

Abstract
Introduction
It has been suggested that obesity may influence Alzheimer's disease (AD) pathogenesis, yet the numerous publications on this topic have inconsistent results and conclusions.
Methods
Our study examined the effect of varying the timing of high‐fat diet (HFD) consumption on AD‐related pathology and cognition in transgenic Tg6799 AD mice.
Results
HFD feeding starting at or before 3 months of age, prior to severe AD pathology, had protective effects in AD mice: reduced extracellular amyloid beta (Aβ) deposition, decreased fibrinogen extravasation into the brain parenchyma, and improved cognitive function. However, delaying HFD consumption until 6 months of age, when AD pathology is ubiquitous, reduced these protective effects in AD mice.
Discussion
Overall, we demonstrate that the timeline of HFD consumption may play an important role in how dietary fats affect AD pathogenesis and cognitive function.
Keywords: Alzheimer's disease, amyloid beta, fibrinogen, high‐fat diet, obesity
1. INTRODUCTION
Alzheimer's disease (AD) is the most prevalent form of dementia and is characterized by extracellular deposits of amyloid beta (Aβ) and intracellular inclusions of hyperphosphorylated tau. Hallmarks of AD also include neuroinflammation, vascular abnormalities, and profound neuronal loss. Cardiovascular risk factors such as obesity, midlife hypertension, and diabetes are thought to increase the risk for developing AD. 1 , 2 , 3 However, the role of obesity as an independent risk factor for AD remains controversial. While some studies show a positive correlation between high body mass index (BMI) and dementia, 4 , 5 a systematic review of several large well‐controlled studies reported that obesity in late life (60+ years) is not associated with higher risk for developing AD. 6 In addition, low BMI in midlife might increase the risk of developing AD later in life. 6 , 7 However, it is unknown whether preventing weight loss in patients with preclinical AD would slow disease progression.
In animal studies, a high‐fat diet (HFD) is often used to induce weight gain and to determine the association between obesity and AD‐related phenotypes. Diet‐induced obesity has been shown to exacerbate AD‐related pathology and further impair cognitive function 8 , 9 or induce memory impairment in AD mice without affecting AD‐related pathologies in the brain. 10 , 11 Conversely, other evidence indicates that HFD might improve blood‐brain barrier (BBB) integrity and spatial learning without affecting Aβ plaque load. 12 Furthermore, others have reported that crossing AD mice with a genetic mouse model of obesity and diabetes leads to an improvement in memory and decreased levels of Aβ. 13 This lack of consistency in results might be due to variations in the AD mouse model used, specific components of the diet, or the time course of HFD consumption.
The goal of our study was to characterize the effect of HFD on AD‐related pathology and cognitive function in mice over various timelines. Tg6799 (AD) mice and their wild type (WT) littermates were fed either HFD or control diet (CON). We determined the diet's effect on Aβ plaque load in the retrosplenial cortex (RSC) and hippocampus of AD mice as well as cognitive function. We also studied the effect of HFD on the extravasation of fibrinogen into the brain, a pro‐inflammatory plasma protein that is involved in AD pathogenesis. 14 We show that when HFD was fed to AD mice early in life, there was less parenchymal Aβ deposition, fibrinogen extravasation into the brain, and cognitive dysfunction compared to AD CON mice. However, this neuroprotection was not observed in AD mice that started consuming a HFD later in life, after AD pathology was already established. These results suggest that at early stages of AD, dietary fats may be protective to the brain and slow AD onset and progression.
2. METHODS
2.1. Animals
Male Tg6799 mice 15 (referred to as AD mice) and their WT littermates were housed with food and water ad libitum, under controlled temperature (20–22℃), humidity (40%–60%), and illumination (12/12‐hour dark cycle). Experimental diets were administered starting at either 1, 3, or 6 months of age. All animal experiments were conducted in accordance with the guidelines of the US National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and with approval from the Animal Care and Use Committee of The Rockefeller University.
2.2. Diet‐induced obesity model
AD mice and their WT littermates were divided into four groups: AD CON, AD HFD, WT CON, and WT HFD. HFD contained 60% kcal from fat, 20% kcal from carbohydrates, and 20% kcal from protein (D12492, Research Diets Inc.), and CON contained 10% kcal from fat, 70% kcal from carbohydrates, and 20% kcal from protein (D12450J, Research Diets Inc.). HFD contained 0.03% cholesterol, while CON contained 0.005% cholesterol. Mice were weighed weekly, and body composition was assessed by EchoMRI‐100H (EchoMRI, LLC). Mice remained on their respective diets during behavioral testing. At the conclusion of diet administration, mice were sacrificed and tissue was collected for analysis.
2.3. Behavioral analysis
For a detailed description, refer to Materials and Methods in supporting information.
RESEARCH IN CONTEXT.
Systematic review: Relevant literature was reviewed using PubMed, which identified a gap in understanding the effect of varied timing onset of high‐fat diet (HFD) consumption on amyloid beta (Aβ) pathology and cognitive function in mouse models of Alzheimer's disease (AD).
Interpretation: Our findings indicate that the timing of HFD consumption plays an important role in the progression of Aβ pathology and cognitive dysfunction in ADmodel mice. Our data show that consumption of a HFD at early ages may slow AD progression, while consumption of HFD after AD pathology has already developed may be detrimental.
Future directions: Future studies will identify specific components of the HFD that protect the brain against Aβ accumulation and blood‐brain barrier permeability. Dietary uptake and measures of metabolic activity will also be analyzed. This research may lead to nutritional approaches to delay or prevent the onset of AD years before patients develop cognitive deficits.
2.4. Immunofluorescence and immunohistochemistry
For a detailed description, refer to Materials and Methods in supporting information.
2.5. Western blot
For a detailed description, refer to Materials and Methods in supporting information.
2.6. Data analysis
Statistical analysis was conducted using GraphPad Prism software. Data are presented as mean ± standard error of the mean. Immunofluorescence results were analyzed using unpaired t‐test, while all other experimental results were analyzed using two‐way analysis of variance. Post hoc analyses were conducted using Sidak's multiple comparison test. Significance threshold was set to P ≤ 0.05.
