

SUMMARY
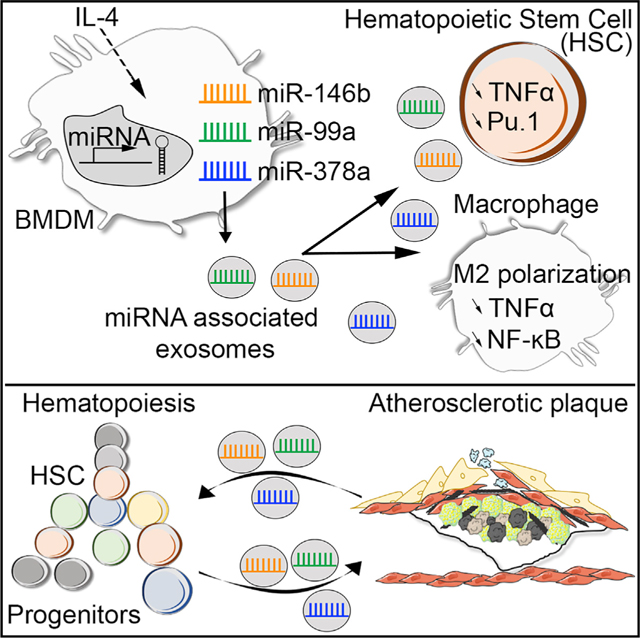
Developing strategies that promote the resolution of vascular inflammation and atherosclerosis remains a major therapeutic challenge. Here, we show that exosomes produced by naive bone marrow-derived macrophages (BMDM-exo) contain anti-inflammatory microRNA-99a/146b/378a that are further increased in exosomes produced by BMDM polarized with IL-4 (BMDM-IL-4-exo). These exosomal microRNAs suppress inflammation by targeting NF-κB and TNF-α signaling and foster M2 polarization in recipient macrophages. Repeated infusions of BMDM-IL-4-exo into Apoe−/− mice fed a Western diet reduce excessive hematopoiesis in the bone marrow and thereby the number of myeloid cells in the circulation and macrophages in aortic root lesions. This also leads to a reduction in necrotic lesion areas that collectively stabilize atheroma. Thus, BMDM-IL-4-exo may represent a useful therapeutic approach for atherosclerosis and other inflammatory disorders by targeting NF-κB and TNF-α via microRNA cargo delivery.
In Brief
Anti-inflammatory properties of M2 macrophages can be communicated as miRNA packaged into exosomes. Bouchareychas et al. show that when tested in the Apoe−/− mouse model of hyperlipidemia, M2 macrophage exosomes reduced hematopoiesis and the inflammatory state of circulating monocytes and macrophages in atherosclerotic plaques.
Graphical Abstract
INTRODUCTION
Cardiovascular events caused by atherosclerosis remain rampant worldwide despite widely available lipid-lowering treatments. The underlying pathogenesis involves an imbalance in plasma lipid metabolism and a chronically activated immune system (Ridker et al., 2017). Therefore, a better understanding of the mechanisms that link inflammation and atherosclerosis is essential to provide avenues to address the residual risk for cardiovascular events through novel therapies (Geovanini and Libby, 2018).
Vascular inflammation results in part from an influx of plasma low-density lipoprotein (LDL) within the intima, where it is susceptible to oxidative modification leading to the recruitment of monocytes, which differentiate into macrophages (Glass and Witztum, 2001). Macrophages accumulate progressively, transform into foam cells, and adopt a phenotype resembling what has been termed M1 polarization in vitro (Moore et al., 2013). The activation of NF-κB in part contributes to macrophage M1 polarization and initiates a complex signaling cascade leading to the production of pro-inflammatory cytokines such as tumor necrosis factor α (TNF-α) and interleukin-1β (IL-1β). The newly synthesized TNF-α further fuels NF-κB activation by binding to TNF receptors (TNFRs) (Hayden and Ghosh, 2014). As the disease progresses, defective efferocytosis and the ensuing accumulation of apoptotic macrophages promote plaque necrosis, frequently found in vulnerable plaques (Bouchareychas et al., 2015; Tabas, 2005).
The advent of new technologies such as single-cell RNA sequencing and mass cytometry has provided new insights into macrophage phenotypic heterogeneity within the arterial wall (Cochain et al., 2018). In mouse models of atherosclerosis regression, lesional macrophages have been shown to be enriched in markers of alternatively activated M2 macrophages (Feig et al., 2011; Rahman et al., 2017). In this regard, new therapeutic strategies in atherosclerosis focusing on inflammation resolution by modulating macrophage polarity toward an M2 phenotype in the arterial wall could provide relief from plaque rupture (Bäck et al., 2019; Rahman and Fisher, 2018).
The discovery of intercellular signaling properties of extracellular vesicles (EVs) has opened new opportunities to modulate macrophage phenotypes that we explored in this study. EVs are subdivided into categories on the basis of their size, subcellular origin, morphology, and methods of collection. Exosomes are a type of EV that originate from within multivesicular endosomal compartments (MVB) and are secreted when the compartments fuse with the plasma membrane (van Niel et al., 2018). Because exosomes generally display a size ranging from 30 to 100 nm, they are a more homogeneous class of EVs compared with other types of EVs.
Exosomes are thought to be generated by all cells and present in biological fluids (Mathieu et al., 2019). They are known to be involved in cell-to-cell communication in health and disease, including within the hematopoietic (Wen et al., 2016) and cardiovascular (Das and Halushka, 2015) systems. They contain numerous cargo molecules, including mRNAs, micro-RNAs, long non-coding RNAs (lncRNAs), proteins, and lipids that participate in intercellular signaling (Boilard, 2018; Jeppesen et al., 2019; Kowal et al., 2016; Valadi et al., 2007). Therefore, exosomes and other EVs are a focus of intense interest in their potential to act as biomarkers and therapeutic agents including in the cardiovascular system (Wu et al., 2019).
Multiple groups have reported that EVs secreted from activated monocytes or macrophages and endothelial cells propagate pro-inflammatory signals through several distinct mechanisms (Hergenreider et al., 2012; Hoyer et al., 2012; Ismail et al., 2013; Njock et al., 2015; Robbins and Morelli, 2014). However, the precise function of these immune cell-derived EVs and the impact of their microRNA cargo, once released into the circulation, remain poorly understood.
Although recent studies support the notion that EVs produced from mouse and human macrophages treated with oxidized LDL (oxLDL) influence macrophage migration in vitro (Nguyen et al., 2018), whether they alter atherosclerosis lesion progression remains unknown. Furthermore, effects of exosomes produced by alternatively activated M2-like macrophages in atherosclerosis have not been reported.
The results of our study demonstrate that exosomes produced by naive and M2-polarized macrophages control inflammatory properties of cultured macrophages. Our findings also show that they control hematopoiesis and thereby myeloid cell numbers in the circulation and vessel wall of Apoe−/− mice via the delivery of microRNA cargo. The favorable properties exerted by naive and M2 macrophage exosomes, along with their ease of production and alteration in microRNA cargo, offer a new therapeutic avenue for the treatment of inflammation, including in the cardiovascular system.
RESULTS
Isolation of Macrophage Exosomes and In Vitro Assessment of Their Cell Signaling Properties
We initiated our studies by isolating exosomes from supernatants of cultured mouse bone marrow-derived macrophages (BMDMs) exposed to the M2-polarizing cytokine IL-4 (Sica and Mantovani, 2012). Exosomes secreted by IL-4-exposed and naive BMDM cultures were purified using our recently described cushioned-density gradient ultracentrifugation (C-DGUC) method (Duong et al., 2019). Subsequently, exosomes were analyzed using nanoparticle tracking analysis (NTA) to quantify and compare their size differences. Data shown in Figure 1A and Figures S1A and S1B demonstrated a similar particle concentration and average mode size of 77.63 ± 2.9 nm for BMDM exosomes (BMDM-exo) and 73.65 ± 2.4 nm for IL-4-stimulated BMDM-exo (BMDM-IL-4-exo), consistent with the 30–100 nm size range expected for these types of EVs (Colombo et al., 2014). We calculated that BMDMs cultured in control medium or when stimulated with IL-4 secreted the same quantity of exosomes in a 24 h period, with averages of 2.81 × 109 and 2.76 × 109 secreted particles per million cells (Figure S1C). Furthermore, examining BMDM-exo using transmission electron microscopy (TEM) revealed the typical and expected round or cup-shaped morphology (Figure 1B).
Figure 1. Isolation of Macrophage Exosomes and In Vitro Assessment of their Cell Signaling Properties.

(A) Representative size and concentration distribution of BMDM-exo or BMDM-IL-4-exo purified from BMDM cell culture supernatants after a 24 h period of culture, determined using nanoparticle tracking analysis.
(B) Electron micrograph of purified exosomes from BMDM cells. Scale bar, 100 nm.
(C) Western blot analysis of Calnexin, GM130, Alix, Flotillin, and CD9 in exosome-free media (EFM), cell lysate, and BMDM-derived exosomes (representative of three independent experiments).
(D) Western blot analysis of IL-4 in cell lysate and BMDM-derived exosomes (representative of two independent experiments).
(E) qRT-PCR analysis of Il1b, Tnf, Arg1, Chil3, and Retnla mRNA expression in BMDMs treated with PBS (control), BMDM-exo, or BMDM-IL-4-exo for 24 h. Results were normalized to B2m and Gapdh mRNA and are presented relative to control (representative of three independent measurements, n = 4 per group).
(F) qRT-PCR analysis of IL1B, TNF, and NFKBIA mRNA expression in human monocyte-derived macrophages treated with PBS (control), BMDM-exo, or BMDM-IL-4-exo for 24 h. Gene expression was normalized against GAPDH mRNA expression and converted to fold change relative to control condition (pool of two independent experiments, n = 7–10 per group). Statistical analysis was performed using the Kruskal-Wallis test and Dunn’s post-test to determine the significant difference among the three groups.
(G) Graph showing representative Seahorse mitochondrial stress tests. O, oligomycin (1 μM); F, FCCP (2 μM); R/AA, rotenone/antimycin A (0.5 μM). One representative experiment out of six experiments is shown; n = 5 per group.
(H) Bar graphs showing quantified cell-normalized mitochondrial OCR from stress tests. Results are presented relative to PBS control; pool of six independent experiments is shown; n = 20–37 in each group.
*p < 0.05, **p < 0.01, and ***p < 0.001 as determined using one-way ANOVA and Holm-Sidak post-test. Data are represented as mean ± SEM.
An assessment of protein concentration also found BMDM-exo and BMDM-IL-4-exo to display similar average concentrations of 36.70 ± 8.7 and 36.05 ± 5.9 μg/mL, respectively (Figure S1D). To validate the purity of our exosome isolates, western blot analyses were performed to examine the presence of confirmed exosomal proteins, including Alix, Flotillin, and CD9, and the absence of cell-associated proteins Calnexin and GM130 (Figure 1C). Because cytokines have been shown to be associated with macrophage-derived exosomes (McDonald et al., 2014), we tested whether IL-4 was present in our BMDM-exo and BMDM-IL-4-exo. Results of western blot detection revealed that IL-4 was not associated with BMDM-derived exosomes (Figure 1D).
In beginning to explore the immune regulatory function of macrophage exosomes, we incubated BMDM-exo and BMDM-IL-4-exo with freshly cultured mouse BMDMs. Our findings revealed that both forms of exosomes reduced the expression of the inflammatory cytokines IL-1β (Il1b) by 2.37-fold and TNF-α (Tnf) by 1.85-fold compared with naive BMDMs treated with control PBS alone using qRT-PCR (Figure 1E). This magnitude of cytokine expression control was similar to the use of IL-4 itself at a concentration of 20 ng/mL in downregulating Il1b and Tnf in BMDMs, at 2.17-fold and 1.56-fold, respectively (Figure S2). We next validated that a similar concentration of IL-4 upregulated the mRNA expression levels of markers commonly associated with M2 macrophages, including Arg1 (by 1,585-fold), Chil3 (by 34-fold), and Retnla (by 146-fold) in BMDMs (Figure S2). Interestingly, we observed that BMDM-exo and BMDM-IL-4-exo also upregulated the expression of the M2 macrophage marker Arg1 by 2.30-fold and 2.40-fold, respectively. However, only BMDM-IL-4-exo further upregulated Chil3 (by 1.85-fold) and Retnla (by 2.40-fold) mRNA expression in BMDMs (Figure 1E). Furthermore, when incubated with human monocyte-derived macrophages, BMDM-IL-4-exo targeted the NF-κB pathway by decreasing TNF expression by 1.65-fold and increasing NF-κB inhibitor alpha by 1.33-fold, although it did not extend to the control of IL-1Β (Figure 1F).
To examine the extent to which BMDM-IL-4-exo induce macrophage polarization, we examined their capacity to induce metabolic reprogramming that is recognized to fuel their effector functions (Galván-Peña and O’Neill, 2014; Van den Bossche et al., 2016). To this end, we quantified changes in the oxygen consumption rate (OCR) in naive BMDMs exposed to either BMDM-exo or BMDM-IL-4-exo (Figure 1G). Interestingly, our data show that naive BMDMs stimulated with BMDM-IL-4-exo displayed enhanced basal and maximal respiration associated with a higher proton leak and ATP production compared with BMDM exposed to PBS or BMDM-exo (Figure 1H). Together, these data provide compelling evidence that exosomes derived from cultured BMDMs display a capacity to communicate anti-inflammatory properties to recipient macrophages and that exosomes produced by M2-like macrophages are more potent.
