psychotherapy Exclusion criteria: · Not being in a generally healthy medical condition as determined by the investigator, having any major acute illness within 90 days before the screening visit, a history of seizure disorder other than a single childhood febrile seizure, a history or current evidence of gastrointestinal disease or surgery known to interfere with absorption or excretion of drugs, or a severe acute or chronic medical or psychiatric condition or laboratory abnormality that may have increased patient risk or interfered with the interpretation of study results · History or presence of major depressive disorder (MDD) with psychotic features or any psychotic disorder, bipolar disorder (or first‐degree relative with bipolar disorder) or manic episodes; current psychoactive substance or alcohol abuse or dependence, post‐traumatic stress disorder, obsessive‐compulsive disorder; current generalised anxiety disorder, panic disorder, social anxiety disorder, or attention‐deficit/hyperactivity disorder considered a primary diagnosis; history or presence of borderline personality disorder, clinically important personality disorder, or history of recurrent, intentional self‐injurious behaviour; or depression associated with the presence of a mental disorder due to a general medical condition, history or presence of anorexia or bulimia · History or current evidence of suicidal behaviour or suicidal ideation associated with actual intent and/or plan at any time in their lifetime based on clinical judgement or Columbia‐Suicide Severity Rating Scale responses at the screening or baseline visit, or first‐degree relative who had committed suicide · Clinically important abnormalities on physical examination, electrocardiography, laboratory tests, or urine drug screen; total bilirubin 2.0 mg/dL (34.2 lmol/L) or greater (unless there was documented history of Gilbert’s syndrome); alanine aminotransferase, aspartate aminotransferase, or alkaline phosphatase three or more times upper limit of normal; prolactin level 40 ng/mL (40 lg/L) or greater; or prerandomisation blood pressure elevation in the 95th percentile or greater for gender, age, and height · Known allergy or hypersensitivity or previous adverse event related to venlafaxine, desvenlafaxine, or fluoxetine, or history of failure to respond to an adequate course of treatment for MDD with fluoxetine, venlafaxine, or desvenlafaxine · History of electroconvulsive therapy · Patients who were pregnant, breastfeeding, or had a positive serum beta human chorionic gonadotropin pregnancy test result · Immediate family members of investigational site staff or Pfizer Inc. employees directly involved in the conduct of the trial, or patients who had a parent or legal guardian who was responsible for another individual enrolled in the study Exclusion of suicidality: History or current evidence of suicidal behaviour or suicidal ideation associated with actual intent and/or plan at any time in their lifetime based on clinical judgement or Columbia‐Suicide Severity Rating Scale responses at the screening or baseline visit, or first‐degree relative who had committed suicide |
|
| Interventions | Intervention group
Drug: Desvenlafaxine
Dosage: 20 to < 35
kg: 25 mg/d; 35 to < 70 kg: 35 mg/d; ≥ 70 kg: 50 mg/d.
Regimen:
Daily
Length of treatment: 8 weeks Intervention group Drug: fluoxetine Dosage: 20 mg/d Regimen: Daily Length of treatment: 8 weeks Control group: Matching placebo capsules administered orally, once daily for 8 weeks (treatment phase), followed by placebo capsules administered once daily as appropriate for 1 week (taper/transition phase) |
|
| Outcomes | Definition and assessment of response: Primary efficacy outcome was change
from baseline CDRS‐R total score at week 8. Secondary efficacy outcome was
change from baseline CGI‐S score; change from baseline CGI‐I score; and
CGI‐I response score ≤ 2 at each visit. Depressive symptoms: Children’s Depression Rating Scale–Revised (CDRS‐R) Functioning: Not assessed Suicidal behaviour: Columbia‐Suicide Severity Rating Scale (C‐SSRS) Other measures: Clinical Global Impressions–Severity (CGI‐S), Clinical Global Impressions–Improvement (CGI‐I) |
|
| Notes | Type of data used for remission/response: intention‐to‐treat | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation and allocation was made via interactive voice response system
(IVRS). Comment: Likely implemented adequate random sequence generation given use of IVRS, no baseline imbalance of major concern, however no statement reported |
| Allocation concealment (selection bias) | Low risk | Randomisation and allocation were made via interactive voice response system (IVRS). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Stated that the study was “double blind”. Placebo delivered in capsules “identical in appearance, color, taste, and smell to study drug” (NCT00849693 document). No information about dropouts due to adverse events |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Potential for unblinding due to TEAEs; significantly more duloxetine 60 mg treated patients experienced at least one TEAE compared with placebo and duloxetine 30 mg treated patients |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: Unclear. 635 were screened and 463 were randomised. Number randomised: 463; duloxetine 60 mg: 105; duloxetine 30 mg: 116; fluoxetine 20 mg: 117; placebo: 122 Number started trial: 463; duloxetine 60 mg: 108; duloxetine 30 mg: 116; fluoxetine 20 mg: 117; placebo: 122 Number of withdrawals: 138; duloxetine 60 mg: 33; duloxetine 30 mg: 35; fluoxetine 20 mg: 33; placebo: 37 Number analysed post‐intervention: 448; duloxetine 60 mg: 105; duloxetine 30 mg: 114; fluoxetine 20 mg: 112; placebo: 117 ITT analysis:; duloxetine 60 mg = 108; duloxetine 30 mg = 116; fluoxetine 20 mg = 117; placebo = 122 ITT analysis: Included the randomised patients with both a baseline and at least 1 post‐baseline value from Visit 3 through 8 Duloxetine 60 mg Adverse events: 12 Lack of efficacy: 1 Physician decision: 2 Parent/caregiver decision: 7 Patient decision: 5 Follow‐up: 5 Protocol violation: 1 Sponsor decision: 0 Duloxetine 30 mg Adverse events: 7 Lack of efficacy: 3 Physician decision: 1 Parent/caregiver decision: 6 Patient decision: 5 Follow‐up: 8 Protocol violation: 5 Sponsor decision: 0 Fluoxetine 20 mg Adverse events: 6 Lack of efficacy: 1 Physician decision: 2 Parent/caregiver decision: 7 Patient decision: 3 Follow‐up: 11 Protocol violation: 2 Sponsor decision: 1 Placebo Adverse events: 4 Lack of efficacy: 2 Physician decision: 1 Parent/caregiver decision: 7 Patient decision: 8 Follow‐up: 9 Protocol violation: 6 Sponsor decision: 0 Statistical analysis: LOCF. The protocol‐specified primary analytic approach for assessing mean changes for all efficacy measures was the recommended restricted maximum likelihood (REML)‐based mixed effects model repeated measures (MMRM) approach using all the longitudinal observations at each post‐baseline visit. The MMRM model included the fixed categorical effects of treatment, pooled investigative site, visit, treatment‐by‐visit interaction, age category (children 7–11, adolescents 12–17), and age category‐by‐visit interaction, as well as the continuous, fixed covariates of the baseline value being analysed and the baseline value of the variable being analysed‐by visit interaction. The baseline value of the variable being analysed and baseline‐by‐visit interaction was included to account for the differing influence over time of the baseline score on the post‐baseline scores. An unstructured covariance structure was used to model the within‐patient errors. A Kenward–Roger correction (Kenward 1997) was used to estimate denominator degrees of freedom. Significance tests were based on least‐squares means (LS means) using a two sided a = 0.05. Additional analyses of continuous efficacy and safety measures were also performed using an analysis of variance (ANOVA) or an analysis of covariance (ANCOVA) model. When an ANOVA model was used, the model contained the main effects of treatment and pooled investigative site. An analysis of covariance (ANCOVA) model, in general, referred to the ANOVA model with baseline values and age category (children 7–11, adolescents 12–17) added as covariates. Type III sum‐of‐squares for the LS mean was used for the statistical comparison of main effects using ANOVA or ANCOVA. Statistical inference for ANOVA or ANCOVA interaction terms was based on type II sum‐of‐squares for the LS mean. A last‐observation‐carried‐forward (LOCF) method was used for these analyses. |
| Selective reporting (reporting bias) | Unclear risk | All outcomes in methods reported? Yes, except that CDRS‐R subscale scores
were not reported as secondary outcomes, as per trial registry. CDRS‐R total
scores were reported, as per trial registry. Data available for use in MA? Unclear if dataset was available. A copy is held by the Center for Drug Evaluation and Research. Access to trial protocol? Trial registry only and no protocol document (NCT00849693 document) with trial registry analysis as well as FDA Statistical Review_F1J‐MC‐HMCK & F1J‐MC‐HMCL synopsis of results and EUCTR2017‐001598‐18 synopsis of results |
| Other bias | Unclear risk | Contact: Clinic visits were scheduled weekly during the screening period,
and at weeks 1, 2, 4, 7, and 10 during the placebo‐controlled acute
treatment period. Screening: 2–4 week screening period An MDD diagnosis, supported by the Mini International Neurospychiatric Interview for children and adolescents (MINI‐KID) and was conducted by two independent evaluators, with at least one evaluator being a psychiatrist. Patients were required to be medically stable based on the physical examination, laboratory tests, and electrocardiogram (ECG) completed at the screening visits. Female patients were required to have a negative serum pregnancy test at screening. Placebo lead‐in: No Baseline imbalance: With the exception of the proportion of males to females in the duloxetine 30 mg treatment arm, there were no significant between‐group differences in baseline demographics or psychiatric profile. |
Keller 2001.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre
Power
calculation: yes
Use of diagnostic criteria (or clear specification of
inclusion criteria): yes
Intervention integrity: yes ‐ "Compliance
with taking trial medication was assessed by recording the amount of drug
dispensed, taken, and returned in the CRF for each patient". Section 3.6
Final report.
Outcome measures described or validated measures used:
yes
Follow‐up assessment points: post‐intervention
No. crossed
over: none Funded by: GlaxoSmithKline |
|
| Participants | Setting of care: outpatient
Recruitment: no information
Mean
age (SD): intervention = 14.8 (1.6); control = 15.1 (1.6)
Age range:
12 to 18 years
Gender (F:M): intervention = 58:35; control:
57:30
Methods used to diagnose: DSM‐IV diagnosis confirmed by K‐SADS‐L
and current duration of episode at least 8 weeks, a score of ≥ 12 on the
HAM‐D, a CGAS score of ≥ 60; screening period of 7 to 14 days, no placebo
run‐in phase
Diagnosis: MDD
Baseline severity of depression:
K‐SADS 9‐item mean depression score; intervention = 28.25; control = 28.84.
C‐GAS mean (SD) score: intervention = 42.7; control = 42.8; CGI not
reported Length of current episode: (mean (months)) intervention: 14 (18); placebo: 13 (17) % first episode: intervention 81%; placebo 77% Comorbidity (intervention): any diagnosis 41; anxiety disorder 19; externalising disorder 25 Comorbidity (control): any diagnosis 50; anxiety disorder 26; externalising disorder 26 Location: USA and Canada Inclusion criteria: MDD of at least 8 weeks duration; at least 80 on the Peabody Picture Completion task; medically healthy Exclusion criteria: bipolar disorder; schizoaffective disorder; eating disorder; alcohol or substance abuse disorder; Obsessive Compulsive Disorder; autism/pervasive developmental disorder; organic brain disorder; PTSD within 12 months of trial entry; current psychotropic drug use; trial of antidepressant medication within 6 months of trial entry Exclusion of suicidality: current suicidal ideation with intent or specific plan; history of suicide attempt by drug overdose |
|
| Interventions | Intervention group
Drug: paroxetine
Dosage: 20 to 40
mg
Regimen: 20 mg daily in week 1 to 4 with optional increase to 30 mg
in week 5 and 40 mg in week 6
Length of treatment: 8 weeks Control group: placebo pill Comparison group: imipramine (gradual upward titration from 200 to 300 mg) |
|
| Outcomes | Definition and assessment of response: we used OC response HAM‐D ≦ 8 or ≥
50% reduction in baseline HAM‐D (they used responders defined as = 8 or less
on HAM‐D or at least 50% decrease from baseline)
Depressive symptoms:
depression items from K‐SADS‐L Functioning: Autonomous Function Checklist Suicidal behaviours: FDA data; no report of continuous measure Adverse events Other measures: HAM‐D; Clinical Global Impressions Scale Improvement (CGI ‐ Improvement); Self Perception Profile; Sickness Impact Scale |
|
| Notes | Addtional data were sought from the authors. They did not have the data
required but provided a contact from GlaxoSmithKline who responded to inform
us of the trial information now published on the web.
MHRA #
329
MHRA contacted for additional data some of which were
provided
Data in MA taken from GlaxoSmithKline Beecham web‐based
report
GlaxoSmithKline web publication Type of data used for remission/response: last‐observation‐carried‐forward |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "computer generated list" pg.764 |
| Allocation concealment (selection bias) | Unclear risk | No statement. GlaxoSmithKline Beecham stated randomisation codes were stored at SB clinical safety department, pg.35. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Tablets overencapsulated in matching supro B locking capsules to preserve medication blinding"; "number of capsules...identical for each...group during forced titration", pg.764 |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No statement |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: 275 Number randomised: paroxetine: 93 placebo: 87 imipramine: 95 total: 275 Number started trial: paroxetine: 93 placebo: 87 imipramine: 95 total: 275 Number of withdrawals: paroxetine: 26 placebo: 21 imipramine: 38 total: 85 Number analysed post‐intervention: paroxetine: 67 placebo: 66 imipramine: 57 total: 190 Reasons for dropout: some information was provided (pg.765) about dropouts, but only about premature trial discontinuation due to adverse effects, which was 6.9% in the placebo group and 9.7% in the placebo group (P = 0.50) and described protocol violation as the most common reason for withdrawal in the placebo group (pg.765) ITT analysis: "efficacy analysis based on patients who were randomised and had at least one post baseline efficacy analysis evaluation" pg.76 Statistical methods: both last‐observation‐carried‐forward (LOCF) and observed case (OC) data analysis undertaken. 2‐factor analysis of variance using general linear models with terms for treatment and investigator. Logistic analysis implemented in the categorical modelling procedure including effects for investigator and treatment |
| Selective reporting (reporting bias) | High risk | Mulitple measurement of depression outcome (HAM‐D, HAM‐D depressed mood item, depression item of K‐SADS‐L and 9‐item depression subscale of the K‐SADS). Response data given as percentages. In a letter to the editor Jureidini 2003 stated that the definition of response was changed so that a significant result could be reported. Overall adverse event rate not described. Kennard 2006 (TADS) stated that Keller had remission definition of HAM‐D < 8, although Keller described this in the Methods section as 'response'. |
| Other bias | High risk | Contact: participants were seen weekly and undertook assessments at each
visit. Supportive case management was provided to all subjects at each visit
(interpersonal or cognitive behavioural psychotherapeutic interventions were
strictly prohibited) (pg.764). Screening: 7‐ to 14‐day screening period with no detail about number of assessments during this screening phase Placebo lead‐in: no Baseline imbalance: treatment groups stated to be similar at baseline for demographic and psychiatric profile (pg.765). These features were described in Table 1. |
Mirtazapine Trial 1.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre
Power
calculation: not stated
Use of diagnostic criteria (or clear
specification of inclusion criteria): yes
Intervention integrity: yes
‐ "Plasma samples, for the purpose of measuring mirtazapine, (Org 3770)
concentrations, were to be collected on trial Days 28 and 56 (or the
subject’s final day of treatment)" pg.4. Company trial report
Outcome
measures described or validated measures used: yes
Follow‐up
assessment points: post‐intervention
No. crossed over: none Funded by: Organon International |
|
| Participants | Setting of care: outpatients
Recruitment: through clinical practice
of investigators, referrals and/or advertisements for volunteers
Mean
age (SD): intervention = 12.3; control = 12.4
Age range: 8 to 18
years
Gender (F:M): intervention = 39:43; control: 25:19
Methods
used to diagnose: DSM‐IV diagnosis confirmed by K‐SADS‐L and baseline score
of ≥ 15 on 1st 17 items of HAM‐D (21‐item), a C‐GAS score of < 70; CDRS‐R
≥ 40; screening period not stated
Diagnosis: MDD
Baseline
severity of depression: CDRS‐R mean (SD) score: intervention = 50.93;
control = 51.93 Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Comorbidity (control): not stated Location: USA Inclusion criteria: current episode of MDD (as defined by DSM‐IV criteria, with a primary diagnosis of major depressive disorder on the K‐SADS PL (Schedule for Affective Disorders and Schizophrenia ‐ Present and Lifetime)) Baseline score of >15 on the 1st 17 items of the Hamilton Scale for Depression, 21 items (HAM‐D 21), < 70 on the Children's Global Assessment Scale (C‐GAS), and a Children's Depression Rating Scale‐Revised (CDRS‐R) score of ≥ 40 Exclusion criteria: concurrent psychiatric diagnosis of anorexia or bulimia, past history of eating disorder, concurrent diagnosis of obsessive compulsive disorder or schizophrenia, bipolar disorder (I or II) or parental history of bipolar I disorder, drug/and or alcohol abuse Exclusion of suicidality: serious suicide attempt during the current major depressive episode, or any previous suicide attempt resulting in hospitalisation |
|
| Interventions | Intervention group
Drug: mirtazapine
Dosage: 15 to 45
mg
Regimen: starting dose 15 mg with increase to 30 to 45 mg in 15 mg
increments during subsequent weeks (to 28 days)
Length of treatment: 8
weeks Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: not stated
Depressive
symptoms: CDRS‐R clinician rating; HAM‐D 21 self‐rating Functioning: C‐GAS used but no report of data Suicidal behaviours: events reported as adverse events; no report of continuous measure Adverse events Other measures: Clinical Global Impressions (CGI), Self Report Childhood Anxiety Related Disorder (SCARED), Connors' Global Index (Parent and Teacher Versions) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No statement |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | MHRA report stated double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | MHRA report stated double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number eligible: not stated Number randomised: mirtazapine: 82; placebo: 44; total: 126 Number started trial: mirtazapine: 82; placebo: 44; total: 126 Number of withdrawals: mirtazapine: 13; placebo: 9; total 22 Number analysed post‐intervention: mirtazapine: 82; placebo: 44; total: 126 Reasons for dropout: MHRA reported dropouts across the 2 mirtazapine trials: 9 (5.3%) patients discontinued due to an adverse event in the mirtazapine group compared with 3 (3.4%) in the placebo‐treated group. The most common adverse treated event leading to discontinuation in the acute phase in the mirtazapine‐treated group was weight gain. Weight gain (31.8% versus 3.4%), somnolence (38.8% versus 6.8%), headache (35% versus 23%), fatigue (19.4% versus 11.4%), increased appetite (8.8% versus 2.3%), urticaria (11.8% versus 6.8%) and hypertriglyceridaemia (2.9% versus 0%) were reported more often for mirtazapine‐treated patients than by placebo‐treated patients. ITT analysis: stated ITT using LOCF was used Statistical methods: not stated |
| Selective reporting (reporting bias) | High risk | Only 1 outcome reported in MHRA report; Rapporteurs report gave safety outcomes in addition. |
| Other bias | High risk | Contact: weekly visits (week 5 and 7 optional); psychotherapy could not be
started during the trial, but ‘supportive care’ as defined in the protocol
was permitted. Screening: unclear Placebo lead‐in: no Baseline imbalance: data not reported Stated it was initially 2 trials that were amalgamated a few months after trial initiation |
Mirtazapine Trial 1 & 2.
| Study characteristics | ||
| Methods | Information provided separately for each trial | |
| Participants | Information provided separately for each trial | |
| Interventions | Information provided separately for each trial | |
| Outcomes | Information provided separately for each trial | |
| Notes | Information provided separately for each trial | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No statement |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | MHRA report stated double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | MHRA report stated double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Reasons for dropout: MHRA reported dropouts across the 2 mirtazapine
trials: 9 (5.3%) patients discontinued due to an adverse event in the
mirtazapine group compared with 3 (3.4%) in the placebo‐treated group. The
most common adverse treated event leading to discontinuation in the acute
phase in the mirtazapine‐treated group was weight gain.
Weight gain
(31.8 versus 3.4%), somnolence (38.8% versus 6.8%), headache (35% versus
23%), fatigue (19.4% versus 11.4%), increased appetite (8.8% versus 2.3%),
urticaria (11.8 versus 6.8%) and hypertriglyceridaemia (2.9 versus 0%) were
reported more often for mirtazapine‐treated patients than by placebo‐treated
patients. ITT analysis: stated ITT using LOCF was used Statistical methods: not stated |
| Selective reporting (reporting bias) | High risk | Only 1 outcome reported in MHRA report; Rapporteurs report gave safety outcomes in addition. |
| Other bias | High risk | Contact: psychotherapy could not be started during the trial, but
‘supportive care’ as defined in the protocol was permitted. Screening: unclear Placebo lead‐in: unclear Baseline imbalance: data not reported Stated it was initially 2 trials that were amalgamated a few months after trial initiation |
Mirtazapine Trial 2.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre
Power
calculation: not stated
Use of diagnostic criteria (or clear
specification of inclusion criteria): yes
Intervention integrity: yes
‐ "Plasma samples, for the purpose of measuring mirtazapine, (Org 3770)
concentrations, were to be collected on trial Days 28 and 56 (or the
subject’s final day of treatment)". Pg.4. Company trial report
Outcome
measures described or validated measures used: yes
Follow‐up
assessment points: post‐intervention
No. crossed over: none Funded by: Organon International |
|
| Participants | Setting of care: outpatients
Recruitment: through clinical practice
of investigators, referrals and/or advertisements for volunteers
Mean
age (SD): intervention = 11.9; control = 12.3
Age range: 8 to 18
years
Gender (F:M): intervention = 46:42; control: 24:21
Methods
used to diagnose: DSM‐IV diagnosis confirmed by K‐SADS‐L and baseline score
of ≥ 15 on 1st 17 items of HAM‐D (21 item), a C‐GAS score of < 70; CDRS‐R
≥ 40; screening period not stated
Diagnosis: MDD
Baseline
severity of depression: CDRS‐R mean (SD) score: intervention = 48.87;
control = 47.57 Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Comorbidity (control): not stated Location: USA Inclusion criteria: current episode of MDD (as defined by DSM‐IV criteria, with a primary diagnosis of major depressive disorder on the K‐SADS P‐L (Schedule for Affective Disorders and Schizophrenia ‐ Present and Lifetime)). Baseline score of > 15 on the 1st 17 items of the Hamilton Scale for Depression, 21 items (HAM‐D 21), < 70 on the Children's Global Assessment Scale (C‐GAS), and a Children's Depression Rating Scale‐Revised (CDRS‐R) score of ≥ 40 Exclusion criteria: serious suicide attempt during the current major depressive episode, or any previous suicide attempt resulting in hospitalisation; concurrent psychiatric diagnosis of anorexia or bulimia, past history of eating disorder, concurrent diagnosis of obsessive compulsive disorder or schizophrenia, bipolar disorder (I or II) or parental history of bipolar I disorder |
|
| Interventions | Intervention group
Drug: mirtazapine
Dosage: 15 to 45
mg
Regimen: starting dose 15 mg with increase to 30 to 45 mg in 15 mg
increments during subsequent weeks (to 28 days)
Length of treatment: 8
weeks Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: not stated
Depressive
symptoms: CDRS‐R clinician rating; HAM‐D 21 self‐rating Functioning: C‐GAS used but no report of data Suicidal behaviours: events reported as adverse events; no report of continuous measure Adverse events Other measures: Clinical Global Impressions (CGI), Self Report Childhood Anxiety Related Disorder (SCARED), Connors' Global Index (Parent and Teacher Versions) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No statement |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | MHRA stated double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | MHRA stated double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number eligible: not stated Number randomised: mirtazapine: 88; placebo: 45; total: 133 Number started trial: mirtazapine: 88; placebo: 45 total: 133 Number of withdrawals: mirtazapine: 19; placebo: 8; total: 27 Number analysed post‐intervention: mirtazapine: 83; placebo: 41; total: 124 Reasons for dropout: MHRA reported dropouts across the 2 mirtazapine trials: 9 (5.3%) patients discontinued due to an adverse event in the mirtazapine group compared with 3 (3.4%) in the placebo‐treated group. The most common adverse treated event leading to discontinuation in the acute phase in the mirtazapine‐treated group was weight gain. Weight gain (31.8% versus 3.4%), somnolence (38.8% versus 6.8%), headache (35% versus 23%), fatigue (19.4% versus 11.4%), increased appetite (8.8% versus 2.3%), urticaria (11.8% versus 6.8%) and hypertriglyceridaemia (2.9% versus 0%) were reported more often for mirtazapine‐treated patients than by placebo‐treated patients. ITT analysis: stated ITT done using LOCF but table of participants showed ITT analysis did not include all randomised patients Statistical methods: not stated |
| Selective reporting (reporting bias) | High risk | Only 1 outcome reported in MHRA report; Rapporteurs report gave safety outcomes in addition. |
| Other bias | Unclear risk | Contact: weekly visits (week 5 and 7 optional); psychotherapy could not be
started during the trial, but ‘supportive care’ as defined in the protocol
was permitted. Screening: unclear Placebo lead‐in: no Baseline imbalance: data not reported Stated it was initially 2 trials that were amalgamated a few months after trial initiation |
NCT02709746.
