

Abstract
Objectives:
Salivary gland tumors are comprised of a diverse group of malignancies with widely varying prognoses. These cancers can be difficult to differentiate, especially in cases with limited potential for immunohistochemistry (IHC)-based characterization. Here, we sought to define the molecular profile of a rare salivary gland cancer called hyalinizing clear cell carcinoma (HCCC), and identify a molecular gene signature capable of distinguishing between HCCC and the histopathologically similar disease, mucoepidermoid carcinoma (MEC).
Materials and Methods:
We performed the first integrated full characterization of five independent HCCC cases.
Results:
We discovered insulin-like growth factor alterations and aberrant IGF2 and/or IGF1R expression in HCCC tumors, suggesting a potential dependence on this pathway. Further, we identified a 354 gene signature that differentiated HCCC from MEC, and was significantly enriched for genes with an ATF1 binding motif in their promoters, supporting a transcriptional pathogenic mechanism of the characteristic EWSR1-ATF1 fusion found in these tumors. Of the differentially expressed genes, IGF1R, SGK1 and SGK3 were found to be elevated in the HCCCs relative to MECs.
Finally, analysis of immune checkpoints and subsequent IHC demonstrated that CXCR4 protein was elevated in several of the HCCC cases.
Conclusion:
Collectively, our data identify an ATF1-motif enriched gene signature that may have clinical utility for molecular differentiation of HCCCs from other salivary gland tumors and discover potential actionable alterations that may benefit the clinical care of recurrent HCCC patients.
Keywords: HCCC, IGF2, EWSR1, ATF1, CXCR4
Introduction
Hyalinizing clear cell carcinoma (HCC) is a rare malignancy that primarily arises in the intraoral minor salivary glands and less commonly in the base of tongue, tonsil, nasopharynx, and lung [1–8]. Despite often multimodal therapy, this disease has a relatively high rate of recurrence [9]. HCCC is morphologically characterized by infiltrating groups of clear cells embedded in hyalinizing stroma. Although pathologically the disease is occasionally confused with variants of related neoplasms such as mucoepidermoid carcinoma and clear cell squamous cell carcinoma [10–13], the presence of one of two different CREB family gene fusions, EWSR1-ATF1 or EWSR1-CREM, provides clear molecular distinction for the majority of HCCCs [14, 15]. Unfortunately, beyond the occurrence of these gene fusions, little else is known about how these fusions regulate the molecular composition of the tumor or the other disruptive molecular events that co-occur to differentiate the molecular profile of HCCC from mucoepidermoid carcinoma.
The EWSR1-ATF1 gene fusion was originally discovered in HCCC in 2011 using fluorescence in situ hybridization (FISH) to show break apart of EWSR1 probes and subsequent 3’ RACE to identify ATF1 as the fusion partner in 13/14 (93%) of cases studied. Since that time, the role of this gene fusion in HCCC molecular oncogenesis has remained uncharacterized.[14] Given this gap in knowledge, we sought to further characterize the molecular composition of HCCC. We performed integrative exome and transcriptome sequencing analysis on two and five independent EWSR1-ATF1 fusion positive HCCC tumors respectively. Because of the rarity of the disease, we postulated that this information could help define novel drivers of HCCC and, when compared to molecular information from a cohort of mucoepidermoid carcinoma, could begin to identify a gene signature characteristic of EWSR1-ATF1 fusion positive HCCC that may have future utility to help distinguish the two tumor types as well as understand the mechanistic role of EWSR1-ATF1 fusions in disease pathogenesis.
Materials and Methods
Clinical specimens and clinical data
Patients with HCCC were identified from the University of Michigan pathology archive using an IRB-approved protocol for next generation sequencing of DNA and RNA (HUM00080561). Clinical, histologic, and outcome data was collected from medical records and the Social Security Death Index as previously described[16]. Following hematoxylin and eosin staining of sections from each block, our head and neck pathologist (J.B.M.) identified blocks with >60% tumor content for coring. DNA and RNA were then simultaneously isolated using the Qiagen AllPrep kit as described [17] and advanced for NGS if it met our previously defined quality standards defined by Qubit and Bioanalyzer analysis [18, 19].
Exome sequencing
Total genomic DNA from each tumor (n=2; HCCC1, HCCC2) and adjacent normal specimen was submit to the University’s DNA sequencing core for library preparation and exome sequencing using the DNA TruSeq Exome Library Preparation kit (Illumina, Catalogue number: FC-150-100x). Libraries were then pooled post-capture and sequenced on a single lane of an Illumina HiSEQ4000 using 150 nucleotide paired end protocol, yielding an average depth of greater than 100x per sample (Supplementary Table 1).
Exome variant calling
We assessed the quality of the sequencing reads using FastQC v.0.11.5 as previously described [20]. Trim galore v0.4.4 was used to remove adapters and trim reads. Reads were aligned to the hg19 reference genome using BWA v0.7.1. Mapping was followed by marking duplicates using PicardTools v1.79. Next, base quality score recalibration was completed using GATK v3.6. Samtools v1.2 was then used to create the pileup files for each tumor-normal pair in the set. We then used Varscan v2.4.1 to call variants from the mpileup files using the somatic mode of the variant caller. Goldex Helix Varseq v1.4.6 was used to annotate variants. All variants in the introns and intergenic regions were filtered out. Variants with more than 30 reads were considered high confidence calls and variants with a minimum of 5 reads supporting the alternate allele in the tumor samples were annotated as low confidence and considered as potential positives for validation by Sanger sequencing.
Copy number analysis
We used the aberration Detection in Tumour Exome (ADTEx) v.2.0 to make copy number estimation calls. A copy number state from 0 to 4 was assigned by the software based on its estimated copy number at each position. State 0 corresponded to a homozygous deletion, 1 corresponds to a heterozygous deletion, normal copy number is denoted by state 2, while states 3 and 4 represent a gain and amplification, respectively. The software was also used to generate representative Manhattan plots for each chromosome of each tumor/normal pair. Genes were annotated using R script (v3.4.0) to list the gene associated with each change.
Microsatellite instability (MSI) detection
We used the MANTIS algorithm to detect somatic MSI from the tumor-normal sample pairs as described [21]. This software package assigns an MSI status to each sample pair based on an instability score calculated for each sample pair.
