

Abstract
We present a new heteroditopic ligand (3pyCCMoid) that contains the typical skeleton of a curcuminoid (CCMoid) decorated with two 3-pyridyl groups. The coordination of 3pyCCMoid with ZnII centres results in a set of novel coordination polymers (CPs) that display different architectures and dimensionalities (from 1D to 3D). Our work analyses how synthetic methods and slight changes in the reaction conditions affect the formation of the final materials. Great efforts have been devoted toward understanding the coordination entities that provide high dimensional systems, with emphasis on the characterization of 2D materials, including analyses of different types of substrates, stability and exfoliation in water. Here, we foresee the great use of CCMoids in the field of CPs and emphasize 3pyCCMoid as a new-born linker.
We forecast the relevance of synthetic curcuminoids (CCMoids) in the creation of coordination polymers (CPs) by showing a set of three novel CPs synthesized with the exclusive use of a CCMoid and ZnII sources under mild conditions.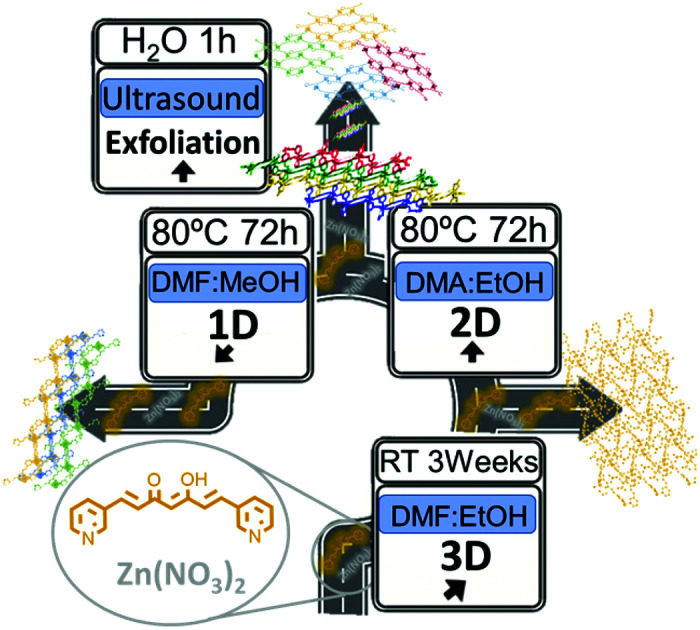
Introduction
Coordination polymers (CPs) comprise a large variety of crystalline metal–organic architectures extending in one, two or three dimensions.1 The potential applications of these materials, as gas storage modules, conducting films, drug delivery vehicles, sensing agents and catalysts, among others,2 have driven intense research concerning the factors that trigger the assembly between inorganic/organic units, ruling topology and final properties.1–3 In addition, the creation of functional materials that combine the diversity of CPs and tailored dimensions is highly desirable, where the tuning of dimensionality and size aims to overcome the present technological challenges exploiting the optical, electronic and/or biological properties of such nanomaterials.4 Toward such goals, special attention has been paid to the design of homoditopic linkers (molecules with identical coordinative sites) with a variety of frameworks (from flexible to rigid)5 and binding sites (e.g.: linkers with O- or N-donor groups).6 Much less information has been gathered about heteroditopic ligands (linkers with more than one type of coordinative ending),7 partly due to the difficulties in achieving crystalline materials and predicting final assemblies. Paradoxically, the latter is also one of the strengths of such multifunctional ligands, capable of generating new topological CPs by binding with one type of metal centre or the combination of a few.7,8
A number of naturally occurring molecules are potential heteroditopic linkers, with great projection in the field, adding beneficial features to CPs, such as low toxicity and biocompatibility.9 In this sense, curcumin (CCM) fulfills all the criteria, as shown in recent publications by Zhu et al.10 and some of us.11 These studies display the two only crystalline ZnMOF/ZnCP structures based exclusively on the ligand CCM.
Encouraged by the available data on CCM derivatives (the so-called curcuminoids, CCMoids), some of us have recently described a new family of CPs containing a natural CCMoid, bisdemethoxycurcumin (BDCM).12 Now, we describe a synthetic linker that displays CCMoid characteristics (diarylheptanoid chain and central keto–enol moiety), with pyridine groups at the ends of the molecule (3pyCCMoid, Fig. 1).