Biodistribution of Macrophage Exosomes and Delivery of Their microRNA Cargo to Tissues of Apoe−/− Mice
In beginning to explore possible in vivo signaling properties of BMDM-exo, we first examined their biodistribution by labeling them with the near-infrared dye DiR, as previously reported (Wiklander et al., 2015). Twenty-five-week-old male Apoe−/− mice fed a Western diet were intraperitoneally (i.p.) injected twice at a 2 day interval with PBS, DiR-labeled BMDM-exo, or BMDM-IL-4-exo for 4 weeks. Perfused organs were collected and subsequently imaged, 12 h after the last injection. Labeled exosomes were detected mainly in the adipose tissue, liver, aorta, hematopoietic compartment, spleen, and bone (Figure 2A). We also observed that isolated peritoneal macrophages (F4/80+ cells) are more efficient than the F4/80− cell population in taking up BMDM-exo (Figure 2B). Immunofluorescence staining also confirmed similar internalization efficiency of both BMDM-exo and BMDM-IL-4-exo by recipient BMDMs (Figure 2C). In testing whether BMDM-exo can deliver microRNA cargo, purified exosomes were loaded with synthetic IRDye-labeled miR-146b and infused into Apoe−/− mice. Interestingly, we observed that the distribution of IRDye-labeled miR-146b-loaded exosome was slightly different than DiR-labeled BMDM-exo. IRDye-labeled miR-146b exosomes distributed mainly to the intestine, lungs, liver, and aorta (Figure 2D) and were also efficiently taken up by peritoneal macrophages (Figure 2E). For all the conditions, no uptake difference was observed between BMDM-exo and BMDM-IL-4-exo. These results show that exosomes can distribute widely among tissues and that the distribution might be different on the basis of their microRNA cargo or by experimental manipulations to load them with RNA cargo.
Figure 2. Biodistribution of Macrophage Exosomes and Delivery of Their microRNA Cargo to Tissues of Apoe−/− Mice.

Exosomes purified from BMDM culture medium were labeled with fluorescent lipophilic tracer DiR and infused into 25-week-old Apoe−/− mice for 4 weeks every 2 days.
(A) Representative images of organs (12 h post-injection) from mice injected i.p. with 1 × 1010 particles (measured using NTA) and quantification using the PBS as background control.
(B) Representative images of peritoneal F4/80− and F4/80+ isolated cells from mice injected with DiR-labeled exosomes. Fluorescence of DiR-labeled BMDM exosomes was detected using the LI-COR Odyssey infrared imaging system. Color scale indicates fluorescence intensity.
(C) Merged images showing internalization of PKH26-labeled BMDM-exo (red) by naive culture BMDMs counterstained with DAPI (blue). BMDMs were co-incubated with 2 × 109 PKH26-labeled exosomes for 2 h at 37°C and washed repeatedly to remove unbound exosomes. All images were acquired using a Zeiss microscope system with a 20× objectives. Scale bars: 100 μm. Statistical analysis of fluorescence intensity was conducted using one-way ANOVA and Tukey’s multiple-comparisons test.
(D and E) Distribution of synthetic IRDye-labeled miR-146b, transfected into BMDM exosomes, 2 h after i.p. injection in 25-week-old Apoe−/− mice (D). Representative images of organs and (E) peritoneal F4/80− and F4/80+ isolated cells from mice injected with IRDye-labeled miR-146b-loaded exosomes detected using the LI-COR Odyssey infrared imaging system.****p < 0.0001 as determined using one-way ANOVA and Tukey’s post-hoc test. Data are represented as mean ± SEM.
Hematopoiesis Control in Apoe−/− Mice Treated with BMDM-IL-4-Exo
To further uncover regulatory properties of BMDM-IL-4-exo in controlling diet-induced myelopoiesis in Apoe−/− mice, we tested whether BMDM-IL-4-exo could alter the expansion of hematopoietic stem and progenitor cells. Although the overall percentage of lineage negative Lin− Sca-1+ cKit+ (LSK) group of hematopoietic progenitor cells was only marginally reduced by BMDM-IL-4-exo (Figures 3A and 3B), distinct populations of these cells were significantly affected. This included a marked contraction among multipotent progenitors (MPPs) that are downstream differentiation products of the hematopoietic stem cell (HSC), containing both myeloid and lymphoid differentiation potentials (Yamamoto et al., 2013). Our data show that BMDM-IL-4-exo reduced the percentage of MPP2 (Flt3−/CD150+/CD48+ LSK), MPP3 (Flt3−/CD150−/CD48+ LSK), and MPP4 (Flt3+/CD150−/CD48+ LSK) populations in the bone marrow compared with PBS-injected mice. Among the more differentiated cKit+ Sca-1− progenitor cell subset examined, only the megakaryocyte-erythroid progenitor (MEP) population was significantly reduced by BMDM-IL-4-exo exposure compared with the PBS-injected group of Apoe−/− mice (Figures 3A and 3B).
Figure 3. Hematopoiesis Control in Apoe−/− Mice Treated with BMDM-IL-4-Exo.

(A) Representative plots of flow cytometric analyses of hematopoietic stem and progenitor cells in the bone marrow of 25-week-old Apoe−/− mice fed with a Western diet injected with PBS, BMDM-exo, or BMDM-IL-4-exo (1 × 1010 particles/mouse every 2 days) for 4 weeks.
(B) Graph showing the percentages of LSK, MPP2, MPP3, MPP4, and MEP in Apoe−/− mice injected with PBS, BMDM-exo, or BMDM-IL-4-exo. One representative experiment out of three experiments is shown; n = 4 or 5 per group. Statistical analysis was performed using Kruskal-Wallis test and Dunn’s post-test to determine the significant difference among the three groups.
(C) HSCs were sorted from 25-week-old Apoe−/− mice fed with a Western diet and incubated for 2 h with DiR-labeled exosomes. The cells were subsequently washed twice with PBS, visualized, and quantified using the Odyssey Infrared Imaging System.
(D) Bone marrow (BM) cells (2 × 104) from 25-week-old Apoe−/− mice were plated in methylcellulose-based medium with recombinant cytokines for colony-forming unit (CFU) and treated every 2 days with PBS, BMDM-exo, or BMDM-IL-4-exo at a dose of 2 × 109 particles/mL. CFU-GEMM, granulocyte-erythroid-monocyte-megakaryocyte mixed CFU; CFU-GM, granulocyte-monocyte CFU were scored on day 12 of culture. Pool of four experiments with four separate preparations of exosome were analyzed.
(E) CD34− LSK cells were sorted from the bone marrow of 10-week-old male C57BL/6J mice and stimulated with PBS, BMDM-exo, or BMDM-IL-4-exo (2 × 109 particles/mL) for 6 or 24 h, and gene expression of Tnf and Pu.1 was assessed using qRT-PCR analysis. One representative experiment out of two experiments is shown; n = 2–4 per group. Statistical analysis was performed using two-way ANOVA with Sidak’s multiple-comparisons post-test.
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Data are represented as mean ± SEM.
Next, we sorted HSCs from Apoe−/− mouse bone marrow and cultured them with DiR-labeled exosomes to test whether BMDM-exo can be taken up by these progenitor cells. We observed an efficient internalization of both BMDM-exo and BMDM-IL-4-exo by recipient HSCs (Figure 3C). In addition, we performed in vitro colony-forming unit (CFU) assays and confirmed a functional decrease of CFU-GM colonies upon treatment with BMDM-exo (Figure 3D). Therefore, to explore a mechanistic basis for our in vivo observations, we tested whether HSCs are sensitive to intercellular signaling from macrophage exosomes. To this end, CD34− LSK cells sorted from mouse bone marrow were incubated with either BMDM-exo or BMDM-IL-4-exo for 6 or 24 h. Our findings show that both types of BMDM-exo reduced the mRNA expression levels of Tnf and the master regulator transcription factor Pu.1 (Scott et al., 1994) (Figure 3E). However, BMDM-IL-4-exo more rapidly communicated a control of Pu.1 mRNA expression in these cells. Together, our results demonstrate that BMDM-IL-4-exo exert profound control over hematopoiesis in the bone marrow of Apoe−/− mice fed a Western diet by targeting a reduction of select hematopoietic progenitor cell subsets that are recognized to contribute to myelopoiesis in conditions of metabolic stress (Pietras et al., 2015). In Apoe−/− mice, hematopoietic stem and progenitor cells progressively migrate from the bone marrow to the splenic red pulp, where they clonally expand and differentiate into Ly-6Chi monocytes that contribute to atherosclerosis (Robbins et al., 2012). Therefore, we tested the ability of the BMDM-exo to modulate extramedullary hematopoiesis. However, no differences were observed in splenic hematopoiesis in mice treated with BMDM exosomes compared with PBS (Figure S3). Hence, bone marrow and splenic hematopoiesis appear to be regulated independently by BMDM-IL-4-exo in this mouse model of hyperlipidemia.
Treatment of Apoe−/− Mice with Macrophage Exosomes Reduces Circulating Myeloid Cells and Induces Macrophage Polarization
Studies have shown that an increase in the count of circulating Ly-6Chi monocytes and neutrophils due to hyperlipidemia-enhanced hematopoiesis correlates with atherosclerotic lesion burden (Murphy et al., 2011; Robbins et al., 2012; Swirski et al., 2007). Therefore, we tested whether the decrease in bone marrow hematopoiesis in Apoe−/− mice treated by BMDM-exo affected the number of mature myeloid cells in the circulation. We analyzed using flow cytometry the numbers of circulating neutrophils and monocytes in Apoe−/− mice injected i.p. with PBS, BMDM-exo, or BMDM-IL-4-exo at a dose of 1 3 1010 particles, three times a week for 4 weeks (Figure 4A). Our findings revealed that injections of BMDM-exo had no measurable impact on altering the number of myeloid cells in the circulation of Apoe−/− mice. In stark contrast, injections of an equal number of BMDM-IL-4-exo produced a 2-fold decrease in the number of neutrophils in the circulation of Apoe−/− mice (Figure 4B). Furthermore, it led to a robust 3-fold reduction in the number of Ly-6Chi monocytes and a 2-fold reduction of Ly-6Clow monocytes in the circulation of these mice compared with levels detected in Apoe−/− mice injected with PBS or BMDM-exo (Figures 4C and 4D). To further address whether BMDM-IL-4-exo affects monocyte phenotypes, we sorted peripheral blood Ly-6Chi monocytes using fluorescence-activated cell sorting (FACS) and performed a transcriptomic analysis using a NanoString gene expression profiling platform. Ly-6Chi monocytes isolated from mice treated with BMDM-IL-4-exo showed reductions of markers involved in cell migration and activation (Ccr7, Flt4, Cd83, Cxcl1) (Figure 4E). In addition, we observed a profound alteration in the expression of inflammatory genes associated with cardiovascular inflammation (Tnf, IL-10, NF-κB) (Tedgui and Mallat, 2006) (Figure 4F). The CCL2/MCP-1 chemokine is well established for its chemoattractant properties for monocytes into atherosclerosis lesion (Gu et al., 1998) but is also suggested to play a role in macrophage polarization (Gschwandtner et al., 2019). Therefore, we quantified Ccl2 mRNA expression in sorted Ly-6Chi monocytes and CCL2 levels in the plasma of Apoe−/− mice injected with PBS, BMDM-exo, or BMDM-IL-4-exo. Our findings showed that Ccl2 mRNA expression in monocytes (Figure 4G) and CCL2 plasma levels (Figure 4H) were decreased in mice injected with BMDM-IL-4-exo compared with control mice.
Figure 4. Treatment of Apoe−/− Mice with Macrophage Exosomes Reduces Circulating Myeloid Cells and Induces Macrophage Polarization.

(A) Representative flow cytometry plots of blood leukocyte subsets from 25-week-old Apoe−/− mice fed with a Western diet and injected with PBS, BMDM-exo, or BMDM-IL-4-exo (1 × 1010 particles/mouse every 2 days for 4 weeks).
(B–D) Quantification of (B) neutrophils, (C) Ly6Chi monocytes, and (D) Ly6Clow monocytes in BMDM-exo or BMDM-IL-4-exo compared with PBS-injected mice. One representative experiment out of three experiments is shown; n = 4 or 5 per group.
(E and F) Heatmap representation of differentially regulated genes involved in (E) cell migration and activation and (F) cardiovascular inflamamtion from Ly-6Chi monocytes sorted from the blood of Apoe−/− mice injected with PBS compared with BMDM-IL-4-exo is depicted. Gene expression was assessed using NanoString (four mice per group).
