

A comprehensive map of the Kir2.1 interactome was generated using the proximity-labeling approach BioID. The map encompasses 218 interactions, the vast majority of which are novel, and explores the variations in the interactome profiles of Kir2.1WT versus Kir2.1Δ314-315, a trafficking deficient ATS1 mutant, thus uncovering molecular mechanisms whose malfunctions may underlie ATS1 disease. PKP4, one of the BioID interactors, is validated as a modulator of Kir2.1-controlled inward rectifier potassium currents.
Keywords: Protein-protein interactions*, cardiovascular disease, cardiovascular function or biology, mass spectrometry, macromolecular complex analysis, BioID, cardiomyopathy, inward rectifier potassium current, Kir2.1, PKP4
Graphical Abstract
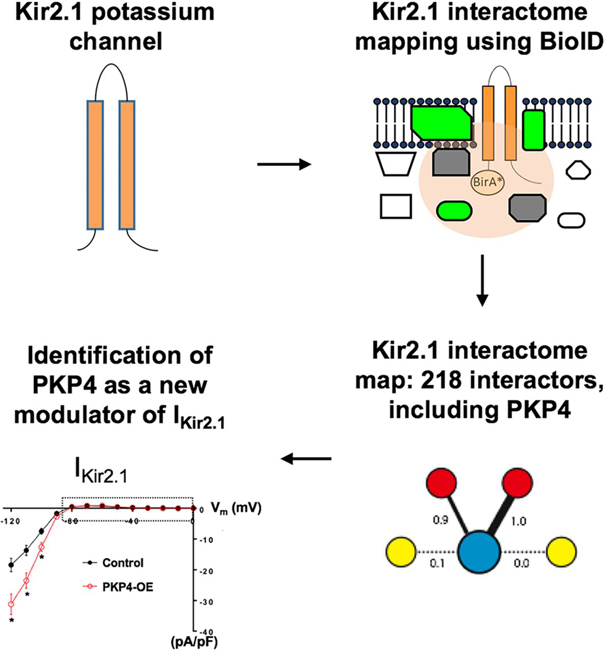
Highlights
-
•
Generation using BioID of a map of the Kir2.1 interactome with 218 interactions.
-
•
Identification of Kir2.1WT- versus Kir2.1Δ314-315-preferred interactors.
-
•
Identification of the desmosome protein PKP4 as a new modulator of IKir2.1 currents.
Abstract
Kir2.1, a strong inward rectifier potassium channel encoded by the KCNJ2 gene, is a key regulator of the resting membrane potential of the cardiomyocyte and plays an important role in controlling ventricular excitation and action potential duration in the human heart. Mutations in KCNJ2 result in inheritable cardiac diseases in humans, e.g. the type-1 Andersen-Tawil syndrome (ATS1). Understanding the molecular mechanisms that govern the regulation of inward rectifier potassium currents by Kir2.1 in both normal and disease contexts should help uncover novel targets for therapeutic intervention in ATS1 and other Kir2.1-associated channelopathies. The information available to date on protein-protein interactions involving Kir2.1 channels remains limited. Additional efforts are necessary to provide a comprehensive map of the Kir2.1 interactome. Here we describe the generation of a comprehensive map of the Kir2.1 interactome using the proximity-labeling approach BioID. Most of the 218 high-confidence Kir2.1 channel interactions we identified are novel and encompass various molecular mechanisms of Kir2.1 function, ranging from intracellular trafficking to cross-talk with the insulin-like growth factor receptor signaling pathway, as well as lysosomal degradation. Our map also explores the variations in the interactome profiles of Kir2.1WTversus Kir2.1Δ314-315, a trafficking deficient ATS1 mutant, thus uncovering molecular mechanisms whose malfunctions may underlie ATS1 disease. Finally, using patch-clamp analysis, we validate the functional relevance of PKP4, one of our top BioID interactors, to the modulation of Kir2.1-controlled inward rectifier potassium currents. Our results validate the power of our BioID approach in identifying functionally relevant Kir2.1 interactors and underline the value of our Kir2.1 interactome as a repository for numerous novel biological hypotheses on Kir2.1 and Kir2.1-associated diseases.
The strong inward rectifier potassium channel Kir2.1 is a key regulator of the resting membrane potential of the cardiomyocyte and plays an important role in controlling ventricular excitation and action potential duration in the human heart (1, 2, 3). Both loss and gain of function mutations in KCNJ2, the gene encoding Kir2.1, result in inheritable cardiac ion channel diseases. For example, several KCNJ2 loss of function mutations are associated with the inheritable type-1 Andersen-Tawil syndrome (ATS1), also known as long QT syndrome type 7, which predisposes patients to cardiac arrhythmias and sudden death (4, 5). On the other hand, KCNJ2 gain-of function mutations give rise to the type-3 variant of the short QT syndrome, which also results in increased risk of sudden cardiac death (6). Understanding the molecular mechanisms that govern the regulation of inward rectifier potassium currents by Kir2.1 in both normal and disease contexts should help uncover novel targets for therapeutic intervention in ATS1 and other Kir2.1-associated channelopathies.
Over the last 20 years, analyses of the Kir2.1 channelosome using genetic, pharmacological, and molecular approaches have greatly contributed to our understanding of its function and its role in cardiac diseases (3, 7, 8). Expression, trafficking, localization, and function of Kir2.1 are regulated by its interactions with multiple proteins. To date, 24 putative Kir2.1 interactors have been identified, out of which 16 are high-confidence Kir2.1 interactors (supplemental Table S1). For example, Leonoudakis et al. showed that the PDZ-binding motif of Kir2.1 interacts with the SAP97, CASK and LIN7C proteins, members of the membrane-associated guanylate kinase (MAGUK) family, which work both as scaffolding proteins for large macromolecular structure and as trafficking regulators (9, 10, 11). Recently, we and others demonstrated that the Kir2.1 channel and the voltage-gate sodium channel, Nav1.5, belong to common multiprotein channelosomes, enabling one to regulate the other's expression (12, 13, 14, 15). It has become apparent that ion channel proteins do not function in isolation but are part of large, multi-protein complexes, comprising not only the ion channels and their auxiliary subunits, but also components of the cytoskeleton, regulatory post-translation modification enzymes, trafficking proteins, extracellular matrix proteins, and even other ion channels (3, 7, 8). Notwithstanding the significance of the aforementioned biochemical studies, the information available on protein-protein interactions involving Kir2.1 channels, with only 16 high-confidence Kir2.1 interactors, remains limited. Additional efforts are necessary to provide a comprehensive map of the Kir2.1 interactome.
The relatively low number of known Kir2.1 interactors described to date in the literature can be attributed to its hydrophobic nature and the technical challenges associated with its biochemical manipulation. Similarly, some members of the protein complexes associated with Kir2.1 may mediate weak, transient interactions that are lost during “standard” biochemical affinity purification in the various lysis, wash, and elution steps. BioID, a cutting-edge proximity-labeling technology that is particularly well-suited to mapping protein interactions for low-solubility proteins, overcomes many of the above barriers imposed by conventional screening methods (16, 17, 18). BioID is based on the fusion of a promiscuous E. coli biotin protein ligase (BirA*) to a bait protein (17). On expression of the BirA*-bait fusion protein in cell and subsequent addition of biotin, proteins that are near-neighbors of the fusion protein are biotinylated in a proximity-dependent manner. Biotinylated proteins are then isolated by affinity capture and identified by MS (MS), thus uncovering the BioID interactome of the bait protein under investigation (17). A key advantage of this proteomic approach resides in the fact that the bait, e.g. the low-solubility channelosome protein Kir2.1, does not need to be biochemically purified in native complex and that transient interactions can be captured (16, 17, 18).
Here, we present the generation of a comprehensive map of the Kir2.1 interactome using BioID. Bait proteins used in our BioID proteomic screens include not only Kir2.1WT but also Kir2.1Δ314-315, an ATS1-associated mutation, which blocks Kir2.1 Golgi export (4, 19, 20). Thus, beyond interactome mapping, our BioID experiments also aim to capture the differences between the protein interaction profiles of Kir2.1WT versus Kir2.1Δ314-315 proteins, and to uncover the molecular mechanisms whose malfunctions underlie ATS1 disease. A total of 218 high-confidence Kir2.1 BioID interactors were identified, including 75 Kir2.1WT-preferred interactors, 66 Kir2.1Δ314-315 mutant-preferred interactors and 77 proteins that interact with both WT and mutant Kir2.1 proteins. Finally, we present patch-clamp analyses of one of our top BioID interactors, PKP4, which validate its functional relevance to modulation of Kir2.1-regulated inward rectifier potassium currents.
EXPERIMENTAL PROCEDURES
Plasmids and Antibodies
For the transmembrane protein control (TM-CTRL), the amino acid sequence IIFRTLFGSLVFAIFLILMIN of the Saccharomyces cerevisiae P25353 protein transmembrane domain, which has no homology to any human proteins, was selected and a humanized codon sequence was synthesized (supplemental Table S2). The protein-encoding Open Reading Frames (ORFs) of Kir2.1WT (NM_000891.2), Kir2.1Δ314-315, and TM-CTRL were first cloned by Gateway recombination cloning from the cDNA plasmids into the Gateway Donor vector pDONR223 to generate Entry clones, as previously described (21) (primer sequences are shown in supplemental Table S2). The Gateway clone of PKP4 (EL733946) was obtained from the Center for Cancer Systems Biology (CCSB, Harvard Medical School, Boston) human ORFeome collection (21). Protein-encoding ORF/s were then subcloned from the Entry clones into the Gateway Destination vectors pDEST-pcDNA5-FRT-BirA*-FLAG-N-term and pDEST-pcDNA3-HA (18, 22). A target sequence of PKP4 for CRISPR/Cas9 (supplemental Table S2) was selected using CHOPCHOP web tool (23) and cloned into pX459 (Addgene, Watertown, MA, 62988), as previously described (24). The following primary and secondary antibodies were used in the Western blot analyses: FLAG (Sigma, Kawasaki, Kanagawa, Japan, A8592) and goat α-rabbit IgG (Cell Signaling, Danvers, MA, 7074).
Cell Culture
Flp-In T-REx 293 cells (ThermoFisher Scientific, Waltham, MA, R78007) were maintained following the manufacturer's guideline and were transfected with the Gateway expression vectors (pcDNA5-FRT-BirA*-FLAG-N-term) of Kir2.1WT, Kir2.1Δ314-315, and TM-CTRL using polyethylenimine (Polysciences, Warrington, PA, 23966-2) (co-transfection with pOG44) as previously described (25). Stable cells were selected using 200 µg/ml hygromycin B (Invitrogen, Carlsbad, CA, 10687010). HEK293T cells were cultivated in DMEM (Invitrogen, 11995) medium supplemented with 10% FBS (Sigma, F0926) and penicillin/streptomycin (Invitrogen, 15140) and were transfected with protein expression vectors using PEI.