| Study characteristics | ||
| Methods | Trial design: Randomised, double‐blind, placebo‐controlled Power calculation: Not fully described, but protocol amendment dated 11 Oct 2015 stated sample size was based on power to detect at least one significant dose with an effect of 4 points on the primary endpoint (CDRS‐R). Also protocol amendment dated 05 Jul 2018 stated that the testing strategy for the primary analysis was modified such that the primary comparison was between the average doses (rather than the individual doses) of vortioxetine to placebo to increase the power of the study. Use of diagnostic criteria (or clear specification of inclusion criteria): Yes. DSM‐5 diagnosis of MDD Intervention integrity: Not described Outcome measures described or validated measures used: Yes. Children’s Depression Rating Scale‐Revised (CDRS‐R) Follow‐up assessment points: Not clearly described, appeared to be weeks 2, 4, 6, 8 No. crossed: None Funded by: H. Lundbeck A/S |
|
| Participants | Setting of care: Not reported Recruitment: Not reported Mean age (SD): Total sample – not reported Vortioxetine 10 mg: 14.8 (1.66) Vortioxetine 20 mg: 14.5 (1.63) Fluoxetine 20 mg: 14.8 (1.6) Placebo: 14.6 (1.6) Age range: 12‐17 years Gender (F:M): 398:218 Vortioxetine 10 mg: 93:54 Vortioxetine 20 mg: 97:65 Fluoxetine 20 mg: 103:50 Placebo: 105:49 Methods used to diagnose: DSM‐5 Diagnosis: MDD Baseline severity of depression: Total sample – not reported Vortioxetine 10 mg: 64.82 (9.38) Vortioxetine 20 mg: 65.29 (9.73) Fluoxetine 20 mg: 64.06 (8.65) Placebo: 64.02 (8.96) Length of current episode: Not reported % first episode: Not reported Comorbidity (intervention): Not reported Comorbidity (control): Not reported Location: United States: 311 Russian Federation: 100 Mexico: 57 Colombia: 32 Serbia: 27 Ukraine: 11 Korea, Republic of: 7 South Africa: 7 Canada: 4 Poland: 69 Spain: 20 United Kingdom: 6 Bulgaria: 23 Estonia: 19 France: 14 Germany: 22 Hungary: 7 Italy: 19 Latvia: 29 Inclusion criteria: Eligible patients from phase A (patients with incomplete improvement). Phase A was a nonrandomised single‐blind treatment period comprising placebo and brief psychosocial intervention (BPI) for 4 weeks. Aged ≥ 12 and ≤ 17 years at screening Major depressive disorder (MDD), diagnosed according to DSM‐5 CDRS‐R total score ≥ 45 and CGI‐S score ≥ 4 at screening and baseline Has provided assent to participation and parent(s)/legal representative(s) signed the Informed Consent Form Exclusion criteria: Has participated in a clinical study < 30 days prior to the screening visit Exclusion of suicidality: Not reported |
|
| Interventions | Intervention group
Drug: vortioxetine, encapsulated,
orally
Dosage: 10 mg or 20 mg
Regimen: Daily
Length of
treatment: 8 weeks. Intervention group Drug: fluoxetine, encapsulated, orally Dosage: 20 mg Regimen: Daily Length of treatment: 8 weeks. Control group: placebo, encapsulated, orally |
|
| Outcomes | Definition and assessment of response: Primary outcome was change from
baseline in Children's Depression Rating Scale‐Revised (CDRS‐R) total score
at week‐8 endpoint. Depressive symptoms: CDRS‐R Functioning: Children’s Global Assessment Scale (C‐GAS), change from baseline to week 8 endpoint Suicidal behaviour: Not reported Other measures: General Behaviour Inventory (GBI) depression subscale Parent Global Assessment–Global Improvement (PGA) Symbol Digit Modalities Test (SDMT) Clinical Global Impression Severity of illness (CGI‐S) Clinical Global Impression ‐ global Improvement (CGI‐I) Pediatric Quality of Life Inventory (PedsQL) Paediatric Quality of Life Enjoyment and Satisfaction Questionnaire (PQ‐LES‐Q) |
|
| Notes | Type of data used for remission/response: Not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described how allocation was made or concealed, aside from stating that participants were randomly assigned 1:1:1:1 |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Trial was described as “double‐blind” and that subject and investigator
roles were blinded. No other information was provided regarding how blinding
was implemented or maintained. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number eligible: 616 (post‐phase A) Number randomised: 616; number started trial: 616; vortioxetine 10 mg: 147; vortioxetine 20 mg: 162; fluoxetine 20 mg: 153; placebo: 154 Number of withdrawals: 74; vortioxetine 10 mg: 21; vortioxetine 20 mg: 22; fluoxetine 20 mg: 15; placebo: 16 (542 completed the intervention phase, but only 539 were analysed, this discrepancy was not explained) Number analysed post‐intervention: 539 Reasons for dropout: vortioxetine 10 mg Protocol deviation 1 Other 6 Non‐compliance with IMP 1 Lack of efficacy 3 Adverse events, non‐fatal 4 Consent withdrawn by subject 2 Enrolled not treated 0 Lost to follow‐up 4 Reasons for dropout: vortioxetine 20 mg Protocol deviation 0 Other 5 Non‐compliance 4 Lack of efficacy 0 Adverse events, non‐fatal 8 Consent withdrawn by subject 2 Enrolled not treated 1 Lost to follow‐up 2 Reasons for dropout: fluoxetine 20 mg Protocol deviation 0 Other 6 Non‐compliance 1 Lack of efficacy 1 Adverse events non‐fatal 5 Consent withdrawn by subject 2 Enrolled not treated 0 Lost to follow‐up 0 Reasons for dropout: placebo Protocol deviation 2 Other 7 Non‐compliance 0 Lack of efficacy 2 Adverse events, non‐fatal 2 Consent withdrawn by subject 1 Enrolled not treated 0 Lost to follow‐up 2 Reasons for dropout: reported for each group; higher noncompliance with IMP in the vortioxetine 20 mg group, more adverse events in the vortioxetine 20 mg group, more lost to follow‐up in the vortioxetine 10 mg group ITT analysis: Not described. Tables appeared to be observed cases. Statistical analysis: A restricted maximum likelihood (REML)‐based mixed model for repeated measures (MMRM). The model included the fixed, categorical effects of treatment, country, and week, the continuous covariate of CDRS‐R total score at randomisation, the treatment‐by‐week interaction, and the CDRS‐R at randomisation‐by‐week interaction. The Kenward‐Roger approximation was used to estimate denominator degrees of freedom. |
| Selective reporting (reporting bias) | High risk | All outcomes in methods reported? Yes; however, the PEDsQL VAS consists of
a number of subscales and there was no description of which were reported;
there was no clear indication of which week assessment of CDRS‐R scores were
going to be reported for and no reporting of the various subscale scores for
the CDRS‐R; and no report of the subscale scores of the GBI. Data available for use in MA? Unknown Access to trial protocol? No, only trial registry |
| Other bias | Unclear risk | Contact: Not clearly described. Contact appeared to have occurred at
baseline and weeks 2, 4, 6, 8. Screening: Phase A of the study occurred prior to baseline and consisted of a nonrandomised single‐blind treatment period comprising placebo and brief psychosocial intervention (BPI) for 4 weeks. Patients with incomplete improvement were then retained for the double‐blind placebo‐controlled treatment phase. 616 of 784 completed this. Placebo lead‐in: See phase A; nonrandomised single‐blind treatment period comprising placebo and brief psychosocial intervention (BPI) for 4 weeks Baseline imbalance: Not described but none apparent |
Paroxetine Trial 1.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multi‐site
Power
calculation: yes
Use of diagnostic criteria (or clear specification of
inclusion criteria): yes
Intervention integrity: yes. Plasma
concentration monitored
Outcome measures described or validated
measures used: yes
Follow‐up assessment points: post‐intervention No. crossed over: none Funded by: GSK |
|
| Participants | Setting of care: unclear
Recruitment: not stated
Mean age:
intervention = 14.4 years (SD = 1.99); placebo = 14.8 years (SD =
2.62)
Age range: 7 to 17 years
Gender (F:M): intervention =
18:9; placebo = 16:13; total female = 34, male = 22
Methods used to
diagnose: DSM‐IV; CDRS‐R score of ≥ 45
Diagnosis: MDD
Baseline
severity of depression: CDRS‐R mean (SD) intervention = 55.4 (7.3); placebo
= 56.8 (8.46) Length of current episode: not stated % first episode: not stated Comorbidity: not stated Location: Japan Inclusion criteria: single episode of MDD or recurrent symptoms of depression or depressed state Exclusion criteria: primary diagnosis of an axis 1 disorder other than MDD, those with a history of psychotic episode or psychotic disorder or bipolar disorder Exclusion of suicidality: not stated |
|
| Interventions | Intervention group
Drug: paroxetine
Dosage: 10 to 40 mg
dependent on age Regimen: 10 mg for 2 weeks and 10 to 20 mg for next 6 weeks for 7 to 11 year olds and 10 to 40 mg for the next 6 weeks for 12 to 17 year olds. The dose described at week 6 was maintained for the last 2 weeks. Length of treatment: 8 weeks Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: Clinical Global Impressions (CGI) of
1 or 2 Depressive symptoms: Change from baseline CDRS‐R Functioning: no report of measure used Suicidal behaviours: events reported as adverse events; no report of continuous data Other: Change from baseline CGI score Incidence of adverse events |
|
| Notes | Type of data used for remission/response: last‐observation‐carried‐forward | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information to make a judgement |
| Allocation concealment (selection bias) | Unclear risk | No information to make a judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind stated but no other details |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind stated but no other details |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number eligible: not stated Number randomised: paroxetine: 29; placebo: 27; total: 56 Number started trial: paroxetine: 29; placebo: 27; total: 56 Number of withdrawals: paroxetine: 4; placebo: 3; total: 7 Number analysed post‐intervention: paroxetine: 29; placebo: 27; total: 56 Reasons for dropout: broad reasons described ITT analysis: yes. Observed case data were used in some secondary analyses. Statistical methods: last‐observation‐carried‐forward (LOCF) analysis was used for the primary outcome, analysis of covariance was used with CDRS‐R total score at week 1 with total score as a covariate. |
| Selective reporting (reporting bias) | High risk | High risk as no published peer‐reviewed data. Drug company report available only |
| Other bias | Unclear risk | Contact: assessment at week 1, 2, 3, 4 and 6, no other details Screening: 2‐week screening, a CDRS‐R score of ≥ 45 at week ‐2 and week 0 Placebo lead‐in: there was a 2‐week placebo lead‐in period. Baseline imbalance: no specific statement, however proportion of female to male was different in the placebo group, and fewer children in the paroxetine group Other: did not reach planned recruitment numbers |
Simeon 1990.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; single site
Power
calculation: not stated
Use of diagnostic criteria (or clear
specification of inclusion criteria): yes
Intervention integrity: yes
‐ assessed by clinical chemistry profile
Outcome measures described or
validated measures used: yes
Follow‐up assessment points: weekly
visits, post‐intervention and long‐term follow‐up on average 24 months
post‐trial termination No. crossed over: none Funded by: not stated |
|
| Participants | Setting of care: outpatient
Recruitment: no information
Mean
age: 16 (group ages not stated)
Age range: actual range not
stated
Gender (total): female = 22; male = 18 (group gender not
stated)
Methods used to diagnose: DSM‐III criteria with HAM‐D score of
≥ 20, 1‐week placebo run‐in period
Diagnosis: MDD
Baseline
severity of depression: not stated for either group Length of current episode: not stated % first episode: not stated Comorbidity: not stated for either group Location: Canada Inclusion criteria: 13 to 18 years; MDD with a HAM‐D score > 20, a Raskin Depression Scale score of > 8, a Raskin Depression Score that must exceed the Covi Anxiety Scale Score, an outpatient Exclusion criteria: history of seizures, schizophrenia or other psychotic illnesses, girls who were sexually active and not using medically accepted means of contraception, patients with recent drug or alcohol abuse Exclusion of suicidality: serious suicidal risk (no further definition) |
|
| Interventions | Intervention group
Drug: fluoxetine
Dosage: 20 to 60
mg
Regimen: initial dose 20 mg daily increased to 40 mg after 4 to 7
days, and up to 60 mg in the second week
Length of treatment: 7
weeks Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: not stated Depressive symptoms: HAM‐D; Raskin Depression Scale Functioning: no report Suicidal behaviours: no report of events or continuous measure Other: Clinical Global Impressions Scale (CGI); Covi Anxiety Scale; Hopkins Symptom Checklist Follow‐up assessment included semi‐structured interviews by a nurse to obtain treatment subsequent to the trial, current activities and functioning with family and peers, and follow‐up interview with parents using the HAM‐D, Raskin, Covi and a DSM‐III checklist for MDD and an adaptive functioning scale |
|
| Notes | Letter requesting additional data sent. Data have not been received. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned" pg.792; no other statement |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind" pg.792 |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No statement |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number eligible: not stated Number randomised: 40, group Ns not stated Number started trial: 40, group Ns not stated Number of withdrawals: 8, group Ns not stated Number analysed post‐intervention: fluoxetine: 16; placebo: 16; total: 32 Reasons for dropout: not stated ITT analysis: not stated Statistical methods: little detail provided; pg.792 stated Wilcoxons Rank Sum Test and Chi2 test used |
| Selective reporting (reporting bias) | High risk | No outcome data were reported. |
| Other bias | High risk | Contact: no details were given of the contact time with clinicians in
either group. Screening: no details of screening procedure given Placebo lead‐in: there was a 1‐week single‐blind placebo lead‐in (pg.792) Baseline imbalance: pg.792 stated there were no significant differences between groups at baseline; however, no demographic or clinical data were provided by group. Other: Hammad 2004 reported that this trial was "terminated early" pg.28. |
TADS 2004.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre Power calculation: yes Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not described for fluoxetine and placebo arms Outcome measures described or validated measures used: yes Follow‐up assessment points: post‐intervention No. crossed over: none Funded by: NIMH | |
| Participants | Setting of care: outpatient
Recruitment: included newspaper, TV and
radio advertising
Mean age (total): 14.6 (SD 1.5)
Age range
(actual): 12 to 18 years
Gender (F:M): 239:200
Methods used to
diagnose:DSM‐IV confirmed using K‐SADS‐PL and a CDRS‐R score of ≥ 45;
assessment (not interview) at consent and baseline Diagnosis: MDD Baseline severity of depression: CDRS‐R raw mean (SD) score: intervention:58.96 (10.16) (T‐score 74.73 (6.74)); control: 61.11 (10.50) (T‐score 76.14 (6.11)): CGI intervention 4.66; CGI placebo 4.84 Length of current episode: (median) intervention 38 weeks; placebo: 35.5 weeks % first episode: 86% of total (not reported by group) Comorbidity (intervention): any 47 ; dysthymia 6; anxiety 26; OCD/tic 2; ADHD 13; substance use 3; disruptive behaviour 25 Comorbidity (control): any 57; dysthymia 12; anxiety 28; OCD/tic 4; ADHD 19; substance use 0; disruptive behaviour 28 Location: USA Inclusion criteria: outpatient; age 12 to 17; full scale IQ > 80; antidepressant‐free before trial Exclusion criteria: bipolar disorder; severe conduct disorder; substance abuse; pervasive developmental disorder; thought disorder; use of psychotropic medication or psychotherapy (stable stimulants permitted for ADHD); 2 previous failed SSRI trials or a failed trial of CBT; confounding medical condition; non‐English speaking Exclusion of suicidality: suicidality or homicidality (patients were excluded for dangerousness to self or others if they had been hospitalised for dangerousness within 3 months of consent or were deemed by a cross‐site panel to be 'high risk' because of a suicide attempt requiring medical attention within 6 months, clear intent or an active plan to commit suicide, or suicidal ideation with a disorganised family unable to guarantee adequate safety monitoring) |
|
| Interventions | Intervention group
Drug: fluoxetine
Dosage: 20 to 40
mg
Regimen: 10 mg daily to start; increase to 20 mg daily in week 1
with increase to a maximum of 40 mg daily thereafter
Length of
treatment: 12 weeks Control group: placebo Comparison group 1: CBT Comparison group 2: CBT plus fluoxetine |
|
| Outcomes | Definition of response and assessment: we used remission CDRS‐R ≤ 28 (they
used a range of outcomes including response and remission, using different
definitions: In the main results paper, they used response defined as a CGI
improvement of 1 or 2)
Depressive symptoms: Children's Depression
Rating Scale ‐ Revised (CDRS‐R) Functioning: C‐GAS Suicidal behaviours: report of events based on Columbia classification; continuous measure using the Suicidal Ideation Questionnaire‐Junior High School Version (SIQ‐JR) Adverse events Other outcomes: Clinical Global Impressions Scale Improvement (CGI‐Improvement); Reynolds Adolescent Depression Scale (RADS) |
|
| Notes | Additional trial information was sought and received from the author. Data
in the MA from the paper
All young people in the trial were included
as adolescents. Type of data used for remission/response: last‐observation‐carried‐forward |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "computer stratified randomisation" pg.808 in 2004 publication |
| Allocation concealment (selection bias) | Low risk | "centralized IVRS service. Eligibility was assessed by same i.e. as did dependent variable assessments. Trial coordinator not independent evaluator interfaced with IVRS and primary clinician for that patient revealed randomization status at Gate C2 after having first confirmed that patient/parent understood and were willing to accept randomization to any TADS treatment" from personal correspondence |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "except in emergencies, participants and clinicians remained blind in fluoxetine alone and placebo" groups (pg.808 in 2004 publication). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "as rated by an independent evaluator" pg.535 in the 2003 publication; "masking was maintained for the primary dependent measures by means of independent evaluators blind to treatment assignment. Specific instructions were provided to parents, participants and the independent evaluator not to disclose treatment assignment" (pg.808 in the 2004 publication). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: 1088 Number randomised: fluoxetine: 109; placebo: 112; total: 439 (including additional 2 trial arms) Number started trial: fluoxetine: 109; placebo: 112; total: 439 (including additional 2 trial arms) Number of withdrawals: fluoxetine: 18; placebo: 23; total: 90 (including additional 2 trial arms) Number analysed post‐intervention: fluoxetine: 109; placebo: 112; total: 439 (including additional 2 trial arms) Reasons for dropout: full table of number of dropouts and reason for dropouts given pg.811. Reasons for dropout were not specific e.g. terminated prematurely. Similar reasons in each group except 10 participants in the placebo group withdrew consent, compared with 5 in the fluoxetine group ITT analysis: "all analyses were conducted using an intent‐to‐treat analysis"; "primary intent to treat, all patients regardless of treatment status return for all scheduled assessments" (pg.535 in the 2003 publication). Statistical methods: for the CDRS‐R results linear random coefficient regression model; used random‐effects for participants and clinical site (but site interaction omitted). Responder (CGI‐I) used logistic regression model for last available assessment point (LOCF) with site as covariate. |
| Selective reporting (reporting bias) | High risk | Percentages given for CGI‐I response rates. Mulitple publications reported varying outcome results that were not consistent across papers. In the 2004 paper presenting the main results, functioning was not reported. |
| Other bias | Unclear risk | Contact: "Patients have one pharmacotherapist throughout the trial who, in
addition to monitoring clinical status and medication effects, offers
general encouragement about the effectiveness of pharmacotherapy for MDD.
