

Summary
Acquired severe aplastic anaemia (SAA) has an immune pathogenesis, and immunosuppressive therapy (IST) with anti-thymocyte globulin and cyclosporine is effective therapy. Eltrombopag (EPAG) added to standard IST was associated with higher overall and complete response rates in patients with treatment-naïve SAA compared to a historical IST cohort. We performed a paediatric subgroup analysis of this trial including all patients aged <18 years who received EPAG plus standard IST (n = 40 patients) compared to a historical cohort (n = 87) who received IST alone. Response, relapse, clonal evolution, event-free survival (EFS), and overall survival were assessed. There was no significant difference in either the overall response rate (ORR) or complete response rate at 6 months (ORR 70% in EPAG group, 72% in historical group, P = 0·78). Adults (≥18 years) had a significantly improved ORR of 82% with EPAG compared to 58% historically (P < 0·001). Younger children had lower response rates than did adolescents. The trend towards relapse was higher and EFS significantly lower in children who received EPAG compared to IST alone. Addition of EPAG added to standard IST did not improve outcomes in children with treatment-naïve SAA. EPAG in the paediatric population should not automatically be considered standard of care. Registration: clinicaltrials.gov (NCT01623167).
Keywords: aplastic anaemia, marrow failure, paediatric aplastic anaemia, paediatric haematology
Introduction
Severe aplastic anaemia (SAA) is characterised by marked pancytopenia with serious clinical sequelae, and disproportionately affects children and young adults. Its aetiology is broadly characterised as either ‘constitutional’ or ‘acquired’. Acquired or immune AA is the most common and due to cytotoxic T-cell destruction of haematopoietic cells.1
Treatment for immune AA is either haematopoietic stem cell transplant (HSCT)2 or immunosuppressive therapy (IST). In children and adults aged <40 years without a fully matched human leucocyte antigen (HLA) sibling donor, IST remains the standard-of-care. Haematological responses to IST in children are favourable,3–5 with an overall response rate (ORR) of 70% and complete response (CR) rate of 23–60%. Overall survival (OS) rates are also high at 80–93%; however, long-term complications such as relapse and clonal evolution result in a long-term event-free survival (EFS) of only 56–62%.
Eltrombopag (EPAG) is an oral thrombopoietin-receptor agonist that is effective in both refractory and treatment-naïve SAA.6–8 Currently, EPAG in combination with IST is approved by the United States Food and Drug Administration (FDA) for children aged ≥2 years as front-line treatment for SAA. Approval followed a National Institutes of Health (NIH) investigator-initiated study showing a significant improvement in both ORR and CR rate compared to a historical IST cohort. Complete response to IST has been previously associated with improved long-term outcomes.9 A total of 19 children included in this initial publication were not reported as a subgroup and a further 21 children have subsequently been enrolled.
Patients and methods
A subgroup analysis was performed of children (aged <18 years) in our ongoing prospective non-randomised Phase I–II study of EPAG added to standard IST for treatment-naïve SAA (clinicaltrials.gov number: NCT01623167).7 A historical paediatric treatment group using standard IST was used for comparison, as were adult patients who received EPAG and IST. Patients in both the EPAG and historical cohorts met clinical criteria for SAA (Table S3). All patients were assessed for: ORR at 3 and 6 months, CR rate, relapse, clonal evolution, EFS, and OS.
This study was approved by the Institutional Review Board of the National Heart, Lung, and Blood Institute (NHLBI). All patients or guardians provided written informed consent.
Paediatric EPAG group
All included patients were treated on protocol before November 2019, aged <18 years when enrolled, and had either reached primary end-point evaluation (6 months) or met off-study criteria prior. Off-study criteria were: protocol completion (5 years), initiation of IST other than cyclosporine (CSA) or steroids (including HSCT), intolerance to EPAG or horse anti-thymocyte globulin (hATG), high-risk clonal evolution (chromosome 7 abnormality, complex cytogenetics or overt myeloid malignancy), pregnancy, and subject choice. Primary end-points were safety and CR rate at 6 months. Patients were evaluated for baseline visits and mandated time-points: 3 and 6 months, 1 year, and yearly thereafter until 5 years. Standard clinical follow-up occurred once off-study. Bone marrow examination with cytogenetics was performed at baseline, all time-points, and as clinically indicated to exclude clonal evolution. All paediatric patients had a diepoxybutane clastogenic stress assay and were screened for germline variants with an inherited bone marrow failure panel (Table S1). Germline variants were further classified for pathogenicity using the Sherloc criteria, which incorporate the American College of Medical Genetics and Genomics (ACMG) criteria.10 Patients were also screened for somatic variants in myeloid cancer genes using a targeted panel at baseline, 6 months, and 2 years. Plasma samples for pharmacokinetic (PK) studies were collected over a 24-h period after a minimum of 7 days of consecutive EPAG for measurement of concentrations at steady-state using a validated liquid chromatography with tandem mass spectrometry method and PK parameters derived by non-compartmental analysis (Phoenix version 6; Certara USA, Inc., Princeton, NJ, USA).
Horse ATG and CSA were administered at standard dosing; hATG at a dose of 40 mg/kg over 4 consecutive days and full-dose CSA at 3 mg/kg twice daily (trough level of 200–400 ng/ml) for 6 months. Due to high relapse rates, the protocol was amended so that responders would continue CSA at a reduced dose (2 mg/kg daily) from 6 months to 2 years; the majority of the patients in the paediatric EPAG group were enrolled after this amendment (n = 33, 83%) to receive CSA for 2 years. EPAG was dosed at 150 mg daily for patients aged ≥12 years, 75 mg for those aged 6–11 years and 2·5 mg/kg for those aged 2–5 years, with standard dosing adjustments made for patients of East Asian and Southeast Asian ancestry based on PK data in immune thrombocytopenic purpura.11 The duration of EPAG administration was either 3 or 6 months, depending on assigned cohorts enrolled consecutively (Figure S1). Responders who relapsed were restarted on CSA and/or EPAG with a duration based on clinical response (Fig 1).9
Fig 1.