Fig. 1. (Left) General molecular structure of 3pyCCMoid. (Right) Picture of 3pyCCMoid from the crystallographic data. H atoms are not labelled for clarity. Colour legend: C, grey; O, red; N, blue; H, light grey.

The choice of the latter confers 3pyCCMoid with: (i) new coordination sites, in comparison with the phenol/methoxide groups of CCM and natural CCMoids10–12 (ii) additional chemical properties, such as weak basicity, variations in the solubility together with the ability to generate supramolecular interactions6,13 and (iii) possible biological contributions, given that pyridines are chemically stable in vivo, present in natural products (vitamins and coenzymes)14 and used as active components in drugs and pesticides.14,15 Connected with these factors, 3pyCCMoid is partially soluble in H2O (Fig. S1†) and has not shown cytotoxic activity against SH-SY5Y cells16 and in in vivo tests with mice,17 properties that encourage its possible use as a biocompatible material. Nevertheless, further applications of this CCMoid exceed the scope of this paper and they will be explored in further work.
Here, we report three novel CPs containing 3pyCCMoid coordinated to ZnII ions. This manuscript explores the use of solvent mixtures in combination with layering and soft solvothermal methods for the creation of 1D, 2D and 3D species (systems 1, 2 and 4, respectively), including crystallographic data for the three systems. We concentrate on the synthesis and physico-chemical characterization of the new species, emphasizing the heteroditopic nature of 3pyCCMoid and its versatile coordination (two/three coordinated sites). These CPs are achieved under mild conditions using straightforward methodologies and purification processes. Furthermore, these systems assert synthetic CCMoids as optimal molecular platforms, being the first examples of the foreseen broad production of CPs based on this family of molecules. In particular, the exfoliation of 2D layered metal–organic hybrid materials,18 such as 2, has attracted attention in recent years due to the different properties shown by a few layers of these materials (nanosheets) when compared to the bulk product. In this work, we present an effective methodology for the exfoliation of 2 in H2O as well as the characterization of the nanosheets.
Results and discussion
Synthesis
3pyCCMoid was prepared in high yield using the classic method described by Pabon.19 Specifically, the intermediate boron complex was isolated and water was used to achieve the free ligand, avoiding this way diluted acidic solutions in the last step that protonate the pyridine moieties and make the extraction of the final system difficult (Fig. 2). Recrystallization with hot MeCN provided the crystals of 3pyCCMoid.
Fig. 2. Scheme of the synthetic path to achieve 3pyCCMoid.

We tested the capability of 3pyCCMoid as a heteroditopic ligand to produce new CPs through the reaction with Zn(NO3)2·6H2O; the latter is a biocompatible metallic source20 that presents a versatile coordination and is well known in the fields of bio-MOFs and CPs.9–12,21Table 1 shows the interplay of solvents as well as the two methodologies studied to afford 1–2 and 4, respectively. A mixture of alcohols (MeOH/EtOH) with high boiling point solvents (DMF/DMA), together with subtle differences in their combinations, provides partial or full coordination of 3pyCCMoid showing penta- or hexacoordination modes for the ZnII centres, as seen in the structures. As a result, the attained crystalline materials display a variety of structural dimensionalities, from 1D to 3D, pointing to the plurality that the design of CCMoids can add in the field of CPs.
Summary of methodologies/conditions used to achieve 1–4 CPs. System 3 was crystalline but not suitable for single-crystal X-ray diffraction; it was found in a mixture together with 1 and has been added to the table for comparative purposes.