Major assessments undertaken at baseline, 12 weeks, 24 weeks and 36 weeks
with minor assessments at 6 weeks, 18 weeks and 30 weeks" (2003 publication
pg.537); "six 20 to 30 minute medication visits spread across 12 weeks of
treatment" (2004 publication pg.809) Screening: phone screening assessment followed by 1 full assessment to determine 'caseness', which on average took 3 weeks (range 2 to 8 weeks) Placebo lead‐in: no Baseline imbalance: for main results paper (2004) there were none reported; no demographic information given by group; Table 1 reported baseline clinical information with no significant differences reported across the four treatment groups. TADS 2005 paper on demographics did not report demographic and clinical characteristics by group. |
VLZ‐MD‐22.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre
Power
calculation: Statistical Analysis Plan (pg.33) stated 400 participants (160
per group for placebo and vilazodone, 80 for fluoxetine) required to achieve
85% power to detect a 4‐point difference between treatment groups on CDRS‐R
change‐from‐baseline score (relative to a pooled SD of 11.1), in the MMRM
analysis, assuming a 0.7 correlation between repeated measures and 17%
dropout rate. At first planned blinded interim analysis (300 participants,
pooled SD 12.3), sample size increased to 455. At the second interim
analysis (427 participants, pooled SD 12.43), sample size increased to 470
(188 per group for placebo and vilazodone, 94 for fluoxetine). Use of diagnostic criteria (or clear specification of inclusion criteria): Primary diagnosis of Major Depressive Disorder (MDD), and CDRS‐R score ≥ 40, and CGI‐S score ≥ 4. Intervention integrity: not reported Outcome measures described or validated measures used: yes, CDRS‐R Follow‐up assessment points: post‐treatment No. crossed: not reported Funded by: Forest Laboratories |
|
| Participants | Setting of care: outpatient
Recruitment: not reported
Mean age
(SD): viladozone = 13 (2.9); fluoxetine = 13.2 (2.8); placebo = 13 (2.9) Age range: 7 to 17 years Gender (F:M): viladozone = 126:61; fluoxetine = 51:46; placebo = 106:80 Methods used to diagnose: DSM‐IV‐TR criteria for MDD using semi‐structured interview; K‐SADS‐PL Diagnosis: MDD Baseline severity of depression, CDRS‐R mean (SD, n): viladozone = 58.3 (9.2, 186); fluoxetine = 58 (8.8, 97); placebo = 57.3 (9.2, 182); total = 57.8 (9.1, 465) Length of current episode: depressive episode ≥ 6 weeks duration at screening % first episode: not reported Comorbidity: not reported Location: 55 sites, United States (n = 53), Canada (n = 2) Inclusion criteria: ‐ Male or female outpatients, 7 to 17 years of age ‐ DSM‐IV‐TR criteria for Major Depressive Disorder (MDD), current episode ≥ 6 weeks duration ‐ Children’s Depression Rating Scale‐Revised (CDRS‐R) score of 40 or greater ‐ Clinical Global Impressions‐Severity (CGI‐S) score of 4 or greater Exclusion criteria: ‐ Current (past 3 months) principal DSM‐IV‐TR‐based diagnosis of an Axis I disorder other than major depressive disorder, that is the primary focus of treatment (comorbid diagnoses of learning disorder, attention deficit disorders, oppositional defiant disorder and anxiety disorders are allowed if these are not the primary reason of treatment) ‐ Conduct disorder ‐ Prior diagnosis of mental retardation, amnesic or other cognitive disorder ‐ A suicide risk, any suicide attempt within past year or a significant risk as judged by the Investigator (psychiatric interview or C‐SSRS) ‐ Pregnant, breastfeeding or planning to become pregnant/breastfeed ‐ Females who are sexually active and not practicing a reliable method of contraception ‐ Not generally healthy medical condition Exclusion of suicidality: yes |
|
| Interventions | Intervention group 1
Drug: viladozone
Dosage: flexible, 15‐30
mg
Regimen: daily, titrated from 5 to 15 mg/day by end of week 1,
flexible increase to 30 mg/day by end of week 3 at investigator
discretion
Length of treatment: 8 weeks acute treatment, 1‐week taper,
completers eligible for 6‐month open‐label extension (VLZ‐MD‐23) Intervention group 2 Drug: fluoxetine Dosage: 20 mg Regimen: daily, 10 mg/day for day 1‐7, increased to 20 mg/day at week 1 visit Length of treatment: 8 weeks acute treatment, 1‐week taper Note: no dosage increases allowed beyond week 3. One dosage decrease allowed for tolerability (to next lower dose) Control group: placebo, 1 capsule and 3 tablets |
|
| Outcomes | Definition and assessment of response: CDRS‐R responders were defined as
patients with a ≥ 40% reduction from baseline in CDRS‐R total score. CDRS‐R
remitters were defined as patients with CDRS‐R ≤ 28. Depressive symptoms: CDRS‐R Functioning: not reported Suicidal behaviour: C‐SSRS (data not reported) Other measures: CGI‐S, CGI‐I (data not reported) |
|
| Notes | Type of data used for remission/response: LOCF was planned (pg.36,
Statistical Analysis Plan) but data not reported. Data sources: Primary source was trial registry entry (NCT02372799 accessed from clinicaltrials.gov on 18 May 2020); contains published Statistical Analysis Plan (dated 5 Oct 2018) and Protocol (dated 15 Apr 2015). Supplementary source was FDA NDA/BLA Multidisciplinary Review and Evaluation (NDA 22567/s021, dated 7 Jul 2019). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Patients … randomized in a ratio of 2:2:1 to … placebo, vilazodone, or
fluoxetine” (pg.23 Protocol) “A list of patient randomization codes will be generated by … [redacted in protocol] … (an electronic version will be stored on a secure server). This list will identify each patient by randomization number and include the patient’s corresponding treatment assignment.” (pg.37 Protocol) ClinicalTrials.gov described a “randomization scheme prepared by Allergen Biostatistics prior to the start of the study”. Randomisation stratified by study center and by country Comment: Likely used adequate random sequence generation, no baseline imbalance of major concern, however no statement reported |
| Allocation concealment (selection bias) | Unclear risk | “Patients … randomized in a ratio of 2:2:1 to … placebo, vilazodone, or
fluoxetine” (pg.23 Protocol) “A list of patient randomization codes will be generated by … [redacted in protocol] … (an electronic version will be stored on a secure server). This list will identify each patient by randomization number and include the patient’s corresponding treatment assignment.” (pg.37 Protocol) Trial registry entry (NCT0237279) stated “Masking: Quadruple (Participant, Care Provider, Investigator, Outcome Assessor)” Comment: Likely implemented adequate allocation concealment, however no statement reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Trial registry entry (NCT0237279) stated “Masking: Quadruple (Participant,
Care Provider, Investigator, Outcome Assessor)” “Matching placebo tablets for viladozone and matching placebo capsules for fluoxetine…” (pg.2 Protocol) “All investigational products taken orally as a single dose of 3 tablets and 1 capsule, once daily at the same time each day, with food” (pg.34 Protocol) Note 1: Fluoxetine daily dosage was 20 mg (1 capsule: 20 mg), viladozone flexible daily dosage was 15‐30 mg (up to 3 tablets: 5, 10, 20 mg), and placebo dosage was 1 matched capsule and 3 matched tablets (Protocol pg.32, 35). Given all investigational products taken … “as single dose of 3 tablets and 1 capsule”, likely participants in both vilazodone and fluoxetine groups were given a mix of active drug allocation and placebo to ensure all groups received 3 tablets and 1 capsule. Note 2: Large difference in reporting of adverse events (not including SAEs) between groups: viladozone = 51.87% (97/187), fluoxetine = 27.84% (27/97), placebo = 29.03% (54/186), see trial registry entry (NCT0237279) Comment: likely participants and personnel were blinded. Unclear if adverse event rate led to unblinding, and if so, whether any differences in provision of or access to care (e.g. other interventions) occurred. No statement on maintenance or evaluation of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial registry entry (NCT0237279) stated “Masking: Quadruple (Participant,
Care Provider, Investigator, Outcome Assessor)” Note 2: Large difference in reporting of adverse events (not including SAEs) between groups: viladozone = 51.87% (97/187), fluoxetine = 27.84% (27/97), placebo = 29.03% (54/186), see trial registry entry (NCT0237279) Comment: likely outcome assessor was blinded; unclear if adverse event rate was detectable by outcome assessor leading to unblinding, and if so, whether outcome assessment was influenced. No statement on maintenance or evaluation of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: 644 screened, 151 ineligible, 14 withdrew consent, 4 lost
to follow‐up Number randomised: 473; viladozone: 187; fluoxetine: 99; placebo: 187 Number started trial: 470; viladozone: 187; fluoxetine: 97; placebo: 186 Number of withdrawals: 84; viladozone: 32; fluoxetine: 17; placebo: 35 Note: 1 placebo and 2 fluoxetine patients added to withdrawals as they were randomised but withdrew prior to start of treatment (excluded from safety population); see FDA review pg.42 Number analysed post‐intervention: 465; viladozone: 186; fluoxetine: 97; placebo: 182 Reasons for dropout: Adverse events: viladozone = 10, fluoxetine = 6, placebo = 3; Insufficient therapeutic response: viladozone = 2, fluoxetine = 0, placebo = 2; Withdrawal by subject: viladozone = 10, fluoxetine = 2, placebo = 11; Lost to follow‐up: viladozone = 5, fluoxetine = 4, placebo = 11; Protocol violation: viladozone = 0, fluoxetine = 1, placebo = 3; Non‐compliance with study drug: viladozone = 4, fluoxetine = 4, placebo = 4; Miscellaneous: viladozone = 1, fluoxetine = 0, placebo = 1. Note: 1 placebo (withdrawal by subject) and 2 fluoxetine patients (protocol violation, lost to follow‐up) have been added to the reasons for dropouts as they were randomised but withdrew prior to start of treatment (excluded from safety population), see FDA review pg.42. Comment: number of withdrawals balanced, however reasons for dropouts were distributed slightly differently, although small numbers; see above. ITT analysis: efficacy analysis was performed on the ITT population, consisting of all patients in the safety population who had baseline and at least one post‐baseline assessment of CDRS‐R total score, corresponding to 465 participants. Statistical analysis: Primary efficacy parameter of change from baseline to week 8 on the CDRS‐R was estimated from Mixed‐effects Model for Repeated Measures (MMRM) using the observed case (OC) approach with fixed effects of treatment group, pooled study centre, visit, and treatment group‐by‐visit interaction and baseline CDRS‐R and baseline CDRS‐R‐by‐visit interaction as covariates. An unstructured covariance matrix modelled the covariance of within‐patient scores. The Kenward‐Roger approximation estimated denominator degrees of freedom. (Statistical Analysis Plan pg.20) |
| Selective reporting (reporting bias) | Unclear risk | All outcomes in methods reported? Protocol‐defined primary (CDRS‐R) and
secondary (CGI‐S) outcomes reported, however CGI‐I, C‐SSRS, and CDRS‐R
remission and response appeared in protocol but have not been reported in
trial registry entry (NCT02372799, accessed 18 May 2020). Data available for use in MA? Yes. Access to trial protocol? Protocol and Statistical Analysis Plan published in trial registry entry (NCT02372799) |
| Other bias | Unclear risk | Contact: screening visits (1 to 5 weeks), baseline, week 1, 2, 3, 4, 6, 8
(post‐treatment) and week 9 (taper) (FDA review pg.38, 39) Screening: 1 week prior to baseline (randomisation), extended up to 5 weeks to accommodate repeat assessments and/or prior medication washout (with prior approval of study physician or designee) (Protocol pg.23) Placebo lead‐in: All participants received placebo (3 tablets, 1 capsule) at screening visit (1 week prior to baseline) to “confirm their ability to swallow investigational product”. Those unable were ineligible for participation (Protocol pg.32, 35). Baseline imbalance: baseline primary outcome (CDRS‐R), age, ethnicity, race, weight, BMI, CGI‐S appeared balanced. Sex was unbalanced with more females in viladozone (67.4%) than placebo (52.6%) and fluoxetine (57%). The following covariates included in primary efficacy analysis: pooled study centre, visit, treatment‐by‐visit interaction, baseline CDRS‐R, baseline‐by‐visit interaction. Other potential covariates (e.g. comorbidities) not reported |
Von Knorring 2006.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre Power calculation: not stated Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: yes ‐ noncompliance assessed by blood levels of citalopram Outcome measures described or validated measures used: yes Follow‐up assessment points: post‐intervention Funded by: pharmaceutical company not stated | |
| Participants | Setting of care: in and outpatient (14% of participants hospitalised at
entry to trial)
Recruitment: no information
Mean age (SD): 16
(1)
Age range: 13 to 18 years
Gender: not stated
Methods
used to diagnose: DSM‐IV including 5‐minute interview with parents. Global
assessment of functioning less than 60 on either symptoms, activities,
relationships or personal care, BDI less than 21 for girls and less than 16
for boys
Diagnosis: MDD
Baseline severity of depression:
K‐SADS‐P mean intervention 32.5; control = 32.3 and totals only for MADRS 30
(SD = 5/6), GAF 55 (SD = 7); CGI not reported Length of current episode: not reported % first episode: intervention 72%; placebo 64% Comorbidity: not stated for either group Location: Denmark, Estonia, Finland, Germany, Norway, Sweden, Switzerland Inclusion criteria: DSM‐IV MDD current episode of greater than 4 weeks but less than 1 year duration; in or outpatient plus score of at least 21 or 16 on BDI and at least 60 on the GAF; 13 to 18 years inclusive; Tanner Stage III (commencement of puberty) Exclusion criteria: bipolar disorder including hypermania; ongoing DSM‐IV attention deficit disorder or disruptive behaviour disorder; DSM‐IV psychotic disorder; progressive neurological disorder; drug or alcohol abuse that influences daily functioning; primary anorexia nervosa or bulimia nervosa; attends special school for mentally retarded; pervasive developmental disorders Exclusion of suicidality: not explicitly stated |
|
| Interventions | Intervention group
Drug: citalopram
Dosage: 10 to 40
mg
Regimen: 10 mg for the first week with dose increases at the end of
the week 1, 2, 5 or 9 weeks of 10 mg if GAF decreased by 10 points or
unchanged to a maximum of 40 mg
Length of treatment: 12 weeks Control group: placebo pill |
|
| Outcomes | Definition of response and assessment: we used OC remission MADRS < 12
(they used responders defined as those with a score of 2 or less on the
K‐SADS‐P depression and anhedonia items or with a reduction of at least 50%
from baseline of the MADRS total score) Functioning: Global Assessment of Functioning (GAF) Suicidal behaviours: FDA data; no report of continuous measure Adverse outcomes Other outcomes:K‐SADS‐P total score; Montgomery‐Asberg Depression Rating Scale (MADRS); Beck Depression Inventory (BDI) |
|
| Notes | MHRA #94404
MHRA contacted for additional data, some of which were
provided We included data on self‐report depression from Von Knorring 2006 assuming the baseline standard deviations from Berard 2006 as the follow‐up standard deviations of Von Knorring 2006. Type of data used for remission/response: last‐observation‐carried‐forward |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized" (pg.311) |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double blind" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No statement |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number eligible: not stated Number randomised: citalopram: 124; placebo: 120; total: 244 Number started trial: citalopram: 121; placebo: 112; total: 233 Number of withdrawals: citalopram: 45; placebo: 46; total: 91 Number analysed post‐intervention: citalopram: 121; placebo: 112; total: 233 Reasons for dropout: full table of number of dropouts but full description of reasons for dropouts not given. More withdrew from the placebo group due to lack of efficacy and more withdrew from the citalopram group due to adverse effects. ITT analysis: efficacy analyses were conducted on an intent‐to‐treat population, which included all randomised patients who took at least 1 dose of double‐blind medication and who had at least 1 valid post‐assessment K‐SADs‐P assessment (pg.312) Statistical methods: primary analysis based on adjusted mean change of observed case data using ANCOVA (analysis of covariance). Dichotomous data analysed using LOCF |
| Selective reporting (reporting bias) | High risk | Error in the von Knorring paper when describing response data where it was reported twice and both times as OC data. Both response and remission data were only reported as percentages and when calculating these out using both the ITT population and the OC population, the whole numbers did not match. Results only (no data) were reported for functioning, depression severity (clinician‐ and self‐rated) |
| Other bias | Unclear risk | Contact: evaluation undertaken at 1, 2, 5, 9 and 12 weeks. Psychotherapy
was allowed and three‐quarters of the participants received it. Screening: there was 1 screening visit and then a baseline visit. Placebo lead‐in: no Baseline imbalance: authors stated that baseline data were similar for the 2 treatment groups, however, much baseline data (e.g. depression severity, age) was not reported by group. There were more patients in the citalopram group hospitalised for a psychiatric disorder and with a first episode. Other: after recruitment of 15% of the population the trialists changed the inclusion criteria to?16 on the BDI for boys and added the MADRS. Post hoc analysis of high versus low baseline scores and of those receiving psychotherapy versus not receiving psychotherapy |
Wagner 2004.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre
Power
calculation: not reported
Use of diagnostic criteria (or clear
specification of inclusion criteria): yes
Intervention integrity: not
described
Outcome measures described or validated measures used:
yes
Follow‐up assessment points: post‐intervention
No. crossed
over: none Funded by: Forest Pharmaceuticals |
|
| Participants | Setting of care: outpatients
Recruitment: no information
Mean
age (SD): intervention = 12.1 (2.8); control = 12.1 (3.1)
Age range: 7
to 17 years
Gender (F:M): intervention = 54:39; control =
43:42
Methods used to diagnose: DSM‐IV confirmed using the Schedule
for Affective Disorders and Schizophrenia for School‐Age Children—Present
and Lifetime Version (K‐SADS PL) and a CDRS‐R score of ≥ 40
Diagnosis:
MDD
Baseline severity of depression: CDRS‐R mean (SD) score:
intervention = 58.8 (10.9); control = 57.8 (11.1); CGI not reported Length of current episode: (mean (months)) intervention: 20.8 (21.4); placebo: 18.6 (16.4) % first episode: intervention 78.7%; placebo 82.4% Comorbidity (intervention): dysthymia 5; enuresis 4; previous ADHD 4 Comorbidity (control): dysthymia 1; enuresis 3; previous ADHD 1 Location: USA Inclusion criteria: MDD of at least 4 weeks' duration; normal physical exam, laboratory tests and electrocardiography (ECG); parent available to accompany child Exclusion criteria: primary psychiatric diagnosis other than MDD; ADHD; PTSD; bipolar disorder; pervasive developmental disorder; mental retardation; CD; ODD; any psychotic features; any personality disorder that would interfere with treatment; alcohol or substance abuse; anorexia or bulimia nervosa; initiation of psychotherapy or behaviour therapy 3 months prior to trial entry; and antidepressant or anxiolytic medication in 2 weeks prior to trial entry; neuroleptic or stimulant medication within 6 months of trial entry Exclusion of suicidality: suicide risk or previous active attempt in previous year or hospitalised due to attempt |
|
| Interventions | Intervention group
Drug: citalopram
Dosage: 20 mg to 40
mg
Regimen: 20 mg daily for 4 weeks with option to increase to 40 mg
daily
Length of treatment: 8 weeks Control group: placebo pill |
|
| Outcomes | Definition of response and assessment: we used what they call response
(called remission in other trials) CDRS‐R ≤ 28 (they used responders defined
as at least = 28 on Children's Depression Rating Scale ‐ Revised
(CDRS‐R))
Depressive symptoms: CDRS‐R Functioning: Children's Global Assessment Scale (C‐GAS) Suicidal behaviours: FDA data; no report of continuous outcome Adverse events Other outcomes: Clinical Global Impressions Scale Improvement (CGI ‐ Improvement); Clinical Global Impressions Scale Severity (CGI ‐ Severity) |
|
| Notes | Additional data were sought from authors. No response was
received.
MHRA # CIT‐MD‐18
MHRA contacted for additional data
some of which were provided Type of data used for remission/response: last‐observation‐carried‐forward |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned" but no statement how |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No statement |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "in a double‐blind fashion" (pg.1080); different colour coating was used for placebo and citalopram pills; 9 patients were dispensed medication that potentially unblinded treatment assignment. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number eligible: 178 Number randomised: citalopram: 93; placebo: 85; total: 178 Number started trial: citalopram: 93; placebo: 85; total: 178 Number of withdrawals: citalopram: 4; placebo: 0; total: 36 Number analysed post‐intervention: citalopram: 89; placebo: 85; total: 174 Reasons for dropout: 4 patients all randomly assigned to citalopram group were lost to follow‐up and did not receive trial medication. "These patients were not included in the Intention‐to‐Treat (ITT) analysis "...of these (ITT population), 18 patients from each group discontinued double‐blind treatment prematurely (pg.1080). Reasons for dropout were not described. ITT analysis: "These (4 patients in the citalopram group who were lost to follow‐up) patients were not included in the Intention‐to‐Treat (ITT) analysis" Statistical methods: analysis of covariance with treatment, trial centre, and age as factors and baseline scores as covariates. Cochrane‐Mantel Haenszel test controlling for centre and age group. Used LOCF |
| Selective reporting (reporting bias) | High risk | Percentages only given for response data. Response in this trial was defined in the same way as remission was defined in many other SSRI trials (Emslie 1997; Emslie 2002; TADS 2004; Emslie 2006) but remission itself was not included as an outcome in this trial. Depression symptom severity means and standard deviations were not reported but represented in a figure with a result only reported (MHRA report, change scores). |
| Other bias | Unclear risk | Contact: evaluation undertaken at 1, 2, 4, 6 and 8 weeks. Psychotherapy was
not allowed (pg.1080). Screening: there was 1 screening visit and then a baseline visit. Placebo lead‐in: 1‐week single‐blind in between screening visit and baseline visit Baseline imbalance: authors reported no significant differences (report data in Table 1) Other: data not reported by child versus adolescent |
Wagner 2006.
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre
Power
calculation: not stated
Use of diagnostic criteria (or clear
specification of inclusion criteria): yes
Intervention integrity: not
described
Outcome measures described or validated measures used:
yes
Follow‐up assessment points: post‐assessment
No. crossed
over: none Funded by: Forest Laboratories, Inc |
|
| Participants | Setting of care: outpatients
Recruitment: no information
Mean
age: intervention = 12.2 (2.9); control = 12.4 (3.0)
Age range: 6 to
17 years
Gender (F:M): intervention = 68:63; control =
69:64
Methods used to diagnose: DSM‐IV confirmed using K‐SADS‐PL and a
CDRS‐R score of ≥ 40; 1‐week placebo run‐in period
Diagnosis:
MDD
Baseline severity of depression: CDRS‐R mean score: intervention =
54.5; control = 56.6; CGI intervention 4.4; CGI placebo 4.2 Length of current episode: (mean (months)) intervention 16.7 (15.3); placebo 15.6 (13.6) % first episode: not reported Comorbidity (intervention): 6 had an ongoing anxiety disorder; none had ADHD Comorbidity (control): 10 had an ongoing anxiety disorder; none had ADHD Location: 25 centres in the USA Inclusion criteria: MDD of at least a 4‐week duration, normal results at screening from physical examination, laboratory tests and electrocardiography Exclusion criteria: any primary psychiatric diagnosis apart from MDD; any psychotic features; any severe personality disorder; met DSM‐IV criteria for attention deficit hyperactivity disorder, post‐traumatic stress disorder, bipolar disorder, pervasive developmental disorder, mental retardation, conduct or oppositional defiant disorder; females not practising or willing to practise a reliable method of birth control; history of anorexia nervosa, bulimia nervosa, substance abuse; initiation of psychotherapy was not allowed during the trial within 3 months before the screening visit; previous treatment failure on SSRI Exclusion of suicidality: suicide risk based on clinical judgement of investigator or ever hospitalised for suicide attempt or had made a suicide attempt within the past year |
|
| Interventions | Intervention group
Drug: escitalopram oxalate
Dosage: fixed
dose of 10 mg for the first 4 weeks; thereafter flexibly dosed from 10 to 20
mg based on clinical response
Regimen: taken daily
Length of
treatment: 8 weeks Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: we used what they called response
(called remission in other trials) CDRS‐R ≤ 2 (they did 2 separate analyses
of response data undertaken using 2 different definitions of response:
CDRS‐R score of less than or equal to 28; or CGI‐I of less than or equal to
2)
Depressive symptoms: Children's Depression Rating Scale ‐ Revised
(CDRS‐R). Functioning: Children's Global Assessment Scale (C‐GAS) Suicidal behaviour: events reported as adverse events; no report of continuous measure Adverse outcomes Other outcomes: Clinical Global Impressions Scale Severity (CGI‐Severity); Clinical Global Impressions Scale Improvement (CGI‐Improvement). |
|
| Notes | Forest pharmaceutical ID was SCT MD 15
Data in the MA from the
web‐based publication. Subsequent to this, Wagner 2006 was published and data checked against this
publication with child and adolescent data added to the MA. Type of data used for remission/response: last‐observation‐carried‐forward |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | (pg.282) computer‐generated randomisation sequence. Patient randomisation numbers were allocated to each site in ascending sequence in blocks of 4. Randomisation was not stratified by age. |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated to be "double blind" with tablets identical indicating participants may be blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No statement but clinicians and subjects completed measures and both of these were probably blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number eligible: 268 Number randomised: escitalopram: 132; placebo: 136; total: 268 Number started trial: escitalopram: 131; placebo: 133; total: 264 Number of withdrawals: escitalopram: 29; placebo: 18; total: 48 Number analysed post‐intervention: escitalopram: 129; placebo: 132; total: 261 Reasons for dropout: full list of dropouts and reasons for dropout figure 1 (pg.283). Trial authors stated no significant differences in specific reasons for premature discontinuation; appeared to be more withdrawing consent from escitalopram group ITT analysis: efficacy analyses were performed on the intent‐to‐treat population, which included all patients in the safety population (i.e. received at least 1 dose of trial medication) who had at least 1 post‐baseline CDRS‐R assessment (pg.282). Statistical methods: LOCF was used (as well as some OC analysis). Analysis of covariance (treatment group and trial centre as factors and baseline scores as covariate). Logistic regression with treatment as the factor and baseline scores as covariates (pg.282) |
| Selective reporting (reporting bias) | Unclear risk | 2 prospective definitions of response were used. A post hoc analysis of suicide‐related outcomes was undertaken (pg.282). Only P values were provided for clinician‐rated depression symptoms. |
| Other bias | Low risk | Contact: evaluations at end of 1, 2, 4, 6 and 8 weeks; psychotherapy was
not allowed (pg.281). Screening: diagnostic criteria had to be met at the screening visit and then again at the baseline visit after the 1‐week placebo lead‐in. Baseline imbalance: authors stated there were no significant differences between the groups. Other: not noted |
Wagner Trial 1.
| Study characteristics | ||
| Methods | See Wagner Trial 1 & 2 (2003) entry | |
| Participants | See Wagner Trial 1 & 2 (2003) entry | |
| Interventions | See Wagner Trial 1 & 2 (2003) entry | |
| Outcomes | See Wagner Trial 1 & 2 (2003) entry | |
| Notes | See Wagner Trial 1 & 2 (2003) entry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "using a computer generated randomisation code" (pg.1034) |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double blind receipt of sertraline or matching placebo" (pg.1034); "trial drug was packaged in identical blister packs...both patients and clinicians were blinded to group assignment" (pg.1035). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No statement |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number randomised: 188 Number of withdrawals: 46 Number analysed post‐intervention: 142 Reasons for dropout: full list of dropouts and reasons for dropouts figure 1 (pg.1036). There were more dropouts due to adverse events reported in the sertraline group. ITT analysis: intention‐to‐treat population was modified... post randomisation efficacy data collected...problems with data collection (pg.1036). Only those who received at least 1 dose of trial medication were included in the efficacy analyses (pg.1036). Statistical methods: used repeated measures mixed‐model analysis with the model including baseline effect as a covariate, random subject effect and fixed‐effect of site, treatment, age group, week and week‐by‐treatment interaction. Response data were analysed using Cochrane‐Mantel Haenszel methods with centres as strata. Last‐observation‐carried‐forward (LOCF) analysis for responder outcome but not clear for Child Depression Rating Scale‐Revised (CDRS‐R). LOCF data were used in analysis of covariance with treatment group, age and baseline effects as covariates. |
| Selective reporting (reporting bias) | High risk | Data were not given separately for each individual trial. The trial reported several response data sets, some weekly data and they looked at individual items in their measures. While the paper did not report on remission as an outcome, the MHRA report did have these data by group. Response data were given as percentages in the paper and these data did not match MHRA data. Denominators for response and remission in the MHRA data were different. They did not report total adverse event rate. |
| Other bias | High risk | Contact: authors stated there were "frequent follow‐up visits" (pg.1039)
and regular measurements taken. They were also allowed to receive therapy
(pg.1035). Screening: diagnostic criteria had to be met at the first and third visits during a 2‐week screening period (total of 3 visits in the screening period). Placebo lead‐in: no Baseline imbalance: authors stated there were no differences between the groups except for gender (more females than males). Other: this was mostly a first episode population; there were 2 studies reported in the one paper; trial 2 had much higher response and remission rates, but data were not reported separately in the published paper. |
Wagner Trial 1 & 2 (2003).
| Study characteristics | ||
| Methods | Trial design: randomised controlled trial; multicentre
Power
calculation: yes
Use of diagnostic criteria (or clear specification of
inclusion criteria): yes
Intervention integrity: not
described
Outcome measures described or validated measures used:
yes
Follow‐up assessment points: post‐intervention
No. crossed
over: none Funded by: Pfizer |
|
| Participants | Setting of care: outpatient
Recruitment: no information
Mean
age: not stated for either group
Age range: 6 to 17 years
Gender
(F:M): intervention = 108:81; control = 84:103
Methods used to
diagnose: DSM‐IV confirmed using K‐SADS‐PL, a CDRS‐R score of ≥ 45 and a
CGI‐S score of ≥ 4
During 2‐week screen, had to meet these criteria at
first and third visit.
Diagnosis: MDD
Baseline severity of
depression: CDRS‐R mean (SD) score intervention = 64.3 (11.0); control =
64.6 (11.0); CGI intervention 4.6 (0.6); CGI placebo 4.5 (0.7) Length of current episode: not reported % first episode: intervention 95%; placebo 95% Comorbidity (intervention and control): 40% of participants had at least 1 comorbid condition; the conditions that occurred in at least 5% of patients included anxiety; phobic disorder; adjustment reaction; ODD Location: USA, India, Canada, Costa Rica, Mexico Inclusion criteria: outpatients; aged 6 to 17; MDD at the first and third visits during a 2‐week screen and current episode had to be of at least 6 weeks duration; illness of at least moderate severity Exclusion criteria: Attention deficit hyperactivity disorder; conduct disorder; obsessive compulsive disorder; panic disorder; history of bipolar or current psychotic features; history of psychotic disorders or autistic spectrum disorders; current anorexia nervosa or bulimia nervosa; drug or alcohol abuse/dependence within 6 months or current positive drug screen; pregnant or breast feeding; abnormal electrocardiography (ECG), laboratory test results, vital signs or body weight; current use of other psychotropic medication; intention to commence psychotherapy; requirement of concomitant psychotropic therapy; previous failed response to an SSRI; additionally, trial 2 stated it excluded those requiring inpatient admission. Exclusion of suicidality: previous suicide attempt or current significant suicidal or homicidal risk |
|
| Interventions | Intervention group
Drug: sertraline
Dosage: flexible dosage 25
to 200 mg
Regimen: 25 mg for 3 days; 50 mg till the end of the second
week; increases as indicated by 50 mg per day to a maximum of 200
mg
Length of treatment: 10 weeks Control group: placebo pill |
|
| Outcomes | Definition and assessment of response: we used OC remission: subjects who
no longer met DSM‐IV criteria for a current major depression episode at
endpoint from MHRA (they used responders defined as at least 40% decrease on
Children's Depression Rating Scale ‐ Revised (CDRS‐R))
Depression
symptoms: Childrens Depression Rating Scale (CDRS‐R) Functioning: Childrens Global Assessment Scale (C‐GAS) Suicidal behaviour: events reported as adverse events; no report of continuous outcome Other outcomes: Clinical Global Impressions Scale Severity (CGI‐Severity); Clinical Global Impressions Scale Improvement (CGI ‐ Improvement); clinician‐rated severity; Multidimensional Anxiety Scale for Children (MASC); Pediatric Quality of Life Enjoyment and Satisfaction Questionnaire (PQ‐LES‐Q); adverse events |
|
| Notes | Additional data were sought from authors. No response was
received.