3. RESULTS
3.1. HFD induced weight gain and affected body composition in 6‐month‐old mice
To investigate the effect of increased dietary fat intake on AD‐related pathology in mice, we placed 4‐ to 5‐week‐old mice on a diet of 60% kcal from fat (HFD) or an isocaloric control diet with 10% kcal from fat (CON) for 20 weeks until the mice reached 6 months of age (Figure 1A). HFD induced significant weight gain in both WT and AD mice (Figure 1B, C). HFD also affected body composition and induced a significant increase in percent body fat mass and a decrease in percent lean mass in HFD‐fed WT and AD mice (Figure 1D, E). HFD impaired the ability to clear circulating glucose after a glucose tolerance test (GTT) in WT mice (Figure S1A, B in supporting information). HFD‐fed AD mice showed an increase in blood glucose after the GTT compared to CON‐fed AD mice (Figure S1A, B). Additionally, HFD induced hyperglycemia after a period of fasting in both WT and AD mice (Figure S1C). Fasting also induced an increase in blood ketones in HFD‐fed WT but not AD mice (Figure S1D). However, after refeeding, both WT HFD and AD HFD mice showed an increase in blood ketones compared to CON‐fed mice (Figure S1E). Plasma levels of cholesterol were significantly increased in HFD‐fed WT and AD mice (Figure S1F). Although HFD induced a small increase in plasma high‐density lipoprotein (HDL) levels, the difference between HFD‐fed and CON‐fed mice was not significant (Figure S1G). HFD did not affect plasma HDL levels in AD mice (Figure S1G). Neither AD genotype nor HFD affected plasma levels of low‐density lipoprotein (Figure S1H).
FIGURE 1.
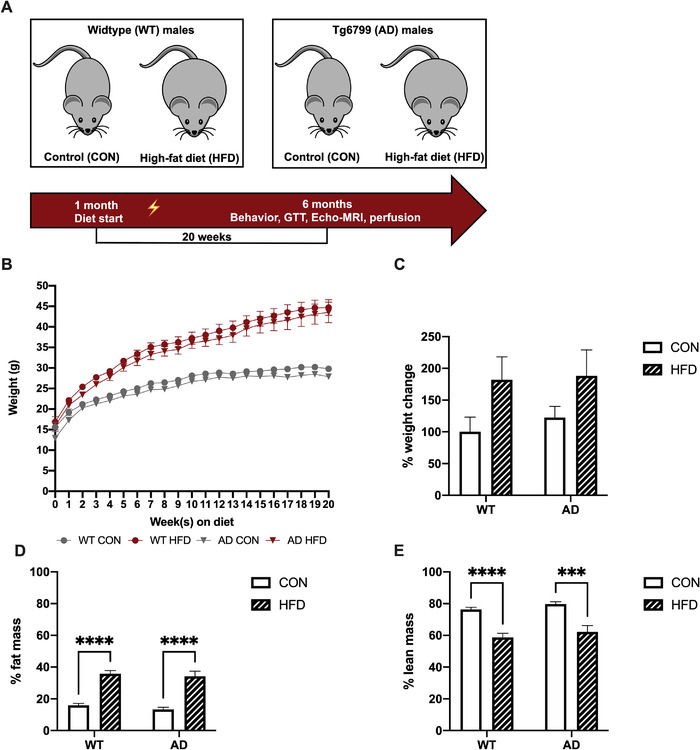
High‐fat diet (HFD) caused significant weight gain and affected body composition similarly in wild‐type (WT) and Alzheimer's disease (AD) mice. A, Schematic shows feeding timeline in WT and AD mice. The lightning icon indicates the onset of AD‐related pathology. B, Male mice (4–5 weeks old) fed a HFD versus control diet (CON) had greater weight gain. C, HFD induced a significant increase in % weight change (P < 0.05), but post hoc analysis did not reveal statistically significant differences between groups. D, HFD increased % fat mass in WT and AD mice. E, HFD decreased % lean mass in WT and AD mice. n = 9–11 mice per group. Results are from one representative experiment. Statistical analysis performed by two‐way analysis of variance. ****P < 0.0001
3.2. HFD consumption starting at 1 month of age reduced Aβ pathology in 6‐month‐old AD mice
HFD significantly reduced percent area covered by Aβ plaques throughout the brains of 6‐month‐old AD mice compared to AD CON mice (Figure 2A, B; Figure S2A in supporting information). Aβ plaque coverage was also specifically quantified in the RSC (boxed area, Figure 2C) and hippocampus at Bregma –2.92 mm of all AD CON and AD HFD mice (Figure 2C). HFD significantly reduced percent area covered by Aβ plaques in the RSC and hippocampus of 6‐month‐old AD mice compared to AD CON mice (Figure 2D, E). WT mice showed no Aβ staining (not shown). Additionally, mouse cortical extracts were analyzed by western blot for expression levels of Aβ (Figure 2F; 6E10), which showed that AD HFD mice had reduced cortical levels of Aβ compared to AD CON (Figure 2G). Furthermore, to investigate other changes in AD‐related pathology induced by dietary fat consumption, mouse cortical sections were examined for the presence of innate immune cells. Compared to AD CON, AD HFD mice exhibited a decrease in CD11b‐positive cells in the RSC (Figure S2B, C) at 6 months of age, consistent with the reduction in Aβ plaques (Figure 2C, D).
FIGURE 2.