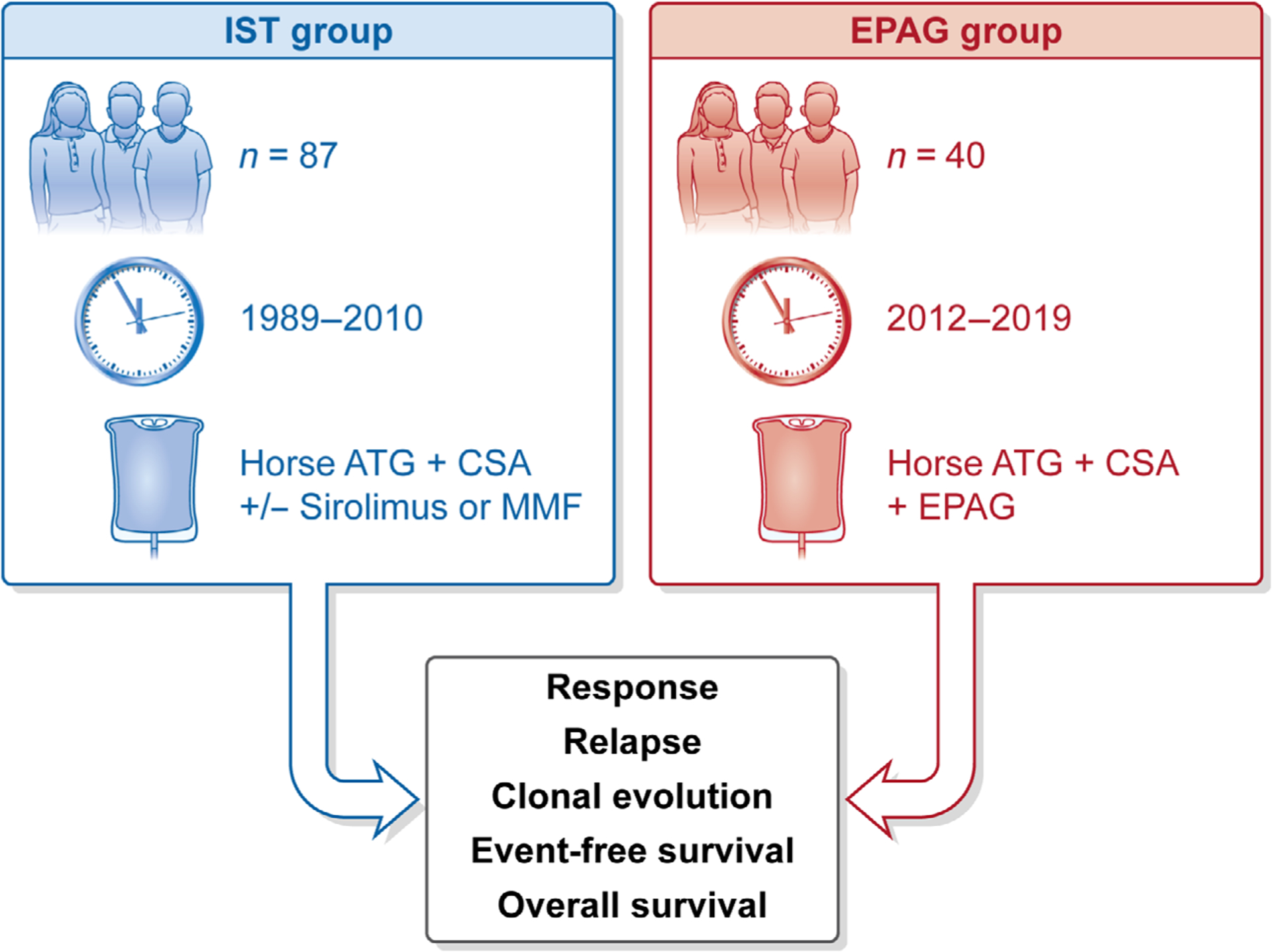
Study design. The study compared two groups: the paediatric EPAG group and the historical paediatric IST group. The paediatric IST group consisted of 87 paediatric patients (aged <18 years) on four different clinical trials (NCT00061360, NCT00001964, NCT00260689 and (no NCT number available: Rosenfeldet al., 20039) taking place at the NIH from 1989 to 2010. The paediatric EPAG group consisted of 40 patients (aged <18 years) enrolled on NCT01623167 from 2012 to 2019; data of 19 patients were previously published.7Analysis was performed using intention to treat. Measured outcomes included: haematological response, relapse, clonal evolution, event-free survival, and overall survival. ATG, anti-thymocyte globulin; CSA, cyclosporine; EPAG, eltrombopag; IST, immunosuppressive therapy; NCT, national clinical trial; NIH, National Institutes of Health; MMF, mycophenolate mofetil.
Paediatric IST group
A historical cohort of 87 children treated at the NIH Clinical Center from 1989 to 2010 was used as a comparison arm (Figure S2). All those aged <18 years with treatment-naïve SAA treated with hATG-based IST were included (Table S3). Patients treated with rabbit ATG,12 alemtuzumab13 or cyclophosphamide14 as front-line treatment were excluded due to study results demonstrating drug inferiority or toxicity. Data from four clinical trials were included: NCT00061360, NCT00001964, NCT00260689 and hATG/CSA (no NCT number).9,14–16 All received hATG at 40 mg/kg over 4 consecutive days and CSA (target 200–400 ng/ml). CSA duration varied slightly among protocols (Table S4), but most patients were enrolled on studies that discontinued CSA at 6 months (n = 63, 72%) and the remainder (n = 24, 28%) on studies with a further CSA taper over 18 months. In two studies, a third investigational immunosuppressive drug was added to standard IST: mycophenolate mofetil (MMF)16 for 18 months (n = 21) or sirolimus15 for 6 months (n = 10). Responses by specific clinical trial included in the historical group are shown in Table S5. Follow-up occurred at 3 months, 6 months, and yearly thereafter until study completion after which they were seen as clinically indicated (Fig 1).
