

Abstract
As one of the most lethal diseases, pancreatic cancer shows a dismal overall prognosis and high resistance to most treatment modalities. Furthermore, pancreatic cancer escapes early detection during the curable period because early symptoms rarely emerge and specific markers for this disease have not been found. Although combinations of new drugs, multimodal therapies, and adjuvants prolong survival, most patients still relapse after surgery and eventually die. Consequently, the search for more effective treatments for pancreatic cancer is highly relevant and justified. As a newly re-discovered mediator of gasotransmission, hydrogen sulfide (H2S) undertakes essential functions, encompassing various signaling complexes that occupy key processes in human biology. Accumulating evidence indicates that H2S exhibits bimodal modulation of cancer development. Thus, endogenous or low levels of exogenous H2S are thought to promote cancer, whereas high doses of exogenous H2S suppress tumor proliferation. Similarly, inhibition of endogenous H2S production also suppresses tumor proliferation. Accordingly, H2S biosynthesis inhibitors and H2S supplementation (H2S donors) are two distinct strategies for the treatment of cancer. Unfortunately, modulation of endogenous H2S on pancreatic cancer has not been studied so far. However, H2S donors and their derivatives have been extensively studied as potential therapeutic agents for pancreatic cancer therapy by inhibiting cell proliferation, inducing apoptosis, arresting cell cycle, and suppressing invasion and migration through exploiting multiple signaling pathways. As far as we know, there is no review of the effects of H2S donors on pancreatic cancer. Based on these concerns, the therapeutic effects of some H2S donors and NO–H2S dual donors on pancreatic cancer were summarized in this paper. Exogenous H2S donors may be promising compounds for pancreatic cancer treatment.
KEY WORDS: Pancreatic cancer, Hydrogen sulfide donor, Sulfur-containing compound, Cell proliferation, Antitumor effect, Signaling pathway
Abbreviations: AMPK, adenosine 5′-monophosphate-activated protein kinase; BCL-2, B-cell lymphoma-2; BITC, benzyl isothiocyanate; BRCA2, breast cancer 2; CAT, cysteine aminotransferase; CBS, cystathionine-β-synthase; CDC25B, cell division cycle 25B; CDK1, cyclin-dependent kinase 1; CHK2, checkpoint kinase 2; CSE, cystathionine-γ-lyase; DATS, diallyl trisulfide; DR4, death receptor; EMT, epithelial–mesenchymal transition; ERK1/2, extracellular signal-regulated kinase; ERU, erucin; FOXM1, forkhead box protein M1; GLUTs, glucose transporters; HDAC, histone deacetylase; HEATR1, human HEAT repeat-containing protein 1; HIF-1α, hypoxia inducible factor; H2S, hydrogen sulfide; iNOS, inducible nitric oxide synthase; ITCs, isothiocyanates; JNK, c-Jun N-terminal kinase; KEAP1‒NRF2‒ARE, the recombinant protein 1-nuclear factor erythroid-2 related factor 2-antioxidant response element; KRAS, kirsten rat sarcoma viral oncogene; 3-MST, 3-mercaptopyruvate sulfurtransferase; NF-κB, nuclear factor kappa B; NO, nitric oxide; OCT-4, octamer-binding transcription factor 4; PARP, poly(ADP-ribose)-polymerase; PDGFRα, platelet-derived growth factor receptor; PEITC, phenethyl isothiocyanate; PI3K/AKT, phosphoinositide 3-kinase/v-AKT murine thymoma viral oncogene; P16, multiple tumor suppressor 1; RASAL2, RAS protein activator like 2; ROS, reactive oxygen species; RPL10, human ribosomal protein L10; SFN, sulforaphane; SHH, sonic hedgehog; SMAD4, mothers against decapentaplegic homolog 4; STAT-3, signal transducer and activator of transcription 3; TRAIL, The human tumor necrosis factor-related apoptosis-inducing ligand; VEGF, vascular endothelial growth factor; XIAP, X-linked inhibitor of apoptosis protein; ZEB1, zinc finger E box-binding protein-1
Graphical abstract
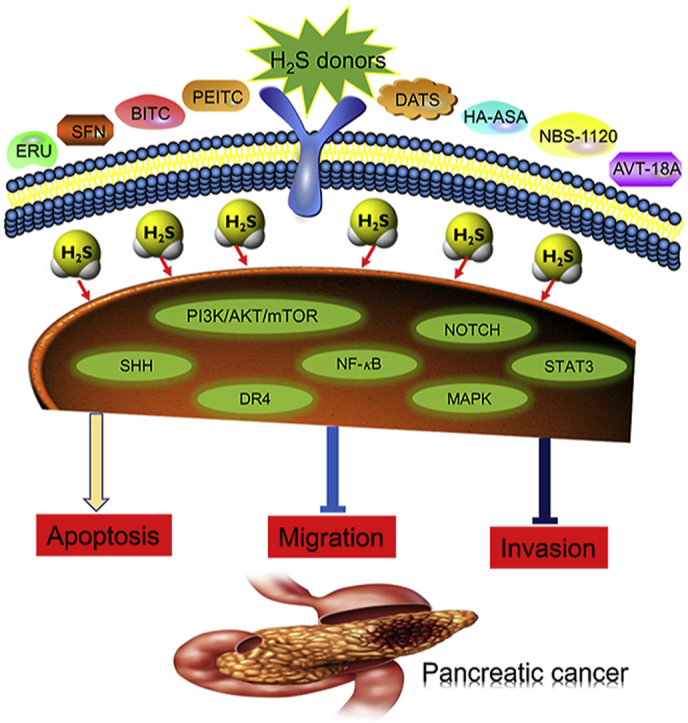
The review summarizes the antitumor mechanisms of H2S donating derivatives against pancreatic cancer by modulating signaling pathways, including PI3K/AKT/mTOR, SHH, NF-κB, MAPK, NOTCH, STAT3, DR4, etc. Perspectives on future directions and challenges are also discussed.
1. Introduction
The increasing number of cases and cancer deaths has critically affected human life and health1. Pancreatic cancer causes the fourth highest mortality rate in developed countries, and is estimated to be the second most deadly neoplasm in the next decade, surpassed only by lung cancer2. Moreover, unhealthy living habits, such as alcohol or nicotine consumption, and pre-existing conditions like type 2 diabetes mellitus, chronic pancreatitis and obesity favor the development of pancreatic cancer3, 4, 5, 6. Frustratingly, the prognosis of this cancer is extremely poor in major malignancies. The lack of early diagnosis, strong invasiveness and early distant metastases are thought to be responsible for the poor outcomes following radical surgical resection7. In addition, the microenvironment of pancreatic cancer is complex8, and the neoplasm is thought to result from the accumulation of multiple genetic and epigenetic mutations, including multiple tumor suppressor 1 (P16), kirsten rat sarcoma viral oncogene (KRAS), mothers against decapentaplegic homolog 4 (SMAD4), breast cancer 2 (BRCA2), human ribosomal protein L10 (RPL10), human HEAT repeat-containing protein 1 (HEATR1) and others9, 10, 11, 12, 13, 14. All these factors lead to <7% 5-year overall survival rate, with most patients expiring within the first year after diagnosis15. Although surgery continues to be employed as first choice of treatment strategies, late diagnosis limits surgical treatment, prompting the need for adjuvant therapy, radiotherapy and chemotherapy16. The chemotherapy drugs contain gemcitabine, and the combination of irinotecan, oxaliplatin and nab-paclitaxel or 5-fluorouracile17,18. Furthermore, radiotherapy and targeted therapies also fail to produce significant clinical benefits19. With intensifying research, new emerging strategies for the treatment of pancreatic cancer have evolved, including immunotherapies20. Molecularly targeted therapies21,22, focusing on the tumor microenvironment as a potential target23, have also been developed. Because these therapies are still far from satisfactory, the search for more drug candidates to treat pancreatic cancer is critical.