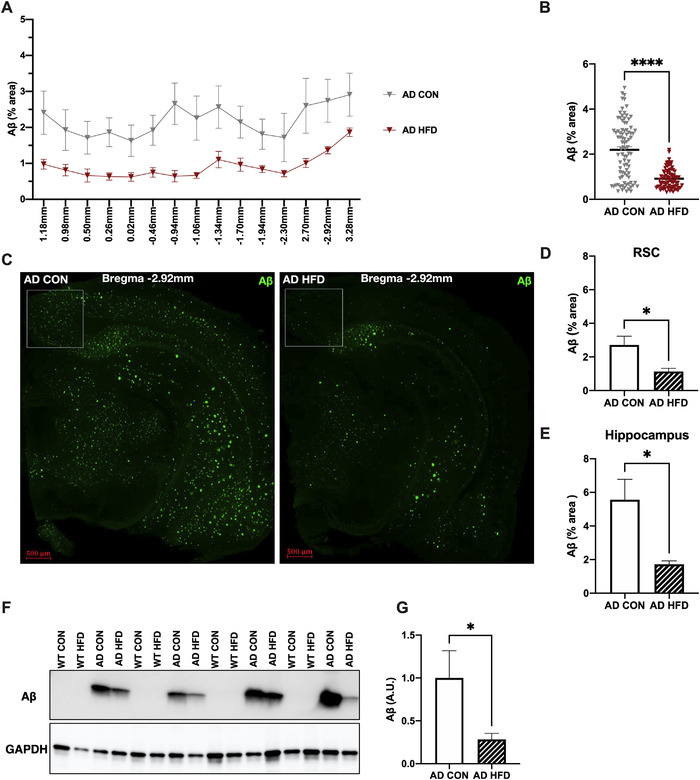
High‐fat diet (HFD) consumption starting at 1 month reduced amyloid beta (Aβ) brain pathology in 6‐month‐old Alzheimer's disease (AD) mice. A, HFD reduced Aβ deposition throughout the brains of AD mice. % area covered in Aβ staining is indicated at each Bregma location. Results are from one representative experiment. B, HFD reduced Aβ deposition at multiple Bregma points in the brains of AD mice; each symbol represents a single Bregma point per mouse imaged. C, Representative images of 6E10 staining used to visualize Aβ plaques in AD mice. Staining was quantified in the retrosplenial cortex (RSC), designated by boxed area, and hippocampus. Wild‐type (WT) mice had no detectable Aβ staining (not shown). D, E, HFD reduced Aβ deposition in the RSC (D) and hippocampus (E) of AD mice. Results are from one representative experiment. F, Representative western blots of cortical protein extracts isolated from 6‐month‐old WT and AD mice, showing expression of human Aβ by 6E10 antibody. GAPDH was used for normalization. G, HFD reduced levels of Aβ in cortical protein extracts isolated from AD mice. Results are from one representative experiment. For (A) and (B), n = 5‐8 mice per group. For (C–E), n = 6 mice per group. For (F) and (G), n = 7 per group. Statistical analysis performed by Student's t test. *P < 0.05, ****P < 0.0001. Scale bar = 500 μm
3.3. Early HFD feeding improved cognitive function in 6‐month‐old AD mice
Memory was assessed in 6‐month‐old AD mice by novel object recognition (NOR), which evaluates the ability of rodents to recognize a novel object in a familiar environment. 16 There were no differences between groups on the training day (Figure 3A), indicating no place preference. As expected, AD CON mice spent less time exploring the novel object on the testing day compared to WT mice (Figure 3B). However, AD HFD mice performed significantly better than AD CON and as well as WT groups (Figure 3B). There was no difference in the total distance moved or velocity during training or testing between any groups (Figure S3 in supporting information), indicating that neither AD genotype nor HFD impaired locomotion. Memory was also assessed in a contextual fear conditioning paradigm and measured as percent time spent freezing, a species‐specific response to fear. Interestingly, while HFD led to a non‐significant decrease in freezing behavior in WT mice, AD HFD mice had a significantly increased freezing response compared to AD CON mice, indicating improved memory of the foot shock (Figure 3C).
FIGURE 3.
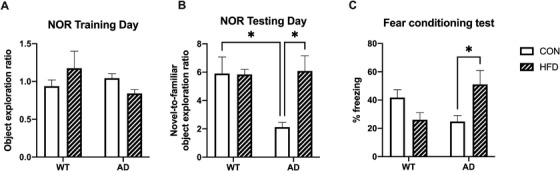
High‐fat diet (HFD) consumption starting at 1 month improved cognition in 6‐month‐old Alzheimer's disease (AD) mice. A, No differences between groups were present on the training day of the novel object recognition (NOR) test, indicating no place preference. B, HFD increased the novel‐to‐familiar object exploration ratio in AD mice on the testing day of the NOR test, indicating improved memory compared to AD control (CON) mice. As expected, the object exploration ratio was lower in AD CON mice compared to wild‐type (WT) groups. Results are from one representative experiment. C, In a contextual fear conditioning test, AD HFD mice spent more time frozen than AD CON mice, indicative of improved memory. Results are from one representative experiment. For (A) and (B), n = 5–6 mice per group. For (C), n = 9–13 mice per group. Statistical analysis performed by two‐way analysis of variance. *P < 0.05
3.4. HFD consumption starting at 1 month of age affected fibrinogen extravasation into the brains of 6‐month‐old AD mice
HFD induces fibrinogen deposition in white adipose tissue (WAT) of mice, 17 so we investigated whether AD genotype affected this result. HFD significantly increased the percentage of fibrinogen‐positive cells in both 6‐month‐old WT and AD mice (Figure 4A, B), indicating that genotype did not affect fibrinogen deposition in WAT. Because AD animal models and human patients show deposition of fibrinogen in their brains, 14 we investigated any change in this pathology after HFD feeding. While AD CON brains showed extensive fibrinogen extravasation from blood vessels into the brain parenchyma, AD HFD brains showed little extravasation (Figure 4C, D). It has also been shown that Aβ and fibrinogen co‐deposit in the brains of AD animal models and patients. 18 Upon investigation, we found minimal Aβ/fibrinogen co‐deposition in the RSC of our experimental AD mice (Figure S4A in supporting information). While there was less Aβ/fibrinogen co‐deposition in the AD HFD mouse brains than AD CON, this difference was not significant (Figure S4B). Our findings showed that while dietary fats consumed at a young age did not alter fibrinogen deposition in WAT of AD mice compared to WT mice, it greatly protected the integrity of the BBB to prevent a blood protein from leaking into the brain tissue, which may aid in preventing Aβ/fibrinogen co‐deposits.
FIGURE 4.