(G–J) qRT-PCR analysis of Ccl2 mRNA expression in isolated Ly-6Chi monocytes and (H) plasma measurement of CCL2 from the blood of Apoe−/− mice injected with either PBS or BMDM-exo or BMDM-IL-4-exo. (I) qRT-PCR analysis of M1-associated genes, including Tnfa, Nos2, and (J) the M2-associated genes Arg1, Chil3, Retnla, and Mrc1 in peritoneal macrophage isolated from Apoe−/− mice injected with PBS, BMDM-exo, or BMDM-IL-4-exo. Genes were normalized against B2m and Gapdh mRNA expression and converted to fold change relative to PBS-injected mice. Pool of three independent experiments is shown; n = 11–15 in each group.
Statistical analysis was performed using the Kruskal-Wallis test and Dunn’s post-test to determine the significant difference among the three groups. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Data are represented as mean ± SEM.
To further address the impact of BMDM-IL-4-exo on mature myeloid cells, we next isolated peritoneal macrophages from Apoe−/− mice injected with BMDM-exo. Our findings showed that BMDM-IL-4-exo potently reduced the expression of pro-inflammatory genes including Tnf and Nos2 in these cells (Figure 4I). Our findings also revealed that such exosomes prominently induced the expression of M2-like macrophage markers, including Arg1, Chil3, Retnla, and Mrc1 in peritoneal macrophages compared with Apoe−/− mice injected with PBS (Figure 4J). Together, our data demonstrate that BMDM-IL-4-exo can profoundly affect myeloid cell phenotypes in mice with hyperlipidemia.
Resolution of Inflammation in Atheroma of Apoe−/− Mice Treated with BMDM-IL-4-Exo
Because myelopoiesis caused by excessive dietary calorie intake contributes directly to atherosclerosis plaque progression in mice (Robbins et al., 2012), we reasoned that BMDM-IL-4-exo might play a role in controlling atherosclerosis. To test this hypothesis, Western diet-fed Apoe−/− mice were treated with BMDM-derived exosomes for 4 weeks. Such injections had no impact on altering either body weight or plasma cholesterol levels (data not shown). An assessment of histological sections prepared from the aortic root of these mice using oil red O staining revealed similar atherosclerotic lesion areas among PBS-, BMDM-exo-, and BMDM-IL-4-exo-treated mice (Figures 5A and 5B). Next, we examined the extent of necrosis by measuring acellular areas in atheroma, as it is recognized as a cardinal feature of advanced and vulnerable atherosclerotic lesions (Tabas, 2005). Our data show that injections of both BMDM-exo and BMDM-IL-4-exo significantly reduced necrotic lesion areas in atheroma of Apoe−/− mice compared with injections of PBS (Figures 5C and 5D). Our findings show that BMDM-IL-4-exo infusions substantially decreased macrophage-positive areas within the atheroma of Apoe−/− mice, whereas this beneficial effect was not observed with BMDM-exo (Figures 5E and 5F). Furthermore, infusions of BMDM-IL-4-exo also led to an increase in CD206 positive surface area among residual lesional macrophages, suggesting their polarization to an M2-like phenotype consistent with our in vitro and in vivo data (Figures 5G and 5H). Together, our data show that infusions of BMDM-IL-4-exo are more effective than BMDM-exo in driving beneficial lesion remodeling and therefore plaque stabilization in mice with dyslipidemia.
Figure 5. Resolution of Inflammation in Atheroma of Apoe−/− Mice Treated with BMDM-IL-4-Exo.

(A and B) Histological analysis (A) and (B) quantification of cross sections of the aortic sinus stained with oil red O (ORO) from 25-week-old Apoe−/− mice fed with a Western diet and injected with PBS, BMDM-exo, or BMDM-IL-4-exo for 4 weeks. Scale bar, 500 μm. n = 13–15 in each group.
(C) Representative cross-sectional view of aortic root stained with DAPI to measure necrosis area from each group of mice. Dashed lines show the boundary of the developing necrotic core. Scale bar, 100 μm.
(D) Quantification of necrotic core area as a percentage of total plaque area.
(E and F) Representative images (E) and (F) quantification of MOMA-2+ macrophages in the atherosclerotic plaques of aortic root areas. Scale bar, 100 μm.
(G) Representative image of CD206 staining in aortic root lesions. Scale bar, 100 μm.
(H) Quantification of CD206 staining as a ratio of macrophage lesion area. Results from a pool of three independent experiments are shown; n = 11–14 in each group.
Statistical analysis was performed using one-way ANOVA and Sidak’s multiple-comparisons post-test. *p < 0.05 and **p < 0.01. Data are represented as mean ± SEM.
Polarizing Macrophages with IL-4 Modulates the Expression and Release of MicroRNA in Exosomes
In looking to uncover the source of the anti-inflammatory properties displayed by BMDM-exo in vitro and in vivo, we examined their microRNA cargo. To this end, we profiled the microRNA transcriptome of RNA extracted from BMDMs and their exosomes by unbiased RNA sequencing, either when they were cultured in control medium or when stimulated with IL-4. As shown in Figure 6A, a total of 71 of the 1,088 microRNAs (6.5%) present in the arrays were differentially expressed between BMDM-exo and their parental cells. Furthermore, we similarly identified 74 microRNAs out of 1,012 (7.3%) that were differentially expressed between IL-4-treated BMDMs and their isolated exosomes (Figure 6B). Importantly, we identified 4 microRNA that were differentially expressed in BMDM-IL-4-exo compared with BMDM-exo. This included miR-100–5p, which was detected at a reduced level in BMDM-IL-4-exo. In contrast, BMDM-IL-4-exo displayed increased levels of miR-99a-5p, miR-146b-5p, and miR-378a-3p (Figure 6C). These changes in microRNA levels were further validated and confirmed when tested using qRT-PCR (Figure 6D). Our data therefore show that IL-4 stimulation modulates microRNA expression in both parental cells and their exosomes.
Figure 6. Polarizing Macrophages with IL-4 Modulates the Expression and Release of MicroRNA in Exosomes.

(A) Heatmap showing the distinct microRNA expression profiles between parental cells (BMDMs; n = 3) and their exosomes (BMDM-exo; n = 4). Venn diagram of unique and shared microRNAs in BMDM and BMDM-exo samples.
(B) Heatmap showing the distinct microRNA expression profiles between BMDMs stimulated with IL-4 (20 ng/mL) during a period of 24 h (BMDM-IL-4; n = 3) and their exosomes (BMDM-IL-4-exo; n = 4). Venn diagram of unique and shared microRNAs in BMDM-IL-4 and BMDM-IL-4-exo samples.
(C) Heatmap illustrating differential expression of microRNAs in BMDM-exo compared with BMDM-IL-4-exo. Red signal and blue signal indicate microRNA expression levels.
(D) qRT-PCR validation of microRNA sequencing. One representative experiment out of three experiments is shown; n = 3 per group.
*p < 0.05 and **p < 0.01 as determined by unpaired Student’s t test. Data are represented as mean ± SEM.
Gene Editing of MicroRNA in Macrophage Exosomes Reveals Their Role in Communicating Anti-inflammatory Signaling
In seeking to test the contribution of our candidate microRNAs in exosome-mediated anti-inflammatory communication, we examined their impact on regulating the expression of their predicted target genes. To this end, we selectively eliminated miR-99a-5p, miR-146b-5p, and miR-378a-3p from exosomes produced by macrophages. We did so by using a CRISPR-Cas9 system previously reported to effectively downregulate the expression of microRNA in cell culture systems (Chang et al., 2016). We thus opted to make use of an immortalized murine bone marrow-derived cell line (iBMDM) expressing the gene editing machinery enabling us to produce a series of microRNA-altered exosomes (Covarrubias et al., 2017).
We designed two sets of single-guide RNAs (sgRNAs) for each of our candidate microRNAs that targeted miR-99a-5p, miR-146b-5p, and miR-378a-3p. For all three microRNAs, we obtained ~99% knockdown relative to control sgRNA (Figure S4A). We next selected the sgRNA with the highest efficiency to selectively eliminate each of the three individual microRNAs in iBMDMs. Interestingly, we observed that the deletion of miR-146b-5p, miR-99a-5p, and miR-378a-3p in iBMDMs increased Tnf, Il1b, and Irak1 mRNA expression in basal condition (Figure S4B), while deletion of miR-99a-5p caused an increase in the expression of Traf6 mRNA expression (Figure S4B). Together, these data validate TNF-α and NF-κB adaptor molecules as predicted targets for miR-99a-5p, miR-146b-5p, and miR-378a-3p in macrophages.
Selective Ablation of MicroRNA in iBMDM-Exo Increases TNF-α and NF-κB Expression
Subsequently we examined whether a loss of any of these three individual microRNA could alter the anti-inflammatory signaling capacity of exosomes produced by iBMDMs. To this end, we isolated exosomes from the cell culture medium of each iBMDM cell line engineered to downregulate levels of miR-99a-5p (sgRNA-miR-99a-exo), miR-146b-5p (sgRNA-miR-146b-exo), miR-378a-3p (sgRNA-miR-378a-exo), or from a control non-targeting sgRNA (sgRNA control-exo) in control conditions (unstimulated) or after a 24 h period of IL-4 stimulation. Data shown in Figures S5A and S5B demonstrated that exosomes produced by Cas9-engineered iBMDMs displayed a similar size range compared with BMDM-exo, with an average mode size of 74.58 nm. Additionally, we calculated that the number of particles secreted by iBMDMs is equivalent to the BMDMs, with an average of 2.4 × 109 secreted particles per million of cells and an average protein concentration in the iBMDM-exo of 42.32 μg/mL (Figures S5C and S5D). Western blot analysis confirmed the presence of the common exosome protein markers Alix and Flotillin and the absence of cell-associated proteins Calnexin and GM130 in the exosome fractions as observed for BMDM-exo (Figure S5E). In addition, we validated that iBMDM-IL-4-exo depleted for either microRNA were taken up by naive BMDMs in vitro (Figure S5F). Taken together, these data support that iBMDM-and BMDM-exo share similar structural and functional features. Importantly, our data show that when stimulated with IL-4, control sgRNA-iBMDMs produced exosomes with increased levels of miR-146b-5p, miR-99a-5p, and miR-378a-3p (Figure 7A). These findings mirror our data observed with exosomes isolated from BMDMs exposed to IL-4 (Figure 6D). We then validated using qRT-PCR that the depletion of select microRNAs in iBMDMs transduced with the three individual sgRNAs also results in a depletion of microRNA in secreted exosomes (Figure 7B). These data demonstrate the successful engineering of exosomes to eliminate select micro-RNA cargo.
Figure 7. Selective Ablation of MicroRNA in iBMDM-Exosomes Increases TNF-α and NF-κB Expression.

(A) qRT-PCR analysis of miR-146b-5p, miR-99a-5p, and miR-378a-3p in iBMDM-exosomes collected from the cell culture supernatant of unstimulated or IL-4-treated iBMDMs. Three separate preparations of exosomes were analyzed.
(B) qRT-PCR analysis of miR-146b-5p, miR-99a-5p, and miR-378a-3p ablation efficiency in iBMDM-exosomes.
(C) Detection of intracellular TNF-α in BMDMs stimulated with iBMDM engineered exosomes for a period of 6 h using flow cytometry. The mean fluorescence intensity (MFI) is normalized to unstimulated iBMDM condition. Results from a pool of two independent experiments are shown; n = 6 or 7 in each group.
(D) Quantification of GFP mean fluorescence intensity using flow cytometry in iBMDMs unstimulated or stimulated simultaneously with LPS (100 ng/mL) and iBMDM-exo for a period of 6 h. The MFI is normalized to unstimulated iBMDMs. Pool of two independent experiments is shown; n = 6–8 in each group.
Statistical analysis was performed using one-way ANOVA and Holm-Sidak post-test to determine the significant difference among the groups. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Data are represented as mean ± SEM.
Finally, we examined the impact of selective microRNA deletion in exosomal anti-inflammatory communication using cultured naive BMDMs with exosomes isolated from iBMDMs exposed to IL-4 or control, as well as from iBMDMs deficient in the expression of miR-146b-5p, miR-99a-5p, and miR-378–3p. After a 6 h incubation period, we noted that iBMDM-exo and iBMDM-IL-4-exo displayed a similar capacity to reduce the level of TNF-α compared with unstimulated BMDMs using flow cytometry (Figure 7C). In addition, we noted that the ablation of miR-146b-5p, miR-99a-5p, or miR-378a-3p in iBMDM-exo impaired their capacity to reduce TNF-α expression in recipient cultured macrophages (Figure 7C). This effect was reduced in miR-146b-depleted iBMDM-IL-4-exo, suggesting that an upregulation of miR-99a-5p and miR-378–3p can partially compensate the loss of miR-146b-5p in BMDM-exo. These findings demonstrated the ability of this set of microRNAs to control the TNF-α pathway in macrophages via exosomal microRNA delivery. We next used an iBMDM-GFP-based NF-κB reporter macrophage cell line to test the possibility that our iBMDM-exo target this central inflammatory pathway. To this end, we stimulated cultured mouse iBMDMs with lipopolysaccharide (LPS) while exposing them to iBMDM-exo for a period of 6 h and quantified the mean fluorescence intensity (MFI) of GFP using flow cytometry. As shown in data presented in Figure 7D, deletion of miR-99a-5p, miR-378–3p, or miR-146b-5p in iBMDM-exo resulted in an increased expression of the NF-κB pathway in these cells compared with control sgRNA-exo and sgRNA-IL-4-exo.