Affinity Purification of Biotinylated Proteins
BioID experiments were performed as previously described (18) with some minor modifications. Triplicate BioID experiments were performed for each of the bait proteins: BirA*-Kir2.1WT, BirA*-Kir2.1Δ314-315 and BirA*-TM-CTRL. Cells were incubated for 24 h with 10 µg/ml tetracycline and 50 μm biotin. After washing with cold PBS, cells (n = 3 × 107) were lysed for 30 min at 4°C with lysis buffer [50 mm Tris pH 7.2, 150 mm NaCl, 10% glycerol, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.2% SDS, protease inhibitor (Roche, Basel, Switzerland, 05 056 489 001), phosphatase inhibitor (Sigma, P0044), and Benzonase (Sigma, E1014)]. The cleared cell lysates were incubated with 30 μL of streptavidin bead slurry (ThermoFisher Scientific, 20361) for 3 h at 4°C. The beads were washed with washing buffer (50 mm Tris pH 7.2, 150 mm NaCl, 10% glycerol, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.2% SDS) four times and then with rinse buffer (50 mm Tris pH 7.2, 150 mm NaCl, 0.5% Nonidet P-40) three times. The beads were washed with fresh 50 mm NH4HCO3 three times and stored at −80°C.
Protein Identification by Mass Spectrometry and BioID Hit Selection
The samples were analyzed in the Proteomics Resource Facility of the Department of Pathology at the University of Michigan for protein identification. Briefly, upon reduction (10 mm DTT) and alkylation (65 mm 2-chloroacetamide) of the cysteines, proteins were digested overnight with 500 ng of sequencing grade, modified trypsin (Promega, Madison, WI, V5111). Resulting peptides were resolved on a nano-capillary reverse phase column (Acclaim PepMap C18, 2 μm, 50 cm, ThermoFisher Scientific, ES803) using 0.1% formic acid/acetonitrile (ACN) gradient at 300 nL/minute (2–25% ACN in 108 min, 25–40% in 20 min, followed by a 90% ACN wash for 10 min and a further 30 min' re-equilibration with 2% ACN) and directly introduced into Orbitrap Fusion Tribrid Mass Spectrometer (ThermoFisher Scientific). MS1 scans were acquired at 120k resolution. Data-dependent high-energy C-trap dissociation MS/MS spectra were acquired with top speed option (3 s) following each MS1 scan (relative CE ∼32%). Proteins were identified by searching the data against Homosapiens database containing both canonical and isoform protein entries (SwissProt v2016-11-30; total number of entries: 42054) using Proteome Discoverer (v2.1, ThermoFisher Scientific, June 2016) with enzyme specificity for trypsin. Search parameters included MS1 mass tolerance of 10 ppm and fragment tolerance of 0.1 Da; two missed cleavages were allowed; carbamidomethylation of cysteine was considered fixed modification and oxidation of methionine, deamidation of asparagine and glutamine were considered as variable modifications. False discovery rate (FDR) was determined using target-decoy strategy and proteins/peptides with a FDR of ≤1% were retained. A stringent set of criteria were used for the selection of the high-confidence Kir2.1 BioID hits: (1) average SpC (spectral counts) in either the Kir2.1WT or the Kir2.1Δ314-315 BioID experiment > 10; (2) SAINT probability (27, 28) > 0.9; and (3) primary fold change FC-A score (28) > 7. The data obtained with the TM-CTRL bait were used as negative controls for the calculations. One of the high-confidence Kir2.1 BioID interactors has been identified with a single peptide, i.e. NACA2. The spectra annotation for NACA2 is provided in the supplemental Fig. S1.
Kir2.1WT versus Kir2.1Δ314-315 Interactome Profiling
We classified the BioID hits as Kir2.1WT-preferred or Kir2.1Δ314-315 -preferred interactors based on the normalized Kir2.1WT/Δ314-315 SpC ratio “R “. For a given protein, “R “ represents the ratio of the SpC observed for this protein in the Kir2.1WT and Kir2.1Δ314-315 BioID experiments. Specifically, R was calculated as follows. First, the number of SpC observed for a given protein in a given triplicate Kir2.1WT or Kir2.1Δ314-315 BioID experiment was normalized to the Kir2.1WT BioID experiment #1 by dividing SpC by the ratio of the Kir2.1 SpC observed in this given experiment to the Kir2.1 SpC observed in the Kir2.1WT BioID experiment #1 (supplemental Table S3, columns X-AE). Second, after adding one to each SpC and Log2-transformation (columns AF-AK), we calculated the average Log2-transformed, normalized SpC for both the Kir2.1WT and Kir2.1Δ314-315 BioID experiments (columns AL and AM). The ratio R was then calculated by performing a power of 2 transformation (column AN). The statistical significances of the difference between the Kir2.1WT and Kir2.1Δ314-315 BioID experiments were computed using a two-sample t test on the Log2-transformed data (column AO). Kir2.1 BioID hits with both R > 2 and p < 0.05 were classified as Kir2.1WT-preferred interactors whereas proteins with both R < 0.5 and p < 0.05, i.e. a 2-fold decrease, were classified as Kir2.1Δ314-315-mutant preferred interactors (column AP). A permutation analysis was performed to estimate the false discovery rate (FDR < 0.007).
Immunofluorescence (IF) Staining Experiments
IF staining experiments were performed utilizing either HEK293 cells or freshly isolated rat ventricular myocytes, as previously described (15) using the following primary and secondary antibodies: PKP4 (ThermoFisher Scientific, PA5-66855, dilution 1/500), Kir2.1 (Alomone Labs, Jerusalem, Israel, AGP-044, dilution 1/20), Actinin (Sigma, SAB4503474, 1/300), GM130 (Novus Biologicals, NBP2-53420; dilution 1/200), FLAG (Sigma-Aldrich; F3165, dilution: 1/50), Alexa Fluor 488 donkey anti-mouse, Alexa Fluor 594 donkey anti-guinea pig and Alexa Fluor 647 donkey anti-rabbit (Jackson ImmunoResearch, West Grove, PA, dilution 1/400).
Patch-Clamping Experiments
HEK293 cells were grown in 60-mm dishes and, upon reaching ∼60–70% confluence, were transiently transfected using X-tremeGENE HP DNA transfection reagent (Sigma, 6366244001) following the supplier's directions. The DNA plasmids used in the patch-clamp experiments were: CRISPR/Cas9 construct pX459-gPKP4 and negative control pX459 and protein expression vectors pcDNA3-HA-PKP4 and “empty vector” negative control pcDNA3-HA. A GFP-expression vector was also co-transfected to visualize the transfected cells during the patch-clamp experiments. Patch-clamp experiments were performed 48 h after transfection, as previously described (12, 13, 15). Inward rectifier potassium currents (IKir2.1) were recorded at room temperature (21–23 °C) using the whole-cell patch-clamp technique and filtered at half the sampling frequency (12, 13, 15). Series resistance was compensated manually and ≥80% compensation was achieved. Under our experimental conditions, no significant voltage errors (<5 mV) because of series resistance were expected with the micropipettes used.
Experimental Design and Statistical Rationale
The total number of samples analyzed and the statistical tests used in the various experiments are indicated throughout the text, e.g. triplicate BioID experiments were performed, the number of transfected cell batches (N) and number of cells (n) used in the patch-clamping experiments are shown, or the statistical significances of the difference between the Kir2.1WT and Kir2.1Δ314-315 BioID experiments were computed using a two-sample t test on the Log2-transformed data.
RESULTS AND DISCUSSION
Kir2.1 Interactome Mapping Using BioID
Using the proximity-labeling technology BioID (16, 17, 18), we generated a map of the Kir2.1 interactome (overall procedure is described in Fig. 1). Both Kir2.1WT and Kir2.1Δ314-315, an ATS-associated mutant protein for which Golgi export is impaired (4, 19, 20), were used as BirA*-tagged BioID baits. We performed patch-clamping analyses of both the BirA*-tagged Kir2.1WT and Kir2.1Δ314-315 bait proteins (supplemental Fig. S2). As expected, HEK293 cells expressing BirA*-tagged Kir2.1WT, but not mutant BirA*-tagged Kir2.1Δ314-315 cells, exhibited the characteristic ability to strongly rectify, that is to pass K+ current in the inward direction much more readily than outward. Moreover, BirA*-tagged Kir2.1WT channels maintain their sensitivity to Ba2+ blocking (supplemental Fig. S2). As a negative control, we used the TM-CTRL construct, a yeast transmembrane domain fused to the BioID BirA* tag. We investigated the subcellular localization of both Kir2.1WT and Kir2.1Δ314-315 bait proteins as well as the TM-CTRL by IF staining (supplemental Fig. S3). After expression of the BirA*-Kir2.1WT, BirA*-Kir2.1Δ314-315 and BirA*-TM-CTRL bait proteins in HEK293 cells (supplemental Fig. S4) and purification of the biotinylated proteins on streptavidin-agarose beads, the purified protein extracts were subjected to MS/MS analysis for identification. The Kir2.1WT, Kir2.1Δ314-315 and TM-CTRL baits were each assessed in triplicate BioID experiments. A stringent set of criteria were used for the selection of the Kir2.1 BioID hits: i) average SpC in either the Kir2.1WT or the Kir2.1Δ314-315 BioID experiments > 10; ii) SAINT probability score (27, 28) > 0.9; and iii) primary fold change FC-A score (28) >7. Our proteomic screen resulted in the identification of 218 high-confidence Kir2.1 BioID hits, the vast majority of which are novel putative Kir2.1 direct or indirect interactors (supplemental Table S3). Interestingly, some of the newly identified BioID interactors include products of genes previously associated with physiological or pathophysiological function of the myocardium, e.g. UTRN (29, 30) and PKP4 (31, 32), as described in detail below. Curation of the literature and of the functional data reported in PubMed for each of these genes led to the identification of a dozen of known protein complexes or functionally related biological modules in our list of 218 high-confidence Kir2.1 BioID hits, e.g. the cadherin adhesome and the COPII complex.
Fig. 1.
