

Abstract
New approaches to cancer immunotherapy have been developed, showing the ability to harness the immune system to treat and eliminate cancer. For many solid tumors, therapy with checkpoint inhibitors has shown promise. For hematologic malignancies, adoptive and engineered cell therapies are being widely developed, using cells such as T lymphocytes, as well as natural killer (NK) cells, dendritic cells, and potentially others. Among these adoptive cell therapies, the most active and advanced therapy involves chimeric antigen receptor (CAR)-T cells, which are T cells in which a chimeric antigen receptor is used to redirect specificity and allow T cell recognition, activation and killing of cancers, such as leukemia and lymphoma. Two autologous CAR-T products have been approved by several health authorities, starting with the U.S. Food and Drug Administration (FDA) in 2017. These products have shown powerful, inducing, long-lasting effects against B cell cancers in many cases. In distinction to the results seen in hematologic malignancies, the field of using CAR-T products against solid tumors is in its infancy. Targeting solid tumors and trafficking CAR-T cells into an immunosuppressive microenvironment are both significant challenges. The goal of this review is to summarize some of the most recent aspects of CAR-T cell design and manufacturing that have led to successes in hematological malignancies, allowing the reader to appreciate the barriers that must be overcome to extend CAR-T therapies to solid tumors successfully.
KEY WORDS: Immunotherapy, T cell therapy, Chimeric antigen receptor (CAR), Genetic engineering
Graphical abstract
This review summarizes some of the most recent aspects of CAR-T cell design and manufacturing that have led to successes in hematological malignancies, allowing the reader to appreciate the barriers that must be overcome to extend CAR-T therapies to solid tumors successfully.
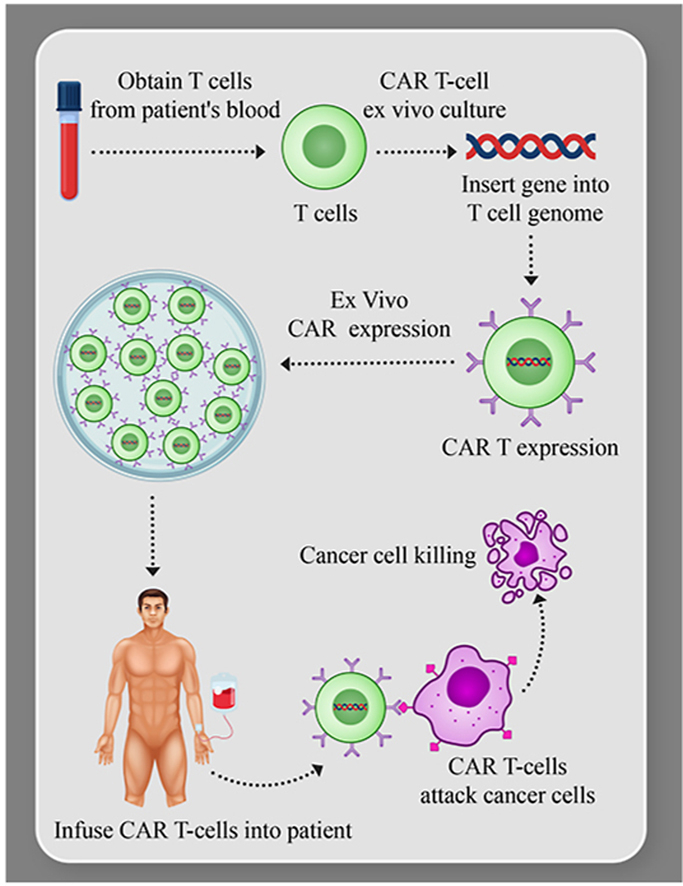
1. Introduction
Examples of anti-cancer immunosurveillance1, the existence of natural T cells with anti-cancer properties, and the ability to genetically engineer cells to target and destroy cancer cells, have taught us that the immune system plays a major therapeutic role in fighting cancer2,3. For decades, there have been four pillars of anti-cancer therapy: surgery, radiation, chemotherapy and immunotherapy, with only some of these active in cancers that have spread beyond the primary site4. We can add to this armamentarium targeted small molecule inhibitors, which have been very effective in specific cancers such as chronic myelogenous leukemia, but this still leaves the field with large areas of unmet medical need5,6. Over the last decade, we have seen significant examples of successful therapy with genetic reprogrammed autologous T cells which utilize a chimeric antigen receptor (CAR)-T cells7, with high remission rates and sustained remissions in B cell acute lymphoblastic leukemia (ALL) and non-Hodgkin lymphoma (NHL)8,9. The autologous products use a complex process that requires collection from each individual patient and “bespoke” manufacturing with full release testing for each individual patient product10,11. The CAR-T approach recognizes antibody-type epitopes on the surface of the cells in an HLA-independent manner. The CAR-T fusion molecule provides antigen recognition and binding (such as the B cell target CD19), coupled with precisely defined T cell activation and co-stimulatory domains (Fig. 1). The results with CAR-T cells have allowed us the hope that we have added a fifth major pillar to anti-cancer therapies12,13.
Figure 1.

Structures of the first through fourth generations of chimeric antigen receptors. All generations of CARs have a typical structure comprised by an extracellular antigen-binding domain (single-chain fragment variable, peptides, nanobodies, cytokines or other ligands), a hinge region (CD28, CD8, IgG1 or IgG4), a transmembrane domain (CD8α, CD4, CD3ζ, CD28, or ICOS) and intracellular signaling domains. The first-generation CARs have only the CD3ζ intracellular activation domain, while an additional co-stimulatory domain was added to second-generation CARs (e.g., CD28, 4-1BB, OX40, ICOS, CD27, KIR2DS2, and MYD88-CD40). The third-generation CAR has two co-stimulatory domains in tandem. The fourth-generation of CAR-T cells, also called armored CAR-T cells, has the same structure of second or third generation CAR. However, their producing vectors were armored with the advantage to secrete some additional molecules that give anti-tumor properties, such as the release of cytokines, chemokines, enzymes, ligands, receptors, peptides or monoclonal antibodies against different therapeutic targets.
2. Milestones leading to the CAR-T cell concept
Immunotherapy comes in many forms. By far the most traditional forms of immunotherapy are monoclonal antibodies (native or conjugated) and allogeneic stem cell transplantation. Other approaches, at various stages of development, include cancer vaccines, exosomes, cytokine therapy (such as INF-γ, IL-2, IL-15, IL-27, etc.), pulsed or genetically modified dendritic cells (DC), natural killer (NK) cells, tumor-infiltrating lymphocytes, cytokine-induced killer cells, and CAR T cells14. NK cells utilize KIR receptors15. DCs can be isolated, differentiated, and propagated ex vivo, although increasing DC numbers ex vivo to clinically useful doses is more of a challenge. DCs can be pulsed with an antigen as a vaccination approach, and can undergo genetic manipulation, which then can allow them to enhance immune activation against tumor antigens. Genetically modified DCs function for the recognition and subsequent immune reaction for the immunological cancer response through signaling via B cells and especially T cells, leading to their activation and greatly enhanced function. The complex formation of modified DCs with T cells relay information involving the antigen response, which can be altered in the self and non-self-recognition process through co-stimulatory molecules as well as peptide antigens on the DCs.
More recently, CAR-T therapy has indeed initiated a new era for cancer treatment. It made gratifying achievements in treating hematologic malignancies like lymphoma and leukaemia, with new data being published in solid tumors such as glioblastomas and neuroblastoma16. Chimeric antigen receptors have been also introduced in NK as another form of cellular immunotherapy, which can also cause cytotoxicity to cancer cells17. Despite some advantages, such as the recognition of tumor cells in both CAR-dependent and CAR-independent manners, with no clonal expansion and no immune rejection, the isolation, purification, and transduction of primary NK cells are complicated and usually produce a heterogeneous population with reduced expansion. As an alternative to the use of primary NKs, it is common the use of an irradiated established tumor cell line (NK-92), which must be injected in the patients17. This further highlights the new platform CAR-T cell therapy has provided.
The concept of CAR-T therapy was developed to overcome significant barriers in cancer immunotherapy related to T cell recognition of cancer antigens. CAR-T cells recognize cell surface antigens and are not compromised by tumor variants with low or absent surface expression of major histocompatibility complex (MHC) antigens18, which would otherwise preclude T cell recognition of tumor antigens. They are not MHC-restricted, thus, simplifying the construction of these therapeutic cells. The interaction with the tumor-associated antigens (TAAs) is driven by a single chain or similar binding domains19. It means that the construction of CAR-T cells with the desired specificity is possible with recognition of any surface structure to which there is an available monoclonal antibody. Another critical advance leading to the first responses in CAR-T clinical trials was the incorporation of co-stimulatory domains like 4-1BB and CD28 into CAR designs (Fig. 1), which has dramatically enhanced the potency of CAR-T cell responses. Alternatively, if recognition of intracellular epitopes is desired, which is a significant fraction of potential TAAs such as cancer testis antigens, this requires engineered T cell receptors (TCRs) and MHC matching with the patient20.
2.1. CAR-T cells in the treatment of hematological tumors
As indicated above, the most significant clinical results have come from trials studying second-generation autologous CD19-specific CAR-T cell therapies, starting with remarkable initial clinical results in recurrent chronic lymphocytic leukemia (CLL)21. Subsequent reports have shown encouraging results using CD19 CAR-T cells in acute lymphoblastic leukemia (ALL)22 and diffuse large B-cell lymphoma (DLBCL)23. CD19 directed CAR-T cell therapy has demonstrated the power of receptor engineering while offering long-term disease control in many cases of hematopoietic cancers24 (Table 125, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40). The long term survivors after anti-CD19 CAR-T also included allogeneic hematopoietic stem cell transplants (allo-HSCT). The vast majority of ALL patients on the ELIANA registration trial for the CD19 CAR-T product CTL019 (tisagenlecleucel) did not go on to allo-HSCT41. Despite being potentially curative, allo-HSCT can lead to fatal complications at rates greater than that seen with CAR-T, including treatment-related mortality and graft vs. host diseases.
Table 1.
A list of major anti-CD19 CAR-T cell therapy trials.