Adult EPAG and adult IST groups
The adult EPAG comparator group comprised adults (aged ≥18 years) at the time of enrolment (n = 131) who received EPAG with IST on protocol (NCT01623167). The adult IST comparator arm comprised adults (aged ≥18 years) at the time of enrolment (n = 286) on the same four protocols as the paediatric IST group. All patients who had either reached primary end-point at 6 months or met off-study criteria prior were included. No age or sex matching was performed.
Response criteria
Response criteria were consistent across all protocols. Overall response was defined as blood counts no longer meeting Camitta criteria for SAA17 with two out of three present: absolute reticulocyte count (ARC) ≥60 × 109/l, platelet count ≥20 × 109/l, or absolute neutrophil count (ANC) ≥0·5 × 109/l. Overall response was further categorised as CR or partial response (PR). Complete responders met the following criteria: haemoglobin (Hb) ≥100 g/l, platelet count ≥100 × 109/l, and ANC ≥1 × 109/l. Additionally, patients had to be transfusion and growth factor independent. Non-responders (NR) failed to achieve a response by 6 months.
Statistics
Analysis was performed on an intention-to-treat (ITT) basis. Summary statistics were used to describe baseline characteristics, response, and laboratory parameters. P values were computed using Student’s t-test for continuous variables and Pearson’s chi-squared test for categorical variables. Kaplan–Meier curves and the Cox proportional-hazards models were used to compare OS, EFS, relapse, and clonal evolution among the paediatric subgroups, and the likelihood ratio test was performed to assess statistical significance between curves. Analysis was performed using competing risk methods for clonal evolution, HSCT and death (Figure S6). As this was an ITT analysis, all patients who were taken off study prior to 6 months were deemed NR. Relapse was defined as a decrease in blood counts requiring AA-specific treatment including the re-initiation of full dose CSA or EPAG, additional IST or HSCT. For EFS, only patients who were responders at 6 months and experienced no events between the start of treatment and the 6-month end-point were included. We defined an event as relapse, clonal evolution, HSCT, or death. The data were analysed using R version 3.5.0 (R Foundation for Statistical Computing, Vienna, Austria) and the Statistical Analysis System (SAS) version 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
Patient characteristics
There were 40 patients in the paediatric EPAG group and 87 in the historical paediatric IST group. There were no significant differences in age or sex, or in baseline ARC, ANC or platelets (Table I). Baseline haemoglobin was significantly lower in the IST group. The median follow-up was 1432·5 days in the EPAG group and 2409 days in the IST group. Adolescents (aged ≥12 years) composed 60% (n = 24) of the EPAG group and 48% (n = 42) of the IST group, with younger children (aged <12 years) at 40% (n = 16) and 52% (n = 45) respectively.
Table I.
Patients’ baseline characteristics.
| Baseline characteristic | IST (n = 87) | EPAG (n = 40) | P |
|---|---|---|---|
| Age at first IST, years, median (range) | 11 (2–17) | 13 (3–17) | 0·42 |
| Sex, n (%) | |||
| Female | 36 (41) | 17 (43) | 0·91 |
| Male | 51 (59) | 23 (58) | |
| Age distribution, n (%) | |||
| <12 years | 45 (52) | 16 (40) | 0·22 |
| ≥12 years | 42 (48) | 24 (60) | |
| Laboratory values, median | |||
| Haemoglobin, g/1 | |||
| All <18 years | 73 | 84 | <0·01 |
| <12 years | 72 | 81 | |
| Absolute reticulocyte count, × 109/l | |||
| All <18 years | 12·1 | 12·9 | 0·63 |
| <12 years | 12·2 | 12·7 | |
| Absolute neutrophil count, × 109/l | |||
| All <18 years | 0·22 | 0·16 | 0·36 |
| <12 years | 0·21 | 0·13 | |
| Platelet count, × 109/l | |||
| All <18 years | 9 | 9 | 0·45 |
| <12 years | 10 | 8 | |
| PNH clone >1% GPI deficient neutrophils, n (%) | |||
| Yes | 14 (29) | 11 (31) | 0·82 |
| No | 34 (71) | 24 (69) | |
| Unknown | n = 39 | n = 5 |
Baseline data for the paediatric IST group are shown on the left and paediatric EPAG group on the right. EPAG, eltrombopag; IST, immunosuppressive therapy; PNH, paroxysmal nocturnal haemoglobinuria; GPI, glycosylphosphatidylinositol.
Haematological response
In the EPAG group, the ORR was 68% at 3 months and 70% at 6 months compared to 63% at 3 months and 72% at 6 months in the IST group (6-month ORR P = 0·78). Complete responders totalled 23% at 3 months and 30% at 6 months in the EPAG group compared to the IST group at 14% and 23% respectively, again without statistical significance (6-month ORR P = 0·42; Table II). In contrast, in adults (aged ≥18 years) there was a significant difference in ORR at 6 months: 58% with IST alone compared with 82% with EPAG (P ≤ 0·001; Table S9). Response rates at 1 year were not significantly different between the EPAG and IST groups; however, patients who failed to achieve a response at 6 months were taken off protocol and not routinely followed, so late responses to treatment may not be captured.
Table II.