Transcriptome sequencing
We submitted up to 500ng of RNA from each tumor (n=5; HCCC1, HCCC2, HCCC4, HCCC5, HCCC7) to the University of Michigan DNA sequencing core for library preparation and subsequent sequencing. Library preparations were completed with the Illumina TruSeq Stranded Total RNA library prep kit (Cat#: RS-122-2201/2) following the manufacturer’s protocol, with only one modification to increase the number of PCR cycles to 14 in order to increase amplification of the library prior to final purification. The samples were then pooled and loaded onto a single lane of an Illumina HiSEQ4000 for paired end sequencing to 75nt. RNA sequencing yielded an average of 62M reads with >88% uniquely mapped for each tumor. A summary of sequencing quality statistics including total unique mapped reads for each sample is provided in Supplementary Table 2.
Transcript quantification and clustering analysis
The quality of RNA sequencing reads was assessed using FastQC v0.11.5. We did not identify any quality issues from either library. We used Star v2.5.2a to generate genome index files based on hg19 version and then generated the transcript annotation file. We used the genome index files to map the sequencing reads against the human genome. Using, Picard v1.79 and samtools v1.2 we then extracted only uniquely mapped reads from the BAM files for quantification. Fragments per kilobase million (FPKM) were then calculated with cufflinks v2.2.0. Since the RNA-seq had a very high depth of coverage, the --max-bundle-frags argument was changed to 100,000,000 from its default value which is 1,000,000. This enabled the calculation of FPKMs at loci with high depth of coverage.
Gene fusion analysis and annotation of potential viral genomes
FusionCatcher (v1.00) was used to identify novel gene fusions as well as validate known gene fusions in our HCCC samples (n=5; HCCC1, HCCC2, HCCC4, HCCC5, HCCC7). In addition to looking for gene fusions, FusionCatcher also provides a list of viral and bacterial reads seen in the samples. This output in addition to HPVDectector [22] was used to confirm that no detectable HPV DNA was identified in the HCCC samples.
Sanger sequencing validation of candidate molecular alterations and fusion genes
Genomic DNA was used to verify somatic alterations including single nucleotide variations. 10ng of total DNA was used for PCR amplified with Platinum Taq DNA Polymerase High Fidelity (Invitrogen) according to manufacturer’s instructions. Primer sequences for each target are listed in Supplementary Table 3. For amplicons greater than 200bp in length, PCR products were PCR purified using the Qiagen PCR purification kit and submit for Sanger sequencing at the University’s DNA sequencing core on the 3730XL DNA Sequencer (Applied Biosystems). For amplicons less than 200bp in length, PCR products were cloned out using pCR8 TOPO vector (Invitrogen) and submitted for Sanger sequencing using forward and reverse primers from the cloning vector. Sequences were aligned using the DNASTAR Lasergene software suite against the RefSEQ annotation from HG19.
Hierarchical clustering of high-variance genes
Sequences were realigned to the human genome (HG19) using Rsubread v1.32.2, to generate raw read counts. Genes with an average raw read count between 100 and 10,000 were included in further analysis as described [23]. Differential expression analysis was performed with DESeq2 v1.18.1, comparing HCCC (n=5; HCCC1, HCCC2, HCCC4, HCCC5, HCCC7) and MEC (n=48) expression. Thus, by comparing to our previously analyzed mucoepidermoid carcinoma cohort (manuscript in review), variance between the HCCC and MEC sample read counts was then calculated using DESeq2 v1.18.1, using a mean fit type, followed by regularized logarithmic transformation of the read counts. The 900 genes with the highest inter-sample variance were included in further analysis, which represents the top 10% of genes initially included in the analysis. Samples underwent hierarchical clustering based on expression of these high-variance genes.
Identifying MEC and HCCC-specific gene signatures
HCCC-specific and MEC-specific gene sets were generated, with an FDR adjusted p value cutoff of 0.1. Transcription factor binding sites associated with MEC- and HCCC-specific gene sets were queried using GenomeRunner db5.00, comparing each gene set to the ENCODE clustered transcription factor binding site database. We queried overlap between transcription factor binding sites and the genes in each gene set, including the gene body and 1000bp upstream of the transcription start site. To define the background, the gene body and 1000bp upstream region of all HG19 genes was used. Transcription factors with binding sites that are significantly enriched in each gene set were identified, using a p value cutoff of 0.01. Data was visualized using Complex Heatmap as previously described [24].
Gene set enrichment analysis
Gene set enrichment analysis was performed using the software GSEA v4.03 from the Broad Institute (http://software.broadinstitute.org/gsea/index.jsp). Pre-ranked gene lists were prepared based on the log2-fold change in FPKM and the gene sets used were selected from the Molecular Signatures Database v7.0, including hallmark gene sets, gene ontology (GO) biological process (BP) gene sets and oncogenic signatures gene sets.
Immunohistochemistry
Immunohistochemical staining was performed (n=7; HCCC1-7) on the DAKO Autostainer (DAKO, Carpinteria, CA) using liquid streptavidin biotin (LSAB+) and diaminobenzadine (DAB) as the chromogen as previously described [25, 26]. De-paraffinized sections were labeled with the CXCR4 (1:500, clone UMB2, AbCam, Cambridge, MA. cat#Ab124824) for 60 minutes at ambient temperature after incubation of the section with background sniper (BioCare Medical, Pacheco, CA) for 30 mins at ambient temperature. Subsequently 10 mM Tris HCl/1 mM EDTA pH9 epitope retrieval was performed prior to staining. Appropriate negative (no primary antibody) and positive controls (kidney) were stained in parallel. Each sample was scored as a percent (0-100%) and intensity (0-3).
Results
Eight patient samples were obtained from our pathology archives and both demographic and clinical variable are shown in Table 1. Patient ages ranged from 41-58 years. Primary sites included base of tongue (BOT), hard palate, nasopharynx, and mandible. Overall stage varied from stage I to stage IV disease and two of the patients in this cohort presented with nodal disease. Two of the samples were initially classified as mucoepidermoid carcinoma and later changed to clear cell carcinoma based on EWSR1-ATF1 fusion status. Mucicarmine stains were performed in two of the samples to differentiate from mucoepidermoid carcinoma and were both negative. All patients in this group underwent surgical excision, one of which was in the salvage setting. Three patients received additional radiation or chemoradiation post-operatively. Two patients recurred (one local and one regional recurrence) and a third had persistent disease after CRT. All patients showed no evidence of disease at their last follow up and total time since therapy ranged from 1 month to 17 years with a median follow up time of 3.4 years.
