

Abstract
Human intestinal peptide transporter PEPT1 is commonly repressed in human colorectal cancer (CRC), yet its relationship with sensitivity to the common CRC treatment ubenimex has not previously been elucidated. In this study, we confirmed PEPT1 suppression in CRC using real-time quantitative polymerase chain reaction and western blotting and then investigated the underlying epigenetic pathways involved using bisulfite sequencing, chromatin immunoprecipitation, siRNA knockdown, and reporter gene assays. We found that PEPT1 transcriptional repression was due to both DNMT1-mediated DNA methylation of the proximal promoter region and HDAC1-mediated histone deacetylation, which blocked P300-mediated H3K18/27Ac at the PEPT1 distal promoter. Finally, the effects of the epigenetic activation of PEPT1 on the CRC response to ubenimex were evaluated using sequential combination therapy of decitabine and ubenimex both in vitro and in xenografts. In conclusion, epigenetic silencing of PEPT1 due to increased DNMT1 and HDAC1 expression plays a vital role in the poor response of CRC to ubenimex.
Subject terms: Molecular biology, Gene silencing
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer and the second most deadly cancer worldwide, with an estimated over 1.9 million new cases and 935,000 deaths recorded in 20201. CRC incidence and mortality have declined with recent screening programs. However, the diagnosis rates of advanced CRC with low overall survival (OS) remain high1. Therefore, it is essential to identify novel biomarkers for the treatment, diagnosis, and prognosis of CRC. Chemotherapy is the treatment of choice to increase the OS of patients with advanced or metastatic CRC2. The resistance of malignant tumor cells to chemotherapy is a crucial reason for poor survival among CRC patients. One explanation for this multidrug resistance (MDR) to cancer chemotherapy is the increased efflux and reduced influx mediated by drug transporters3. For example, our laboratory’s previous studies have demonstrated that the repression of uptake transporters resulted in decreased drug concentrations in tumors4,5.
The peptide transporter PEPT1 (encoded by SLC15A1) is a prototypical member of the SLC15 family6. PEPT1 is predominantly responsible for the absorption of di/tripeptides and is mainly located on brush border membranes of small intestinal epithelia. In addition to that in the intestine, PEPT1 has been detected in tissues such as the nasal epithelium, kidney, biliary duct, and macrophages7,8. Besides, PEPT1 is overexpressed in the colon of inflammatory bowel disease patients and prostate cancer cells, which provides novel insight into the pathogenesis and tumor-specific drug delivery of this diseases9,10. However, there are inconsistent reports on the expression levels of PEPT1 mRNA and protein in the colon11. While one study reported low PEPT1 expression in the colon12, it could not be detected in other studies9,13. Another recent study showed that PEPT1 is highly expressed in the distal colon14. These inconsistent results may be explained at least partially by the different research groups measuring expression in different areas of the colon. PEPT1 has considerable substrate specificity for oligopeptides produced after the digestion of dietary or body proteins and structurally related drugs, such as the anticancer agent ubenimex (UBEN) and peptidomimetic prodrugs15. Ubenimex (UBEN), more commonly known as Bestatin, is a drug with immune-modulatory and anti-tumor activities16. At present, it has been widely used to treat acute myelocytic leukemia17 and it was once reportedly, preferably delivered into tumor cells overexpressing PEPT118. Therefore, PEPT1 is increasingly becoming a potential target for modulating the efficacy of various chemotherapeutic agents.
Until now, the transcriptional regulation of PEPT1 has not been widely investigated. Only a few transcription factors (sp1, cdx2, nrf2) have been shown to regulate the transcriptional activity of the PEPT1 gene promoter19–21. Epigenetic regulation of gene transcription, which involves dynamic modifications such as DNA methylation, histone acetylation, and noncoding RNAs, has been associated with several physiological and pathological processes. Although DNA methylation typically involved in gene repression is performed by DNA methyltransferase (DNMT1, DNMT3a, and DNMT3b), DNMT1 is primarily responsible for maintenance methylation by converting hemimethylated duplexes into symmetrically methylated CpG dinucleotides during DNA replication22,23. Alterations in DNA methylation caused by abnormalities in DNMT1 have been shown to drive tumorigenesis in several studies24,25. Histone acetylation primarily occurs at numerous lysine residues at the N terminus of histones, including H3K9Ac, H3K18Ac, and H3K27Ac, which are enriched around the transcription start site (TSS) and are generally associated with gene activation26,27. Acetyl groups are added at these specific histone sites by histone acetyltransferases (HATs) and removed by histone deacetylases (HDACs)28. The four main groups of HATs are the GCN5, MYST, CBP/P300, and SRC/p160 nuclear receptor coactivator families, and the HDAC family comprises eighteen different isoforms (HDAC1-11 and SIRT1-7)28. Similar to DNA methylation, histone acetylation at various gene transcriptional regulatory elements has been correlated with tumor development29. Epigenetic instability in CRC occurs early and manifests more frequently than genetic modifications30. For example, DNMT1-mediated methylation of Cdknla promoted cell proliferation in a carcinogen-administered CRC mouse model31. In addition, P300 was shown to promote PHF5A acetylation at K29 during cellular stress, which consequently contributed to colon carcinogenesis32 in the CRC xenograft model. Here, we determined the expression level of PEPT1 in CRC compared to normal tissues and analyzed the epigenetic mechanisms deregulating the expression of PEPT1 in CRC. We then designed a combination treatment of decitabine (DAC) and UBEN to sensitize CRC cells to UBEN. These findings highlight the potential clinical usefulness of various epigenetic modifications as biomarkers for the early diagnosis and pharmacological treatment of CRC patients.
Results
PEPT1 is repressed in CRC
Boxplot analysis on GEPIA revealed that PEPT1 transcription in CRC tumor tissues was markedly decreased compared to that in the normal colon (Fig. 1A). To confirm the mRNA expression levels of PEPT1, RT-qPCR was performed on 58 CRC tumor tissues and matched normal tissues (Fig. 1B, C). PEPT1 expression was dramatically reduced in most CRC tissue samples (43/58). Next, we evaluated the protein expression of PEPT1 via western blotting (Fig. 1D). Thirteen of the 14 CRC patient samples displayed low PEPT1 expression (Fig. 1B), whereas 1 sample (CRC47) displayed no significant change in protein expression. Moreover, no significant correlation was found between PEPT1 expression levels and gender, age, TNM stage, location (Fig. S1A–D, Table S1). These data together demonstrate that PEPT1 is downregulated at both the mRNA and protein levels in most CRC tumors, regardless of gender, age, and TNM stage. These results imply that the expression of PEPT1 is an important factor for the diagnosis of CRC.
Fig. 1. The mRNA and protein expression levels of PEPT1 are frequently downregulated in CRC.

A PEPT1 expression data were provided by TCGA in GEPIA. Box plots showed PEPT1 mRNA expression in CRC tumor tissues (T) and normal tissues (N). *P < 0.05, one-tailed unpaired t-test. B The mRNA expression of PEPT1 was detected in 58 pairs of CRC and corresponding adjacent non-tumorous tissues by RT-qPCR. Data are shown as means ± SD, n = 58. C The fold change log2(T/N) of PEPT1 expression between 58 pairs of CRC and adjacent normal tissues were summarized. <−1 is downregulation, −1 to 1 is unchanged and >1 is upregulation. D Representative images of western blotting analysis for the expression of PEPT1 in 14 paired samples of CRC. GAPDH was used as a loading control.