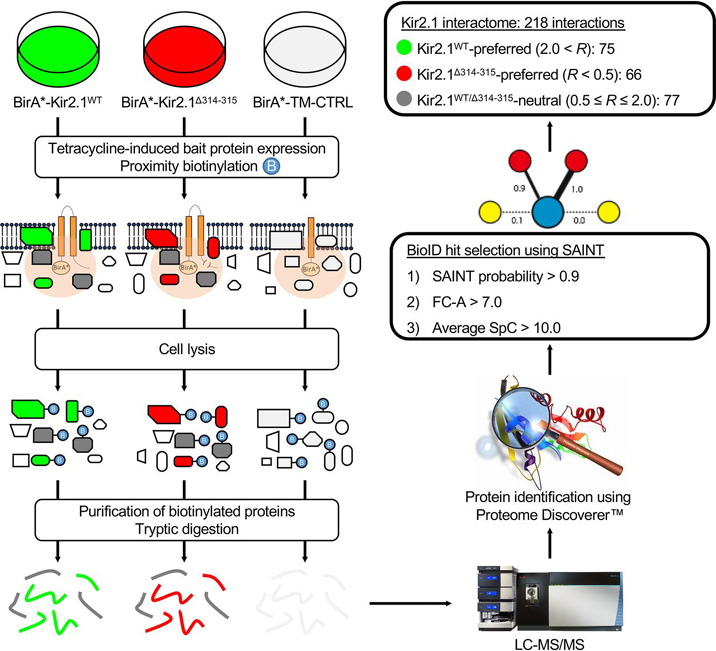
Overall procedure to generate the Kir2.1 BioID interactome map. Stable cells expressing BirA*-tagged Kir2.1WT, Kir2.1Δ314-315 or TM-CTRL bait proteins were generated using the Flp-In T-Rex 293 cell line. Expression of the bait proteins was induced by tetracycline and cells were treated with Supplemental biotin for 24 h. After cell lysis, biotinylated proteins were purified on streptavidin-agarose beads and digested with trypsin. Tryptic peptides were analyzed using LC–MS/MS and proteins were identified using Proteome Discoverer. After applying a stringent set of criteria, we identified 218 high-confidence Kir2.1 BioID hits. Using the normalized Kir2.1WT/Δ314-315 SpC ratio “R ”, we classified the interactors in three categories: 75 Kir2.1WT-preferred interactors, 66 Kir2.1Δ314-315-preferred interactors and 77 Kir2.1WT/Δ314-315-neutral interactors. CTRL: control; LC–MS/MS: liquid chromatography with tandem MS; SAINT: Significance Analysis of INTeractome; SpC: spectral counts; TM: transmembrane; WT: WT.
To assess the sensitivity of our BioID proteomic screen, we measured the proportion of the previously described Kir2.1 interactors that are also present in our list of 218 high-confidence Kir2.1 BioID interactors. A review of several protein interaction repositories [BioGRID (33), IntAct (34), HPRD (35), HuRI (36, 37)] identified 24 putative Kir2.1 protein interactions. After literature curation, we selected 16 high-confidence Kir2.1 interactions that had been observed in at least two experiments and/or validated for their functional relevance (supplemental Table S1). We note, though, that some of these interactions have been studied using the Mus musculus or Rattus norvegicus proteins, but not the human proteins (columns I–J in supplemental Table S1). Out of all the 24 previously reported putative Kir2.1 interactors, 10 (∼42%) were identified in our BioID screen. Similarly, out of the 16 high-confidence Kir2.1 interactions, 7 (∼44%) were identified in our BioID screen, i.e. SAP97, CASK, CAV1, LIN7C, AKAP5, Kir2.6 and Kir2.3 (supplemental Table S1 and S3). This analysis suggests a high sensitivity of >40% for our BioID assay. By comparison, the sensitivity of high-throughput protein interaction mapping assays used on a proteome-scale ranges from ∼20 to ∼35% (38).
Kir2.1WT versus Kir2.1Δ314-315 Interactome Profiling
Understanding the molecular mechanisms that govern the regulation of inward rectifier potassium currents by Kir2.1 in both normal and disease contexts should help uncover novel targets for therapeutic intervention in ATS1 and other Kir2.1-associated channelopathies. Beyond interaction mapping, our proteomic experiments also aims at capturing the differences between the interaction profiles of Kir2.1WT and Kir2.1Δ314-315. Using the normalized Kir2.1WT/Δ314-315 SpC ratio “R” (supplemental Table S3, columns AN-AP; see Methods for details about the calculation, normalization and statistical analysis), we classified the 218 high-confidence Kir2.1 BioID hits: 75 tend to interact preferentially with Kir2.1WT, 66 interact preferentially with Kir2.1Δ314-315 and the remaining 77 BioID interactors interact with both the Kir2.1WT and Kir2.1Δ314-315 proteins (FDR < 0.007; Fig. 2). For example, the normalized SpC counts for UTRN in the BioID experiments are: 86.0 SpC for Kir2.1WT and 0.9 SpC for Kir2.1Δ314-315, i.e. r = 56.0 and p = 8 × 10−4. UTRN is thus classified as a Kir2.1WT-preferred interactor.
Fig. 2.
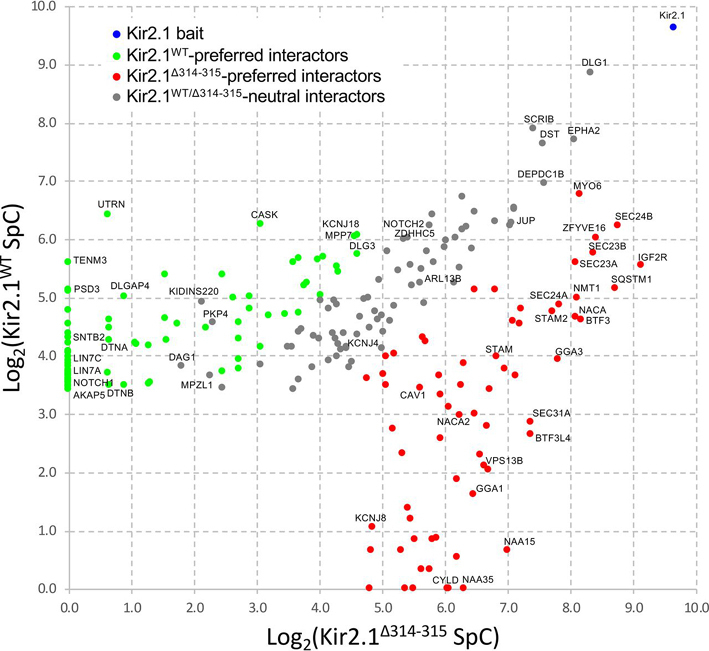
Kir2.1WTversus Kir2.1Δ314-315 interactome profiling. Variations in the Kir2.1WT and Kir2.1Δ314-315 interactome profiles are visualized in a scatter plot representing the average Log2-transformed, normalized SpC counts for both the Kir2.1WT (y axis) and Kir2.1Δ314-315 (x axis) bait proteins. The Kir2.1 bait, Kir2.1WT-preferred interactors, Kir2.1Δ314-315-preferred interactors and Kir2.1WT/Δ314-315-neutral interactors are represented as blue, green, red and gray dots, respectively.
The ATS1-associated mutation Kir2.1Δ314-315 blocks Kir2.1 Golgi export (4, 19, 20). Hence, Kir2.1WT-preferred interactors may encompass both: i) protein interactors that are directly involved in the normal trafficking of Kir2.1, and ii) protein interactors that, as an indirect consequence of the Golgi-trapping, do not interact with Kir2.1Δ314-315 simply because it is not present at the plasma membrane. Interestingly, out of the 75 Kir2.1WT-preferred interactors, 13 of them have been associated (mutation and/or GWAS hit) with one or several heart-related traits or diseases, e.g. atrial fibrillation (AF) or systolic/diastolic blood pressure (supplemental Table S3, column AR).
Most ATS mutants, including Kir2.1Δ314-315, exert a dominant-negative effect on Kir2.1WT channels, as oppose to an haploinsufficiency effect (20). Accordingly, protein interactions involving Kir2.1Δ314-315-preferred interactors may also encompass molecular mechanisms that are not properly regulated in the presence of Kir2.1Δ314-315. For example, the Kir2.1Δ314-315 mutation may result in the gain of an interaction (or increased binding affinity) between Kir2.1Δ314-315 and a protein present in the Golgi that perturbs normal trafficking and results in the trapping of Kir2.1Δ314-315. On one hand, the increased ability for Kir2.1Δ314-315 to interact with some of these Kir2.1Δ314-315-mutant preferred interactors may be an indirect consequence of the Δ314-315 mutation and these Kir2.1Δ314-315-mutant preferred interactors are thus likely to have low relevance to the disease. On the other hand, the increased ability for Kir2.1Δ314-315 to interact with some of these Kir2.1Δ314-315-mutant preferred interactors may be a direct consequence of the Δ314-315 mutation and some of these Kir2.1Δ314-315-mutant preferred interactors may thus be possible direct contributing factors to the disease. Interestingly, out of the 66 Kir2.1Δ314-315-preferred interactors, 10 of them have been associated (mutation and/or GWAS hit) with one or several heart-related traits or diseases, e.g. atrial fibrillation (AF) or systolic/diastolic blood pressure (supplemental Table S3, column AR). Below, we discuss in details the potential implications associated with some of these Kir2.1WT-preferred interactors, e.g. UTRN, and Kir2.1Δ314-315-preferred interactors, e.g. NAC and IGF1R.
The analysis of the variations in the interactome profiles of Kir2.1WT and Kir2.1Δ314-315 allows us to identify protein complexes or groups of functionally related proteins that, for the most part, tend to interact primarily with either Kir2.1WT or Kir2.1Δ314-315 (Fig. 3). For example, most of the proteins in the MAGUK complex interact preferentially with Kir2.1WT (colored in green in Fig. 3). In contrast, Kir2.1 interactors in the Nascent polypeptide-associated complex (NAC) bind preferentially to Kir2.1Δ314-315 (colored in red in Fig. 3). Interestingly, the “Channels-Transporters” group encompasses proteins that interact preferentially with either cytoplasmic membrane-bound Kir2.1WT, Golgi-trapped Kir2.1Δ314-315 or that are Kir2.1WT/Δ314-315-neutral interactors (colored in gray in Fig. 3). This observation indicates that Kir2.1 not only interacts with other channels-transporters at the cytoplasmic membrane but that the co-trafficking of these proteins in the endoplasmic reticulum (ER) and Golgi apparatus is an important aspect of their function. Thus, the previously described dynamic reciprocity for the regulation of Nav1.5 sodium and Kir2.1 potassium channel expression, which controls cardiac excitability and arrhythmia (12), likely extends to other channelosome protein complexes beyond Nav1.5.