Haematological response in paediatric IST group versus EPAG group.
| IST group (n = 87) | EPAG group (n = 40) | P | |
|---|---|---|---|
| 3-month response | |||
| Response, n (%) | |||
| Overall response | 55 (63) | 27 (68) | 0·64 |
| CR | 12 (14) | 9 (23) | 0·26 |
| PR | 43 (49) | 18 (45) | |
| NR | 29 (33) | 9 (23) | |
| Off study | 3 (3) | 4 (10) | |
| 6-month response | |||
| Response, n (%) | |||
| Overall response | 63 (72) | 28 (70) | 0·78 |
| CR | 20 (23) | 12 (30) | 0·42 |
| PR | 43 (49) | 16 (40) | |
| NR | 19 (22) | 4 (10) | |
| Off study | 5 (6) | 8 (20) | |
| 1-year response | |||
| Response, n (%) | |||
| Overall response | 52 (60) | 20 (50) | 0·38 |
| CR | 31 (36) | 16 (40) | 0·57 |
| PR | 21 (24) | 4 (10) | |
| NR | 0 (0) | 0 (0) | |
| Relapsed | 5 (6) | 7 (18) | |
| Off study | 30 (34) | 12 (30) | |
| Not reached | 0 (0) | 1 (3 |
Rates of haematological response are shown for the paediatric IST group on the left and paediatric EPAG group on the right. Overall response is further broken down into CR or PR. Patients who were off study at 1 year were for many different reasons including: NR at 6 months, alternate treatment such as repeat IST or HSCT, death, or were lost to follow-up. Patients who were deemed non-responders at 6 months were not routinely monitored for late response beyond 6 months as they were taken off study. CR, complete response; EPAG, eltrombopag; IST, immunosuppressive therapy; NR, no response; PR, partial response.
Younger children had lower response rates than adolescents with the addition of EPAG, having an ORR of 63% compared with 78% for IST (P = 0·29) and a CR rate of just 6% compared to 24% with IST (P = 0·049), but patient numbers were low in this group. In contrast, adolescents achieved a CR rate of 46% with EPAG compared to 21% with IST (P = 0·052), and overall responses of 75% versus 67% (P = 0·48) respectively (Table III).
Table III.
Haematological response at 6 months in younger children versus adolescents.
| IST group | EPAG group | P | |
|---|---|---|---|
| Younger children aged <12 years | |||
| Response, n (%) | n = 45 | n = 16 | |
| Overall response | 35 (78) | 10 (63) | 0·29 |
| CR | 11 (24) | 1 (6) | 0·049* |
| PR | 24 (53) | 9 (56) | |
| NR | 9 (20) | 2 (13) | |
| Off study | 1 (2) | 4 (25) | |
| Adolescents aged >12 years | |||
| Response, n (%) | n = 42 | n = 24 | |
| Overall response | 28 (67) | 18 (75) | 0·48 |
| CR | 9 (21) | 11 (46) | 0·052 |
| PR | 19 (45) | 7 (29) | |
| NR | 10 (24) | 2 (8) | |
| Off study | 4 (10) | 4 (17) |
Rates of haematological response are shown for the paediatric IST group on the left and paediatric EPAG group on the right. Younger children (aged <12 years) are shown on the top and adolescents on the bottom. Overall response is further broken down into CR or PR. CR, complete response; EPAG, eltrombopag; IST, immunosuppressive therapy; NR, no response; PR, partial response.
Note, significance uncertain given low patient numbers.
A higher proportion of patients went off study at or before 6 months in the EPAG group than in the IST group (6%, n = 5 in IST vs. 20%, n = 8 in EPAG) (Table S7). In the EPAG group, all patients pursued HSCT; two had clonal evolution (monosomy 7), four pursued HSCT due to urgent clinical necessity related to severe neutropenia and two underwent elective HSCT due to lack of response at 3 months. In the IST group, four died prior to 6 months and one pursued urgent HSCT.
Relapse and EFS
Of a total of 28 responders in the paediatric EPAG group, 43% relapsed (n = 12) compared to 27% (17 of 63 responders) in the paediatric IST group (P = 0·0661, Fig 2A). The median time to relapse was 349 days in the EPAG group and 585 days in the IST group. Relapses in the EPAG group peaked after 6 months and again at 2 years. Relapse in the EPAG group was initially treated with the re-initiation of full-dose CSA and/or EPAG; five of 11 (46%) patients recovered bloods counts and did not ultimately require further IST or HSCT, and one was lost to follow-up. Five children deemed 6-month responders received a 3-month course of sirolimus starting at the 2-year time-point on a separate clinical protocol (NCT02979873) investigating the use of sirolimus for relapse prevention in patients with stable blood counts and on low-dose CSA; two remain in CR, one PR, and two had relapsed prior to enrolment. Children who received EPAG had a similar rate of relapse (43%) as adults who received EPAG (41%) (likelihood ratio P = 0·56; Fig 2B). The EFS at 1432·5 days (the median follow-up time in the EPAG group), in 6-month responders, was significantly lower in the EPAG group (57%) compared with the IST group (69%, likelihood ratio P = 0·0499; Fig 2C). All events prior to 6 months were excluded in the main EFS analysis; EFS for all patients (including non-responders) from time 0 can be seen in Figure S3.
Fig 2.