| CP | Ligand | Metal salt | Method | Solvent | Conditions |
|---|---|---|---|---|---|
| 1 | 3pyCCMoid | Zn(NO3)2·6H2O | Solvothermal | DMF : MeOH (1 : 4) | 80 °C |
| 0.018 mmol | 0.054 mmol | 72 h | |||
| 2 | 3pyCCMoid | Zn(NO3)2·6H2O | Solvothermal | DMA : EtOH (1 : 4) | 80 °C |
| 0.018 mmol | 0.054 mmol | 72 h | |||
| 1 & 3 | 3pyCCMoid | Zn(NO3)2·6H2O | Solvothermal | DMF : EtOH (1 : 4) | 80 °C |
| 0.018 mmol | 0.054 mmol | 72 h | |||
| 4 | 3pyCCMoid | Zn(NO3)2·6H2O | Layering | DMF : EtOH (3 : 7) | RT |
| 0.108 mmol | 0.215 mmol | 2–3 weeks |
Structural descriptions
The single crystal X-ray crystallographic resolution of 1, 4 and 3pyCCMoid required the use of a synchrotron source, while for 2 routine equipment could be used. Table S1† shows the general crystalline structural data for all the species and Fig. S2–S4† show the enlarged views, for clarity, of the images shown in this section. The selected bond lengths and angles for each system are listed in Tables S2, S3 and S4,† respectively. Compound 3 did not provide suitable crystals for crystallographic resolution, which is the reason why the analysis of its structure is still elusive. This compound was achieved together with 1 and provides some insights into the effects of solvents and the synthetic methodology. Fig. S5† shows the X-ray diffraction (XRD) patterns generated from the three single crystal datasets compared with those measured from the powdered samples, except for 4, with reproducible synthesis but low yield, not enough to record the experimental XRD data, therefore only the simulated ones from single X-ray diffraction are shown.
3pyCCMoid
This CCMoid (Fig. 1) crystallizes in the monoclinic space group P21/c. The molecule presents a diarylheptanoid skeleton that contains alternating conjugated bonds on both sides of the β-diketone moiety (C C and C–C distances range between 1.336–1.385 Å and 1.454–1.469 Å, respectively). Distances of 1.315 Å and 1.281 Å were found for the C–OH and C O groups, in this order, and the intramolecular hydrogen bond displayed an angle of 158.68°, similar to alike systems in the literature.10,12,22 The molecule is almost flat, and only the pyridine arms show slight deviations from the plane that contains the linear framework (6.78°).23 Such units are placed at the ends, having the N atoms at the meta-positions and facing opposite directions with respect to each other. A rich supramolecular environment leads the packing arrangement (not shown). The pseudo-hydrogen bonds between each pyridine moiety and neighbours appear to be weak (Cpy–H⋯N) but numerous, displaying angles of 152.83° and 158.84° and distances of 3.384 Å and 3.441 Å. Additionally, the C O⋯H–Cpy interactions assist the network extension, with a distance of 3.350 Å and an angle of 150.45°. The final assembly displays negligible π–π interactions.
[Zn0.5(3pyCCMoid)]n (1)
1 crystallizes in the monoclinic space group C2/c. The asymmetric unit includes one 3pyCCMoid molecule and one-half of the Zn ion. The ZnII centres are hexacoordinated, where each unit binds to two 3pyCCMoids through their β-diketone moieties (Zn–O distances between 2.026 and 2.132 Å) and two pyridine groups (Zn–N distances of 2.211 Å) from neighbouring CCMoids (Fig. 3a). In this arrangement, the β-diketone groups are contained in crossed planes, disposing in a cis configuration the two pyridine moieties (Fig. 3a). Regarding each 3pyCCMoid ligand, one of the pyridine moieties remains always free (Fig. 3a–c). Compared with the free CCMoid, here the CCMoid skeletons are slightly bent (23.16°, Fig. S6†)23 with the pyridine groups facing in opposite directions as seen before. The framework spreads forming loops through the coordination of the adjacent unit, similar to Fig. 3a, by pyridine endings (Zn⋯Zn distance 9.271 Å, Fig. 3b and c). In addition, the ZnII centres act as nodes among the loops, displaying them in a nearly perpendicular way (angles of 87.52° and 93.77°) and as a result, the system evolves into a staircase shaped 1D net. The chains are aligned and efficiently packed (Fig. 3d), with Zn⋯Zn distances of 14.5 Å and 12.5 Å among the closest neighbours.
Fig. 3. Crystal structure of 1: (a) Representation of the coordination sphere of a ZnII centre and the coordination modes of 3pyCCMoid molecules; (b) representation of two loops connected through the intermediate Zn(ii) centre; (c) extended view of one chain; (d) arrangement of four chains in the crystal. Colour legend: Zn, pink; C, grey; O, red; N, blue; H atoms are omitted for clarity.