Hydrogen sulfide (H2S) is the third gasotransmitter possessing a regulatory effect on vascular function and intracellular signaling. It is generated from l-cysteine mainly through the catalysis of cysteine aminotransferase (CAT), cystathionine-β-synthase (CBS), 3-mercaptopyruvate sulfurtransferase (3-MST) and cystathionine-γ-lyase (CSE) in the mammalian tissues including pancreas, liver, kidney, intestine, heart and central nervous system24, 25, 26, 27, 28, 29. Great interest has been shown in H2S due to the extensive pharmacological and pathological activities in human30. As brief examples, in the cardiovascular system, H2S mitigates oxidative stress and myocardial injury connected with ischemia‒reperfusion events31,32. For the central nervous system, H2S exerts the protective effects against neurodegenerative diseases by the antioxidant properties33,34. In the immune system, H2S presents anti-inflammatory effects through binding with zinc, ferric iron or copper residues of metalloproteins and enabling sulfidation of protein cysteine residues35,36. In addition, H2S functions in homeostatic mechanisms and affects pancreatic β-cells by inhibiting the release of insulin and reducing cellular stress caused by glucose37,38. It can also function in repairing the damage of inflammation and is used as to protect gastrointestinal mucosa39. It is well documented that H2S regulates the kidney excretory capacity on renal tubular cells by inhibiting sodium transporters40. Many studies have documented that the extensive physiological functions of H2S are mediated by numerous molecular mechanisms, including the persulfidation of target proteins, interactions with ion channels and others41. Persulfidation, a key post-translational modification, regulates the function of the proteins. H2S is oxidized to polysulfide, or the target cysteine is oxidized to sulfonic acid or disulfide to form a cysteine persulfide, which then accesses to the cytosol to affect the activity of the protein42. Moreover, H2S also interacts with the metal center of the target protein, especially heme protein, by inducing the covalent modification of heme to form sulfheme43,44. Finally, H2S targets several ion channels and receptors to modulate the functions of different systems45. For example, H2S protects the heart from ischemia and reperfusion injury through activating ATP-sensitive K+ channels46. It contracts vascular smooth muscle by inhibiting T-type Ca2+ channels47 and exerts protective effects on oxidative neurons through actions on Cl‒ channels48. Over the last decade, researchers have turned their attention to the antitumor effects of H2S49, 50, 51. Considerable evidence has shown that H2S regulates oxidative stress, interacts with free radicals and inactivates tumorigenic pathways52. However, it is noteworthy that H2S exhibits a bell-shaped pattern in cancer therapy (Fig. 1). Thus, endogenous or low concentrations of exogenous H2S possess tumor-promoting effects by stimulating angiogenesis with increased cell proliferation and metastasis53, 54, 55. In contrast, high-dose exogenous H2S administration or inhibition of endogenous H2S production enables cancer cell death through inhibiting proliferation, inducing apoptosis and arresting cell cycle56, 57, 58. The paradoxical role indicates that H2S biosynthesis inhibitors (CBS, CBE and 3-MST inhibitors) and H2S supplementation (H2S donors) open up two distinct strategies for the treatment of cancer.
Figure 1.

The schematic diagram of bell-shaped effects of the exogenous and endogenous H2S on cancer. In short, endogenous or low concentrations of exogenous H2S promotes cancer growth by stimulating angiogenesis, and promoting cell proliferation and metastasis. Inhibition of endogenous H2S production or high-dose exogenous H2S administration enables cancer cell death through inhibiting proliferation, inducing apoptosis and DNA damage, and arresting cell cycle. The models suggest that the inhibitors of H2S biosynthetic enzymes and H2S donors represent two strategies to treat cancer.
Exogenous H2S donors belong to a class of sulfur-containing compounds. For centuries, sulfur has widely existed in natural products and continues to maintain its status as the dominating heteroatom, due to various biological activities and extremely important functions in the pharmaceutical industry59. Currently, more than 250 U.S. Food and Drug Administration approved sulfur-based small molecule drugs are available in the market, which are widely used to treat various types of diseases. Devimistat, venetoclax, trabadectin, bleomycin and others are used to treat cancer60, 61, 62. Specifically, devimistat has passed the European Medicines Agency orphan drug designation for the treatment of metastatic pancreatic cancer by triggering reactive oxygen species (ROS)-associated apoptosis, increasing autophagy and repressing lipid metabolism through activating the AMPK signaling63. It is speculated that the therapeutic effect may be partly the result of released H2S during drug metabolism or the effect of the drugs on the production of endogenous H2S64. Although reviews have been published on the role of H2S and its donors in some kinds of cancers, including prostate cancer, colorectal cancer, kidney cancer, bladder cancer and triple-negative breast cancer65, 66, 67, 68, no systematic review of the effects on pancreatic cancer have thus far been reported. Therefore, the therapeutic effects of H2S donors on pancreatic cancer are reviewed in this paper.
2. Antitumor mechanisms of H2S donors against pancreatic cancer
Natural isothiocyanates (ITCs) isolated from Brassicaceae plants, including erucin (ERU), sulforaphane (SFN), benzyl isothiocyanate (BITC) and phenethyl isothiocyanate (PEITC, Fig. 2), slowly release H2S in biological environments69, 70, 71. Due to H2S release, the relatively high concentrations of ITCs exhibit significant inhibitory effects on cancer cell growth by inducing apoptosis, arresting cell cycle, interacting with the recombinant protein 1-nuclear factor erythroid-2 related factor 2-antioxidant response element (KEAP1‒NRF2‒ARE) pathway and by up-regulating phase II detoxification enzymes72,73. Notably, ITCs have high bioavailability and are easily absorbed, making them potential compounds for anticancer therapy73.
Figure 2.

The chemical structures of H2S donors and NO–H2S dual donor derivatives with potential anti-pancreatic cancer activity.
Recently, H2S donors were found to possess potential in the treatment of pancreatic cancer. In 2019, Citi et al.74 reported that ERU (Fig. 2) could cross cell membranes of AsPC-1 and release H2S at the intracellular level. Furthermore, high concentration of ERU (30–100 μmol/L) inhibited cell proliferation by reducing phosphorylated extracellular signal regulated kinase (ERK1/2) levels, and inducing cell apoptosis through up-regulating the apoptosis marker caspase-3 and caspase-7 expression (Fig. 3). As shown in Fig. 3, the mitogen-activated protein kinase (MAPK) pathway mainly contains three groups: ERK, P38, and c-Jun NH2 terminal kinase (JNK), which play vital roles in cell proliferation, differentiation, survival and apoptosis75. In addition, pancreatic cancer cell lines are characterized by mutations in the KRAS gene, which leads to a KRAS hyperactivation with a resulting hyperphosphorylation of the downstream kinases ERK1/276. FAS and binding of the human tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to the death receptor (DR4) lead to the recruitment of an adaptor protein FAS-associated death domain (FADD), which functions as a molecular bridge to caspase-8. This activated caspase-8 directly cleaves and activates caspase-3 and -7, which further cleaves the downstream substrates poly ADP ribose polymerase (PARP) to stimulate apoptosis77,78.
Figure 3.

Possible signaling pathways of ITCs and DATS involved in the antiproliferation of pancreatic cancer. ITCs, including ERU, SFN, BITC and PEITC, belong to natural H2S donors. They slowly release H2S in biological environments69, 70, 71. The relatively high concentrations of ITCs and DATS exhibit antiproliferative effects on pancreatic cancer by inducing apoptosis, arresting cell cycle and suppressing invasion and migration of tumor cells72,73. In brief, these donors suppressed cell proliferation by inhibiting SHH86, PI3K/AKT/mTOR92, SPTFs94, STAT394, RAC190, and NOTCH95, and activating AMPK79 and RASAL282 signaling pathways. Moreover, they also inhibited early metastasis by down-regulating ZEB186, β-CATENIN85, TWIST-185, and vimentin85. Further, they arrested cell cycle by up-regulating P2188, activating CHK288, and down-regulating cyclin B1/CDK1102 and CDC25B87. In addition, they induced apoptosis through activating caspase-374, caspase-774, BAK95, BAX102, P3875, JNK75, DR477 and ERK76, and inhibiting BCL-285, BCL-XL95, NF-κB110 and PARP78.