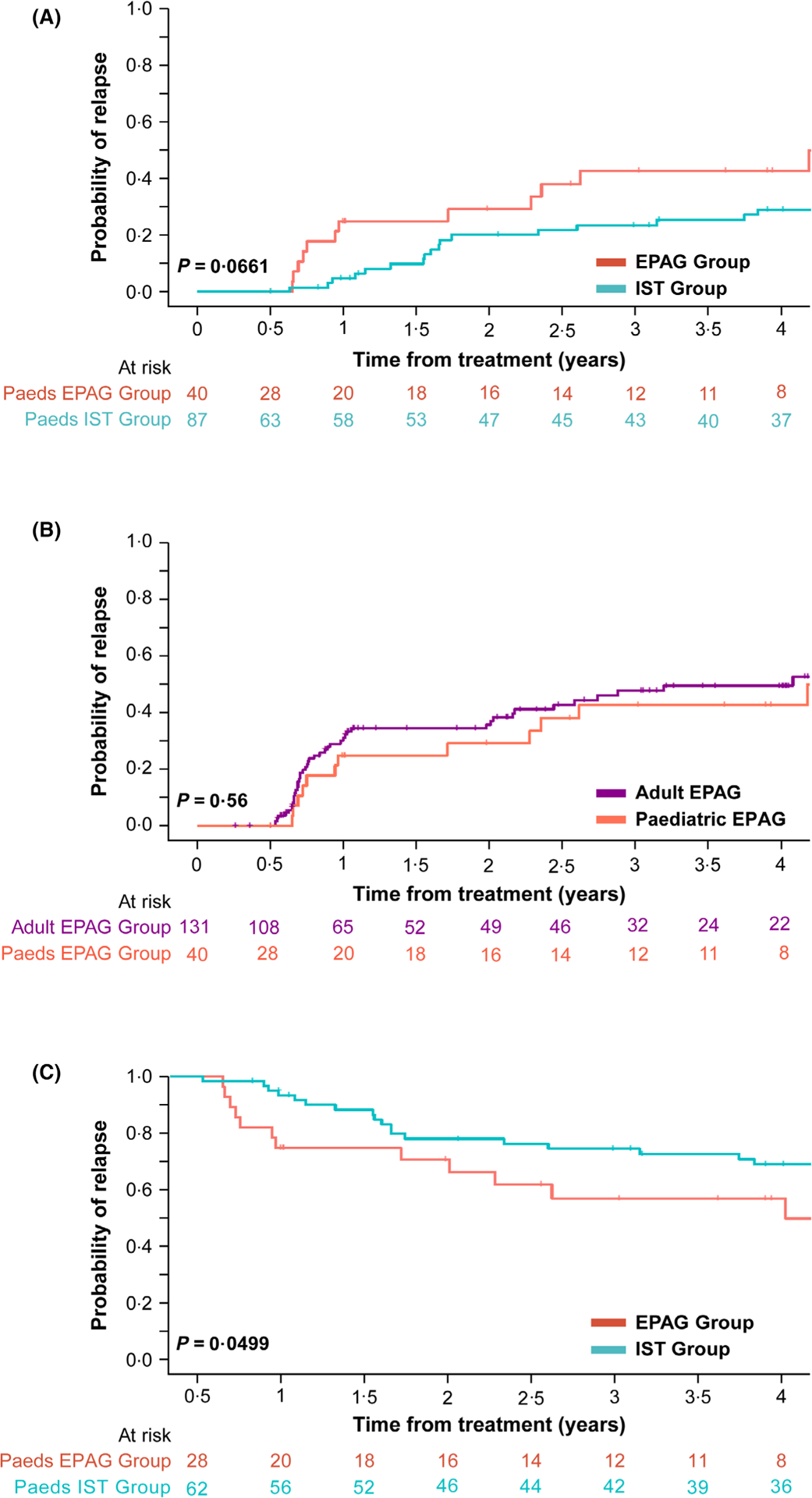
Relapse and event-free survival (A) Kaplan–Meier curve comparing relapse in all paediatric responders; the paediatric ISTversuspaediatric EPAG group. The median time to relapse was 349 days in the EPAG group and 585 days in the IST group. Relapse was 43% in the EPAG group and 27% in the IST group. This difference was not significant (likelihood ratioP = 0·0661). (B) Kaplan–Meier curve comparing relapse between children and adults who received EPAG. The median time to relapse was 349 days in children and 277 in adults. There was no significant difference in relapse seen with 41% of adults relapsing compared with 43% of children (likelihood ratioP = 0·56). (C) Kaplan–Meier curve comparing EFS in paediatric responders in the ISTversusEPAG group. At 1432·5 days (median follow-up time in the EPAG group) EFS was significantly lower in the EPAG group (57%) compared with the IST group (69%) (likelihood ratioP = 0·0499). The median follow-up time was 2409 days (6·6 years) in the paediatric IST group and 1432·5 days (3·9 years) in the paediatric EPAG group. EFS, event-free survival; EPAG, eltrombopag; IST, immunosuppressive therapy.
Clonal evolution
Clonal evolution was not significantly different (13% in the paediatric EPAG group vs. 9% with IST, likelihood ratio P = 0·215). High-risk clonal evolution was defined as a chromosome 7 abnormality, complex karyotype, or overt myeloid malignancy, with all other isolated chromosomal abnormalities considered low risk. Five children (13%) evolved in the EPAG group; three were high risk (all monosomy 7) and two low risk (one del 5q and one translocation 5;12). Two patients who developed monosomy 7 did so rapidly at 3 months. One high-risk patient was off protocol at the time of evolution; a non-responder who received further IST (alemtuzumab) and was on long-term CSA and EPAG when found to have acute myeloid leukaemia (AML) with monosomy 7. All those with high-risk evolution ultimately proceeded to transplant. In the IST group, 9% of children (eight of 87) had clonal evolution with four at high risk and four at low risk (Table IV).
Table IV.
Clonal evolution in paediatric patients.