MHRA contacted for additional data for #1001 and 1017, some
of which were provided
MHRA data used in MA as it gave data for each
separate trial and separately for child and adolescents Type of data used for remission/response: last‐observation‐carried‐forward |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "using a computer generated randomisation code" (pg.1034) |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double blind receipt of sertraline or matching placebo" (pg.1034); "trial drug was packaged in identical blister packs...both patients and clinicians were blinded to group assignment" (pg.1035). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No statement |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number eligible: 376 Number randomised: sertraline: 189; placebo: 187; total: 376 Number started trial: sertraline: 189; placebo: 187; total: 376 Number of withdrawals: sertraline: 46; placebo: 31; total: 77 Number analysed post‐intervention: sertraline: 185; placebo: 179; total: 364 Trial 1 Number randomised: 188 Number of withdrawals: 46 Number analysed post intervention: 142 Trial 2 Number randomised: 188 Number of withdrawals: 31 Number analysed post intervention: 157 Reasons for dropout: full list of dropouts and reasons for dropout figure 1 (pg.1036). There were more dropouts due to adverse events reported in the sertraline group. ITT analysis: intention‐to‐treat population was modified... post‐randomisation efficacy data collected...problems with data collection (pg.1036). Only those who received at least one dose of trial medication were included in the efficacy analyses (pg.1036). Statistical methods: used repeated measures mixed‐model analysis with the model including baseline effect as a covariate, random subject effect and fixed‐effect of site, treatment, age group, week and week‐by‐treatment interaction. Response data were analysed using Cochrane‐Mantel Haenszel methods with centres as strata. Last‐observation‐carried‐forward (LOCF) analysis for responder outcome but not clear for Child Depression Rating Scale‐Revised (CDRS‐R). LOCF data were used in analysis of covariance with treatment group, age and baseline effects as covariates. |
| Selective reporting (reporting bias) | High risk | Data were not given separately for each individual trial. The trial reported several response data sets, some weekly data and they looked at individual items in their measures. While the paper did not report on remission as an outcome, the MHRA report did have these data by group. Response data were given as percentages in the paper and these data did not match MHRA data. Denominators for response and remission in the MHRA data were different. They did not report total adverse event rate. |
| Other bias | High risk | Contact: authors stated there were "frequent follow‐up visits" (pg.1039)
and regular measurements taken. They were also allowed to receive therapy
(pg.1035). Screening: diagnostic criteria has to be met at the first and third visits during a 2‐week screening period (total of 3 visits in the screening period). Placebo lead‐in: no Baseline imbalance: authors stated there were no differences between the groups except for gender (more females than males). Other: this was mostly a first‐episode population; there were 2 studies reported in the 1 paper; trial 2 had much higher response and remission rates, but data were not reported separately in the published paper. |
Wagner Trial 2.
| Study characteristics | ||
| Methods | See Wagner Trial 1 & 2 (2003) entry | |
| Participants | See Wagner Trial 1 & 2 (2003) entry | |
| Interventions | See Wagner Trial 1 & 2 (2003) entry | |
| Outcomes | See Wagner Trial 1 & 2 (2003) entry | |
| Notes | See Wagner Trial 1 & 2 (2003) entry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "using a computer generated randomisation code" (pg.1034) |
| Allocation concealment (selection bias) | Unclear risk | No statement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double blind receipt of sertraline or matching placebo" (pg.1034); "trial drug was packaged in identical blister packs...both patients and clinicians were blinded to group assignment" (pg.1035). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No statement |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number randomised: 188 Number of withdrawals: 31 Number analysed post intervention: 157 Reasons for dropout: full list of dropouts and reasons for dropout figure 1 (pg.1036). There were more dropouts due to adverse events reported in the sertraline group. ITT analysis: intention‐to‐treat population was modified... post‐randomisation efficacy data collected...problems with data collection (pg.1036). Only those who received at least one dose of trial medication were included in the efficacy analyses (pg.1036). Statistical methods: used repeated measures mixed‐model analysis with the model including baseline effect as a covariate, random subject effect and fixed‐effect of site, treatment, age group, week and week‐by‐treatment interaction. Response data were analysed using Cochrane‐Mantel Haenszel methods with centres as strata. Last‐observation‐carried‐forward (LOCF) analysis for responder outcome but not clear for Child Depression Rating Scale‐Revised (CDRS‐R). LOCF data were used in analysis of covariance with treatment group, age and baseline effects as covariates. |
| Selective reporting (reporting bias) | High risk | Data were not given separately for each individual trial. The trial reported several response data sets, some weekly data and they looked at individual items in their measures. While the paper did not report on remission as an outcome, the MHRA report did have these data by group. Response data were given as percentages in the paper and these data did not match MHRA data. Denominators for response and remission in the MHRA data were different. They did not report total adverse event rate. |
| Other bias | High risk | Contact: authors stated there were "frequent follow‐up visits" (pg.1039)
and regular measurements taken. They were also allowed to receive therapy
(pg.1035). Screening: diagnostic criteria has to be met at the first and third visits during a 2‐week screening period (total of 3 visits in the screening period). Placebo lead‐in: no Baseline imbalance: authors stated there were no differences between the groups except for gender (more females than males). Other: this was mostly a first‐episode population; there were 2 studies reported. |
Weihs 2018.
| Study characteristics | ||
| Methods | Trial design: Multicentre, randomised, double‐blind placebo‐controlled
trial, parallel group Power calculation: power of 85% to detect 5‐point difference in the primary endpoint between the desvenlafaxine and placebo groups Use of diagnostic criteria (or clear specification of inclusion criteria): Yes. DSM‐IV‐TR criteria for Major Depressive Disorder as the primary diagnosis Intervention integrity: High ‐ 12% of the intention‐to‐treat population discontinued early Outcome measures described or validated measures used: Yes. The primary efficacy outcome was change from baseline in the Children’s Depression Rating Scale‐R total score at week 8. The key secondary efficacy outcome was change from baseline in Clinical Global Impressions‐Severity score; other secondary efficacy outcomes were change from baseline in CGI Scale–Improvement (CGI‐I) score and CGI‐I response at each visit. Follow‐up assessment points: Efficacy assessments were administered at weeks 1, 2, 3, 4, 6, 8, and/or at early termination in the double‐blind phase. A week‐9 assessment was administered after taper or after transition as the baseline assessment for the extension study for those who were continuing. No. crossed: None Funded by: Pfizer Inc. |
|
| Participants | Setting of care: Outpatient
Recruitment: On‐site
psychiatrist
Mean age (SD): Children ‐ intervention = 9.3 (1.4);
reference = 9.6 (1.3) ; placebo = 9.4 (1.3) Adolescents ‐ intervention = 15.0 (1.5); reference = 14.7 (1.6) ;placebo = 14.6 (1.5) Age range: Children: 7‐11 years, adolescents: 12‐18 years Gender (F:M): Children ‐ intervention = 20:23; reference = 14:31; placebo = 23:19 Adolescents ‐ intervention = 43:29; reference = 43:24; placebo = 41:29 Methods used to diagnose: Children’s Depression Rating Scale – Revised Diagnosis: Major depressive disorder Baseline severity of depression: Children – Intervention mean = 56.4 (10.9), reference = 55.0 (8.7); placebo = 57.0 (8.6) Adolescents ‐ Intervention mean = 56.3 (8.8), reference = 57.0 (8.1);placebo = 57.1 (9.1) Length of current episode: Duration of most recent episode Children – Intervention = 8 months (1‐71); reference = 6 months (1‐42); control = 11 months (1‐57) Adolescents ‐ Intervention = 7 months (1‐61); reference = 7 months (1‐96); control = 8 months (1‐69) % first episode: Not reported Comorbidity: information available only for other psychiatric conditions Intervention: 31.3% Reference: 25.9% Placebo: 33.9% Location: US (35 sites) and Mexico (2 sites) Inclusion criteria: Yes. · male and female outpatients · aged 7 to < 18 years · weighed at least 20 kg at the screening and baseline visits and met criteria for major depressive disorder as the primary diagnosis, had depressive symptoms of at least moderate severity for at least 30 days, and did not require concomitant psychotherapy Exclusion criteria: history or presence of MDD with psychotic features or any psychotic disorder, bipolar disorder (or first‐degree relative with bipolar disorder) or manic episodes or comorbid primary psychiatric condition other than MDD, or a history of or current significant risk of suicide, or first‐degree relative who had committed suicide Exclusion of suicidality: Yes |
|
| Interventions | Intervention group
Drug: Desvenlafaxine
Dosage: Based on the
patient’s body weight at the baseline visit, with 50 mg/d as the highest
dose, as follows: 20 to < 35 kg: 25 mg/d; 35 to < 70 kg: 35 mg/d; and
>= 70 kg: 50mg/day Regimen: Daily Length of treatment: 9 weeks (including 1‐week transition phase or taper phase) Control group: Matching placebo, length of treatment: 9 weeks ‐ once daily for 8 weeks during treatment phase), once daily as appropriate for 1 week during taper/transition phase |
|
| Outcomes | Definition and assessment of response: 5‐point difference in CDRS‐S total
in the primary endpoint between desvenlafaxine and placebo groups (baseline
to 8 weeks). Secondary efficacy outcome ‐ change from baseline CGI‐S score;
change from baseline CGI‐I score; and CGI‐I response score ≤ 2 at each
visit Depressive symptoms: CDRS‐R total score Functioning: not reported Suicidal behaviour: Suicidal ideation or behaviour (C‐SSRS results) Other measures: CGI‐S, CGI‐I and other measures ‐ blood pressure, pulse and weight; lab tests (ALT, AST, bilirubin, cholesterol, triglycerides, prolactin, haematocrit, haemoglobin, leukocytes, ketones, urine protein) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Allocated in a 1:1:1 ratio to the three arms, stratified by age (children [baseline age 7‐11 years] and adolescents [baseline age 12‐17 years] at 1:1 ratio) and country (FDA Statistical Review). Information about the randomisation process not reported |
| Allocation concealment (selection bias) | Unclear risk | Information about allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded – participants and carers. Matching placebo tablets (unclear what it they were matched for) were administered orally, once daily for 8 weeks. Dropout due to adverse events not different across groups |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study used an objective outcome measure – CDRS‐R total score but no
information if the site personnel administering the outcome assessments were
blinded to the intervention arm (only information about receiving intensive training as assessors) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 520 patients screened. Number eligible: 341 (including one person who was screened but not randomised) Number randomised: 340 (excluding one person who was screened but not randomised); desvenlafaxine: 115; fluoxetine group: 113; placebo: 112 Number started trial: desvenlafaxine: 115; fluoxetine group: 112; placebo: 112 Number of withdrawals: 42; desvenlafaxine: 16; fluoxetine: 13; placebo: 13 Number analysed post‐intervention: 337; desvenlafaxine: 115; fluoxetine:110; placebo: 112 Reasons for dropout a. Adverse events Desvenlafaxine: 2 Fluoxetine:1 Placebo: 2 b. Lack of efficacy Desvenlafaxine: 1 Fluoxetine: 0 Placebo: 3 c. Lost‐to‐follow‐up Desvenlafaxine: 6 Fluoxetine: 5 Placebo: 4 d. Protocol violation Desvenlafaxine: 3 Fluoxetine: 0 Placebo: 1 e. No longer willing to participate Desvenlafaxine: 2 Fluoxetine: 7 Placebo: 2 f. Other Desvenlafaxine: 2 Fluoxetine: 0 Placebo: 1 ITT analysis: 337 (130 children and 207 adolescents) ‐ all patients who were randomly assigned to treatment and received at least one dose of study medication and had a baseline and at least one post‐baseline primary efficacy assessment Statistical analysis: Mixed‐effects model for repeated measures (MMRM). Also, the primary and secondary efficacy analyses were all analysed using an ANCOVA models based on the last‐observation‐carried‐forward (LOCF), an ANCOVA model based on the observed cases (OC), and a pattern‐mixture model as sensitivity analyses. |
| Selective reporting (reporting bias) | Unclear risk | All outcomes in methods reported? All outcomes in the methods were
reported, although in the primary paper CDRS‐R scores at weeks 1, 2, 3, 4, 6
were only presented in a graph; more detail was provided in the FDA
Statistical Review. Data available for use in MA? Unclear Access to trial protocol? Access to trial registry document but not protocol (NCT01372150); results synopses available from trial registry and B2061014_Pfizer_Public Disclosure Synopsis and EUCTR2008‐002063‐13 and FDA Statistical Review_B2061014 |
| Other bias | Low risk | Contact: For assessments at weeks 1, 2, 3, 4, 6, 8 Screening: Screening phase up to 28 days prior to baseline/day 1 visit. The MDD diagnosis was confirmed by a psychiatrist at the study site and supported by the K‐SADS‐PL. Placebo lead‐in: No Baseline imbalance: No |
ADD: attention deficit disorder ADHD: attention deficit hyperactivity disorder AE: Adverse event AN: anorexia nervosa ANCOVA: Analysis of Covariance ANOVA: Analysis of Variance ALT:Alanine Aminotransferase AST:Aspartate Aminotransferase BDI: Beck Depression Inventory BMI: Body Mass Index BN: bulmia nervosa BPI: Brief pain inventory BPRS‐C: Brief Psychiatry Rating Scale ‐ Children's β‐hCG: betya human chorionic gonadotrophin CBT: Cognitive behavioural therapy CD: conduct disorder CDI: Children's Depression Inventory CDRS‐R: Children's Depression Rating Scale ‐ Revised C‐GAS: Children's Global Assessment Scale CGI: Clinical Global Impressions Scale CGI‐I: Clinical Global Impressions Scale ‐ Improvement CGI‐S: Clinical Global Impressions Scale ‐ Severity CI: Confidence interval CNS: central nervous system CRO: Contract research organisation C‐SSRS: Columbia Suicide Severity Rating Scale DICA: Diagnostic Interview for Children and Adolescents DSM‐II‐RK‐SADS: Diagnostic and Statistical Manual of Mental Disorder II DSM‐IV‐TR: Diagnostic and Statistical Manual of Mental Disorders IV ‐ Text Revision DSDR: Depression Self Rating Scale DSRS: Depression Self Assessment Scale ECG:Electrocardiogram ECT: electroconvulsive therapy ER: Extended release FDA: US Food and Drug Administration FSIQ: Full Scale Intelligence Quotient GAS: Global Assessment Scale GAD: generalised anxiety disorder GAF: Global Assessment of Functioning GBI: General Behaviour Inventory GSK: GlaxoSmithKline HAMA: Hamilton Anxiety Rating Scale HAM‐D: Hamilton Rating Scale for Depression HARS: Hamilton Anxiety Rating Scale HDRS: Hamilton Depression Rating Scale ITT: intention‐to‐treat IVRS: Interactive voice response systems KADS: Kutcher Adolescent Depression Scale K‐SADS(‐PL): Kiddie Schedule for Affective Disorders and Schizophrenia for School Aged Children (Present and Lifetime version) LOCF: last‐observation‐carried‐forward LS: Least square MA: meta‐analysis MADRS: Montgomery‐Asberg Depression Rating Scale MAOI: Monoamine oxidase inhibitors MASC: Multidimensional Anxiety Scale for Children MC‐SSRS: Modified Columbia Suicide Severity Rating Scale MDD: major depressive disorder MHRA: Committee on Safety of Medicines, Medicines and Healthcare Products Regulatory Agency MINI KIDS: Mini International Neuropsychiatric Interview for children and adolescents MMRM: Mixed model repeated measures NIMH: National Institutes for Mental Health OC: observed case OCD: obsessive–compulsive disorder ODD: oppositional defiant disorder PDD: pervasive developmental disorder PedsQL: Pediatric Quality of Life PGA: Physician Global Assessm PQ‐LES‐Q: Pediatric Quality of Life Enjoyment and Satisfaction Scale PTSD: post‐traumatic stress disorder QD: quaque die: once a day RADS: Reynolds Adolescent Depression Scale REML: Restricted Maximum Likelihood SAE: Serious adverse event SAS: Statistical analysis software SCARED: Screen for Child Anxiety Related Disorders SD: standard deviation SDMT: Symbol digit modalities test SE: standard error SIQ‐JR: Suicidal Ideation Questionnaire‐Junior High School Version SNRI: Serotonin‐norepinephrine reuptake inhibitor SSRI: Selective serotonin reuptake inhibitors TADS: Treatment for Adolescents with Depression (study) TEAE: Treatment emergent adverse effects WSAS: Weinberg Screening Affective Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Atkinson 2018 extension phase | Uncontrolled extension‐phase trial (for acute phase see included studies: Atkinson 2018 and Weihs 2018) |
| Braconnier 2003 | Comparison was not placebo; paroxetine was compared with clomipramine |
| Cheung 2016 | Discontinuation‐phase trial. Acute phase was uncontrolled, open‐label (no placebo/antidepressant comparator) |
| Cornelius 2009 | No pure fluoxetine or placebo treatment arm |
| Cornelius 2010 | No pure fluoxetine or placebo treatment arm |
| Cosgrove 1994 | Case trial design |
| Emslie 2008 discontinuation phase | Discontinuation‐phase trial. Acute phase was uncontrolled, open‐label (no placebo/antidepressant comparator) |
| ePOD‐SSRI | Mixed sample of major depressive disorder and/or anxiety disorder |
| Findling 2009 | Focus of the intervention was comorbid substance use rather than depression |
| Henkel 2010 | Wrong age (adult) |
| JPRN‐UMIN000016192 | Wrong comparator (CBT plus duloxetine vs CBT alone) |
| Kennard 2018 | Discontinuation‐phase trial. Acute phase was uncontrolled, open‐label (no placebo/antidepressant comparator) |
| Kennedy 2014 | Wrong age (adult) |
| Liebowitz 2008 | Wrong age (adult) |
| Mandoki 1997 | Comparison of venlafaxine plus psychotherapy with placebo and psychotherapy |
| NCT00005015 | Primary diagnosis of bipolar disorder; trial discontinued |
| NCT00249886 | Maintenance‐phase trial. Acute phase was uncontrolled, open‐label (no placebo/antidepressant comparator) |
| NCT00508859 | Maintenance‐phase trial. Acute phase uncontrolled, open‐label (no placebo/antidepressant comparator) |
| NCT02871297 | Uncontrolled extension‐phase trial (for acute phase, see included and ongoing studies: NCT02709746 and NCT02709655) |
| NCT03436173 | Single‐dose fluoxetine versus peppermint syrup; no depression outcomes planned |
| Riggs 2007 | No pure fluoxetine or placebo treatment arm |
| Sallee 1997 | Antidepressant not on our list of included compounds |
| Tashakori 1997 | Not a RCT |
| Walker 2017 | Primary diagnosis was bipolar disorder |
| Wohlfarth 2007 | Not a RCT |
CBT: Cognitive Behavioural Therapy RCT: randomised controlled trial
Characteristics of studies awaiting classification [ordered by study ID]
Haneveld 2003.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | could not obtain full text |
Characteristics of ongoing studies [ordered by study ID]
EUCTR2015‐002181‐23.
| Study name | EUCTR2015‐002181‐23 |
| Methods | Trial design: randomised placebo‐ and active‐reference controlled trial;
multicentre, individually randomised, 4 parallel groups Power calculation: not stated Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not stated Outcome measures described or validated measures used: yes Follow‐up assessment points: unclear, at least weeks 2, 4, 8 and 12, plus end of treatment follow‐up period (undefined) No. crossed: not stated Funded: Servier (Institut de Recherches Internationales Servier, France) |
| Participants | Setting of care: outpatient and inpatient Recruitment: not stated Mean age (SD): not stated Age range: 7 to 18 years Gender (F:M): not stated Methods used to diagnose: DSM‐IV defined MDD, confirmed by K‐SADS‐PL interview Diagnosis: MDD Baseline severity of depression: not stated Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Comorbidity (control): not stated Location: Bulgaria, Finland, Germany (withdrawn), Hungry, Poland, Romania, Russian Federation, Serbia, South Africa, Ukraine Inclusion criteria:
Exclusion criteria:
Exclusion of suicidality: yes, current suicide risk according to the clinical opinion of the investigator and based on the information obtained during the evaluation of the C‐SSRS‐C |
| Interventions | Intervention group 1
Drug: Agomelatine 10 mg
Dosage: 10
mg
Regimen: Daily: 2.5 mL oral solution (placebo, at wake), 1 tablet
(agomelatine 10 mg, bedtime). Increased to 5 mL (placebo) from week 2 if
insufficient improvement
Length of treatment: 12 weeks. Intervention group 2 Drug: Agomelatine 25 mg Dosage: 25 mg Regimen: Daily: 2.5mL oral solution (placebo, at wake), 1 tablet (agomelatine 25 mg, bedtime). Increased to 5 mL (placebo) from week 2 if insufficient improvement Length of treatment: 12 weeks Intervention group 3 Drug: fluoxetine 10‐20 mg (active reference) Dosage: 10‐20 mg Regimen: Daily: 2.5 mL oral solution (10 mg fluoxetine, at wake), 1 tablet (placebo, bedtime). Increased to 5 mL (20 mg fluoxetine) from week 2 if insufficient improvement Length of treatment: 12 weeks Control group: placebo: 2.5 mL oral solution (at wake), 1 tablet (at bedtime). Increased to 5 mL from week 2 if insufficient improvement |
| Outcomes | Definition and assessment of response: not stated Depressive symptoms: CDRS‐R raw score, change from baseline to week 12 (primary outcome), ADRS (adolescents only, secondary outcome) Functioning: C‐GAS Suicidal behaviour: C‐SSRS Other measures: CGI‐I, CGI‐S, Pediatric Adverse Event Rating Scale (PAERS) |
| Starting date | First enrolment: 23 Feb 2016 Current status: complete (14 Jan 2020), results posted (https://www.clinicaltrialsregister.eu/ entry for 2015‐002181‐23 last updated 24 Jul 2020) |
| Contact information | Email: clinicaltrials@servier.com |
| Notes | Results first available: 24 Jul 2020 (https://www.clinicaltrialsregister.eu/ entry for 2015‐002181‐23).
To be assessed for inclusion in subsequent review update. Servier study ID: CL3‐20098‐076 |
Glod 2004.
| Study name | Glod 2004 |
| Methods | Trial design: randomised controlled trial
Power calculation: not
stated
Use of diagnostic criteria (or clear specification of inclusion
criteria): yes
Intervention integrity: not stated.