Fig. 3.
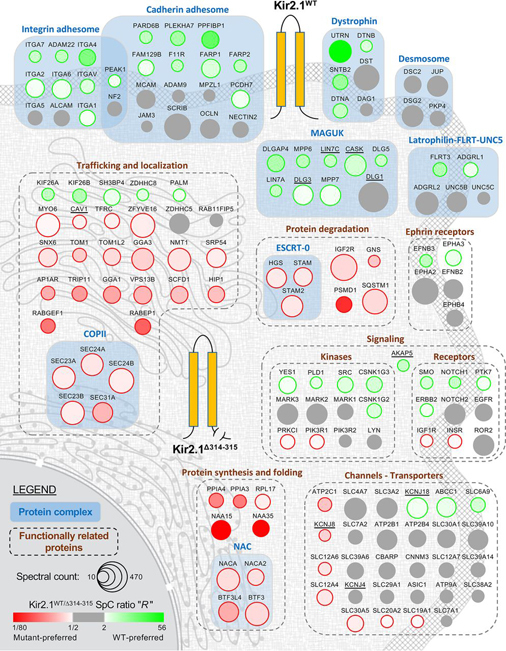
Graphical representation of the Kir2.1 BioID interactome. Protein complexes and groups of functionally related proteins encompassing 152 out of the 218 high-confidence Kir2.1 BioID hits are depicted in a cell. Major organelles in the cell (nucleus, endoplasmic reticulum, Golgi apparatus and cytoplasmic membrane) are shown (light gray) in the background to roughly indicate the approximate subcellular localization of the proteins in the cell. The Kir2.1WT-preferred interactors, Kir2.1Δ314-315-preferred interactors and Kir2.1WT/Δ314-315-neutral interactors are represented as green, red and gray circles, respectively. The color intensity of each circle is an indicator of the strength of the normalized Kir2.1WT/Δ314-315 SpC ratio “R” value. The size of each circle represents the average SpC counts observed in either the Kir2.1WT or Kir2.1Δ314-315 BioID experiment (whichever is the largest is represented in the figure).
Enrichment Analyses
To further extend our analysis of the protein complexes and the groups of genes that interact with Kir2.1, we performed a gene set enrichment analysis using DAVID (39) (supplemental Table S4). We observed that Kir2.1WT-preferred interactors are enriched for protein families involved in cell adhesion [GO:0007155, Fold Enrichment (FE) = 8, p = 2 × 10−8], including integrin complex proteins (GO:0008305, FE = 55, p = 6 × 10−8) and cadherin adhesome proteins (GO:0098641, FE = 6.1, p = 9 × 10−4). In addition to the protein domains characteristic of integrin proteins, e.g. InterPro domain IPR018184 (FE = 94, p = 3 × 10−9), Kir2.1WT-preferred interactors also tend to contain PDZ domains (IPR001478, FE = 15, p = 2 × 10−7), further highlighting the previously described key role of PDZ domain-scaffolding proteins in the modulation of Kir2.1 channelosomes (8). As expected, top KEGG pathways enriched in Kir2.1WT-preferred interactors include arrhythmogenic right ventricular cardiomyopathy (hsa05412), dilated cardiomyopathy (hsa05414) and hypertrophic cardiomyopathy (hsa05410) (FE > 17, p < 2 × 10−5).
In contrast, Kir2.1Δ314-315-preferred interactors are enriched for protein families involved in many aspects of intracellular protein transport (GO:0006886, FE = 16, p = 3 × 10−12), including COPII (GO:0030127, FE = 145, p = 3 × 10−8) and ER to Golgi transport (GO:0012507, FE = 22, p = 7 × 10−4). On the same note, seven proteins containing the multipurpose docking adapter VHS domain, e.g. TOM1 and STAM, which are associated with vesicular trafficking, are enriched among the Kir2.1Δ314-315-preferred interactors (IPR002014, FE = 222, p = 1 × 10−13). Thus, in agreement with the previous reports (4, 19, 20), our data show that ATS1 pathogenesis associated with the Kir2.1Δ314-315 mutation underlies malfunctions in the mechanisms governing Kir2.1 trafficking from the Golgi to the cytoplasmic membrane.
Kir2.1Δ314-315-preferred interactors are also linked to ESCRT-0 (GO:0033565, FE = 289, p = 3 × 10−5) and lysosome (hsa04142, FE = 7, p = 0.02), suggesting that this ATS1 mutation leads to increased targeting of Kir2.1 to lysosomal degradation. These observations agree with the previous report by Kolb et al. that Kir2.1 degradation is primarily lysosomal dependent and requires ESCRT (40). However, using a pharmacological approach to inhibit either proteasomal or vacuolar proteases, Kolb et al. also observed that Kir2.1Δ314-315 mutation targets the channel for proteasomal degradation rather than vacuolar-dependent degradation (40). Though we indeed note the presence of PSMD1, the 26S proteasome nonATPase regulatory subunit 1, among the Kir2.1Δ314-315-preferred interactors, our enrichment analysis does not provide support to the “proteasome hypothesis” but instead, strongly suggests that vacuolar-dependent degradation remains an important route responsible for the increased Kir2.1Δ314-315 protein turnover.
Strikingly, all the aforementioned functional attributes and the vast majority of the gene sets are specifically enriched in either Kir2.1WT-preferred interactors or Kir2.1Δ314-315-preferred interactors, but not both. For example, out of the top 30 gene sets enriched for Kir2.1WT-preferred interactors (p < 1 × 10−3), only one was also enriched in the Kir2.1Δ314-315-preferred interactors: basolateral plasma membrane (GO:0033565, FE = 6, p = 0.02) (supplemental Table S5). Similarly, out of the top 34 gene sets enriched for Kir2.1Δ314-315-preferred interactors (p < 1 × 10−3), only two were also enriched in the Kir2.1WT-preferred interactors (supplemental Table S5). This observation further underscores the fact that the Kir2.1WT and Kir2.1Δ314-315 proteins obey to dramatically different fates in the cell.
Kir2.1 BioID Interactome: A Repository for Novel Biological Hypotheses
The Kir2.1 BioID interactome data set represents a repository for numerous, novel biological hypotheses for genes and molecular mechanisms implicated in Kir2.1-associated cardiomyopathies. For example, out of the 218 high-confidence Kir2.1 interactors identified in our BioID screen, 37 of them have been associated (mutation and/or GWAS hit) with one or several heart-related traits or diseases, e.g. atrial fibrillation (AF) or systolic/diastolic blood pressure (supplemental Table S3, column AR). Below, we describe a few examples: UTRN, NACA, IGF1R and PKP4.
UTRN was identified in our BioID screen as a Kir2.1WT-preferred interactor (normalized SpC counts: Kir2.1WT: 86.0 and Kir2.1Δ314-315: 0.9; supplemental Table S3). Utrophin deficiency worsens cardiac contractile dysfunction present in dystrophin-deficient mice (29). Interestingly, a race-stratified genome-wide gene-environment interaction association study recently identified polymorphic variations in the UTRN gene locus as potential risk factors in peripheral arterial disease (30). UTRN is part of the Dystrophin‐associated proteins (DAP) complex (41) and several other DAP proteins interact with and modulate both sodium and potassium channelosome protein complexes (8, 11, 42). This novel Kir2.1-UTRN interaction further underlines the role of DAP in regulating these complexes and malfunction of these molecular mechanisms may contribute to pathophysiological conditions of the myocardium.
Nascent-Polypeptide-Associated Complex (NAC)
Our BioID screen identified four NAC-associated proteins as Kir2.1Δ314-315-preferred interactors, i.e. NACA, NACA2, BTF3 and BTF3L4. For example, the normalized SpC counts for NACA are as follows: Kir2.1WT: 24.4 and Kir2.1Δ314-315: 270.7 (supplemental Table S3). The heterodimeric NAC protein complex binds to newly synthesized polypeptide chains that lack a signal peptide motif as they emerge from the ribosome, blocking interaction with the signal recognition particle (SRP) and preventing mistranslocation to the endoplasmic reticulum (43). Potential roles for NAC complex thus range from protein folding chaperone to negative regulator of translocation into the endoplasmic reticulum (43). We note, though, that NACA has also been described as a transcription factor (44), e.g. in collaboration with the heart-specific HDAC-dependent repressor SMYD1 (45). The roles of NACA and NAC in the cell remain to be comprehensively characterized.
Several observations link NACA to heart biology and diseases. For example, GWAS studies have identified NACA gene variants as potential risk factors in atrial fibrillation (AF) (46, 47), or as a regulator of myocardial mass (48). In both Drosophila melanogaster flies and Danio rerio zebrafish, the knock-down of NACA, i.e. fly NACα and zebrafish skNAC, results in severe cardiac muscle defects (48, 49). In mouse, Naca deficiency results in embryonic lethality with cardiac developmental defects (50). Park et al. originally linked this phenotype to the role of NACA as a transcription factor and as a major binding partner for SMYD1 in the developing heart (50). The Kir2.1-NACA interaction suggests a more complex role for NACA in the myocardium. Indeed, Kir2.1 deficiency results not only in the loss of inward rectifier potassium currents but also in developmental defects and dysmorphic features, e.g. low-set ears, wide-set eyes, small mandible, clinodactyly and syndactyly (4, 5, 51, 52, 53). The molecular mechanism underlying these dysmorphic features is poorly understood. The Kir2.1-NACA interaction provides a biophysical bridge between the respective roles of these two proteins during biological development in general and cardiomyogenesis in particular.
Cross-talks between Kir2.1 and Signal Transduction Pathways
The Kir2.1 interactor data set as a whole is enriched for proteins involved in various aspects of signal transduction, i.e. enrichment is observed for all three classes of Kir2.1 interactors (WT, mutant and neutral; GO:0007165, FE > 2.4, p < 0.01). Each class of interactors is enriched with different signaling pathways, though. Examples of signaling pathways over-represented in our Kir2.1WT-preferred interactor data set include the ephrin receptor signaling pathway (GO:0048013, FE = 12, p = 5 × 10−3) and the Hedgehog signaling pathway (KEGG hsa04340, FE = 26, p = 5 × 10−3).