| Patient | Time to clonal evolution, days | Clonal evolution | Karyotype | Risk category | SAMD9/SAMD9L |
|---|---|---|---|---|---|
| Paediatric EPAG group | |||||
| EPAG2 | 1470 | Del 5q | 46,XX,del(5)(q15q31)[2]/46,XX[19] | Low | Not assessed |
| EPAG9 | 733 | Translocation (5;12) | 46,XY,t(5;12)(p10;p10)[2]/46,XY[18] | Low | Not assessed |
| EPAG15 | 88 | Monosomy 7 | 45,XY,−7[6]/46,XY[14] | High | Not assessed |
| EPAG28 | 1049 | AML with Monosomy 7 | 45,XX,−7[7]/46,XX[13] | High | No |
| EPAG40 | 97 | Monosomy 7 | 45,XY,−7[12]/46,XY[8] | High | No |
| Paediatric IST group | |||||
| IST46 | 2528 | AML | 46,XY[20] | High | |
| IST57 | 990 | 13q del | 46,XX,del(13)(q12q14)[5] | Low | |
| IST20 | 185 | Trisomy 8 | 47,XX,+8[2]/46,XX[19] | Low | |
| IST29 | 2715 | MDS-MLD | Unavailable | High | |
| IST78 | 193 | 13q del | 46,XY,del(13)(q12q14)[3]/46,XY[1] | Low | |
| IST92 | 955 | Translocation (6;14) | 46,XY,t(6;14)(q24;q24)[3] 46,XY[17] | Low | |
| IST97 | 359 | Monosomy 7 | 45,XY,−7[4/20] | High | |
| 45,XY,−7,inv(12)(q15q24.1)[1/20] | |||||
| 46,XY,inv (12)(q15q24.1)[2/20] | |||||
| 46,XY[13/20] | |||||
| IST135 | 1853 | Monosomy 7 | Unavailable | High | |
All patients in the paediatric IST and paediatric EPAG group who underwent clonal evolution are listed. Included is the type of clonal evolution, the karyotyping results, and whether it was deemed high or low risk. High risk was defined as a chromosome 7 abnormality, complex karyotype, or morphologically overt myelodysplastic syndrome or other myeloid malignancy with all other types of clonal evolution being deemed low risk. Assessment for variants in SAMD9/SAMD9L was performed only in EPAG 28 and 40. Germline mutations were not assessed in the IST group. AML, acute myeloid leukaemia; EPAG, eltrombopag; IST, immunosuppressive therapy; MDS-MLD, myelodysplastic syndrome with multilineage dysplasia; SAMD9/L, sterile alpha motif domain containing 9 like.
Genomics
No patients had a pathogenic or likely pathogenic germline variant in genes related to inherited bone marrow failure. Although some patients were found with variants of unknown significance in genes associated with telomere diseases (EPAG9, 15, 20, and 27) or in sterile alpha motif domain containing 9/-like (SAMD9/L) (EPAG14 and 11), most evidence marks these variants as not contributing to disease; normal telomere length, lack of clinical manifestations or relevant family history, in silico predictions, and lack of bone marrow dysplasia or development of monosomy 7 in SAMD9/L patients (Table S2). Although unlikely, we cannot definitively exclude a modulating effect in these patients’ disease.
All children who received EPAG were screened for somatic variants in myeloid cancer genes at different time-points. Clonal haematopoiesis is prevalent in SAA; variants in phosphatidylinositol glycan anchor biosynthesis class A (PIGA), BCL-6 co-repressor (BCOR) and BCL-6 co-repressor-like protein 1 (BCORL1) are associated with an improved response to IST, and unfavourable mutations as a group including in DNA methyltransferase 3α (DNMT3A) and additional sex combs like-1 (ASXL1) genes have been correlated with a poorer prognosis.18
At baseline, only one patient had a somatic variant, in ASXL1 [variant allele fraction (VAF) of 6·2%]. Five patients developed somatic mutations having had none at baseline; four at the 6-month time-point [two with alpha thalassaemia/mental retardation syndrome X-linked (ATRX) variants at VAF 3·5% and 13%, and two with ASXL1 variants at VAF 9% and 24%] as well as one with a Runt-related transcription factor 1 (RUNX1) variant (VAF 7%) at 21 months (Figure S4). None had morphological dysplasia or abnormal cytogenetics at the time of detection. Of patients with ASXL1 variants, all were deemed 6-month NR; one (with baseline mutation) underwent HSCT for non-response, one responded to EPAG monotherapy restarted off protocol, and one developed clonal evolution off protocol (AML with monosomy 7). Two patients with ATRX variants remained in a CR and PR when last seen, and the patient with a RUNX1 variant was lost to follow-up.
Survival
The OS at 1432·5 days was not significantly different between the paediatric EPAG group and paediatric IST group at 94% and 84% respectively (likelihood ratio P = 0·092; Figure S6).
Pharmacokinetics
Pharmacokinetic parameters of EPAG at steady state were available in 11 paediatric patients with SAA (Table S4). Peak concentrations (Cmax) in plasma were achieved between 4 and 6 h (Tmax) after the morning dose and the overall variability of parameters was moderate, consistent with previous clinical studies. The Cmax, area under the plasma concentration–time curve from, time zero to time of last measurable concentration (AUClast), and trough plasma concentration (Ctrough) normalised to the starting dose of 75 mg in patients aged 6–11 years were 39·5 μg/ml, 640 μg × h/ml, and 19·2 μg/ml respectively; these were similar (Cmax and AUC) or slightly lower (Ctrough, −25%) than in older (aged ≥12 years) patients receiving 150 mg as the initial dose. PK data in adolescents and adults were similar, the Cmax, AUClast and Ctrough were 41·6 μg/ml, 767 μg × h/ml and 25·6 μg/ml respectively in the group aged 12–17 years, and 40·3 μg/ml, 785 μg × h/ml and 27·1 μg/ml in the group aged ≥18 years.
Safety
Safety data have been published.7 There were three severe adverse events (SAEs) in the paediatric EPAG group that required cessation of study drug; two cutaneous eruptions (Grade 2 and 3), and one Grade 3 hyperbilirubinaemia. In all, 10 patients had reversible liver function abnormalities possibly attributable to EPAG that was reported as an AE (Grades 2–3 rise in bilirubin, aspartate aminotransferase and alanine transaminase).
No deaths occurred in the EPAG group while enrolled on protocol. Two patients later died of infection after HSCT.
Discussion
Optimal first-line treatment for children with SAA who lack a matched-sibling donor is not settled. For those treated with IST, responses are good but relapse and clonal evolution remain concerns, particularly in children who have the most benefit to gain over time. Improvements in transplant-related morbidity and mortality have made matched unrelated donor (MUD)19,20 and alternative donor transplant21–23 attractive options. In adults, the addition of EPAG has offered significant improvements in response rates to >80%, but this result was not replicated in children.