| CD19-positive B-cell malignancy | n | LD chemotherapy | Adverse effect | Long-term EFS | PFS | CAR-T cell dose/kg | OS | Outcome | Best response duration | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| FL | 2 | FLU (post T-cell infusion) | Lymphopenia | ND | ND | 100–200 × 107/m2 total T cells | ND | No responses | ND | 25 |
| CLL, ALL |
9 | None or (CTX) | B cell aplasia, fever, hypotension, death | ND | ND | 0.4–3 × 107 | 1 PR, 2 SD, 1 CR, 4 NR, 1 death | PR up to 12 week | 26 | |
| NHL, CLL, SMZL |
8 | CTX, FLU | B cell aplasia, CRS | ND | ND | 0.3–3 × 107 | 6 PR, 1 CR, 1 NE The total SOFA: serum IFNγ and serum TNF levels versus time (P = 0.02 for IFNγ and P = 0.001 for TNF |
CR > 18 months | 27 | |
| CLL | 14 | CTX, FLU | B cell aplasia, CRS, TLS, neutropenia |
ND | 18-month PFS 28.6% | 1.4–113 × 107 | 29 months; 18-month, OS 71% | 4 CR, 4 PR The peak value of CTL019 was statistically associated with response (P = 0.008), higher peak expansion of CTL019 associated with CRS, and CRS was associated with clinical response (P < 0.05) |
CR up to 53 months | 28, 29 |
| ALL | 16 | CTX | Severe CRS | ND | ND | 0.14–0.3 × 107 | 14 CR, 12 MRD The threshold where patients are at high risk for clinical complications secondary to sCRS. ∗P < 0.05, unpaired t tests at the corresponding time point. Specific P values for the time points are as follows: Day 2, P = 0.035 (n = 13); Day 4, P = 0.025 (n = 12); Day 5, P = 0.019 (n = 11); and Day 9, P = 0.01 (n = 8). |
CR up to 3 months | 30, 31 | |
| CLL, NHL |
20 | Allo-HSCT preparative regimen, DLI; none | B cell aplasia, CRS, hypotension, TLS |
39% at 6 months (Brundo et al.) | ND | 0.04–0.8 × 107 | 2 PR, 6 CR, 8 SD, 4 PD For CD4+ T cells, the difference in PD-1 expression between CAR+ cells and CAR– cells was statistically significant (P = 0.03) |
CR up to 30 months | 32, 33 | |
| ALL | 30 | None or VP/CTX | CRS, CNS toxicity | 67% at 6 months | ND | 0.2–1.2 × 107 | 6-month overall survival was 78% | 27 CR, 22 MRD Patients who had severe cytokine-release syndrome had higher peak levels of interleukin-6 (P < 0.001), higher peak levels of C-reactive protein (P = 0.02), ferritin (P = 0.005), interferon-γ (P < 0.001), and soluble interleukin-2 receptor (P < 0.001), patients with severe cytokine-release syndrome also had higher levels of CTL019-positive CD8 cells (P = 0.012) and CTL019-positive CD3 cells (P = 0.026). |
CR up to 24 months | 2, 34 |
| ALL, CLL |
8 | Allo-HSCT preparative regimen; none immediately before T-cell infusion | None | ND | ND | 1.9–11 × 107 | ND | 1 CR, 1 PR, 1 SD, 2 cCR, 3 NR | CR up to 3 months | 35 |
| DLBCL, PMBCL, follicular |
22 | CTX, FLU | Neurologic toxicities, myelodysplastic syndrome, vision loss |
ND | 12-month PFS 63.3% | 0.1–0.6 × 107 | ND | 4 PR, 12 CR, 4 PD, 2 SD There was an association between the percentage of central memory T cells among the infused CAR-T cells and peak blood CAR T-cell levels (P < 0.001; Spearman r = 0.7) |
CR > 24 months | 36 |
| ALL, NHL |
21 | CTX, FLU | CRS, B cell aplasia | 78.8% at 4.8 months | ND | 0.3–0.003 × 107, 2 × 107 |
51.6% at 9.7 months | 14 CR, 13 MRD The concentrations of CSF CD19-CAR T cells were higher in patients who developed neurotoxicity than in those who did not (P = 0·0039) |
CR up 19 months | 37 |
| NHL | 32 | CTX, FLU | Severe neurotoxicity, CRS | ND | ND | 2 × 105, 2 × 106, 2 × 107 EGFRt+cells/kg | 10 CR, 19 ORR of 30 evaluable | 38 | ||
| ALL | 45 | CTX, FLU | Severe neurotoxicity, CRS | 50.8% at 12 months | ND | 0,5 × 106, 1 × 106, 5 × 106, 10 × 106 EGFRt+cells/kg | 69.5% at 12 months | MRD-negative CR 93% There was no effect of dose level (P = 0.32), disease burden (P = 0.93), CD19 antigen load (P = 0.23), or fludarabine and cyclophosphamide lymphodepletion (P = 0.32) on the occurrence of severe neurotoxicity; however, the presence of severe CRS was predictive of subsequent severe neurotoxicity (P = 0.01) |
27 months | 39 |
| DLBCL | 7 | CTX, FLU | Severe neurotoxicity, CRS | ND | ND | 2 × 106 | ND | CR 57%, ORR 71% | 12 months | 40 |
cCR, continuous complete remission; CR, complete remission; CRS, cytokine release syndrome; CTX, cotrimoxazole; EFS, event free survival; FLU, fludarabine; HSCT, hematopoietic stem cell transplantation; LD, low dose; MRD, minimal residual disease; ND, not detected; NE, not evaluable; NR, no response; ORR, overall response rate; OS, overall survival; PD, progressive disease; PFS, progression-free survival; PR, partial remission; SD, stable disease.
Unfortunately, resistance to Anti-CD19 CAR-T therapy is a reality, and bispecific CAR-T cells, like anti-CD19/CD22 and anti-CD19/CD20 are promising options that are currently in clinical trials (NCT03448393 and NCT03271515, respectively).
2.2. CAR-T cells in the treatment of solid tumors
Clinically, the implementation of CAR-T therapy in solid tumors is still at the very earliest stages. In addition to the issues listed above, the tumor microenvironment adapts to limit T cell trafficking and function, using mechanisms such as: (a) hypoxia, nutritional depletion, oxidative stress42; (b) the presence of soluble suppressive factors, cytokines, and suppressive immune cells such as myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophage (TAM)/tumor-associated neutrophils (TAN)43; (c) prevention of trafficking and the penetration of T cells into a solid tumor mass (physical barriers)44; and (d) the presence of negative regulators of T cell function (for example, upregulation of cytoplasmic and surface inhibitory receptors such as programmed death 1 (PD-1) and T lymphocyte-associated antigen 4 (CTLA-4)45. To overcome these limitations, advancements in cell engineering, such as armored CAR-T cells (Fig. 1), and further investigations into tumor immunology are being undertaken.
In a clinical trial, where neuroblastoma patients were treated with GD2 ganglioside specific CAR-CTLs, four of the eight patients with an evaluable disease had evidence of tumor necrosis or regressions, including one sustained CR. None developed detectable antibodies to CAR-CTLs, and there were no adverse events attributable to the genetically modified T cells in the 11 subjects followed for up to 24 months46,47. In another interesting study, Brown et al.48 demonstrated the clinical response of a patient with recurrent multifocal glioblastoma who received multiple infusions of anti-IL-13Rα2 CAR-T cells over 220 days through two intracranial delivery routes (infusions into the resected tumor cavity followed by infusions into the ventricular system). Their data showed that intracranial infusions of anti-IL-13Rα2 CAR-T cells were not associated with any high-grade toxic effects (≥grade 3). After CAR-T cell therapy, regression of all intracranial and spinal tumors, along with corresponding increases in levels of cytokines and immune cells in the cerebrospinal fluid, was observed. This clinical response was continued for 7.5 months after the initiation of CAR-T cell therapy48. These results further proved that CAR-T cells could drive clinical responses in advanced cancer patients. Looking more broadly, several clinical trials have been conducted or are ongoing, as shown in Table 2.
Table 2.
Various clinical trials using CAR-T cell therapy in solid tumors (https://www.clinicaltrials.gov).
| Disease | Antigen | Phase | Clinicaltrials.gov identifier |
|---|---|---|---|
| Neuroblastoma, ganglioneuroblastoma | CD171 | I | NCT02311621 |
| Adenocarcinoma | CEA | I | NCT00004178 |
| Colorectal carcinoma | CEA | I | NCT00673322 |
| Breast cancer | CEA | I | NCT00673829 |
| Solid tumor | CEA | I | NCT01212887 |
| Liver metastases | CEA | I | NCT01373047 |
| CEA+ solid tumors | CEA | I | NCT02349724 |
| Glioblastoma | EGFRvIII | I | NCT02209376 |
| Advanced glioma | EGFR | I | NCT02331693 |
| Ovarian cancer | FR | I | NCT00019136 |
| Neuroblastoma | GD2 | I | NCT01822652 |
| Neuroblastoma | GD2 | I | NCT01460901 |
| Neuroblastoma | GD2 | I | NCT00085930 |
| Neuroblastoma | GD2 | NCT02439788 | |
| Non-neuroblastoma, GD2+ solid tumors | GD2 | I | NCT02107963 |
| Hepatocellular carcinoma | GPC3 | I | NCT02395250 |
| Lung malignancy | HER2 | I | NCT00889954 |
| Metastasized HER2+ cancer | HER2 | I | NCT00924287 |
| Brain and CNS tumors | IL 13 zetakine | I | NCT00730613 |
| Glioma, neoplasm | IL13Rα2 | I | NCT02208362 |
| Pancreatic cancer | Mesothelin | I | NCT02465983 |
| Ovarian cancer Mesothelioma |
Mesothelin | I | NCT02159716 |
| Prostate cancer | PSMA | I | NCT00664196 |
| Head and neck cancer | ErbB | I | NCT01818323 |
| Glioblastoma | EGFRvIII | I/II | NCT01454596 |
| EGFR positive advanced solid tumors | EGFR | I/II | NCT01869166 |
| Solid tumors | HER2 | I/II | NCT01935843 |
In contrast to hematological malignancies, solid tumors not only lack conventional co-stimulatory molecules, which may be expressed on malignant and normal B lymphocytes but also have evolved mechanisms to suppress the immune system actively49, 50, 51. Tumor cells produce several immunosuppressive molecules and/or recruit multiple immunosuppressive cells to evade or misdirect a tumor-specific immune response52,53. The prostaglandin 2 (PGE2)/EP2/protein kinase A (PKA) signaling pathway combined with adenosine mediates immunosuppression, activates PKA, and blocks TCR activation. A small peptide called the “regulatory subunit I anchoring disruptor” (RIAD) diminishes the negative effects of PKA on TCR activation—a function that investigators employed to improve T cell function. By the generation of CAR-T cells expressing RIAD, Newick et al.54 showed that blocking the upstream immunosuppressive mediators of PKA activation, such as PGE2 and adenosine, could improve TCR signaling, increase cytokine release, and increase CAR-T cell infiltration, leading to greater tumor cell killing. Chronic inflammation activity is a hallmark of the tumor microenvironment and generates higher levels of reactive oxygen species (ROS) that can abrogate anti-tumor activity. Ligtenberg et al.55 proposed that CAR-T cells overexpressing catalase (CAT) would perform better compared to conventional CAR-T cells. Their data indicate that CAT–CAR-T cells produce more intracellular CAT, resulting in a reduced oxidative state with less ROS accumulation in both the basal and activated states. The CAT–CAR-T cells maintained an anti-tumor activity even within a H2O2 hostile environment55,56. T cell exhaustion is often found in solid tumors, where the immune checkpoint molecules are usually overexpressed, e.g., programmed cell death ligand 1 (PD-L1). The release of immune checkpoint blockade antibodies by armored CAR-T cells into the tumor microenvironment is another important effort to improve CAR-T function. Anti-PD-L1 releasing CAR-T cells, for example, were able to decrease tumor growth in five times when compared with the CAR-T cells alone in a humanized mice model of renal cell carcinoma, with remarkable immune checkpoint blockade57. Some anti-PD-1, anti-PD-L1, or anti-CTLA-4 antibodies-expressing CAR-T cells and PD-1 knockout-engineered CAR-T cells for different targets are currently under clinical trials. Altogether, it seems that modification of CAR-T cells by different means (e.g., gene-editing tools, pharmacological inhibitors, and others) will likely be the best way to open new avenues to achieve results in solid tumors similar to those seen in hematologic malignancies58, 59, 60.
3. Limitations of CAR-T therapy and counteracting strategies
3.1. On-target/off-tumor toxicity
Unique, tumor-specific antigens are rare, so the approach depends on the identification and use of TAAs. TAAs are self-antigens and can be expressed on normal tissues (CD19 is an example of this), requiring careful selection and de-risking of possible targets. TAAs can also be derived from subtle modifications of self-molecules, and there are potential strategies that can leverage differences in antigen density between tumor cells and normal cells61. However, heterogeneity can be an issue, as TAAs can be expressed at lower levels or even be missing from subpopulations of tumor cells. Most importantly, tumor cell populations are quite adept at evading immune detection and employ a large array of mechanisms to do so62. These include the downregulation of the expression of TAAs themselves, production of immunosuppressive molecules and/or recruiting immunosuppressive cells, and a tumor microenvironment metabolically hostile to T cell activation and functionality53. An ideal tumor antigen would allow tumor cell targeting while restricting this targeting to the tumor cell. A CAR-T cell TAA may also be expressed in healthy tissues, which presents the risk of “on-target/off-tumor” toxicity63. Thus, most CAR-T-targeted antigens are not tumor ‘specific’ but tumor ‘associated’: even though the antigens are expressed on tumors at high levels, they exist in normal cells as well. As a result, “on-target/off tumor” recognition may result in toxicity to normal tissues64.