Table 1:
Patient Demographics
| Age at Diagnosis | Gender | Site | Overall Stage | TNM stage | Special IHC stains | Smoking Status |
Adjuvant Treatment |
Recurrence | Disease Status |
Time from treatment |
|---|---|---|---|---|---|---|---|---|---|---|
| 55 | F | Hard palate | IV | T4aN0M0 | none | Former | no | no | AWOD | 8 years |
| 41 | F | Hard palate | I | T1N0M0 | none | Never | no | no | NED at last follow up | 4 years |
| 56 | M | Mandible | unknown initial stage |
N1 recurrence | none | Current | Second surgery for nodal recurrence |
Regional | NED at last follow up | 17 yrs from initial disease 5 yrs since nodal recurrence |
| 41 | F | BOT | IV | T4aN2bM0 | none | Current | CRT | no | AWOD | 1.8 years |
| 58 | F | Nasopharynx | III | T3N0M0 | none | Never | salvage surgery after CRT | no | AWOD | 1.6 years |
| 54 | M | BOT | II | T2N0M0 | Positive for cytokeratin, EMA and vimentin (focally). PAS stain positive and mucicarmine negative | Never | no | Local | AWOD | 17 years from initial disease 8 months local recurrence |
| 57 | F | BOT, FOM, and oral tongue | IV | T3N2bM0 | none | Never | RT | no | AWOD | 1 month |
| 48 | M | Nasopharynx | I | T1N0M0 | Positive for p63 and focally/weakly for CAM5.2. Negative for calponin, C-kit, smooth muscle actin and S100. Mucicarmine stain is negative for intracytoplasmic mucin |
Never | no | no | NED at last follow up | 2.7 years |
Of the 8 total cases, two HCCC specimens had DNA sufficient for integrated NGS based analysis (Supplementary Table 4), thus, we sequenced the exomes of both of these HCCC cases to greater than 100x depth and identified 142 high confidence alterations (79 somatic mutations and 63 INDELs) in HCCC1 and 28 high confidence alterations (10 somatic mutations and 18 INDELs) in HCCC2. Detailed annotation of the alterations are provided in Supplementary Tables 5 and 6, respectively. Surprisingly, we did not identify any recurrent molecular alterations or alterations to commonly mutated and cancer associated genes (e.g. TP53, PIK3CA, KRAS) in either tumor, but did identify an alteration of NOTCH2NL in HCCC1 (NM_001278267:c.5358-31971delC), supporting a potential disruptive role of NOTCH signaling in HCCC similar to the previously reported disruptive role in HNSCC [27], as well as an A299D mutation in TNK1, tyrosine kinase, non-receptor, 1.
We then performed functional annotation and gene set enrichment analysis of the altered genes in each tumor using the database for annotation, visualization and integrated discovery (DAVID). While this analysis did not identify any strong molecular enrichments in the genomic alterations of HCCC1, in HCCC2, the analysis revealed an enrichment of alterations to the p53 signaling pathway, with disruptive splice region variants in both CCNB1 (cyclin B1, NM_004701:c.25-3delC) and COP1 (NM_022457:c.1142-8delT). These genes are known to regulate sustained G2 arrest and negative feedback regulation of p53, respectively. Similarly, this tumor had an enrichment of disruptive mutations to ubiquitin kinases including: COP1, DCAF5, PDZRN3, CUL2 and UBR2.
Next, we analyzed genomic copy number changes in the exome data and found that overall copy number was relatively stable in each tumor (Figure 1, Supplementary Tables 7 and 8). We did observe a few genes in HCCC1 that had amplifications and several with single copy number gains including IGF2R, TERT, AKT2 and AURKC, while HCCC2 had no genes with amplifications and several genes with single copy gains including IGF2, HRAS and RPTOR. Together these data suggested a potential role of IGF and PI3K/mTOR signaling in these tumors. In contrast, the tumors both had single copy deletions of ATRX and BRWD3, while HCCC1 also had a single copy deletions of CHEK2, RBL1 and BRCA1 suggesting a potential defect in either DNA damage repair and/or cell cycle regulation.
Figure 1. Identification of IGF2R and IGF2 copy number amplifications in two Hyalinizing Clear Cell Carcinomas.
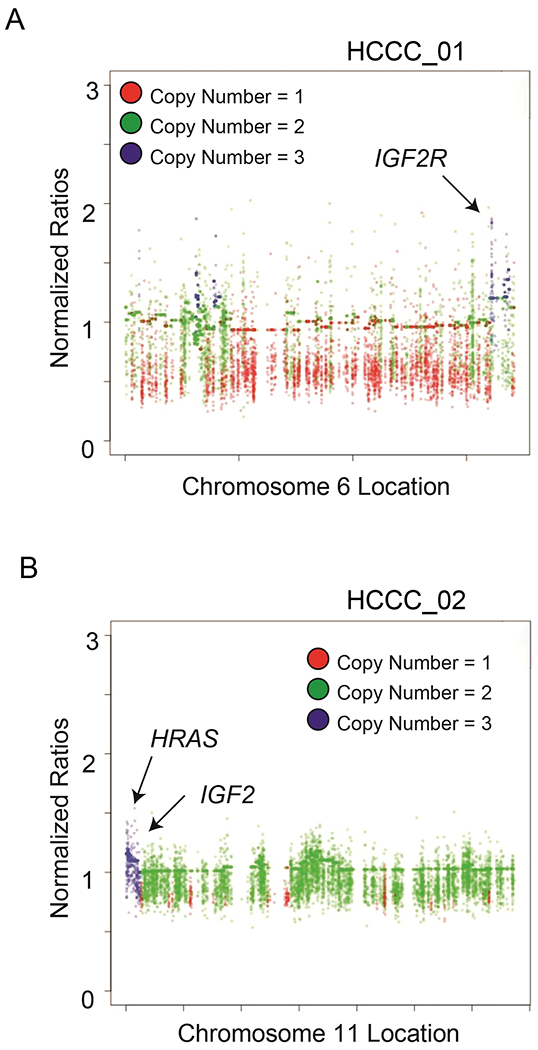
A) Manhattan plot shows copy number amplifications or deletions across the HCCC1 exome sequencing data for Chromosome 6, with amplifications highlighted by blue dots and deletions annotated as red dots as indicated. Functionally prioritized genetic lesions are shown with respect to their genomic location on the plot as indicated. B) As in (A), with Manhattan plot for HCCC2 Chromosome 11.