Likewise, the Kir2.1Δ314-315-preferred interactors are enriched for proteins involved in the insulin-like growth factor receptor signaling pathway, e.g. IGF1R and INSR (GO:0048009, FE = 57, p = 1 × 10−3). The normalized SpC counts for IGF1R and INSR are as follows: Kir2.1WT: 12.1 and 15.7, and for Kir2.1Δ314-315: 31.6 and 35.4, respectively (supplemental Table S3). GWAS studies have identified IGF1R gene variants as potential risk factors in atrial fibrillation (AF) (46, 47), as well as a regulator of myocardial mass (48) and global electrical heterogeneity ECG (54). We hypothesize that the Kir2.1-IGF1R interaction is part of a molecular mechanism that underlies the functional associations between IGF1R and cardiac physiology.
Could the Kir2.1-IGF1R interaction also help better understand the potential role of Kir2.1 in diabetes? A functional link between Kir2.1 and the insulin-like growth factor receptor signaling pathway has been previously proposed. As early as 1998, Wischmeyer et al. observed that treatment of Xenopus oocytes with insulin was found to robustly suppress IKir2.1 (55). Electrical activity plays a central role in glucose-stimulated insulin secretion from human pancreatic β-cells. We note that Kir2.1 is expressed in human pancreatic islets (56) and that functional Kir2.1 currents are present in human β-cells (57). Riz et al. predicted that blocking Kir2.1 channels increases the rate of insulin secretion and that hyperactive Kir2.1 channels may lead to reduced insulin secretion (57). Similarly, in a recent mathematical model, Kir2.1 was predicted to be a critical actor in the regulation of oscillations in cellular activity that underlie insulin pulsatility in pancreatic islets cells lacking K(ATP) channels (58). On the same note, several publications link abnormal QT interval regulation and diabetes (59, 60, 61). Moreover, patients with long QT syndrome associated to potassium channel deficiency, e.g. KCNQ1 mutants, suffer from over-secretion of insulin, hyperinsulinemia, and symptomatic hypoglycemia (62). Our BioID data suggest that these functional associations between Kir2.1 and the insulin pathway need not be indirect but may also be mediated by biophysical interactions between Kir2.1 channels and insulin pathway proteins.
Desmosome
Several desmosome-associated proteins interact with both Kir2.1WT and Kir2.1Δ314-315, e.g. JUP, DSG2, DSC2 and PKP4 (supplemental Table S3). Desmosomes act as mechanical cell-cell adhesion junctions maintaining the structural integrity of the tissue. Mutations in the JUP, DSG2 and DSC2 genes have been associated with Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) (63). Actually, with ∼half of ARVC patients carrying a mutation in one of the genes encoding desmosomal proteins expressed in the heart, ARVC can be considered a disease of the desmosome (63). Mutations in the JUP genes have also been associated with Naxos disease, a diffuse nonepidermolytic palmoplantar keratoderma with wooly hair and cardiomyopathy (64). We also note that JUP mRNA is highly differentially expressed in AF (65).
Functional Validation of PKP4 by Immunofluorescence and Patch-Clamp Analyses
PKP4 is one of the desmosome proteins identified in our BioID experiment as interacting with both Kir2.1WT and Kir2.1Δ314-315 (normalized SpC counts: Kir2.1WT: 23.3 and Kir2.1Δ314-315: 7.6). PKP4 is a multifunctional armadillo protein coordinating cell adhesion with cytoskeletal organization that also has a role in vesicle transport processes (66). Though less frequent than the ones observed for the JUP, DSG2 and DSC2 genes, mutations in the PKP4 gene have been observed in ARVC patients (31, 32). Similarly to many other Kir2.1 interactors (8), PKP4 carries a PDZ binding site at its carboxyl terminus. We hypothesize that the Kir2.1-PKP4 interaction contributes to the regulation of Kir2.1.
As a first step toward validating the role of PKP4 as a regulator of Kir2.1, we investigated the subcellular localization of both proteins by immunofluorescence (IF) staining in freshly isolated adult rat ventricular myocytes. We observed that Kir2.1 and PKP4 co-localize at intercalated disks and at z-disks near the cardiac sarcomeres (Pearson correlation coefficient = 0.85) (Fig. 4).
Fig. 4.
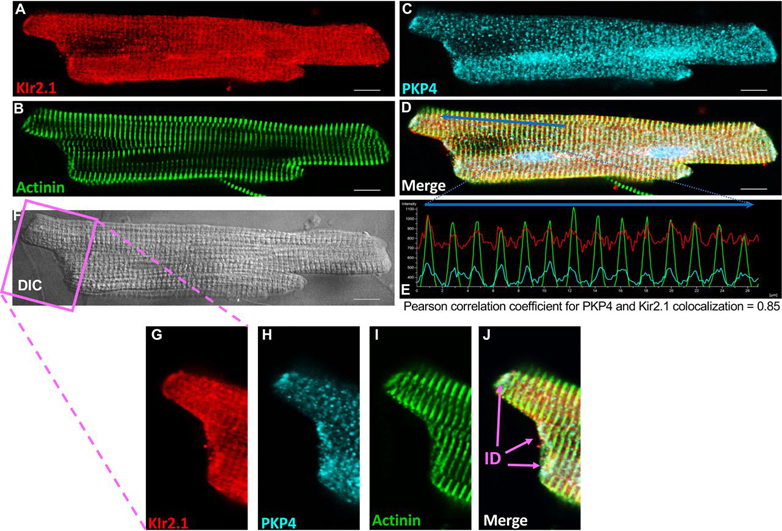
Kir2.1 and PKP4 co-localize in adult ventricular myocytes. Immunofluorescence (IF) staining analyses of the subcellular localization of Kir2.1 (A, red), Actinin (B, green) and PKP4 (C, light blue) in a freshly isolated rat adult ventricular myocytes. (D) Merge image. (E) Pixel intensity profile of PKP4, Kir2.1 and actinin along a line in the merge image, i.e. blue arrow shown in (D) showing the striated co-localization of the three proteins at the z-disks near the cardiac sarcomeres. (F) Differential interference contrast (DIC) image of the myocyte. (G–J) Zoomed in images of the intercalated disks. Scale bars: 10 μm. ID: intercalated disk.
Finally, to assess the functional relevance of PKP4 to the modulation of IKir2.1, we performed patch-clamp in the voltage clamp configuration (12, 13, 15) in a stable HEK293 cell line expressing Kir2.1 upon genetic perturbation of PKP4 (Fig. 5). Upon PKP4 overexpression, we observed a gain of IKir2.1 density (Fig. 5A). Accordingly, upon CRISPR/Cas9-mediated depletion of PKP4, we observed a loss of IKir2.1 density (Fig. 5B). Therefore, in addition to interacting with Kir2.1 (Fig. 2) and to being mutated in an ARVC patients (31, 32), PKP4 is a positive regulator of IKir2.1 density.
Fig. 5.
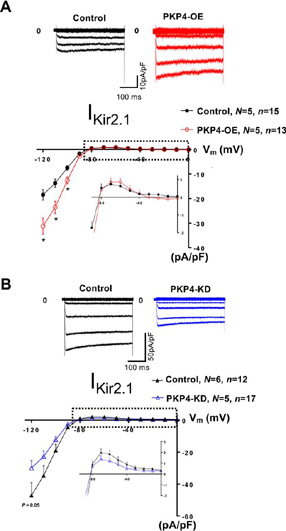
PKP4 is a positive regulator of IKir2.1. Patch-clamping analyses in HEK293 cells upon genetic perturbation of PKP4, i.e. (A) upon overexpression of PKP4 and (B) upon CRISPR/Cas9-mediated depletion of PKP4. *P < 0.05 versus control. N: number of transfected cell batches; n: number of cells; OE: overexpression; KD: knock-down.
CONCLUSION
Evidence accumulated over the last 20 years strongly indicates that potassium channels function depends on a complex and dynamic ensemble of molecular events occurring in various organelles in the cell that help in the assembly and delivery of the right channel subunits to the right place in the cell membrane at the right time. Understanding such molecular events should help uncover novel insights into how channelosome proteins and accessory proteins participate in the control of cardiac excitability and the mechanisms of arrhythmias. Using a BioID proteomic approach, we have generated the most comprehensive Kir2.1 interactome map known to date with 218 high-confidence Kir2.1 BioID hits, the vast majority of which are novel putative Kir2.1 interactors. Moreover, our approach allowed us to identify novel proteins that interact preferentially with either Kir2.1WT or the ATS1 mutant Kir2.1Δ314-315, thus uncovering novel molecular mechanisms underlying Kir2.1 function/dysfunction in ATS1. Our Kir2.1 interactome map represents a repository for numerous novel biological hypotheses. Using patch-clamping, we verified the functional relevance of one of our hits, PKP4, to the modulation of IKir2.1, thus providing a molecular mechanism by which PKP4 may be involved in ARVC. This result validates the power of our BioID interactome approach in identifying functionally relevant Kir2.1 interactors and modulators of IKir2.1.
DATA AVAILABILITY
The MS raw data and Proteome Discoverer protein identification have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (26) with the data set identifier PXD011004.
Acknowledgments
We thank Drs. Anne Claude Gingras (The Lunenfeld-Tanenbaum Research Institute, University of Toronto, Canada) and Brian Raught (Princess Margaret Cancer Center, University of Toronto, Canada) for providing us with BioID reagents. We thank Dr. Robin Kunkel for her help with the cell illustration shown in Fig. 3. We thank Dr. M. Vidal and members of the CCSB (Harvard Medical School, Boston) for sharing the PKP4 ORFeome clone.
HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI) (R01HL122352) to José Jalife
HHS | NIH | National Institute of General Medical Sciences (NIGMS) (R01GM094231) to Alexey I. Nesvizhskii
HHS | NIH | National Cancer Institute (NCI) (U24CA210967) to Alexey I. Nesvizhskii
HHS | NIH | National Cancer Institute (NCI) (P30CA046592) to Rork Kuick
Footnotes
This article contains supplemental data.
Funding and additional information—This work was supported by the National Institutes of Health (NIH) through the National Heart, Lung, and Blood Institute (NHLBI) grant R01HL122352 awarded to J.J., as well as the National Institute of General Medical Sciences (NIGMS) grant R01GM094231 and the National Cancer Institute (NCI) grant U24CA210967 awarded to A.I.N. R.K. is supported by the NCI support grant P30CA046592 awarded to the University of Michigan Rogel Cancer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of interest—Authors declare no competing interests.