DNMT1 mediates the suppression of PEPT1
The silencing of gene expression by CpG methylation is one of the most frequent epigenetic inactivation events. Therefore, we next investigated whether DNA methylation at the PEPT1 promoter contributes to altered PEPT1 transcription in CRC. As shown in Fig. 2A, we found that treatment with DAC, a demethylation reagent that blocks cellular DNA methyltransferases (DNMTs), activated PEPT1 transcription in SW480 and SW620 cells in a dose-dependent manner. Western blot analysis confirmed that DAC induced PEPT1 expression at the protein level in both SW480 and SW620 cells (Fig. 2B). These data together imply that PEPT1 expression is controlled by DNA demethylation. As DNA methylation is catalyzed by DNMTs, including DNMT1, DNMT3a, and DNMT3b, we next investigated which DNMTs repressed PEPT1 transcription in CRC. Among the three DNMTs, only DNMT1 was upregulated in CRC tissues compared with normal tissues (Figs. 2C, S2), which indicated that DNMT1 might be a crucial factor responsible for PEPT1 repression. To investigate this further, we designed siRNAs against all 3 DNMTs to determine their role in regulating PEPT1. Interestingly, after transient transfection with siRNA, we found that PEPT1 mRNA and protein expression was upregulated by siDNMT1, but not siDNMT3a or siDNMT3b, in both SW480 and SW620 cells (Figs. 2D and E, S3). Furthermore, the ChIP results demonstrated that DNMT1 was absent at the PEPT1 promoter after DAC treatment (Fig. 2F). As a control, DAC treatment showed no effect on the DNMT1 signature at the GAPDH promoter (Fig. S4A). Overall, our results indicate that DNMT1 is associated with transcriptional repression of PEPT1 in CRC cells.
Fig. 2. The inhibition of DNMT1 increases PEPT1 in CRC.

A The mRNA expression of PEPT1 in SW480 and SW620 cells. Cells were treated with DMSO, 2.5 μM DAC, or 5 μM DAC for 72 h. Data are shown as means ± SD, n = 3, two-tailed unpaired t-test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, significantly different from DMSO. B Immunoblotting confirmed DAC treatment in SW480 and SW620 cells. C DNMT1 mRNA levels in CRC tumors (T) and normal tissues (N) from the TCGA in GEPIA. *P < 0.05, one-tailed unpaired t-test. D Knockdown of DNMT1 activated PEPT1 mRNA expression in SW480 and SW620 cells, respectively. SiNC, cells transfected with negative control siRNA, siDNMT1#1, siDNMT1#2, two siRNAs for DNMT1. Data are shown as means ± SD, n = 3, two-tailed unpaired t-test, **P < 0.01, ***P < 0.001. E PEPT1 and DNMT1 protein expression after DNMT1 knockdown in SW480 and SW620 cells. F ChIP-qPCR analyses of DNMT1 enrichment at the proximal promoter of PEPT1 in SW480 and SW620 cells after DAC (5 μM for 72 h) treatment. Data are shown as means ± SD, two-tailed unpaired t-test, *P < 0.05, ***P < 0.001.
DNA hypermethylation of PEPT1
As shown in Fig. 3A, the upstream region around the TSS of PEPT1 contains a putative CpG island (CGI, 900 bp). Using TCGA analysis, we found that promoter methylation of PEPT1 is significantly increased in CRC tissues compared to normal tissues (Fig. 3B). The upstream CGI at the PEPT1 proximal promoter region (−264 bp to +36 bp) contains 35 CpG sites. To directly determine whether the promoter region of PEPT1 is subject to DNA hypermethylation, we next examined methylation status at the PEPT1 promoter by BSP. To rule out unbiased amplification, the PCR reaction was optimized by combined bisulfite restriction analysis (Fig. S5). Eleven pairs of CRC and matched adjacent non-tumor tissues were used in the BSP. CRC tissues with the repression of PEPT1 had higher levels of DNA methylation in the sequenced region (−264 bp to +36 bp) compared with adjacent non-tumor colonic tissue samples, especially in the 25th to 30th CpG sites, suggesting that hypermethylation occurs at the PEPT1 promoter in CRC (Fig. 3C). Next, we examined whether exogenous DNA demethylation affects DNA methylation of the PEPT1 promoter in CRC cells. The PEPT1 promoter was hypermethylated in SW480 and SW620 cells (Fig. 3D). Upon treatment with DAC, globally inhibiting DNA methylation, CRC cells had decreased DNA methylation levels in the 25th to 30th CpG sites. In addition, a similar effect on the DNA methylation levels after DNMT1 siRNA transfection was also observed (Fig. 3E). As shown in Fig. 3F, knockdown of DNMT1 could also decrease the relative luciferase activity, indicating that DNMT1 binds directly to the promoter of PEPT1. Thus, we concluded that DNA hypermethylation mediated by DNMT1 represses PEPT1 in CRC.
Fig. 3. Repression of PEPT1 expression via DNMT1 mediated DNA hypermethylation.

A PEPT1 promoter region (±2.5 kb) around 5’ regions adjacent to the TSS. CGI, CpG islands. BSP, bisulfite-sequencing PCR. B DNA methylation data of PEPT1 promoter in cancerous and adjacent non-tumorous tissues from the TCGA database (***P < 0.001). C BSP analysis of PEPT1 CGI in cancerous and adjacent non-tumorous tissues (n = 11). Methylation percentages of the 35 CpG loci in the sequenced region were calculated. 11 pairs of CRC tissues belong to tissues in Fig. 1B (CRC17, 19, 22, 26, 41, 47, 49, 50, 51, 52, 54). The y axis indicates the average methylation percentage of each CpG site calculated from patient tissues with PEPT1 repression. D–E Calculation of methylation percentages of PEPT1 promoter after DAC and siDNMT1 treatment in CRC cells. F Luciferase assay in DNMT1 knockdown CRC cells. Mock, cells transfected with negative control siRNA and PEPT1 promoter constructs. Si-DNMT1, cells transfected with siDNMT1#2 and PEPT1 promoter constructs. Data are shown as means ± SD, two-tailed unpaired t-test, *P < 0.05, **P < 0.01.
Histone hypoacetylation represses PEPT1 in CRC
Histone acetylation is another important form of epigenetic regulation. We next attempted to decipher how histone acetylation contributes to PEPT1 repression in CRC. We found that treatment with the HDAC inhibitors TSA and SAHA also strongly increased the mRNA and protein expression of PEPT1 (Fig. 4A, B). Next, we treated cells with siRNAs targeting all HDAC family classes and observed significant upregulation of PEPT1 only after knockdown of HDAC1 (Figs. 4C and D, S3). These data suggest that hypoacetylation at the PEPT1 promoter region caused by HDAC1 subsequently leads to the repression of PEPT1 in CRC. In addition, ChIP-qPCR analyses demonstrated that the activating signals H3K18Ac and H3K27Ac increased after SAHA treatment (Figs. 4E, S4B). To confirm this result, we analyzed the occupancy of H3K18Ac and H3K27Ac to the promoter region of PEPT1 in CRC samples. In five CRC tissues with repressed PEPT1, both H3K18Ac and H3K27Ac were decreased at the promoter region (Figs. 4F and G, S4C). Collectively, these results suggest that the absence of H3K18Ac and H3K27Ac combined with HDAC1-mediated deacetylation results in histone hypoacetylation at the PEPT1 promoter region and transcriptional silencing of PEPT1 in CRC.
Fig. 4. Histone hypoacetylation around PEPT1 promoter in CRC.