We then performed high depth transcriptome sequencing on RNA from 5 of the HCCC tumors, including both HCCC1 and HCCC2, to an average depth of >50M reads (Supplementary Table 9). Fusion status was determined in all five cases using the FusionCatcher algorithm. This confirmed that HCCC1, HCCC2, HCCC5, and HCCC7 harbored the canonical EWSR1-ATF1 gene fusion, and identified two different fusion forms (in HCCC1 and HCCC2), which we confirmed by Sanger sequencing (Figure 2A and 2B). Surprisingly, additional clinically relevant and previously described gene fusions common to lung cancer were also identified by the FusionCatcher in HCCC5 including IGFBP5-ALK, and EML4-ALK (Supplementary Table 10). [28–30] To then determine the relationship of gene expression in HCCC relative mucoepidermoid carcinomas (MEC), we performed unsupervised hierarchical clustering on the 5 HCCC cases with 48 MEC tumors that we previously sequenced (Figure 3A). Importantly, the 5 HCCC cases clustered together and could be differentiated from the MEC tumors by a 354 gene signature (189 upregulated and 165 downregulated genes, Supplementary Tables 11 and 12, respectively). To validate the molecular signatures, we analyzed gene signatures with altered expression specifically evaluating for transcription factor enrichments, hypothesizing that genes upregulated in the HCCC tumors would be enriched for ATF1 binding sites. For this analysis, the gene body and region 1000bp upstream of the transcription start site of each gene in each respective gene list were included in the enrichment analysis, and we utilized the ENCODE clustered transcription factor binding sites to define binding enrichment. Importantly, in the HCCC upregulated gene set, ATF1 binding sites were significantly overrepresented (FDR adjusted p=5.6 x 10−28), compared to background expression of all genes. These data strongly support the accuracy of our gene signature and suggest that additional analysis of our gene sets may help to identify pivotal genes and/or effectors of the EWSR1-ATF1 fusion.
Figure 2. De novo gene fusion discovery confirms the EWSR1-ATF1 gene fusion structures in HCCC1 and HCCC2.
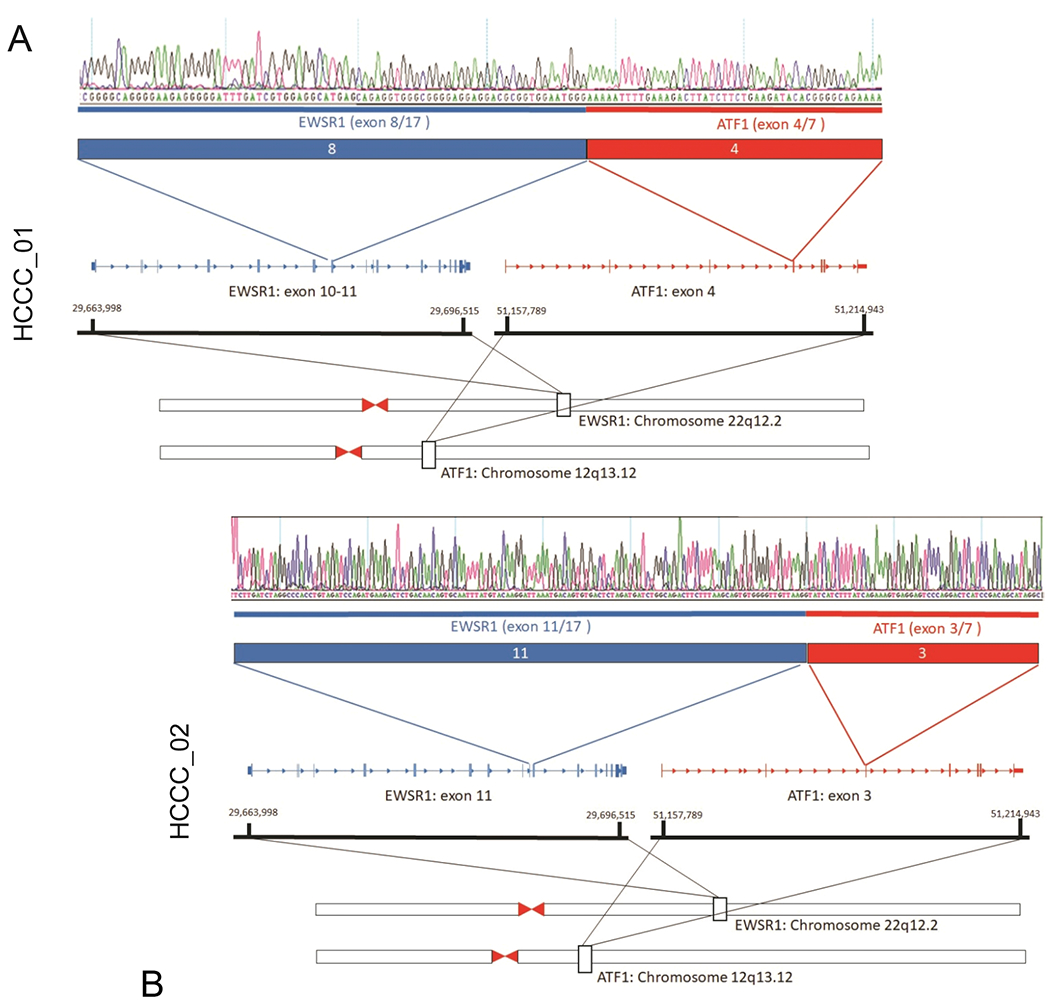
A) RNAseq data from HCCC1 was analyzed using the FusionCatcher bioinformatics algorithm, which nominated an EWSR1-ATF1 fusion gene junction in this tumor. The junction was validated by PCR amplification of tumor-derived cDNA, followed by Sanger sequencing (top panel), structure of the fusion shown below. B) HCCC2 also harbored a characteristic EWSR1-ATF1 fusion gene, but with a unique structure from HCCC1.