Altogether, our data demonstrate that exosomes produced by cultured macrophages have a capacity to communicate anti-inflammatory signaling. Furthermore, our findings also show that IL-4-stimulated macrophages produce exosomes with an enhanced capacity to resolve inflammation via anti-inflammatory microRNA cargo, including in atherosclerotic lesions.
DISCUSSION
In this study, we have shown that exosomes isolated from the conditioned medium of cultured mouse BMDMs can exert intercellular communication to modulate aspects of systemic and vascular inflammation via delivery of microRNA cargo in mice with hyperlipidemia. Because our overarching goal is to develop new therapies to control the pathogenesis of atherosclerosis, our focus was to study properties of exosomes isolated from naive and IL-4-stimulated macrophages.
Our rationale stemmed from studies reporting that exosomes secreted by macrophages stimulated with LPS lead to transcriptional activation of NF-κB (McDonald et al., 2014), while their stimulation with oxLDL results in exosomes that communicate impaired migratory capacity to naive macrophages via micro-RNA communication (Nguyen et al., 2018). Furthermore, exosomes isolated from macrophages infected with an intracellular pathogen induced IL-12p40 and TNF-α production, which can play a key role in cellular recruitment in vivo upon infection (Bhatnagar et al., 2007; Singh et al., 2012). On the basis of such reported findings, we reasoned that exosomes produced by M2 macrophages could in contrast communicate anti-inflammatory and pro-resolving signaling to control atherosclerosis in mice with hyperlipidemia.
In testing this hypothesis, we first examined the distribution in vivo of i.p. infused BMDM-exo and their microRNA cargo through the delivery of DiR-labeled exosomes or with exosomes loaded with synthetic IRDye-labeled miR-146b. Although we noted no difference in the uptake of either BMDM-exo or BMDM-IL-4-exo by cells and tissues, we observed differences in the biodistribution of exosomes labeled with either DiR and exosomes loaded with labeled microRNA. The distribution of exosomes in vivo might differ because of numerous variables, including the type of originating cells, vesicle sizes, route of administration, exosome injection dose, protocol for EV isolation, and methods used to label the exosomes, combined with the limited tissue penetration depth of fluorescent probes and their half-life in vivo (Wiklander et al., 2015; Yi et al., 2020). In our study, the different time points of detection, methods of exosome labeling and cargo loading, as well as microRNA distribution and fate in the recipient cells might all account for the differences observed in biodistribution (Figures 2A, 2B, 2D, and 2E).
Although no apparent biophysical differences tested seemed to distinguish BMDM-IL-4-exo from BMDM-exo in vitro, their functional ability to communicate anti-inflammatory properties in vivo was more apparent. Indeed, both forms of exosomes had similar potency in suppressing the expression of the inflammatory cytokines IL-1β and TNF-α and in raising the expression level of the classical M2 marker gene Arginase-1. However, only BMDM-IL-4-exo caused a further upregulation of other M2 marker genes, demonstrating their capacity to more profoundly reprogram macrophage polarity. Our observed anti-inflammatory properties displayed by naive macrophage exosomes extend findings from Bisgaard et al. (2016), who reported M2-like features in cultured naive macrophage. Importantly, treatment of human monocyte-derived macrophages with BMDM-IL-4-exo significantly downregulated TNF and upregulated NFKBIA mRNA expression levels, reproducing the benefits of BMDM-IL-4-exo observed in mice. The findings further highlighted the value of BMDM-exo in human inflammation control.
Numerous studies have shown that inflammatory M1 macrophages display enhanced aerobic glycolysis and reduced mitochondrial activity. In contrast, anti-inflammatory M2 macrophages use mitochondrial oxidative phosphorylation, characterized by an enhanced spare respiratory capacity (Galván-Peña and O’Neill, 2014; Van den Bossche et al., 2016). Remarkably, our results revealed that BMDM-IL-4-exo potently communicated cellular reprograming by enhancing mitochondrial respiration and energy metabolism in naive macrophages.
Our findings also revealed that BMDM-IL-4-exo exerted control over hyperlipidemia-driven hematopoiesis by restricting the expansion of MPPs, suggesting that they regulate HSC self-renewal and/or differentiation. Our in vitro studies further confirmed a capacity for BMDM-exo and BMDM-IL-4-exo to be taken up by hematopoietic progenitors, profoundly regulating colony formation and modulating the mRNA expression of Pu.1 and Tnf. Interestingly, a recent study showed that TNF-α-driven p65-NF-κB signaling protects HSC from necroptosis and promotes myeloid priming (Yamashita and Passegué, 2019). By downregulating the TNF-α/NF-κB signaling pathway, it is possible that BMDM-IL-4-exo affect HSC necroptosis, which could in part explain our observed decrease in the number of MPP cells in the bone marrow. Furthermore, miR-99a has been described as a potent suppressor of HSC differentiation through HOXA1 (Khalaj et al., 2017), which might be another potential target of hematopoiesis control to explore in future studies. Alternatively, it is possible that macrophage exosomes target MPPs indirectly through the stem cell niche, which generates mobilizing cytokines and hormonal signals that regulate HSC self-renewal, quiescence, and differentiation (Pietras et al., 2015; Yamazaki et al., 2011). The spleen is a major site for extramedullary hematopoiesis in response to inflammation and calorie excess (Murphy et al., 2011; Robbins et al., 2012; Swirski et al., 2009). In the present study, we observed that BMDM-derived exosomes do not affect splenic hematopoiesis in Apoe−/− mice. This finding also fostered the idea that BMDM-IL-4-exo could target the bone marrow stem cell niche in addition to HSCs themselves or that splenic hematopoiesis may in fact be regulated through different mechanisms than bone marrow hematopoiesis.
As expected, on the basis of the control of hematopoietic progenitor cell expansion in Apoe−/− mice, the numbers of circulating neutrophils and monocytes of both subsets were decreased only by infusions of BMDM-IL-4-exo. Furthermore, we observed that beyond exerting a control over hematopoiesis and myelopoiesis, BMDM-IL-4-exo profoundly reprogramed Ly-6Chi monocytes toward an anti-inflammatory phenotype. BMDM-IL-4-exo increased the expression of NF-κB inhibitors, including Chuk and Ikbke, in circulating Ly-6Chi monocytes, suggesting control of NF-κB activity in these leukocytes (Hayden and Ghosh, 2004). Furthermore, we observed a modulation of genes implicated in the TNF-α (Tnfrsf1b), IL-1β (Slc7a5), and IL-10 (IL-10ra, Eif4ebp1) signaling pathways as well as those involved in monocyte and macrophage polarization (Fam129c, Lilrb4a) toward resolution of inflammation (Pros1) (Lumbroso et al., 2018). However, the relevance in the expression of those genes in monocytes remains poorly understood. Of note,
Irf3, which was downregulated by BMDM-IL-4-exo treatment, has been associated with a pro-atherogenic phenotype by increasing inflammation and the instability of atherosclerotic plaques. Furthermore, Irf3 deficiency in endothelial cells is associated with a reduction of ICAM-1 and VCAM-1 secretion and macrophage infiltration (Okon et al., 2017). Interestingly, we also observed that BMDM-IL-4-exo treatment decreased the expression of genes associated with monocyte activation (cd83, Flt4), mobilization, and recruitment to atherosclerotic vessels (Ccr7, Cxcl1, and Ccl2) (Soehnlein et al., 2013). Prior studies of Apoe−/− CCL2−/− mice showed reduced atherosclerosis with reduced macrophage accumulation in lesions (Combadière et al., 2008). Altogether, the reduced number of bone marrow progenitors, circulating myeloid cells, CCL2 plasma levels, and reduced plaque macrophage allow us to argue that BMDM-IL-4-exo could influence monocyte recruitment into plaques.
Beyond their benefit in limiting hematopoiesis, BMDM-IL-4-exo modulated the size of the necrotic core and the absolute number of macrophages in lesions, two important features that have been documented to enhance the stabilization of atheroma. The observed benefits could have occurred through direct exosomal reprograming of inflamed M1-like lesional macrophages to an M2-like phenotype. This possibility is supported by our observation of BMDM-IL-4-exo action on driving M2 polarity among peritoneal macrophages collected from these mice. Support for this possibility also come from our data demonstrating that lesion macrophages in BMDM-IL-4-exo infused Apoe−/− mice adopted M2-like features by displaying more pronounced staining for the M2 marker CD206. Therefore, newly alternatively activated lesional macrophages could have enhanced their capacity for the efferocytotic clearance of apoptotic cells and cellular debris contributing to the reduced necrotic cores observed in our histological specimens. It is noteworthy to mention that lesion stabilization via infusions of BMDM-IL-4-exo occurred despite sustained Western diet consumption and hyperlipidemia that is recognized to drive the process of hematopoiesis (Murphy et al., 2011). Thus, it is reasonable to suspect that BMDM-IL-4-exo could produce more profound lesion remodeling after shifting mice to chow diet to favor a plaque regression phenotype.
Finally, a source for the anti-inflammatory signaling by BMDM-exo was revealed by selectively deleting three candidate micro-RNAs found to be enriched in these exosomes. The loss of miR-99a-5p, miR-146b-5p, or miR-378–3p in exosomes produced by cultured macrophage exposed to IL-4 significantly impaired their capacity to reduce TNF-α expression and NF-κB signaling in recipient cultured macrophages. Such findings support that an intercellular communication of these three microRNAs by BMDM-exo is central to our observations. Further supporting this hypothesis, an upregulation of miR-99a in M2 macrophages has been shown to control TNF-α, which is implicated in M1 macrophage polarization (Jaiswal et al., 2019). Furthermore, miR-99a has been reported to control self-renewal by targeting HOXA1 (Khalaj et al., 2017).
Similarly, microRNA-146b has been recognized to suppress M1 pro-inflammatory activity, including by targeting the expression of the Toll-like receptor adaptor molecules IL-1R-associated kinase (IRAK1) and TNFR-associated factor (TRAF6), two key adaptor molecules in the TLR/NF-κB pathway that are known to contribute to atherosclerosis progression. Finally, microRNA-378a-3p was shown to be induced upon stimulation with IL-4 to control macrophage proliferation (Rückerl et al., 2012). Collectively, these data strongly support the idea that each one of these three microRNAs carried in BMDM-IL-4-exo participate in the regulation of inflammatory responses by targeting different gene sets that converge on controlling TNF-α/NF-κB signaling pathways.
The ability of exosomal-associated microRNA to mediate intercellular communication is still under debated because of stoichiometric analyses showing that on average, exosomes contain fewer than one copy of their most abundant microRNAs (Chevillet et al., 2014; Wei et al., 2017). However, multiple reports including our study have shown that exosomes carry and transfer microRNAs between cells, mediating target gene repression both in vitro and in vivo (Alexander et al., 2015; Squadrito et al., 2014; Wei et al., 2017). One hypothesis advanced by Chevillet et al. (2014) is the low-occupancy/high-microRNA concentration model, in which there are rare exosomes in the population carrying many copies of a given microRNA. In addition, massive and/or highly selective exosome uptake might be required to affect signaling in the recipient cells via their micro-RNA cargo. Therefore, our data suggest that BMDM-exo may be highly concentrated in miR-99a-5p, miR-146b-5p, or miR-378–3p and greatly target HSCs and myeloid cells to exert anti-inflammatory response. In addition, Squadrito et al. (2014) demonstrated that microRNA sorting to exosomes is controlled by the cellular levels of their targeted transcripts. This would suggest that miR-99a-5p, miR-146b-5p, or miR-378–3p is enriched in BMDM-IL-4-exo because of the reduction of their endogenous targets TNF-α and NF-κB signaling molecules in IL-4-stimulated BMDMs.
Finally, although our study focused on microRNAs, it is possible that the proteins, lipids, and other RNAs species of the BMDM-exo might affect cellular communication differently than microRNAs. Future studies will be required to fully understand the contribution of each component of the BMDM-exo in controlling inflammation. Results from our study and those of others showed that immune cell-derived EVs could be used to modulate and shape the immune system to a desired inhibitory or inflammatory setting (Veerman et al., 2019). In this study, we also demonstrated that modulating parental cell culture conditions can profoundly modulate exosomal cargo. Therefore, exosomes designed for drug delivery could be loaded with therapeutic cargo directly or by modulating parental cells with cargo precursors. However, there are still many challenges ahead, including a need for large-scale production of exosomes, appropriate cell types to use for exosome derivation, loading techniques, and determination of their potency and toxicology in vivo (Burnouf et al., 2019). Despite these challenges, exosomes remain an attractive new avenue for therapeutic applications, including in the cardiovascular system.
In conclusion, the present study demonstrates that exosomes produced by alternatively activated macrophages could represent a natural source of immune modulation to resolve inflammation. Furthermore, we have demonstrated that exosomes produced by IL-4-induced M2-like macrophages can serve to control inflammatory disorders including atherosclerosis in mice. Engineering the tissue targeting and microRNA cargo of such exosomes could in future studies result in new anti-inflammatory therapies in the cardiovascular system and beyond.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Robert L. Raffai (robert.raffai@ucsf.edu).