The explanation for our present results showing lower efficacy of EPAG in children is unclear. Children historically have had better response rates to IST than adults and they tolerate more intensive IST regimens.3,24 The difference in response between younger children and adolescents may hint at an underlying biological difference. PKs do not appear to be responsible, as comparable EPAG exposures were achieved across age groups, although the number tested was low in the younger age group (more data will be acquired in an ongoing clinical trial, NCT03025698). There was no clear relationship between exposure and haematological response at the tested dose in each group. Inherited marrow failure disorders were excluded as much as was feasible in the EPAG arm with no definite pathogenic variants found, as unknown germline dispositions25 would affect response to either regimen.
Relapse was higher and EFS significantly lower in the EPAG group. A peak in relapse at 6 months and 2 years, when drug modifications occur (EPAG discontinued and CSA dose stopped or reduced), was obvious. Haematological responses may in some patients be dependent on either marrow stimulation or ongoing IST. One series of 11 children showed longer-term administration of EPAG may be efficacious.26
There was a trend towards longer survival in the paediatric EPAG group, but this is likely related to disparate time-periods (1989–2010 for the IST group, 2012–2019 for the EPAG group) as survival in SAA has increased over time due to supportive care.27 Transplant has become more widely utilised due to decreased transplant-related morbidity and mortality, and wider availability of alternative donors; more patients went off study in the EPAG group to pursue HSCT. Additionally, a paediatric subgroup analysis was not part of the initial trial design.
The safety profile of EPAG is good; no patients died or had serious morbidity as a result of EPAG. We noted that clonal evolution was more frequent in children who received EPAG, although the difference from the IST group was not statistically significant. EPAG is expensive and significantly increases the cost of IST for SAA,28,29 a burden both to patients and to healthcare systems.
The data presented in this non-randomised subgroup analysis do not support the addition of EPAG to IST for treatment-naïve SAA in children, although this regimen has been FDA approved in patients aged ≥2 years. Results from randomised trials in progress and accumulated experience at paediatric haematology centres will be important in definitively addressing the role of thrombopoietin mimetics in children.
Supplementary Material
Table S1. Inherited bone marrow failure panel from University of Chicago.
Table S2. Germline variants identified in our cohort in genes associated with inherited bone marrow failure by targeted sequencing panel.
Table S3. Haematological criteria for trial inclusion in both EPAG and IST groups.
Table S4. Administration of cyclosporine on different trial protocols.
Table S5. Haematological response at 6 months in patients who received MMF and sirolimus in the paediatric IST group.
Table S6. PNH clones in patients in paediatric EPAG and IST groups.
Table S7. Off study patients (prior to 6 months).
Table S8. Causes of death for all patients.
Table S9. Haematological response at 6 months: adults versus children.
Table S10. Summary of PK parameters at steady state normalised to the planned starting dose for eltrombopag by age category.
Fig S1. Study design for eltrombopag trial.
Fig S2. Components of the historical paediatric IST group.
Fig S3. EFS of all patients (including non-responders).
Fig S4. Somatic variants in patients from the EPAG cohort at baseline and over the course of treatment.
Fig S5. Clonal evolution in paediatric patients.
Fig S6. Overall survival in paediatric patients.
Fig S7. Competing risk analysis for clonal evolution, HSCT and death.
Acknowledgements
The authors would like to thank the patients and families who participated in these studies. Support for this trial was provided by the Intramural Research Program of the NHLBI as well as GlaxoSmithKline and Novartis, the manufacturers of EPAG, who also supplied the study drug.
Footnotes
Conflict of interest
Neal S. Young has a cooperative research and development agreement (CRADA) with Novartis that provides research funding. Subsequent to her involvement with this research, Danielle M. Townsley became an employee of AstraZeneca but had no competing financial interests while at NIH. Daniela Baldoni and Annie St. Pierre are employees of Novartis. Novartis provided PK data for this manuscript and reviewed the manuscript prior to submission. All other authors have no conflicts of interest to declare.
Data sharing
De-identified patient data will be shared indefinitely with other researchers upon reasonable request to the corresponding author (emma.groarke@nih.gov) or last author (youngns@nhlbi.nih.gov). This will require a proposal to the study investigators and a data transfer agreement. The research protocol and patient consent will be made freely available to other researchers upon request to the corresponding author.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- 1.Young NS. Aplastic anemia. N Engl J Med. 2018;379:1643–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bejanyan N, Kim S, Hebert KM, Kekre N, Abdel-Azim H, Ahmed I, et al. Choice of conditioning regimens for bone marrow transplantation in severe aplastic anemia. Blood Adv. 2019;3:3123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheinberg P, Wu CO, Nunez O, Young NS. Long-term outcome of pediatric patients with severe aplastic anemia treated with antithymocyte globulin and cyclosporine. J Pediatr. 2008;153:814–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogers ZR, Nakano TA, Olson TS, Bertuch AA, Wang W, Gillio A, et al. Immunosuppressive therapy for pediatric aplastic anemia: a North American Pediatric Aplastic Anemia Consortium study. Haematologica. 2019;104:1974–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida N, Kobayashi R, Yabe H, Kosaka Y, Yagasaki H, Watanabe KI, et al. First-line treatment for severe aplastic anemia in children: bone marrow transplantation from a matched family donor versus immunosuppressive therapy. Haematologica. 2014;99:1784–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desmond R, Townsley DM, Dumitriu B, Olnes MJ, Scheinberg P, Bevans M, et al. Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. Blood. 2014;123:1818–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Townsley DM, Scheinberg P, Winkler T, Desmond R, Dumitriu B, Rios O, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376:1540–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winkler T, Fan X, Cooper J, Desmond R, Young DJ, Townsley DM, et al. Treatment optimization and genomic outcomes in refractory severe aplastic anemia treated with eltrombopag. Blood. 2019;133:2575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289:1130–5. [DOI] [PubMed] [Google Scholar]