Outcome
measures described or validated measures used: yes
Follow‐up
assessment points: not stated
No. crossed over: not stated Funded by: not stated |
| Participants | Setting of care: outpatient
Recruitment: not stated
Mean age:
15.5 years (1.9)
Age range: 12 to 19 years
Gender (F:M):
12:6
Methods used to diagnose: semi‐structured clinical interview
(K‐SADS‐E)
Diagnosis: DSM‐IV‐defined MDD
Baseline severity of
depression: 20.3 (3.7) on the Hamilton Depression Rating
Scale
Comorbidity intervention and control: not stated Location: not stated Inclusion criteria: MDD; no further details stated Exclusion criteria: not stated |
| Interventions | Intervention group: citalopram Drug arm 1: not stated Dosage: not stated Regimen: not stated Length of treatment: 8 weeks Drug arm 2: bupropion Dosage: not stated Regimen: not stated Length of treatment: 8 weeks Control group: placebo Evaluations at baseline: not stated |
| Outcomes | Depressive symptoms: change in Hamilton Depression Rating Scale score |
| Starting date | |
| Contact information | |
| Notes | Dr Carol Glod contacted on 28 November 2011 for additional information |
IRCT138901093607N1.
| Study name | IRCT138901093607N1 |
| Methods | Trial design: Randomised active reference‐controlled trial; 2 parallel
groups Power calculation: not stated Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not stated Outcome measures described or validated measures used: yes Follow‐up assessment points: weeks 2, 4 and 8 No. crossed: not stated Funded by: Kashan University of Medical Sciences |
| Participants | Setting of care: inpatient Recruitment: not stated Mean age (SD): not stated Age range: 8 to 18 years Gender (F:M): not stated Methods used to diagnose: DSM‐IV‐defined MDD Diagnosis: MDD Baseline severity of depression: not stated Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Comorbidity (control): not stated Location: Iran Inclusion criteria: IQ more than 70 characterised by psychologist and psychiatrist via clinical interview, age 8‐17 years, having major depression without psychotic features based on DSM‐IV Exclusion criteria: severe disabling physical illness, other medical problems that counteract with drug use or other antidepressant drugs, severe mental disorders such as psychotic, bipolar disorders, catatonia symptoms, substance abuse Exclusion of suicidality: not stated |
| Interventions | Intervention group 1
Drug: Fluvoxamine
Dosage: 25
mg
Regimen: Daily
Length of treatment: 8 weeks Intervention group 2 Drug: fluoxetine (active reference) Dosage: 10 mg Regimen: Daily Length of treatment: 8 weeks Control group: none (no placebo comparator) |
| Outcomes | Definition and assessment of response: not stated Depressive symptoms: CDI Functioning: C‐GAS Suicidal behaviour: not stated Other measures: not stated |
| Starting date | First enrolment: estimated 23 Aug 2009 Current status: recruitment complete, no results posted (Iranian Registry of Clinical Trials https://en.irct.ir/ entry for IRCT138901093607N1 last updated 4 Jun 2010) |
| Contact information | Public contact: Zahra Sepehrmanesh, MD, Kashan University of Medical
Sciences Email: sepehrmanesh‐z@kaums.ac.ir |
| Notes | Trial registered with IRCT (https://en.irct.ir/) while recruitment was underway IRCT study ID: 3704 |
JPRN‐JapicCTI‐194585.
| Study name | JPRN‐JapicCTI‐194585 |
| Methods | Trial design: randomised placebo‐controlled trial; multicentre,
individually randomised, 2 parallel groups Power calculation: not stated Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not stated Outcome measures described or validated measures used: yes Follow‐up assessment points: not stated No. crossed: not stated Funded by: Mochida Pharmaceutical Co., Ltd |
| Participants | Setting of care: not stated Recruitment: not stated Mean age (SD): not stated Age range: 12 to 17 years Gender (F:M): not stated Methods used to diagnose: DSM‐5‐defined MDD Diagnosis: MDD Baseline severity of depression: not stated Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Co‐morbidity (control): not stated Location: Japan Inclusion criteria: Patients with a primary diagnosis of MDD or persistent depressive disorder completely meeting the criteria of major depressive episode throughout the preceding 1 year according to DSM‐5 Exclusion criteria: Patients with a complication or history of bipolar (and related disorders) or schizophrenia spectrum (and other psychotic disorders) Exclusion of suicidality: yes. Patients with a history of suicidal behavior (suicide attempt, interrupted suicide attempts) based on C‐SSRS within one year prior to screening |
| Interventions | Intervention group 1
Drug: Escitalopram
Dosage: not
stated
Regimen: not stated
Length of treatment: not
stated
Control group: placebo |
| Outcomes | Definition and assessment of response: Time to relapse (no definition
stated; primary outcome) Depressive symptoms: change in CDRS‐R total score (time point not stated; secondary outcome) Functioning: not stated Suicidal behaviour: C‐SSRS Other measures: not stated |
| Starting date | First enrolment: 24 Jan 2019 Current status: active, recruiting (https://www.clinicaltrials.jp/ entry for JapicCTI‐194585 last updated 10 Jul 2020) |
| Contact information | Email: clinical.trials.contact@mochida.co.jp |
| Notes | Mochida study ID: MLD5511P31 |
NCT00353028.
| Study name | NCT00353028 |
| Methods | Trial design: randomised controlled trial; multicentre
Power
calculation: not stated
Use of diagnostic criteria (or clear
specification of inclusion criteria): yes
Intervention integrity:
NA
Outcome measures described or validated measures used:
yes
Follow‐up assessment points: 8 weeks
No. crossed over: not
stated Funded by: Solvay Pharmaceuticals |
| Participants | Setting of care: not stated
Recruitment: not stated
Mean age:
not stated
Age range: 8 to 18 years
Gender (F:M):
NA
Methods used to diagnose: the Japanese Version of the Structured
Interview Guide for the Hamilton Depression Rating Scale (JSIGH‐D) 17‐item
total score
Diagnosis: depression or depressive state
Baseline
severity of depression: not stated
Comorbidity intervention and
control: NA Location: Japan Inclusion criteria: a minimum total score of 18 on the JSIGH‐D, weight within the standard weight ± 2 SD based on the standard weight for each age in the School Health Statistical Survey Exclusion criteria: predominant psychiatric diagnosis ‐ schizophrenia, or previously been treated with fluvoxamine maleate |
| Interventions | Intervention group: Drug arm 1: fluvoxamine maleate Dosage: 25 mg to 150 mg (1 to 6 tablets) Regimen: once daily Length of treatment: 8 weeks Control group: placebo Evaluations at baseline, 8 weeks |
| Outcomes | Depressive symptoms: time of onset of 50% decrease from baseline in the
JSIGH‐D Functioning: the Clinical Global Impression scale (CGI) |
| Starting date | |
| Contact information | |
| Notes | Contacted Toshiaki Yamaguchi, trial director at Solvay Pharmaceuticals on 13 October 2011 regarding trial status, however no reply received at time of publication |
NCT00426946.
| Study name | NCT00426946 |
| Methods | Trial design: randomised, active reference‐controlled trial; 2 parallel
groups Power calculation: not stated Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not stated Outcome measures described or validated measures used: yes Follow‐up assessment points: weeks 2, 4, 6, 8 (end of treatment), 12 (4 weeks post‐treatment) No. crossed: not stated Funded by: unclear, study sponsor: Geha Mental Health Center (Israel) |
| Participants | Setting of care: not stated Recruitment: not stated Mean age (SD): not stated Age range: 6 to 18 years Gender (F:M): not stated Methods used to diagnose: DSM‐IV‐TR defined MDD or dysthymic disorder, confirmed by psychiatric assessment by a child and adolescent psychiatrist Diagnosis: MDD or dysthymic disorder Baseline severity of depression: not stated Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Comorbidity (control): not stated Location: Israel Inclusion criteria:
Exclusion criteria:
Exclusion of suicidality: not stated |
| Interventions | Intervention group 1
Drug: Reboxetine
Dosage: 4
mg
Regimen: Daily
Length of treatment: 8 weeks (dosage was
either maintained at 4 mg/day or increased to 8 mg/day following a week‐4
assessment) Intervention group 2 Drug: fluoxetine (active reference) Dosage: 20 mg Regimen: Daily Length of treatment: 8 weeks (dosage was either maintained at 20 mg/day or increased to 40 mg/day following a week‐4 assessment) Control group: none (no placebo comparator) |
| Outcomes | Definition and assessment of response: not stated Depressive symptoms: CDI, CDRS‐R Functioning: not stated Suicidal behaviour: SIQ‐SV Other measures: CGI‐I, CGI‐S, RCMAS, DSM‐IV ADHD scale |
| Starting date | First enrolment: Jan 2005 Current status: unknown, no results posted (https://clinicaltrials.gov/ entry for NCT00426946 last updated 25 Jan 2007) |
| Contact information | Paz Toren, Geha Mental Health Center, Israel Email: ptoren@post.tau.ac.il |
| Notes | Newly identified publication reporting partial or complete study
results: Toren 2019. To be
assessed for inclusion in subsequent review update Other study ID: TACMHC1 |
NCT01185977.
| Study name | NCT01185977 |
| Methods | Trial design: randomised placebo‐controlled trial; individually randomised,
2 parallel groups Power calculation: not stated Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not stated Outcome measures described or validated measures used: yes Follow‐up assessment points: weeks 1, 2, 4, and 8 No. crossed: not stated Funded by: unclear, study sponsor: University of California, Los Angeles |
| Participants | Setting of care: outpatient Recruitment: not stated Mean age (SD): not stated Age range: 14 to 18 years Gender (F:M): not stated Methods used to diagnose: DSM‐IV‐defined MDD, confirmed by K‐SADS‐PL Diagnosis: MDD Baseline severity of depression: not stated Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Comorbidity (control): not stated Location: United States Inclusion criteria:
Exclusion criteria: subjects will have no unstable medical illness that would prevent completion of participation in the trial (determined as needed from physical examination, ECG, laboratory safety tests, and review of systems). Other specific exclusionary criteria also are based on the BRITE‐MD parameters, and include:
Exclusion of suicidality: yes. Patients with suicidal ideation are eligible only if the thoughts of death or of life not being worth living are not accompanied by a plan or intention for self‐harm. |
| Interventions | Intervention group 1
Drug: fluoxetine.
Dosage: 20 mg (10 mg/day
for 4 days, then 20 mg/day thereafter)
Regimen: Daily
Length of
treatment: 8 weeks (plus 1‐week single‐blind placebo lead‐in phase) Control group: placebo pill, one pill daily for 4 days, then two pills daily thereafter |
| Outcomes | Definition and assessment of response: not stated Depressive symptoms: CDRS‐R (primary outcome), HAM‐D (secondary outcome) Functioning: not stated Suicidal behaviour: measured at each visit, but not stated Other measures: electroencephalography (EEG) |
| Starting date | First enrolment: Apr 2010 Current status: complete Oct 2011, no results published (https://clinicaltrials.gov/ entry for NCT01185977 last updated 3 Jul 2014) |
| Contact information | Ian A. Cook, M.D., University of California, Los Angeles Email: icook@ucla.edu |
| Notes | Other study ID: 090713201 |
NCT02129751.
| Study name | NCT02129751 |
| Methods | Trial design: randomised placebo‐controlled trial; multicentre,
individually randomised, 2 parallel groups Power calculation: not stated Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not stated Outcome measures described or validated measures used: yes Follow‐up assessment points: unclear; stated measurement timeframe is baseline to 2 years. No. crossed: not stated Funded by: Bausch Health Americas, Inc. |
| Participants | Setting of care: outpatient Recruitment: not stated Mean age (SD): not stated Age range: 7 to 17 years Gender (F:M): not stated Methods used to diagnose: DSM‐IV‐TR‐defined MDD (K‐SADS‐PL interview) Diagnosis: MDD Baseline severity of depression: not stated Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Comorbidity (control): not stated Location: United States (multicentre) Inclusion criteria:
Exclusion criteria:
Exclusion of suicidality: yes, previous history of attempted suicide excluded |
| Interventions | Intervention group 1
Drug: Bupropion hydrobromide
(Aplenzin)
Dosage: not stated
Regimen: not stated
Length
of treatment: unclear (measurement timeframe stated as baseline to 2
years) Control group: placebo |
| Outcomes | Definition and assessment of response/remission: Response = proportion of patients with ≥ 40% improvement from baseline to end‐of‐treatment on total CDRS‐R raw score (secondary outcome) Remission = proportion of patients with total CDRS‐R raw score < 29 at end‐of‐treatment (secondary outcome) Depressive symptoms: CDRS‐R total score, change from baseline to end‐of‐treatment (primary outcome) Functioning: not stated Suicidal behaviour: C‐SSRS Other measures: CGI, adverse events, vital signs and blood/urine laboratory panels, ECG, sleep subscale of the CDRS‐R |
| Starting date | First enrolment: estimated Apr 2021 Current status: active, not yet recruiting (https://clinicaltrials.gov/ entry for NCT02129751 last updated 3 Sept 2020) |
| Contact information | Study Director: Johnson Varughese, Bausch Health Americas, Inc. Public contact: Sandra Narain Email: sandra.narain@bauschhealth.com |
| Notes | Bausch Health Americas Study ID: V01‐BUPA‐401 |
NCT02431806.
| Study name | NCT02431806 |
| Methods | Trial design: randomised placebo‐ and active reference‐controlled trial;
multicentre, individually randomised, 4 parallel groups Power calculation: yes, including interim analysis and updated sample size calculation (see protocol pg.59‐60) Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: yes, dispensed medication blister cards returned at next study visit and number of capsules taken noted in medical record Outcome measures described or validated measures used: yes Follow‐up assessment points: week 8 No. crossed: not stated Funded by: Forest Laboratories (Allergan affiliate) |
| Participants | Setting of care: outpatient Recruitment: not stated Mean age (SD): not stated Age range: 12 to 17 years Gender (F:M): not stated Methods used to diagnose: DSM‐IV‐TR‐defined MDD, confirmed by K‐SADS‐PL interview Diagnosis: MDD Baseline severity of depression: not stated Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Comorbidity (control): not stated Location: Puerto Rico, United States Inclusion criteria:
Exclusion criteria:
Exclusion of suicidality: yes. Significant suicide risk excluded: suicide attempt within the past year OR investigator judgement (based on psychiatric interview and C‐SSRS) |
| Interventions | Intervention group 1
Drug: Levomilnacipran 40 mg
Dosage: 40
mg
Regimen: Daily (1 levomilnacipran capsule, 1 dose‐matched placebo
capsule)
Length of treatment: 8 weeks (plus 1‐week taper) Intervention group 2 Drug: Levomilnacipran 80 mg Dosage: 80 mg Regimen: Daily (2 levomilnacipran capsules) Length of treatment: 8 weeks (plus 1‐week taper) Intervention group 3 Drug: fluoxetine (active reference) Dosage: 20 mg Regimen: Daily (1 fluoxetine 10 mg capsule or 20 mg tablet plus 1 dose‐matched placebo capsule) Length of treatment: 8 weeks (plus 1‐week taper) Control group: placebo, 2 dose‐matched over‐encapsulated placebo capsules |
| Outcomes | Definition and assessment of response: not stated Depressive symptoms: CDRS‐R total score, change from baseline to week 8 (primary outcome) Functioning: not stated Suicidal behaviour: C‐SSRS Other measures: CGI‐S, CGI‐I |
| Starting date | First enrolment: 23 Jun 2015 Current status: complete (19 Aug 2019), results posted (https://clinicaltrials.gov/ entry for NCT02431806 last updated 7 Sept 2020) |
| Contact information | Study director: Daniel Radecki, Forrest Laboratories, Allergan Email: clinicaltrials@allergan.com |
| Notes | Results first available: 7 Sept 2020 (https://clinicaltrials.gov/ entry
for NCT02431806). To be assessed for inclusion in subsequent
update Study Protocol: https://clinicaltrials.gov/ProvidedDocs/06/NCT02431806/Prot_001.pdf Statistical Analysis Plan: https://clinicaltrials.gov/ProvidedDocs/06/NCT02431806/SAP_000.pdf Forest Laboratories/Allergan study ID: LVM‐MD‐11 |
NCT02709655.
| Study name | NCT02709655 |
| Methods | Trial design: randomised placebo‐ and active reference‐controlled trial;
multicentre, individually randomised, 4 parallel groups Power calculation: not stated Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not stated Outcome measures described or validated measures used: yes Follow‐up assessment points: week 12 No. crossed: not stated Funded by: H. Lundbeck A/S |
| Participants | Setting of care: outpatient Recruitment: not stated Mean age (SD): not stated Age range: 7 to 11 years Gender (F:M): not stated Methods used to diagnose: DSM‐5‐defined MDD Diagnosis: MDD Baseline severity of depression: not stated Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Comorbidity (control): not stated Location: Austria, Belgium, Bulgaria, Canada, Colombia, Denmark, Estonia, Finland, France, Germany, Hungary, Ireland, Israel, Italy, Republic of Korea, Latvia, Lithuania, Mexico, Netherlands, Poland, Romania, Russian Federation, Serbia, Slovakia, South Africa, Spain, Sweden, Ukraine, United Kingdom, United States Inclusion criteria:
Exclusion criteria:
Other protocol defined inclusion and exclusion criteria may apply. Exclusion of suicidality: not stated |
| Interventions | Intervention group 1
Drug: vortioxetine 10 mg
Dosage: 10
mg
Regimen: Daily
Length of treatment: 12 weeks. Intervention group 2 Drug: vortioxetine 20 mg Dosage: 20 mg Regimen: Daily Length of treatment: 12 weeks Intervention group 3 Drug: fluoxetine (active reference) [NB a decision has been taken to stop recruitment into this treatment arm]. Dosage: 20 mg Regimen: Daily Length of treatment: 12 weeks Control group: placebo, encapsulated tablet, orally |
| Outcomes | Definition and assessment of response/remission: Response = 50% reduction in CDRS‐R total score from baseline to week 12 Remission = CDRS‐R total score ≤ 28 at each assessment visit (to 12 weeks) Depressive symptoms: CDRS‐R total score, change from baseline to week 12 (primary outcome) Functioning: C‐GAS Suicidal behaviour: not stated Other measures: CGI‐S, CGI‐I, GBI, PGA, PedsQL‐VAS, PQ‐LES‐Q |
| Starting date | First enrolment: May 2016 Current status: active, recruiting (https://clinicaltrials.gov/ entry for NCT02709655 last updated 9 Feb 2021) |
| Contact information | Lundbeck A/S +4536301311 LundbeckClinicalTrials@Lundbeck.com |
| Notes | Protocol changes: a decision was made to stop recruitment into the
fluoxetine arm (change first appears in 6 Jan 2020 version of https://clinicaltrials.gov/ entry
for NCT02709655) Lundbeck study ID: 12709A EU clinical trials registry EudraCT Number: 2008‐005353‐38 |
NCT03315793.
| Study name | NCT03315793 |
| Methods | Trial design: randomised placebo‐controlled trial; multicentre,
individually randomised, 2 parallel groups Power calculation: estimated 66 subjects per group (total 132) required to achieve a detection power of 80% or higher in a two‐sample t‐test at a two‐sided significance level of 0.05. Predicting 10% dropout, target increased to 74 subjects per group (total 148). Assumptions: 5.9 point difference on change‐from‐baseline to week 6 on CDRS‐R between duloxetine and placebo. SD of change = 12 for both groups, estimated effect size of 0.49. (protocol pg.52) Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not stated Outcome measures described or validated measures used: yes Follow‐up assessment points: weeks 1, 2, 3, 5 and 6 (plus 1‐week taper) No. crossed: not stated Funded by: Eli Lilly and Company, and Shionogi Inc. (co‐sponsors) |
| Participants | Setting of care: outpatient and inpatient Recruitment: not stated Mean age (SD): not stated Age range: 9 to 17 years Gender (F:M): not stated Methods used to diagnose: DMS‐5‐defined MDD or persistent depressive disorder, confirmed by Mini International Neuropsychiatric Interview for Children and Adolescents (MINI‐KID) Diagnosis: MDD or persistent depressive disorder Baseline severity of depression: not stated Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Comorbidity (control): not stated Location: Japan Inclusion criteria:
Exclusion criteria:
Exclusion of suicidality: yes, either (i) suicidal ideation and/or suicidal attempt within 1 year before visit 1; or (ii) at visits 1 and 2, answer “Yes” on questions 4 and/or 5 about suicidal ideation, and/or any of the questions about suicidal behaviours (except those about non‐suicidal self‐injurious behavior), on the C‐SSRS |
| Interventions | Intervention group 1
Drug: duloxetine
Dosage: flexible, 40‐60
mg
Regimen: Daily
Length of treatment: 6 weeks (plus 1‐3 weeks
screening, 1‐week tapering period and 1‐week follow‐up period) Control group: placebo |
| Outcomes | Definition and assessment of response: 30% response = proportion of
subjects with CDRS‐R total score decreases of 30% or more from baseline to
week 6 (secondary outcome); 50% response = proportion of subjects with
CDRS‐R total score decreases of 50% or more from baseline to week 6
(secondary outcome); remission rate = proportion of subjects with CDRS‐R
total score of 28 or lower (secondary outcome) Depressive symptoms: CDRS‐R, change from baseline to week 6 (primary outcome) Functioning: not stated Suicidal behaviour: C‐SSRS Other measures: CGI‐S |
| Starting date | First enrolment: 4 Dec 2017 Current status: complete (Nov 2019), results posted (https://clinicaltrials.gov/ entry for NCT03315793 last updated 5 Jan 2021) |
| Contact information | Email: shionogiclinicaltrials-admin@shionogi.co.jp |
| Notes | Results first available: 5 Jan 2021 from https://clinicaltrials.gov/ entry
NCT03315793. To be assessed for inclusion in subsequent update. Protocol: https://clinicaltrials.gov/ProvidedDocs/93/NCT03315793/Prot_000.pdf Statistical Analysis Plan: https://clinicaltrials.gov/ProvidedDocs/93/NCT03315793/SAP_001.pdf Eli Lilly study ID: F1J‐JE‐B058 Shionogi study ID: 1701A3631 Other study ID: 14937 |
NCT03569475.
| Study name | NCT03569475 |
| Methods | Trial design: Randomised placebo‐ and active reference‐controlled trial;
multicentre, individually randomised, 3 parallel groups Power calculation: not stated Use of diagnostic criteria (or clear specification of inclusion criteria): yes Intervention integrity: not stated Outcome measures described or validated measures used: yes Follow‐up assessment points: Week 8 No. crossed: not stated Funded by: Allergan. |
| Participants | Setting of care: outpatient Recruitment: not stated Mean age (SD): not stated Age range: 7 to 17 years Gender (F:M): not stated Methods used to diagnose: DSM‐5‐defined MDD, confirmed by K‐SADS‐PL Diagnosis: MDD Baseline severity of depression: not stated Length of current episode: not stated % first episode: not stated Comorbidity (intervention): not stated Comorbidity (control): not stated Location: United States (47 sites) Inclusion criteria:
Exclusion criteria:
Other exclusion criteria:
Exclusion of suicidality: Significant suicide risk screening or baseline visit judged by the investigator based on psychiatric interview or information collected from the C‐SSRS, or suicide attempt within the past year |
| Interventions | Intervention group 1
Drug: Levomilnacipran ER
Dosage: 40
mg
Regimen: Daily. (10 mg/day for days 1–3, then 20 mg/day for days
4–7, then 40 mg/day from week 2 through 8. Dose increase to 80 mg/day
permitted at week 3 through 8 based on therapeutic response and
tolerability)
Length of treatment: 8 weeks (plus 1‐week taper‐down
period) Intervention group 2 Drug: fluoxetine (active reference) Dosage: 20 mg Regimen: Daily. (10 mg/day for days 1–7, then 20 mg/day from week 2 through 8) Length of treatment: 8 weeks (plus 1‐week taper‐down period) Control group: placebo capsules, once daily |
| Outcomes | Definition and assessment of response: not stated Depressive symptoms: CDRS‐R, change from baseline to week 8 (primary outcome) Functioning: not stated Suicidal behaviour: C‐SSRS Other measures: CGI‐S |
| Starting date | First enrolment: 6 Jul 2018 Current status: complete, no results posted (https://clinicaltrials.gov/ entry for NCT03569475 last updated 1 Mar 2021) |
| Contact information | Study director: Daniel Radecki, PhD, Allergan Email: not provided |
| Notes | Allergan Study ID: LVM‐MD‐14 |
ADHD: Attention deficit hyperactivity disorder ADRS: Adolescent Depression Rating Scale ALP: Alkaline phosphates ALT: Alanine Aminotransferase
AST: Aspartate Aminotransferase
BRITE‐MD: Biomarkers of Antidepressant Treatment in Adolescents with Major Depression CBT: Cognitive Behavioural Therapy CDI: Children's Depression Inventory CDRS‐R: Children's Depression Rating Scale ‐ Revised CGI: Clinical Global Impressions Scale C‐GAS: Clinical Global Assessment Scale CGI‐I: Clinical Global Impression‐Improvement Scale CGI‐S: Clinical Global Impression‐Severity Scale C‐SSRS(‐C): Columbia‐Suicide Severity Rating Scale DSM‐IV‐TR: Diagnostic and Statistical Manual of Mental Disorders IV ‐ Text Revision DSM‐5: Diagnostic and Statistical Manual of Mental Disorders ‐ 5 ECG: Electrocardiogram ECT: Electroconvulsive therapy EEG: Electroencephalogram ER: Extended release FLX: Fluoxetine GBI: General Behaviour Inventory HAM‐D: Hamilton Depression Rating Scale HIPPA: Health insurance portability and accountability Act IQ: Intelligence Quotient JSIGH‐D: Japanese Version of the Structured Interview Guide for the Hamilton Depression Rating Scale K‐SADS(‐PL)(‐E): Schedule for Affective Disorders and Schizophrenia for School Aged Children (Present and Lifetime version)(Epidemiologic) MAOI: Monoamine oxidase inhibitors MDD: major depressive disorder MINI‐KID: Mini International Neuropsychiatric Interview for Children and Adolescents NA: not applicable OC: observed case PAERS: Pediatric Adverse Event Rating Scale PedsQL‐VAS: PedsQL Present Functioning Visual Analogue Scales PGA: Physician Global Assessment PQ‐LES‐Q: Paediatric Quality of Life Enjoyment and Satisfaction Questionnaire QEEG: Quantitative Electroencephalogram RCMAS: Revised Children's Manifest Anxiety Scale RCT: Randomised controlled trial SD: standard deviation SIQ‐SV: Suicide Ideation Questionnaire‐Short Version SSRI: Selective serotonin reuptake inhibitors SNRI: Serotonin and norepinephrine reuptake inhibitors TADS: Treatment of Adolescent Depression Study ULN: Upper limit of normal
Differences between protocol and review
In the protocol, we had originally only set equivalence ranges for outcomes included in the summary of findings. But to aid consistency of interpretation and reported results we set equivalence ranges post‐hoc for all other outcomes.
In our 'Summary of findings' tables, to aid interpretation, we presented estimates of risk difference, in addition to odds ratios. We had intended to present estimates of risk difference for a range of comparator group rates (lowest, highest and median rate derived from comparator groups of included studies). However, for most interventions there were insufficient data; therefore, we only used median rates to derive estimates of risk difference.