CD19 is a favorable target in this regard, which is why it was chosen in so many early CAR-T trials. CD19 is expressed only on normal and malignant B cells. CD19 is expressed on nearly 100% of newly diagnosed ALL and B cell lymphomas. Off-tumor cytotoxicity results in normal B cell aplasia, which in turn results in hypogammaglobulinemia, the principal long term toxicity of a persistent CD19 CAR-T product like CTL01965,66. In addition to the favorable target profile of the TAA CD19, there are other positive aspects of B cell malignancies and CD19 as CAR-T targets67. Liquid tumors, such as B cell ALL, provide what may be an optimal environment for CAR-T function, without negative regulatory factors such as high local levels of transforming growth factor-beta and hypoxia, which are usually associated with solid tumor microenvironments68. The CAR-T cells have immediate access to all or most of the ALL cells after infusion, leading to rapid responses and, possibly, more rapid toxicity. Interestingly, DLBCL can be thought of as an “intermediate” between a liquid tumor (like ALL) and a true solid epithelial cancer, since lymphoma cells are often found organized into nodal type masses. This may account for why CRs occur in ALL by 30 days, while full responses in DLBCL can take three months or longer9,69. One downside of CD19 is that it can be susceptible to editing or downregulation, which can lead to relapse as a result of CD19 antigen escape70,71.
Other TAAs can present more significant challenges, especially in the solid tumor setting. In a study conducted by Morgan et al.72, targeting ERBB2 overexpressing tumors has led to the failure of a patient because of respiratory distress secondary to localization of a large number of CAR-T cells to the lung immediately following infusion and, as a consequence, release of inflammatory cytokines. This was thought to occur as a result of the recognition of low levels of ERBB2 on lung epithelial cells. These and similar results emphasize the importance of antigen selection for overcoming on-target/off-tumor toxicity of CAR-T cells. Considerable effort is being expended currently to de-risk solid tumor TAAs before going into clinical trials, but some interactions and some side effects only become apparent in the clinical setting. Altogether, the intensity of reported events ranges anywhere from manageable lineage depletion to severe toxicity, including fatal outcomes in a small number of patients.
Upon overexpression of targets in tumors, affinity-tuned CARs have shown increased tumor specificity73,74. Moreover, to raise safety and specificity, various dual targeting strategies have been generated. One such strategy is the modification of T cells with two CARs, where CAR number 1 initiates killing by providing the CD3ζ signal, and CAR number 2 delivers the co-stimulatory signal75. When both of the antigens are recognized by the CARs, activation of the CAR-T cell is also achieved. Furthermore, inhibitory CARs recognize antigens expressed on non-tumor tissues, and CTLA-4 or PD-1 are able to harness signals exerted by natural T cell inhibition. This method, in preclinical mouse models, has shown to protect healthy tissue from damage caused by off-target effects76.
3.2. Tumor heterogeneity
Antigen heterogeneity is a significant limitation for all proposed tumor-associated antigens in solid tumors. Heterogeneity is variability in the expression of the antigen on the cells within a given tumor77. Some cells can have low expression of the TAA, while some cells may be negative for the TAA entirely. Because of this, as a first step in developing a solid tumor, CAR-T is to identify an appropriate and safe, solid TAA, but the second step may be to deal with this issue of antigen heterogeneity78. For example, although mesothelin is expressed on >90% of malignant epithelial mesothelioma tumor cells, it is also expressed on lower percentages of tumor cells in lung, ovarian, and breast cancer79. Some CARs can bind to and be activated by antigens that are expressed at very low levels. This is one way of dealing with heterogeneity, but also increases the risk of “on-target/off-tumor” toxicity. Furthermore, most tumor cells escape host immunity by the elimination of immunogenic epitopes. Hence, for solid tumor treatment, identifying immunogenic and specific tumor antigens could be necessary. The ability of CAR-T cells to trigger antigen spreading and/or induce indirect tumor killing is a key question highly relevant to the heterogeneity problem left unanswered80.
Several surface antigens are variably expressed by tumor cells, so TAA expression levels can be correlated with a failure of achieving durable responses81. Epidermal growth factor receptor (EGFR) is a transmembrane protein with cytoplasmic kinase activity that belongs to the oncogene family ErbB and is expressed in normal and tumor cells. In the clinical setting, EGFR upregulation is associated with poor prognosis82. EGFRvIII is specifically expressed on malignant tumor cells and contains a deletion of the extracellular amino acids 6–273, which leads to constitutive tyrosine kinase activity, eventually promoting tumor metastasis83,84. EGFRvIII-CAR-T cells have the ability to kill EGFRvIII+ tumor cells and also produce effector cytokines, such as interferon-γ. EGFRvIII-CAR-T cells were shown to eliminate glioma cells85. Sampson et al.86 showed that following CAR-T cell therapy directed against EGFRvIII in syngeneic mice after the treatment of brain tumors, the cured mice were resistant to the re-introduction of EGFRvIII-negative tumors which represents the generation of host immunity against additional tumor antigens. In a clinical study of anti-EGFRvIII CAR-T cell therapy for glioblastoma, O'Rourke et al.87 noted a wide regional variation of EGFRvIII expression in tumor samples after CAR-T cell therapy. Most patients had a loss or decreased expression of EGFRvIII in tumors despite no change in the degree of EGFR amplification or other tumor mutations88. The study poses the question of whether targeting EGFRvIII alone can provide durable clinical benefits or whether antigen escape will negate the clinical impact79. This may or may not be the case; therefore, the enrollment criteria for CAR-T cell therapy trials need to include prescreening for the intensity and percentage of target antigen expression on tumor cells by immunohistochemistry and/or immunofluorescent techniques. However, these criteria are hard to define prospectively and may result in the need for a companion diagnostic, so the argument can be made to allow patients with lower TAA expression to enroll and then assess clinical responses. Moreover, targeting of various tumor antigens concurrently or in combination (above) could result in a better killing effect and potentially blunt the appearance of target antigen-negative tumor variants.
At this point, at least 30 solid tumor antigens have been evaluated; these include developmental or oncofetal antigens, neoantigens (for example, mutated sequences), endogenous tumor-specific antigens, and tumor-selective antigens (for example, the low-level basal expression on normal cells, but the enriched expression on neoplastic cells)89. Many of the tumor-associated antigens, for instance, EGFR/EGFRvIII, HER2, mesothelin, IL-13Rα2, Carcinoembryonic antigen (CEA), CD171, and GD2 identified are shown to be highly expressed by solid tumors, thus further allowing for the use of CARs targeting multiple antigens as combination therapies (Table 390, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109). Antigens resulting from somatic mutations on tumor cells are neoantigens, which can be unique for each patient110 and serve as tumor-specific antigens instead of TAAs. Since their expression is restricted to tumor cells, they are highlighted as an ideal target for CAR-T cell therapy, further minimizing the risk of destroying healthy tissue. Researchers may design CARs targeting abnormal tumor-specific oncogenic mutations or modified tumor-associated antigens. For instance, a study introduced a new CAR unusually targeting glycosylated tumor-associated MUC1111. In this case, the Tn glycoform of MUC1 on tumor cells was specifically recognized; however, these CAR-T cells did not bind to glycosylated MUC1 normally. These studies showed that anti-MUC1-CAR-T cells did not exhibit any cytotoxicity against normal human primary cells112. Similar to this research was a study in ovarian carcinomas reporting that CAR-T cells target MUC16, routinely overexpressed in these types of cancer113. CEA is a 180-kD cell surface glycoprotein expressed during developmental growth; however, it is restricted in transformed cells and normal adult tissues. Evidence of tumor eradication in mice has been reported by CEA-CAR-T cells114. Preclinical studies demonstrate that anti-CEA CAR-T cells can effectively target CEA expressed in colorectal adenocarcinoma115 and pancreatic tumors116. Another promising target for brain malignancy is the IL-13 receptor α2 (IL-13Rα2), which binds IL-13 with high affinity and modulates its response. Mirzaei et al.79 and other researchers have developed ligand-based CARs for the preferential recognition of IL-13Rα2 over the more ubiquitously expressed IL13Rα1, through the use of membrane-bound IL-13 muteins.
Table 3.
A summary of various cancer antigens expressed by different cancers.
| Cancer | Antigen |
|---|---|
| Ovarian cancer | B7H690, natural killer group 2 member D (NKG2D)91, human epidermal growth factor receptor 2 (HER2)92, alpha folate receptor (alpha FR)93, TNF-related apoptosis-inducing ligand (TRAIL) receptor 1 (TRAIL-receptor 1)94, adhesion molecule L1-CAM (L1-CAM) (CD171)95 |
| Glioblastoma | Interleukin-13Ra2 (IL13Ra2)96, AC13397, type III variant of the epidermal growth factor (EGFR)98, erythropoietin-producing hepatocellular A2 (EphA2)99, |
| Prostate cancer | Prostate-specific membrane antigen (PSMA)100 |
| Pancreatic cancer | Prostate stem cell antigen (PSCA)101, carcinoembryonic antigen (CEA)102 |
| Neuroblastoma | Disialoganglioside 2 (GD2), L1-CAM (CD171)95 |
| Breast carcinoma | MUC-1103, chondroitin sulfate proteoglycan-4 (CSPG4)104 |
| Lung cancer | EphA2105 |
| Malignant pleural mesotheliomas | Mesothelin106 |
| Colon cancer | TRAIL-receptor 1107 |
| Hepatocellular carcinoma | Glypican-3 (GPC3)108 |
| Renal cell carcinoma | Carbonic anhydrase IX (CAIX)57,109 |
3.3. CAR-T cell trafficking
There is a specific interconnection between tumors and surrounding cells, characterizing the organization of tumor tissue. This tumor–tissue connection includes the immune system, for example, the lymphoid and myeloid compartments, the extracellular matrix, fibroblasts, vasculature, and signaling molecules. Fibroblasts, blood vessels, and low nutrients conditions (such as low oxygen and low pH) in surrounding tumors make therapeutic delivery extremely difficult89,117.
Another important obstacle in CAR-T cell therapy of solid tumors is the suboptimal trafficking of CAR-T cells to the tumor site118. This trafficking is a dynamic process managed by an interconnected network68. CAR-T cells require penetration into the tumor tissue in order to kill tumor cells. Endogenous T-cells are usually excluded from the tumor microenvironment; therefore, a lack of penetration of CAR-T cells into the tumor has been seen with many solid tumor CARs.
Local delivery of CAR-T cells into the tumor can enhance their effectivity in therapy; however this is impractical in patients with many metastatic lesions119. The successful application of CAR-T cells in hematological malignancies is associated with the fact that effector and tumor cells share hematopoietic origins, therefore, causing a higher tendency for them to migrate to lymph nodes and bone marrow120. In contrast, solid tumors secrete factors, including chemokines (such as CXCL12 and CXCL5, growth factors, and cytokines), which prevent effector T cells from infiltrating into the tumors. Hence, whereas CAR-T cells are transferred intravenously to gain access to hematologic malignancies in the periphery, most solid tumors are not as easy to reach.
After infusion, CAR-T cells require migration to the blood circulation, extravasation, and infiltration to the tumor site to initiate their functions121. The extravasation of T cells is dependent on the transendothelial migration of leukocytes and chemokines, their interaction with specific adhesion molecules, firm adhesion, mediating capture, and rolling behavior122. For effective therapy using CAR-T cells and the enhancement of their migration to tumor sites, various studies have shown the expression of receptors-matched with tumor-secreted chemokines on CAR-T cells. Moon and colleagues123 revealed that the overexpression of CCR2b in anti-mesothelin CAR-T cells led to a more than a 12.5-fold increase in CAR-T cells homing toward mesothelin+ malignant pleural mesothelioma in mice, leading to improved anti-tumor effects. Another study described that the expression of CCR2b on GD2-CAR-T cells resulted in a more than a 10-fold increase in CAR-T cells migrating to CCL2 secreting neuroblastoma cells124.