Figure 3. Identification of differential genes and pathways between HCCC and Mucoepidermoid Carcinoma.
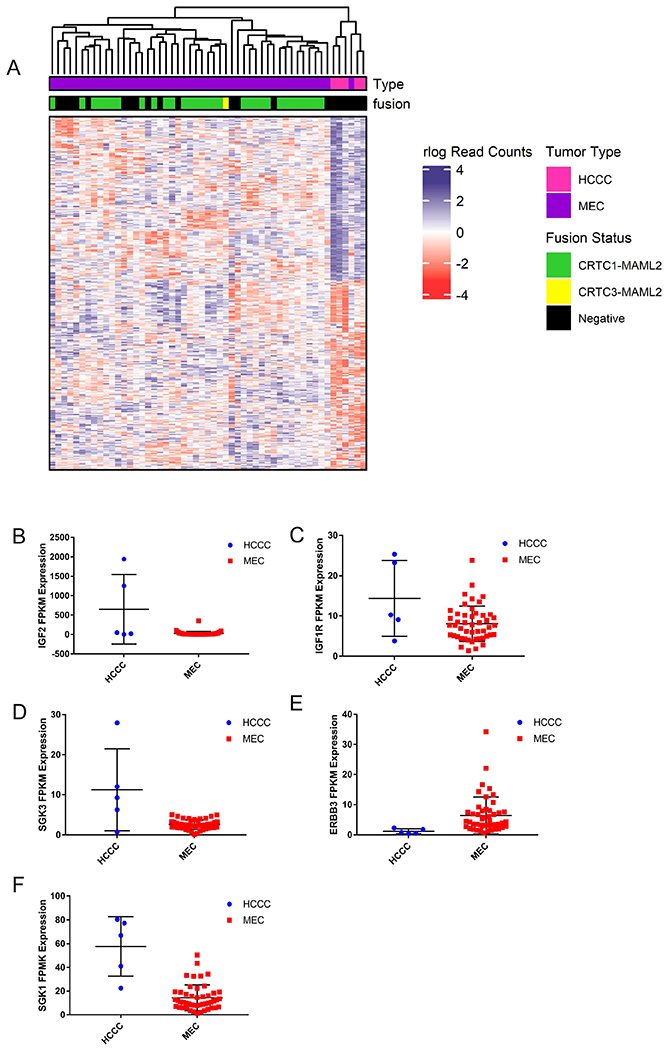
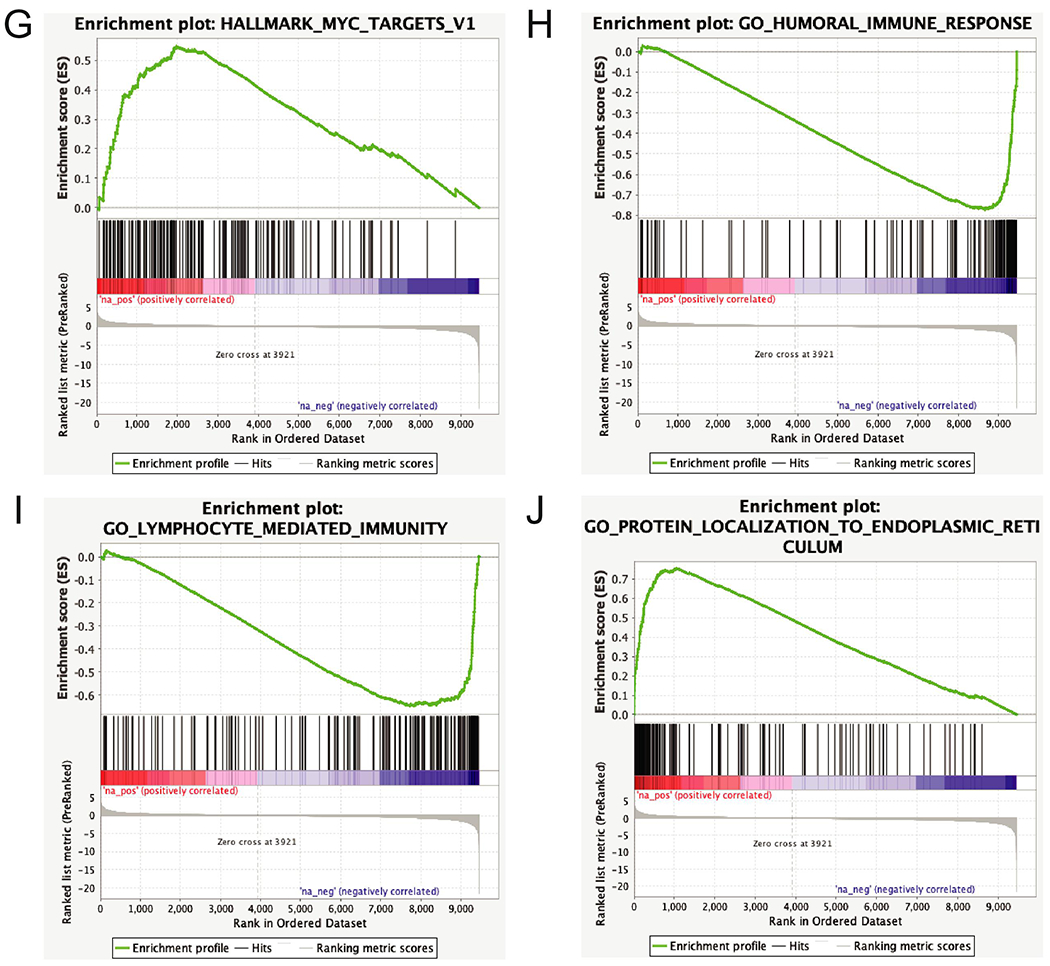
A) RNAseq data from HCCC and 48 MECs were clustered using DESEQ2 to identify genes differentially expressed between the two tumor types, and complex heatmap was used to visualize the differential gene signature. B) Dot plot shows relative expression of IGF2 in the HCCC and MEC cases as indicated, C-F) As in (B), with IGF1R, ERBB3, SGK3, and SGK1 expression, G-J) Representative pathway enrichment analysis of gene sets differentially expressed between HCCC and MEC.