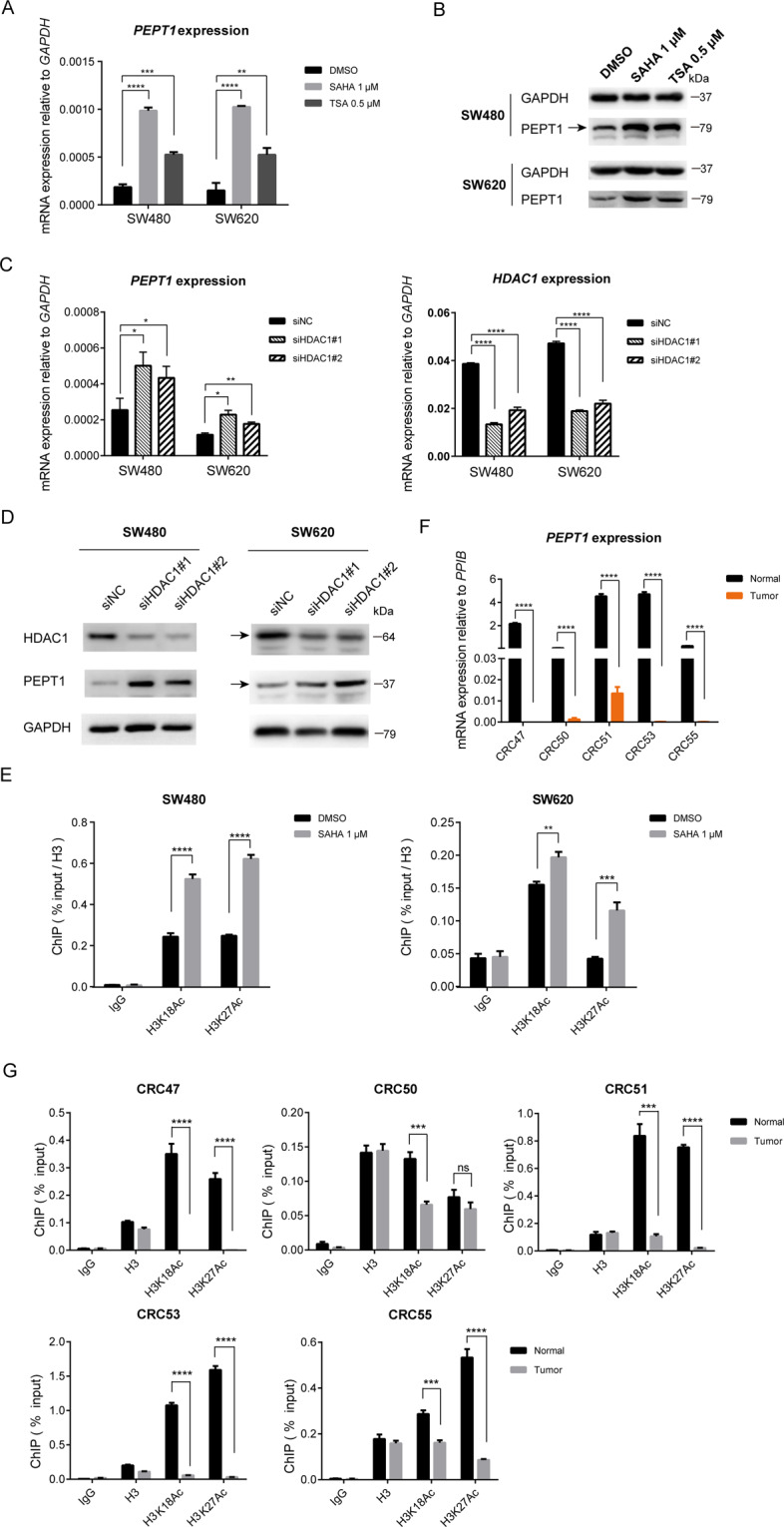
A The mRNA expression of PEPT1 in SW480 and SW620 cells after HDAC inhibitors treatment. Cells were treated with DMSO, 1 μM SAHA for 48 h or 0.5 μM TSA for 24 h. Data are shown as means ± SD, n = 3, two-tailed unpaired t-test, ns, no significance, **P < 0.01, ***P < 0.001, ****P < 0.0001. B Immunoblotting confirmed HDAC inhibitors treatment in SW480 and SW620 cells. C–D Knockdown of HDAC1 activated PEPT1 mRNA and protein expression in SW480 and SW620 cells, respectively. SiNC, cells transfected with negative control siRNA, siHDAC1#1, siHDAC1#2, two siRNAs for HDAC1. Data are shown as means ± SD, n = 3, two-tailed unpaired t-test, *P < 0.05, **P < 0.01, ****P < 0.0001. E ChIP-qPCR analyses of H3K18/H3K27Ac occupancy at PEPT1 promoter in SW480 and SW620 cells after SAHA (1 μM for 48 h) treatment. Data are shown as means ± SD, two-tailed unpaired t-test, **P < 0.01, ***P < 0.001, ****P < 0.0001. F PEPT1 mRNA expression in CRC47, 50, 51, 53, 55. Data are shown as means ± SD, two-tailed paired t-test, **P < 0.01, ***P < 0.001. G ChIP-qPCR analyses of H3K18/K27Ac occupancy at the PEPT1 promoter in five paired CRC tissues. Data are shown as means ± SD, two-tailed unpaired t-test, ***P < 0.001, ****P < 0.0001.
PEPT1 repression via the HDAC1-CBP/P300 axis
We next attempted to determine the role of positive and negative regulators of histone acetylation in regulating PEPT1. The acetyl group is usually added to lysine residues by histone acetyltransferases and removed by one of the HDAC enzymes. We performed dual-luciferase gene reporter assays to study whether histone acetyltransferases could bind to the UTR of PEPT1 and regulate its expression. We transiently co-transfected CRC cells with five PEPT1 promoter constructs based on the pGL3 luciferase reporter (Fig. 5A) and siRNAs targeting the histone acetyltransferases CBP/P300, which are specifically required for H3K18Ac and H3K27Ac33,34. We observed that inhibition of P300, but not CBP, reduced luciferase reporter activity by approximately 65% specifically at the region from −1750 to +129 bp in both SW480 and SW620 cells (Figs. 5B and S6). The luciferase assay results showed that P300 is a crucial factor for PEPT1 regulation and that P300 trans-activation of the PEPT1 promoter depends on the GGGAGTG sequence35, a consensus DNA-binding sequence for P300 (Fig. 5A, B). In addition, significantly lower expression levels of PEPT1 were found after P300 depletion in SW480 cells. (Fig. 5C, D). ChIP analysis also revealed that, after SAHA treatment, H3K18Ac and H3K27Ac were unoccupied in P300-silenced SW480 cells but highly enriched in non-targeting siRNA-expressing cells (Figs. 5E, S4D). In addition, H3K18Ac and H3K27Ac around the PEPT1 promoter were induced following knockdown of HDAC1 (Figs. 5F, S4E). These results together provide evidence that the HDAC1-CBP/P300 axis catalyzes H3K18Ac and H3K27Ac modification at the PEPT1 promoter.
Fig. 5. PEPT1 repression via HDAC1-CBP/P300 axis.

A Promoter constructs of PEPT1 in the luciferase assay. GGGAGTG, a consensus DNA binding sequence for p300. B Luciferase assay in P300 knockdown CRC cells. Mock cells transfected with negative control siRNA and promoter constructs. Si-P300, cells transfected with siP300#2 and promoter constructs. Data are shown as means ± SD, n = 3, two-tailed unpaired t-test, ns, no significance, *P < 0.05, **P < 0.01. C–D Inhibition of P300 downregulated PEPT1. SiNC, cells transfected with negative control siRNA, siP300#1, siP300#2, two siRNAs for P300. Data are shown as means ± SD, n = 3, two-tailed unpaired t-test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. E ChIP-qPCR analysis showed the effect of P300 expression on H3K18/K27Ac occupancy at the PEPT1 promoter. Cells were treated with DMSO or 1 μM SAHA for 48 h. SiNC, cells transfected with negative control siRNA, siP300, cells transfected with siP300#2. Data are shown as means ± SD, two-tailed unpaired t-test, ns, no significance, **P < 0.01, ***P < 0.001, ****P < 0.0001. F ChIP-qPCR analysis showed the effect of HDAC1 expression on H3K18/K27Ac occupancy at the PEPT1 promoter. SiNC, cells transfected with negative control siRNA, siHDAC1, cells transfected with siHDAC1#2. Data are shown as means±SD, two-tailed unpaired t-test, ***P < 0.001, ****P < 0.0001.