Myeloid cells and tumor fibroblasts have the ability to contribute to pro-tumoral fibrotic extracellular matrix development which may result in the interference of the penetration of T-cells118. Also, T lymphocytes during extravasation constantly degrade extracellular matrix (ECM) components as well as sub-endothelial basement membranes125. This includes heparan sulfate proteoglycans (HSPGs), which constitutes the majority of the extracellular matrix126. To facilitate CAR-T infiltration into the tumor stroma, Caruana et al.127 developed CAR-T cells to overexpress the heparanase enzyme to degrade heparan sulfate proteoglycans (HSPGs). The ECM is an integral component of the stroma, and therefore, CAR-T cells attacking stroma-rich solid tumors must be armed with enzymes to degrade HSPGs in order to infiltrate readily into tumor stroma and exert anti-tumor effects. The authors found that engineered CAR-T cells expressing heparanase promote more efficacious anti-tumor activity127. These studies support the concept that arming of CAR-T cells with a chemokine receptor or ECM-degrading enzyme(s) could facilitate infiltration into the tumor stroma and enhance anti-tumor efficacy.
Moreover, methods have been developed in the context of non-CAR-T therapy, which may become beneficial in infiltrating the physical barrier and supplementing CAR-T cell trafficking. In polarized epithelial tumor cells, manipulation of JO-1 transiently separated tight junctions, which caused an increased efficiency in monoclonal antibody (mAb) treatments in xenograft tumor models128.
Tumor endothelial cells express a variety of molecules, including tumor necrosis factor (TNF)-related apoptosis-inducing ligands, PD-L1, IL-10, transforming growth factor beta (TGF-β), and indoleamine-pyrrole 2,3-dioxygenase (IDO), which may contribute to immune suppression. Previous studies have highlighted that vascular targeting drugs can decrease endothelial expression of immunosuppressive molecules and can specifically reverse endothelial cell-induced exhaustion and further improve CAR-T cell therapies.
3.4. CAR-T cell persistence
The administration of anti-HER2 CAR-T cells to patients with sarcoma (up to 108 cells/m2) has demonstrated no evident toxic effects but limited anti-tumor activity and T cell persistence. To improve the persistence of adoptively transferred CAR-T cells, Pule at al47. generated virus-specific CAR-T cells. By conducting a phase I dose-escalation study, the team examined the safety and efficacy of autologous anti-HER2 CAR virus-specific T cells in 17 patients with progressive glioblastoma. Although the CAR-T cells did not expand, the cells were detectable in the peripheral blood for up to 12 months. Of eight patients, one had a partial response and seven had a stable disease. The median overall survival was 11.1 months after CAR-T cell infusion and 24.5 months after diagnosis47. The results highlight the need for the improvement in expansion and persistence of CAR-T cells. To boost CAR-T cell survival, various studies have demonstrated that the local production of IL-15, IL-7 and IL-12 could improve CAR-T cell expansion, induce expansion of CAR-T memory stem cells and prolong persistence in vivo by increasing the expression of antiapoptotic molecules, such as BCL-2129. Koneru et al.130 showed that secretion of IL-12 by CAR-T cells could eradicate human ovarian xenografts. The authors showed that IL-12-secreting CAR-T cells display improved anti-tumor efficacy as described by increased survival, prolonged persistence of T cells, and higher systemic IFN-γ. However, it should be pointed out that the authors have measured the secretion of human IL-12 (P70) in the serum of CAR-T cell-treated SCID-Beige mice (with normal macrophages and dendritic cells populations) with established ovarian tumors. Since SCID-Beige mice have normal macrophages and DCs populations, and these cells are endogenous sources of mouse IL-12 production upon tumor challenge, these mice could, in fact, produce IL-12 after tumor challenge. The total concentration of serum IL-12, therefore, consisted of both endogenous and exogenous IL-12, and it may not solely reflect IL-12 production by IL-12-secreting CAR-T cells131.
3.5. Metabolic and physical barriers
Nutrient starvation is a second key factor inhibiting the proliferation of T-cells and the production of cytokines, due to glucose limitation, other metabolites and increased levels of lactate (leading to acidosis). The deficiency of nutrients, especially amino acids (such as arginine, lysine, and tryptophan) are able to activate the integrated stress response, further regulating T cell activity132. Though tryptophan cannot be synthesized, it is essential for many biological functions. Tryptophan metabolism is catalyzed by IDO. Myeloid cells and tumor cells can express IDO in the tumor microenvironment, leading to T cell anergy, death, and also T-reg accumulation. Researchers recently showed that IDO expression could also inhibit CAR-T cells. In particular, Ninomiya et al.133 demonstrated that CD19 CAR-T cells were not able to manage the improvement of CD19+ IDO-expressing tumors. In addition, they showed that cyclophosphamide and fludarabine administration improved CAR-T cell effectiveness by decreasing IDO expression. For malignancies resistant to immunotherapy and chemotherapy, the combination of IDO inhibitors and CAR-T cells may represent a valuable option.
3.6. Tumor-derived soluble factors and cytokines
In sera, published work has shown that the tissue extracts and ascites fluids from cancer patients contain soluble immunosuppressive factors. PKA is the downstream effector of two immunosuppressive factors: 1, PGE2 and 2, adenosine.
Many studies have reported the role of PGE2 and adenosine molecules as key inhibitors of both the proliferation and activity of T cells. Furthermore, various studies have reported the subversion of CD8 differentiation through PGE as well as CD4 suppression. Both these molecules elicit immunosuppressive effects through the signaling of G-coupled receptors, further activating protein kinase A134. TGF-β is a significant cytokine that negatively regulates the immune response. Cytokines involved in inflammatory reactions in a tumor can act as a double-edged sword, either inhibiting or bolstering the anti-tumor reaction. The ability of TGF-β to enhance matrix production, promotes epithelial-to-mesenchymal transition, manipulate immune responses towards a Th2 phenotype, and finally promote metastasis, has direct negative effects on the functions of effector T-cells135.
The tumor microenvironment can either suppress or overall inactivate CAR-T cells, even upon successful trafficking to tumor sites. So, in CAR-T cell studies, strategies named TRUCK (T cells Redirected for Universal Cytokine Killing) are being developed.
The aim is for CAR-T cells to gain the capability of remodeling tumor microenvironment's suppressive function by secreting anti-cancer cytokines. Recently, several cytokines, including interleukin (IL)-12136, IL-18137, and TNFRSF14 have been utilized in TRUCK designs. IL-12 is an inflammatory cytokine able to induce a Th1 (CD4+) response, improve T-cell activation, be cytotoxic to T cell (CD8+) clonal expansion, and has effector function. The ability of TRUCKs to change the tumor microenvironment and their utilization to deliver a range of cytokines warrants further exploration138.
3.7. Immunosuppressive immune cells
Regulatory T cells (T-regs) are T cells that suppress immune activity139 and regulate immune responses, including type 1 T-regs and CD4+CD25+ T-regs, and its absence results in lethal autoimmunity. FOXP3, a regulator of CD4+CD25+ T-reg transcription factor, is highly expressed on T-regs, while TGF-β is essential for its function140. Generally, T-regs are highly expressed at tumor sites and in inflammation, where an immune reaction is regulated141. Furthermore, T-regs can produce many immunomodulatory cytokines such as TGF-β and IL-10 in order to suppress the activity of T cells142. CD4+/FOXP3+ T-regs are known suppressors of T cell activity functioning through contact-dependent mechanisms or IL-10 and TGF-β secretion. Since selectively depleting T-regs are difficult, studying the effects of them on therapy by CAR-T cells has been difficult. However, some studies have been performed using either the adoptive transfer of T-regs genetically or depletion approaches with CAR-T cells143. Tumor regression in the tumor microenvironment was accompanied by a decrease of myeloid-derived suppressor cells. However, the role of MDSCs is in the modulation and mechanism of increased anti-tumor activity remains to be seen144.
Altogether, it seems that modification of CAR-T cells by different means such as using gene-editing tools, pharmacological inhibitors and concomitant targeting of immunosuppressive mechanisms will open new avenues for cancer immunotherapy particularly in the treatment of solid tumors.
4. Production of CAR-T cells
The protocol to generate CAR-T cells follows specific quality controlled steps, which must be performed with precision and under Good Manufacturing Practice (GMP) conditions145. The production of CAR-T cells is a multi-step process, including leukapheresis to collect cells from the patient, T cell stimulation, transfection or transduction, expansion, and extensive release testing. After successful manufacturing, the patient undergoes lymphodepleting chemotherapy to maximize cell engraftment and expansion, followed by infusion146. Initially, leukapheresis removes blood from the patient while separating the leukocytes, which include T cells. Separation of T cells, where needed, can be performed by bead selection or size/density centrifugation approaches (Fig. 2). The next phase is the activation of T cells, harnessing signals which mimic T cell activation by antigen-presenting cells (APCs). T cell activation consists of two signals, signal 1 (the primary specific activation signal delivered by the T cell receptor) and signal 2 (co-stimulatory signals such as those mediated by CD28, 4-1BB, or OX-40). Initially, ex vivo culture systems involved anti-CD3 antibody and IL-2147. More recently, the APC role can be more completely replaced by substrates or beads coated with anti-CD3 and anti-CD28 monoclonal antibodies, which provide both activation signals and better preserve the proliferative phenotype required for in vivo CAR-T function. Another T cell activation approach is the use of artificial antigen-presenting cells (AAPCs). The AAPCs or beads are removed from the T cells at the end of the culture through magnetic separation148. The selection and generation of a favorable manufacturing procedure for (GMP)-grade AAPC lines, due to its complexity, requires additional resources. Currently, CAR-T manufacturing introduces the CAR gene using viral gene transfer systems. In a separate phase of manufacturing, the viral vector encoding the CAR is incubated with T cells. Eventually, the vector is diluted out of the culture and/or washed away through the change of medium, and the vector is not infused into the patient. For clinical applications, the vectors routinely used for stable gene expression are γ-retroviral vectors and lentiviral vectors. Lentiviral vectors compared to retroviruses are used with more reliability and also transduce T cells more effectively; however, the production of GMP lentiviral vectors is expensive, with limited GMP manufacturing capacity currently available worldwide. Various non-viral approaches, such as the Sleeping Beauty transposon/transposase system, or mRNA transfection methods, can also be used149,150. While the Sleeping Beauty transposon system has been used in early phase clinical trials and is cost-effective, their efficiency remains an issue. Transient mRNA transfection is used for CAR-T cell generation; however, several rounds of CAR-T cell infusion is required for this method to be executed properly. During culture, the goal is to activate and expand the gene-modified cells. In GMP manufacturing, culture mixing is required to grow cells in large numbers, and optimal gas exchange is a must. For this, bioreactor culture systems have been designed, such as the GE WAVE, which is widely used for expansion. This bioreactor uses a rocking platform further to expand the CD19-targeted CAR-T cells151. For the expansion of cells from low seeding densities, a relatively new platform is used called G-Rex. The G-Rex is basically a cell culture flask containing a gas-permeable membrane. The advantages of G-Rex include the one time upfront feeding regimen, allowing for the reduction of volume at harvest time, its low seeding density, and the ease of using an incubator for cell growth152. Newer and more modular technologies (such as the CliniMACS Prodigy) afford a single device used for CAR-T cell manufacturing that accomplishes all phases altogether153. Owing to the multifaceted nature of CAR-T cell products, a cautiously planned list of in-process and release tests is essential to afford sufficient evidence of identity, safety, purity, and potency. The identity of CAR-T cell products is usually characterized by CAR surface expression143,154. Safety requires a lack of unsafe contaminations, such as endotoxin, mycoplasma, replication-competent retrovirus or lentivirus (RCR or RCL), and prevention of genotoxicity by properly limiting the level of transgene integration. The purity of the product depends, in part, on specified levels of CD3+ and CAR+ T cells. The impurities incorporated by initial patient material and ancillary elements, such as undesired cell types, tumor burden, and residual beads, have to be below certain specific levels approved by the U.S. Food and Drug Administration (FDA)155. Up to now, the potency of CAR-T cells is frequently determined by in vitro cytotoxic T lymphocyte assays or interferon-γ production. Based on the nature of the specific product, further testing may be essential. Setting up of a complete testing panel with the guidance of the FDA in the early development phase is necessary for the final commercialization of the product. Table 4113,149,151 recaps selected CAR-T cell release tests using different genetic modification methods. After the cells reach the numbers required for clinical uses, they are collected, may be cryopreserved, and transferred back to the clinical center for patient infusion.
Figure 2.