Abbreviations—The abbreviations used are:
- ACN
- acetonitrile
- AF
- atrial fibrillation
- ARVC
- arrhythmogenic right ventricular cardiomyopathy
- ATS1
- type-1 Andersen-Tawil syndrome
- BirA*
- promiscuous biotin ligase
- CTRL
- control
- DAP
- dystrophin‐associated proteins
- DIC
- differential interference contrast
- ER
- endoplasmic reticulum
- EV
- empty vector control
- FDR
- false discovery rate
- IKir2.1
- inward rectifier potassium current
- IF
- immunofluorescence
- KD
- knock-down
- LC-MS/MS
- liquid chromatography with tandem mass spectrometry
- MAGUK
- membrane-associated guanylate kinase
- MS
- mass spectrometry
- NAC
- nascent polypeptide-associated complex
- OE
- overexpression
- ORF
- open reading frame
- PEI
- polyethylenimine
- R
- the normalized Kir2.1WT/Δ314-315 SpC ratio
- SAINT
- significance analysis of interactome
- SpC
- spectral count
- SRP
- signal recognition particle
- Tet
- tetracycline
- TM
- transmembrane
- TM-CTRL
- transmembrane protein control
- WB
- western blot
- WT
- wild type.
Author contributions—J.F.R. and J.J. conceived and directed the project. S.S.P., J.Y., D.M., K.P.C., V.B. and A.I.N. designed and performed the BioID experiments. R.K. performed the interactome profiling and enrichment analyses. D.P.B. and G.G.S. designed and performed the patch-clamping and the IF analyses. J.F.R. wrote the manuscript, with contributions from other coauthors.
Contributor Information
José Jalife, Email: jjalife@umich.edu.
Jean-François Rual, Email: jrual@umich.edu.
Supplementary Material
REFERENCES
- 1.Wang Z., Yue L., White M., Pelletier G., Nattel S. Differential distribution of inward rectifier potassium channel transcripts in human atrium versus ventricle. Circulation. 1998;98:2422–2428. doi: 10.1161/01.cir.98.22.2422. [DOI] [PubMed] [Google Scholar]
- 2.Nichols C.G., Makhina E.N., Pearson W.L., Sha Q., Lopatin A.N. Inward rectification and implications for cardiac excitability. Circ. Res. 1996;78:1–7. doi: 10.1161/01.res.78.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Hibino H., Inanobe A., Furutani K., Murakami S., Findlay I., Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 4.Plaster N.M., Tawil R., Tristani-Firouzi M., Canún S., Bendahhou S., Tsunoda A., Donaldson M.R., Iannaccone S.T., Brunt E., Barohn R., Clark J., Deymeer F., George A.L., Fish F.A., Hahn A., Nitu A., Ozdemir C., Serdaroglu P., Subramony S.H., Wolfe G., Fu Y.H., Ptácek L.J. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell. 2001;105:511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen H.L., Pieper G.H., Wilders R. Andersen-Tawil syndrome: clinical and molecular aspects. Int J Cardiol. 2013;170:1–16. doi: 10.1016/j.ijcard.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 6.Priori S.G., Pandit S.V., Rivolta I., Berenfeld O., Ronchetti E., Dhamoon A., Napolitano C., Anumonwo J., di Barletta M.R., Gudapakkam S., Bosi G., Stramba-Badiale M., Jalife J. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 2005;96:800–807. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 7.Abriel H., Rougier J.S., Jalife J. Ion channel macromolecular complexes in cardiomyocytes: roles in sudden cardiac death. Circ. Res. 2015;116:1971–1988. doi: 10.1161/CIRCRESAHA.116.305017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willis B.C., Ponce-Balbuena D., Jalife J. Protein assemblies of sodium and inward rectifier potassium channels control cardiac excitability and arrhythmogenesis. Am. J. Physiol. Heart Circ Physiol. 2015;308:H1463–H1473. doi: 10.1152/ajpheart.00176.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leonoudakis D., Conti L.R., Radeke C.M., McGuire L.M., Vandenberg C.A. A multiprotein trafficking complex composed of SAP97, CASK, Veli, and Mint1 is associated with inward rectifier Kir2 potassium channels. J. Biol. Chem. 2004;279:19051–19063. doi: 10.1074/jbc.M400284200. [DOI] [PubMed] [Google Scholar]
- 10.Leonoudakis D., Mailliard W., Wingerd K., Clegg D., Vandenberg C. Inward rectifier potassium channel Kir2.2 is associated with synapse-associated protein SAP97. J. Cell Sci. 2001;114:987–998. doi: 10.1242/jcs.114.5.987. [DOI] [PubMed] [Google Scholar]
- 11.Leonoudakis D., Conti L.R., Anderson S., Radeke C.M., McGuire L.M., Adams M.E., Froehner S.C., Yates J.R., 3rd, Vandenberg C.A. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J. Biol. Chem. 2004;279:22331–22346. doi: 10.1074/jbc.M400285200. [DOI] [PubMed] [Google Scholar]
- 12.Milstein M.L., Musa H., Balbuena D.P., Anumonwo J.M., Auerbach D.S., Furspan P.B., Hou L., Hu B., Schumacher S.M., Vaidyanathan R., Martens J.R., Jalife J. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc. Natl. Acad. Sci. U S A. 2012;109:E2134–E2143. doi: 10.1073/pnas.1109370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matamoros M., Pérez-Hernández M., Guerrero-Serna G., Amorós I., Barana A., Núñez M., Ponce-Balbuena D., Sacristán S., Gómez R., Tamargo J., Caballero R., Jalife J., Delpón E. Nav1.5 N-terminal domain binding to alpha1-syntrophin increases membrane density of human Kir2.1, Kir2.2 and Nav1.5 channels. Cardiovasc. Res. 2016;110:279–290. doi: 10.1093/cvr/cvw009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Utrilla R.G., Nieto-Marín P., Alfayate S., Tinaquero D., Matamoros M., Pérez-Hernández M., Sacristán S., Ondo L., de Andrés R., Díez-Guerra F.J., Tamargo J., Delpón E., Caballero R. Kir2.1-Nav1.5 Channel Complexes Are Differently Regulated than Kir2.1 and Nav1.5 Channels Alone. Front Physiol. 2017;8:903. doi: 10.3389/fphys.2017.00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ponce-Balbuena D., Guerrero-Serna G., Valdivia C.R., Caballero R., Diez-Guerra F.J., Jiménez-Vázquez E.N., Ramírez R.J., Monteiro da Rocha A., Herron T.J., Campbell K.F., Willis B.C., Alvarado F.J., Zarzoso M., Kaur K., Pérez-Hernández M., Matamoros M., Valdivia H.H., Delpón E., Jalife J. Cardiac Kir2.1 and NaV1.5 Channels Traffic Together to the Sarcolemma to Control Excitability. Circ. Res. 2018;122:1501–1516. doi: 10.1161/CIRCRESAHA.117.311872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li P., Li J., Wang L., Di L.J. Proximity labeling of interacting proteins: Application of BioID as a discovery tool. Proteomics. 2017;17:1700002. doi: 10.1002/pmic.201700002. [DOI] [PubMed] [Google Scholar]
- 17.Roux K.J., Kim D.I., Raida M., Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012;196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambert J.P., Tucholska M., Go C., Knight J.D., Gingras A.C. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J Proteomics. 2015;118:81–94. doi: 10.1016/j.jprot.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma D., Taneja T.K., Hagen B.M., Kim B.Y., Ortega B., Lederer W.J., Welling P.A. Golgi export of the Kir2.1 channel is driven by a trafficking signal located within its tertiary structure. Cell. 2011;145:1102–1115. doi: 10.1016/j.cell.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bendahhou S., Donaldson M.R., Plaster N.M., Tristani-Firouzi M., Fu Y.-H., Ptácek L.J. Defective potassium channel Kir2.1 trafficking underlies Andersen-Tawil syndrome. J. Biol. Chem. 2003;278:51779–51785. doi: 10.1074/jbc.M310278200. [DOI] [PubMed] [Google Scholar]
- 21.Rual J.F., Hirozane-Kishikawa T., Hao T., Bertin N., Li S., Dricot A., Li N., Rosenberg J., Lamesch P., Vidalain P.O., Clingingsmith T.R., Hartley J.L., Esposito D., Cheo D., Moore T., Simmons B., Sequerra R., Bosak S., Doucette-Stamm L., Le Peuch C., Vandenhaute J., Cusick M.E., Albala J.S., Hill D.E., Vidal M. Human ORFeome version 1.1: a platform for reverse proteomics. Genome Res. 2004;14:2128–2135. doi: 10.1101/gr.2973604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu T., Park S.S., Giaimo B.D., Hall D., Ferrante F., Ho D.M., Hori K., Anhezini L., Ertl I., Bartkuhn M., Zhang H., Milon E., Ha K., Conlon K.P., Kuick R., Govindarajoo B., Zhang Y., Sun Y., Dou Y., Basrur V., Elenitoba-Johnson K.S., Nesvizhskii A.I., Ceron J., Lee C.Y., Borggrefe T., Kovall R.A., Rual J.F. RBPJ/CBF1 interacts with L3MBTL3/MBT1 to promote repression of Notch signaling via histone demethylase KDM1A/LSD1. EMBO J. 2017;36:3232–3249. doi: 10.15252/embj.201796525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Labun K., Montague T.G., Gagnon J.A., Thyme S.B., Valen E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016;44:W272–W276. doi: 10.1093/nar/gkw398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Longo P.A., Kavran J.M., Kim M.S., Leahy D.J. Transient mammalian cell transfection with polyethylenimine (PEI) Methods Enzymol. 2013;529:227–240. doi: 10.1016/B978-0-12-418687-3.00018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deutsch E.W., Csordas A., Sun Z., Jarnuczak A., Perez-Riverol Y., Ternent T., Campbell D.S., Bernal-Llinares M., Okuda S., Kawano S., Moritz R.L., Carver J.J., Wang M., Ishihama Y., Bandeira N., Hermjakob H., Vizcaíno J.A. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;45:D1100–D1106. doi: 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi H., Larsen B., Lin Z.Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z.S., Tyers M., Gingras A.C., Nesvizhskii A.I. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods. 2011;8:70–73. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y., Halim V.A., Bagshaw R.D., Hubner N.C., Al-Hakim A., Bouchard A., Faubert D., Fermin D., Dunham W.H., Goudreault M., Lin Z.Y., Badillo B.G., Pawson T., Durocher D., Coulombe B., Aebersold R., Superti-Furga G., Colinge J., Heck A.J., Choi H., Gstaiger M., Mohammed S., Cristea I.M., Bennett K.L., Washburn M.P., Raught B., Ewing R.M., Gingras A.C., Nesvizhskii A.I. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janssen P.M., Hiranandani N., Mays T.A., Rafael-Fortney J.A. Utrophin deficiency worsens cardiac contractile dysfunction present in dystrophin-deficient mdx mice. Am. J. Physiol. Heart Circ Physiol. 2005;289:H2373–H2378. doi: 10.1152/ajpheart.00448.2005. [DOI] [PubMed] [Google Scholar]