Of the genes differentially expressed between HCCC and MEC, IGF2 was upregulated exclusively in HCCC1, and IGF1R (Insulin growth factor receptor 1, adjusted p=0.0042) was significantly upregulated in all HCCCs relative to the MECs. Accordingly, SGK3 (Serine/Threonine Protein kinase 3, adjusted p=1.45x10−11) and SGK1 (Serine/Threonine Protein kinase 1, adjusted p=1.07x10−5) were significantly upregulated in the HCCCs compared to our cohort of MECs (Figure 3B). Of note, ERBB3 (Epidermal Growth Factor Receptor 3, or HER3, adjusted p=6.64x10−7) was significantly downregulated in the HCCCs compared to MECs (Figure 3C–F). Subsequent pathway analysis of the differential gene sets with GSEA (Broad) demonstrated that our HCCC gene signature was enriched for 29 positively associated and 44 negatively associated concepts from the broad database with FDR q-value < 0.05 (Supplementary Table 13). Positively enriched concepts included HALLMARK_MYC_TARGETS_V1 and several concepts associated with protein targeting to the endoplasmic reticulum (e.g. GO_PROTEIN_LOCALIZATION_TO_ENDOPLASMIC_RETICULUM), while negatively enriched concepts were associated with decreased humoral (GO_HUMORAL_IMMUNE_RESPONSE) and lymphocytic (GO_LYMPHOCYTIC_MEDIATED_IMMUNITY) immune expression relative to the MEC cohort (Figure 3G–J).
Despite these findings and given the recent clinical success of immune checkpoint inhibitors, we evaluated the expression of known immune checkpoints in the RNAseq data (Figure 4A). This identified HLA-G as modestly expressed at the RNA level in 3/5 of the HCCC tumors. Finally, because we noted strong enrichment of SGK3 in the HCCCs, which is known to be a strong positive regulator of CXCR4 protein expression [31] through prevention of ubiquitin-mediated degradation of the checkpoint, and given the recent clinical success of CXCR4 antibodies, we sought to evaluate CXCR4 protein expression in our HCCC tumors. Immunohistochemistry confirmed expression of CXCR4 in 5/7 (71%) of our evaluable HCCC tumors, with high intensity staining in 2/7 (29%) cases (Figure 4B–H), suggesting that this may be an important immune escape mechanism for some HCCCs.
Figure 4. Characterization of immune checkpoint expression in HCCC.
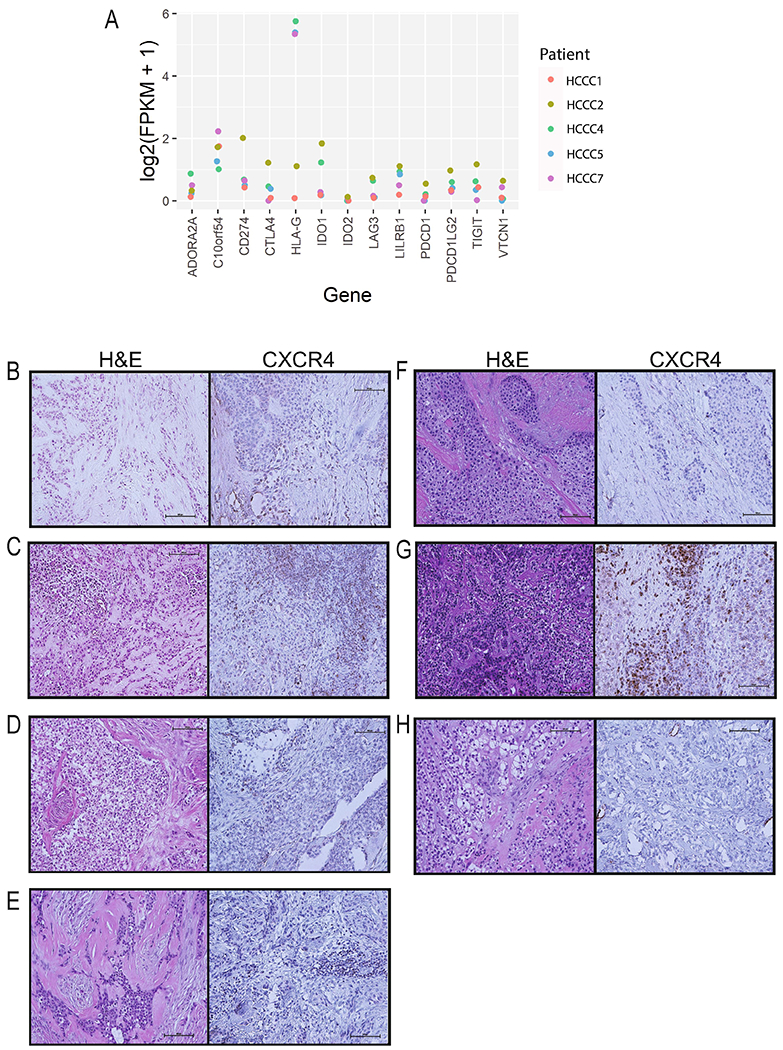
A) RNA expression (FPKM) of immune checkpoints in our HCCC cohort. B-H) CXCR4 immunohistochemistry was performed on 7 different HCCC cases, and representative photographs were taken as indicated and are shown next to the representative hematoxylin and eosin stain. Each sample is scored as a percent (0-100%) and intensity (0-3). Images shown at 40x. B) HCCC1 - 10%, 1. C) HCCC2 - 50%, 3. D) HCCC3 - 15%, 2. E) HCCC4 - 20%, 1. F) HCCC5 - 0%, 0. G) HCCC6 - 50%, 3. H) HCCC7 - 0%, 0.
Finally, using HPVDetector, we assessed both the exomes and transcriptomes for the presence of reads mapping to any HPV type, and found that both of these tumors (HCCC1 and HCCC2) were HPV negative (data not shown). Likewise, MANTIS analysis demonstrated that both of these HCCC tumors were microsatellite stable (data not shown).