Sensitization of CRC cells to UBEN by PEPT1 activation
UBEN, a dipeptide analog, has been demonstrated to have diverse antitumor and immunomodulatory effects36,37. As UBEN is a typical PEPT1 substrate and appears to be transported into cells predominantly by PEPT138,39, the effects of UBEN are mediated by this oligopeptide transport activity18. Our earlier results showed that transcriptional repression of PEPT1 was associated with DNA methylation and that the demethylation reagent DAC induced PEPT1 expression. We, therefore, examined whether a combination treatment of DAC and UBEN enhanced cell death relative to either agent alone in vitro and in vivo. As shown in Fig. 6A, DAC greatly enhanced the cytotoxic effect of UBEN. Based on the Chou-Talalay method40, the sequential combination of DAC and UBEN resulted in a synergistic effect, with a combination index (CI) value less than 0.8 (Fig. 6B). DAC pretreatment lowered the IC50 values for UBEN from 106.82 to 2.09 μM (51-fold improvement) in SW480, and from an undetermined IC50 to 0.38 μM in SW620 (Fig. 6C). This result gives a new insight into how understanding PEPT1 epigenetic regulation allows a targeted approach to the synergistic drug combination of DAC and UBEN. Finally, the in vivo effects of combination therapy with DAC and UBEN were analyzed in SW480 and SW620 xenograft models. The timeline of the three cycles of combination chemotherapy is shown in Fig. 6D. PEPT1 expression was induced in xenograft tumors 7 days after the first DAC pretreatment (Fig. S7A and B). The administration of DAC resulted in weak tumor-suppressive effects. However, the sequential combination of DAC and UBEN led to strong tumor suppression effects, with a 50% reduction in tumor weight (Figs. 6E, F and S7D). In contrast, there were no significant changes in body weight between the treated and untreated groups (Fig. S7C). Collectively, these results demonstrate that DAC treatment, which induces epigenetic alteration of PEPT1, enhanced cellular accumulation and increased cytotoxicity of UBEN both in vitro and in vivo.
Fig. 6. Activation of PEPT1 promotes the antitumor effects of UBEN in CRC.

A Dose-effect curves of DAC, UBEN, and combination (DAC + UBEN) treatment in SW480 and SW620 cells. Cells were treated as indicated in Supplemental Table S2 and were subsequently analyzed using CCK8 assay. Data are shown as means ± SD, n = 6, nonlinear regression (curve fit) analysis. B Combination index (CI)–fraction affected (Fa) plots of DAC and UBEN combination were calculated by CompuSyn software in CRC cells. CI value is defined as follows: <0.8 is synergistic effect, from 0.8 to 1.2 is additive effect and >1.2 is antagonistic effect. C IC50 values of UBEN in CRC cells receiving combination treatment compared with UBEN alone. D Drug administration timeline and dosing schedule for xenograft models of CRC cells. E Relative tumor volume (RTV) curves in SW480 and SW620 xenograft models. Data represent the mean ± SD (n = 5). NC, UBEN, DAC, and DAC + UNEN indicate mice treated with sterile saline, ubenimex alone, decitabine alone, and decitabine-ubenimex combination, respectively. F Tumor weight of mice bearing SW480 and SW620 xenografts. Data represent means ± SD (n = 5), two-tailed unpaired t-test, ns, no significance, *P < 0.05, **P < 0.01.
Discussion
UBEN, a CD13/aminopeptidase N inhibitor, has been used in adjuvant chemotherapy as an excellent anticancer immunopotentiator and has been found to have cytotoxic effects in several cancer-cell lines41–44. Previous studies reported that combining UBEN with anticancer drugs, such as 5-FU, CDDP, and DXR, can reverse the resistance of various cancer cells to anticancer drugs by increasing intracellular ROS levels45. However, further mechanisms or signaling pathways underlying resistance to UBEN have not been clearly elucidated. Cancer cells have been shown to defend themselves against some chemotherapeutics, at least in part, by repression of the transporters responsible for their uptake4. In this study, we attempted to reverse drug resistance by targeting the epigenetic mechanisms that alter the expression of the transporter responsible for the uptake of UBEN into cancer cells.
PEPT1 is increasingly recognized as an important determinant of drug efficacy and a promising and attractive target in prodrug design15,46. In this study, our present data suggesting that the mRNA and protein levels of PEPT1 are decreased in CRC. Importantly, we found that DAC induced PEPT1 and enhanced the cytotoxicity of UBEN against human CRC cells, suggesting that PEPT1 plays a crucial role in chemotherapy resistance. It is also possible that other peptide transporters, such as PHT1, PHT2, and in particular PEPT2, which has a structural resemblance to PEPT1, may contribute to multidrug resistance in CRC47. However, PEPT2 is not found in the intestine and is instead highly expressed in renal proximal tubular cells47,48. In addition, DAC did not alter the expression level of PEPT2 in CRC cells (Fig. S8), indicating that UBEN is primarily transported by PEPT1, consequently resulting in high accumulation and increased cytotoxicity of UBEN after DAC treatment in CRC cells.
Our investigations into the epigenetic mechanisms underlying PEPT1 repression in CRC have indicated that both DNA methylation and histone acetylation regulate PEPT1 transcription (Fig. 7). We showed that a hypomethylated CpG island at the proximal promoter and occupancy of H3K18/27Ac at distal promoters leads to transcriptional activation of PEPT1 in the normal colorectum. CREB-binding protein (CBP) and its paralog P300 are histone acetyltransferases capable of acetylating H3K18, H3K27, H3K56, H3K14, and H3K2327,49. Previous studies have demonstrated that CBP and P300 act as histone acetyltransferase complexes due to their conserved sequence regions50,51. Some evidence also indicates that CBP and P300 perform unique functions52. For example, P300, but not CBP, independent of its histone acetyltransferase activity, is required to induce Tripartite motif 22 in IFNγ-mediated antiviral activity53. In mouse embryonic stem cells, only P300, and not CBP, is a critical factor for maintaining H3K27Ac at specific promoter regions of the genome54. In contrast, CBP (but not P300 or PCAF) is responsible for the hyperacetylation of DDX21, which impairs its helicase activity and leads to the accumulation of R loops and DNA damage55. Consistent with this, our results indicate that P300 is predominantly responsible for H3K18/27ac around the PEPT1 promoter. In addition, P300 contains an intrinsic and conserved DNA-binding domain, which shows a preferential affinity for the sequence GGGAGTG35. Our results confirmed that the element GGGAGTG (−1706 to 1701 bp) at the PEPT1 promoter contributed to the transcription of PEPT1. Future studies on P300 catalytic core domains such as PHD fingers may further decipher the details of histone acetylation at the PEPT1 promoter. Furthermore, we found that PEPT1 promoter repression in CRC is associated with a hypermethylated CpG island at the proximal promoter mediated by DNMT1 and the absence of H3K18/27Ac around the distal promoter due to HDAC1. Aberrant DNMT1 expression has been detected in CRC31,56 and, consistently, our results showed that enrichment of DNMT1 at the proximal promoter region of PEPT1 was responsible for PEPT1 hypermethylation. In summary, our data suggest that alteration of DNA methylation, as well as histone acetylation, contribute to the repression of PEPT1, regardless of gender, age, TNM stage, and location in CRC. Promoter hyper-methylation is an early event in CRC carcinogenesis. Commercial Cologuard® Kit57, a multi-target stool DNA test that measures two methylation biomarkers (BMP3, NDRG4), is available for screening CRC. Aberrant DNA methylation at the proximal promoter region of the PEPT1 gene (−264 to +36 bp) in stool, plasma, and serum might be beneficial for the diagnosis of CRC.
Fig. 7. PEPT1 transcription machinery in Colorectal cancer.