[Zn2(3pyCCMoid)2(NO3)2]n (2)
System 2 crystallizes in the monoclinic space group P21/n. The asymmetric unit cell contains two [Zn(3pyCCMoid)(NO3)] fragments, each presenting 3pyCCMoid molecules with slightly different distances and conformations. As in 1, the ZnII ions adopt a pseudo-octahedral geometry, but now there is only one 3pyCCMoid chelating the metal centre together with one NO3− anion. The two remaining sites, within the ZnII unit, are occupied by pyridine moieties from nearby CCMoids. Here again, the disposition of the chelating groups, CCMoid and NO3−, is in the cis-configuration (Fig. 4a). The 3pyCCMoid ligand is now fully coordinated, bringing into play its three coordinative sites. Each 3pyCCMoid coordinates through the β-diketone moiety to one of the ZnII ions in the fragment, binding the pyridine groups to two neighbouring ZnII centres (Fig. 4a). The assembly of two asymmetric units forms loops too (Zn⋯Zn distance of 9.028 Å, Fig. 4b). Within such dimers, everything relates by an inversion centre, making the NO3− groups point out in opposite directions. The connection of four of these loops through the remaining pyridine groups creates a bigger cavity (Fig. 4c). Now the void displays a rhomboid shape, being the longest Zn⋯Zn distance of 22.060 Å. This implies a greater twist of the skeletons of the CCMoids (60.42° and 75.52°),23 where the pyridine moieties face almost perpendicularly allowing the extension in two dimensions and forming infinite layers (Fig. 4b and c). Despite the presence of several cavities in each single layer, the material presents a dense arrangement, where two NO3− groups, from adjacent layers, are inserted in the bigger voids resulting in a compact material (Fig. 4d).
Fig. 4. Crystal structure of 2: (a) Representation of the coordination sphere of a ZnII centre and the coordination mode of 3pyCCMoid; (b) picture of the smallest loop in the layer; (c) single 2D layer; (d) framework consisting of packed 2D layers (each layer represented by a different colour). Colour legend: Zn, pink; C, grey; O, red; N, blue; H atoms are omitted for clarity.

[Zn(3pyCCMoid)(NO3)(EtOH)2]n (4)
System 4 crystallizes in the orthorhombic P212121 space group. The asymmetric unit is formed by one [Zn(3pyCCMoid)(EtOH)]·NO3·EtOH fragment. Here, the ZnII ions are pentacoordinated adopting a trigonal bipyramidal geometry (τ = 0.85). Their coordination sphere chemically resembles the previous one, with every metallic centre coordinated to one β-diketone group from one CCMoid and two pyridine moieties from two other neighbours. However, the remaining position is now occupied by an EtOH molecule leaving uncoordinated the NO3− ion (Fig. 5a). In the structure, the CCMoid shows deviations from planarity (35.25°)23 but less pronounced than for compound 2, having here the pyridine groups oriented in opposite directions as in the pristine 3pyCCMoid and in 1. A remarkable difference with previous CP structures is the absence of loops. The shortest Zn⋯Zn distance is now 9.31 Å. In this case, the pyridine groups of the neighbouring CCMoids do not promote the formation of dimers, exhibiting their rotational freedom and spreading of the net (Fig. 5b) assisted by the ZnII centres. This, together with the fact that all the active sites of the 3pyCCMoid ligands are coordinated, favours the formation of a compact 3D structure (Fig. 5c). The new CP displays small voids occupied by one extra molecule of EtOH and the NO3− ion (Fig. 5c), both promoting supramolecular interactions with adjacent CCMoid residues.
Fig. 5. Crystal structure of 4: (a) Representation of the coordination sphere of a ZnII centre and the coordination mode of 3pyCCMoid; (b) picture of the connection of two neighbouring CCMoids; (c) 3D structure with molecules of the solvent and magnification of their disposition in the cavity. (Zn, pink; C, grey; O, red; N, blue; H atoms are omitted for clarity).