In the protocol, regarding subgroup analyses, we stated we would estimate a common regression coefficient in our meta‐regression analyses. However, in the review we decided it would be more appropriate to estimate regression coefficients by comparison rather than using a common regression co‐efficient. Second, in the protocol, we stated that we would assess the extent to which the meta‐regression model reduced the between‐trial variance, but we have not included this as this option is not available in the Stata package we used to analyse the data.
Contributions of authors
SH: Co‐ordinated preparation of the protocol and the review. Drafted the protocol, drafted the discussion and conclusions, abstract and PLS JM: Methodological and statistical oversight of protocol and review write‐up, including interpretation of analyses, assisted with 'Risk of bias' ratings and did CINeMA rating AB: Screening, data extraction, reviewed write‐up of review VS: Screening, data extraction, reviewed write‐up of review CM: Screening, data extraction, reviewed write‐up of review PB: Screening, data extraction, reviewed write‐up of review GC: Screening, data extraction, reviewed write‐up of review SM: Clinical oversight of protocol and review write‐up, including interpretation of analyses NM: Helped to draft the protocol, assisted with 'Risk of bias' ratings and did CINeMA ratings, undertook the analysis, refined write‐up of review
Sources of support
Internal sources
University of Auckland, New Zealand
Monash University, Australia
The University of Melbourne, Australia
External sources
-
Auckland Medical Research Foundation, New Zealand
SH salary is supported by a Douglas Goodfellow Repatriation Grant
-
A Better Start, National Science Challenge, Ministry of Business, Innovation & Employment, New Zealand
SH salary is support by A Better Start, National Science Challenge (UOAX1901)
-
Cure Kids, New Zealand
SH salary is supported by Cure Kids and SM holds the Cure Kids Duke Family Chair in Child and Adolescent Mental Health
-
National Health and Medical Research Council (NHMRC), Australia
JM is supported by a NHMRC Career Development Fellowship (1143429)
-
National Institute for Health Research (NIHR), UK
NM contribution to this review is supported by Cochrane Infrastructure funding to the Common Mental Disorders Cochrane Review Group
Declarations of interest
SH: is a PI on the YoDA‐C study — a trial of CBT and fluoxetine versus CBT and placebo. She is the Joint Co‐ordinating Editor of the Cochrane Common Mental Disorders Group. She is funded by the Auckland Medical Research Foundation and CureKids. JM: no conflicts of interest AB: no conflicts of interest VS: no conflicts of interest CM: no conflicts of interest PB: no conflicts of interest GC: no conflicts of interest SM: led the development of SPARX, a computerised intervention for depression in young people. In the event of successful commercialisation of SPARX, Dr Merry would receive a portion of the profits. NM: is the Deputy Co‐ordinating Editor of the Cochrane Common Mental Disorders Group.
New
References
References to studies included in this review
Almeida‐Montes 2005 {published data only}
- Almeida-Montes LG, Friederichsen A. Treatment of major depressive disorder with fluoxetine in children and adolescents. A double-blind, placebo-controlled study [Tratamiento del trastorno depresivo mayor con fluoxetina en ninos y adolescentes. Estudio doble ciego, controlado con placebo]. Psiquiatria Biologica 2005;12(5):198-205. [3331112] [Google Scholar]
Atkinson 2014 {published data only}
- Atkinson SD, Prakash A, Zhang Q, Pangallo BA, Bangs ME, Emslie GJ, et al. A double-blind efficacy and safety study of duloxetine flexible dosing in children and adolescents with major depressive disorder. Journal of Child and Adolescent Psychopharmacology 2014;26(4):180-9. [DOI: 10.1089/cap.2013.0146] [DOI] [PubMed] [Google Scholar]
- Bliznak L, Berg R, Hage A, Dittmann RW. High rate of non-eligibility: methodological factors impacting on recruitment for a multicentre, double-blind study of paediatric patients with major depressive disorder. Pharmacopsychiatry 2013;46(1):23-8. [3331228] [DOI] [PubMed] [Google Scholar]
- Emslie GJ, Wells TG, Prakash A, Zhang Q, Pangallo BA, Bangs ME, et al. Acute and longer-term safety results from a pooled analysis of duloxetine studies for the treatment of children and adolescents with major depressive disorder. Journal of Child and Adolescent Psychopharmacology 2015;25(4):293-305. [DOI] [PubMed] [Google Scholar]
- EUCTR2008-006492-71-FI. A double-blind, efficacy and safety study of duloxetine versus placebo in the treatment of children and adolescents with major depressive disorder. clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2008-006492-71 (first received 2 December 2008).
- Lobo ED, Quinlan T, Prakash A. Pharmacokinetics of orally administered duloxetine in children and adolescents with major depressive disorder. Clinical Pharmacokinetics 2014;53(8):731-40. [DOI] [PubMed] [Google Scholar]
- NCT00849901. A double-blind, efficacy and safety study of duloxetine versus placebo in the treatment of children and adolescents with major depressive disorder. clinicaltrials.gov/ct2/show/NCT00849901 (first received 24 February 2009). [3331229]
Atkinson 2018 {published data only}
- Atkinson S, Lubaczewski S, Ramaker S, England RD, Wajsbrot DB, Abbas R, et al. Desvenlafaxine versus placebo in the treatment of children and adolescents with major depressive disorder. Journal of Child and Adolescent Psychopharmacology 2018;28(1):55-65. [DOI: 10.1089/cap.2017.0099] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01371734. A study of DVS SR In treatment of children and adolescent outpatients with MDD. clinicaltrials.gov/show/NCT01371734 (first received 20 March 2017).
Berard 2006 {published data only}
- Berard R, Fong R, Carpenter DJ, Thomason C, Wilkinson C. An international, multicentre, placebo-controlled trial of paroxetine in adolescents with major depressive disorder. Journal of Child and Adolescent Psychopharmacology 2004;16(1-2):59-75. [3331114] [DOI] [PubMed] [Google Scholar]
- GlaxoSmithKline. A double-blind, multicentre placebo controlled study of paroxetine in adolescents with unipolar major depression [Study 377]. www.gsk.com/media/paroxetine.htm (accessed 16 August 2004). [3331115]
- Medicines and Healthcare products Regulatory Agency (MHRA). Paroxetine Study 2 ID#377. medicines.mhra.gov.uk (accessed 20 June 2004). [3331116]
- Milin R, Simeon J, Spenst W. Double-blind study of paroxetine in adolescents with unipolar major depression. Unpublished manuscript; found via pharmaceutical webpage. [3331117]
Durgam 2018 {published data only}
- Durgam S, Chen C, Migliore R, Prakash C, Edwards J, Findling RL. A phase 3, double-blind, randomized, placebo-controlled study of vilazodone in adolescents with major depressive disorder. Pediatric Drugs 2018;20:353-63. [DOI: 10.1007/s40272-018-0290-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01878292. Safety and efficacy of vilazodone in adolescent patients with major depressive disorder. ClinicalTrials.gov/show/NCT01878292 (first received 14 June 2013).
Emslie 1997 {published data only}
- Emslie GJ, Rush AJ, Weinberg WA, Kowatch RA, Carmody T, Mayes TL. Fluoxetine in child and adolescent depression: acute and maintenance treatment. Depression and Anxiety 1998;7:32-9. [3331119] [DOI] [PubMed] [Google Scholar]
- Emslie GJ, Rush AJ, Weinberg WA, Kowatch RA, Hughes CW, Carmody T. A double-blind placebo controlled trial of fluoxetine in depressed children and adolescents. In: Sixth World Congress of Biological Psychiatry; 1997 June 22-27; Nice, France. 1997. [3331120] [DOI] [PubMed]
- Emslie GJ, Rush R, Weinberg WA, Kowatch WA, Hughes CW, Carmody T, et al. A double blind, randomized, placebo-controlled trial of fluoxetine in children and adolescents with depression. Archives of General Psychiatry 1997;54(11):1031-7. [3331121] [DOI] [PubMed] [Google Scholar]
- Emslie GJ, Weinberg WA, Mayes TL. Treatment of children with antidepressants: focus on selective serotonin reuptake inhibitors. Depression and Anxiety 1998;8(Suppl 1):13-7. [3331122] [PubMed] [Google Scholar]
- Hughes CW, Emslie G, Kowatch R, Weinberg W, Rintelmann J, Rush AJ. Clinician, parent, and child prediction of medication or placebo in double-blind depression study. Neuropsychopharmacology 2000;23(5):591-4. [3331123] [DOI] [PubMed] [Google Scholar]
- Kowatch RA, Carmody TJ, Emslie GJ, Rintelmann JW, Hughes CW, Rush AJ. Prediction of response to fluoxetine and placebo in children and adolescents with major depressive disorder: a hypothesis generating study. Journal of Affective Disorders 1999;54:269-76. [3331124] [DOI] [PubMed] [Google Scholar]
- Medicines and Healthcare products Regulatory Agency (MHRA). Fluoxetine Study 1 ID# X065. medicines.mhra.gov.uk (accessed 20 June 2004). [3331125]
- Rintelmann JW, Emslie GJ, Rush AJ, Varghese T, Gullion CM, Kowatch RA. The effects of extended evaluation on depressive symptoms in children and adolescents. Journal of Affective Disorders 1996;41:149-56. [3331126] [DOI] [PubMed] [Google Scholar]
Emslie 2002 {published data only}
- Emslie GJ, Heiligenstein JH, Hoog SL, Judge R, Brown EB, Nilsson M. Fluoxetine for acute treatment of depression in children and adolescents: a placebo-controlled, randomized clinical trial. In: 39th Annual Meeting of the American College of Neuropsychopharmacology; 2000 Dec 10-14; San Juan, Puerto Rico. 2000:172. [3331128]
- Emslie GJ, Heiligenstein JH, Wagner KD, Hoog SI, Ernest DE, Brown E, et al. Fluoxetine for acute treatment of depression in children and adolescents: a placebo-controlled randomized clinical trial. Journal of the American Academy of Child and Adolescent Psychiatry 2002;41(10):1205-15. [3331129] [DOI] [PubMed] [Google Scholar]
- Jain S, Carmody TJ, Trivedi MH, Hughes C, Bernstein IH, Detal M. A psychometric evaluation of the CDRS and MADRS in assessing depressive symptoms in children. Journal of the American Academy of Child & Adolescent Psychiatry 2007;46(9):1204-12. [3331130] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicines and Healthcare products Regulatory Agency (MHRA). Fluoxetine Study 2 ID# HCJE. medicines.mhra.gov.uk (accessed 20 June 2004). [3331131]
- Nilsson M, Joliat MJ, Miner CM, Brown EB, Heiligenstein JH. Safety of subchronic treatment with fluoxetine for major depressive disorder in children and adolescents. Journal of Child and Adolescent Psychopharmacology 2004;14(3):412-7. [3331132] [DOI] [PubMed] [Google Scholar]
Emslie 2006 {published data only}
- Brooks SJ, Krulewicz MA, Kutcher S. The Kutcher Adolescent Depression Scale: assessment of its evaluative properties over the course of an 8-week pediatric pharmacotherapy trial. Journal of Child and Adolescent Psychopharmacology 2003;13(3):227-349. [3331134] [DOI] [PubMed] [Google Scholar]
- Emslie GJ, Wagner KD, Kutcher S, Krulewicz S, Fong R, Carpenter DJ, et al. Paroxetine treatment in children and adolescents with major depressive disorder: a randomized, multicentre, double-blind, placebo-controlled trial. Journal of the American Academy of Child and Adolescent Psychiatry 2006;45(6):709-19. [3331135] [DOI] [PubMed] [Google Scholar]
- GlaxoSmithKline. A randomized multicentre, 8-week, double-blind, placebo-controlled flexible dose study to evaluate the efficacy and safety of paroxetine in children and adolescents with major depressive disorder (29060/701). www.gsk.com/media/paroxetine.htm (accessed 16 August 2004). [3331136]
- Medicines and Healthcare products Regulatory Agency (MHRA). Paroxetine Study 3 ID#701. medicines.mhra.gov.uk (accessed 20 June 2004). [3331137]
Emslie 2007 {published data only}
- Emslie GJ, Finding RL, Yeung PP, Kunz NR, Li Y, Durn BL. Efficacy and safety of venlafaxine ER in children and adolescents with major depressive disorder (poster presentation). In: American Psychiatric Association; 2004 May 1-6; New York. 2004. [3331139]
- Emslie GJ, Findling RL, Yeung PP, Kunz NR, Li Y. Venlafaxine ER for the treatment of pediatric subjects with depression: results of two placebo-controlled trials. Journal of the American Academy of Child and Adolescent Psychiatry 2007;46(4):479-88. [3331140] [DOI] [PubMed] [Google Scholar]
- Emslie GJ, Yeung PR, Kunz NR, Li Y. Long-term efficacy and safety of venlafaxine ER in children and adolescents with major depressive disorder. In: American Psychiatric Association; 2004 May 1-6; New York. 2004. [3331141]
Emslie 2007 Trial 1 {published data only}
- Wyeth. Double-blind, placebo-controlled study of venlafaxine ER in children and adolescents with major depression. (Protocol 0600B1-382-US (Study Period: October 1997 to September 2000)). clinicalstudyresults.org/documents/company-study_44_0.pdf 2004 (accessed 24 November 2011). [3331143]
Emslie 2007 Trial 2 {published data only}
- Wyeth. Double-blind, placebo-controlled study of venlafaxine ER in children and adolescents with major depressive disorder (Protocol 0600B1-394-US) (Study Period: August 2000 to August 2001). clinicalstudyresults.org/documents/company-study_2252_0.pdf 2004 (accessed 24 November 2011). [3331145]
Emslie 2009 {published data only}
- Emslie GJ, Ventura D, Korotzer A, Tourkodimitris S. Escitalopram in the treatment of adolescent depression: a randomized placebo-controlled multisite trial. Journal of the American Academy of Child and Adolescent Psychiatry 2009;48(7):721-9. [3331147] [DOI] [PubMed] [Google Scholar]
Emslie 2014 {published data only}
- Emslie GJ, Prakash A, Zhang Q, Pangallo BA, Bangs ME, March JS. A double-blind efficacy and safety study of duloxetine fixed doses in children and adolescents with major depressive disorder. Journal of Child and Adolescent Psychopharmacology 2014;24(4):170-9. [DOI: 10.1089/cap.2013.0096] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emslie GJ, Wells TG, Prakash A, Zhang Q, Pangallo BA, Bangs ME, et al. Acute and longer-term safety results from a pooled analysis of duloxetine studies for the treatment of children and adolescents with major depressive disorder. Journal of Child and Adolescent Psychopharmacology 2015;25(4):293-305. [DOI] [PubMed] [Google Scholar]
- Lobo ED, Quinlan T, Prakash A. Pharmacokinetics of orally administered duloxetine in children and adolescents with major depressive disorder. Clinical Pharmacokinetics 2014;53(8):731-40. [DOI] [PubMed] [Google Scholar]
- NCT00849693. A double-blind, efficacy and safety study of duloxetine versus placebo in the treatment of children and adolescents with major depressive disorder. clinicaltrials.gov/ct2/show/NCT00849693 (first received 19 April 2012). [3331224]
Keller 2001 {published data only}
- GlaxoSmithKline. A multi-centre, double-blind, placebo controlled study of paroxetine and imipramine in adolescents with unipolar major depression - acute phase (29060/329). gsk.com/media/paroxetine.htm (accessed 16 August 2004). [3331149]
- Jureidini J, Tonkin A. Paroxetine in major depression. Journal of the American Academy of Child and Adolescent Psychiatry 2003;42(5):514. [3331150] [DOI] [PubMed] [Google Scholar]
- Keller MB, Ryan ND, Birmaher B, Klein RG, Strober M, Wagner K. Paroxetine and imipramine in the treatment of adolescent depression [Abstract NR206]. In: 151st Annual Meeting of the American Psychiatric Association: New Research Program and Abstracts; 1998 May 30 - June 4; Toronto, Ontario, Canada. 1998:123. [3331151]
- Keller MB, Ryan ND, Strober M, Klein RG, Kutcher SP, Birmaher B, et al. Efficacy of paroxetine in the treatment of adolescent major depression: a randomised controlled trial. Journal of the American Academy of Child and Adolescent Psychiatry 2001;40(7):762-72. [3331152] [DOI] [PubMed] [Google Scholar]
- Le Noury J, Nardo JM, Healy D, Jureidini J, Raven M, Tufanaru C, et al. Restoring Study 329: efficacy and harms of paroxetine and imipramine in treatment of major depression in adolescence. British Medical Journal 2015;351:h4320. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicines and Healthcare products Regulatory Agency (MHRA). Paroxetine Study 1 ID#329. medicines.mhra.gov.uk (accessed 20 June 2004). [3331153]
Mirtazapine Trial 1 {published data only}
- Anonymous. Rapporteurs public assessment report for paediatric data in EU worksharing procedure: Mirtazapine (Remeron) UK/H/0016/pdWS/001. hma.eu/fileadmin/dateien/Human_Medicines/CMD_h_/Paediatric_Regulation/Assessment_Reports/Article_45_work-sharing/Mirtazapine_2010_07_45PaedPAR.pdf) 2010 (accessed prior to 12 March 2021). [3331155]
- Food and Drug administration. Pediatric supplement SE05-11 Executive Summary. FDA, 2001. fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM164066.pdf (accessed prior to 1 November 2012). [3331156]
Mirtazapine Trial 1 & 2 {published data only}
- Anonymous. Rapporteurs public assessment report for paediatric data in EU worksharing procedure: Mirtazapine (Remeron) UK/H/0016/pdWS/001. hma.eu/fileadmin/dateien/Human_Medicines/CMD_h_/Paediatric_Regulation/Assessment_Reports/Article_45_work-sharing/Mirtazapine_2010_07_45PaedPAR.pdf 2010 (accessed prior to 12 March 2021). [3331159]
- Food and Drug administration. Pediatric supplement SE05-11 Executive Summary. FDA, 2001. fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM164066.pdf (accessed prior to 1 November 2012). [3331160]
- Organon PUSAI. A multicenter, randomized, double-blind, placebo-controlled, efficacy and safety study of Remeron® in outpatient children and adolescents with major depressive disorder [Studies 1 and 2]. clinicalstudyresults.org/documents/company-study_51_0.pdf 2001 (accessed 24 November 2011). [3331158]
Mirtazapine Trial 2 {published data only}
- Anonymous. Rapporteurs public assessment report for paediatric data in EU worksharing procedure: Mirtazapine (Remeron) UK/H/0016/pdWS/001. hma.eu/fileadmin/dateien/Human_Medicines/CMD_h_/Paediatric_Regulation/Assessment_Reports/Article_45_work-sharing/Mirtazapine_2010_07_45PaedPAR.pdf 2010 (accessed prior to 12 March 2021). [3331162]
- Food and Drug administration. Pediatric supplement SE05-11 Executive Summary. FDA, 2001. www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM164066.pdf (accessed prior to 1 November 2012). [3331163]
NCT02709746 {published data only}
- 2008-005354-20. Interventional, randomised, double-blind, placebo-controlled,active reference (fluoxetine), fixed-dose study of vortioxetine in paediatric patients aged 12 to 17 years, with Major Depressive Disorder (MDD). clinicaltrialsregister.eu/ctr-search/search?query=2008-005354-20 (first received 15 March 2016). [2008-005354-20]
- NCT02709746. Active reference (fluoxetine) fixed-dose study of vortioxetine in paediatric patients aged 12 to 17 years with major depressive disorder (MDD). ClinicalTrials.gov/show/NCT02709746 (first received 16 March 2016).
Paroxetine Trial 1 {published data only}
- GlaxoSmithKline. A randomised, double-blind, placebo controlled, parallel group, flexible dose study to evaluate the efficacy and safety of Paxil® tablets in children and adolescents with major depressive disorder (post-marketing clinical study). gsk-clinicalstudyregister.com (first received 1 February 2011). [3331165]
Simeon 1990 {published data only}
- Simeon JG, Dinicola VF, Ferguson HB, Copping W. Adolescent depression: a placebo controlled fluoxetine treatment study and follow-up. Progress in Neuro-Psychopharmacology and Biological Psychiatry 1990;14:791-5. [3331167] [DOI] [PubMed] [Google Scholar]
TADS 2004 {published data only}
- Emslie G, Kratochvil C, Vitiello B, Silva S, Mayes T, McNulty S, et al. Treatment for Adolescents with Depression Study (TADS): safety results. Journal of the American Academy of Child Adolescent Psychiatry 2006;45(12):1440-55. [3331169] [DOI] [PMC free article] [PubMed] [Google Scholar]
- March J, Silva S, Petrycki S, Curry J, Wells K, Fairbank J, et al. Fluoxetine, cognitive-behavioral therapy, and their combination for adolescents with depression: Treatment for Adolescents with Depression Study (TADS) randomized controlled trial. JAMA 2004;292:807-20. [3331170] [DOI] [PubMed] [Google Scholar]
- Treatment Adolescents with Depression Study Team. Treatment for Adolescents with Depression Study (TADS): rationale, design, and methods. Journal of the American Academy of Child and Adolescent Psychiatry 2003;42:531-42. [3331171] [DOI] [PubMed] [Google Scholar]
- Vitiello B, Silva S, Rohde P, Kratochvil C, Kennard B, Reinecke M. Suicidal events in the Treatment for Adolescents with Depression Study (TADS). Journal of Clinical Psychiatry 2009;70(5):741–7. [3331172] [DOI] [PMC free article] [PubMed] [Google Scholar]
VLZ‐MD‐22 {published data only}
- Findling RL, McCusker E, Strawn JR. A randomized, double-blind, placebo-controlled trial of vilazodone in children and adolescents with major depressive disorder with twenty-six-week open-label follow-up. Journal of Child and Adolescent Psychopharmacology 2020;30(6):355-65. [DOI: 10.1089/cap.2019.0176] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT02372799. Safety and efficacy of vilazodone in pediatric patients with major depressive disorder (VLZ-MD-22). ClinicalTrials.gov/show/NCT02372799 (first received 1 October 2019).
Von Knorring 2006 {published data only}
- Medicines and Healthcare products Regulatory Agency (MHRA). Citalopram Study 2 ID#94404. medicines.mhra.gov.uk (accessed 20 June 2004). [3331174]
- Von Knorring A, Olsson GI, Thomson PH, Lemming OM, Hulten A. A randomized, double blind, placebo controlled study of citalopram in adolescents with major depressive disorder. Journal of Clinical Psychopharmacology 2006;26(3):311-5. [3331175] [DOI] [PubMed] [Google Scholar]
Wagner 2004 {published data only}
- CIT-MD-20. A randomized, double-blind, placebo-controlled evaluation of the safety and efficacy of citalopram in children and adolescent outpatients diagnosed with major depressive disorders (MDD) [CIT-MD-18]. Forest Clinical Trial Registry (www.forestclinicaltrials.com/CTR/CTRController/CTRViewPdf?_file_id=scsr/SCSR_CIT-MD-18final.pdf ) 2005 (accessed 24 November 2011). [3331177]
- Medicines and Healthcare Products Regulatory Agency (MHRA). Citalopram Study 1 ID# CIT-MD-18. medicines.mhra.gov.uk (accessed 20 June 2004). [3331178]
- Wagner KD, Robb AS, Findling R, Tiseo P. Citalopram is effective in the treatment of major depressive disorder in children and adolescents: results of a placebo-controlled trial. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):161. [3331179] [Google Scholar]
- Wagner KD, Robb AS, Findling RL, Jin J, Gutierrez MM, Heydorn WE. A randomized, placebo-controlled trial of citalopram for the treatment of major depression in children and adolescents. American Journal of Psychiatry 2004;161:1079-83. [3331180] [DOI] [PubMed] [Google Scholar]
- Wagner KD, Robb AS, Findling RL, Jin J, Tiseo PJ. Efficacy of citalopram in the treatment of MDD in children and adolescents [Abstract NR328]. In: 155th Annual Meeting of the American Psychiatric Association: New Research Program and Abstracts; 2002 May 18-23; Philadelpheia, PA, USA. 2002:90. [3331181]
Wagner 2006 {published data only}
- Forest LL. Forest announces results of recently completed Lexapro pediatric depression clinical trial. healthyplace.com/Communities/Depression/news/children_antidepressants_lexapro_2.asp OR frx.com/ (accessed October 2004). [3331183]
- Wagner KD, Jonas J, Findling RL, Ventura D, Saikali K. A double-blind, randomized, placebo-controlled trial of escitalopram in the treatment of pediatric depression. Journal of the American Academy of Child and Adolescent Psychiatry 2006;45(3):280-8. [3331184] [DOI] [PubMed] [Google Scholar]
Wagner Trial 1 {published data only}
- Pfizer PI. Study A050100. A multicenter 10-week randomized double-blind placebo-controlled flexible dose outpatient study of sertraline in children and adolescents with major depressive disorder. clinicalstudyresults.org/documents/company-study_98_0.pdf 2004 (accessed 24 November 2011). [3331186]
Wagner Trial 1 & 2 (2003) {published data only}
- Medicines and Healthcare products Regulatory Agency (MHRA). Sertraline Study 1 ID#1001. medicines.mhra.gov.uk (accessed 20 June 2004). [3331188]
- Medicines and Healthcare products Regulatory Agency (MHRA). Sertraline Study 2 ID # 1017. medicines.mhra.gov.uk (accessed 20 June 2004). [3331189]
- Wagner KD, Ambrosini P, Rynn M, Wohlberg C, Yang R, Greenbaum MS, et al. Efficacy of sertraline in the treatment of children and adolescents with major depressive disorder. JAMA 2003;290(8):1033-41. [3331190] [DOI] [PubMed] [Google Scholar]
- Wagner KD, Wohlberg C. Efficacy and safety of sertraline for treatment of pediatric major depressive disorder [Abstract NR327]. In: 155th Annual Meeting of the American Psychiatric Association; 2002 May 18-23; Philadelphia, PA. 2002. [3331191]
- Wagner KD, Wohlberg CJ, Yang R. Efficacy of sertraline in the treatment of children and adolescents with major depressive disorder: Reply. JAMA 2004;291(1):42. [3331192] [DOI] [PubMed] [Google Scholar]
Wagner Trial 2 {published data only}
- Pfizer PI. Study A0501017A. Multicenter 10-week randomized double-blind placebo-controlled flexible dose outpatient study of sertraline in children and adolescents with major depressive disorder. clinicalstudyresults.org/documents/company-study_1981_0.pdf 2004 (accessed 24 November 2011). [3331194]
Weihs 2018 {published data only}
- NCT01372150. A study of DVS SR in treatment of children and adolescent outpatients with MDD. clinicaltrials.gov/show/NCT01372150 (first received 13 June 2011).