Collectively, our informatic results confirm the presence of the characteristic EWSR1-ATF1 fusion and defines a unique ATF-enriched molecular signature that significantly differentiates our HCCC cases from a cohort of mucoepidermoid carcinomas.
Discussion
Our study overcomes a significant gap in knowledge by providing the first detailed view of the molecular composition of EWSR1-ATF1 fusion positive HCCC cases. Here, we identified a unique 354 gene signature enriched in EWSR1-ATF1 fusion positive HCCC compared to a cohort of CRTC1-MAML2 positive, CRTC3-MAML2 positive and CRTC1/3-MAML2 negative mucoepidermoid carcinomas. Because of the rarity of HCCC, we are not yet positioned to validate the clinical utility of this signature. Our hope is that future molecular studies will be able to use this data set as an initiation point to refine and validate our gene signature to identify the minimal gene set capable of differentiating the two diseases.
While occurrence of EWSR1-ATF1 gene fusions have been well documented in HCCC, our study is also the first to demonstrate the presence of an ATF1-enriched gene signature in the disease, which confirms that this fusion drives aberrant transcriptional activation of ATF1-regulated genomic loci in this disease. This is the first study to utilize clinical samples to confirm that ATF1-regulated genes are deregulated in HCCC, and supports development of further mechanistic studies to identify how these genes may play a role in disease pathogenesis. Indeed, these data supports mechanistic analogies to similar EWSR1-transcription factor fusions found in other cancers. For example, similar studies seeking to identify functionally pivotal effectors that are transcriptionally regulated by EWSR1-FLI1 or EWSR1-ERG in Ewing’s Sarcoma have success identifying targets critical for Ewing’s Sarcoma pathogenesis [32, 33].
Additionally, we also identified two potentially druggable targets in our samples. The DNA aberrations in the IGF/IGF1R signaling pathway and subsequent gene set enrichment analysis that identified an upregulation of PI3K/mTOR/FOXO pathway support a potential role for IGF1R inhibitor and several small molecule inhibitors targeting this pathway have already been advanced for other cancers [34, 35]. Similarly, we identified a strong overexpression of CXCR4 in a subset of the tumors also representing a clear therapeutic target for which several therapies are being clinically evaluated [36–38]. Interestingly, CXCR4 is associated with metastasis, which is not a strong clinical phenotype associated with HCCC; however, in tissues with elevated expression of the CXCR4 chemokine ligand CXCL12, CXCR4 expression tends to be associated with increased angiogenesis and tumor growth [38], which may be a critical mechanism for HCCC pathogenesis. Finally, we also discover the presence of additional fusions including EML4-ALK which has possible clinical implications as non-small cell lung cancer tumors harboring these fusions have shown to be sensitive to ALK inhibition.[28]
HCCC is an extremely rare cancer associated with fairly good prognostic outcomes, but a disease that can be difficult to identify without additional staining in some cases. Our study addresses the challenge of using molecular sequencing to differentiate HCCC samples and identifies a gene signature that is significantly different from MEC, and that can be evaluated in future studies containing larger case series of HCCCs. Although our study was limited to just seven samples, due to the extreme rarity of the disease, this comprehensive molecular analysis represents the first step forward in understanding the pathogenesis of disease. Our hope is that in the future, the molecular information learned in this study can serve as the basis for improved molecular annotation of these rare diseases in order to differentiate those associated with aggressive clinical phenotypes from those like HCCC with less aggressive clinical phenotypes that may benefit from de-escalated therapeutic strategies.
Supplementary Material
This work provides the first full integrated sequencing of hyalinizing clear cell carcinoma
HCCC samples contained insulin-like growth factor alterations and aberrant IGF2 and/or IGF1R expression
We demonstrate a unique gene signature that separates HCCC from MEC
The HCCC signature is enriched for genes with an ATF1 binding motif supporting a functional role of EWSR1-ATF1 fusion
Grant Support:
M.E.H received funding from the NIH (T32 DC005356) and J.C.B. received funding from the NIH (Grants U01-DE025184 and R01-CA194536). Funding sources had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations:
- HCCC
Hyalinizing clear cell carcinoma
- MEC
mucoepidermoid carcinoma
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability - The data that supports the findings of this study are available in the supplementary material of this article
Conflict of interest statements – The authors have nothing to disclose
References:
- 1.Jeffus SK, et al. , Hyalinizing Clear Cell Carcinoma of the Lung: Case Report and Review of the Literature. Am J Clin Pathol, 2017. 148(1): p. 73–80. [DOI] [PubMed] [Google Scholar]
- 2.Gushi E, Seki U, and Orhan K, Hyalinizing Clear Cell Carcinoma of Maxilla. J Clin Diagn Res, 2017. 11(7): p. ZL01–ZL02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burgess B, Ananthanarayanan V, and Charous S, Hyalinizing Clear Cell Carcinoma of the Tonsil: A Case Report. Head Neck Pathol, 2017. 11(4): p. 580–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shahi M, Dolan M, and Murugan P, Hyalinizing Clear Cell Carcinoma of the Bronchus. Head Neck Pathol, 2017. 11(4): p. 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fukuda A, et al. , Nasopharyngeal hyalinizing clear cell carcinoma with EWSR1 rearrangements diagnosed by fluorescence in situ hybridization. Auris Nasus Larynx, 2015. 42(5): p. 412–5. [DOI] [PubMed] [Google Scholar]
- 6.Tang SK, Wan SK, and Chan JK, Hyalinizing clear cell carcinoma of salivary gland: report of a case with multiple recurrences over 12 years. Am J Surg Pathol, 1995. 19(2): p. 240–1. [DOI] [PubMed] [Google Scholar]
- 7.Batsakis JG, el-Naggar AK, and Luna MA, Hyalinizing clear cell carcinoma of salivary origin. Ann Otol Rhinol Laryngol, 1994. 103(9): p. 746–8. [DOI] [PubMed] [Google Scholar]