- 30.Ward-Caviness C.K., Neas L.M., Blach C., Haynes C.S., LaRocque-Abramson K., Grass E., Dowdy E., Devlin R.B., Diaz-Sanchez D., Cascio W.E., Lynn Miranda M., Gregory S.G., Shah S.H., Kraus W.E., Hauser E.R. Genetic Variants in the Bone Morphogenic Protein Gene Family Modify the Association between Residential Exposure to Traffic and Peripheral Arterial Disease. PLoS ONE. 2016;11:e0152670. doi: 10.1371/journal.pone.0152670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gandjbakhch E., Vite A., Gary F., Fressart V., Donal E., Simon F., Hidden-Lucet F., Komajda M., Charron P., Villard E. Screening of genes encoding junctional candidates in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Europace. 2013;15:1522–1525. doi: 10.1093/europace/eut224. [DOI] [PubMed] [Google Scholar]
- 32.Xu T., Yang Z., Vatta M., Rampazzo A., Beffagna G., Pilichou K., Pillichou K., Scherer S.E., Saffitz J., Kravitz J., Zareba W., Danieli G.A., Lorenzon A., Nava A., Bauce B., Thiene G., Basso C., Calkins H., Gear K., Marcus F., Towbin J.A., Multidisciplinary Study of Right Ventricular Dysplasia Investigators Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2010;55:587–597. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chatr-Aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N.K., O'Donnell L., Oster S., Theesfeld C., Sellam A., Stark C., Breitkreutz B.J., Dolinski K., Tyers M. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45:D369–D379. doi: 10.1093/nar/gkw1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N.H., Chavali G., Chen C., del-Toro N., Duesbury M., Dumousseau M., Galeota E., Hinz U., Iannuccelli M., Jagannathan S., Jimenez R., Khadake J., Lagreid A., Licata L., Lovering R.C., Meldal B., Melidoni A.N., Milagros M., Peluso D., Perfetto L., Porras P., Raghunath A., Ricard-Blum S., Roechert B., Stutz A., Tognolli M., van Roey K., Cesareni G., Hermjakob H. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keshava Prasad T.S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., Balakrishnan L., Marimuthu A., Banerjee S., Somanathan D.S., Sebastian A., Rani S., Ray S., Harrys Kishore C.J., Kanth S., Ahmed M., Kashyap M.K., Mohmood R., Ramachandra Y.L., Krishna V., Rahiman B.A., Mohan S., Ranganathan P., Ramabadran S., Chaerkady R., Pandey A. Human Protein Reference Database–2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rolland T., Taşan M., Charloteaux B., Pevzner S.J., Zhong Q., Sahni N., Yi S., Lemmens I., Fontanillo C., Mosca R., Kamburov A., Ghiassian S.D., Yang X., Ghamsari L., Balcha D., Begg B.E., Braun P., Brehme M., Broly M.P., Carvunis A.-R., Convery-Zupan D., Corominas R., Coulombe-Huntington J., Dann E., Dreze M., Dricot A., Fan C., Franzosa E., Gebreab F., Gutierrez B.J., Hardy M.F., Jin M., Kang S., Kiros R., Lin G.N., Luck K., MacWilliams A., Menche J., Murray R.R., Palagi A., Poulin M.M., Rambout X., Rasla J., Reichert P., Romero V., Ruyssinck E., Sahalie J.M., Scholz A., Shah A.A., Sharma A., Shen Y., Spirohn K., Tam S., Tejeda A.O., Trigg S.A., Twizere J.-C., Vega K., Walsh J., Cusick M.E., Xia Y., Barabási A.-L., Iakoucheva L.M., Aloy P., De Las Rivas J., Tavernier J., Calderwood M.A., Hill D.E., Hao T., Roth F.P., Vidal M. A proteome-scale map of the human interactome network. Cell. 2014;159:1212–1226. doi: 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rual J.F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G.F., Gibbons F.D., Dreze M., Ayivi-Guedehoussou N., Klitgord N., Simon C., Boxem M., Milstein S., Rosenberg J., Goldberg D.S., Zhang L.V., Wong S.L., Franklin G., Li S., Albala J.S., Lim J., Fraughton C., Llamosas E., Cevik S., Bex C., Lamesch P., Sikorski R.S., Vandenhaute J., Zoghbi H.Y., Smolyar A., Bosak S., Sequerra R., Doucette-Stamm L., Cusick M.E., Hill D.E., Roth F.P., Vidal M. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 38.Braun P., Tasan M., Dreze M., Barrios-Rodiles M., Lemmens I., Yu H., Sahalie J.M., Murray R.R., Roncari L., de Smet A.S., Venkatesan K., Rual J.F., Vandenhaute J., Cusick M.E., Pawson T., Hill D.E., Tavernier J., Wrana J.L., Roth F.P., Vidal M. An experimentally derived confidence score for binary protein-protein interactions. Nat. Methods. 2009;6:91–97. doi: 10.1038/nmeth.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 40.Kolb A.R., Needham P.G., Rothenberg C., Guerriero C.J., Welling P.A., Brodsky J.L. ESCRT regulates surface expression of the Kir2.1 potassium channel. Mol. Biol. Cell. 2014;25:276–289. doi: 10.1091/mbc.E13-07-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tommasi di Vignano A., Di Zenzo G., Sudol M., Cesareni G., Dente L. Contribution of the different modules in the utrophin carboxy-terminal region to the formation and regulation of the DAP complex. FEBS Lett. 2000;471:229–234. doi: 10.1016/s0014-5793(00)01400-9. [DOI] [PubMed] [Google Scholar]
- 42.Matamoros M., Pérez-Hernández M., Guerrero-Serna G., Amorós I., Barana A., Núñez M., Ponce-Balbuena D., Sacristán S., Gómez R., Tamargo J., Caballero R., Jalife J., Delpón E. Nav1.5 N-terminal domain binding to alpha1-syntrophin increases membrane density of human Kir2.1 Kir2.2 and Nav1.5 channels. Cardiovascular Research. 2016;110:279–290. doi: 10.1093/cvr/cvw009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rospert S., Dubaquie Y., Gautschi M. Nascent-polypeptide-associated complex. Cell Mol Life Sci. 2002;59:1632–1639. doi: 10.1007/PL00012490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yotov W.V., St-Arnaud R. Differential splicing-in of a proline-rich exon converts alphaNAC into a muscle-specific transcription factor. Genes Dev. 1996;10:1763–1772. doi: 10.1101/gad.10.14.1763. [DOI] [PubMed] [Google Scholar]
- 45.Sims R.J., 3rd, Weihe E.K., Zhu L., O'Malley S., Harriss J.V., Gottlieb P.D. m-Bop, a repressor protein essential for cardiogenesis, interacts with skNAC, a heart- and muscle-specific transcription factor. J. Biol. Chem. 2002;277:26524–26529. doi: 10.1074/jbc.M204121200. [DOI] [PubMed] [Google Scholar]
- 46.Morales J., Welter D., Bowler E.H., Cerezo M., Harris L.W., McMahon A.C., Hall P., Junkins H.A., Milano A., Hastings E., Malangone C., Buniello A., Burdett T., Flicek P., Parkinson H., Cunningham F., Hindorff L.A., MacArthur J.A.L. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 2018;19:21. doi: 10.1186/s13059-018-1396-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roselli C., Chaffin M.D., Weng L.C., Aeschbacher S., Ahlberg G., Albert C.M., Almgren P., Alonso A., Anderson C.D., Aragam K.G., Arking D.E., Barnard J., Bartz T.M., Benjamin E.J., Bihlmeyer N.A., Bis J.C., Bloom H.L., Boerwinkle E., Bottinger E.B., Brody J.A., Calkins H., Campbell A., Cappola T.P., Carlquist J., Chasman D.I., Chen L.Y., Chen Y.I., Choi E.K., Choi S.H., Christophersen I.E., Chung M.K., Cole J.W., Conen D., Cook J., Crijns H.J., Cutler M.J., Damrauer S.M., Daniels B.R., Darbar D., Delgado G., Denny J.C., Dichgans M., Dorr M., Dudink E.A., Dudley S.C., Esa N., Esko T., Eskola M., Fatkin D., Felix S.B., Ford I., Franco O.H., Geelhoed B., Grewal R.P., Gudnason V., Guo X., Gupta N., Gustafsson S., Gutmann R., Hamsten A., Harris T.B., Hayward C., Heckbert S.R., Hernesniemi J., Hocking L.J., Hofman A., Horimoto A., Huang J., Huang P.L., Huffman J., Ingelsson E., Ipek E.G., Ito K., Jimenez-Conde J., Johnson R., Jukema J.W., Kaab S., Kahonen M., Kamatani Y., Kane J.P., Kastrati A., Kathiresan S., Katschnig-Winter P., Kavousi M., Kessler T., Kietselaer B.L., Kirchhof P., Kleber M.E., Knight S., Krieger J.E., Kubo M., Launer L.J., Laurikka J., Lehtimaki T., Leineweber K., Lemaitre R.N., Li M., Lim H.E., Lin H.J., Lin H., Lind L., Lindgren C.M., Lokki M.L., London B., Loos R.J.F., Low S.K., Lu Y., Lyytikainen L.P., Macfarlane P.W., Magnusson P.K., Mahajan A., Malik R., Mansur A.J., Marcus G.M., Margolin L., Margulies K.B., Marz W., McManus D.D., Melander O., Mohanty S., Montgomery J.A., Morley M.P., Morris A.P., Muller-Nurasyid M., Natale A., Nazarian S., Neumann B., Newton-Cheh C., Niemeijer M.N., Nikus K., Nilsson P., Noordam R., Oellers H., Olesen M.S., Orho-Melander M., Padmanabhan S., Pak H.N., Pare G., Pedersen N.L., Pera J., Pereira A., Porteous D., Psaty B.M., Pulit S.L., Pullinger C.R., Rader D.J., Refsgaard L., Ribases M., Ridker P.M., Rienstra M., Risch L., Roden D.M., Rosand J., Rosenberg M.A., Rost N., Rotter J.I., Saba S., Sandhu R.K., Schnabel R.B., Schramm K., Schunkert H., Schurman C., Scott S.A., Seppala I., Shaffer C., Shah S., Shalaby A.A., Shim J., Shoemaker M.B., Siland J.E., Sinisalo J., Sinner M.F., Slowik A., Smith A.V., Smith B.H., Smith J.G., Smith J.D., Smith N.L., Soliman E.Z., Sotoodehnia N., Stricker B.H., Sun A., Sun H., Svendsen J.H., Tanaka T., Tanriverdi K., Taylor K.D., Teder-Laving M., Teumer A., Theriault S., Trompet S., Tucker N.R., Tveit A., Uitterlinden A.G., Van Der Harst P., Van Gelder I.C., Van Wagoner D.R., Verweij N., Vlachopoulou E., Volker U., Wang B., Weeke P.E., Weijs B., Weiss R., Weiss S., Wells Q.S., Wiggins K.L., Wong J.A., Woo D., Worrall B.B., Yang P.S., Yao J., Yoneda Z.T., Zeller T., Zeng L., Lubitz S.A., Lunetta K.L., Ellinor P.T. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet. 2018;50:1225–1233. doi: 10.1038/s41588-018-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Harst P., van Setten J., Verweij N., Vogler G., Franke L., Maurano M.T., Wang X., Mateo Leach I., Eijgelsheim M., Sotoodehnia N., Hayward C., Sorice R., Meirelles O., Lyytikainen L.P., Polasek O., Tanaka T., Arking D.E., Ulivi S., Trompet S., Muller-Nurasyid M., Smith A.V., Dorr M., Kerr K.F., Magnani J.W., Del Greco M.F., Zhang W., Nolte I.M., Silva C.T., Padmanabhan S., Tragante V., Esko T., Abecasis G.R., Adriaens M.E., Andersen K., Barnett P., Bis J.C., Bodmer R., Buckley B.M., Campbell H., Cannon M.V., Chakravarti A., Chen L.Y., Delitala A., Devereux R.B., Doevendans P.A., Dominiczak A.F., Ferrucci L., Ford I., Gieger C., Harris T.B., Haugen E., Heinig M., Hernandez D.G., Hillege H.L., Hirschhorn J.N., Hofman A., Hubner N., Hwang S.J., Iorio A., Kahonen M., Kellis M., Kolcic I., Kooner I.K., Kooner J.S., Kors J.A., Lakatta E.G., Lage K., Launer L.J., Levy D., Lundby A., Macfarlane P.W., May D., Meitinger T., Metspalu A., Nappo S., Naitza S., Neph S., Nord A.S., Nutile T., Okin P.M., Olsen J.V., Oostra B.A., Penninger J.M., Pennacchio L.A., Pers T.H., Perz S., Peters A., Pinto Y.M., Pfeufer A., Pilia M.G., Pramstaller P.P., Prins B.P., Raitakari O.T., Raychaudhuri S., Rice K.M., Rossin E.J., Rotter J.I., Schafer S., Schlessinger D., Schmidt C.O., Sehmi J., Sillje H.H.W., Sinagra G., Sinner M.F., Slowikowski K., Soliman E.Z., Spector T.D., Spiering W., Stamatoyannopoulos J.A., Stolk R.P., Strauch K., Tan S.T., Tarasov K.V., Trinh B., Uitterlinden A.G., van den Boogaard M., van Duijn C.M., van Gilst W.H., Viikari J.S., Visscher P.M., Vitart V., Volker U., Waldenberger M., Weichenberger C.X., Westra H.J., Wijmenga C., Wolffenbuttel B.H., Yang J., Bezzina C.R., Munroe P.B., Snieder H., Wright A.F., Rudan I., Boyer L.A., Asselbergs F.W., van Veldhuisen D.J., Stricker B.H., Psaty B.M., Ciullo M., Sanna S., Lehtimaki T., Wilson J.F., Bandinelli S., Alonso A., Gasparini P., Jukema J.W., Kaab S., Gudnason V., Felix S.B., Heckbert S.R., de Boer R.A., Newton-Cheh C., Hicks A.A., Chambers J.C., Jamshidi Y., Visel A., Christoffels V.M., Isaacs A., Samani N.J., de Bakker P.I.W. 52 Genetic loci influencing myocardial mass. J. Am. Coll. Cardiol. 2016;68:1435–1448. doi: 10.1016/j.jacc.2016.07.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H., Randall W.R., Du S.J. skNAC (skeletal Naca), a muscle-specific isoform of Naca (nascent polypeptide-associated complex alpha), is required for myofibril organization. Faseb J. 2009;23:1988–2000. doi: 10.1096/fj.08-125542. [DOI] [PubMed] [Google Scholar]
- 50.Park C.Y., Pierce S.A., von Drehle M., Ivey K.N., Morgan J.A., Blau H.M., Srivastava D. skNAC, a Smyd1-interacting transcription factor, is involved in cardiac development and skeletal muscle growth and regeneration. Proc. Natl. Acad. Sci. U S A. 2010;107:20750–20755. doi: 10.1073/pnas.1013493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tristani-Firouzi M., Jensen J.L., Donaldson M.R., Sansone V., Meola G., Hahn A., Bendahhou S., Kwiecinski H., Fidzianska A., Plaster N., Fu Y.H., Ptacek L.J., Tawil R. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome) J. Clin. Invest. 2002;110:381–388. doi: 10.1172/JCI15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tawil R., Ptacek L.J., Pavlakis S.G., DeVivo D.C., Penn A.S., Ozdemir C., Griggs R.C. Andersen's syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann. Neurol. 1994;35:326–330. doi: 10.1002/ana.410350313. [DOI] [PubMed] [Google Scholar]
- 53.Yoon G., Oberoi S., Tristani-Firouzi M., Etheridge S.P., Quitania L., Kramer J.H., Miller B.L., Fu Y.H., Ptácek L.J. Andersen-Tawil syndrome: prospective cohort analysis and expansion of the phenotype. Am. J. Med. Genet. A. 2006;140:312–321. doi: 10.1002/ajmg.a.31092. [DOI] [PubMed] [Google Scholar]
- 54.Tereshchenko L.G., Sotoodehnia N., Sitlani C.M., Ashar F.N., Kabir M., Biggs M.L., Morley M.P., Waks J.W., Soliman E.Z., Buxton A.E., Biering-Sorensen T., Solomon S.D., Post W.S., Cappola T.P., Siscovick D.S., Arking D.E. Genome-Wide Associations of Global Electrical Heterogeneity ECG Phenotype: The ARIC (Atherosclerosis Risk in Communities) Study and CHS (Cardiovascular Health Study) J. Am. Heart Assoc. 2018;7 doi: 10.1161/JAHA.117.008160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wischmeyer E., Doring F., Karschin A. Acute suppression of inwardly rectifying Kir2.1 channels by direct tyrosine kinase phosphorylation. J. Biol. Chem. 1998;273:34063–34068. doi: 10.1074/jbc.273.51.34063. [DOI] [PubMed] [Google Scholar]
- 56.Kutlu B., Burdick D., Baxter D., Rasschaert J., Flamez D., Eizirik D.L., Welsh N., Goodman N., Hood L. Detailed transcriptome atlas of the pancreatic beta cell. BMC Med. Genomics. 2009;2:3. doi: 10.1186/1755-8794-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Riz M., Braun M., Wu X., Pedersen M.G. Inwardly rectifying Kir2.1 currents in human beta-cells control electrical activity: characterisation and mathematical modelling. Biochem. Biophys. Res. Commun. 2015;459:284–287. doi: 10.1016/j.bbrc.2015.02.099. [DOI] [PubMed] [Google Scholar]
- 58.Yildirim V., Vadrevu S., Thompson B., Satin L.S., Bertram R. Upregulation of an inward rectifying K+ channel can rescue slow Ca2+ oscillations in K(ATP) channel deficient pancreatic islets. PLoS Comput. Biol. 2017;13:e1005686. doi: 10.1371/journal.pcbi.1005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kobayashi S., Nagao M., Asai A., Fukuda I., Oikawa S., Sugihara H. Severity and multiplicity of microvascular complications are associated with QT interval prolongation in patients with type 2 diabetes. J. Diabetes Investig. 2018;9:946–951. doi: 10.1111/jdi.12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Veglio M., Sivieri R., Chinaglia A., Scaglione L., Cavallo-Perin P. QT interval prolongation and mortality in type 1 diabetic patients: a 5-year cohort prospective study. Neuropathy Study Group of the Italian Society of the Study of Diabetes, Piemonte Affiliate. Diabetes Care. 2000;23:1381–1383. doi: 10.2337/diacare.23.9.1381. [DOI] [PubMed] [Google Scholar]
- 61.Suys B., Heuten S., De Wolf D., Verherstraeten M., de Beeck L.O., Matthys D., Vrints C., Rooman R. Glycemia and corrected QT interval prolongation in young type 1 diabetic patients: what is the relation? Diabetes Care. 2006;29:427–429. doi: 10.2337/diacare.29.02.06.dc05-1450. [DOI] [PubMed] [Google Scholar]
- 62.Torekov S.S., Iepsen E., Christiansen M., Linneberg A., Pedersen O., Holst J.J., Kanters J.K., Hansen T. KCNQ1 long QT syndrome patients have hyperinsulinemia and symptomatic hypoglycemia. Diabetes. 2014;63:1315–1325. doi: 10.2337/db13-1454. [DOI] [PubMed] [Google Scholar]
- 63.Swope D., Li J., Radice G.L. Beyond cell adhesion: the role of armadillo proteins in the heart. Cell Signal. 2013;25:93–100. doi: 10.1016/j.cellsig.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cabral R.M., Liu L., Hogan C., Dopping-Hepenstal P.J.C., Winik B.C., Asial R.A., Dobson R., Mein C.A., Baselaga P.A., Mellerio J.E., Nanda A., Boente M.D.C., Kelsell D.P., McGrath J.A., South A.P. Homozygous mutations in the 5' region of the JUP gene result in cutaneous disease but normal heart development in children. J. Investigative Dermatol. 2010;130:1543–1550. doi: 10.1038/jid.2010.7. [DOI] [PubMed] [Google Scholar]
- 65.Ou F., Rao N., Jiang X., Qian M., Feng W., Yin L., Chen X. Analysis on differential gene expression data for prediction of new biological features in permanent atrial fibrillation. PLoS ONE. 2013;8:e76166. doi: 10.1371/journal.pone.0076166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keil R., Schulz J., Hatzfeld M. p0071/PKP4, a multifunctional protein coordinating cell adhesion with cytoskeletal organization. Biol. Chem. 2013;394:1005–1017. doi: 10.1515/hsz-2013-0114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS raw data and Proteome Discoverer protein identification have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (26) with the data set identifier PXD011004.