Manufacture of autologous CAR-T immunotherapy for B-cell ALL and NHL: T cells engineered to express a CAR, enabling CAR-armed T cells to attack tumors.
Table 4.
CAR-T cell release tests in different CAR-T cell production methods.
| Parameter | Retroviral and lentiviral vector-based method | Transposon/transposase-based method | mRNA electroporation-based method |
|---|---|---|---|
| Safety | #Gram stain, 7–14 day culture#Mycoplasma (qPCR) #Endotoxin level (cell lines PTC) #Copies of transgene insertion (qPCR) #RCR/RCL (for RCR: marker-rescue cell culture assay; for RCL: co-culture on C8166 cells with amplification and indicator phases) |
##Gram stain, 7–14 day culture #Mycoplasma (qPCR) #Endotoxin level (LAL test) |
#Gram stain, 7–14 day culture #Mycoplasma (qPCR) #Endotoxin level (LAL test) |
| Purity | #%CD3+T cells (flow cytometry) #%CAR-T cells (flow cytometry) #Residual tumor burden (flow cytometry) #Residual beads (microscopy) |
#%CD3+T cells (flow cytometry) #%CAR-T cells (flow cytometry) #Residual AAPCs (flow cytometry) |
#%CD3+T cells (flow cytometry) |
| Identity | #%CAR+T cells (flow cytometry) | ND | ND |
| Potency | #In vitro CTL or IFN-γ secretion (chromium release assay & ELISA) | ND | ND |
| References | 151 | 149 | 113 |
AAPC, artificial antigen-presenting cells; CAR, chimeric antigen receptor; CTL, cytotoxic T lymphocyte; IFN, interferon; RCL, replication-competent lentivirus; RCR, replication-competent retrovirus, PTC, point to consider; qPCR, quantitative PCR; LAL, limulus amoebocyte lysate; AAPCs, artificial antigen-presenting cells.
ND, not detected.
5. TCR-T therapy
In the same way as CAR-T cell therapy, T-cell receptor-engineered T cell (TCR-T) is a type of adoptive cell therapy that uses genetically altered T cells to treat tumors156,157. In the case of TCR-T, T cells are adapted to express a TCR capable of interacting with a cancer-specific peptide-MHC (P-MHC). This peptide can be a tumor-associated antigen (TAA) overexpressed by the tumor or a neo-antigen, which differs from a wild-type antigen through mutations that cannot be recognized through self-tolerance mechanisms158,159. TCRs must be compatible with human leukocyte antigen (HLA), recognizing P-MHCs, and promoting cancer cell death160. While CAR-T cells act by binding their receptor to the surface antigens of cancer cells, comprising only about 28% of the antigens available, TCR-T can bind to peptides originally secreted or located inside the cell, expanding the range of tumor targets The selection of a specific TCR can be performed by different approaches, usually involving single-cell sequencing or clonal T cell analysis as the final steps161, 162, 163.
Clinical trials indicate that TCR-T cells are more effective than CAR-T cell therapy in solid tumors157,160,164. However, some disadvantages are the limited number of tumor-specific TCRs, the possibility of TCR chain cross pairing between the introduced and endogenous TCR chains, and the need to match the patient's HLA157,159. Although CAR-T cells have a limited number of antigens that can be targeted, they do not have this type of limitation and can be used in all patients. In addition, the downregulation or loss of HLA can be a strategy of immune evasion by malignant cells that can be a limitation of the therapy with TCR-T, but not of CAR-T cells157. As an advantage, it appears that TCR-T cells mediate the release of lower levels of cytokines, decreasing the consequent risk to developing cytokine release syndrome when compared to CAR-T cells165,166. Also, CARs do not have a self-regulatory mechanism, while engineered TCRs and native TCRs, are subjected to the same mechanisms of activation and auto regulation157. New exciting efforts have been applied to produce non-MHC restricted TCR-T cells. The monomorphic MHC class I-related protein (MR1)-restricted TCR that recognizes cancer cells of different origins, being inert to non-tumor cells167 and the anti-cancer γδ TCRs that recognizes metabolites overproduced by cancer cells168, are attractive candidates to be used as “off-the-shelf” TCRs.
In a systematic review, Zhang and Wang164 showed that there is a general trend of increasing in the number of clinical trials using TCR-T cells from 2004 to 2018. It is also clear from the review that TCR-T cell therapy is a therapeutic tool relatively new, compared to CAR-T cell160 and that there is still a long way to the clinical application of this therapy. The vast majority (84%) of the clinical trials examined by Zhang and Wang164 were performed on solid tumors, possibly because CAR-T has already proven its efficiency in the treatment of hematological malignancies, while, as evidenced by several studies, its effects on solid tumors have not been so successful.
6. Conclusions and future directions
Cancer immunotherapy has been evolving on several fronts, most heading in the direction of immune checkpoint blockade and CAR T-cell therapy. CAR-T was the first genic therapy to be approved by the FDA, and has an undeniable potency, especially for the treatment of hematological tumors. Nevertheless, multiple factors impact the efficacy of CAR-T therapy, including the type of CAR structure, the quality of T cells infused and host-specific factors, such as tumor heterogeneity and the tumor microenvironment.
The variable genotypic and phenotypic profiles presented by malignant cells make their complete eradication a real challenge. For a therapeutic strategy to be promising, in addition to its high potency and low toxicity, it must act concomitantly on different targets or regulating different mechanisms that favor tumor regression. Therefore, the use of multi-specific CAR-T cells and armored CAR-T cells, especially the ones able to induce on-target immunomodulation, have more chances to induce robust clinical outcomes. The importance of this approach can be seen by the increasing number clinical protocols available based on CAR-T cells that can release immune checkpoint blockade antibodies. The release of bispecific T-cell engager (BiTE) by CAR-T cells is another attractive alternative to improve CAR-T cells function with low toxicity169. BiTE is a bispecific protein that has two linked scFvs, one targeting a cell-surface molecule on T cells (for example, CD3ε) and the other targeting a tumor antigen on malignant cells, resulting in T-cell responses and killing of tumor cells170. There are some technologies, such as XTEN, where bispecific T-cell engagers are bounded to a “polyethylene glycol-like” tail, which can be cleaved by proteases present only in the inflammatory tumor milieu, allowing the local delivery of BiTEs with lower toxicity171. A possible release of bifunctional checkpoint-inhibitory T cell engagers (CiTEs) by CAR-T cells is also an exciting alternative to improve the functionality of CAR-T cells172. We can also have an even more personalized therapy since there are some clinical trials where the immunophenotyping of the patient's tumor defines the type of CAR-T cells that will be produced (NCT03423992).
Patients treated with CAR-T cells can have some serious adverse effects, such as tumor lysis syndrome and cytokines release syndrome (CRS) that, despite manageable, can become severe complications if not appropriately handled. On-target off-tumor toxicity is also a concern, and some current approaches allow specific CAR-T cell ablation, if necessary. Among them, we can cite the insertion of a suicide gene into the CAR, for example, the inducible caspase-9, which can be activated by a small drug called rimiducid (AP1903), leading to apoptosis of CAR-T cells173 or the co-expression of CD20 or EGFRt by the CAR, sensitizing it to the treatment with rituximab or cetuximab, respectively174,175. Some designed CAR-T cells can be inhibited when they bind to a specific antigen expressed only by healthy cells, despite the simultaneous binding with their TAA175, blocking off-target effects. A decrease in the dose used in CAR-T studies is also a possible strategy to minimize off-target effects, and a mathematical model has shown that using one-tenth and even one percent of the current CAR-T doses used in pre-clinical trials can lead to similar results176. Therefore, lower CAR-T doses must be tested in pre-clinical and clinical trials.
The resistance to CAR-T cell therapies is a significant limitation, commonly caused by loss or downregulation of TAA expression. A recent paper has shown that CARs can induce reversible antigen loss through a process called trogocytosis. In this process, the TAA is transferred to T cells, impairing target density on tumor cells, and promoting T cell killing and exhaustion177. The reversion of the antigen loss processes must be explored to counteract the resistance to CAR-T cell therapies. Besides, circulating tumor cells can be collected together with lymphocytes by apheresis, and the CAR can be transduced into these tumor cells, binding in cis to the target antigen expressed by the same cell, leading to tumor cell escape. This effect was seen in a patient that relapsed nine months after CD19-targeted CAR-T cell therapy178.
The treatment of solid tumors with CAR-T cells is a challenge due to the immunosuppressive microenvironment of these tumors, and the difficult CAR-T trafficking and persistence. CAR-T cells have been adapted to express chemokines and cytokines, such as CCR2123 or CCR2b124 alone, CCL19 associated with IL-7179, improving transendothelial migration and intratumoral trafficking, with prolonged survival and complete tumor regression in different solid tumors. Besides, some enzymes that can, for example, degrade the extracellular matrix compounds, such as heparanase127, were also coupled in the second cassette of CAR constructs, improving T cell infiltration and anti-tumor activity; however, this approach raises some concerns due to its intrinsic nonspecificity.
The costs and time involved in autologous CAR-T cell manufacturing is another big issue, and the use of allogeneic universal CAR-T cells made from healthy donors could make these therapies more accessible to the majority of the population. This option could also be useful in patients with lymphopenia or dysfunctional T cells. The use of CRISPR or other gene-editing nucleases to knock out HLA, as well as the endogenous TCR, could eliminate rejection, graft-versus-host disease, and cross-reactivity, dramatically reducing costs and time with a functional allogeneic universal CAR T cell therapy180.
Most of the drawbacks of CAR-T therapy, including their safety and efficacy, can be improved by using gene-editing technologies and synthetic biology, which also implicate in risks. The safety of the viral vector used for CAR-T cells must be considered, since the integration of viral vectors can induce insertional mutagenesis, disrupting tumor-suppressor genes or activating proto-oncogenes, with ability to transform healthy CAR-T cells in malignant clones. The use of non-viral vectors, such as Sleeping Beauty transposons—especially the cassettes vectorized as minicircles associated with mRNA transposase—can be an alternative to improve both safety and costs181,182.
Moreover, the growing amount of information resulting from the several clinical trials performed with CAR-T cells can also address important questions regarding the safety and efficacy of genic therapy, setting up new directions and opportunities for this field. There is a very significant investment into CAR-T approaches by large pharma, biotech startups, and many academic medical centers, much of which has been devoted to solving the current limitations of CAR T cell therapies. As rapidly as this field is moving, the one prediction we can make for certain is that 5–10 years from now, the field will look entirely different. We are just at the beginning.
7. Practice points
-
•
In the clinical setting, CAR-T cells have shown particular effectiveness in the treatment of B cell malignancies, and some clinical trials using anti-B cell maturation antigen CAR-T cells have shown remarkable activity for the treatment of multiple myeloma.
-
•
The expression rates of the TAA target for CAR-T cell therapy must be confirmed since they determine the therapeutic efficacy.
-
•
Lymphodepletion ameliorates CAR-T cell performance in most cases.
-
•
Second generation CAR-T cells are the best studied in clinical trials until now, and the 4-1BB co-stimulatory domain is slightly superior for long-term results compared to CD28. However, third-generation CARs combining both co-stimulatory domains are superior in most pre-clinical studies.
-
•
Armored CAR-T cells able to release immune checkpoint blockade antibodies, BiTEs, CiTEs, enzymes, cytokines or chemokines have been promising in pre-clinical and clinical trials, improving the therapeutic efficacy of CAR-T cells against solid tumors.
-
•
The choice of non-viral vectors can be a safer and cheaper alternative for CAR-T cell production.
-
•
CAR-T cells are beyond cancer, with additional power to treat autoimmune diseases and some viral infections, such as HIV, hepatitis B, hepatitis C, cytomegalovirus as well as some opportunistic fungus infections. Anti-HIV CAR-T cell therapy is already on clinical trials183.
-
•
The use of all-in-one closed systems, such as CliniMACS Prodigy184 or similar for CAR-T cell production in clinical scale may improve the feasibility of this therapeutic approach.
8. Research agenda
-
•
Limited clinical efficacy has been seen for the treatment of solid tumors with CAR-T cells, and the research in this field must be continually refined and improved.