- 8.Milchgrub S, et al. , Hyalinizing clear cell carcinoma of salivary gland. Am J Surg Pathol, 1994. 18(1): p. 74–82. [DOI] [PubMed] [Google Scholar]
- 9.Yang XH, et al. , Hyalinizing clear cell carcinoma of salivary gland origin in the head and neck: clinical and histopathological analysis. Int J Oral Maxillofac Surg, 2018. 47(6): p. 692–698. [DOI] [PubMed] [Google Scholar]
- 10.Roby BB, Pambuccian SE, and Khariwala SS, Pathology quiz case 2. Hyalinizing clear cell carcinoma. Arch Otolaryngol Head Neck Surg, 2012. 138(2): p. 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Sullivan-Mejia ED, et al. , Hyalinizing clear cell carcinoma: report of eight cases and a review of literature. Head Neck Pathol, 2009. 3(3): p. 179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai G, et al. , The role of immunohistochemistry in the diagnosis of hyalinizing clear cell carcinoma of the minor salivary gland: a case report. Eur J Histochem, 2008. 52(4): p. 251–4. [DOI] [PubMed] [Google Scholar]
- 13.Milchgrub S, et al. , Hyalinizing clear-cell carcinoma of salivary glands in fine-needle aspiration. Diagn Cytopathol, 2000. 23(5): p. 333–7. [DOI] [PubMed] [Google Scholar]
- 14.Antonescu CR, et al. , EWSR1-ATF1 fusion is a novel and consistent finding in hyalinizing clear-cell carcinoma of salivary gland. Genes Chromosomes Cancer, 2011. 50(7): p. 559–70. [DOI] [PubMed] [Google Scholar]
- 15.Chapman E, et al. , Molecular Profiling of Hyalinizing Clear Cell Carcinomas Revealed a Subset of Tumors Harboring a Novel EWSR1-CREM Fusion: Report of 3 Cases. Am J Surg Pathol, 2018. 42(9): p. 1182–1189. [DOI] [PubMed] [Google Scholar]
- 16.Birkeland AC, et al. , Pathogenetic Analysis of Sinonasal Teratocarcinosarcomas Reveal Actionable beta-catenin Overexpression and a beta-catenin Mutation. J Neurol Surg B Skull Base, 2017. 78(4): p. 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birkeland AC, et al. , Correlation of Crtc1/3-Maml2 fusion status, grade and survival in mucoepidermoid carcinoma. Oral Oncol, 2017. 68: p. 5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birkeland AC, et al. , Identification of Targetable ERBB2 Aberrations in Head and Neck Squamous Cell Carcinoma. JAMA Otolaryngol Head Neck Surg, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tillman BN, et al. , Fibroblast growth factor family aberrations as a putative driver of head and neck squamous cell carcinoma in an epidemiologically low-risk patient as defined by targeted sequencing. Head Neck, 2016. 38 Suppl 1: p. E1646–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith J, et al. , Whole-Exome Sequencing of Sinonasal Small Cell Carcinoma Arising within a Papillary Schneiderian Carcinoma In Situ. Otolaryngol Head Neck Surg, 2018: p. 194599818774004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kautto EA, et al. , Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget, 2017. 8(5): p. 7452–7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandrani P, et al. , NGS-based approach to determine the presence of HPV and their sites of integration in human cancer genome. Br J Cancer, 2015. 112(12): p. 1958–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ludwig ML, et al. , The genomic landscape of UM-SCC oral cavity squamous cell carcinoma cell lines. Oral Oncol, 2018. 87: p. 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swiecicki PL, et al. , A multi-center phase II trial evaluating the efficacy of palbociclib in combination with carboplatin for the treatment of unresectable recurrent or metastatic head and neck squamous cell carcinoma. Invest New Drugs, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mann JE, et al. , Analysis of tumor-infiltrating CD103 resident memory T-cell content in recurrent laryngeal squamous cell carcinoma. Cancer Immunol Immunother, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoesli R, et al. , Proportion of CD4 and CD8 tumor infiltrating lymphocytes predicts survival in persistent/recurrent laryngeal squamous cell carcinoma. Oral Oncol, 2018. 77: p. 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J, et al. , Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A, 2013. 110(50): p. 20224–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camidge DR, et al. , Updated Efficacy and Safety Data and Impact of the EML4-ALK Fusion Variant on the Efficacy of Alectinib in Untreated ALK-Positive Advanced Non-Small Cell Lung Cancer in the Global Phase III ALEX Study. J Thorac Oncol, 2019. 14(7): p. 1233–1243. [DOI] [PubMed] [Google Scholar]
- 29.Haimes JD, et al. , Uterine Inflammatory Myofibroblastic Tumors Frequently Harbor ALK Fusions With IGFBP5 and THBS1. Am J Surg Pathol, 2017. 41(6): p. 773–780. [DOI] [PubMed] [Google Scholar]
- 30.Soda M, et al. , Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature, 2007. 448(7153): p. 561–6. [DOI] [PubMed] [Google Scholar]
- 31.Slagsvold T, et al. , CISK attenuates degradation of the chemokine receptor CXCR4 via the ubiquitin ligase AIP4. EMBO J, 2006. 25(16): p. 3738–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charville GW, et al. , EWSR1 fusion proteins mediate PAX7 expression in Ewing sarcoma. Mod Pathol, 2017. 30(9): p. 1312–1320. [DOI] [PubMed] [Google Scholar]
- 33.Camoes MJ, et al. , Potential downstream target genes of aberrant ETS transcription factors are differentially affected in Ewing’s sarcoma and prostate carcinoma. PLoS One, 2012. 7(11): p. e49819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qu X, et al. , Update of IGF-1 receptor inhibitor (ganitumab, dalotuzumab, cixutumumab, teprotumumab and figitumumab) effects on cancer therapy. Oncotarget, 2017. 8(17): p. 29501–29518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yee D, Anti-insulin-like growth factor therapy in breast cancer. J Mol Endocrinol, 2018. 61(1): p. T61–T68. [DOI] [PubMed] [Google Scholar]
- 36.Eckert F, et al. , Potential Role of CXCR4 Targeting in the Context of Radiotherapy and Immunotherapy of Cancer. Front Immunol, 2018. 9: p. 3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu C, et al. , CXCR4 in breast cancer: oncogenic role and therapeutic targeting. Drug Des Devel Ther, 2015. 9: p. 4953–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okuyama Kishima M, et al. , Immunohistochemical expression of CXCR4 on breast cancer and its clinical significance. Anal Cell Pathol (Amst), 2015. 2015: p. 891020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.