Chen et al.79 explored the in vitro and in vivo antitumor effect of SFN (Fig. 2) on pancreatic cell lines PANC-1 and MIA PaCa-2. The results showed that SFN inhibited cancer cells proliferation, colony formation, migration and invasion. Moreover, after SFN treatment, excessively generated ROS activated adenosine 5′-monophosphate-activated protein kinase (AMPK), and subsequently elevated the NRF2 nuclear translocation, which inhibited pancreatic cancer cell proliferation (Fig. 3). In a pancreatic cancer transgenic mouse model, SFN treatment (50 mg/kg, i.p.) inhibited tumor growth, consistent with the antiproliferative effects of SFN through ROS activated AMPK signaling pathway and NRF2 nuclear translocation. Kallifatidis et al.80 verified that SFN induced apoptosis of pancreatic AsPC-1, MIA PaCa-2, Capan-1 and BxPC-3 cells by the repression of nuclear factor kappa B (NF-κB) through inhibiting the subunit c-Rel activity and down-regulation of anti-apoptotic genes XIAP and cIAP1 expression. MIA PaCa-2 cells were xenografted subcutaneously into nude mice, and tumor growth was measured in untreated mice or upon treatment with SFN, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), or both. The results demonstrated that SFN (4.4 mg/kg) alone or combined with TRAIL potentially reduced tumor growth by inhibiting NF-κB activity, tumor angiogenesis, proliferation and inducing apoptosis without exhibiting toxicity to normal tissue. Soon afterwards, the same research group81 reported that SFN induced the expression of miR-365a-3p to restrain NF-κB activity through down-regulating c-Rel, eventually leading to the apoptosis of pancreatic cancer cells AsPC-1, BxPC-3, PANC-1 and MIA PaCa-2. After the miR-365a-5p functional effects were established in vitro, xenograft egg model studies were performed, and lipofected BxPC-3 cells were transplanted to the chorioallantoic membrane in chick development. The findings revealed that the miR-365a-3p expression induced by SFN were 6-fold higher after xenotransplantation than those in control cells. Moreover, the xenografts tumor sizes with high miR-365a-3p expression were significantly decreased. Antiproliferative effects were also reported on AsPC-1, BxPC-3 and PANC-1 cells, and tumor growth in the BxPC-3 xenograft egg model was strongly inhibited by SFN through inducing miR-135b-5p expression, followed by enhanced expression of the RAS protein activator like 2 (RASAL2) protein (Fig. 3)82. Subsequently, Li et al.83,84 revealed that SFN inhibited the proliferation of PANC-1 and MIA PaCa-2 cells by disrupting HSP90–P50Cdc37, and by directly interacting with the amino acid residues of HSP90. Srivastava et al.85 reported that SFN induced pancreatic cancer cell apoptosis by inhibiting X-linked inhibitor of apoptosis protein (XIAP) and the phosphorylation of FKHR, alterations in the levels of B-cell lymphoma-2 (BCL-2), and activation of caspase-3 in MIA PaCa-2, BxPC-3, AsPC-1 and PANC-1 cells. Additionally, it also inhibited early metastasis by down-regulating the epithelial–mesenchymal transition (EMT) related proteins vimentin, β-CATENIN, TWIST-1 and zinc finger E box-binding protein-1 (ZEB1, Fig. 3). Li et al.86 observed that SFN significantly inhibited proliferation and angiogenesis of stem cells of mice NOD/SCID/IL2Rγ with CD133+/CD44+/CD24+/ESA+ immunophenotype through blocking the sonic hedgehog (SHH) pathway and its downstream target gene expression, including transcription factors NANOG, platelet-derived growth factor receptor (PDGFRα), octamer-binding transcription factor 4 (OCT-4) and vascular endothelial growth factor (VEGF, Fig. 3). Further, SFN induced apoptosis and inhibited early metastasis by inhibiting the expression of XIAP and BCL-2, and down-regulating ZEB1 levels, respectively.
Srivastava et al.87, 88, 89 studied the role of the natural isothiocyanate compound BITC (Fig. 2) in pancreatic cancer cell lines and animal models. As early as 2004, they found that BITC dose-dependently inhibited BxPC-3 cells proliferation through inactivating NF-κB by down-regulating the protein levels of IκBα and cyclin D1. The treatment of BITC down-regulated the protein levels of cyclin B1, cyclin-dependent kinase 1 (CDK1), and cell division cycle 25B (CDC25B) and arrested cell cycle at G2/M stage (Fig. 3). Further, BITC also induced cell apoptosis through enhancing the BAX/BCL-2 ratio, the DNA fragmentation, and PARP and procaspase-3 cleavage (Fig. 3)87. In 2006, BITC was suggested to induce DNA damage by causing phosphorylation of H2A.X in Capan-2 cells. BITC caused cell cycle arrest at G2/M phase through up-regulating the levels of cyclin dependent kinase inhibitor P21 and checkpoint kinase 2 (CHK2)88. In 2009, these authors showed that BITC treatment led to ROS generation, which was orchestrated by depleting the level of reduced glutathione. The subsequent G2/M arrest was caused through the activation of ERK, whereas apoptosis was induced by activating JNK, ERK and P38 in MIA PaCa-2 and Capan-2 cells (Fig. 3)89. In the same year, another research group, Basu et al.90 reported that the proapoptotic and antiproliferative properties of BITC were related to the down-regulation of RAC1 and the activation of DR4 in CFPAC-1, HS766T and BxPC-3 cells. In 2010, they proposed that the inhibition of histone deacetylase (HDAC) 1/3 and NF-κB by BITC might suppress the growth of BxPC-3 and Capan-2 cells in vitro. In BxPC-3 pancreatic tumor xenografts in athymic nude mice, oral BITC administration (90 mg/kg) significantly suppressed the growth of pancreatic tumor xenograft by down-regulating expression of NF-κB, cyclin D1, HDAC 1 and HDAC 391. In 2011, Boreddy et al.92 reported that BITC induced apoptosis by blocking the phosphoinositide 3-kinase/v-AKT murine thymoma viral oncogene (PI3K/AKT) pathway and mainly up-regulating their downstream FOXO1 pathway in PANC-1 and BxPC-3 cells (Fig. 3). In BxPC-3 tumor-bearing mice with the tumor size of approximately 70 mm3, the results showed that oral gavage of 12 μmol/L BITC significantly reduced the tumor growth, and tumor volume in the treated group (266.7 ± 35.4 mm3) was reduced as compared with control groups (465.8 ± 30.8 mm3). Furthermore, there was no significant difference in mice weight, indicating no apparent systemic toxicity in BITC-treated mice. Subsequently the levels of PI3K and AKT were examined in the tumor lysates by Western blot, confirming that BITC suppressed tumor growth by inducing apoptosis which was related to the inhibition of PI3K/AKT pathway. This group then showed that BITC impeded cell proliferation by inhibiting tumor angiogenesis through the signal transducer and activator of transcription 3 (STAT-3)-dependent pathway by suppressing vascular endothelial growth factor (VEGFR-2) phosphorylation and hypoxia inducible factor (HIF-1α) levels, and increased expression of Rho-GTPases (Fig. 3). Following these results, in vivo tumor xenograft experiments were conducted in female athymic nude mice. Treatment of mice with 12 μmol/L BITC markedly suppressed BxPC-3 tumor growth, and BITC treated tumor xenografts showed 61% reduced hemoglobin content, compared with untreated xenografts. Tumors were then analyzed by Western blot, which showed that the phosphorylation of STAT-3, VEGR-2 and the expression of HIF-1α and RhoC in the tumors of mice treated with BITC were significantly reduced, consistent with the in vitro results93. In 2016, Kasiappan et al.94 revealed that BITC inhibited cell growth and invasion in PANC-1, L3.6pl and MIA PaCa-2 through enhancing the generation of ROS to down-regulate the expression of SP transcription factors, STAT3 and c-MYC (Fig. 3). These findings are consistent with the results of L3.6pl xenograft mice, which showed that treatment of mice with 20 mg/kg BITC by i.p. injection remarkably inhibited tumor growth and did not affect body weights. In addition, Western blot analysis of tumor lysates affirmed that STAT3 levels were decreased in tumors from BITC-treated mice.