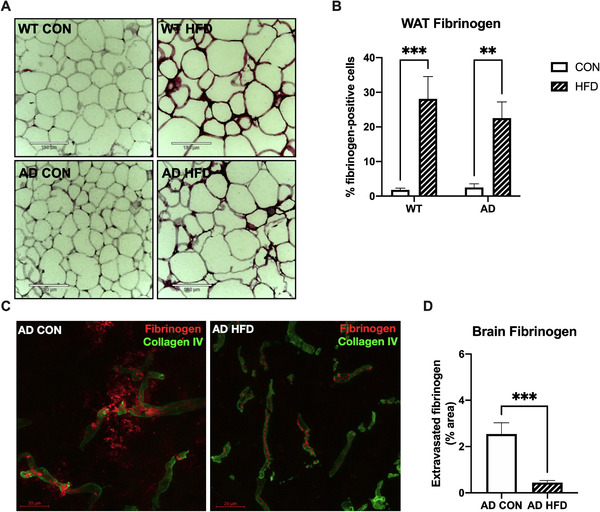
High‐fat diet (HFD) consumption starting at 1 month reduced fibrinogen extravasation into the retrosplenial cortex (RSC) of 6‐month‐old mice. A, Representative images of white adipose tissue (WAT) fibrinogen staining. Scale bar = 130 μm. B, HFD increased percent fibrinogen‐positive cells in WAT of wild‐type (WT) and Alzheimer's disease (AD) mice. Results are combined from two independent experiments. C, Representative images of fibrinogen staining (red) show fibrinogen extravasation from collagen IV‐positive blood vessels (green) into the RSC of AD control (CON), but not AD HFD mice. Scale bar = 20 μm. D, There was significantly less fibrinogen staining outside of blood vessels in RSC of AD HFD mice, indicating strong blood‐brain barrier integrity. For (A) and (B), n = 9–10 mice per group, three sections per animal. For (C) and (D), n = 6–7 mice per group, three sections per animal. Results are combined from two independent experiments. Statistical analysis performed by two‐way analysis of variance (A and B) or Student's t‐test (C and D). **P < 0.01, ***P < 0.001
3.5. HFD consumption starting at 3 months, but not 6 months, reduced Aβ pathology and improved cognitive function in AD mice
In Tg6799 mice, Aβ plaque deposition starts as early as at 2 months of age. By 6 months of age, Tg6799 mice display abundant parenchymal Aβ plaque deposition and memory deficits. 18 We investigated whether the timing of HFD feeding was related to the improvement in AD‐related pathology and cognition in our AD mouse cohorts. Therefore, we delayed the onset of HFD administration until 3 months of age (Cohort I) or 6 months of age (Cohort II), after AD‐related pathology begins to develop. The diet regimen continued until Cohort I mice were 8 months and Cohort II mice were 11 months old (Figure 5A).
FIGURE 5.
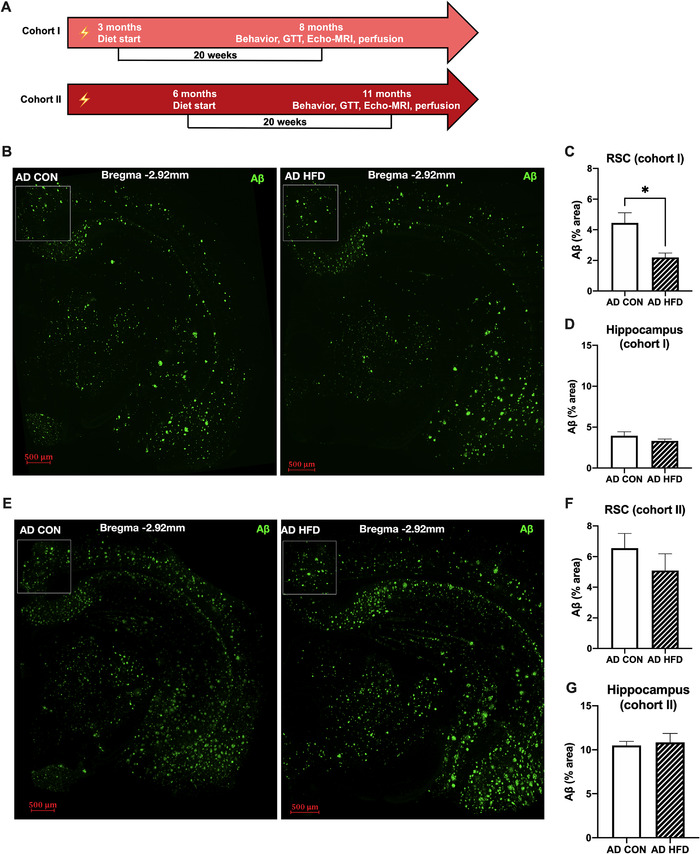
High‐fat diet (HFD) consumption starting at 3 months, but not 6 months, reduced amyloid beta (Aβ) pathology in the retrosplenial cortex (RSC) of Alzheimer's disease (AD) mice. A, Schematics show HFD feeding timeline in cohorts I and II. Lightning icon indicates the onset of AD‐related pathology. B, Representative images of Aβ staining in whole brain slices of 8‐month‐old AD mice. Staining was quantified in the RSC, designated by boxed area, and hippocampus. Wild‐type (WT) mice had no detectable Aβ deposits (not shown). Scale bar = 500 μm. C, D, In cohort I, delaying the onset of HFD to 3 months of age reduced Aβ deposition in the RSC but not the hippocampus of 8‐month‐old AD mice. E, Representative images of Aβ staining in 11‐month‐old mice. Staining was quantified in the RSC and hippocampus. WT mice had no detectable Aβ deposits (not shown). Scale bar = 500 μm. F, G, In cohort II, delayed HFD feeding until 6 months of age did not significantly decrease Aβ plaque deposition in the RSC or hippocampus. For B‐D, n = 5–6 mice per group. For E‐G, n = 8 mice per group. Results are from one representative experiment. Statistical analysis performed by Student's t‐test. *P < 0.05
In Cohort I, HFD consumption starting at 3 months of age significantly reduced Aβ plaque burden across the entire brain (Figure 5B and S5A, B in supporting information). More specifically, there was significantly less Aβ staining in the RSC of AD HFD versus AD CON mice, yet no difference was detected in the hippocampus (Figure 5C, D). Neither genotype nor diet affected object exploration or locomotion on NOR training day (not shown). However, during testing, HFD increased the novel‐to‐familiar object exploration ratio in AD mice compared to AD CON mice (Figure S5C), indicating that HFD consumption starting at 3 months, after the onset of AD‐like pathology, still led to improved cognitive function in 8‐month‐old AD mice. HFD had a similar effect on AD‐related pathology and cognitive function in 6‐month‐old and 8‐month‐old mice, despite the delayed onset of high‐fat feeding (1 month vs. 3 months). Therefore, consuming dietary fats prior to significant AD pathology may be protective and delay disease progression. AD CON mice showed an increase in total distance moved and velocity compared to WT CON mice during NOR testing (Figure S5D, E), indicating that AD genotype caused hyperactivity in 8‐month‐old mice. Interestingly, HFD consumption corrected this phenotype, restoring locomotion to WT levels in Cohort I AD mice (Figure S5D, E).