-
•
The loss or downregulation of the TAA on tumor cells remains an essential hurdle to the sustained effects of CAR-T-based therapies and strategies to block trogocytosis, and other mechanisms related to antigen loss or downregulation might increase the durability of CAR-T therapies, with consequent better outcomes.
-
•
The search for more specific TAAs must go on.
-
•
Better manufacturing processes to reduce time and costs for CAR-T cell production are necessary.
Author contributions
Stephan A. Grupp, Ghanbar Mahmoodi Chalbatani and Hassan Dana designed the study. Hassan Dana, Ghanbar Mahmoodi Chalbatani, Seyed Amir Jalali and Eloah Rabello Suarez wrote the manuscript Hamid Reza Mirzaei and Catarina Rapôso helped supervise the project. Stephan A. Grupp and Thomas J. Webster approved final version.
Conflicts of interest
Stephan A. Grupp receives study support from Novartis, Kite, Servier, and Vertex. He consults for Novartis, Roche, GSK, Humanigen, CBMG, and Janssen/JnJ. He is on study steering committees or scientific advisory boards for Novartis, Allogene, Jazz, Adaptimmune, TCR2, Cellectis, Juno, and Vertex/CRISPR. He has a patent (Toxicity management for anti-tumor activity of CARs, WO 2014011984 A1) that is managed according to the University of Pennsylvania patent policy. The other authors declare no conflicts of interests in this work.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
References
- 1.Swann J.B., Smyth M.J. Immune surveillance of tumors. J Clin Invest. 2007;117:1137–1146. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gun S.Y., Lee S.W.L., Sieow J.L., Wong S.C. Targeting immune cells for cancer therapy. Redox Biol. 2019;25:101174. doi: 10.1016/j.redox.2019.101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arruebo M., Vilaboa N., Sáez-Gutierrez B., Labea J., Tres A., Valladares M. Assessment of the evolution of cancer treatment therapies. Cancers (Basel) 2011;3:3279–3330. doi: 10.3390/cancers3033279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imai K., Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Canc. 2006;6:714–727. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- 6.Woessner D.W., Lim C.S., Deininger M.W. Development of an effective therapy for chronic myelogenous leukemia. Canc J. 2011;17:477–486. doi: 10.1097/PPO.0b013e318237e5b7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li D., Li X., Zhou W.L., Huang Y., Liang X., Jiang L. Genetically engineered T cells for cancer immunotherapy. Signal Transduct Target Ther. 2019;4:35. doi: 10.1038/s41392-019-0070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiNofia A.M., Maude S.L. Chimeric antigen receptor T-cell therapy clinical results in pediatric and young adult B-ALL. Hemasphere. 2019;3:e279. doi: 10.1097/HS9.0000000000000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuster S.J., Svoboda J., Chong E.A., Nasta S.D., Mato A.R., Anak O. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377:2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang K., Liu Y., Li J., Wang B., Bishop R., White C. A multiscale simulation framework for the manufacturing facility and supply chain of autologous cell therapies. Cytotherapy. 2019;21:1081–1093. doi: 10.1016/j.jcyt.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Harrison R.P., Zylberberg E., Ellison S., Levine B.L. Chimeric antigen receptor-T cell therapy manufacturing: modelling the effect of offshore production on aggregate cost of goods. Cytotherapy. 2019;21:224–233. doi: 10.1016/j.jcyt.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Whilding L.M., Maher J. CAR T-cell immunotherapy: the path from the by-road to the freeway?. Mol Oncol. 2015;9:1994–2018. doi: 10.1016/j.molonc.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu D. CAR-T "the living drugs", immune checkpoint inhibitors, and precision medicine: a new era of cancer therapy. J Hematol Oncol. 2019;12:113. doi: 10.1186/s13045-019-0819-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galluzzi L., Vacchelli E., Pedro J.M.B.S., Buqué A., Senovilla L., Baracco E.E. Classification of current anticancer immunotherapies. Oncotarget. 2014;5:12472–12508. doi: 10.18632/oncotarget.2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anfossi N., André P., Guia S., Falk C.S., Roetynck S., Stewart C.A. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–342. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 16.Sha H.H., Wang D.D., Yan D.L., Hu Y., Yang S.J., Liu S.W. Chimaeric antigen receptor T-cell therapy for tumour immunotherapy. Biosci Rep. 2017;37 doi: 10.1042/BSR20160332. BSR20160335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu W., Wang G., Huang D., Sui M., Yibing X. Cancer immunotherapy based on natural killer cells: current progress and new opportunities. Front Immunol. 2019;10:1205. doi: 10.3389/fimmu.2019.01205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filley A.C., Henriquez M., Dey M. CART immunotherapy: development, success, and translation to malignant gliomas and other solid tumors. Front Oncol. 2018;8:453. doi: 10.3389/fonc.2018.00453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long N.E., Sullivan B.J., Ding H., Doll S., Ryan M.A., Hitchcock C.L. Linker engineering in anti-TAG-72 antibody fragments optimizes biophysical properties, serum half-life, and high-specificity tumor imaging. J Biol Chem. 2018;293:9030–9040. doi: 10.1074/jbc.RA118.002538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walseng E., Köksal H., Sektioglu I.M., Fane A., Skorstad G., Kvalheim G. A TCR-based chimeric antigen receptor. Sci Rep. 2017;7:10713. doi: 10.1038/s41598-017-11126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu X., Sun Q., Liang X., Chen Z., Zhang X., Zhou X. Mechanisms of relapse after CD19 CAR T-cell therapy for acute lymphoblastic leukemia and its prevention and treatment strategies. Front Immunol. 2019;10:2664. doi: 10.3389/fimmu.2019.02664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan W., Hu H., Tang B. Advances of chimeric antigen receptor T cell therapy in ovarian cancer. OncoTargets Ther. 2019;12:8015–8022. doi: 10.2147/OTT.S203550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hopfinger G., Jäger U., Worel N. CAR-T cell therapy in diffuse large B cell lymphoma: hype and hope. Hemasphere. 2019;3:e185. doi: 10.1097/HS9.0000000000000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charrot S., Hallam S. CAR-T cells: future perspectives. Hemasphere. 2019;3:e188. doi: 10.1097/HS9.0000000000000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen M.C., Popplewell L., Cooper L.J., DiGiusto D., Kalos M., Ostberg J.R. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brentjens R.J., Rivière I., Park J.H., Davila M.L., Wang X., Stefanski J. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kochenderfer J.N., Dudley M.E., Feldman S.A., Wilson W.H., Spaner D.E., Maric I. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porter D.L., Hwang W.T., Frey N.V., Lacey S.F., Shaw P.A., Loren A.W. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brentjens R.J., Davila M.L., Riviere I., Park J., Wang X., Cowell L.G. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davila M.L., Riviere I., Wang X., Bartido S., Park J., Curran K. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kochenderfer J.N., Dudley M.E., Carpenter R.O., Kassim S.H., Rose J.J., Telford W.G. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122:4129–4139. doi: 10.1182/blood-2013-08-519413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brudno J.N., Kochenderfer J.N. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cruz C.R.Y., Micklethwaite K.P., Savoldo B., Ramos C.A., Lam S., Ku S. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122:2965–2973. doi: 10.1182/blood-2013-06-506741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kochenderfer J.N., Somerville R.P.T., Lu T., Shi V., Bot A., Rossi J. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol. 2017;35:1803. doi: 10.1200/JCO.2016.71.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gardner R.A., Finney O., Annesley C., Brakke H., Summers C., Leger K. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Locke F.L., Neelapu S.S., Bartlett N.L., Siddiqi T., Chavez J.C., Hosing C.M. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25:285–295. doi: 10.1016/j.ymthe.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vairy S., Garcia J.L., Teira P., Bittencourt H. CTL019 (tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia. Drug Des Dev Ther. 2018;12:3885–3898. doi: 10.2147/DDDT.S138765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siska P.J., Rathmell J.C. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015;36:257–264. doi: 10.1016/j.it.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ostrand-Rosenberg S., Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. 2018;200:422–431. doi: 10.4049/jimmunol.1701019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia A.L., Wang X.C., Lu Y.J., Lu X.J., Sun B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget. 2017;8:90521–90531. doi: 10.18632/oncotarget.19361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seidel J.A., Otsuka A., Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86. doi: 10.3389/fonc.2018.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilham D.E., Debets R., Pule M., Hawkins R.E., Abken H. CAR-T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med. 2012;18:377–384. doi: 10.1016/j.molmed.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 47.Pule M.A., Savoldo B., Myers G.D., Rossig C., Russell H.V., Dotti G. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knochelmann H.M., Smith A.S., Dwyer C.J., Wyatt M.M., Mehrotra S., Paulos C.M. CAR T cells in solid tumors: blueprints for building effective therapies. Front Immunol. 2018;9:1740. doi: 10.3389/fimmu.2018.01740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen L., Linsley P.S., Hellstrom K.E. Costimulation of T cells for tumor immunity. Immunol Today. 1993;14:483–486. doi: 10.1016/0167-5699(93)90262-J. [DOI] [PubMed] [Google Scholar]
- 51.Zhong X.S., Matsushita M., Plotkin J., Riviere I., Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beatty G.L., Gladney W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Canc Res. 2015;21:687–692. doi: 10.1158/1078-0432.CCR-14-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rabinovich G.A., Gabrilovich D., Sotomayor E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Newick K., O'Brien S., Sun J., Kapoor V., Maceyko S., Lo A. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol Res. 2016;4:541–551. doi: 10.1158/2326-6066.CIR-15-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ligtenberg M.A., Mougiakakos D., Mukhopadhyay M., Witt K., Lladser A. Coexpressed catalase protects chimeric antigen receptor-redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J Immunol. 2016;196:759–766. doi: 10.4049/jimmunol.1401710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weinberg F., Ramnath N., Nagrath D. Reactive oxygen species in the tumor microenvironment: an overview. Cancers (Basel) 2019;11:1191. doi: 10.3390/cancers11081191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suarez E.R., Chang D.K., Sun J., Sui J., Freeman G.J., Signoretti S. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7:34341–34355. doi: 10.18632/oncotarget.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mirzaei H.R., Pourghadamyari H., Rahmati M., Mohammadi A., Nahand J.S., Rezaei A. Gene-knocked out chimeric antigen receptor (CAR) T cells: tuning up for the next generation cancer immunotherapy. Canc Lett. 2018;423:95–104. doi: 10.1016/j.canlet.2018.03.010. [DOI] [PubMed] [Google Scholar]
- 59.Jung I.Y., Lee J. Unleashing the therapeutic potential of CAR-T cell therapy using gene-editing technologies. Mol Cell. 2018;41:717–723. doi: 10.14348/molcells.2018.0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miliotou A.N., Papadopoulou L.C. CAR T-cell therapy: a new era in cancer immunotherapy. Curr Pharmaceut Biotechnol. 2018;19:5–18. doi: 10.2174/1389201019666180418095526. [DOI] [PubMed] [Google Scholar]
- 61.Chen N., Xiaoyu L., Chintala N.K., Tano Z.E., Adusumilli P.S. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr Opin Immunol. 2018;51:103–110. doi: 10.1016/j.coi.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marcus A., Gowen B.G., Thompson T.W., Iannello A., Ardolino M., Deng W. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol. 2014;122:91–128. doi: 10.1016/B978-0-12-800267-4.00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Minutolo N.G., Hollander E.E., Powell D.J., Jr. The emergence of universal immune receptor T cell therapy for cancer. Front Oncol. 2019;9:176. doi: 10.3389/fonc.2019.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han X., Wang Y., Wei J., Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol. 2019;12:128. doi: 10.1186/s13045-019-0813-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia; chimeric antigen receptor-modified T cells for acute lymphoid leukemia; chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2016;374:998. [Google Scholar]