In related studies, Stan et al.95 demonstrated that PEITC (Fig. 2) also dose-dependently inhibited the viability of MIA PaCa-2, PL-45 and BxPC-3 cells through apoptosis. The treatment of PEITC up-regulated BAK and down-regulated BCL-2 and BCL-XL expression (Fig. 3). PEITC also suppressed cell proliferation by reducing the levels of NOTCH 1 and 2 (Fig. 3). In a MIA PaCa-2 xenograft mice model with a tumor size of approximately 673.8 mm3, oral administration of 12 μmol/L PEITC suppressed growth, with 37% lower tumor volume compared with control. Notably, PEITC treatment was relatively safe and did not cause organ damage.
In another approach, Fortunato et al.96 exploited the large amount of sugar consumption of cancer cells by overexpressing the membrane glucose transporters (GLUTs)97, especially glucose and fructose, to develop a series of H2S-releasing glycoconjugates. These agents delivered high amounts of H2S to cytoplasm with a resulting antitumor effect. More importantly, the aqueous solubility of the derivatives was improved by the glycoconjugation with H2S-releasing unit (isothiocyanate portions). Among them, glycoconjugated H2S donors 1 and 2 (Fig. 2) released H2S in AsPC-1 cells which altered the cell cycle and showed potent inhibitory effects on cell viability.
In other related work, diallyl trisulfide (DATS, Fig. 2) was shown to react with thiols in the cell, then crossed the cell membranes and interacted with intracellular glutathione to generate H2S98, 99, 100, which contributed to the antitumor effect101. Ma et al.102 showed that DATS inhibited Capan-2 cell proliferation by arresting cell cycle and inducing apoptosis. Western blot results indicated that cell cycle retention was related to increased P21 expression and decreased levels of cyclin B1. BCL-2 down-regulation and the increase of P53, FAS and BAX protein levels were involved in the apoptosis process (Fig. 3).
HS-aspirin (HS-ASA, Fig. 2) is composed of the non-steroidal anti-inflammatory drug aspirin and ADT-OH (the dithiolethione unit). In vitro studies found that HS-ASA inhibited the growth of MIA PaCa-2 and BxPC-3 cells with the IC50 values of 2.1 and 1.9 μmol/L, respectively. Moreover, HS-ASA inhibited proliferation, arrested the cell cycle at G0/G1 phase, and induced apoptosis103.
Like H2S, nitric oxide (NO), as a gasotransmitter, possesses a wide range of physiological and pathological effects104. Accumulating evidence indicates that high concentrations of NO, either biologically generated by inducible nitric oxide synthase (iNOS) or by NO donors, exert antitumor activities by suppressing cell proliferation, improving the sensitivity of tumor cells to radiotherapy, chemotherapy and immune-toxicities, and also inhibiting metastasis and EMT. Mechanically, NO-mediated effects mainly regulate the NF-κB/SNAIL/RKIP/PTEN signaling pathway in cancer cells105. In pancreatic cancer, NO plays a significant role in the invasion of PANC-1 cells by a JAK independent and MEK-ERK-dependent mechanism106. In addition, excessive production of NO can lead to epigenetic and genetic changes in pancreatic cancer107,108. Recently, Kashfi et al.109 reported a series of ASA hybrids consisting of both NO and H2S-releasing moieties. Among them, NOSH-1 (NBS-1120, Fig. 2) was the most potent one in MIA PaCa-2 and BxPC-3 cells with the IC50 values of 0.047 and 0.057 μmol/L, respectively. Moreover, the lactate dehydrogenase release assay indicated that NBS-1120 possessed a remarkable degree of safety In-depth study showed that NBS-1120 inhibited proliferation, arrested cell cycle and eventually led to increased apoptosis, which was associated with the increased levels of caspase-3 and ROS. Furthermore, in MIA PaCa-2 xenograft mice model with the tumor size of approximately 70 mm3, 100 mg/kg NBS-1120-treated mice by gavage showed a significant reduction in tumor volume (330 ± 95 mm3), compared with control mice (3265 ± 520 mm3). Tumor mass was also inhibited in NBS-1120-treated mice (0.62 ± 0.25 g) vs. controls (2.47 ± 0.24 g). Immunohistochemical examination of tumor sections demonstrated inhibition of tumor growth, accompanied by the induction of apoptosis, cell cycle arrest by increasing the expression of P53, and decreased expression of NF-κB and forkhead box protein M1 (FOXM1). In short, these signaling pathways contributed to NBS-1120 mediated pancreatic cancer inhibition in vitro and in vivo110.
Subsequently, Kashfi et al.111 developed another compound (NOSH-sulindac, AVT-18A, Fig. 2) which could also release both NO and H2S. The antiproliferative activity test results showed that AVT-18A possessed potent effects on inhibiting BxPC-3 and MIA PaCa-2 growth with the IC50 values of 0.098 and 0.12 μmol/L, respectively, which was at least 8000-fold more potent than sulindac. AVT-18A also inhibited proliferation through inducing apoptosis and G2/M phase cell cycle arrest.
3. Conclusions and perspectives
Despite advances in therapy strategy, pancreatic cancer remains a fatal disease, due to the late diagnosis, poor prognosis, and extensive resistance to most treatment modalities. To date, the mortality caused by pancreatic cancer still gradually increases, which shows the increased need for effective therapeutic drugs.
Currently, H2S is a rapidly developing field in biomedical research. H2S donors which release H2S under different conditions are constantly emerging. Although the pathways involved in endogenous H2S production have not been clearly elucidated in pancreatic cancer, current data suggest that some H2S-donors including mainly ITCs, DATS and ADT–OH–aspirin hybrids have been shown to have beneficial effects. For example, many chemotherapeutic drugs exert drug resistance by inhibiting FOXO proteins or activating AKT, but BITC boosts FOXO nuclear shuttling by inhibiting AKT phosphorylation to weaken the resistance of tumor cells and induce cell apoptosis. BITC-mediated generation of ROS and inhibition of NF-κB cause pancreatic cancer cell cycle arrest and apoptosis. It also restrains tumor angiogenesis and proliferation through blocking HIF-1α/VEGF/Rho-GTPases pathways. Moreover, the synergistic interaction of H2S and NO maintains and enhances the expression of cGMP to promote angiogenesis and vasodilation112,113. Furthermore, the crosstalk of H2S and NO under physiological conditions has been proved to exert antitumor activity114. Based on these considerations, the dual H2S and NO donor drugs (NBS-1120 and AVT-18A) are currently being developed, and various pharmacological activities are evaluated, particularly for the anti-pancreatic cancer actions by suppressing cell proliferation, inducing apoptosis, and arresting cell cycle through increasing ROS, iNOS and P53, and decreasing NF-κB and FOXM1. As previously noticed, H2S donors are expected to be further developed in pancreatic cancer. Yet some problems also need to be addressed urgently, such as, how to specifically deliver therapeutic concentrations of H2S to pancreatic cancer cells, how to preserve a reasonable H2S concentration in pancreatic cancer tissues, how to measure accurate content of H2S in pancreatic cancer cells, and how to clarify the function of each of the two gaseous transmitters in dual H2S/NO donors. With progress in the understanding of molecular mechanisms of the pancreatic cancer driver genes, molecular targeted drugs are being applied to pancreatic cancer treatments, yet the overall benefit is limited115,116. Despite the lack of targeted therapy drugs, it is still a promising direction that deserves research and exploration. Recent research shows that ROS-activated, pH-controlled and photo-controllable H2S donors can be applied based on the special tumor microenvironment117, 118, 119. Last but not least, H2S donors can be used to combine with the marketed pancreatic cancer drugs or drug candidates, hoping to increase the efficacy and selectivity, and reduce drug resistance and side effects.