- Weihs KL, Murphy W, Abbas R, Chiles D, England RD, Ramaker S, et al. Desvenlafaxine versus placebo in a fluoxetine-referenced study of children and adolescents with major depressive disorder. Journal of Child and Adolescent Psychopharmacology 2018;28(1):36-46. [DOI: 10.1089/cap.2017.0100] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihs KL, Wajsbrot DB, Chiles D, Ramaker S, Chappell P. Letter to the editor: Re: "desvenlafaxine versus placebo in a fluoxetine-referenced study of children and adolescents with major depressive disorder: design, definitions, and ongoing challenges for child and adolescent psychopharmacology research" (Strawn JR, Croarkin PE, Journal of Child & Adolesent Psychopharmacoly 2018;28:(5)363). Journal of Child and Adolescent Psychopharmacology 2019;29(3):245-6. [DOI: 10.1089/cap.2018.0163] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Atkinson 2018 extension phase {published data only}
- Atkinson S, Thurman L, Ramaker S, Buckley G, Jones SR, England R, et al. Safety, tolerability, and efficacy of desvenlafaxine in children and adolescents with major depressive disorder: results from two open-label extension trials. CNS Spectrums 2019;24(5):496-506. [DOI] [PubMed] [Google Scholar]
Braconnier 2003 {published data only}
- Braconnier A, Coent R, Cohen D. Paroxetine versus clomipramine in adolescents with severe major depression: a double blind, randomized, multicentre trial. Journal of the American Academy of Child and Adolescent Psychiatry 2003;42(1):22-9. [3331196] [DOI] [PubMed] [Google Scholar]
Cheung 2016 {published data only}
- Cheung A, Levitt A, Cheng M, Santor D, Kutcher S, Dubo E, et al. A pilot study of citalopram treatment in preventing relapse of depressive episode after acute treatment. Journal de L'academie Canadienne de Psychiatrie de L'enfant ET de L'adolescent [Journal of the Canadian Academy of Child and Adolescent Psychiatry] 2016;25(1):11-16. [PMC free article] [PubMed] [Google Scholar]
Cornelius 2009 {published data only}
- Cornelius JR, Bukstein OG, Wood DS, Kirisci L, Douaihy A, Clark DB. Double-blind placebo-controlled trial of fluoxetine in adolescents with comorbid major depression and an alcohol use disorder. Addictive Behaviors 2009;34(10):905-9. [3331198] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelius JR, Clark DB, Bukstein OG, Salloum IM, Matta J, Wood DS. Alcohol use but not cannabis use reported to contribute to depression in treatment trial of comorbid adolescents. In: Proceedings of the 68th Annual Scientific Meeting of the College on Problems of Drug Dependence; June 17-22 2006. Scottsdale, Arizona, USA, 2006. [3331199]
- NCT00027378. Pharmacological Intervention Project (Fluoxetine). clinicaltrials.gov/show/NCT00027378 (first received 1 December 2001).
Cornelius 2010 {published data only}
- Cornelius JR, Bukstein OG, Douaihy AB, Clark DB, Chung TA, Daley DC, et al. Double-blind fluoxetine trial in comorbid MDD-CUD youth and young adults. Drug and Alcohol Dependence 2010;112(1-2):39-45. [3331201] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelius JR, Clark DB, Bukstein OG, Salloum IM, Matta J, Wood DS. Alcohol use but not cannabis use reported to contribute to depression in treatment trial of comorbid adolescents. In: Proceedings of the 68th Annual Scientific Meeting of the College on Problems of Drug Dependence; June 17-22. Scottsdale, Arizona, USA, 2006. [3331202]
Cosgrove 1994 {published data only}
- Cosgrove PVF. Fluvoxamine in the treatment of depressive illness in children and adolescents. Journal of Psychopharmacology 1994;8(2):118-23. [3331204] [DOI] [PubMed] [Google Scholar]
Emslie 2008 discontinuation phase {published data only}
- Emslie GJ. Predictors and moderators of relapse in depressed youth. Neuropsychiatrie de L'enfance et de L'adolescence 2012;60(5 suppl. 1):S168.
ePOD‐SSRI {published data only}
- Bottelier MA, Schrantee AGM, Wingen GA, Ruhe HG, Reneman L. Treatment with fluoxetine in adolescents may aggravate emotional dysregulation: A power analysis for future studies. European Neuropsychopharmacology 2015;25:S461-2.
Findling 2009 {published data only}
- Findling RL, Pagano ME, McNamara NK, Stansbrey RJ, Faber JE, Lingler J, et al. The short-term safety and efficacy of fluoxetine in depressed adolescents with alcohol and cannabis use disorders: a pilot randomized placebo-controlled trial. Child and Adolescent Psychiatry and Mental Health 2009;3(1):11. [3331206] [DOI] [PMC free article] [PubMed] [Google Scholar]
Henkel 2010 {published data only}
- Henkel V, Mergl R, Allgaier Ak, Hautzinger M, Kohnen R, Coyne Jc, et al. Treatment of atypical depression: post-hoc analysis of a randomized controlled study testing the efficacy of sertraline and cognitive behavioural therapy in mildly depressed outpatients. European Psychiatry : The Journal of the Association of European Psychiatrists 2010;25(8):491-8. [DOI] [PubMed]
JPRN‐UMIN000016192 {published data only}
- JPRN-UMIN000016192. Investigation for the effect of duloxetine on adolescent depression; Ver. 2. upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000018778 (first received 4 August 2011).
Kennard 2018 {published data only}
- Kennard BD, Mayes TL, Chahal Z, Nakonezny PA, Moorehead A, Emslie G J. Predictors and moderators of relapse in children and adolescents with major depressive disorder. Journal of Clinical Psychiatry 2018;79(2):5m10330. [DOI: 10.4088/JCP.15m10330] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kennedy 2014 {published data only}
- Kennedy S H, Avedisova A, Belaidi C, Picarel-Blanchot F, Bodinat C. Sustained efficacy of agomelatine 10 mg, 25 mg, and 25-50 mg on depressive symptoms and functional outcomes in patients with major depressive disorder. A placebo-controlled study over 6 months. European Neuropsychopharmacology 2016;26(2):378-89. [DOI] [PubMed] [Google Scholar]
- Kennedy S H, Avedisova A, Gimenez-Montesinos N, Belaidi C, de Bodinat C - Agomelatine Study Group. A placebo-controlled study of three agomelatine dose regimens (10 mg, 25 mg, 25-50 mg) in patients with major depressive disorder. European Neuropsychopharmacology 2014;24(4):553-63. [DOI] [PubMed] [Google Scholar]
Liebowitz 2008 {published data only}
- Liebowitz MR, Manley AL, Padmanabhan SK, Ganguly R, Tummala R, Tourian KA. Efficacy, safety, and tolerability of desvenlafaxine 50 mg/day and 100 mg/day in outpatients with major depressive disorder. Current Medical Research and Opinion 2008;24(7):1877-90. [DOI] [PubMed] [Google Scholar]
Mandoki 1997 {published data only}
- Mandoki MW, Tappia MR, Tapia MA, Sumner GS, Parker JL. Veanlafaxine in the treatment of children and adolescents with major depression. Psychopharmacology Bulletin 1997;33(1):149-54. [3331208] [PubMed] [Google Scholar]
NCT00005015 {published data only}
- NCT00005015. Fluoxetine for the treatment of major depression in youth with bipolar disorder [Study numbers N01-MH-70008A; N01-MH-70008]. clinicaltrials.gov/ct2/show/NCT00005015 (first received 3 April 2000). [3331210]
NCT00249886 {published data only}
- ISRCTN42386710. A double blind placebo controlled discontinuation of citalopram in adolescents with major depression. isrctn.com/ISRCTN42386710 (first received 5 September 2005).
NCT00508859 {published data only}
- NCT00508859. Maintenance Study for Adolescent Depression. ClinicalTrials.gov/show/NCT00508859 (first received 30 July 2007).
NCT02871297 {published data only}
- EUCTR2008-005356-25-LV. A study to evaluate the long-term safety and tolerability of a product named vortioxetine (used in depression) in children and adolescents. www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2008-005356-25 (first received 6 April 2016).
NCT03436173 {published data only}
- NCT03436173. OxSYPan: Oxford study with young people on antidepressants. clinicaltrials.gov/show/nct03436173 (first received 19 February 2018).
Riggs 2007 {published data only}
- Riggs PD, Lohman M, Davies R, Mikulich-Gilbertson S, Laudenslager M, Klein C. Randomized controlled trial of fluoxetine/placebo and cognitive behavioral treatments in depressed adolescents with substance use disorders. In: Proceedings of the 67th Annual Scientific Meeting of the College on Problems of Drug Dependence; June 19-23 2005. Orlando, Florida, USA, 2005. [3331212]
- Riggs PD, Mikulich-Gilbertson SK, Davies RD, Lohman M, Klein C, Stover SK. A randomized controlled trial of fluoxetine and cognitive behavioral therapy in adolescents with major depression, behavior problems, and substance use disorders. Archives of Pediatrics and Adolescent Medicine 2007;161(11):1026-34. [3331213] [DOI] [PubMed] [Google Scholar]
- Thurstone C, Riggs PD, Orton HD, Libby AM. Fluoxetine for cannabis withdrawal in depressed adolescents. In: Proceedings of the 67th Annual Scientific Meeting of the College on Problems of Drug Dependence; June 19-23 2005. Orlando, Florida, USA, 2005. [3331214]
Sallee 1997 {published data only}
- Chabrol H, Peresson G. Pulse clomipramine for depressed adolescents [letter]. American Journal of Psychiatry 1998;155(7):995. [3331216] [DOI] [PubMed] [Google Scholar]
- Sallee FR, Vrindavanam NS, Deas-Nesmith D, Carson SW, Sethuraman G. Pulse intravenous clomipramine for depressed adolescents: double-blind, controlled trial. American Journal of Psychiatry 1997;154(5):668-73. [3331217] [DOI] [PubMed] [Google Scholar]
- Sallee FR. Dr. Sallee replies [letter]. American Journal of Psychiatry 1998;155(7):995. [3331218] [Google Scholar]
Tashakori 1997 {published data only}
- Tashakori A, Arabgol F, Panaghi L, Davari R. The effect of reboxetine in the treatment of depression in children and adolescents. Tehran University Medical Journal 2007;65(8):40-8. [3331220] [Google Scholar]
Walker 2017 {published data only}
- Walker DJ, DelBello MP, Landry J, D'Souza DN, Detke HC. Quality of life in children and adolescents with bipolar I depression treated with olanzapine/fluoxetine combination. Child & Adolescent Psychiatry & Mental Health [Electronic Resource] 2017;11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wohlfarth 2007 {published data only}
- Wohlfarth T, Boer F, den Brink W. Withdrawal of attention rather than pharmacological treatment affects suicide rates in depressed children and adolescents. American Journal of Psychiatry 2007;164(12):1908-10. [3331222] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Haneveld 2003 {published data only}
- Haneveld JK. Two studies confirm the efficacy of sertraline for the treatment of depression in children and adolescents. Pharmaceutisch Weekblad 2003;138(46):1616-7. [Google Scholar]
References to ongoing studies
EUCTR2015‐002181‐23 {published data only}
- EUCTR2015-002181-23-FI. Efficacy and safety of 2 doses of agomelatine (10mg, 25mg) given orally in children (from 7 to less than 12 years) and adolescents (from 12 to less than 18 years) with moderate to severe Major Depressive Disorder. www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2015-002181-23 (first received 14 December 2015).
Glod 2004 {published data only}
- Glod CA, Lynch A, Berkowitz C, Hennen J, Baldessarini RJ. Bupropion versus citalopram versus placebo in adolescents with major depression [Abstract NR484]. 157th Annual Meeting of the American Psychiatric Association; May 1-6 2004 2004. [3331231]
IRCT138901093607N1 {published data only}
- IRCT138901093607N1. Comparing the effect of Fluoxetine and Fluvoxamine on child and adolescent depression admitted to Akhavan hospital, Kashan. www.who.int/trialsearch/Trial2.aspx?TrialID=IRCT138901093607N1 (first received 4 June 2010).
JPRN‐JapicCTI‐194585 {published data only}
- JPRN-JapicCTI-194585. A clinical trial of MLD-55 in pediatric patients with depression. www.who.int/trialsearch/Trial2.aspx?TrialID=JPRN-JapicCTI-194585 (first received 6 January 2016).
NCT00353028 {published data only}
- Yamaguchi T. SME3110 (fluvoxamine maleate) in the treatment of depression/depressive state: a post-marketing clinical study in children and adolescents (8 through 18 years of age) - a double-blind, randomized, placebo-controlled study. www.clinicaltrials.gov/ct2/show/NCT00353028 (first received 17 July 2006). [3331233]
NCT00426946 {published data only}
- NCT00426946. Reboxetine Treatment in Depressed Children and Adolescents an 8-Week, Open Study. ClinicalTrials.gov/show/NCT00426946 (first received 25 January 2007).
NCT01185977 {published data only}
- NCT01185977. Biomarkers of Antidepressant Treatment in Adolescents With Major Depression (The Adolescents MDD Study). clinicaltrials.gov/show/NCT01185977 (first received 20 August 2010).
NCT02129751 {published data only}
- NCT02129751. Efficacy and Safety of Bupropion Hydrobromide in Adolescents and Children With Major Depressive Disorder. clinicaltrials.gov/show/NCT02129751 (first received 2 May 2014).
NCT02431806 {published data only}
- NCT02431806. Safety and Efficacy of Levomilnacipran ER in Adolescent Patients With Major Depressive Disorder. ClinicalTrials.gov/show/NCT02431806 (first received 1 May 2015).
NCT02709655 {published data only}
- EUCTR2008-005353-38-EE. Interventional, randomised, double-blind, placebo-controlled, active reference (fluoxetine), fixed-dose study of vortioxetine in paediatric patients aged 7 to 11 years, with Major depressive disorder (MDD). clinicaltrialsregister.eu/ctr-search/search? Query=eudract_number:2008-005353-38 (first received 25 November 2015).
- NCT02709655. Active Reference (Fluoxetine) Fixed-dose Study of Vortioxetine in Paediatric Patients Aged 7 to 11 Years With Major Depressive Disorder (MDD). ClinicalTrials.gov/show/NCT02709655 (first received 16 March 2016).
NCT03315793 {published data only}
- JPRN-JapicCTI-173734. A phase 3 clinical trial of duloxetine in children and adolescents with depressive disorder. www.who.int/trialsearch/Trial2.aspx?TrialID=JPRN-JapicCTI-173734 (first received 13 October 2017).
- NCT03315793. A Study of Duloxetine (LY248686) in Japanese Children and Adolescents With Depressive Disorder. clinicaltrials.gov/show/nct03315793 (first received 20 October 2017).
NCT03569475 {published data only}
- NCT03569475. Efficacy, Safety, and Tolerability of Levomilnacipran ER in Pediatric (7-17 Years) With Major Depressive Disorder. clinicaltrials.gov/show/nct03569475 (first received 26 June 2018).
Additional references
AACAP 2007
- American Academy of Child and Adolescent Psychiatry (AACAP). Practice parameters for the assessment and treatment of children and adolescents with depressive disorders. Journal of the American Academy of Child and Adolescent Psychiatry 2007;46(11):1503-26. [DOI] [PubMed] [Google Scholar]
Altman 2006
- Altman DG, Royston P. The cost of dichotomising continuous variables. BMJ 2006;332(7549):1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Angold 1999
- Angold A, Costello EJ, Erkanli A. Comorbidity. Journal of Child Psychology and Psychiatry 1999;40(1):57-87. [DOI: 10.1111/1469-7610.00424] [DOI] [PubMed] [Google Scholar]
APA 1980
- American Psychiatic Association. Diagnostic and Statistical Manual of Mental Disorders. 3rd edition. Washington, DC: American Psychiatric Association, 1980. [Google Scholar]
APA 1987
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 3rd edition. Text Revision. Washington, DC: American Psychiatric Association, 1987. [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th edition. Washington, DC: American Psychiatic Association, 1994. [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostical and Statistical Manual of Mental Disorders. 4th edition. Text Revision. Washington DC: APA Press, 2000. [Google Scholar]
APA 2013
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th edition. Arlington, VA: American Psychiatric Association, 2013. [Google Scholar]
Birmaher 1996
- Birmaher B, Ryan ND, Williamson DE, Brent DA, Kaufman J, Dahl RE, et al. Childhood and adolescent depression: a review of the past 10 years: Part 1. Journal of the American Academy ofChild and Adolescent Psychiatry 1996;35(11):1427-39. [DOI] [PubMed] [Google Scholar]
Birmaher 2007
- Birmaher B, Brent D. Practice parameter for the assessment and treatment of children and adolescents with depressive disorders. Journal of the American Academy of Child and Adolescent Psychiatry 2007;46(11):1503-26. [DOI] [PubMed] [Google Scholar]
Björkholm 2016
- Björkholm C, Monteggiab LM. BDNF – a key transducer of antidepressant effects. Neuropharmacology 2016;102:72-9. [DOI: 10.1016/j.neuropharm.2015.10.034] [DOI] [PMC free article] [PubMed] [Google Scholar]
Brooks 2001
- Brooks SJ, Kutcher S. Diagnosis and measurement of depression: a review of commonly utilized instruments. Journal of Child and Adolescent Psychopharmacology 2001;11(4):341-76. [DOI] [PubMed] [Google Scholar]
Cash 2009
- Cash SJ, Bridge JA. Epidemiology of youth suicide and suicidal behavior. Current Opinion in Pediatrics 2009;21(5):613-9. [DOI: 10.1097/MPO.0b013e32933063e1] [DOI] [PMC free article] [PubMed] [Google Scholar]
Castrén 2005
- Castrén E. Is mood chemistry? Nature Reviews Neuroscience 2005;6(3):241-6. [DOI] [PubMed] [Google Scholar]
Castrén 2014
- Castrén E. Neurotrophins and psychiatric disorders. In: Lewin G, Carter B, editors(s). Neurotrophic Factors. Handbook of Experimental Pharmacology. Vol. 220. Berlin, Heidelberg: Springer, 2014:461-79. [DOI] [PubMed] [Google Scholar]
Castrén 2017
- Castrén E, Kojima M. Brain-derived neurotrophic factor in mood disorders and antidepressant treatments. Neurobiology of Disease 2017;97(Pt B):119-26. [DOI: 10.1016/j.nbd.2016.07.010] [DOI] [PubMed] [Google Scholar]
Chaimani 2012
- Chaimani A, Salanti G. Using network meta-analysis to evaluate the existence of small-study effects in a network of interventions. Research Synthesis Methods 2012;3(2):161-76. [DOI: 10.1002/jrsm.57] [DOI] [PubMed] [Google Scholar]
Chaimani 2013
- Chaimani A, Higgins JP, Mavridis D, Spyridonos P, Salanti G. Graphical tools for network meta-analysis in STATA. Plos One 2013;8(10):e76654. [DOI: 10.1371/journal.pone.0076654] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chaimani 2015
- Chaimani A, Salanti G. Visualizing assumptions and results in network meta-analysis: the network graphs package. Stata Journal 2015;15(4):905-50. [Google Scholar]
Chambers 1985
- Chambers WJ, Puig-Antich J, Hirsch M, Paez P, Ambrosini PJ, Tabrizi MA, et al. The assessment of affective disorders in children and adolescents by semi-structured interview: test-retest reliability of the Schedule for Affective Disorders and Schizophrenia for school-age children. Present episode version. Archives of General Psychiatry 1985;42(7):696-702. [DOI] [PubMed] [Google Scholar]
Cheung 2018
- Cheung AH, Zuckerbrot RA, Jensen PS, Laraque D, Stein REK, GLAD-PC Steering Group. Guidelines for adolescent depression in primary care (GLAD-PC): II treatment and ongoing management. Pediatrics 2018;141(3):e20174082. [DOI: ] [DOI] [PubMed] [Google Scholar]
Churchill 2010
- Churchill R, Caldwell D, McGuire H, Barbui C, Cipriani A, Furukawa TA, et al. Reboxetine versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2010, Issue 6. Art. No: CD007852. [DOI: 10.1002/14651858.CD007852] [DOI] [Google Scholar]
CINeMA 2017 [Computer program]
- CINeMA: Confidence in Network Meta-Analysis [Computer program]. Bern: Institute of Social and Preventive Medicine, University of Bern, 2017.
Cipriani 2005
- Cipriani A, Brambilla P, Furukawa T, Geddes J, Gregis M, Hotopf M, et al. Fluoxetine versus other types of pharmacotherapy for depression. Cochrane Database of Systematic Reviews 2005, Issue 5. Art. No: CD004185. [DOI: 10.1002/14651858.CD004185.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2007
- Cipriani A, Furukawa TA, Veronese A, Watanabe N, Churchill R, McGuire H, et al. Paroxetine versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews 2007, Issue 2. Art. No: CD006531. [DOI: 10.1002/14651858.CD006531] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2009a
- Cipriani A, Signoretti A, Furukawa TA, Churchill R, Tomelleri S, Omori IM, et al. Venlafaxine versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews 2009, Issue 3. Art. No: CD006530. [DOI: 10.1002/14651858.CD006530] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2009b
- Cipriani A, Santilli C, Furukawa TA, Signoretti A, Nakagawa A, McGuire H, et al. Escitalopram versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2009, Issue 2. Art. No: CD006532. [DOI: 10.1002/14651858.CD006532.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2010
- Cipriani A, La Ferla T, Furukawa TA, Signoretti A, Nakagawa A, Churchill R, et al. Sertraline versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2010, Issue 4. Art. No: CD006117. [DOI: 10.1002/14651858.CD006117.pub4] [DOI] [PubMed] [Google Scholar]
Cipriani 2016
- Cipriani A, Zhou X, Giovane CD, Hetrick SE, Qin B, Whittington C, et al. Comparative efficacy and tolerability of antidepressants for major depression in children and adolescents: a network meta-analysis. The Lancet 2016;388(10047):881-90. [DOI] [PubMed] [Google Scholar]
Cohen 2004
- Cohen D, Gerardin P, Mazet P, Purper-Ouakil D, Flament MF. Pharmacological treatment of adolescent major depression. Journal of Child and Adolescent Psychopharmacology 2004;14(1):19-31. [DOI] [PubMed] [Google Scholar]
Cooper 1988
- Cooper GL. The safety of fluoxetine - an update. British Journal of Psychiatry. Supplement 1988;3:77-86. [PubMed] [Google Scholar]
Copeland 2015
- Copeland WE, Wolke D, Shanahan L, Costello EJ. Adult functional outcomes of common childhood psychiatric problems: a prospective, longitudinal study. JAMA Psychiatry 2015;72(9):892-9. [DOI: 10.1001/jamapsychiatry.2015.0730] [DOI] [PMC free article] [PubMed] [Google Scholar]
Costello 2006
- Costello JE, Erkanli A, Angold A. Is there an epidemic of child or adolescent depression? Journal of Child Psychology and Psychiatry, and Allied Disciplines 2006;47(12):1263-71. [DOI] [PubMed] [Google Scholar]
Cousins 2015
- Cousins L, Goodyer IM. Antidepressants and the adolescent brain. Journal of Psychopharmacology 2015;29(5):545-5. [DOI: ] [DOI] [PubMed] [Google Scholar]
CSM 2004
- Medicines and Healthcare Products Regulatory Agency Committee on Safety of Medicines (CSM). Report of the CSM Expert Working Group on the Safety of Selective Serotonin Reuptake Inhibitor Antidepressants. MHRA; December 2004. Available at mhra.gov.uk/home/groups/pl-p/documents/drugsafetymessage/con019472.pdf.