Materials Availability
BMDM-derived exosomes generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
The accession number for the RNA-seq data reported in this paper is EXRNA-KJENS1DIAB available within the exRNA atlas.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mice were housed and bred in specific pathogen–free conditions in the Animal Research Facility at the San Francisco Veterans Affairs Medical Center. All animal experiments were approved by the Institutional Animal Care and Use Committee at the VA Medical Center. Wild-type C57BL/6J and Apoetm1Unc/J (Apoe–/–) mice were purchased from Jackson Laboratory (Sacramento, CA). Male mice were used for both in vivo and in vitro experiments. Eight-week-old male Apoe–/– mice on C57BL/6 background were fed a western diet (Research Diets) for 17 weeks and then were infused intraperitoneally (IP) three times a week for 4 weeks with saline (PBS) or with 1 × 1010 BMDM-derived exosomes. Mice that appeared unhealthy (body-weight < 2 SD below average) or with skin inflammation were excluded.
Primary cells used in this study
Bone marrow-derived macrophage (BMDM) and hematopoietic stem cells (HSC) were derived from male C57BL/6J mice ranging from 8 to 25 weeks of age that in some instances were deficient in ApoE expression.
Human monocyte-derived macrophages were obtained from unidentified healthy male blood donors at the VA Medical Center San Francisco.
Immortalized murine bone marrow derived macrophage (iBMDM) cell line was reported by Covarrubias et al. (2017).
METHOD DETAILS
Cell culture
Murine BMDM were obtained as described previously (Bouchareychas et al., 2017). Briefly, bone marrow cells were flushed from the tibia and femurs of 8- to 12-week-old C57BL/6 mice. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (Corning) supplemented with 10% fetal bovine serum (GIBCO), 1% GlutaMax (GIBCO), and 1% penicillin-streptomycin (GIBCO) and differentiated with 25 ng/ml mouse M-CSF (Peprotech) for 6 days. Cell cultures used to isolate exosomes were grown in exosome-depleted media prepared by ultracentrifugation for 18 hours at 100,000 × g (Type 45 Ti rotor, Beckman Coulter) and filtration (0.2 μm). For exosomes isolation, BMDM were washed two times with PBS and cultured in EV-free media (EFM) for 24 hours with or without IL-4 (Peprotech) at 20 ng/ml. For in vitro experiments, BMDM were dispensed into 12-well culture plates (Corning) at a concentration of 3 × 105 cells/well and stimulated with BMDM–exo or BMDM–IL-4-exo for 24h at a concentration of 2 × 109 particles per ml.
Human blood was obtained from healthy donors and processed within 30 min after the collection. Ten milliliters of blood were collected by standard venipuncture into EDTA treated tubes and placed on ice. Each tube was diluted with an equal volume of PBS, and 15 mL of blood preparation were overlaid onto 10 mL of Ficoll-Paque Plus (GE Healthcare Systems). Tubes were centrifuged at 400 × g for 40 min, the plasma layer was removed, and the mononuclear cell layer was diluted with 3 volume of PBS and centrifuged at 400 × g for 10 min. The number of PBMC was determined using a TC20 automated cell counter (Bio-Rad). A positive selection of CD14+ cells was performed using CD14 MicroBeads (Miltenyi Biotec) according to the manufacturer instructions. The purity of CD14+ cells was evaluated at 94% using flow cytometry. CD14+ cells were cultured in DMEM medium supplemented with 10% FBS, 1% Penicillin-Streptomycin and 1% Glutamax (GIBCO) and differentiated with 50 ng/ml human M-CSF (Peprotech) for 7 days. For exosome stimulation experiments, monocyte-derived macrophages were dispensed into 12-well culture plates (Corning) at a concentration of 1 × 105 cells/well and stimulated with BMDM–exo or BMDM–IL-4-exo for 24h at a concentration of 2 × 109 particles per ml. HSC culture in vitro was performed as previously reported (Wilkinson et al., 2020). Suspensions of bone marrow (BM) cells from the femurs, tibiae, and iliac crest were stained and sorted following the gating strategy depicted in Figure 3A on a FACSAria IIU cell sorter (BD Biosciences). For qPCR analysis, fifty HSC (CD34− LSK) were sorted from 10-week-old C57BL/6 male mice and cultured into a 96-well fibronectin-coated plate (Corning) containing Ham’s F-12 Nutrient Mix liquid medium (GIBCO), 1% Insulin-Transferrin-Selenium-Ethanolamine, 1% Penicillin-Streptomycin-Glutamine, 10 mM HEPES (all from Life Technologies), 100 ng/ml of mouse TPO (Peprotech), 10 ng/ml of mouse SCF (Peprotech) and 0.1% polyvinyl alcohol (Sigma). After 3–4 days, HSC (300–500) were transferred into a 96 well-round bottom plate and stimulated with PBS, BMDM–exo or BMDM–IL-4-exo for 6 or 24 hours at a concentration of 2 × 109 particles per ml. For the CFUs assay, 2 × 104 total bone marrow cells were isolated from the legs and hips of 25-week-old male Apoe–/– mice and cultured in Methylcellulose-based medium with recombinant cytokine (StemCell Technologies). Cells were treated with PBS, BMDM–exo or BMDM–IL-4-exo every two days at a dose of 2 × 109 particles/ml. Number and morphology of the colonies were analyzed after 12 days of culture.
Murine iBMDMs were infected with an NF-κB reporter construct and clonally selected to maximize NF-κB-GFP induction. These cells were lentivirally infected with a Cas9 or CRISPRi construct and were selected with blasticidin for > 2 weeks to obtain Cas9-expressing cells (validated by GFP knock down). High activity clones were generated by limited dilution for both Cas9 and CRISPRi iBMDM–NF-κB cells. iBMDM were stimulated with LPS (100 ng/ml, Sigma-Aldrich) for 6 hours to induce NF-κB-GFP signaling. All cells were cultured at 37°C and 5% CO2.
Exosome isolation and nanoparticle tracking analysis
Exosomes were isolated from conditioned cell culture medium by Cushioned-Density Gradient Ultracentrifugation (C-DGUC) as previously described (Duong et al., 2019). Bone marrow cells were plated onto 150 mm round dishes at a density of 5 × 106 cells per dish and 1 × 106 cells per dish for iBMDM. Cell culture supernatant (20 ml) was harvested from an average of 20 plates with 5 × 106 cells per plate and an average viability of 95% for BMDM. iBMDM culture supernatant was harvested from 5 plates per condition with an average of 3.5 × 107 cells per plate and an average viability of 95%. The supernatant was centrifuged at 400 × g for 10 min at 4 °C to pellet dead cells and debris followed by centrifugation at 2000 × g for 20 min at 4 °C to eliminate debris and larges vesicles. The supernatant was then filtered (0.2 μm) and centrifuged on a 60% iodixanol cushion (Sigma-Aldrich) at 100,000 × g for 3 hours (Type 45 Ti, Beckman Coulter). OptiPrep density gradient (5%, 10%, 20% w/v iodixanol) was employed to further purify exosomes at 100 000 × g for 18 hours at 4°C (SW 40 Ti rotor). Afterward, twelve 1 mL fractions were collected starting from the top of the tube. Fraction 7 of the gradient was dialyzed in PBS with the Slide-A-Lyzer MINI Dialysis Device (Thermo Fisher Scientific) and used for subsequent experiments and analyses. Particles in Fraction 7 was subjected to size and concentration measurement by NanoSight LM14 (Malvern Instruments, Westborough, MA) and performed with a 488-nm detection wavelength. The analysis settings were optimized and kept identical for each sample, with a detection threshold set at 3, three video of 1min each were analyzed to give the mean, mode, median, and estimated concentration for each particle size. Samples were diluted in 1:100 or 1:200 PBS and measured in triplicates. Data were analyzed with the NTA 3.2 software. All the exosome samples were used fresh (less than a month after isolation) and store at 4°C. We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV200042) (EV-Track Consortium et al., 2017).
Exosome labeling
Fluorescently detectable BMDM and iBMDM-derived exosomes were generated using the lipid dye PKH26 (Sigma-Aldrich) or with DiR (DiIC18(7) (1,1’-Dioctadecyl-3,3,3′,3′ Tetramethylindotricarbocyanine Iodide) (Invitrogen) according to the manufacturer’s instructions. Briefly, PKH26 or DiR dye was added to the 3mL iodixanol cushion layer containing exosomes or to 3ml of PBS to achieve a final concentration of 3.5 μM for PKH26 and 1 μM for DiR and incubated for 5 min and 20 min respectively at room temperature. Next, we loaded the labeled cushion layer below an iodixanol step gradient as described in the Exosome Isolation section. Free dye and non-specific protein-associated dye was eliminated from labeled exosomes or from PBS control during this separation step. Fluorescence intensity of the PKH26+ cells was measured by using ImageJ. To quantify the delivery of exosomal miRNA cargo to target cells, BMDM-exosomes were transfected with synthetic miR-146b, labeled with IRDye® 800 (IDTDNA, Inc) as previously described with minor modifications (Manca et al., 2018). For transfection, 3 × 1011 exosomes (or an equivalent volume of PBS) were incubated with 1nmoles/ml of microRNA, 1mM calcium chloride and 40% ethanol in a volume of 4 ml for 15 min. Free dyes and extra-exosomal synthetic microRNAs were removed by two PBS washes (120,000 × g for 90 min), and 1 × 1010 exosomes were administered IP in 25-week-old Apoe–/– mice. DiR and IRDye® 800 fluorescence signals were visualized and quantified using the Odyssey Infrared Imaging System and Image Studio software.
Immunoblotting
Each fraction of the C-DGUC purified exosomes (37.5 μL sample) was mixed with 12.5 μL of4 × Laemmli buffer (Bio-Rad) and boiled at 95°C for 5 minutes. Samples were then loaded on a 10% SDS-PAGE gel and transferred onto PVDF membrane (Bio-Rad). The membranes were blocked with 5% non-fat milk dissolved in PBS for one hour and then were probed with primary antibodies overnight at 4°C (primary antibodies: anti-CD9 (1:100, Abcam), anti-Flotillin (1:500, Cell Signaling), anti-Alix (1:100, Santa Cruz Biotechnology), anti-Calnexin (1:500, Abcam), anti-GM130 (1:250, BD Biosciences), anti-IL-4 (1:150, Abcam). After 4 washes in PBS containing 0.1% Tween (PBST), membranes were incubated with corresponding HRP-conjugated secondary antibodies: anti-Mouse IgG-HRP (1:1000, Santa Cruz Biotechnology), anti-Rabbit IgG-HRP (1:1000, Thermo Fisher Scientific) or anti-Rat IgG-HRP (1:1000, Thermo Fisher Scientific) for 1h and washed in PBST. Signals were visualized after incubation with Amersham ECL Prime substrate and imaged using an ImageQuant LAS 4000.
Transmission electron microscopy
An assessment of exosome morphology was assessed by Electron microscopy by loading 7 × 108 exosomes onto a glow discharged 400 mesh Formvar-coated copper grid (Electron Microscopy Sciences). The nanoparticles were left to settle for two minutes, and the grids were washed four times with 1% Uranyl acetate. Excess Uranyl acetate was blotted off with filter paper. Grids were then allowed to dry and subsequently imaged at 120kV using a Tecnai 12 Transmission Electron Microscope (FEI).
RNA extraction and qRT-PCR
Exosome preparations were treated with 0.4 mg/ml of RNase A/T1 Mix (Thermo Fisher Scientific) for 20 min at 37°C before RNA extraction. Total RNA isolated from cells and exosomes was purified using the miRNeasy Mini Kit (QIAGEN) according to the manufacturer’s protocol. RNA was quantified using Nanodrop or Quant-iT RiboGreen RNA Assay Kit (Thermo Fisher Scientific) and reverse transcribed using the iScript Reverse Transcription Supermix (Bio-Rad) for mRNA or the miRCURY LNA RT Kit (QIAGEN) for microRNA analysis. PCR reactions were performed using the Fast SYBR Green Master Mix (Applied Biosystems) for mRNA or the miRCURY LNA SYBR Green PCR Kit (QIAGEN) for microRNA and run on a QuantStudio 7 Flex Real-Time PCR System. Ct values were normalized to the housekeeping gene Gapdh and B2m for mouse and GAPDH for human mRNA. For microRNA expression, U6 snRNA, miR-16–5p, miR-21–5p (QIAGEN) were used as reference genes. All reactions were done in triplicate. Primers used for qRT-PCR are listed in Table S1.