Acknowledgments
This paper was financially supported by Fok Ying Tung Education Foundation (171035, China), State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources (CMEMR2018-B04, Guangxi Normal University, China); High-level Innovative Project in Shenyang (Young and Middle-aged Technological Innovative Support Plan, RC190483, China) and Career Development Support Plan in Shenyang Pharmaceutical University, China.
Author contributions
Xu Hu designed and wrote the paper. Yan Xiao revised the manuscript according to the reviewers’ suggestions. Jianan Sun, Bao Ji, Shanshan Luo, Bo Wu, Chao Zheng, Peng Wang and Keguang Cheng wrote and revised the original manuscript. Fanxing Xu, Huiming Hua, and Dahong Li were responsible for the conception and design of the review.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Fanxing Xu, Email: fanxing0011@163.com.
Huiming Hua, Email: huimhua@163.com.
Dahong Li, Email: lidahong0203@163.com.
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. 2020. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L., Smith B.D., Aizenberg R., Rosenzweig A.B., Fleshman J.M., Matrisian L.M. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Can Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 3.Kleeff J., Korc M., Apte M., La Vecchia C., Johnson C.D., Biankin A.V. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. doi: 10.1038/nrdp.2016.22. [DOI] [PubMed] [Google Scholar]
- 4.Alexandrov L.B., Ju Y.S., Haase K., Van Loo P., Martincorena I., Nik-Zainal S. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618–622. doi: 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tramacere I., Scotti L., Jenab M., Bagnardi V., Bellocco R., Rota M. Alcohol drinking and pancreatic cancer risk: a meta-analysis of the dose-risk relation. Int J Canc. 2010;126:1474–1486. doi: 10.1002/ijc.24936. [DOI] [PubMed] [Google Scholar]
- 6.Pothuraju R., Rachagani S., Junker W.M., Chaudhary S., Saraswathi V., Kaur S. Pancreatic cancer associated with obesity and diabetes: an alternative approach for its targeting. J Exp Clin Canc Res. 2018;37:319. doi: 10.1186/s13046-018-0963-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neoptolemos J.P., Dunn J.A., Stocken D.D., Almond J., Link K., Beger H. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: a randomised controlled trial. Lancet. 2001;358:1576–1585. doi: 10.1016/s0140-6736(01)06651-x. [DOI] [PubMed] [Google Scholar]
- 8.Ho W.J., Jaffee E.M., Zheng L. The tumour microenvironment in pancreatic cancer-clinical challenges and opportunities. Nat Rev Clin Oncol. 2020;17:527–540. doi: 10.1038/s41571-020-0363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hruban R.H., Adsay N.V., Albores-Saavedra J., Anver M.R., Biankin A.V., Boivin G.P. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Can Res. 2006;66:95–106. doi: 10.1158/0008-5472.CAN-05-2168. [DOI] [PubMed] [Google Scholar]
- 10.Hezel A.F., Kimmelman A.C., Stanger B.Z., Bardeesy N., Depinho R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 11.Nelson S.R., Walsh N. Genetic alterations featuring biological models to tailor clinical management of pancreatic cancer patients. Cancers (Basel) 2020;12:1233. doi: 10.3390/cancers12051233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mann K.M., Ying H., Juan J., Jenkins N.A., Copeland N.G. KRAS-related proteins in pancreatic cancer. Pharmacol Ther. 2016;168:29–42. doi: 10.1016/j.pharmthera.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Yang J., Chen Z., Liu N., Chen Y. Ribosomal protein L10 in mitochondria serves as a regulator for ROS level in pancreatic cancer cells. Redox Biol. 2018;19:158–165. doi: 10.1016/j.redox.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Y., Wang K., Zhou Y., Li T., Yang M., Wang R. HEATR1 deficiency promotes pancreatic cancer proliferation and gemcitabine resistance by up-regulating Nrf2 signaling. Redox Biol. 2020;29:101390. doi: 10.1016/j.redox.2019.101390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel R.L., Miller K.D., Jemal A. Cancer statistics. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 16.Neoptolemos J.P., Stocken D.D., Friess H., Bassi C., Dunn J.A., Hickey H. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350:1200–1210. doi: 10.1056/NEJMoa032295. [DOI] [PubMed] [Google Scholar]
- 17.Conroy T., Desseigne F., Ychou M., Bouche O., Guimbaud R., Becouarn Y. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 18.Von Hoff D.D., Ervin T., Arena F.P., Chiorean E.G., Infante J., Moore M. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neuzillet C., Tijeras-Raballand A., Bourget P., Cros J., Couvelard A., Sauvanet A. State of the art and future directions of pancreatic ductal adenocarcinoma therapy. Pharmacol Ther. 2015;155:80–104. doi: 10.1016/j.pharmthera.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Knudsen E.S., Kumarasamy V., Chung S., Ruiz A., Vail P., Tzetzo S. Targeting dual signalling pathways in concert with immune checkpoints for the treatment of pancreatic cancer. Gut. 2021;70:127–138. doi: 10.1136/gutjnl-2020-321000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosein A.N., Brekken R.A., Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. 2020;17:487–505. doi: 10.1038/s41575-020-0300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su T., Yang B., Gao T., Liu T., Li J. Polymer nanoparticle-assisted chemotherapy of pancreatic cancer. Ther Adv Med Oncol. 2020;12 doi: 10.1177/1758835920915978. 1758835920915978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y., Feng M., Chen H., Yang G., Qiu J., Zhao F. Mechanistic target of rapamycin in the tumor microenvironment and its potential as a therapeutic target for pancreatic cancer. Canc Lett. 2020;485:1–13. doi: 10.1016/j.canlet.2020.05.003. [DOI] [PubMed] [Google Scholar]
- 24.Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 25.Kaneko Y., Kimura Y., Kimura H., Niki I. L-Cysteine inhibits insulin release from the pancreatic beta-cell: possible involvement of metabolic production of hydrogen sulfide, a novel gasotransmitter. Diabetes. 2006;55:1391–1397. doi: 10.2337/db05-1082. [DOI] [PubMed] [Google Scholar]
- 26.Geng B., Yang J., Qi Y., Zhao J., Pang Y., Du J. H2S generated by heart in rat and its effects on cardiac function. Biochem Biophys Res Commun. 2004;313:362–368. doi: 10.1016/j.bbrc.2003.11.130. [DOI] [PubMed] [Google Scholar]
- 27.Stipanuk M.H., Beck P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel P., Vatish M., Heptinstall J., Wang R., Carson R.J. The endogenous production of hydrogen sulphide in intrauterine tissues. Reprod Biol Endocrinol. 2009;7:10. doi: 10.1186/1477-7827-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warenycia M.W., Goodwin L.R., Benishin C.G., Reiffenstein R.J., Francom D.M., Taylor J.D. Acute hydrogen sulfide poisoning. Demonstration of selective uptake of sulfide by the brainstem by measurement of brain sulfide levels. Biochem Pharmacol. 1989;38:973–981. doi: 10.1016/0006-2952(89)90288-8. [DOI] [PubMed] [Google Scholar]
- 30.Wallace J.L., Wang R. Hydrogen sulfide-based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat Rev Drug Discov. 2015;14:329–345. doi: 10.1038/nrd4433. [DOI] [PubMed] [Google Scholar]
- 31.Polhemus D.J., Lefer D.J. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ Res. 2014;114:730–737. doi: 10.1161/CIRCRESAHA.114.300505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donnarumma E., Trivedi R.K., Lefer D.J. Protective actions of H2S in acute myocardial infarction and heart failure. Comp Physiol. 2017;7:583–602. doi: 10.1002/cphy.c160023. [DOI] [PubMed] [Google Scholar]