In Cohort II, in which HFD consumption was delayed until 6 months of age, there was an overall increase in Aβ brain staining in AD HFD mice compared to that of AD CON (Figure S5F, G). When specifically comparing Aβ staining in the RSC of AD mice, there was no longer a protective effect of the HFD (Figure 5E, F). There was no effect of HFD feeding on Aβ deposition in the hippocampus (Figure 5G) or cognitive function by NOR (Figure S5H). These results indicate that delaying HFD feeding until after moderate AD‐related pathology was developed failed to slow AD‐like progression. Neither genotype nor diet affected object exploration or locomotion on NOR training day (not shown). HFD had no effect on locomotion during NOR testing (Figure S5I, J) in 11‐month‐old mice. Therefore, our findings suggest that delaying the onset of HFD feeding until 6 months, when AD pathology is widespread, does not alleviate AD‐related pathology or cognitive dysfunction in mice.
3.6. Delayed onset of HFD consumption until 3 and 6 months of age reduced fibrinogen extravasation into the brains of AD mice
In Cohort I, in which the 20‐week HFD regimen was delayed until mice were 3 months old, HFD significantly increased the percentage of fibrinogen‐positive cells in the WAT of both WT and AD mice (Figure S6A, B in supporting information). In parallel with our findings in 6‐month‐old AD mice, HFD also significantly reduced fibrinogen extravasation from blood vessels into the brain parenchyma of 8‐month‐old AD mice in this cohort (Figure 6A, B). Therefore, despite the delayed onset of feeding, HFD helped improve BBB integrity.
FIGURE 6.
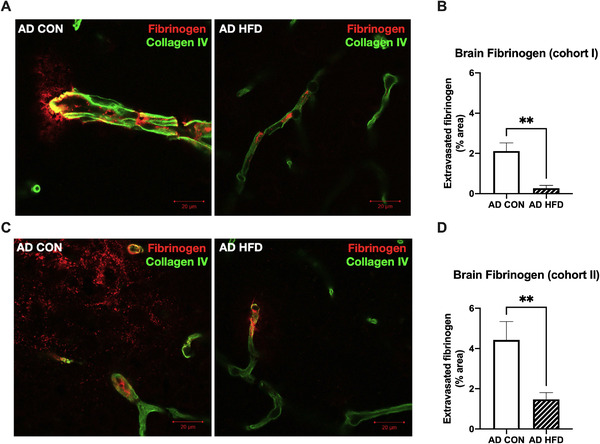
Delayed onset of high‐fat diet (HFD) consumption until 3 and 6 months of age reduced cortical fibrinogen extravasation in 8‐ and 11‐month‐old Alzheimer's disease (AD) mice. A, C, Representative images of fibrinogen staining (red) show fibrinogen extravasation from collagen IV‐positive blood vessels (green) into the retrosplenial cortex (RSC) of (A) 8‐month‐old and (C) 11‐month‐old AD control (CON) mice, but not AD HFD mice. Scale bar = 20 μm. B, D, HFD significantly reduced fibrinogen staining outside of blood vessels in RSC of 8‐ and 11‐month‐old AD mice. n = 8 mice per group, three sections per animal. Results are from one independent experiment. Statistical analysis performed by Student's t‐test. **P < 0.01
In Cohort II, in which the onset of HFD was delayed until 6 months, HFD did not significantly affect the percentage of fibrinogen‐positive cells in the WAT of 11‐month‐old WT and AD mice (Figure S6C, D). Additionally, we found evidence of fibrinogen deposition in the WAT of CON‐fed WT and AD mice (Figure S6C), indicating that fibrinogen deposition in peripheral tissues might accompany aging in the absence of metabolic insults. Interestingly, HFD still significantly reduced fibrinogen extravasation into the brain parenchyma of 11‐month‐old AD mice (Figure 6C, D), indicating that delayed‐onset HFD improved BBB integrity even after AD‐like pathology had begun but was unable to reverse or slow Aβ deposition.
HFD did not affect Aβ/fibrinogen co‐deposition in the RSC of 8‐month‐old AD mice (Cohort I; Figure S6E, F) or 11‐month‐old AD mice (Cohort II; Figure S6G, H), although a small non‐significant decrease in Aβ/fibrinogen co‐localization was observed in AD HFD mice in both cohorts. Overall, these results suggest that HFD improved BBB integrity and reduced fibrinogen extravasation into the brain parenchyma, although it had little effect on Aβ/fibrinogen co‐deposits, which have been shown to exacerbate AD‐related pathology.
4. DISCUSSION
The goal of our study was to determine the effect of varied HFD feeding onset on AD‐related pathology and cognitive function in Tg6799 AD mice. In this rapid‐progression animal model of AD, extracellular plaque deposition begins at 2 months and is readily detectable throughout the cortex and hippocampus by 6 months. 15 Furthermore, Tg6799 mice display evidence of spatial memory deficits as early as 4 months of age. 15
We initially administered HFD to Tg6799 AD mice before the onset of extracellular Aβ deposition or cognitive deficits. Contrary to some previous reports, HFD consumption significantly reduced the percent area covered by Aβ plaques in the RSC and hippocampus as well as throughout the whole brain of 6‐month‐old AD mice compared to their CON‐fed AD littermates. HFD also reduced the number of CD11b‐positive microglial cells surrounding extracellular Aβ deposits. Importantly, HFD improved object recognition memory and conditioned fear memory in AD mice, which indicates an improvement in cognitive function. Surprisingly, when we delayed the onset of HFD until 3 months of age, after Aβ starts accumulating, HFD similarly reduced Aβ plaque deposition in the RSC and improved cognitive function in 8‐month‐old AD mice. However, HFD did not improve Aβ pathology or cognitive function in 11‐month‐old AD mice when HFD consumption started at 6 months of age. In fact, Aβ pathology was exacerbated across the entire brain when HFD feeding occurred after AD pathology had already begun. Thus, the timing of HFD consumption is critical for improving AD‐related pathology and cognition, as HFD does little to rescue AD‐related phenotypes after Aβ pathology has already spread throughout the brain.