Transcriptional profiling of inflammatory gene expression in Ly-6Chi monocytes
Ly-6Chi monocytes were sorted from the blood of Apoe–/– mice infused IP, three times a week for 4 weeks with PBS or with 1 × 1010 BMDM-derived exosomes. RNA was isolated with miRNeasy Mini Kit (QIAGEN), quantified using Quant-iT RiboGreen RNA Assay Kit and 10ng of RNA was amplified using the nCounter Low RNA Input Amplification Kit before the RNA expression analysis with the nCounter analysis system (Nanostring Technologies). The mRNA detection procedure was performed according to the manufacturer’s instructions using the autoimmune profiling pathways panel. Number of mRNA molecules counted was imported into NSolver Analysis Software 4.0 (Nanostring Technologies) with default settings, corrected and normalized against the reference genes annotated in the kit that were found to be stable (Tubb5, Ppia, Cyc1, Hprt, G6pdx, Gusb, Polr2a, Oaz1, Sdha, Nmt1, Pgk1, Rpl4). Gene data with a ratio significantly different between PBS and BMDM–IL-4-exo were further imported into R version 15.6.0 to generate Heatmaps using the pheatmap package version 1.0.10. Expression levels are shown as z-scores of the normalized gene counts. The result was considered statistically significant when the p value is ≤ 0.05. Normalized data are presented in Table S2.
BIOO Small RNA library preparation and sequencing
Small RNA libraries were generated using NEXTflex Small RNA Library Prep Kit v3 following the manufacturer’s instructions with 16 cycles of PCR amplification and 50% adaptor. Following PCR amplification, libraries between 140 and 160bp in size were gel purified using 6% TBE gels followed by ethanol precipitation and resuspension in 11ul of ultra-pure water. Gel purified libraries were quantified using Agilent 2100 Bioanalyzer with High-Sense DNA chips. Equimolar amounts of libraries were pooled and quantified by Bioanalyzer with High-Sense DNA chips. Pooled libraries were normalized and denatured at a working concentration range of 6–8pM with 5% PhiX spike-in for flow cell cluster generation.
RNA-Sequencing data analysis
The raw sequence image files from the Illumina HiSeq in the form of bcl were converted to the fastq format using bcltofastq v. 2.19.1.403 and checked for quality to ensure the quality scores did not deteriorate at the read ends. The Bioo Scientific NEXTflex adapters and random 4bp ends were clipped from the reads using cutadapt v.1.14. Reads shorter than 15 nts were discarded and after adaptor trimming, the 3′ bases below a quality score of 30 were also trimmed. The reads are first mapped to a library of UniVec contaminants, a collection of common vector, adaptor, linker and PCR primer sequences collated by the NCBI. They are then mapped to mouse rRNA sequences obtained from NCBI. Next, reads are mapped to the mouse genome (GRCm38) and transcriptome, where the transcriptome contains all ensembl genes plus annotations for microRNAs, as obtained for miRBase (v.21). Differential expression was conducted using the DESeq2 package (version 1.20.0) in R (version 3.5.0) for all microRNAs. The raw read counts for the samples were normalized using the median ratio method (default in DESeq2). The significant differentially expressed microRNAs (by Benjamini-Hochberg adjusted p values) are reported in the paper. Heatmaps and Venn diagrams were created using the pheatmap (v.1.0.10) and VennDiagram (v.1.6.20) packages in R.
Primary cell preparation and purification
Mice were anesthetized with isoflurane (Forane, Baxter) and peripheral blood was collected by retro-orbital bleeding with heparinized micro-hematocrit capillary (Fisher Scientific) in tubes containing 0.5M EDTA. Red blood cells were lysed in RBC lysis buffer (BioLegend). Nonspecific binding was blocked with TruStain FcX Ab (BioLegend) for 10 min at 4°C in FACS buffer (Ca2+/Mg2+-free PBS with 2% FBS and 0.5 mM EDTA) before staining with appropriate Abs: CD11b (clone M1/70), Ly-6C (clone HK1.4), CD115 (clone AFS98) CD45 (clone 30-F11) (all BioLegend) for 30 min at 4°C. The antibody dilutions ranged from 1:200 to 1:100. Blood Ly-6Chi monocytes from Apoe–/– mice were sorted using a FACSAria IIU cell sorter (BD Biosciences) after 4 weeks of saline, BMDM–exo or BMDM–IL-4-exo treatment. Peritoneal cells were harvested by lavage with 10 ml cold PBS using a 16-G needle. Cells were then plated in 6-well plates and harvested 6h later. For macrophage enrichment, peritoneal single cell suspensions were magnetically labeled with anti-F4/80 microbeads UltraPure (Miltenyi Biotec) and then positively isolated using MACS LS column according to the manufacturer’s protocols. Bone marrow cells were collected by flushing bones with PBS. Cells were centrifuged at 300 × g, 5 min at 4°C, resuspended in red cell lysis buffer for 5 min and run through a 40 μm strainer. BM cells obtained from C57BL/6 mice or Apoe–/– mice were stained as previously described (Yamamoto et al., 2013) with a lineage-marker cocktail of biotinylated anti-CD4 (RM4–5), -CD8 (53–6.7), -B220/CD45RA (RA3–6B2), -TER-119 (TER-119), -Gr-1 (RB6–8C5), and -CD127 (IL-7Rα/A7R34) antibodies (all from BioLegend). These cells were then stained with anti-CD34 (RAM34, eBioscience), anti-CD150 (TC15–12F12.2, BioLegend), anti-CD48 (Invitrogen), anti-Sca-1 (D7, Invitrogen) anti-CD135 (A2F10, Invitrogen) anti-c-Kit (2B8, Life Technologies), anti-CD16/32 (93, BioLegend), anti-CD41 (MWReg30, BioLegend) and streptavidin-BV786 (BD Biosciences) to detect biotinylated antibodies. Sca1+ c-Kit+ (LSK) Flt3–CD34– was used as HSC-gating parameters. MPP1 population was defined as CD34+ LSK CD150+ CD48– cells, MPP2 as CD34+ LSK CD150+ CD48+ cells, MPP3 as CD34+ LSK CD150– CD48+ cells and MPP4 as CD34+ LSK Flt3+ CD150– CD48+ cells. Common myeloid progenitors (CMPs) was gated as Sca-1– c-Kit+ CD41– CD34+ CD16/32–. The granulocyte/macrophage progenitors (GMPs) was defined as Sca-1– c-Kit+ CD41– CD34+ CD16/32+ and megakaryocyte/erythrocyte progenitors (MEPs) as Sca-1– c-Kit+ CD41– CD34– CD16/32–. Doublets, dead cells, and Lin– cells were excluded prior gating analysis.
For the detection of intracellular TNFα, the cells were pretreated with Golgiplug (1ug/ml) and iBMDM–exo for 6 h. Then, the cells were harvested, fixed and permeabilized according to the manufacturer’s instructions using Fixation/Permeabilization Solution Kit (BD Biosciences) before staining with APC-conjugated rat anti-mouse TNFα antibody (MP6-XT22, BD Biosciences). Absolute numbers of cells were calculated using CountBright Absolute Counting Beads (Thermo Fisher Scientific). Data were acquired on an LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo v10.0.7 (Tree Star, Inc.).
Seahorse extracellular flux analysis
BMDM were plated at 50 000 cells / well into XFe24 cell culture microplates (Agilent) and incubated overnight at 37°C and 5% CO2. The following day, BMDM were stimulated with PBS, BMDM–exo or BMDM–IL-4-exo for 24 hours. Before analysis, cells were washed with Seahorse XF assay buffer (Agilent) supplemented with 10 mM glucose (Sigma-Aldrich), 1 mM pyruvate (Agilent) and 2 mM glutamine (GIBCO) and incubated for 1 h at 37°C without CO2. OCR and ECAR were measured using the mitochondrial stress test kit (Agilent) in response to 1 μM Oligomycin, 2 μM Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and 0.5 μM Rotenon/Antimycin A (R/AA) with the Seahorse XFe-24 Bioanalyzer (Agilent) and were analyzed using XFe Wave software. All OCR measurements were normalized to cell number and used to calculate various mitochondrial parameters, including basal mitochondrial OCR, proton leak, ATP synthesis, maximum OCR and spare respiratory capacity.
Histological quantification of atherosclerosis
Aortic root sections were stained and quantified as previously described (Bouchareychas et al., 2015). Beginning at the base of the aortic root, 120 sections per mice were cut at 10 μm, and arranged in 4 sections per slide. Atherosclerotic lesions in the aortic root were quantified by staining with oil red O (Sigma-Aldrich) to reveal neutral lipids in 20 cross-sections, 50 μm apart starting at the coronary ostium and extending through the base of the aortic valve. All slides were counterstained with modified Mayer’s hematoxylin (Thermo Fisher Scientific). Lesion area was quantified by averaging six sections that were spaced 50 mm apart, starting from the base of the aortic root. Plaque necrosis was quantified by measuring the area of DAPI negative acellular areas in the intima, as described previously (Bouchareychas et al., 2015). For macrophage staining, sections were labeled with a primary rat anti-mouse MOMA-2 antibody (Cedarlane labs, NC) and detected with an Alexa Fluor 488 anti-rat IgG (H+L) antibody (Life Technologies). For the detection of the M2 macrophage marker CD206, the adjacent frozen sections were fixed and permeabilized before incubation with BLOXALL blocking solution (Vector Laboratories) to inhibit endogenous peroxidase activity. The sections were then incubated with 5% normal rabbit serum to inhibit non-specific binding for 2 hours. Sections were incubated with a rat anti-mouse CD206 antibody (MR5D3, Bio-Rad) at 4°C overnight and then incubated with the rabbit anti-rat biotinylated secondary antibody (1:500) at 37°C for 60 min and with the VECTASTAIN Elite ABC reagent (Vector Laboratories). The labeling of the CD206 macrophages were visualized using peroxidase substrate solution (Vector Laboratories). Sections were counterstain with modified Mayer’s hematoxylin (Thermo Fisher Scientific). Positive labeling (red) was defined by the application of a color threshold mask, and the same threshold was applied to all sections. The results are expressed as a ratio of CD206 positive area / macrophage positive area. Images were captured with a Nikon Eclipse Ni microscope and analyzed using NIS-Elements Software and ImageJ. Analyses were performed in a blinded manner.