- 10.Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, et al. Sherloc: a comprehensive refinement of the ACMG–AMP variant classification criteria. Genet Med. 2017;19:1105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wire MB, Li X, Zhang J, Sallas W, Aslanis V, Ouatas T. Modeling and simulation support eltrombopag dosing in pediatric patients with immune thrombocytopenia. Clin Pharmacol Ther. 2018;104:1199–207. [DOI] [PubMed] [Google Scholar]
- 12.Scheinberg P, Nunez O, Weinstein B, Scheinberg P, Biancotto A, Wu CO, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365:430–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheinberg P, Nunez O, Weinstein B, Scheinberg P, Wu CO, Young NS. Activity of alemtuzumab monotherapy in treatment-naive, relapsed, and refractory severe acquired aplastic anemia. Blood. 2012;119:345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheinberg P, Townsley D, Dumitriu B, Scheinberg P, Weinstein B, Daphtary M, et al. Moderate-dose cyclophosphamide for severe aplastic anemia has significant toxicity and does not prevent relapse and clonal evolution. Blood. 2014;124:2820–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheinberg P, Wu CO, Nunez O, Scheinberg P, Boss C, Sloand EM, et al. Treatment of severe aplastic anemia with a combination of horse antithymocyte globulin and cyclosporine, with or without sirolimus: a prospective randomized study. Haematologica. 2009;94:348–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheinberg P, Nunez O, Wu C, Young NS. Treatment of severe aplastic anaemia with combined immunosuppression: anti-thymocyte globulin, ciclosporin and mycophenolate mofetil. Br J Haematol. 2006;133:606–11. [DOI] [PubMed] [Google Scholar]
- 17.Camitta B, Rozman C, Marin P, Nomdedeu B, Montserrat E. Criteria for severe aplastic anaemia. Lancet. 1988;331:303–4. [DOI] [PubMed] [Google Scholar]
- 18.Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samarasinghe S, Steward C, Hiwarkar P, Saif MA, Hough R, Webb D, et al. Excellent outcome of matched unrelated donor transplantation in paediatric aplastic anaemia following failure with immunosuppressive therapy: a United Kingdom multicentre retrospective experience. Br J Haematol. 2012;157:339–46. [DOI] [PubMed] [Google Scholar]
- 20.Clesham K, Dowse R, Samarasinghe S. Upfront matched unrelated donor transplantation in aplastic anemia. Hematol Oncol Clin North Am. 2018;32:619–28. [DOI] [PubMed] [Google Scholar]
- 21.Kim H, Im HJ, Koh KN, Kang SH, Yoo JW, Choi ES, et al. Comparable outcome with a faster engraftment of optimized haploidentical hematopoietic stem cell transplantation compared with transplantations from other donor types in pediatric acquired aplastic anemia. Biol Blood Marrow Transplant. 2019;25:965–74. [DOI] [PubMed] [Google Scholar]
- 22.Xu LP, Zhang XH, Wang FR, Mo XD, Han TT, Han W, et al. Haploidentical transplantation for pediatric patients with acquired severe aplastic anemia. Bone Marrow Transplant. 2017;52:381–7. [DOI] [PubMed] [Google Scholar]
- 23.Choi YB, Yi ES, Lee JW, Sung KW, Koo HH, Yoo KH. Immunosuppressive therapy versus alternative donor hematopoietic stem cell transplantation for children with severe aplastic anemia who lack an HLA-matched familial donor. Bone Marrow Transplant. 2017;52:47–52. [DOI] [PubMed] [Google Scholar]
- 24.Gamper CJ, Takemoto CM, Chen AR, Symons HJ, Loeb DM, Casella JF, et al. High-dose cyclophosphamide is effective therapy for pediatric severe aplastic anemia. J Pediatr Hematol Oncol. 2016;38:627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bluteau O, Sebert M, Leblanc T, Peffault de Latour R, Quentin S, Lainey E, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood. 2018;131:717–32. [DOI] [PubMed] [Google Scholar]
- 26.Filippidou M, Avgerinou G, Tsipou H, Tourkantoni N, Katsibardi K, Vlachou A, et al. Longitudinal evaluation of eltrombopag in paediatric acquired severe aplastic anaemia. Br J Haematol. 2020;190:e157–9. [DOI] [PubMed] [Google Scholar]
- 27.Valdez JM, Scheinberg P, Nunez O, Wu CO, Young NS, Walsh TJ. Decreased infection-related mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011;52:726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai B, Said Q, Li X, Li FY, Arcona S. Healthcare resource use and direct costs in severe aplastic anemia (SAA) patients before and after treatment with eltrombopag. J Med Econ. 2020;23:243–51. [DOI] [PubMed] [Google Scholar]
- 29.Tremblay G, Said Q, Roy AN, Cai B, Ashton Garib S, Hearnden J, et al. Budget impact of eltrombopag as first-line treatment for severe aplastic anemia in the United States. Clinicoecon Outcomes Res. 2019;11:673–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Inherited bone marrow failure panel from University of Chicago.
Table S2. Germline variants identified in our cohort in genes associated with inherited bone marrow failure by targeted sequencing panel.
Table S3. Haematological criteria for trial inclusion in both EPAG and IST groups.
Table S4. Administration of cyclosporine on different trial protocols.
Table S5. Haematological response at 6 months in patients who received MMF and sirolimus in the paediatric IST group.
Table S6. PNH clones in patients in paediatric EPAG and IST groups.
Table S7. Off study patients (prior to 6 months).
Table S8. Causes of death for all patients.
Table S9. Haematological response at 6 months: adults versus children.
Table S10. Summary of PK parameters at steady state normalised to the planned starting dose for eltrombopag by age category.
Fig S1. Study design for eltrombopag trial.
Fig S2. Components of the historical paediatric IST group.
Fig S3. EFS of all patients (including non-responders).
Fig S4. Somatic variants in patients from the EPAG cohort at baseline and over the course of treatment.
Fig S5. Clonal evolution in paediatric patients.
Fig S6. Overall survival in paediatric patients.
Fig S7. Competing risk analysis for clonal evolution, HSCT and death.