In this study, consumption of HFD at an early age led to region‐specific as well as widespread reduction of Aβ deposition throughout the brains of AD mice. One of the specific brain area affected by dietary fat consumption was the RSC, which has important reciprocal connections with several brains regions, including the hippocampus, 19 visuospatial cortex, 20 prefrontal cortex, 21 and posterior secondary motor cortex. 22 The connectivity pattern of the RSC is consistent with its role in cognitive function. 23 , 24 , 25 , 26 The RSC is thought to play a critical role in AD pathophysiology as it is one of the first regions to undergo pathological changes in patients. 27 , 28 Pathological changes in the RSC are also present in mouse models of AD. Tg2576 AD mice show aberrant changes in the markers of cellular activity in RSC before the formation of overt Aβ pathology. 29 In Tg6799 AD mice, increased accumulation of Aβ in the RSC is accompanied by an impairment in object recognition memory as early as at 4 months of age. 30 Taken together, these results indicate that pathological changes in the RSC may reflect some of the earliest AD‐related processes and impact cognitive function in mouse models of AD. In our study, HFD consumption reduced Aβ deposition in the RSC and improved object recognition memory in 6‐ and 8‐month‐old AD mice. However, delaying the onset of HFD consumption until after mice display overt Aβ accumulation and cognitive deficits did not improve object recognition memory. Furthermore, HFD increased freezing behavior in the contextual fear conditioning test in 6‐month‐old AD mice, indicating improvement in fear learning and memory. While WT HFD mice exhibited a decreased freezing response compared to WT CON mice, this difference was not statistically significant. HFD consumption did not affect WT mouse learning and memory behaviors in NOR nor were there any changes observed in brain pathology of WT mice. Therefore, our results indicate that early consumption of HFD led to a reduction in Aβ accumulation in the RSC and hippocampus, which contributed to an improvement in cognitive performance in AD mice.
In addition to hallmark AD pathologies, AD is also associated with vascular abnormalities, 31 , 32 such as parenchymal deposition of fibrinogen, a critical component of the blood coagulation cascade. 14 , 33 , 34 , 35 Fibrinogen is a glycoprotein found in large quantities in blood. 36 In response to injury, fibrinogen is converted to fibrin, the major component of blood clots. Fibrinogen is normally excluded from the brain by the BBB. However, AD patients and mouse models show a loss of BBB integrity, 37 , 38 , 39 , 40 , 41 which promotes fibrinogen extravasation into the brain. We have previously shown that fibrinogen extravasation correlates with the degree of Aβ pathology in AD mice and patients. 33 , 42 , 43 , 44 Due to its potent pro‐inflammatory properties and direct interaction with Aβ, 18 , 45 , 46 , 47 extravascular fibrinogen may contribute to and/or promote neuroinflammatory responses and neuronal dysfunction. 34 , 48 In fact, fibrin deposition is present in areas of the brain with prominent synaptic degeneration. 33 In this study, we found extravasated fibrinogen in the RSC of 6‐month‐old AD mice, while their WT littermates showed none. However, chronic HFD consumption reduced fibrinogen extravasation in AD mice. Surprisingly, 20‐week HFD consumption also reduced fibrinogen extravasation in the RSC of 8‐ and 11‐month‐old AD mice (Cohorts I and II), but HFD only reduced Aβ deposition in the RSC of 8‐, but not 11‐, month‐old mice. This result indicates that a reduction in extravascular fibrinogen does not directly reflect a reduction in Aβ. In addition, we found minimal Aβ/fibrinogen co‐deposits in the RSC of AD HFD mice at 6, 8, and 11 months of age. This result also suggests that Aβ accumulation occurs prior to BBB disruption and fibrinogen extravasation into the brain parenchyma. Therefore, the Aβ/fibrinogen interaction resulting in their co‐deposition in the brain might represent a later stage in the development of AD‐related pathology. Additionally, HFD failed to induce an improvement in object recognition memory in 11‐month‐old AD mice. This result suggests that chronic HFD consumption is not sufficient to preserve cognitive function in older AD mice if HFD feeding starts after significant Aβ deposition, despite potential neuroprotective effects that accompany reduced BBB permeability to fibrinogen. 33 Future studies will determine whether HFD‐induced reduction in fibrinogen extravasation protects the brains of AD mice from synaptic degeneration with HFD feeding starting at 1, 3, or 6 months of age.
Our results are consistent with previous studies that have reported HFD‐induced improvement in BBB function in AD mice. For example, in rodent MRI studies HFD significantly reduces BBB permeability to the contrast agent and decreases the volume of lateral ventricles in Tg2576 AD mice, indicating a reduction in brain atrophy. 12 This reduction in BBB leakage is accompanied by a significant improvement in learning in HFD‐fed AD mice. However, the protective effect of HFD on BBB function might depend on HFD composition. For example, Theriault et al. reported that a sucrose‐rich “Western diet” containing 42% kcal from fat and 42.7% kcal from carbohydrates does not affect BBB permeability and exacerbates AD‐related cognitive decline in APPswe/PS1 mice. 49 In addition, another Western diet high in saturated fat and sucrose increases Aβ plaque load and microglial density in APP/PS1 mice without affecting short‐term memory. 50 These results indicate that a high‐fat, low‐carbohydrate diet—like the one used in our study—may be protective against AD‐induced BBB damage, while high‐fat diets containing high carbohydrates or high sucrose have deleterious effects on AD pathophysiology.