- 66.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyerr M., Bittencourt H. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu B., Yan L., Zhou M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am J Cancer Res. 2019;9:228–241. [PMC free article] [PubMed] [Google Scholar]
- 68.D'Aloia M.M., Zizzari I.G., Sacchetti B., Pierelli L., Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. 2018;9:282. doi: 10.1038/s41419-018-0278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sotillo E., Barrett D.M., Black K.L., Bagashev A., Oldridge D., Wu G. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Canc Discov. 2015;5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Orlando E.J., Han X., Tribouley C., Wood P.A., Leary R.J., Riester M. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24:1504–1506. doi: 10.1038/s41591-018-0146-z. [DOI] [PubMed] [Google Scholar]
- 72.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chu F., Cao J., Neelalpu S.S. Versatile CAR T-cells for cancer immunotherapy. Contemp Oncol (Pozn) 2018;22:73–80. doi: 10.5114/wo.2018.73892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu X., Jiang S., Fang C., Yang S., Olalere D., Paquignot E.C. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Canc Res. 2015;75:3596–3607. doi: 10.1158/0008-5472.CAN-15-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Q.Y., Yang J.D., Wang Y.S. Current strategies to improve the safety of chimeric antigen receptor (CAR) modified T cells. Immunol Lett. 2017;190:201–205. doi: 10.1016/j.imlet.2017.08.018. [DOI] [PubMed] [Google Scholar]
- 76.Jaspers J.E., Brentjens R.J. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacol Ther. 2017;178:83–91. doi: 10.1016/j.pharmthera.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khan J.F., Khan A.S., Brentjens R.J. Application of CAR T cells for the treatment of solid tumors. Prog Mol Biol Transl Sci. 2019;164:293–327. doi: 10.1016/bs.pmbts.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 78.Ma S., Li X., Wang X., Cheng L., Li Z., Zhang C. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci. 2019;15:2548–2560. doi: 10.7150/ijbs.34213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mirzaei H.R., Rodriguez A., Shepphird J., Brown C.E., Badie B. Chimeric antigen receptors T cell therapy in solid tumor: challenges and clinical applications. Front Immunol. 2017;8:1850. doi: 10.3389/fimmu.2017.01850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Benmebarek M.R., Karches C.H., Cadilha B.L., Lesch S., Endres S., Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. 2019;20:1283. doi: 10.3390/ijms20061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abbott R.C., Cross R.S., Jenkins M.R. Finding the keys to the CAR: identifying novel target antigens for T cell redirection immunotherapies. Int J Mol Sci. 2020;21:151. doi: 10.3390/ijms21020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arteaga C.L. Epidermal growth factor receptor dependence in human tumors: more than just expression? Oncol. 2002;7(Suppl 4):31–39. doi: 10.1634/theoncologist.7-suppl_4-31. [DOI] [PubMed] [Google Scholar]
- 83.Heimberger A.B., Suki D., Yang D., Shi W., Aldape K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J Transl Med. 2005;3:38. doi: 10.1186/1479-5876-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morgan R.A., Johnson L.A., Davis J.L., Zheng Z., Woolard K.D., Reap E.A. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23:1043–1053. doi: 10.1089/hum.2012.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ren P.P., Li M., Li T.F., Han S.Y. Anti-EGFRvIII chimeric antigen receptor-modified T cells for adoptive cell therapy of glioblastoma. Curr Pharmaceut Des. 2017;23:2113–2116. doi: 10.2174/1381612823666170316125402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sampson J.H., Choi B.D., Sanchez-Perez L., Suryadevara C.M., Snyder D.J., Flores C.T. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Canc Res. 2014;20:972–984. doi: 10.1158/1078-0432.CCR-13-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.O'Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lorimer I.A. Mutant epidermal growth factor receptors as targets for cancer therapy. Curr Cancer Drug Targets. 2002;2:91–102. doi: 10.2174/1568009023333926. [DOI] [PubMed] [Google Scholar]
- 89.Newick K., Moon E., Albelda S.M. Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther Oncolytics. 2016;3:16006. doi: 10.1038/mto.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu M.R., Zhang T., DeMars L.R., Sentman C.L. B7H6-specific chimeric antigen receptors lead to tumor elimination and host antitumor immunity. Gene Ther. 2015;22:675–684. doi: 10.1038/gt.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spear P., Barber A., Rynda-Apple A., Sentman C.L. NKG2D CAR T-cell therapy inhibits the growth of NKG2D ligand heterogeneous tumors. Immunol Cell Biol. 2013;91:435–440. doi: 10.1038/icb.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lanitis E., Dangaj D., Hagemann I.S., Song D.G., Best A., Sandaltzopoulos R. Primary human ovarian epithelial cancer cells broadly express HER2 at immunologically-detectable levels. PLoS One. 2012;7 doi: 10.1371/journal.pone.0049829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Song D.G., Ye Q., Carpenito C., Poussin M., Wang L.P., Ji C. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB) Canc Res. 2011;71:4617–4627. doi: 10.1158/0008-5472.CAN-11-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kobayashi E., Kishi H., Ozawa T., Hamana H., Nakagawa H., Jin A. A chimeric antigen receptor for TRAIL-receptor 1 induces apoptosis in various types of tumor cells. Biochem Biophys Res Commun. 2014;453:798–803. doi: 10.1016/j.bbrc.2014.10.024. [DOI] [PubMed] [Google Scholar]
- 95.Hong H., Stastny M., Brown C., Chang W.C., Ostberg J.R., Forman S.J. Diverse solid tumors expressing a restricted epitope of L1-CAM can be targeted by chimeric antigen receptor redirected T lymphocytes. J Immunother. 2014;37:93–104. doi: 10.1097/CJI.0000000000000018. [DOI] [PubMed] [Google Scholar]
- 96.Krebs S., Chow K.K.H., Yi Z., Rodriguez-Cruz T., Hegde M., Gerken C. T cells redirected to interleukin-13Ralpha2 with interleukin-13 mutein—chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Ralpha1. Cytotherapy. 2014;16:1121–1131. doi: 10.1016/j.jcyt.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Campos B., Zeng L., Daotrong P.H., Eckstein V., Unterberg A., Mairbäurl H. Expression and regulation of AC133 and CD133 in glioblastoma. Glia. 2011;59:1974–1986. doi: 10.1002/glia.21239. [DOI] [PubMed] [Google Scholar]
- 98.Miao H., Choi B.D., Suryadevara C.M., Sanchez-Perez L., Yang S., Leon G.D. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS One. 2014;9 doi: 10.1371/journal.pone.0094281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kakarla S., Chow K.K.H., Mata M., Shaffer D.R., Song X.T., Wu M.F. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21:1611–1620. doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stephan M.T., Ponomarev V., Brentjens R.J., Chang A.H., Dobrenkov K.V., Heller G. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13:1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 101.Abate-Daga D., Lagisetty K.H., Tran E., Zheng Z., Gattinoni L., Yu Z. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther. 2014;25:1003–1012. doi: 10.1089/hum.2013.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chmielewski M., Rappl G., Hombach A.A., Abken H. T cells redirected by a CD3zeta chimeric antigen receptor can establish self-antigen-specific tumour protection in the long term. Gene Ther. 2013;20:177–186. doi: 10.1038/gt.2012.21. [DOI] [PubMed] [Google Scholar]
- 103.Wilkie S., Picco G., Foster J., Davies D.M., Julien S., Cooper L. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180:4901–4909. doi: 10.4049/jimmunol.180.7.4901. [DOI] [PubMed] [Google Scholar]
- 104.Geldres C., Savoldo B., Hoyos V., Caruana I., Zhang M., Yvon E. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Canc Res. 2014;20:962–971. doi: 10.1158/1078-0432.CCR-13-2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chow K.K., Naik S., Kakarla S., Brawley V.S., Shaffer D.R., Yi Z. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21:629–637. doi: 10.1038/mt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beatty G.L., Haas A.R., Maus M.V., Torigian D.A., Soulen M.C., Plesa G. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kretz A.L., Trauzold A., Hillenbrand A., Knippschild U., Henne-Bruns D., Karstedt S.V. TRAILblazing strategies for cancer treatment. Cancers (Basel) 2019;11:456. doi: 10.3390/cancers11040456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Di S., Li Z. Treatment of solid tumors with chimeric antigen receptor-engineered T cells: current status and future prospects. Sci China Life Sci. 2016;59:360–369. doi: 10.1007/s11427-016-5025-6. [DOI] [PubMed] [Google Scholar]
- 109.Lamers C.H., Sleijfer S., Steenbergen S.V., Elzakker P.V., Krimpen B.V., Groot C. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Klebanoff C.A., Rosenberg S.A., Restifo N.P. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med. 2016;22:26–36. doi: 10.1038/nm.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maher J., Wilkie S., Davies D.M., Arif S., Picco G., Julien S. Targeting of tumor-associated glycoforms of MUC1 with CAR T cells. Immunity. 2016;45:945–946. doi: 10.1016/j.immuni.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 112.Posey A.D., Jr., Clausen H., June C.H. Distinguishing truncated and normal MUC1 glycoform targeting from Tn-MUC1-specific CAR T cells: specificity is the key to safety. Immunity. 2016;45:947–948. doi: 10.1016/j.immuni.2016.10.015. [DOI] [PubMed] [Google Scholar]
- 113.Chekmasova A.A., Rao T.D., Nikhamin Y., Park K.J., Levine D.A., Spriggs D.R. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin Canc Res. 2010;16:3594–3606. doi: 10.1158/1078-0432.CCR-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chmielewski M., Hahn O., Rappl G., Nowak M., Schmidt-Wolf I.H., Hombach A.A. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology. 2012;143:1095–1107. doi: 10.1053/j.gastro.2012.06.037. e2. [DOI] [PubMed] [Google Scholar]
- 115.Sur D., Havasi A., Cainap C., Samasca G., Burz C., Balacescu O. Chimeric antigen receptor T-cell therapy for colorectal cancer. J Clin Med. 2020;9:182. doi: 10.3390/jcm9010182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Springuel L., Lonez C., Alexandre B., Cutsem E.V., Machiels J.P.H., Eynde M.V.D. Chimeric antigen receptor-T cells for targeting solid tumors: current challenges and existing strategies. BioDrugs. 2019;33:515–537. doi: 10.1007/s40259-019-00368-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Trédan O., Galmarini C.M., Patel K., Tannock Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441–1454. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 118.Yong C.S.M., Dardalhon V., Devaud C., Taylor N., Darcy P.K., Kershaw M.H. CAR T-cell therapy of solid tumors. Immunol Cell Biol. 2017;95:356–363. doi: 10.1038/icb.2016.128. [DOI] [PubMed] [Google Scholar]
- 119.Adusumilli P.S., Cherkassky L., Villena-Vargas J., Colocos C., Servais E., Plotkin J. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Enblad G., Karlsson H., Loskog A.S. CAR T-cell therapy: the role of physical barriers and immunosuppression in lymphoma. Hum Gene Ther. 2015;26:498–505. doi: 10.1089/hum.2015.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ramachandran M., Dimberg A., Essand M. The cancer-immunity cycle as rational design for synthetic cancer drugs: novel DC vaccines and CAR T-cells. Semin Canc Biol. 2017;45:23–35. doi: 10.1016/j.semcancer.2017.02.010. [DOI] [PubMed] [Google Scholar]
- 122.Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. 2015;15:692–704. doi: 10.1038/nri3908. [DOI] [PubMed] [Google Scholar]