A hypomethylated CpG island at the proximal promoter and occupancy of H3K18/27Ac at distal promoters leads to transcriptional activation of PEPT1 in the normal colorectum. P300, but not CBP, is mainly responsible for H3K18/27ac around PEPT1 promoter and the element GGGAGTG (−1706 to 1701 bp) at PEPT1 promoter contributed to the basic transcription of PEPT1. Furthermore, the repressive PEPT1 promoter in CRC is characterized by a hypermethylated CpG island at the proximal promoter mediated by DNMT1 and the absence of H3K18/27Ac around the distal promoter due to HDAC1. The red star represents the dipeptide anti-cancer drug.
HDAC1-10 are essential transcriptional cofactors that lead to low acetylation. Interestingly, HDAC1 was also found to be overexpressed in CRC58 and promoted tumor angiogenesis by activating HIF1α/VEGFA59. A novel HDAC1 inhibitor, CBUD-1001, exerted anticancer effects by modulating apoptosis and EMT in CRC cells60. In our study, HDAC1 led to histone hypoacetylation at site H3K27, which was acetylated by P300. Moreover, DNMT1 itself was associated with deacetylase activity61, and when isolated from nuclear extracts, was found to be associated with HDAC1, as well as purified methyltransferase activity. It is therefore possible that the DNMT1/HDAC1 complex is required for maintaining the expression of PEPT1. Further research into the mechanisms of DNA methylation and its crosstalk with histone acetylation will be required in future studies.
Conclusion
Thus, our study improves our understanding of the epigenetic repression of PEPT1 promoters in CRC, we successfully applied this knowledge to design an epigenetic combination therapy sensitizing CRC to UBEN in vitro and in vivo.
Materials and methods
Patients
Fifty-eight fresh paired primary CRC specimens (Table S1) were prospectively collected for the study in one year. Of these, 14 paired CRC specimens (14/58) were obtained for the analysis of the PEPT1 protein level by western blotting. All tissue samples were obtained immediately after surgical resection from Hangzhou Cancer Hospital, with the patients’ written informed consent and approval from the Institutional Review Board of Hangzhou Cancer Hospital (Permit Number: HZCH-2017-09).
Cell culture and drug treatment
The human CRC cell lines SW620 and SW480 were purchased from the cell bank of the Chinese Academy of Sciences (Shanghai, China, with STR detection ok) and were maintained in Leibovitz’s L-15 medium (Corning, USA) supplemented with 10% (v/v) fetal bovine serum (Gibco, USA) and 1% penicillin-streptomycin solution (New Cell & Molecular Biotech, China). Both cell lines were cultured at 37 °C in a humidified incubator without CO2 according to the instructions of the American Type Culture Collection (ATCC). UBEN was purchased from National Drug Reference Standards (purity 99.5%, Beijing, China). The DNMT inhibitor DAC and HDAC inhibitors trichostatin A (TSA) and SAHA (vorinostat) were purchased from Selleck (Houston, TX, USA) and dissolved in DMSO as a 50 mM stock solution. For drug treatment, cells at a confluency of 20–30% were treated with medium containing the indicated doses of DAC for 72 h, with fresh medium replacement every 24 h. Treatments with SAHA (48 h) and TSA (24 h) were performed similarly at cell densities of 40–50% and 60%, respectively.
CCK8 assay
SW620 and SW480 cells were seeded into 96-well plates at a density of 1000 or 2000 cells per well, respectively, and pre-cultured overnight at 37 °C. In this study, we designed three groups of drug treatments, as listed in Table S2. For CCK8 assays, cells were treated with the drug treatments indicated for 72 h and subsequently maintained in a drug-free medium for 24 h. Next, 100 μL of fresh medium containing 10 µL CCK8 solution was added to each well of the 96-well plate, and the absorbance at 450 nm and 650 nm was read by a microplate reader (BioTek, USA). For drug synergy analysis, the combination index (CI) calculation was performed using Compusyn software (ComboSyn, Inc.) as described previously40. The CI values indicated the drug relationships as follows: <0.8 for synergism, 0.8 to 1.2 for additivity, and >1.2 for antagonism.
Real-time quantitative polymerase chain reaction analysis
Total RNA was extracted from tissues and cells using the RNA mini-prep kit (Tiangen, China) and multisource total RNA mini-prep kit (Axygen, China), respectively. For gene expression studies, a reverse-transcription reaction was performed using PrimeScript RT Master Mix (Takara, Japan) to acquire cDNA, followed by a real-time quantitative polymerase chain reaction (RT-qPCR). The detection of amplification products conducted using TB Green Premix Ex Taq (Takara, Japan) in a 7500 Real-Time PCR instrument (Applied Biosystems). Relative gene expression values were calculated by the 2−ΔΔCT method. All data were normalized to the reference genes PPIB and GAPDH for tissues and cell lines, respectively. The specific primers are listed in Table S3.
Western blotting analysis
Whole-cell lysates of CRC tissues and cells were prepared in radioimmunoprecipitation assay (RIPA) buffer (Beyotime, China) supplemented with protease inhibitor cocktail (Beyotime, China), followed by centrifugation at 20,000×g for 10 min at 4 °C. The protein supernatants were collected and their concentration was quantified using a BCA Protein Assay Kit (Beyotime, China) to allow the dilution to similar concentrations with PBS and 5× protein loading buffer (Sangon, China). Samples were boiled for 10 min at 100 °C to allow denaturation and then SDS-PAGE analysis was performed immediately. Equal quantities of protein were loaded to each well and separated as follows: 70 V for 30 min in a 5% stacking gel, and then 150 V for 1.5 h in a 10% separating gel. The isolated protein was then transferred electrophoretically at 200 mA for 1.5 h from the gels onto PVDF membranes, followed by blocking in 5% skim milk at room temperature. Primary antibodies [anti-SLC15A1 (1:500, Abcam Cat# ab78020), anti-DNMT1 (1:1000, Abcam Cat# ab13537), anti-HDAC1 (1:2000, Abcam Cat# ab7028), anti-KAT3B/p300 (1;1000, Abcam Cat# ab14984), and anti-GAPDH (1:3000, Multi Sciences, Cat# Mab5465)] were incubated with PVDF membranes overnight at 4 °C. After washing, anti-rabbit IgG(H + L) and anti-mouse IgG(H + L) HRP-conjugated secondary antibodies (Multi-Science, China) were diluted 1:5000 and applied for 2 h at room temperature. Finally, the blots were washed three times with 1× TBST, and the immunoreactive protein bands were detected using ultra-sensitive ECL chemiluminescence substrate (4 A Biotech, China) in a C428-Odyssey-SA-GBOX Biosystem (LICOR, USA).
Small interfering RNA transfection
The small interfering RNAs (siRNAs) for CBP/P300, DNMTs, and HDACs listed in Table S4 were synthesized by GenePharm (Shanghai, China) and transfected into CRC cells using Lipofectamine 3000 (Life Technologies, Waltham, MA) according to the manufacturer’s instructions. In brief, a mixture of Opti-MEM (Gibco, USA), 4 µl siRNA, and 5 µl Lipofectamine 3000 (Thermo Fisher, USA) was pre-incubated for 15 min at room temperature and then added to 70-80% confluent cells in a 6-well plate with fresh L-15 medium. A negative control siRNA was transiently transfected in the same way, and 48 h was the optimal sampling time for gene expression assays.
Luciferase assays
The PGL3 Basic vector (Promega) was recombined with different sheared PEPT1 promoter fragments to construct reporter plasmids. The promoter fragments of PEPT1 were obtained by PCR with the specific primers listed in Table S3, with cell genomic DNA as an amplifying template. SW480 and SW620 CRC cells at 70–90% confluency in 24-well plates were transfected with siRNAs (as described in section 2.6) 24 h before plasmid transfection. After transfection for 48 h, cells were harvested and subjected to luciferase assay using the Dual-Luciferase Reporter Assay System (Promega, USA) according to the manufacturer’s protocol. The relative light unit (RLU) was calculated as the firefly luciferase activity from the promoter construct normalized to the corresponding Renilla luciferase activity of the same sample.