Comparative structural analysis of 3pyCCMoid
Compounds 1, 2 and 4 show that 3pyCCMoid is an excellent heteroditopic ligand that always binds through the β-diketone group with partial or total coordination of the pyridine moieties. It is worth noting that the description of our experimental combinations, regarding the metallic salt, solvents, and methods, was restricted for comparative purposes in contrast to the variety of results. Still, we focus on 3pyCCMoid, where the reasonable study of all variables would be highly complex. Nevertheless, some facts are highlighted for the general overview.
Briefly, solvents have a regulative role in the assembly process of different CPs, acting as orchestrators of the final coordination of the metal and/or as guest molecules, although it is difficult to assert the specifics of their performance.12,24,25 In this regard, the solvents used in this work (high boiling points, DMA and DMF, and alcoholic solvents, EtOH and MeOH) present excellent coordination abilities (DMF > MeOH > DMA > EtOH)26 and the synthetic procedures of the three systems, 1–2 and 4, display different combinations of them. This coordination ability is even more remarkable in 4, where EtOH forms part of the structure. Considering the metal centre, the ZnII ions can show a variety of coordination numbers (from 4 to 6) optimal to stabilize the coordination segments that the latter can connect and expand, increasing the dimensionality.25,27 Nevertheless, the arrangement among the units (Table S5†) is responsible for the final dimension (4 is a 3D system that displays pentacoordinated ZnII ions, while 1 and 2 show hexacoordinated metal centres, being 1D and 2D systems, respectively). With reference to the methodology, the achievement of a mixture of crystals, 1 & 3, instead of 4 (Table 1) is a clear example of the effects of temperature, pressure and time on the creation of fragments that grow and organize. System 3, although crystalline, could not be resolved. This together with the achievement of 1, with two different mixtures of solvents, indicates that independent of the methodology, similar coordination entities may be possible or even coexist in solution, the solvent mixture being responsible for favouring one over the others.
On the basis of the CCMoid structures, Fig. 6 shows the crystallographic variations among the coordinated 3pyCCMoid in the CPs compared with the pristine ligand. The uncoordinated 3pyCCMoid (in grey) shows a quasi-planar disposition that appears slightly disturbed in 1 and 4 (Fig. 6, magenta and blue, respectively). System 2 shows the highest distortion, having two crystallographically different CCMoids with twisted skeletons (yellow and green, Fig. 6), showing a clear predisposition towards the perpendicular orientations of the pyridine groups in the same ligand (green). Comparing 1 and 2, it can be observed that the dimers that provide small loops do not relate to the framework differences, although the higher distortion observed in 2 allows the connection among dimers creating additional bigger loops (Fig. 4d).
Fig. 6. Superimposition of 3pyCCMoid in the asymmetric units of the different compounds, compared to pristine CCMoid: 3pyCCMoid (grey), 1(purple), 2 (yellow and green) and 4 (blue).

Hence, the gentle conformational changes in the CCMoid skeleton, together with the rotation of the pyridine groups, allow further coordination to the ZnII units, increasing the variety of possible arrays and, therefore, the dimensionality. In this context, the N atoms in their pyridine groups appear always facing opposite directions.
Studies on the 2D material (compound 2)
Encouraged by the known biocapabilities of 3pyCCMoid and that the synthesis of nano-sized 2D CPs is highly desired for a number of applications, including membrane sensors and biomedicine,10,28,29 we further characterized our 2D system (compound 2). In this way, exfoliation was pursued towards the achievement of few-layered sheets from the hybrid material, together with the analysis of their thickness and stability. The physico-chemical properties of 2 were studied, together with the assessment of the number of layers after exfoliation, among others.30,31 Regarding this matter, compound 2 is an appealing system due to its arrangement and composition, although limitations such as constrain, thermal and/or solvent instability had to be tested.32 The powder XRD spectrum of 2 before (crystals) and after grinding (fine powder) displays a consistent pattern (Fig. S7†). STA(TG-DSC) experiments (Fig. S8†) show that 1 and 2 start decomposing at 325/262 °C, temperatures higher than the one found for the neat 3pyCCMoid, which starts to show a remarkable loss of weight at 185 °C. Regarding the two CPs, the lower thermal stability of 2 with respect to 1 could be related to the lability of the NO3− ions and further instability of the remaining system.