In sum, we show that chronic HFD consumption reduced AD‐related pathology, including Aβ accumulation, fibrinogen extravasation, and cognitive dysfunction in 6‐ and 8‐month‐old AD mice when HFD intake started at or before 3 months of age (before or early‐stage disease). Delaying the onset of HFD until 6 months (moderate disease pathology) reduced this protective effect, as HFD did not reduce Aβ plaque deposition or aid in cognitive performance in 11‐month‐old AD mice. However, the delayed onset of HFD feeding still reduced the extravasation of fibrinogen into the brain parenchyma, thus raising the possibility that a diet high in fat improved BBB function even after AD mice developed overt Aβ pathology and cognitive deficits. HFD‐fed 6‐ and 8‐month‐old AD mice also showed a decrease in BBB permeability, suggesting that improved cognition in these animals might be due to the decrease in synaptic damage induced by the alleviated Aβ load and reduced fibrinogen extravasation into the brain parenchyma.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
Supporting information
Supplementary information
ACKNOWLEDGMENTS
We thank Dr. Sidney Strickland and his laboratory for fruitful discussion of experimental results and editorial suggestions to the manuscript; Drs. Jeffrey Friedman and Kristina Hedbacker for their assistance with EchoMRI experiments; Dr. Paul Cohen for helpful discussion of experimental strategy; Caroline Jiang and Neha Singh for their guidance on statistical analysis; and Dr. Dorit Farfara for the invaluable help with tissue collection. This work was supported by NIH NS106668, Rudin Family Foundation, The Rockefeller University Sackler Center for Biomedicine & Nutrition Research, The Rockefeller University Women & Science Initiative, Studienstiftung des Deutschen Volkes, and Mr. John A. Herrmann Jr.
Amelianchik A, Merkel J, Palanisamy P, et al. The protective effect of early dietary fat consumption on Alzheimer's disease–related pathology and cognitive function in mice. Alzheimer's Dement. 2021;7:e12173. 10.1002/trc2.12173
Anna Amelianchik and Jonathan Merkel contributed equally to this study.
There are no human subjects involved in this work, so consent was not necessary.
REFERENCES
- 1. Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61:661‐666. [DOI] [PubMed] [Google Scholar]
- 2. Gustafson D, Rothenberg E, Blennow K, Steen B, Skoog I. An 18‐year follow‐up of overweight and risk of Alzheimer disease. Arch Intern Med. 2003;163:1524‐1528. [DOI] [PubMed] [Google Scholar]
- 3. Yaffe K, Kanaya A, Lindquist K, et al. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA. 2004;292:2237‐2242. [DOI] [PubMed] [Google Scholar]
- 4. Beydoun MA, Lhotsky A, Wang Y, et al. Association of adiposity status and changes in early to mid‐adulthood with incidence of Alzheimer's disease. Am J Epidemiol. 2008;168:1179‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xu WL, Atti AR, Gatz M, Pedersen NL, Johansson B, Fratiglioni L. Midlife overweight and obesity increase late‐life dementia risk: a population‐based twin study. Neurology. 2011;76:1568‐1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pedditzi E, Peters R, Beckett N. The risk of overweight/obesity in mid‐life and late life for the development of dementia: a systematic review and meta‐analysis of longitudinal studies. Age Ageing. 2016;45:14‐21. [DOI] [PubMed] [Google Scholar]
- 7. Qizilbash N, Gregson J, Johnson ME, et al. BMI and risk of dementia in two million people over two decades: a retrospective cohort study. Lancet Diabetes Endocrinol. 2015;3:431‐436. [DOI] [PubMed] [Google Scholar]
- 8. Herculano B, Tamura M, Ohba A, Shimatani M, Kutsuna N, Hisatsune T. beta‐alanyl‐L‐histidine rescues cognitive deficits caused by feeding a high fat diet in a transgenic mouse model of Alzheimer's disease. J Alzheimers Dis. 2013;33:983‐997. [DOI] [PubMed] [Google Scholar]
- 9. Ho L, Qin W, Pompl PN, et al. Diet‐induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J. 2004;18:902‐904. [DOI] [PubMed] [Google Scholar]
- 10. Knight EM, Martins IV, Gumusgoz S, Allan SM, Lawrence CB. High‐fat diet‐induced memory impairment in triple‐transgenic Alzheimer's disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol Aging. 2014;35:1821‐1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sah SK, Lee C, Jang JH, Park GH. Effect of high‐fat diet on cognitive impairment in triple‐transgenic mice model of Alzheimer's disease. Biochem Biophys Res Commun. 2017;493:731‐736. [DOI] [PubMed] [Google Scholar]
- 12. Elhaik Goldman S, Goez D, Last D, et al. High‐fat diet protects the blood‐brain barrier in an Alzheimer's disease mouse model. Aging Cell. 2018;17:e12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang L, Fernandez‐Kim SO, Beckett TL, et al. The db mutation improves memory in younger mice in a model of Alzheimer's disease. Biochim Biophys Acta Mol Basis Dis. 2019;1865:2157‐2167. [DOI] [PubMed] [Google Scholar]
- 14. Cortes‐Canteli M, Paul J, Norris EH, et al. Fibrinogen and beta‐amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer's disease. Neuron. 2010;66:695‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oakley H, Cole SL, Logan S, et al. Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129‐10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Antunes M, Biala G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn Process. 2012;13:93‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kopec AK, Abrahams SR, Thornton S, et al. Thrombin promotes diet‐induced obesity through fibrin‐driven inflammation. J Clin Invest. 2017;127:3152‐3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahn HJ, Zamolodchikov D, Cortes‐Canteli M, Norris EH, Glickman JF, Strickland S. Alzheimer's disease peptide beta‐amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci U S A. 2010;107:21812‐21817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kononenko NL, Witter MP. Presubiculum layer III conveys retrosplenial input to the medial entorhinal cortex. Hippocampus. 2012;22:881‐895. [DOI] [PubMed] [Google Scholar]