Bisulfite sequencing analysis
Genomic DNA was extracted from CRC tissues and matched para-cancerous normal tissues, or CRC cell lines, using the QIAamp DNA Mini Kit (Qiagen, Germany). Sodium bisulfite conversion of genomic DNA and the subsequent cleanup of the converted DNA was carried out using the EpiTect Fast DNA Bisulfite Kit (Qiagen, Germany) according to the manufacturer’s instructions. The Bisulfite sequencing PCR (BSP) primers used were designed using the software MethPrimer62 and are listed in Table S3. The bisulfite-treated DNA was further amplified by PCR using Zymo Taq PreMix (Zymo Research, USA). PCR was performed under the following conditions: pre-denaturation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 53 °C for 30 s, and extension at 72 °C for 1 min, followed by a final extension at 72 °C for 7 min. The annealing temperature used in the PCR reaction was optimized by combined bisulfite restriction analysis (COBRA)63 to eliminate biased amplification. In brief, the promoter region of PEPT1 (−2223 bp to +129 bp) was amplified and used as a reference template. The template was treated with (methylated) or without (mock-treated) CpG Methyltransferase M.SssI (New England Biolabs, USA) and S-adenosyl-L-methionine (SAM), followed by digestion with the methylation-sensitive restriction enzyme HpaII (New England Biolabs, USA) to identify the methylation efficiency. The methylated and mock-treated templates were converted and purified as described above. Subsequently, a 1:1 mixture of the fully methylated and mock-methylated samples was used as the template for gradient PCR. The PCR products were treated with MbiI (Thermo Fisher, USA), which only digested PCR products generated from methylated templates (as selected by Snake Charmer software). After restriction digestion, the DNA was analyzed using agarose gel electrophoresis. The annealing temperature at which both unmethylated and fully methylated DNA was amplified in an unbiased way was used in the BSP experiments. The purified PCR products were ligated with the pMD19-T vector using the DNA Ligation Kit Ver. 2.1 (Takara, Japan) and subsequently transformed into E. coli DH5α for sequencing. At least 10 clones for each sample were sequenced, and data analysis was performed using BiQ Analyzer.
Chromatin immunoprecipitation (ChIP) assay
To further elucidate the epigenetic regulatory mechanisms of the target gene, we performed a ChIP assay adapted from that described previously64, with optimal modifications. The genomic DNA was broken into different-sized fragments, with 200–1000 bp DNA ladders interacting with histone acetylation marks, and 1000–2000 bp fragments being used for detection of transcription factor binding. CRC paired tissues and cells treated with drugs were crosslinked with PBS containing 1.1% formaldehyde (Thermo Forma, USA) on a rotating wheel before chromatin extraction, and then the chromatin was sheared by sonication. For tissue chromatin, an extra treatment with micrococcal nuclease (Sangong, China) was included to loosen chromatin before sonication. A 1% agarose gel was used to check the shearing efficiency. Proper sheared chromatin, together with ChIP-grade antibody and TrueBlot® Anti-Rabbit Ig IP Agarose Beads (Rockland, USA), was incubated overnight at 4 °C with gentle shaking. After washing the antibody/chromatin/bead complexes with ice-cold LiCl washing buffer and TE buffer, the eluted antibody/chromatin in the supernatant was collected for reversion crosslinking in a 65 °C water bath overnight. Finally, following incubation with DNase-free RNase A (Tiangen, China) and proteinase K (Tiangen, China), the purified chromatin was extracted with phenol: chloroform: isoamyl alcohol (25:24:1, v/v) for subsequent RT-qPCR. Specific primers used in ChIP-qPCR are listed in Table S3. The enrichment of ChIP was indicated as the percentage (%) of input. ChIP-grade antibodies used in this study were as follows: anti-H3 (Abcam, Cat# ab1791), anti-H3Ac (Millipore, Cat# 06-599), anti-H3K9Ac (Abcam, Cat# ab4441), anti-H3K18Ac (Abcam, Cat# ab1191), anti-H3K27Ac (Abcam, Cat# ab4729), anti-DNMT1 (Abcam, Cat# 19905), and normal rabbit IgG (CST, Cat# 2729) as a negative control. Considering the difference in yield and purity between tumor tissues and adjacent tissues, as well as cells treated with drugs, the histone acetylation marks were normalized to the signal obtained with H365.
Animals and experimental design
Four-week-old female immune-deficient nude mice (BALB/c nude) were purchased from GemPharmatech (Jiangsu, China) and maintained under specific pathogen-free conditions with access to food and water ad libitum and under a constant temperature, humidity, and light cycle (12 h/12 h). All mouse experiments followed the relevant guidelines of Animal Welfare and were approved by the Zhejiang University Animal Care and Use Committee (Ethics Code: ZJU20200062). For experiments, 2.5 × 106 SW620 or 1 × 107 SW480 cells were suspended in 100 µL PBS and implanted subcutaneously into the right axilla of each mouse. When the tumor size approached 50 mm3, approximately one week later, mice were randomly divided into four treatment groups (n = 5): DAC (5 mg/kg b.w., i.p.), UBEN (15 mg/kg b.w., i.p.), combination (DAC and UBEN, i.p.), and negative control (blank solvent, sterile saline, i.p.), according to tumor volume. The doses of DAC and UBEN were based on a previous study66. The drug administration cycle comprised of three sequential DAC inductions on day 0, followed by 3-day UBEN treatment. The mouse weights and tumor sizes were measured and recorded every day. Tumor volumes were calculated using the formula L × W2/2, where L and W represent the longest and shortest dimensions, respectively. Relative tumor volume (RTV) and relative body weight (RBW) were normalized to the tumor volume and body weight on the first day of treatment with drugs, respectively. Mice were euthanized if the tumor size reached 2000 mm3 At the end of the experimental period (approximately 22 days), all animals were euthanized, and the primary xenograft tumors were extracted, weighed, and collected for further analysis.
Statistical analysis
All results were expressed as the means ± SD and analyzed using GraphPad Prism 6 (GraphPad Software, USA). The statistical significance of two sets was calculated by unpaired t-test (one or two-tailed) as specifically mentioned, while the differences among groups were monitored by one-way analysis of variance (ANOVA), as per the significant level at p < 0.05. Drug IC50 calculations were performed using Prism version 6.0 with nonlinear regression (curve fit). Sample sizes of patient tissues and animal models were pre-calculated from our experience. Tissue samples were excluded from analysis when ∆Ct of housekeeping gene in paired tissues was more than 2. Animals were same-sex and age but were randomly divided into different groups. The investigator was not blinded to the group allocation during the experiment. Replicate of each experiment was listed in the figure legend.
Supplementary information
Appendix A. Supplementary figures and Supplementary tables.
Appendix B. Original image files and descriptions of western blotting in figures.
Acknowledgements
We would like to thank Editage (www.editage.cn) for English language editing.
Author contributions
X.Z. and L.Y. conceived and designed the experiments. Y.W. and X.Z. wrote the manuscript. Y.W. and L.Q. performed the majority of the experiments and data analysis. J.W. and L.Y. conducted some experiments about Bisulfite sequencing analysis. Y.H., S.W. and S.Z. supervised and validated the paper. All authors have read and approved the final manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 81702801), China Postdoctoral Science Foundation (2020M680130), Zhejiang Provincial Natural Sciences Foundation of China (No. LY18H310012), Hangzhou City Scientific Technology Research Foundation of Zhejiang Province, China (No. 20180533B67), National Natural Science Foundation of China (No. 81872935).
Data availability
Chemical compounds studied in this article Decitabine (PubChem CID: 451668); Vorinostat (PubChem CID: 5311); Trichostatin A (PubChem CID: 444732); Ubenimex (PubChem CID: 72172). Key gene targets expressed in this article are hyperlinked to corresponding entries in the GEPIA2021 database (http://gepia2.cancerpku.cn/#analysis), which provides RNA sequencing (RNA-seq) data from 9736 tumors of The Cancer Genome Atlas (TCGA) database and 8587 normal samples of the Genotype-Tissue Expression (GTEx) database and offers tools for differential analysis, survival analysis, similar gene analysis, correlation analysis, and principal component analysis.