- 33.Tabassum R., Jeong N.Y., Jung J. Protective effect of hydrogen sulfide on oxidative stress-induced neurodegenerative diseases. Neural Regen Res. 2020;15:232–241. doi: 10.4103/1673-5374.265543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karki R., Kodamullil A.T., Hofmann-Apitius M. Comorbidity analysis between Alzheimer's disease and type 2 diabetes mellitus (T2DM) based on shared pathways and the role of T2DM drugs. J Alzheimers Dis. 2017;60:721–731. doi: 10.3233/JAD-170440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sivarajah A., Collino M., Yasin M., Benetti E., Gallicchio M., Mazzon E. Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock. 2009;31:267–274. doi: 10.1097/SHK.0b013e318180ff89. [DOI] [PubMed] [Google Scholar]
- 36.Fagone P., Mazzon E., Bramanti P., Bendtzen K., Nicoletti F. Gasotransmitters and the immune system: mode of action and novel therapeutic targets. Eur J Pharmacol. 2018;834:92–102. doi: 10.1016/j.ejphar.2018.07.026. [DOI] [PubMed] [Google Scholar]
- 37.Okamoto M., Ishizaki T., Kimura T. Protective effect of hydrogen sulfide on pancreatic beta-cells. Nitric Oxide. 2015;46:32–36. doi: 10.1016/j.niox.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 38.Beltowski J., Wojcicka G., Jamroz-Wisniewska A. Hydrogen sulfide in the regulation of insulin secretion and insulin sensitivity: implications for the pathogenesis and treatment of diabetes mellitus. Biochem Pharmacol. 2018;149:60–76. doi: 10.1016/j.bcp.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 39.Chan M.V., Wallace J.L. Hydrogen sulfide-based therapeutics and gastrointestinal diseases: translating physiology to treatments. Am J Physiol Gastrointest Liver Physiol. 2013;305:G467–G473. doi: 10.1152/ajpgi.00169.2013. [DOI] [PubMed] [Google Scholar]
- 40.Cao X., Bian J.S. The role of hydrogen sulfide in renal system. Front Pharmacol. 2016;7:385. doi: 10.3389/fphar.2016.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 42.Greiner R., Pálinkás Z., Bäsell K., Becher D., Antelmann H., Nagy P. Polysulfides link H2S to protein thiol oxidation. Antioxidants Redox Signal. 2013;19:1749–1765. doi: 10.1089/ars.2012.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boubeta F.M., Bieza S.A., Bringas M., Palermo J.C., Boechi L., Estrin D.A. Hemeproteins as targets for sulfide species. Antioxidants Redox Signal. 2020;32:247–257. doi: 10.1089/ars.2019.7878. [DOI] [PubMed] [Google Scholar]
- 44.Rios-Gonzalez B.B., Roman-Morales E.M., Pietri R., Lopez-Garriga J. Hydrogen sulfide activation in hemeproteins: the sulfheme scenario. J Inorg Biochem. 2014;133:78–86. doi: 10.1016/j.jinorgbio.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang G., Wu L., Wang R. Interaction of hydrogen sulfide with ion channels. Clin Exp Pharmacol Physiol. 2010;37:753–763. doi: 10.1111/j.1440-1681.2010.05351.x. [DOI] [PubMed] [Google Scholar]
- 46.Hess R.M., Niu Y., Garrud T.A.C., Botting K.J., Ford S.G., Giussani D.A. Embryonic cardioprotection by hydrogen sulphide: studies of isolated cardiac function and ischaemia‒reperfusion injury in the chicken embryo. J Physiol. 2020;598:4197–4208. doi: 10.1113/JP279978. [DOI] [PubMed] [Google Scholar]
- 47.Sun Y.G., Cao Y.X., Wang W.W., Ma S.F., Yao T., Zhu Y.C. Hydrogen sulphide is an inhibitor of L-type calcium channels and mechanical contraction in rat cardiomyocytes. Cardiovasc Res. 2008;79:632–641. doi: 10.1093/cvr/cvn140. [DOI] [PubMed] [Google Scholar]
- 48.Kimura Y., Kimura H. Hydrogen sulfide protects neurons from oxidative stress. Faseb J. 2004;18:1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- 49.Szabo C. Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat Rev Drug Discov. 2016;15:185–203. doi: 10.1038/nrd.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hellmich M.R., Szabo C. Hydrogen sulfide and cancer. Handb Exp Pharmacol. 2015;230:233–241. doi: 10.1007/978-3-319-18144-8_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao X., Ding L., Xie Z.Z., Yang Y., Whiteman M., Moore P.K. A review of hydrogen sulfide synthesis, metabolism, and measurement: is modulation of hydrogen sulfide a novel therapeutic for cancer?. Antioxidants Redox Signal. 2019;31:1–38. doi: 10.1089/ars.2017.7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kajimura M., Fukuda R., Bateman R.M., Yamamoto T., Suematsu M. Interactions of multiple gas-transducing systems: hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxidants Redox Signal. 2010;13:157–192. doi: 10.1089/ars.2009.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szabo C., Coletta C., Chao C., Módis K., Szczesny B., Papapetropoulos A. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci U S A. 2013;110:12474–12479. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu D., Si W., Wang M., Lv S., Ji A., Li Y. Hydrogen sulfide in cancer: friend or foe?. Nitric Oxide. 2015;50:38–45. doi: 10.1016/j.niox.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 55.Untereiner A.A., Pavlidou A., Druzhyna N., Papapetropoulos A., Hellmich M.R., Szabo C. Drug resistance induces the upregulation of H2S-producing enzymes in HCT116 colon cancer cells. Biochem Pharmacol. 2018;149:174–185. doi: 10.1016/j.bcp.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai W.J., Wang M.J., Ju L.H., Wang C., Zhu Y.C. Hydrogen sulfide induces human colon cancer cell proliferation: role of Akt, ERK and p21. Cell Biol Int. 2010;34:565–572. doi: 10.1042/CBI20090368. [DOI] [PubMed] [Google Scholar]
- 57.Wu D., Li M., Tian W., Wang S., Cui L., Li H. Hydrogen sulfide acts as a double-edged sword in human hepatocellular carcinoma cells through EGFR/ERK/MMP-2 and PTEN/AKT signaling pathways. Sci Rep. 2017;7:5134. doi: 10.1038/s41598-017-05457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang S.S., Chen Y.H., Chen N., Wang L.J., Chen D.X., Weng H.L. Hydrogen sulfide promotes autophagy of hepatocellular carcinoma cells through the PI3K/Akt/mTOR signaling pathway. Cell Death Dis. 2017;8:e2688. doi: 10.1038/cddis.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feng M., Tang B., Liang S.H., Jiang X. Sulfur containing scaffolds in drugs: synthesis and application in medicinal chemistry. Curr Top Med Chem. 2016;16:1200–1216. doi: 10.2174/1568026615666150915111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao C., Rakesh K.P., Ravidar L., Fang W.Y., Qin H.L. Pharmaceutical and medicinal significance of sulfur (SVI)-containing motifs for drug discovery: a critical review. Eur J Med Chem. 2019;162:679–734. doi: 10.1016/j.ejmech.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scott K.A., Njardarson J.T. Analysis of US FDA-approved drugs containing sulfur atoms. Top Curr Chem (Cham) 2018;376:5. doi: 10.1007/s41061-018-0184-5. [DOI] [PubMed] [Google Scholar]
- 62.Ilardi E.A., Vitaku E., Njardarson J.T. Data-mining for sulfur and fluorine: an evaluation of pharmaceuticals to reveal opportunities for drug design and discovery. J Med Chem. 2014;57:2832–2842. doi: 10.1021/jm401375q. [DOI] [PubMed] [Google Scholar]