CRISPR/Cas9-Mediated Targeted Deletion and lentivirus generation
Guide RNAs targeting miR-99a-5p, miR-146b-5p and miR-378a-3p were designed (2 guides per microRNA) using the UCSC Genome Browser CRISPR track, targeting the 5′ arm for miR-99a-5p and miR-146b-5p and targeting the 3′arm for miR-378a-3p. The primers used in plasmid construction are listed in Table S1. The forward/reverse oligos corresponding to each guide were annealed and cloned via the AaRI site of a gRNA vector, which also contained an EF1a promoter driving puro-T2A-cherry (Covarrubias et al., 2017). Virus were obtained through the UCSF ViraCore. Briefly, HEK293T cells were seeded at 70,000 cells per cm2 in 15 cm tissue culture dishes in 20 mL media (DMEM, 10% FBS) and incubated overnight at 37°C, 5% CO2. Twenty-four hours after plating, 12 μg of lentiviral transfer vector was transfected alongside 7 μg psPAX2 (Addgene), and 3 μg pMD2.G (Addgene) with 50 μL jet-PRIME transfection reagent (Polyplus) according to manufacturer’s protocol. Seventy-two hours post-transfection, lentiviral supernatant was collected, passed through 0.45 μm filters (Millipore) and aliquots were stored at −80°C. Filtered lentiviral supernatant was concentrated by ultracentrifugation, 90,000 × g for 120 min, on a Beckman L8–80M ultracentrifuge and resuspended in 100 μL of sterile PBS. Lentivirus supernatant was used (MOI = 0.3) to transduce 1 × 105 iBMDM–NF-κB-Cas9 cells. Two days post transduction, puromycin (5ug/ml) was added to select for gRNA-expressing cells (cherry positive).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism v7 or v8, and the tests used were unpaired, two-tailed, Student’s t test (two groups) and one-way or two-way analysis of variance (ANOVA) with post-tests as indicated in figure legends for multiple groups. *p < 0.05, **p < 0.01, ***p < 0.001 ****p < 0.0001. All error bars represent the mean ± the standard error of the mean (SEM unless stated). All experiments were repeated at least twice or performed with independent samples.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Immunoblotting: Rabbit monoclonal anti-CD9 | Abcam | Cat# ab92726; RRID: AB_10561589 |
| Immunoblotting: Rabbit polyclonal anti-Calnexin | Abcam | Cat# ab10286; RRID: AB_2069009 |
| Immunoblotting: Mouse monoclonal anti-GM130 | BD Biosciences | Cat# 610823; RRID: AB_398142 |
| Immunoblotting: Rabbit monoclonal anti-Flotillin-1 | Cell Signaling Technology | Cat# 18634; RRID: AB_2773040 |
| Immunoblotting: Mouse monoclonal anti-Alix | Santa Cruz Biotechnology | Cat# sc-53540; RRID: AB_673819 |
| Immunoblotting: Rat monoclonal anti-IL-4 | Abcam | Cat# ab11524; RRID: AB_298129 |
| Immunoblotting: rabbit anti-Rat IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat# 61–9520; RRID: AB_2533945 |
| Immunoblotting: mouse IgG kappa binding protein (m-IgGκ BP)-HRP | Santa Cruz Biotechnology | Cat# sc-516102; RRID: AB_2687626 |
| Immunoblotting: F(ab)2-Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, HRP | Thermo Fisher Scientific | Cat# A10547; RRID: AB_2534046 |
| Flow Cytometry: Biotin anti-mouse/human CD45R/B220 antibody | BioLegend | Cat# 103204; RRID: AB_312989 |
| Flow Cytometry: Biotin anti-mouse CD4 antibody | Biolegend | Cat# 100508; RRID: AB_312711 |
| Flow Cytometry: Biotin anti-mouse CD8a antibody | Biolegend | Cat# 100704; RRID: AB_312743 |
| Flow Cytometry: Brilliant Violet 421 anti-mouse CD16/32 antibody | Biolegend | Cat# 101332; RRID: AB_2650889 |
| Flow Cytometry: Biotin anti-mouse Ly-6G/Ly-6C (Gr-1) antibody | Biolegend | Cat# 108404; RRID: AB_313369 |
| Flow Cytometry: PE/Cy7 anti-mouse CD150 (SLAM) antibody | Biolegend | Cat# 115914; RRID: AB_439797 |
| Flow Cytometry: Biotin anti-mouse TER-119/Erythroid Cells antibody | Biolegend | Cat# 116204; RRID: AB_313705 |
| Flow Cytometry: Brilliant Violet 510 anti-mouse CD41 antibody | Biolegend | Cat# 133923; RRID: AB_2564013 |
| Flow Cytometry: Biotin anti-mouse CD127 (IL-7Rα) antibody | Biolegend | Cat# 135006; RRID: AB_2126118 |
| Flow Cytometry: PE anti-mouse CD115 (CSF-1R) antibody | Biolegend | Cat# 135505; RRID: AB_1937254 |
| Flow Cytometry: FITC anti-mouse Ly-6C antibody | Biolegend | Cat# 128006; RRID: AB_1186135 |
| Flow Cytometry: PerCP/Cyanine5.5 anti-mouse/human CD11b antibody | Biolegend | Cat# 101228; RRID: AB_893232 |
| Flow Cytometry: APC anti-mouse CD45 antibody | Biolegend | Cat# 103112; RRID: AB_312977 |
| Flow Cytometry: c-Kit Monoclonal Antibody (2B8), APC-Cyanine7 | Thermo Fisher Scientific | Cat# A15423; RRID: AB_2534436 |
| Flow Cytometry: CD34 Monoclonal Antibody (RAM34), FITC | Thermo Fisher Scientific | Cat# 11-0341-85; RRID: AB_465022 |
| Flow Cytometry: Ly-6A/E (Sca-1) Monoclonal Antibody (D7), PE | Thermo Fisher Scientific | Cat# 12-5981-83; RRID: AB_466087 |
| Flow Cytometry: CD48 Monoclonal Antibody (HM48-1), APC | Thermo Fisher Scientific | Cat# 17-0481-82; RRID: AB_469408 |
| Flow Cytometry: CD135 (Flt3) Monoclonal Antibody (A2F10), PerCP-eFluor 710 | Thermo Fisher Scientific | Cat# 46-1351-82; RRID: AB_10733393 |
| Flow Cytometry: APC Rat Anti-Mouse TNF (MP6-XT22) | BD Biosciences | Cat# 554420; RRID: AB_398553 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21208; RRID: AB_2535794 |
| Anti-Mouse Macrophages / Monocytes Purified (Clone MOMA-2) (rat IgG2b) | Cedarlane | Cat# CL89154; RRID: AB_10086520 |
| Rat anti-mouse CD206 | Bio-Rad Laboratories | Cat# MCA2235; RRID: AB_324622 |
| TruStain FcX Antibody | Biolegend | Cat# 101320; RRID: AB_1574975 |
| BV786 Streptavidin | BD Biosciences | Cat# 563858 |
| CD14 MicroBeads, human | Miltenyi Biotec | Cat#130-118-906; RRID: AB_2665482 |
| Anti-F4/80 MicroBeads, mouse | Miltenyi Biotec | Cat#130-110444 |
| Anti-APC MicroBeads, mouse | Miltenyi Biotec | Cat# 130-090-855; RRID: AB_244367 |
| Chemicals, Peptides and Recombinant Proteins | ||
| DiR (DiIC18(7) (1,1’-Dioctadecyl-3,3,3′,3′- Tetramethylindotricarbocyanine Iodide)) |
Invitrogen | Cat# D12731 |
| Fast SYBR Green Master Mix | Applied Biosystems | Cat# 4385614 |
| RBC Lysis Buffer (10X) | BioLegend | Cat# 420301 |
| iScript Reverse Transcription Supermix | Bio-Rad Laboratories | Cat# 1708841 |
| 10% Mini-PROTEAN TGX Gels | Bio-Rad Laboratories | Cat# 4561034 |
| 4x Laemmli Sample Buffer | Bio-Rad Laboratories | Cat# 1610747 |
| RIPA buffer (10x) | Cell Signaling Technology | Cat# 9806 |
| Amersham ECL Prime Western Blotting Detection Reagent | GE Healthcare | Cat# RPN2232 |
| Penicillin-Streptomycin | GIBCO | Cat# 15140122 |
| L-Glutamine | GIBCO | Cat# 25030-081 |
| Recombinant Murine IL-4 | Peprotech | Cat# 214-14 |
| Recombinant Murine IL-4 | Abcam | Cat# ab9729 |
| Recombinant Murine M-CSF | Peprotech | Cat# 315-02 |
| Recombinant Human M-CSF | Peprotech | Cat# 300–25 |
| Animal-Free Recombinant Murine TPO | Peprotech | Cat# AF-315-14 |
| Animal-Free Recombinant Murine SCF | Peprotech | Cat# AF-250-03 |
| Poly (vinyl alcohol) | Sigma-Aldrich | Cat# P8136 |
| miRCURY LNA RT Kit | QIAGEN | Cat# 339340 |
| miRCURY LNA SYBR Green PCR Kit | QIAGEN | Cat# 339347 |
| RD Western Diet | Research Diets | Cat# D12079B |
| OptiPrep density gradient medium | Sigma-Aldrich | Cat# D1556-250ML |
| Oil Red O | Sigma-Aldrich | Cat# O1391 |
| Mayer’s Hematoxylin | Thermo Fisher Scientific | Cat# 72804 |
| BLOXALL Blocking Solution | Vector Laboratories | Cat# SP-6000 |
| ImmPACT NovaRED Peroxidase (HRP) Substrate | Vector Laboratories | Cat# SK-4805 |
| D-Glucose solution | Sigma-Aldrich | Cat# G8769-100ML |
| Lipopolysaccharides from Escherichia coli O55:B5 | Sigma-Aldrich | Cat# L2880 |
| Tissue-Tek O.C.T Compound | Sakura FineTek | Cat# 4583 |
| Sucrose, 20% Sterile Solution | VWR | Cat# E543-100ML |
| 10X Tris-EDTA, pH 7.4 | Fisher Scientific | Cat# BP24771 |
| CountBright Absolute Counting Beads | Thermo Fisher Scientific | Cat# C36950 |
| RNase A/T1 Mix | Thermo Fisher Scientific | Cat# EN0551 |
| jetPRIME transfection reagent | Polyplus | Cat# 114-07 |
| VECTASHIELD Antifade Mounting Medium with DAPI | Vector Laboratories | Cat# H-1200 |
| Seahorse XF base medium | Agilent | Cat# 103335-100 |
| Seahorse XF 100 mM pyruvate solution | Agilent | Cat# 103578–100 |
| Novex 6% TBE Gels | Thermo Fisher | Cat# EC6265BOX |
| TruSeq SR Cluster Kit v3 - cBot - HS | Illumina | Cat# GD-401-3001 |
| TruSeq SBS Kit v3 - HS | Illumina | Cat# FC-401-3002 |
| T4 DNA Ligase | NEB | Cat# M0202 |
| Trypsin-EDTA (0.05%) | GIBCO | Cat# 25300054 |
| Opti-MEM | GIBCO | Cat# 31985–062 |
| Glutamax | GIBCO | Cat# 35050061 |
| Ficoll-Plaque Plus | GE Healthcare | Cat#17-1440-02 |
| Critical Commercial Assays | ||
| miRNeasy Mini Kit | QIAGEN | Cat# 217004 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Quant-iT RiboGreen RNA Assay Kit | Thermo Fisher Scientific | Cat# R11490 |
| Slide-A-Lyzer MINI Dialysis Device, 10K MWCO | Thermo Fisher Scientific | Cat# 88404 |
| LS Columns | Miltenyi Biotec | Cat# 130-042-401 |
| Subcloning Efficiency DH5a Competent Cells | Thermo Fisher Scientific | Cat# 18265017 |
| Zyppy Plasmid Miniprep | Zymo Research | Cat# D4036 |
| Seahorse XFe24 FluxPaks | Agilent | Cat# 102340-100 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat# 103015-100 |
| BioAnalyzer High Sensitivity DNA Analysis | Agilent | Cat# 5067-4626 |
| NEXTFLEX Small RNA Sequencing kit for Illumina Platforms | Perkin Elmer | Cat# NOVA-5132-06 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat# 554714 |
| Qubit Protein Assay Kit | Thermo Fisher Scientific | Cat# Q33211 |
| PKH26 Red Fluorescent cell Linker kit | Sigma | Cat# PKH26GL-1KT |
| Exosome Spin Columns | Invitrogen | Cat# 448449 |
| Seahorse XFe24 Cell Culture Microplates | Agilent | Cat# 100777-004 |
| Vectastain Elite ABC Kit, Peroxidase (Rat IgG) | Vector Laboratories | Cat# PK-6104 |
| nCounter® Low RNA Input Kit | Nanostring Technologies | Cat# LOW-RNA-48 |
| Autoimmune Profiling Panel | Nanostring Technologies | Cat# XT-CSO-MAIPI1-12 |
| V-Plex Mouse Custom Cytokine Kit MCP-1 | Meso Scale Discovery | Cat# K152A0H-1 |
| MethoCult GF | StemCell Technologies | Cat#GF M3434 |
| Deposited Data | ||
| miRNA-Seq | This paper | exRNA atlas: EXRNA-KJENS1DIAB, https://exrna-atlas.org/exat/datasets/EXR-KJENS1Lz9cll-AN |
| Experimental Models: Cell Lines | ||
| iBMDM-NFKB-Cas9 Cells | Covarrubias et al., 2017 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129P2-Apoetm1Unc/J | Jackson Laboratories | JAX:002052 |
| Mouse: C57BL6/J | Jackson Laboratories | JAX:000664 |
| Oligonucleotides | ||
| hsa-miR-146b-5p miRCURY LNA miRNA PCR Assay | QIAGEN | Cat# YP00204553 |
| hsa-miR-16-5p miRCURY LNA miRNA PCR Assay | QIAGEN | Cat# YP00205702 |
| hsa-miR-21-5p miRCURY LNA miRNA PCR Assay | QIAGEN | Cat# YP00204230 |
| hsa-miR-99a-5p miRCURY LNA miRNA PCR Assay | QIAGEN | Cat# YP00204521 |
| mmu-miR-378a-3p miRCURY LNA miRNA PCR Assay | QIAGEN | Cat# YP00204179 |
| U6 snRNA (hsa, mmu) miRCURY LNA miRNA PCR Assay | QIAGEN | Cat# YP00203907 |
| UniSp6 miRCURY LNA miRNA PCR Assay | QIAGEN | Cat# YP00203954 |
| 5′-IRD 800-labeled miR-146b | IDT | N/A |
| Primers for qRT-PCR and plasmid construction: see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| psPAX2 | Addgene | RRID: Addgene_12260 |
| pMD2.G | Addgene | RRID: Addgene_12259 |
| Software and Algorithms | ||
| FlowJo | FlowJo | https://www.flowjo.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Photoshop CC | Adobe | https://www.adobe.com/products/photoshop.html |
| Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| 2100 Expert Bioanalyzer | Agilent | https://www.agilent.com/en/product/automated-electrophoresis/bioanalyzer-systems/bioanalyzer-software/2100-expert-software-228259 |
| XFe Wave software | Agilent | https://www.agilent.com/en/products/cell-analysis/cell-analysis-software/data-analysis/wave-desktop-2–6 |
| NTA 3.2 | Malvern Panalytical | https://www.malvernpanalytical.com/en/support/product-support/software/NanoSight-NTA-software-update-v3–2 |
| NIS Elements BR 4.3 | Nikon | https://www.nikon.com/products/microscope-solutions/support/download/software/imgsfw/nis-br_v4300164.htm |
| nSolver Analysis Software | Nanostring Technologies | https://www.nanostring.com/products/analysis-software/nsolver |
Highlights.
M2 macrophage exosomes reprogram inflammation and energy metabolism in recipient cells
Specific miRNAs enriched in M2 macrophage exosomes reduce TNF-α and NF-κB signaling
M2 exosomes control atherosclerosis by reducing hematopoiesis and myeloid inflammation
ACKNOWLEDGMENTS
We thank the staff at the University of California, Berkeley, Electron Microscope Laboratory for advice and assistance with electron microscopy sample preparation and imaging. We also thank the Parnassus Flow Cytometry Core for assistance, which is in part supported by DRC Center Grant P30 DK063720 from the National Institutes of Health (NIH), and the University of California, San Francisco (UCSF), and Veterans Affairs Medical Center (VAMC) skeletal core facility, supported by NIH grant P30 AR066262. This work was supported by grants from the NIH, including from the Extracellular RNA Communication Consortium Common Fund (grants U19CA179512 and U19CA179513 to R.L.R. and M.T.M.), and grant R01HL133575 to R.L.R.; U.S. Department of Veterans Affairs Merit Award grant 1I01BX003928-01A2 to R.L.R.; and American Heart Association Grant-in-Aid 16GRNT27640007 to R.L.R. L.B. was supported by a postdoctoral fellowship from the American Heart Association (19POST34380672).