Ethics statement
This project was approved by the Institutional Review Board of Hangzhou Cancer Hospital (Permit Number: HZCH-2017-09). All animal experiments were approved by the Zhejiang University Animal Care and Use Committee (Ethics Code: ZJU20200062).
Conflict of interest
The authors declare no competing interests.
Footnotes
Edited by A. Woloszynska
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yanhong Wang, Jiaqi Wang
Contributor Information
Lushan Yu, Email: yuls@zju.edu.cn.
Xiaoli Zheng, Email: gezixll@hotmail.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41419-021-03814-5.
References
- 1.Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. caac.21660 10.3322/caac.21660 (2021). [DOI] [PubMed]
- 2.Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019;394:1467–1480. doi: 10.1016/S0140-6736(19)32319-0. [DOI] [PubMed] [Google Scholar]
- 3.Li W, et al. Overcoming ABC transporter-mediated multidrug resistance: molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updat. 2016;27:14–29. doi: 10.1016/j.drup.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, et al. Epigenetic activation of the drug transporter OCT2 sensitizes renal cell carcinoma to oxaliplatin. Sci. Transl. Med. 2016;8:348ra97–348ra97. doi: 10.1126/scitranslmed.aaf3124. [DOI] [PubMed] [Google Scholar]
- 5.Ye C, et al. Inhibition of histone deacetylase 7 reverses concentrative nucleoside transporter 2 repression in colorectal cancer by up-regulating histone acetylation state: CNT2 repression in colorectal cancer. Br. J. Pharmacol. 2018;175:4209–4217. doi: 10.1111/bph.14467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daniel H, Kottra G. The proton oligopeptide cotransporter family SLC15 in physiology and pharmacology. Pflug. Arch. 2004;447:610–618. doi: 10.1007/s00424-003-1101-4. [DOI] [PubMed] [Google Scholar]
- 7.Agu R, et al. Proton-coupled oligopeptide transporter (POT) family expression in human nasal epithelium and their drug transport potential. Mol. Pharm. 2011;8:664–672. doi: 10.1021/mp100234z. [DOI] [PubMed] [Google Scholar]
- 8.Smith DE, et al. Tubular localization and tissue distribution of peptide transporters in rat kidney. Pharm. Res. 1998;15:1244–1249. doi: 10.1023/A:1011996009332. [DOI] [PubMed] [Google Scholar]
- 9.Merlin D, et al. Colonic epithelial hPepT1 expression occurs in inflammatory bowel disease: transport of bacterial peptides influences expression of MHC class 1 molecules. Gastroenterology. 2001;120:1666–1679. doi: 10.1053/gast.2001.24845. [DOI] [PubMed] [Google Scholar]
- 10.Sun D, et al. Expression of proton-coupled oligopeptide transporter (POTs) in prostate of mice and patients with benign prostatic hyperplasia (BPH) and prostate cancer (PCa) Prostate. 2013;73:287–295. doi: 10.1002/pros.22568. [DOI] [PubMed] [Google Scholar]
- 11.Wang C-Y, Liu S, Xie X-N, Tan Z-R. Regulation profile of the intestinal peptide transporter 1 (PepT1) Drug Des. Dev. Ther. 2017;11:3511–3517. doi: 10.2147/DDDT.S151725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziegler TR, et al. Distribution of the H+/peptide transporter PepT1 in human intestine: up-regulated expression in the colonic mucosa of patients with short-bowel syndrome. Am. J. Clin. Nutr. 2002;75:922–930. doi: 10.1093/ajcn/75.5.922. [DOI] [PubMed] [Google Scholar]
- 13.Sai Y, et al. Immunolocalization and pharmacological relevance of oligopeptide transporter PepT1 in intestinal absorption of beta-lactam antibiotics. FEBS Lett. 1996;392:25–29. doi: 10.1016/0014-5793(96)00778-8. [DOI] [PubMed] [Google Scholar]
- 14.Wuensch T, et al. The peptide transporter PEPT1 is expressed in distal colon in rodents and humans and contributes to water absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2013;305:G66–G73. doi: 10.1152/ajpgi.00491.2012. [DOI] [PubMed] [Google Scholar]
- 15.Minhas GS, Newstead S. Structural basis for prodrug recognition by the SLC15 family of proton-coupled peptide transporters. Proc. Natl Acad. Sci. USA. 2019;116:804–809. doi: 10.1073/pnas.1813715116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ota K. Review of ubenimex (Bestatin): clinical research. Biomed. Pharmacother. 1991;45:55–60. doi: 10.1016/0753-3322(91)90123-B. [DOI] [PubMed] [Google Scholar]
- 17.Sekine K, Fujii H, Abe F. Induction of apoptosis by bestatin (ubenimex) in human leukemic cell lines. Leukemia. 1999;13:729–734. doi: 10.1038/sj.leu.2401388. [DOI] [PubMed] [Google Scholar]
- 18.Nakanishi T, Tamai I, Takaki A, Tsuji A. Cancer cell-targeted drug delivery utilizing oligopeptide transport activity. Int. J. Cancer. 2000;88:274–280. doi: 10.1002/1097-0215(20001015)88:2<274::AID-IJC20>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 19.Shimakura J, Terada T, Katsura T, Inui K-I. Characterization of the human peptide transporter PEPT1 promoter: Sp1 functions as a basal transcriptional regulator of human PEPT1. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;289:G471–G477. doi: 10.1152/ajpgi.00025.2005. [DOI] [PubMed] [Google Scholar]
- 20.Shimakura J, Terada T, Shimada Y, Katsura T, Inui K. The transcription factor Cdx2 regulates the intestine-specific expression of human peptide transporter 1 through functional interaction with Sp1. Biochem. Pharmacol. 2006;71:1581–1588. doi: 10.1016/j.bcp.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 21.Geillinger KE, et al. Nrf2 regulates the expression of the peptide transporter PEPT1 in the human colon carcinoma cell line Caco-2. Biochim. Biophys. Acta. 2014;1840:1747–1754. doi: 10.1016/j.bbagen.2013.12.026. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z-M, et al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature. 2018;554:387–391. doi: 10.1038/nature25477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009;10:805–811. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sen GL, Reuter JA, Webster DE, Zhu L, Khavari PA. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature. 2010;463:563–567. doi: 10.1038/nature08683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robert M-F, et al. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet. 2003;33:61–65. doi: 10.1038/ng1068. [DOI] [PubMed] [Google Scholar]
- 26.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin Q, et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30:249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015;16:258–264. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Z, Shilatifard A. Epigenetic modifications of histones in cancer. Genome Biol. 2019;20:245. doi: 10.1186/s13059-019-1870-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okugawa Y, Grady WM, Goel A. Epigenetic alterations in colorectal cancer: emerging biomarkers. Gastroenterology. 2015;149:1204–1225. doi: 10.1053/j.gastro.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo Y, et al. Intestinal PPARα protects against colon carcinogenesis via regulation of methyltransferases DNMT1 and PRMT6. Gastroenterology. 2019;157:744–759. doi: 10.1053/j.gastro.2019.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z, et al. Acetylation of PHF5A modulates stress responses and colorectal carcinogenesis through alternative splicing-mediated upregulation of KDM3A. Mol. Cell. 2019;74:1250–1263. doi: 10.1016/j.molcel.2019.04.009. [DOI] [PubMed] [Google Scholar]
- 33.Raisner R, et al. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep. 2018;24:1722–1729. doi: 10.1016/j.celrep.2018.07.041. [DOI] [PubMed] [Google Scholar]