- 20. Passarelli L, Rosa MGP, Bakola S, et al. Uniformity and Diversity of cortical projections to precuneate areas in the Macaque monkey: what defines area PGm?. Cereb Cortex. 2018;28:1700‐1717. [DOI] [PubMed] [Google Scholar]
- 21. Aggleton JP. Understanding retrosplenial amnesia: insights from animal studies. Neuropsychologia. 2010;48:2328‐2338. [DOI] [PubMed] [Google Scholar]
- 22. Yamawaki N, Radulovic J, Shepherd GM. A corticocortical circuit directly links retrosplenial cortex to M2 in the mouse. J Neurosci. 2016;36:9365‐9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mitchell AS, Czajkowski R, Zhang N, Jeffery K, Nelson AJD. Retrosplenial cortex and its role in spatial cognition. Brain Neurosci Adv. 2018;2:2398212818757098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Powell AL, Vann SD, Olarte‐Sanchez CM, et al. The retrosplenial cortex and object recency memory in the rat. Eur J Neurosci. 2017;45:1451‐1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robinson S, Todd TP, Pasternak AR, et al. Chemogenetic silencing of neurons in retrosplenial cortex disrupts sensory preconditioning. J Neurosci. 2014;34:10982‐10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Landeta AB, Pereyra M, Medina JH, Katche C. Anterior retrosplenial cortex is required for long‐term object recognition memory. Sci Rep. 2020;10:4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nestor PJ, Fryer TD, Ikeda M, Hodges JR. Retrosplenial cortex (BA 29/30) hypometabolism in mild cognitive impairment (prodromal Alzheimer's disease). Eur J Neurosci. 2003;18:2663‐2667. [DOI] [PubMed] [Google Scholar]
- 28. Pengas G, Hodges JR, Watson P, Nestor PJ. Focal posterior cingulate atrophy in incipient Alzheimer's disease. Neurobiol Aging. 2010;31:25‐33. [DOI] [PubMed] [Google Scholar]
- 29. Poirier GL, Amin E, Good MA, Aggleton JP. Early‐onset dysfunction of retrosplenial cortex precedes overt amyloid plaque formation in Tg2576 mice. Neuroscience. 2011;174:71‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim DH, Kim HA, Han YS, Jeon WK, Han JS. Recognition memory impairments and amyloid‐beta deposition of the retrosplenial cortex at the early stage of 5XFAD mice. Physiol Behav. 2020;222:112891. [DOI] [PubMed] [Google Scholar]
- 31. Govindpani K, McNamara LG, Smith NR, et al. Vascular dysfunction in Alzheimer's disease: a prelude to the pathological process or a consequence of it?. J Clin Med. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Strickland S. Blood will out: vascular contributions to Alzheimer's disease. J Clin Invest. 2018;128:556‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cortes‐Canteli M, Mattei L, Richards AT, Norris EH, Strickland S. Fibrin deposited in the Alzheimer's disease brain promotes neuronal degeneration. Neurobiol Aging. 2015;36:608‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cortes‐Canteli M, Zamolodchikov D, Ahn HJ, Strickland S, Norris EH. Fibrinogen and altered hemostasis in Alzheimer's disease. J Alzheimers Dis. 2012;32:599‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hultman K, Cortes‐Canteli M, Bounoutas A, Richards AT, Strickland S, Norris EH. Plasmin deficiency leads to fibrin accumulation and a compromised inflammatory response in the mouse brain. J Thromb Haemost. 2014;12:701‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weisel JW. Fibrinogen and fibrin. Adv Protein Chem. 2005;70:247‐299. [DOI] [PubMed] [Google Scholar]
- 37. Kelly P, McClean PL, Ackermann M, Konerding MA, Holscher C, Mitchell CA. Restoration of cerebral and systemic microvascular architecture in APP/PS1 transgenic mice following treatment with Liraglutide. Microcirculation. 2015;22:133‐145. [DOI] [PubMed] [Google Scholar]
- 38. Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer's disease. J Exp Med. 2007;204:1999‐2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sagare AP, Bell RD, Zhao Z, Ma Q, et al. Pericyte loss influences Alzheimer‐like neurodegeneration in mice. Nat Commun. 2013;4:2932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40. Sweeney MD, Sagare AP, Zlokovic BV. Blood‐brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Viggars AP, Wharton SB, Simpson JE, et al. Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer‐type pathology: a study in the MRC‐CFAS population neuropathology cohort. Neurosci Lett. 2011;505:25‐30. [DOI] [PubMed] [Google Scholar]
- 42. Cullen KM, Kocsi Z, Stone J. Pericapillary haem‐rich deposits: evidence for microhaemorrhages in aging human cerebral cortex. J Cereb Blood Flow Metab. 2005;25:1656‐1667. [DOI] [PubMed] [Google Scholar]
- 43. Lipinski B, Sajdel‐Sulkowska EM. New insight into Alzheimer disease: demonstration of fibrin(ogen)‐serum albumin insoluble deposits in brain tissue. Alzheimer Dis Assoc Disord. 2006;20:323‐326. [DOI] [PubMed] [Google Scholar]
- 44. Ryu JK, McLarnon JG. A leaky blood‐brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer's disease brain. J Cell Mol Med. 2009;13:2911‐2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol. 2012;34:43‐62. [DOI] [PubMed] [Google Scholar]
- 46. Soria J, Mirshahi S, Mirshahi SQ, et al. Fibrinogen alphaC domain: its importance in physiopathology. Res Pract Thromb Haemost. 2019;3:173‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zamolodchikov D, Berk‐Rauch HE, Oren DA, et al. Biochemical and structural analysis of the interaction between beta‐amyloid and fibrinogen. Blood. 2016;128:1144‐1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ahn HJ, Chen ZL, Zamolodchikov D, Norris EH, Strickland S. Interactions of beta‐amyloid peptide with fibrinogen and coagulation factor XII may contribute to Alzheimer's disease. Curr Opin Hematol. 2017;24:427‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Theriault P, ElAli A, Rivest S. High fat diet exacerbates Alzheimer's disease‐related pathology in APPswe/PS1 mice. Oncotarget. 2016;7:67808‐67827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bracko O, Vinarcsik LK, Cruz Hernandez JC, et al. High fat diet worsens Alzheimer's disease‐related behavioral abnormalities and neuropathology in APP/PS1 mice, but not by synergistically decreasing cerebral blood flow. Sci Rep. 2020;10:9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information