Footnotes
DECLARATION OF INTERESTS
L.B. and R.L.R. have filed an invention disclosure related to some aspects of this work with the Department of Veterans Affairs.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107881.
REFERENCES
- Alexander M, Hu R, Runtsch MC, Kagele DA, Mosbruger TL, Tolmachova T, Seabra MC, Round JL, Ward DM, and O’Connell RM (2015). Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat. Commun. 6, 7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäck M, Yurdagul A Jr., Tabas I, Öörni K, and Kovanen PT (2019). Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat. Rev. Cardiol. 16, 389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar S, Shinagawa K, Castellino FJ, and Schorey JS (2007). Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood 110, 3234–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgaard LS, Mogensen CK, Rosendahl A, Cucak H, Nielsen LB, Rasmussen SE, and Pedersen TX (2016). Bone marrow-derived and peritoneal macrophages have different inflammatory response to oxLDL and M1/M2 marker expression - implications for atherosclerosis research. Sci. Rep. 6, 35234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boilard E. (2018). Extracellular vesicles and their content in bioactive lipid mediators: more than a sack of microRNA. J. Lipid Res. 59, 2037–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchareychas L, Pirault J, Saint-Charles F, Deswaerte V, Le Roy T, Jessup W, Giral P, Le Goff W, Huby T, Gautier EL, and Lesnik P. (2015). Promoting macrophage survival delays progression of pre-existing atherosclerotic lesions through macrophage-derived apoE. Cardiovasc. Res. 108, 111–123. [DOI] [PubMed] [Google Scholar]
- Bouchareychas L, Grössinger EM, Kang M, Qiu H, and Adamopoulos IE (2017). Critical role of LTB4/BLT1 in IL-23-induced synovial inflammation and osteoclastogenesis via NF-κB. J. Immunol. 198, 452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnouf T, Agrahari V, and Agrahari V. (2019). Extracellular vesicles as nanomedicine: hopes and hurdles in clinical translation. Int. J. Nanomedicine 14, 8847–8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Yi B, Ma R, Zhang X, Zhao H, and Xi Y. (2016). CRISPR/Cas9, a novel genomic tool to knock down microRNA in vitro and in vivo. Sci. Rep. 6, 22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevillet JR, Kang Q, Ruf IK, Briggs HA, Vojtech LN, Hughes SM, Cheng HH, Arroyo JD, Meredith EK, Gallichotte EN, et al. (2014). Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. U S A 111, 14888–14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, and Zernecke A. (2018). Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res. 122, 1661–1674. [DOI] [PubMed] [Google Scholar]
- Colombo M, Raposo G, and Théry C. (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 30, 255–289. [DOI] [PubMed] [Google Scholar]
- Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, and Mallat Z. (2008). Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117, 1649–1657. [DOI] [PubMed] [Google Scholar]
- EV-Track Consortium; Van Deun J, Mestdagh P, Agostinis P, Akay O, Anand S, Anckaert J, Martinez ZA, Baetens T, Beghein E, et al. (2017). EV-TRACK: transparent reporting and centralizing knowledge in extra-cellular vesicle research. Nat. Methods 14, 228–232. [DOI] [PubMed] [Google Scholar]
- Covarrubias S, Robinson EK, Shapleigh B, Vollmers A, Katzman S, Hanley N, Fong N, McManus MT, and Carpenter S. (2017). CRISPR/Cas-based screening of long non-coding RNAs (lncRNAs) in macrophages with an NF-κB reporter. J. Biol. Chem. 292, 20911–20920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, and Halushka MK (2015). Extracellular vesicle microRNA transfer in cardiovascular disease. Cardiovasc. Pathol. 24, 199–206. [DOI] [PubMed] [Google Scholar]
- Duong P, Chung A, Bouchareychas L, and Raffai RL (2019). Cushioned-density gradient ultracentrifugation (C-DGUC) improves the isolation efficiency of extracellular vesicles. PLoS ONE 14, e0215324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig JE, Parathath S, Rong JX, Mick SL, Vengrenyuk Y, Grauer L, Young SG, and Fisher EA (2011). Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation 123, 989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galván-Peña S, and O’Neill LA (2014). Metabolic reprograming in macrophage polarization. Front. Immunol. 5, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geovanini GR, and Libby P. (2018). Atherosclerosis and inflammation: overview and updates. Clin. Sci. (Lond.) 132, 1243–1252. [DOI] [PubMed] [Google Scholar]
- Glass CK, and Witztum JL (2001). Atherosclerosis. the road ahead. Cell 104, 503–516. [DOI] [PubMed] [Google Scholar]
- Gschwandtner M, Derler R, and Midwood KS (2019). More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front. Immunol. 10, 2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, and Rollins BJ (1998). Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell 2, 275–281. [DOI] [PubMed] [Google Scholar]
- Hayden MS, and Ghosh S. (2004). Signaling to NF-kappaB. Genes Dev. 18, 2195–2224. [DOI] [PubMed] [Google Scholar]
- Hayden MS, and Ghosh S. (2014). Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 26, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergenreider E, Heydt S, Tréguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, et al. (2012). Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 14, 249–256. [DOI] [PubMed] [Google Scholar]
- Hoyer FF, Giesen MK, Nunes França C, Lütjohann D, Nickenig G, and Werner N. (2012). Monocytic microparticles promote atherogenesis by modulating inflammatory cells in mice. J. Cell. Mol. Med. 16, 2777–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail N, Wang Y, Dakhlallah D, Moldovan L, Agarwal K, Batte K, Shah P, Wisler J, Eubank TD, Tridandapani S, et al. (2013). Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood 121, 984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal A, Reddy SS, Maurya M, Maurya P, and Barthwal MK (2019). MicroRNA-99a mimics inhibit M1 macrophage phenotype and adipose tissue inflammation by targeting TNFα. Cell. Mol. Immunol. 16, 495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, Liebler DC, Ping J, Liu Q, Evans R, et al. (2019). Reassessment of exosome composition. Cell 177, 428–445.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalaj M, Woolthuis CM, Hu W, Durham BH, Chu SH, Qamar S, Armstrong SA, and Park CY (2017). miR-99 regulates normal and malignant hematopoietic stem cell self-renewal. J. Exp. Med. 214, 2453–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, and Théry C. (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. U S A 113, E968–E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumbroso D, Soboh S, Maimon A, Schif-Zuck S, Ariel A, and Burstyn-Cohen T. (2018). Macrophage-derived protein S facilitates apoptotic polymorphonuclear cell clearance by resolution phase macrophages and supports their reprogramming. Front. Immunol. 9, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca S, Upadhyaya B, Mutai E, Desaulniers AT, Cederberg RA, White BR, and Zempleni J. (2018). Milk exosomes are bioavailable and distinct microRNA cargos have unique tissue distribution patterns. Sci. Rep. 8, 11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu M, Martin-Jaular L, Lavieu G, and Thé ry C. (2019). Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 21, 9–17. [DOI] [PubMed] [Google Scholar]
- McDonald MK, Tian Y, Qureshi RA, Gormley M, Ertel A, Gao R, Aradillas Lopez E, Alexander GM, Sacan A, Fortina P, and Ajit SK (2014). Functional significance of macrophage-derived exosomes in inflammation and pain. Pain 155, 1527–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Sheedy FJ, and Fisher EA (2013). Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 13, 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, et al. (2011). ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Invest. 121, 4138–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MA, Karunakaran D, Geoffrion M, Cheng HS, Tandoc K, Perisic Matic L, Hedin U, Maegdefessel L, Fish JE, and Rayner KJ (2018). Extracellular Vesicles Secreted by Atherogenic Macrophages Transfer MicroRNA to Inhibit Cell Migration. Arterioscler. Thromb. Vasc. Biol. 38, 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njock MS, Cheng HS, Dang LT, Nazari-Jahantigh M, Lau AC, Boudreau E, Roufaiel M, Cybulsky MI, Schober A, and Fish JE (2015). Endothelial cells suppress monocyte activation through secretion of extracellular vesicles containing antiinflammatory microRNAs. Blood 125, 3202–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okon I, Ding Y, and Zou MH (2017). Ablation of interferon regulatory factor 3 promotes the stability of atherosclerotic plaques. Hypertension 69, 407–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Reynaud D, Kang YA, Carlin D, Calero-Nieto FJ, Leavitt AD, Stuart JM, Göttgens B, and Passegué E. (2015). Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell 17, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman K, and Fisher EA (2018). Insights from pre-clinical and clinical studies on the role of innate inflammation in atherosclerosis regression. Front. Cardiovasc. Med. 5, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, et al. (2017). Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J. Clin. Invest. 127, 2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. ; CANTOS Trial Group (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Robbins PD, and Morelli AE (2014). Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 14, 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, Etzrodt M, Weber GF, Ueno T, van Rooijen N, et al. (2012). Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation 125, 364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rückerl D, Jenkins SJ, Laqtom NN, Gallagher IJ, Sutherland TE, Duncan S, Buck AH, and Allen JE (2012). Induction of IL-4Rα-dependent microRNAs identifies PI3K/Akt signaling as essential for IL-4-driven murine macrophage proliferation in vivo. Blood 120, 2307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EW, Simon MC, Anastasi J, and Singh H. (1994). Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 265, 1573–1577. [DOI] [PubMed] [Google Scholar]
- Sica A, and Mantovani A. (2012). Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PP, Smith VL, Karakousis PC, and Schorey JS (2012). Exosomes isolated from mycobacteria-infected mice or cultured macrophages can recruit and activate immune cells in vitro and in vivo. J. Immunol. 189, 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soehnlein O, Drechsler M, Döring Y, Lievens D, Hartwig H, Kemmerich K, Ortega-Gómez A, Mandl M, Vijayan S, Projahn D, et al. (2013). Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol. Med. 5, 471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squadrito ML, Baer C, Burdet F, Maderna C, Gilfillan GD, Lyle R, Ibberson M, and De Palma M. (2014). Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep. 8, 1432–1446. [DOI] [PubMed] [Google Scholar]
- Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, and Pittet MJ (2007). Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest. 117, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, et al. (2009). Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325, 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I. (2005). Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler. Thromb. Vasc. Biol. 25, 2255–2264. [DOI] [PubMed] [Google Scholar]
- Tedgui A, and Mallat Z. (2006). Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol. Rev. 86, 515–581. [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, and Lötvall JO. (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659. [DOI] [PubMed] [Google Scholar]
- Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, Luque-Martin R, Chen HJ, Boshuizen MC, Ahmed M, et al. (2016). Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 17, 684–696. [DOI] [PubMed] [Google Scholar]
- van Niel G, D’Angelo G, and Raposo G. (2018). Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228. [DOI] [PubMed] [Google Scholar]
- Veerman RE, Güçlüler Akpinar G, Eldh M, and Gabrielsson S. (2019). Immune cell-derived extracellular vesicles—functions and therapeutic applications. Trends Mol. Med. 25, 382–394. [DOI] [PubMed] [Google Scholar]
- Wei Z, Batagov AO, Schinelli S, Wang J, Wang Y, El Fatimy R, Rabinovsky R, Balaj L, Chen CC, Hochberg F, et al. (2017). Coding and non-coding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun. 8, 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen S, Dooner M, Cheng Y, Papa E, Del Tatto M, Pereira M, Deng Y, Goldberg L, Aliotta J, Chatterjee D, et al. (2016). Mesenchymal stromal cell-derived extracellular vesicles rescue radiation damage to murine marrow hematopoietic cells. Leukemia 30, 2221–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiklander OP, Nordin JZ, O’Loughlin A, Gustafsson Y, Corso G, Mäger I, Vader P, Lee Y, Sork H, Seow Y, et al. (2015). Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 4, 26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson AC, Ishida R, Nakauchi H, and Yamazaki S. (2020). Long-term ex vivo expansion of mouse hematopoietic stem cells. Nat. Protoc. 15, 628–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Gao W, Yao K, and Ge J. (2019). Roles of exosomes derived from immune cells in cardiovascular diseases. Front. Immunol. 10, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, Ema H, and Nakauchi H. (2013). Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154, 1112–1126. [DOI] [PubMed] [Google Scholar]
- Yamashita M, and Passegué E. (2019). TNF-α coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell 25, 357–372.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, Taketo MM, Karlsson S, Iwama A, and Nakauchi H. (2011). Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 147, 1146–1158. [DOI] [PubMed] [Google Scholar]
- Yi YW, Lee JH, Kim SY, Pack CG, Ha DH, Park SR, Youn J, and Cho BS (2020). Advances in analysis of biodistribution of exosomes by molecular imaging. Int. J. Mol. Sci. 21, 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is EXRNA-KJENS1DIAB available within the exRNA atlas.