- 34.Peng J, et al. p300/CBP inhibitor A-485 alleviates acute liver injury by regulating macrophage activation and polarization. Theranostics. 2019;9:8344–8361. doi: 10.7150/thno.30707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rikitake Y, Moran E. DNA-binding properties of the E1A-associated 300-kilodalton protein. Mol. Cell. Biol. 1992;12:2826–2836. doi: 10.1128/MCB.12.6.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, et al. Role of ubenimex as an anticancer drug and its synergistic effect with Akt inhibitor in human A375 and A2058 cells. OncoTargets Ther. 2018;11:943–953. doi: 10.2147/OTT.S157480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang Y, et al. Discovery of a novel chimeric ubenimex-gemcitabine with potent oral antitumor activity. Bioorg. Med. Chem. 2016;24:5787–5795. doi: 10.1016/j.bmc.2016.09.033. [DOI] [PubMed] [Google Scholar]
- 38.Wang L, et al. PEPT1- and OAT1/3-mediated drug-drug interactions between bestatin and cefixime in vivo and in vitro in rats, and in vitro in human. Eur. J. Pharm. Sci. 2014;63:77–86. doi: 10.1016/j.ejps.2014.06.019. [DOI] [PubMed] [Google Scholar]
- 39.Jia Y, et al. Enhancement effect of resveratrol on the intestinal absorption of bestatin by regulating PEPT1, MDR1 and MRP2 in vivo and in vitro. Int. J. Pharm. 2015;495:588–598. doi: 10.1016/j.ijpharm.2015.09.042. [DOI] [PubMed] [Google Scholar]
- 40.Chou T-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 41.Otsuki, T. et al. Aminopeptidase N/CD13 as a potential therapeutic target in malignant pleural mesothelioma. Eur. Respir. J. 51, 1701610 (2018). [DOI] [PubMed]
- 42.Terauchi M, et al. Inhibition of APN/CD13 leads to suppressed progressive potential in ovarian carcinoma cells. BMC Cancer. 2007;7:140. doi: 10.1186/1471-2407-7-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Umezawa H, Aoyagi T, Suda H, Hamada M, Takeuchi T. Bestatin, an inhibitor of aminopeptidase B, produced by actinomycetes. J. Antibiot. 1976;29:97–99. doi: 10.7164/antibiotics.29.97. [DOI] [PubMed] [Google Scholar]
- 44.Muskardin DT, Voelkel NF, Fitzpatrick FA. Modulation of pulmonary leukotriene formation and perfusion pressure by bestatin, an inhibitor of leukotriene A4 hydrolase. Biochem. Pharm. 1994;48:131–137. doi: 10.1016/0006-2952(94)90232-1. [DOI] [PubMed] [Google Scholar]
- 45.Yamashita M, et al. A CD13 inhibitor, ubenimex, synergistically enhances the effects of anticancer drugs in hepatocellular carcinoma. Int. J. Oncol. 2016;49:89–98. doi: 10.3892/ijo.2016.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson BR, Shi J, Zhu H-J, Smith DE. Pharmacokinetics of gemcitabine and its amino acid ester prodrug following intravenous and oral administrations in mice. Biochem. Pharmacol. 2020;180:114127. doi: 10.1016/j.bcp.2020.114127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newstead S. Recent advances in understanding proton coupled peptide transport via the POT family. Curr. Opin. Struct. Biol. 2017;45:17–24. doi: 10.1016/j.sbi.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu X, et al. Human oligopeptide transporter 2 (PEPT2) mediates cellular uptake of polymyxins. J. Antimicrob. Chemother. 2016;71:403–412. doi: 10.1093/jac/dkv340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–117. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manickavinayaham S, et al. E2F1 acetylation directs p300/CBP-mediated histone acetylation at DNA double-strand breaks to facilitate repair. Nat. Commun. 2019;10:4951. doi: 10.1038/s41467-019-12861-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H-M, et al. A potent CBP/p300-Snail interaction inhibitor suppresses tumor growth and metastasis in wild-type p53-expressing cancer. Sci. Adv. 2020;6:eaaw8500. doi: 10.1126/sciadv.aaw8500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henry RA, Kuo Y-M, Andrews AJ. Differences in specificity and selectivity between CBP and p300 acetylation of histone H3 and H3/H4. Biochemistry. 2013;52:5746–5759. doi: 10.1021/bi400684q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao B, et al. p300, but not PCAF, collaborates with IRF-1 in stimulating TRIM22 expression independently of its histone acetyltransferase activity. Eur. J. Immunol. 2013;43:2174–2184. doi: 10.1002/eji.201343308. [DOI] [PubMed] [Google Scholar]
- 54.Martire S, Nguyen J, Sundaresan A, Banaszynski LA. Differential contribution of p300 and CBP to regulatory element acetylation in mESCs. BMC Mol. Cell Biol. 2020;21:55. doi: 10.1186/s12860-020-00296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song C, Hotz-Wagenblatt A, Voit R, Grummt I. SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev. 2017;31:1370–1381. doi: 10.1101/gad.300624.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xie J, et al. Metformin selectively inhibits metastatic colorectal cancer with the KRAS mutation by intracellular accumulation through silencing MATE1. Proc. Natl Acad. Sci. USA. 2020;117:13012–13022. doi: 10.1073/pnas.1918845117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Imperiale TF, et al. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014;370:1287–1297. doi: 10.1056/NEJMoa1311194. [DOI] [PubMed] [Google Scholar]
- 58.Lutz L, et al. Histone modifiers and marks define heterogeneous groups of colorectal carcinomas and affect responses to HDAC inhibitors in vitro. Am. J. Cancer Res. 2016;6:664–676. [PMC free article] [PubMed] [Google Scholar]
- 59.Chen C, et al. The histone deacetylase HDAC1 activates HIF1α/VEGFA signal pathway in colorectal cancer. Gene. 2020;754:144851. doi: 10.1016/j.gene.2020.144851. [DOI] [PubMed] [Google Scholar]
- 60.Kim, S. L., La, M. T., Shin, M. W., Kim, S.-W. & Kim, H.-K. A novel HDAC1 inhibitor, CBUD-1001, exerts anticancer effects by modulating the apoptosis and EMT of colorectal cancer cells. Int. J. Oncol. 10.3892/ijo.2020.5109 (2020). [DOI] [PubMed]
- 61.Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000;24:88–91. doi: 10.1038/71750. [DOI] [PubMed] [Google Scholar]
- 62.Li L-C, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 63.Eads, C. A. & Laird, P. W. Combined Bisulfite Restriction Analysis (COBRA). In DNA Methylation Protocols (eds. Mills, K. I. & Ramsahoye, B. H.) 71–86 (Springer, New York, 2002). [DOI] [PubMed]
- 64.Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc. 2006;1:729–748. doi: 10.1038/nprot.2006.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haring M, et al. Chromatin immunoprecipitation: optimization, quantitative analysis and data normalization. Plant Methods. 2007;3:11. doi: 10.1186/1746-4811-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao S, et al. Inhibition of LTA4H by bestatin in human and mouse colorectal cancer. EBioMedicine. 2019;44:361–374. doi: 10.1016/j.ebiom.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix A. Supplementary figures and Supplementary tables.
Appendix B. Original image files and descriptions of western blotting in figures.
Data Availability Statement
Chemical compounds studied in this article Decitabine (PubChem CID: 451668); Vorinostat (PubChem CID: 5311); Trichostatin A (PubChem CID: 444732); Ubenimex (PubChem CID: 72172). Key gene targets expressed in this article are hyperlinked to corresponding entries in the GEPIA2021 database (http://gepia2.cancerpku.cn/#analysis), which provides RNA sequencing (RNA-seq) data from 9736 tumors of The Cancer Genome Atlas (TCGA) database and 8587 normal samples of the Genotype-Tissue Expression (GTEx) database and offers tools for differential analysis, survival analysis, similar gene analysis, correlation analysis, and principal component analysis.