Solvent studies
2 is stable in EtOH at RT for long periods of time (>1 month). The powder XRD patterns, shown in Fig. S9,† demonstrate that the treatment with this solvent, which includes washing and conservation, does not affect its general structure, although the intensity of some peaks changes which is especially noticeable in the peak around 6.4°. These small changes can be related to the labile nitrate group or changes in the preferential growth direction of the crystals. However, when neat DMA is used to keep the crystals, the material loses crystallinity and the solution becomes yellowish, indicating partial re-dissolution. The remaining solid and solution were studied in parallel. First, in an attempt to identify the components of the yellowish solution, DMF-d7 was used to partially dissolve the crystals and the filtered solution was studied by 1H NMR to compare with the spectrum of the pristine ligand (Fig. S10†). The spectrum of 2 displayed a set of well-defined shifts that agreed with the presence of one fragment different from the free ligand, pointing to the existence of a ZnII-CCMoid compound, similar to other mononuclear ZnII species previously published.22 Nevertheless, the putative structure is still elusive (e.g.: mononuclear or some sort of dinuclear/short polynuclear systems). Beyond the singularity of its NMR in DMF, ESI studies in DMA did not show traceable molecular weight patterns (Fig. S11†). In addition, unfiltered solutions of 2 in DMA and the remaining solid were studied by SEM and AFM after deposition on different substrates (drop casting on SiO2/Si wafers, HOPG and mica). EDX experiments corroborated the existence of Zn and expected C and N atoms in the samples (Fig. S12†). Drop casting of the unfiltered solution after the complete evaporation of the solvent on SiO2/Si shows flakes of 2 and aggregation (Fig. 7, right). Here, there is a significant effect on the final morphology depending on the surface. Similar organization is observed for HOPG, although on mica the molecules spread out in a distinct manner, presenting less aggregates and, therefore, affinity for the substrate and higher disorder in their disposition (Fig. S13†). The morphologies found in all the cases for the unfiltered solution are dramatically different from those found for 3pyCCMoid under the same conditions and substrates (Fig. S14†), emphasizing, once more, the idea that the dissolved systems from DMA do not relate to the free ligand. On the other hand, the remaining solid from DMA is dispersed on the different substrates in a similar manner, displaying homogeneous cubes with an average size of 1 μm, (Fig. 7, left and S14†). Related types of shapes are reported studying the effect of solvent and surfaces on some CP dispersions, showing that crystals are formed by domains that can disaggregate.33
Fig. 7. SEM images of different substrates of 2 after treatment with DMA on silicon wafer: (left) solid sample and (right) solution.

AFM topographic images, using the tapping mode for the SiO2/Si wafers, are shown in Fig. 8. The so-called flakes from solution appear as amorphous aggregates of different heights (average 1 μm, Fig. 8, top). In contrast, the AFM images of the remaining solid show the robustness of the cubes with dimensions of 1 to 1.5 μm and a height of 0.5 μm (Fig. 8, bottom). Within the cubes, no further information regarding the orientation of the sheets with respect to the surface was extracted; further work may require functionalization of the cubes/surfaces to promote their alignment with the working surfaces. Overall, the cubes appear as intermediate forms between the crystals and the amorphous flakes that segregate, at the same time, in the units observed by NMR; overall, these experiments show the steps into the formation/aggregation of the 2D system.
Fig. 8. AFM images of the cubes of 2 (left) and 3D representations (right) obtained with the tapping mode for solution samples (top) and solid (bottom).

Exfoliation studies
Several sets of experiments in H2O were performed with 2, by varying sonication and resting times, as well as sample collection modes. A complete description of the most reproducible methodology is provided in the Experimental section. The Tyndall effect (Fig. S15†) proved the presence of nanosheets in suspension. Statistical analysis by AFM of the height profile using 100 measurements of these nanosheets deposited on SiO2/Si (Fig. 9) shows that approximately 70 of the measured nanosheet heights were between 11–50 nm (approximately: 12% of 11–20 nm, 21% of 21–30, 22% of 31–40 nm and 15% of 41–50 nm) followed by 15 measures that show values under 10 nm and in the same number of nanosheets higher than 50 nm.