Davey 2019
- Davey CG, Chanen AM, Hetrick SE, Cotton SM, Ratheesh A, Amminger GP, et al. The addition of fluoxetine to cognitive behavioural therapy for youth depression: The YoDA-C randomised clinical trial. Lancet Psychiatry 2019;6(9):735-44. [DOI: 10.1016/S2215-0366(19)30215-9] [DOI] [PubMed] [Google Scholar]
Delgado 2000
- Delgado PL. Depression: The case for a monoamine deficiency. Journal of Clinical Psychiatry 2000;61(Suppl 6):7-11. [PubMed] [Google Scholar]
Dubicka 2006
- Dubicka B, Hadley S, Roberts C. Suicidal behaviour in youths diagnosed with depression treated with new-generation antidepressants. British Journal of Psychiatry 2006;189:393-8. [DOI] [PubMed] [Google Scholar]
Dubitsky 2004
- Dubitsky GM. Review and Evaluation of Clinical Data: Placebo-Controlled Antidepressant Studies In Pediatric Patients. Medicines and Healthcare Products Regulatory Agency 2004.
Dunlop 2010
- Dunlop BW, Vaughan CL. Survey of investigators opinions on the acceptability of interactions with patients participating in clinical trials. Journal of Clinical Psychopharmacology 2010;30(3):323-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dunn 2006
- Dunn V, Goodyer IA. Longitudinal investigation into childhood- and adolescence- onset depression: psychiatric outcome in early adulthood. British Journal of Psychiatry 2006;188:216-22. [DOI: 10.1192/bjp.188.3.216] [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta-analyses involving cross-over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140-9. [DOI] [PubMed] [Google Scholar]
EMA 2005
- European Medicines Agency. European Medicines Agency finalises review of antidepressants in children and adolescents; April 2005. www.ema.europa.eu/en/documents/referral/european-medicines-agency-finalises-review-antidepressants-children-adolescents_en.pdf (accessed 23 March 2021).
FDA 2004
- US Food and Drug Administration Public Health Advisory. Suicidality in children and adolescents being treated with antidepressant medications. October 15, 2004. www.fda.gov/cder/drug/antidepressants/SSRIPHA200410.htm (accessed prior to 1 November 2012).
FDA 2018
- US Food and Drug Administration Public Health Advisory. Suicidality in Children and Adolescents Being Treated with Antidepressant Medications. www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/suicidality-children-and-adolescents-being-treated-antidepressant-medications (accessed 5 May 2018).
Fergusson 2007
- Fergusson DM, Boden JM, Horwood LJ. Recurrence of major depression in adolescence and early adulthood, and later mental health, educational and economic outcomes. British Journal of Psychiatry 2007;191:335-42. [DOI: ] [DOI] [PubMed] [Google Scholar]
Folstein 1975
- Folstein MF, Folstein SE, McHugh. Minii-Mental State: A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research 1975;12:189-98. [DOI] [PubMed] [Google Scholar]
Frank 1991
- Frank E, Prien RF, Jarrett RB, Keller MB, Kupfer DJ, Lavori PW, et al. Conceptualization and rationale for consensus definitions of terms in major depressive disorder. Remission, recovery, relapse, and recurrence. Archives of General Psychiatry 1991;48(9):851-5. [DOI: 10.1001/archpsyc.1991.01810330075011] [DOI] [PubMed] [Google Scholar]
Gibbons 2007
- Gibbons RD, Brown CH, Hur K, Marcus SM, Bhaumik DK, Erkens JA, et al. Early evidence on the effects of regulators' suicidality warnings on SSRI prescriptions and suicide in children and adolescents. American Journal of Psychiatry 2007;164:1356-63. [DOI] [PubMed] [Google Scholar]
Gore 2011
- Gore FM, Bloem PJ, Patton GC, Ferguson J, Joseph V, Coffey C, et al. Global burden of disease in young people aged 10-24 years: a systematic analysis. Lancet 2011;377(9783):2093-102. [DOI] [PubMed] [Google Scholar]
Guaiana 2010
- Guaiana G, Gupta S, Chiodo D, Davies SJC. Agomelatine versus other antidepressive agents for major depression. Cochrane Database of Systematic Reviews 2010, Issue 11. Art. No: CD008851. [DOI: 10.1002/14651858.CD008851] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guy 1976
- Guy W. ECDEU Assessment Manual for Psychopharmacology. 2nd edition. Washington, DC: US Government Printing Office, 1976. [Google Scholar]
Hammad 2004
- Hammad TA. Relationship between psychotropic drugs and pediatric suicidality. Review and evaluation of clinical data. US Food and Drug Administration (FDA) 16 August 2004.
Hammad 2006
- Hammad TA, Laugren T, Racoosin J. Suicidality in pediatric patients treated with antidepressant drugs. Archives of General Psychiatry 2006;63(3):332-9. [DOI] [PubMed] [Google Scholar]
Harmer 2017
- Harmer CJ, Duman RS, Cowen PJ. How do antidepressants work? New perspectives for refining future treatment approaches. Lancet Psychiatry 2017;4(5):409-18. [DOI: 10.1016/S2215-0366(17)30015-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
Harrington 1990
- Harrington R, Fudge H, Rutter M, Pickles A, Hill J. Adult outcomes of childhood and adolescent depression, I: psychiatric status. Archives of General Psychiatry 1990;47(5):465-73. [PMIMD: 2184797] [DOI] [PubMed] [Google Scholar]
Hazell 2002
- Hazell P, O'Connell D, Heathcote D, Henry D. Tricyclic drugs for depression in children and adolescents. Cochrane Database of Systematic Reviews 2002, Issue 2. Art. No: CD002317. [DOI: 10.1002/14651858.CD002317] [DOI] [PubMed] [Google Scholar]
Healy 2003
- Healy D. Lines of evidence on the risks of suicide with selective serotonin reuptake inhibitors. Psychotherapy and Psychosomatics 2003;72(2):71-9. [DOI] [PubMed] [Google Scholar]
Hetrick 2007
- Hetrick SE, Merry SN, McKenzie JE, Proctor M, Simmons M. Selective serotonin reuptake inhibitors (SSRIs) for depressive disorders in children and adolescents. Cochrane Database of Systematic Reviews 2007, Issue 3. Art. No: CD004851. [DOI: 10.1002/14651858.CD004851.pub2] [DOI] [PubMed] [Google Scholar]
Hetrick 2012
- Hetrick SE, McKenzie JE, Cox GE, Simmons MB, Merry SN. Newer generation antidepressants for depressive disorders in children and adolescents. Cochrane Database of Systematic Reviews 2012, Issue 11. Art. No: CD004851. [DOI: 10.1002/14651858.CD004851.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2009
- Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2009]. The Cochrane Collaboration, 2009. Available from training.cochrane.org/handbook/archive/v5.0.2/.
Higgins 2012
- Higgins JPT, Jackson D, Barrett JK, Lu G, Ades AE, White IR. Consistency and inconsistency in network meta-analysis: Concepts and models for multi-arm studies. Research Synthesis Methods 2012;3:98-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2019
- Higgins JP, Eldridge S, Li T (editors). Chapter 23: Including variants on randomized trials. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Hollis 1999
- Hollis S, Campbell F. What is meant by intention to treat analysis? Survey of published randomised controlled trials. BMJ 1999;319:670-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hosenbocus 2011
- Hosenbocus S, Chahal R. SSRIs and SNRIs: A review of the discontinuation syndrome in children and adolescents. Journal de l'Academie Canadienne de Psychiatrie de l'Enfant et de l'Adolescent [Journal of the Canadian Academy of Child and Adolescent Psychiatry] 2011;20(1):60-7. [PMC free article] [PubMed] [Google Scholar]
Hyde 2008
- Hyde JS, Mezulis AH, Abramson LY. The ABCs of depression: integrating affective, biological, and cognitive models to explain the emergence of the gender difference in depression. Psychological Review 2008;115(2):291-313. [DOI] [PubMed] [Google Scholar]
Imperadore 2007
- Imperadore G, Cipriani A, Signoretti A, Furukawa TA, Watanabe N, Churchill R, et al. Citalopram versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews 2007, Issue 2. Art. No: CD006534. [DOI: 10.1002/14651858.CD006534] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ioannidis 2008
- Ioannidis JP. Effectiveness of antidepressants: an evidence myth constructed from a thousand randomized trials? Philosophy Ethics and Humanities in Medicine 2008;3:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
John 2016
- John A, Marchant AL, Fone DL, McGregor JI, Dennis MS, Tan JO, et al. Recent trends in primary-care antidepressant prescribing to children and young people: an e-cohort study. Psychological Medicine 2016;46(16):3315-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jureidini 2003
- Jureidini J, Tonkin A. Paroxetine in major depression (letter). Journal of the American Academy of Child and Adolescent Psychiatry 2003;42(5):514. [DOI] [PubMed] [Google Scholar]
Jureidini 2004
- Jureidini JN, Doecke CJ, Mansfield PR, Haby MM, Menkes DB, Tonkin AL. Efficacy and safety of antidepressants for children and adolescents. BMJ 2004;328:879-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Keller 2003
Kennard 2006
- Kennard B, Silva S, Vitiello B, Ginsberg G, Emslie G, March J. Remission and residual symptoms after short-term treatment in the Treatment of Adolescents With Depression Study (TADS). Journal of the American Academy of Child and Adolescent Psychiatry 2006;45:1404-11. [DOI] [PubMed] [Google Scholar]
Kenward 1997
- Kenward M, Roger J. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics 1997;53:983-97. [PubMed] [Google Scholar]
Kessler 2001
- Kessler RC, Avenevoli S, Ries Merikangas K. Mood disorders in children and adolescents: an epidemiologic perspective. Biological Psychiatry 2001;49(12):1002-14. [PMID: 11430842] [DOI] [PubMed] [Google Scholar]
Kessler 2005
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey replication. Archives of General Psychiatry 2005;62(6):593-602. [DOI] [PubMed] [Google Scholar]
Kieser 2004
- Kieser M, Rohmel J, Friede T. Power and sample size determination when assessing the clinical relevance of trial results by 'responder analyses'. Statistics in Medicine 2004;23(21):3287-305. [DOI] [PubMed] [Google Scholar]
Kirsch 2008
- Kirsch I, Deacon BJ, Heudo-Medina TB, Scoboria A, Moore TJ, Johnson BT. Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Medicine 2008;5(2):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kovacs 1989
- Kovacs M, Gatsonis C, Paulauskas SL, Richards C. Depressive disorders in childhood: IV. A longitudinal study of comorbidity with and risk for anxiety disorders. Archives of General Psychiatry 1989;46(9):776-82. [DOI] [PubMed] [Google Scholar]
Krause 2021
- Krause KR, Chung S, Adewuya AO, Albano AM, Babins-Wagner R, Birkinshaw L, et al. International consensus on a standard set of outcome measures for child and youth anxiety, depression, obsessive-compulsive disorder, and post-traumatic stress disorder. Lancet Psychiatry 2021;8:76–86. [DOI] [PubMed] [Google Scholar]
Le Noury 2015
- Le Noury J, Nardo JM, Healy D, Jureidini J, Raven M, Tufanaru C, et al. Restoring Study 329: efficacy and harms of paroxetine and imipramine in treatment of major depression in adolescence. BMJ 2015;351:h4320. [DOI: 10.113 6/bmj.h4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lenox 2008
- Lenox RH, Frazer A. Section 8, Chapter 79: Mechanism of action of antidepressants and mood stabilizers: In: Davis KL, Charney D, Coyle JT, Nemeroff C (editors) Neuropsychopharmacology – 5th Generation of Progress. Available at acnp.org/wp-content/uploads/2017/11/CH79_1139-1164.pdf (accessed prior to 20 May 2021).
Lewinsohn 1998
- Lewinsohn PM, Rohde P, Seeley JR. Major depressive disorder in older adolescents: prevalence, risk factors and clinical implications. Clinical Psychology Review 1998;18(7):765-94. [DOI] [PubMed] [Google Scholar]
Lexchin 2003
- Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003;326(7400):1167-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Locher 2017
- Locher C, Koechlin H, Zion SR, Werner C, Pine DS, Kirsch I, et al. Efficacy and safety of selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and placebo for common psychiatric disorders among children and adolescents: a systematic review and meta-analysis. JAMA Psychiatry 2017;74(10):1011-20. [DOI: 10.1001/jamapsychiatry.2017.2432] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lu 2014
- Lu C, Zhang F, Lakoma MD, Madden JM, Rusinak D, Penford RB, et al. Changes in antidepressant use by young people and suicidal behavior after FDA warnings and media coverage: a quasi-experimental study. BMJ 2014;348:g3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lundh 2017
- Lundh A, Lexchin J, Mintzes B, Schroll JB, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. Art. No: MR000033. [DOI: 10.1002/14651858.MR000033.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
MacQueen 2011
- MacQueen G, Frodl T. The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Molecular Psychiatry 2011;16(3):252-64. [DOI: 10.1038/mp.2010.80] [PMID: 20661246] [DOI] [PubMed] [Google Scholar]
March 2004
- March J, Silva S, Petrycki S, Curry J, Wells K, Fairbank J, et al. Fluoxetine, cognitive-behavioral therapy, and their combination for adolescents with depression: Treatment for Adolescents with Depression Study (TADS) randomized controlled trial. Journal of the American Medical Association 2004;292:807-20. [DOI] [PubMed] [Google Scholar]
Maughan 2013
- Maughan B, Collinshaw S, Stringaris A. Depression in childhood and adolescence. Journal of the Canadian Academcy of Child and Adolescent Psychiatry 2013;22(1):35-40. [PMCID:PMC3565713] [PMID:23390431] [PMC free article] [PubMed] [Google Scholar]
Mavridis 2016
- Mavridis D, Efthimiou O, Leucht S, Salanti G. Publication bias and small-study effects magnified effectiveness of antipsychotics but their relative ranking remained invariant. Journal of Clinical Epidemiology 2016;69:161-9. [DOI] [PubMed] [Google Scholar]
McDermott 2011
- McDermott B, Baigent M, Chanen A, Fraser L, Graetz B, Hayman N, et al. Beyond Blue Expert Working Committee (2010) Clinical practice guidelines: Depression in adolescents and young adults. www.beyondblue.org.au/health-professionals/clinical-practice-guidelines (accessed prior to 1 December 2012).
Merry 2017
- Merry SN, Hetrick SE, Stasiak K. Effectiveness and safety of antidepressants for children and adolescents: Implications for clinical practice. JAMA Psychiatry 2017;74(10):985-6. [DOI] [PubMed] [Google Scholar]
MHRA 2014
- Medicines & Healthcare Products Regulatory Agency (MHRA). Selective serotonin reuptake inhibitors (SSRIs) and serotonin and noradrenaline reuptake inhibitors (SNRIs): use and safety; 18 December 2018. Available at: www.gov.uk/government/publications/ssris-and-snris-use-and-safety/selective-serotonin-reuptake-inhibitors-ssris-and-serotonin-and-noradrenaline-reuptake-inhibitors-snris-use-and-safety.
Moher 2010
- Moher D, Hopewell S, Schulz KF, Montori V, Gotzsche PC, Devereaux PJ, et al. CONSORT 2010 Explanation and Elaboration: Updated guidelines for reporting parallel group randomised trials. Journal of Clinical Epidemiology 2010;63(8):e1-37. [DOI] [PubMed] [Google Scholar]
Moncrieff 2001
- Moncrieff J, Churchill R, Drummond C, McGuire H. Development of a quality assessment instrument for trials of treatments for depression and neurosis. International Journal of Methods in Psychiatric Research 2001;10(3):126-33. [Google Scholar]
Moncrieff 2004
- Moncrieff J, Wessely S, Hardy R. Active placebo versus antidepressants for depression. Cochrane Database of Systematic Reviews 2005, Issue 1. Art. No: CD003012. [DOI: 10.1002/14651858.CD003012.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Murphy 2009
- Murphy SE, Norbury R, O’Sullivan U, Cowen PJ, Harmer CJ. Effect of a single dose of citalopram on amygdala response to emotional faces. British Journal of Psychiatry 2009;194(6):535-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nakagawa 2009
- Nakagawa A, Watanabe N, Omori IM, Barbui C, Cipriani A, McGuire H, et al. Milnacipran versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2009, Issue 3. Art. No: CD006529. [DOI: 10.1002/14651858.CD006529.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2019
- National Institute for Health and Clinical Excellence (NICE). Depression in children and young people: identification and management. NICE guideline [NG134]; 25 June 2019. Available at nice.org.uk/guidance/ng134. [PubMed]
Nikolakopoulou 2020
- Nikolakopoulou A, Higgins JP, Papakonstantinou T, Chaimani A, Del Giovane C, Egger M et al. CINeMA: An approach for assessing confidence in the results of a network meta-analysis. PLoS Medicine 2020;17(4):e1003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nosè 2007
- Nosè M, Cipriani A, Furukawa TA, Omori IM, Churchill R, McGuire H et al. Duloxetine versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews 2007, Issue 2. Art. No: CD006533. [DOI: 10.1002/14651858.CD006533] [DOI] [PMC free article] [PubMed] [Google Scholar]
Olver 2001
- Olver JS, Burrows GD, Norman TR. Third-generation antidepressants - do they offer advantages over the SSRIs? CNS Drugs 2001;15(12):941-54. [DOI] [PubMed] [Google Scholar]
Omori 2010
- Omori I, Watanabe N, Nakagawa A, Cipriani A, Barbui C, McGuire C, et al. Fluvoxamine versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews 2010, Issue 3. Art. No: CD006114. [DOI: 10.1002/14651858.CD006114.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Page 2019
- Page MJ, Higgins JP, Sterne JA. Chapter 13: Assessing risk of bias due to missing results in a synthesis. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Petti 1985
- Petti TA. Scales of potential use in the psychopharmacological treatment of depressed children and adolescents. Psychopharmacology Bulletin 1985;21(4):951-77. [PubMed] [Google Scholar]
Plöderl 2019
- Plöderl M, Hengartner MP. Antidepressant prescription rates and suicide attempt rates from 2004 to 2016 in a nationally representative sample of adolescents in the USA. Epidemiology and Psychiatric Sciences 2019;28:589-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Poznanski 1996
- Poznanski EO, Mokros HB. Children's Depression Rating Scale Revised. Los Angeles, CA: WPS, 1996. [Google Scholar]
Price 2010
- Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology 2010;35(1):192-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Reynolds 1987
- Reynolds WM. Suicidal Ideation Questionnaire. Odessam FL: Psychological Assessment Resources, 1987. [Google Scholar]
Rhode 2013
- Rhode P, Lewinsohn PM, Klein DN, Seeley JR, Gau JM. Key characteristics of major depressive disorder occurring in childhood, adolescence, emerging adulthood, adulthood. Clinical Psychological Science 2013;1(1):10.1177/2167702612457599. [DOI: 10.1177/2167702612457599] [DOI] [PMC free article] [PubMed]
Rucker 2012
- Rucker G. Network meta-analysis, electrical networks and graph theory. Research Synthesis Methods 2012;3:112-24. [DOI] [PubMed] [Google Scholar]
Rucker 2015
- Rucker G, Schwarzer G. Ranking treatments in frequentist network meta-analysis works without resampling methods. BMC Medical Research Methodology 2015;15:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rush 2006
- Rush AJ, Kraemer HC, Sackeim HA, Fava M, Trivedi MH, Frank E, et al. Report by the ACNP Task Force on response and remission in major depressive disorder. Neuropsychopharmacology 2006;31(9):1841-53. [DOI] [PubMed] [Google Scholar]
Salanti 2014
- Salanti G, Del Giovane C, Chaimani A, Caldwell DM, Higgins JPT. Evaluating the quality of evidence from a network meta-analysis. Plos One 2014;9(7):ee9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Santesso 2020
- Santesso N, Glenton C, Dahm P, Garner P, Akl EA, Alper B, et al GRADE Working Group. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of Clinical Epidemiology 2020;119:126-35. [DOI] [PubMed] [Google Scholar]
Shaffer 1985
- Shaffer D, Gould MS, Brasic J, Ambrosini P, Fisher P, Bird H, et al. A children's global assessment scale (CGAS). Archives of General Psychiatry 1985;40:1228-31. [DOI] [PubMed] [Google Scholar]
Simmons 2017
- Simmons MB, Elmes A, McKenzie JE, Travena L, Hetrick SE. Right time, Right choice: Evaluation of an online decision aid for youth depression. Health Expectations 2017;20:714-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sismondo 2008
- Sismondo S. Pharmaceutical company funding and its consequences: a qualitative systematic review. Contemporary Clinical Trials 2008;29:109–13. [DOI] [PubMed] [Google Scholar]
Stallwood 2021
- Stallwood E, Monsour A, Rodrigues C, Monga S, Terwee C, Offringa M, et al. Systematic review: the measurement properties of the Children’s Depression Rating Scale−Revised in adolescents with major depressive disorder. Journal of the American Academy of Child and Adolescent Psychiatry 2021;60(1):119-33. [DOI] [PubMed] [Google Scholar]
Stata 2019 [Computer program]
- Stata. Version 16. College Station, TX, USA: StataCorp, 2019. Available at www.stata.com.
Sterne 2009
- Sterne JA, White IR, Carlin JB, Spratt M, Royston P, Kenward MG, et al. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ 2009;338:b2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Thapar 2012
- Thapar A, Collishaw S, Pine DS, Thapar AK. Depression in adolescence. Lancet 2012;379(9820):1056-67. [DOI: 10.1016/S0140-6736(11)60871-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Toren 2019
- Toren P, Goldstein G, Ben-Amitay G, Ben-Sheetrit J, Slone M. Reboxetine: a randomized controlled open-label study in children and adolescents with major depression. Israeli Journal of Psychiatry 2019;56(1):26-33. [Google Scholar]
Tsapakis 2008
- Tsapakis EM, Soldani F, Tondo L, Baldessarini RJ. Efficacy of antidepressants in juvenile depression: meta-analysis. British Journal of Psychiatry 2008;193:10-7. [DOI] [PubMed] [Google Scholar]
Turner 2008
- Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. New England Journal of Medicine 2008;358:252-60. [DOI] [PubMed] [Google Scholar]
Vitiello 2006
- Vitiello B, Zuvekas SH, Norquist GS. National estimates of antidepressant medication use among US children. Journal of the American Academy of Child and Adolescent Psychiatry 2006;45(3):271-9. [DOI] [PubMed] [Google Scholar]
Watanabe 2011
- Watanabe N, Omori IM, Nakagawa A, Cipriani A, Barbui C, Churchill R, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2011, Issue 12. Art. No: CD006528. [DOI: 10.1002/14651858.CD006528.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weersing 2006
- Weersing VR, Brent DA. Cognitive behavioral therapy for depression in youth. Child and Adolescent Psychiatric Clinics of North America 2006;15(4):939-57. [DOI] [PubMed] [Google Scholar]
Weissman 1999
- Weissman MM, Wolk S, Goldstein RB, Moreau D, Adams P, Greenwald S, et al. Depressed adolescents grown up. Journal of the American Medical Association 1999;281(18):1707-13. [PMID: 10328070] [DOI] [PubMed] [Google Scholar]
Weisz 2006
- Weisz JR, McCarty CA, Valeri SM. Effects of psychotherapy for depression in children and adolescents: a meta-analysis. Psychological Bulletin 2006;132(1):132-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Weisz 2017
- Weisz J, Kuppens S, Ng MY, Eckshtain D, Ugueto AM, Vaughn-Coaxum R, et al. What five decades of research tells us about the effects of youth psychological therapy: A multilevel meta-analysis and implications for science and practice. American Psychologist 2017;72(2):79-117. [DOI] [PubMed] [Google Scholar]
Weller 2000
- Weller EB, Weller RA. Treatment options in the management of adolescent depression. Journal of Affective Disorders 2000;61(Suppl):23-8. [DOI] [PubMed] [Google Scholar]
White 2012
- White IR, Barrett JK, Jackson D, Higgins JP. Consistency and inconsistency in network meta-analysis: model estimation using multivariate meta-regression. Research Synthesis Methods 2012;3(2):111-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
White 2015
- White IR. Network meta-analysis. The Stata Journal 2015;15:951-85. [Google Scholar]
Whitely 2020
- Whitely M, Raven M, Jureidini J. Antidepressant Prescribing and Suicide/Self-Harm by Young Australians: Regulatory Warnings, Contradictory Advice, and Long-Term Trends. Frontiers in Psychiatry 2020;11:478. [DOI: 10.3389/fpsyt.2020.00478] [DOI] [PMC free article] [PubMed] [Google Scholar]
Whittington 2004
- Whittington CJ, Kendall T, Fonagy P, Cottrell D, Cotgrove A, Boddington E. Selective serotonin reuptake inhibitors in childhood depression: systematic review of published versus unpublished data. Lancet 2004;363(9418):1341-5. [DOI] [PubMed] [Google Scholar]
WHO 1992
- World Health Organisation (WHO). ICD-10 Chapter V: Classification of Mental and Behavioural Disorders; 1992. Available at www.who.int/classifications/icd/en/bluebook.pdf.
WHO 2019
- World Health Organisation. International Statistical Classification of Diseases and Related Health Problems 10th Revision; 2019. Available at icd.who.int/browse10/2019/en.
Yepes‐Nunez 2019
- Yepes-Nunez JJ, Li SA, Guyatt G, Jack SM, Brozek JL, Beyene J, et al. Development of the summary of findings table for network meta-analysis. Journal of Clinical Epidemiology 2019;115:1-13. [DOI] [PubMed] [Google Scholar]
Zisook 2007
- Zisook S, Less I, Steward JW, Wisniewski SR, Balasuubramani GK, Gilmer WS, et al. Effect of age at onset of the course of major depressive disorder. American Journal of Psychiatry 2007;164(10):1539-46. [DOI: 10.1176/appi.ajp.2007.06101757] [DOI] [PubMed] [Google Scholar]