- 63.Gao L., Xu Z., Huang Z., Tang Y., Yang D., Huang J. CPI-613 rewires lipid metabolism to enhance pancreatic cancer apoptosis via the AMPK-ACC signaling. J Exp Clin Canc Res. 2020;39:73. doi: 10.1186/s13046-020-01579-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zaorska E., Tomasova L., Koszelewski D., Ostaszewski R., Ufnal M. Hydrogen sulfide in pharmacotherapy, beyond the hydrogen sulfide-donors. Biomolecules. 2020;10:323. doi: 10.3390/biom10020323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H., Xu F., Gao G., Gao X., Wu B., Zheng C. Hydrogen sulfide and its donors: novel antitumor and antimetastatic therapies for triple-negative breast cancer. Redox Biol. 2020;34:101564. doi: 10.1016/j.redox.2020.101564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akbari M., Sogutdelen E., Juriasingani S., Sener A. Hydrogen sulfide: emerging role in bladder, kidney, and prostate malignancies. Oxid Med Cell Longev. 2019;2019:2360945. doi: 10.1155/2019/2360945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu M., Wu L., Montaut S., Yang G. Hydrogen sulfide signaling axis as a target for prostate cancer therapeutics. Prostate Cancer. 2016;2016:8108549. doi: 10.1155/2016/8108549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo F.F., Yu T.C., Hong J., Fang J.Y. Emerging roles of hydrogen sulfide in inflammatory and neoplastic colonic diseases. Front Physiol. 2016;7:156. doi: 10.3389/fphys.2016.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clarke J.D., Hsu A., Riedl K., Bella D., Schwartz S.J., Stevens J.F. Bioavailability and inter-conversion of sulforaphane and erucin in human subjects consuming broccoli sprouts or broccoli supplement in a cross-over study design. Pharmacol Res. 2011;64:456–463. doi: 10.1016/j.phrs.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Citi V., Martelli A., Testai L., Marino A., Breschi M.C., Calderone V. Hydrogen sulfide releasing capacity of natural isothiocyanates: is it a reliable explanation for the multiple biological effects of Brassicaceae?. Planta Med. 2014;80:610–613. doi: 10.1055/s-0034-1368591. [DOI] [PubMed] [Google Scholar]
- 71.Lucarini E., Micheli L., Trallori E., Citi V., Martelli A., Testai L. Effect of glucoraphanin and sulforaphane against chemotherapy-induced neuropathic pain: Kv7 potassium channels modulation by H2S release in vivo. Phytother Res. 2018;32:2226–2234. doi: 10.1002/ptr.6159. [DOI] [PubMed] [Google Scholar]
- 72.Lin Y., Yang X., Lu Y., Liang D., Huang D. Isothiocyanates as H2S donors triggered by cysteine: reaction mechanism and structure and activity relationship. Org Lett. 2019;21:5977–5980. doi: 10.1021/acs.orglett.9b02117. [DOI] [PubMed] [Google Scholar]
- 73.Fofaria N.M., Ranjan A., Kim S.H., Srivastava S.K. Mechanisms of the anticancer effects of isothiocyanates. Enzymes. 2015;37:111–137. doi: 10.1016/bs.enz.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 74.Citi V., Piragine E., Pagnotta E., Ugolini L., Di Cesare Mannelli L., Testai L. Anticancer properties of erucin, an H2S-releasing isothiocyanate, on human pancreatic adenocarcinoma cells (AsPC-1) Phytother Res. 2019;33:845–855. doi: 10.1002/ptr.6278. [DOI] [PubMed] [Google Scholar]
- 75.Wada T., Penninger J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 76.Deer E.L., Gonzalez-Hernandez J., Coursen J.D., Shea J.E., Ngatia J., Scaife C.L. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pirnia F., Schneider E., Betticher D.C., Borner M.M. Mitomycin C induces apoptosis and caspase-8 and -9 processing through a caspase-3 and Fas-independent pathway. Cell Death Differ. 2002;9:905–914. doi: 10.1038/sj.cdd.4401062. [DOI] [PubMed] [Google Scholar]
- 78.Gibson S.B., Oyer R., Spalding A.C., Anderson S.M., Johnson G.L. Increased expression of death receptors 4 and 5 synergizes the apoptosis response to combined treatment with etoposide and TRAIL. Mol Cell Biol. 2000;20:205–212. doi: 10.1128/mcb.20.1.205-212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen X., Jiang Z., Zhou C., Chen K., Li X., Wang Z. Activation of Nrf2 by sulforaphane inhibits high glucose-induced progression of pancreatic cancer via AMPK dependent signaling. Cell Physiol Biochem. 2018;50:1201–1215. doi: 10.1159/000494547. [DOI] [PubMed] [Google Scholar]
- 80.Kallifatidis G., Rausch V., Baumann B., Apel A., Beckermann B.M., Groth A. Sulforaphane targets pancreatic tumour-initiating cells by NF-kappaB-induced antiapoptotic signalling. Gut. 2009;58:949–963. doi: 10.1136/gut.2008.149039. [DOI] [PubMed] [Google Scholar]
- 81.Yin L., Xiao X., Georgikou C., Yin Y., Liu L., Karakhanova S. MicroRNA-365a-3p inhibits c-Rel-mediated NF-κB signaling and the progression of pancreatic cancer. Canc Lett. 2019;452:203–212. doi: 10.1016/j.canlet.2019.03.025. [DOI] [PubMed] [Google Scholar]
- 82.Yin L., Xiao X., Georgikou C., Luo Y., Liu L., Gladkich J. Sulforaphane induces miR135b-5p and its target gene, RASAL2, thereby inhibiting the progression of pancreatic cancer. Mol Ther Oncolytics. 2019;14:74–81. doi: 10.1016/j.omto.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li Y., Karagöz G.E., Seo Y.H., Zhang T., Jiang Y., Yu Y. Sulforaphane inhibits pancreatic cancer through disrupting Hsp90–p50Cdc37 complex and direct interactions with amino acids residues of Hsp90. J Nutr Biochem. 2012;23:1617–1626. doi: 10.1016/j.jnutbio.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Y., Zhang T., Schwartz S.J., Sun D. Sulforaphane potentiates the efficacy of 17-allylamino 17-demethoxygeldanamycin against pancreatic cancer through enhanced abrogation of Hsp90 chaperone function. Nutr Canc. 2011;63:1151–1159. doi: 10.1080/01635581.2011.596645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Srivastava R.K., Tang S.N., Zhu W., Meeker D., Shankar S. Sulforaphane synergizes with quercetin to inhibit self-renewal capacity of pancreatic cancer stem cells. Front Biosci (Elite Ed) 2011;3:515–528. doi: 10.2741/e266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li S.H., Fu J., Watkins D.N., Srivastava R.K., Shankar S. Sulforaphane regulates self-renewal of pancreatic cancer stem cells through the modulation of Sonic hedgehog-GLI pathway. Mol Cell Biochem. 2013;373:217–227. doi: 10.1007/s11010-012-1493-6. [DOI] [PubMed] [Google Scholar]
- 87.Srivastava S.K., Singh S.V. Cell cycle arrest, apoptosis induction and inhibition of nuclear factor kappa B activation in anti-proliferative activity of benzyl isothiocyanate against human pancreatic cancer cells. Carcinogenesis. 2004;25:1701–1709. doi: 10.1093/carcin/bgh179. [DOI] [PubMed] [Google Scholar]
- 88.Zhang R., Loganathan S., Humphreys I., Srivastava S.K. Benzyl isothiocyanate-induced DNA damage causes G2/M cell cycle arrest and apoptosis in human pancreatic cancer cells. J Nutr. 2006;136:2728–2734. doi: 10.1093/jn/136.11.2728. [DOI] [PubMed] [Google Scholar]
- 89.Sahu R.P., Zhang R., Batra S., Shi Y., Srivastava S.K. Benzyl isothiocyanate-mediated generation of reactive oxygen species causes cell cycle arrest and induces apoptosis via activation of MAPK in human pancreatic cancer cells. Carcinogenesis. 2009;30:1744–1753. doi: 10.1093/carcin/bgp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Basu A., Haldar S. Anti-proliferative and proapoptotic effects of benzyl isothiocyanate on human pancreatic cancer cells is linked to death receptor activation and RasGAP/Rac1 down-modulation. Int J Oncol. 2009;35:593–599. doi: 10.3892/ijo_00000370. [DOI] [PubMed] [Google Scholar]