- 123.Moon E.K., Carpenito C., Sun J., Wang L.C.S., Kapoor V., Predina J. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Canc Res. 2011;17:4719–4730. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Craddock J.A., Lu A., Bear A., Pule M., Brenner M.K., Rooney C.M. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Edsparr K., Basse P.H., Goldfarb R.H., Albertsson P. Matrix metalloproteinases in cytotoxic lymphocytes impact on tumour infiltration and immunomodulation. Cancer Microenviron. 2011;4:351–360. doi: 10.1007/s12307-010-0057-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stewart M.D., Sanderson R.D. Heparan sulfate in the nucleus and its control of cellular functions. Matrix Biol. 2014;35:56–59. doi: 10.1016/j.matbio.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Caruana I., Savoldo B., Hoyos V., Weber G., Liu H., Kim E.S. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524–529. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li J., Li W., Huang K., Zhang Y., Kupfer G., Zhao Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: lessons learned and strategies for moving forward. J Hematol Oncol. 2018;11:22. doi: 10.1186/s13045-018-0568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yang S., Ji Y., Gattinoni L., Zhang L., Yu Z., Restifo N.P. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol Immunother. 2013;62:727–736. doi: 10.1007/s00262-012-1378-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Koneru M., Purdon T.J., Spriggs D., Koneru S., Brentjens R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. OncoImmunology. 2015;4 doi: 10.4161/2162402X.2014.994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mirzaei H.R., Hadjati J. Commentary: IL-12-secreting tumor-targeted chimeric antigen receptor T cells: an unaddressed concern on Koneru et al. (2015) OncoImmunology. 2016;5 doi: 10.1080/2162402X.2015.1100792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Howie D., Waldmann H., Cobbold S. Nutrient sensing via mTOR in T cells maintains a tolerogenic microenvironment. Front Immunol. 2014;5:409. doi: 10.3389/fimmu.2014.00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ninomiya S., Narala N., Huye L., Yagyu S., Savoldo B., Dotti G. Tumor indoleamine 2,3-dioxygenase (Ido) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. 2015;125:3905–3916. doi: 10.1182/blood-2015-01-621474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Su Y., Huang X., Raskovalova T., Zacharia L., Lokshin A., Jackson E. Cooperation of adenosine and prostaglandin E2 (PGE2) in amplification of cAMP-PKA signaling and immunosuppression. Cancer Immunol Immunother. 2008;57:1611–1623. doi: 10.1007/s00262-008-0494-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Massague J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kerkar S.P., Muranski P., Kaiser A., Boni A., Sanchez-Perez L., Yu Z. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Canc Res. 2010;70:6725–6734. doi: 10.1158/0008-5472.CAN-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Avanzi M.P., Leeuwen D.G.V., Li X., Cheung K., Park H., Purdon T.J. IL-18 secreting CAR T cells enhance cell persistence, induce prolonged B cell aplasia and eradicate CD19+ tumor cells without need for prior conditioning. Blood. 2016;128:816. [Google Scholar]
- 138.Chmielewski M., Abken H. TRUCKs: the fourth generation of CARs. Expet Opin Biol Ther. 2015;15:1145–1154. doi: 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- 139.Haribhai D., Lin W., Edwards B., Ziegelbauer J., Salzman N.H., Carlson M.R. A central role for induced regulatory T cells in tolerance induction in experimental colitis. J Immunol. 2009;182:3461–3468. doi: 10.4049/jimmunol.0802535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fontenot J.D., Gavin M.A., Rudensky A.Y. Pillars article: Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. J Immunol. 2017;198:986–992. [PubMed] [Google Scholar]
- 141.Sakaguchi S., Yamaguchi T., Nomura T., Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 142.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 143.Levine B.L., Miskin J., Wonnacott K., Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017;4:92–101. doi: 10.1016/j.omtm.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Scarfo I., Maus M.V. Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. J Immunother Cancer. 2017;5:28. doi: 10.1186/s40425-017-0230-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Levine B.L. Performance-enhancing drugs: design and production of redirected chimeric antigen receptor (CAR) T cells. Canc Gene Ther. 2015;22:79–84. doi: 10.1038/cgt.2015.5. [DOI] [PubMed] [Google Scholar]
- 146.Allegra A., Innao V., Gerace D., Vaddinelli D., Musolino C. Adoptive immunotherapy for hematological malignancies: current status and new insights in chimeric antigen receptor T cells. Blood Cells Mol Dis. 2016;62:49–63. doi: 10.1016/j.bcmd.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 147.Yun Y.S., Hargrove M.E., Ting C.C. In vivo antitumor activity of anti-CD3-induced activated killer cells. Canc Res. 1989;49:4770–4774. [PubMed] [Google Scholar]
- 148.East J.E., Sun W., Webb T.J. Artificial antigen presenting cell (aAPC) mediated activation and expansion of natural killer T cells. J Vis Exp. 2012;70:4333. doi: 10.3791/4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Singh H., Manuri P.R., Olvares S., Dara N., Dawson M.J., Huls H. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Canc Res. 2008;68:2961–2971. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Boissel L., Betancur M., Wels W.S., Tuncer H., Klingemann H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res. 2009;33:1255–1259. doi: 10.1016/j.leukres.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hollyman D., Stefanski J., Przybylowski M., Bartido S., Borquez-Ojeda O., Taylor C. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 2009;32:169. doi: 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bajgain P., Mucharla R., Wilson J., Welch D., Anurathapan U., Liang B. Optimizing the production of suspension cells using the G-Rex "M" series. Mol Ther Methods Clin Dev. 2014;1:14015. doi: 10.1038/mtm.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kaiser A.D., Assenmacher M., Schröder B., Meyer M., Orentas R., Bethke U. Towards a commercial process for the manufacture of genetically modified T cells for therapy. Canc Gene Ther. 2015;22:72–78. doi: 10.1038/cgt.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chang Z.L., Silver P.A., Chen Y.Y. Identification and selective expansion of functionally superior T cells expressing chimeric antigen receptors. J Transl Med. 2015;13:161. doi: 10.1186/s12967-015-0519-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang X., Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;3:16015. doi: 10.1038/mto.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mo Z., Du P., Wang G., Wang Y. The multi-purpose tool of tumor immunotherapy: gene-engineered T cells. J Canc. 2017;8:1690–1703. doi: 10.7150/jca.18681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Getts D., Hofmeister R., Quintás-Cardama A. Synthetic T cell receptor-based lymphocytes for cancer therapy. Adv Drug Deliv Rev. 2019;141:47–54. doi: 10.1016/j.addr.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 158.Kass I., Buckle A.M., Borg N.A. Understanding the structural dynamics of TCR-pMHC complex interactions. Trends Immunol. 2014;35:604–612. doi: 10.1016/j.it.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 159.Crowther M.D., Svane I.M., Met Ö. T-cell gene therapy in cancer immunotherapy: why it is no longer just CARs on the road. Cells. 2020;9:1588. doi: 10.3390/cells9071588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Garber K. Driving T-cell immunotherapy to solid tumors. Nat Biotechnol. 2018;36:215–219. doi: 10.1038/nbt.4090. [DOI] [PubMed] [Google Scholar]
- 161.Shitaoka K., Hamana H., Kishi H., Hayakawa Y., Kobayashi E., Sukegawa K. Identification of tumoricidal TCRs from tumor-infiltrating lymphocytes by single-cell analysis. Cancer Immunol Res. 2018;6:378–388. doi: 10.1158/2326-6066.CIR-17-0489. [DOI] [PubMed] [Google Scholar]
- 162.Parkhurst M., Gros A., Pasetto A., Prickett T., Crystal J.S., Robbins P. Isolation of T-cell receptors specifically reactive with mutated tumor-associated antigens from tumor-infiltrating lymphocytes based on CD137 expression. Clin Canc Res. 2017;23:2491–2505. doi: 10.1158/1078-0432.CCR-16-2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Janssen A., Hidalgo J.V., Beringer D.X., Dooremalen S.V., Fernando F., Diest E.V. γδ T-cell receptors derived from breast cancer-infiltrating T lymphocytes mediate antitumor reactivity. Cancer Immunol Res. 2020;8:530–543. doi: 10.1158/2326-6066.CIR-19-0513. [DOI] [PubMed] [Google Scholar]
- 164.Zhang J., Wang L. The emerging world of TCR-T cell trials against cancer: a systematic review. Technol Canc Res Treat. 2019;18 doi: 10.1177/1533033819831068. 1533033819831068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Xu Y., Yang Z., Horan L.H., Zhang P., Liu L., Zimdahl B. A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release. Cell Discov. 2018;4:62. doi: 10.1038/s41421-018-0066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Harris D.T., Hager M.V., Smith S.N., Cai Q., Stone J.D., Kruger P. Comparison of T cell activities mediated by human TCRs and CARs that use the same recognition domains. J Immunol. 2018;200:1088–1100. doi: 10.4049/jimmunol.1700236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Crowther M.D., Dolton G., Legut M., Caillaud M.E., Lloyd A., Attaf M. Author correction: genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat Immunol. 2020;21:695. doi: 10.1038/s41590-019-0578-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Legut M., Dolton G., Mian A.A., Ottmann O.G., Sewell A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood. 2018;131:311–322. doi: 10.1182/blood-2017-05-787598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Choi B.D., Yu X., Castano A.P., Bouffard A.A., Schmidts A., Larson R.C. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. 2019;37:1049–1058. doi: 10.1038/s41587-019-0192-1. [DOI] [PubMed] [Google Scholar]
- 170.Slaney C.Y., Wang P., Darcy P.K., Kershaw M.H. CARs versus BiTEs: a comparison between T cell-redirection strategies for cancer treatment. Canc Discov. 2018;8:924–934. doi: 10.1158/2159-8290.CD-18-0297. [DOI] [PubMed] [Google Scholar]
- 171.Podust V.N., Balan S., Sim B.C., Coyle M.P., Ernst U., Peters R.T. Extension of in vivo half-life of biologically active molecules by XTEN protein polymers. J Control Release. 2016;240:52–66. doi: 10.1016/j.jconrel.2015.10.038. [DOI] [PubMed] [Google Scholar]
- 172.Goebeler M.E., Bargou R.C. T cell-engaging therapies—BiTEs and beyond. Nat Rev Clin Oncol. 2020;7:418–434. doi: 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 173.Gargett T., Brown M.P. The inducible caspase-9 suicide gene system as a "safety switch" to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014;5:235. doi: 10.3389/fphar.2014.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tasian S.K., Kenderian S.S., Shen F., Ruella M., Shestova O., Kozlowski M. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood. 2017;129:2395–2407. doi: 10.1182/blood-2016-08-736041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yu S., Yi Ming, Qin S., Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Canc. 2019;18:125. doi: 10.1186/s12943-019-1057-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Levin A.G., Kronik N., Shiloach T., Waks T., Eshhar Z., Vainstein V. Less is more: reducing the number of administered chimeric antigen receptor T cells in a mouse model using a mathematically guided approach. Cancer Immunol Immunother. 2020;69:1165–1175. doi: 10.1007/s00262-020-02516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hamieh M., Dobrin A., Cabriolu A., Stegen S.J.C., Giavridis T., Mansilla-Soto J. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568:112–116. doi: 10.1038/s41586-019-1054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ruella M., Xu J., Barrett D.M., Fraietta J.A., Reich T.J., Ambrose D.E. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24:1499–1503. doi: 10.1038/s41591-018-0201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Adachi K., Kano Y., Nagai T., Okuyama N., Sakoda Y., Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36:346–351. doi: 10.1038/nbt.4086. [DOI] [PubMed] [Google Scholar]
- 180.Ren J., Zhao Y. Advancing chimeric antigen receptor T cell therapy with CRISPR/Cas9. Protein Cell. 2017;8:634–643. doi: 10.1007/s13238-017-0410-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chicaybam L., Abdo L., Carneiro M., Peixoto B., Viegas M., Sousa P. CAR T cells generated using Sleeping Beauty transposon vectors and expanded with an EBV-transformed lymphoblastoid cell line display antitumor activity in vitro and in vivo. Hum Gene Ther. 2019;30:511–522. doi: 10.1089/hum.2018.218. [DOI] [PubMed] [Google Scholar]
- 182.Hudecek M., Izsvák Z., Johnen S., Renner M., Thumann G., Ivics Z. Going non-viral: the Sleeping Beauty transposon system breaks on through to the clinical side. Crit Rev Biochem Mol Biol. 2017;52:355–380. doi: 10.1080/10409238.2017.1304354. [DOI] [PubMed] [Google Scholar]
- 183.Seif M., Einsele H., Loffler J. CAR T cells beyond cancer: hope for immunomodulatory therapy of infectious diseases. Front Immunol. 2019;10:2711. doi: 10.3389/fimmu.2019.02711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mock U., Nickolay L., Philip B., Cheung G.W.K., Zhan H., Johnston I.C.D. Automated manufacturing of chimeric antigen receptor T cells for adoptive immunotherapy using CliniMACS prodigy. Cytotherapy. 2016;18:1002–1011. doi: 10.1016/j.jcyt.2016.05.009. [DOI] [PubMed] [Google Scholar]