- 91.Batra S., Sahu R.P., Kandala P.K., Srivastava S.K. Benzyl isothiocyanate-mediated inhibition of histone deacetylase leads to NF-kappaB turnoff in human pancreatic carcinoma cells. Mol Canc Therapeut. 2010;9:1596–1608. doi: 10.1158/1535-7163.MCT-09-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boreddy S.R., Pramanik K.C., Srivastava S.K. Pancreatic tumor suppression by benzyl isothiocyanate is associated with inhibition of PI3K/AKT/FOXO pathway. Clin Canc Res. 2011;17:1784–1795. doi: 10.1158/1078-0432.CCR-10-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boreddy S.R., Sahu R.P., Srivastava S.K. Benzyl isothiocyanate suppresses pancreatic tumor angiogenesis and invasion by inhibiting HIF-α/VEGF/Rho-GTPases: pivotal role of STAT-3. PLoS One. 2011;6 doi: 10.1371/journal.pone.0025799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kasiappan R., Jutooru I., Karki K., Hedrick E., Safe S. Benzyl isothiocyanate (BITC) induces reactive oxygen species-dependent repression of STAT3 protein by down-regulation of specificity proteins in pancreatic cancer. J Biol Chem. 2016;291:27122–27133. doi: 10.1074/jbc.M116.746339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stan S.D., Singh S.V., Whitcomb D.C., Brand R.E. Phenethyl isothiocyanate inhibits proliferation and induces apoptosis in pancreatic cancer cells in vitro and in a MIAPaca2 xenograft animal model. Nutr Canc. 2014;66:747–755. doi: 10.1080/01635581.2013.795979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fortunato S., Lenzi C., Granchi C., Citi V., Martelli A., Calderone V. First examples of H2S-releasing glycoconjugates: stereoselective synthesis and anticancer activities. Bioconjugate Chem. 2019;30:614–620. doi: 10.1021/acs.bioconjchem.8b00808. [DOI] [PubMed] [Google Scholar]
- 97.Macheda M.L., Rogers S., Best J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005;202:654–662. doi: 10.1002/jcp.20166. [DOI] [PubMed] [Google Scholar]
- 98.Benavides G.A., Squadrito G.L., Mills R.W., Patel H.D., Isbell T.S., Patel R.P. Hydrogen sulfide mediates the vasoactivity of garlic. Proc Natl Acad Sci U S A. 2007;104:17977–17982. doi: 10.1073/pnas.0705710104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liang D., Wu H., Wong M.W., Huang D. Diallyl trisulfide is a fast H2S donor, but diallyl disulfide is a slow one: the reaction pathways and intermediates of glutathione with polysulfides. Org Lett. 2015;17:4196–4199. doi: 10.1021/acs.orglett.5b01962. [DOI] [PubMed] [Google Scholar]
- 100.Cai Y.R., Hu C.H. Computational study of H2S release in reactions of diallyl polysulfides with thiols. J Phys Chem B. 2017;121:6359–6366. doi: 10.1021/acs.jpcb.7b03683. [DOI] [PubMed] [Google Scholar]
- 101.Zhang F., Jin H., Wu L., Shao J., Zhu X., Chen A. Diallyl trisulfide suppresses oxidative stress-induced activation of hepatic stellate cells through production of hydrogen sulfide. Oxid Med Cell Longev. 2017;2017:1406726. doi: 10.1155/2017/1406726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ma H.B., Huang S., Yin X.R., Zhang Y., Di Z.L. Apoptotic pathway induced by diallyl trisulfide in pancreatic cancer cells. World J Gastroenterol. 2014;20:193–203. doi: 10.3748/wjg.v20.i1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chattopadhyay M., Kodela R., Nath N., Dastagirzada Y.M., Velazquez-Martinez C.A., Boring D. Hydrogen sulfide-releasing NSAIDs inhibit the growth of human cancer cells: a general property and evidence of a tissue type-independent effect. Biochem Pharmacol. 2012;83:715–722. doi: 10.1016/j.bcp.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 104.Lowenstein C.J., Dinerman J.L., Snyder S.H. Nitric oxide: a physiologic messenger. Ann Intern Med. 1994;120:227–237. doi: 10.7326/0003-4819-120-3-199402010-00009. [DOI] [PubMed] [Google Scholar]
- 105.Bonavida B., Garban H. Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics. Redox Biol. 2015;6:486–494. doi: 10.1016/j.redox.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fujita M., Somasundaram V., Basudhar D., Cheng R.Y.S., Ridnour L.A., Higuchi H. Role of nitric oxide in pancreatic cancer cells exhibiting the invasive phenotype. Redox Biol. 2019;22:101158. doi: 10.1016/j.redox.2019.101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang L., Xie K. Nitric oxide and pancreatic cancer pathogenesis, prevention, and treatment. Curr Pharmaceut Des. 2010;16:421–427. doi: 10.2174/138161210790232194. [DOI] [PubMed] [Google Scholar]
- 108.Wang J., Hussain S.P. NO• and pancreatic cancer: a complex interaction with therapeutic potential. Antioxidants Redox Signal. 2017;26:1000–1008. doi: 10.1089/ars.2016.6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kodela R., Chattopadhyay M., Kashfi K. NOSH-aspirin: a novel nitric oxide-hydrogen sulfide-releasing hybrid: a new class of anti-inflammatory pharmaceuticals. ACS Med Chem Lett. 2012;3:257–262. doi: 10.1021/ml300002m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chattopadhyay M., Kodela R., Santiago G., Le TTC, Nath N., Kashfi K. NOSH-aspirin (NBS-1120) inhibits pancreatic cancer cell growth in a xenograft mouse model: modulation of FoxM1, p53, NF-κB, iNOS, caspase-3 and ROS. Biochem Pharmacol. 2020;176:113857. doi: 10.1016/j.bcp.2020.113857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kashfi K., Chattopadhyay M., Kodela R. NOSH-sulindac (AVT-18A) is a novel nitric oxide- and hydrogen sulfide-releasing hybrid that is gastrointestinal safe and has potent anti-inflammatory, analgesic, antipyretic, anti-platelet, and anti-cancer properties. Redox Biol. 2015;6:287–296. doi: 10.1016/j.redox.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Coletta C., Papapetropoulos A., Erdelyi K., Olah G., Modis K., Panopoulos P. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci U S A. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bruce King S. Potential biological chemistry of hydrogen sulfide (H2S) with the nitrogen oxides. Free Radic Biol Med. 2013;55:1–7. doi: 10.1016/j.freeradbiomed.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cortese-Krott M.M., Kuhnle G.G., Dyson A., Fernandez B.O., Grman M., DuMond J.F. Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl. Proc Natl Acad Sci U S A. 2015;112:E4651–E4660. doi: 10.1073/pnas.1509277112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Barati Bagherabad M., Afzaljavan F., ShahidSales S., Hassanian S.M., Avan A. Targeted therapies in pancreatic cancer: promises and failures. J Cell Biochem. 2019;120:2726–2741. doi: 10.1002/jcb.26284. [DOI] [PubMed] [Google Scholar]
- 116.Zhu L., Staley C., Kooby D., El-Rays B., Mao H., Yang L. Current status of biomarker and targeted nanoparticle development: the precision oncology approach for pancreatic cancer therapy. Canc Lett. 2017;388:139–148. doi: 10.1016/j.canlet.2016.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhao Y., Henthorn H.A., Pluth M.D. Kinetic insights into hydrogen sulfide delivery from caged-carbonyl sulfide isomeric donor platforms. J Am Chem Soc. 2017;139:16365–16376. doi: 10.1021/jacs.7b09527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kang J., Li Z., Organ C.L., Park C.M., Yang C.T., Pacheco A. pH-Controlled hydrogen sulfide release for myocardial ischemia-reperfusion injury. J Am Chem Soc. 2016;138:6336–6339. doi: 10.1021/jacs.6b01373. [DOI] [PubMed] [Google Scholar]
- 119.Venkatesh Y., Das J., Chaudhuri A., Karmakar A., Maiti T.K., Singh N.D.P. Light triggered uncaging of hydrogen sulfide (H2S) with real-time monitoring. Chem Commun (Camb) 2018;54:3106–3109. doi: 10.1039/c8cc01172a. [DOI] [PubMed] [Google Scholar]