Fig. 9. AFM height profile of three different nanosheets showing the most abundant height ranges and the statistical analysis of the nanosheet thickness.

In contrast, the extension of the nanosheets is dramatically large (average 1–5 μm, Fig. 10). Nanosheet deposition on the TEM grids showed transparent layers and continuous surfaces (Fig. 10a and b). After three days of sample deposition, the setups displayed breaks on the edges and folding of the sheets (live phenomenon in the ESI† video), due to surface tension effects and the low thickness of the material (Fig. 10c). Attempts to decrease the height of the sheets were made by applying longer resting periods, from 7 to 12 h. However, as a result, degradation of the material was observed, where the TEM grids presented black aggregates together with nanosheets that displayed evident holes. We postulate that at the interface between the nanosheets and the solvent, due to the labile nature of the NO3− groups coordinated to the ZnII centres, together with sonication and long periods in contact with H2O molecules, must trigger the segregation of the units which in H2O deteriorate, creating the holes in the nanosheets.
Fig. 10. TEM images of nanosheets (a) after the evaporation of the drop on the TEM grid, (b) magnification of (a), (c) after 3 days in the TEM grid, and (d) after the addition of more water.

A similar nanosheet appearance is observed when more water is added to the nanosheets after 6 hours of resting, proving the important role that this solvent plays in the degradation (Fig. 10d). The composition of the nanosheets deposited on the TEM grids was analysed by EDX showing the same elements as the original crystals (Fig. S16†).
Solid state fluorescence studies
The luminescence properties of 3pyCCMoid together with compounds 1 and 2 were analysed in the solid state at room temperature (Fig. S17†). 3pyCCMoid shows an emission band at 527 nm, expected in part from the CCMoid dye nature. However, compound 1 does not show fluorescence, and 2 compared with the neat ligand displays an attenuated band at the same wavelength. These results were somehow unexpected, taking into account that the chelation enhancement of fluorescence (CHEF)34 is normally observed when ZnII ions coordinate to dyes, which is our past experience with different Zn-CCMoid coordination compounds, although coordination in the latter was exclusively through the β-diketone group.22 Conversely, here the outcome indicates that the coordination of ZnII units to the pyridine moieties of the skeleton may have an adverse effect on the fluorescence.
Experimental section
Materials and methods
Experiments were carried out using commercial HPLC grade solvents. The chemicals were purchased from Sigma and used as received. The oven used for the CP synthesis is an Ecocell comfort.
Synthesis
Synthesis of 3pyCCMoid.35 0.7 g of acac (7 mmol) and 0.35 g of B2O3 (5 mmol) were dissolved in 8 mL of EtOAc. The reaction mixture was heated at 70 °C for 30 min until a white paste was formed. Then, a solution of 1.5 g of 3-pyridine carboxyaldehyde (14 mmol) and 3.22 g of tributyl borate (14 mmol) in 3 mL of EtOAc was added. The mixture was stirred and refluxed (70 °C) for an additional 4 h. After cooling down, a solution of n-butylamine (0.4 mL, 4 mmol) in EtOAc (1 mL) was added dropwise, and the final reaction mixture was kept stirring at room temperature overnight. The reaction mixture turned yellow with the precipitate on the next day. This suspension was filtered, washed with EtOAc and dried to afford a yellow solid. Then, the solid was suspended in 30 mL of H2O and stirred for 3 h at room temperature, followed by filtering, washing with H2O and finally vacuum drying. Pure 3pyCCMoid was achieved by recrystallization from hot MeCN. Yield: 1.56 g, 80%. 1H NMR (400 MHz, DMSO-d6) δ 15.96 (s, 1H), 8.91 (d, J = 1.7 Hz, 2H), 8.59 (dd, J = 4.8, 1.7 Hz, 2H), 8.19 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 16.1 Hz, 2H), 7.48 (dd, J = 7.9, 4.8 Hz, 2H), 7.13 (d, J = 16.1 Hz, 2H), 6.22 (s, 1H). Elemental analysis (%) calculated for C17H14N2O2·0.15 H2O: C 72.66, H 5.13, N 9.97; found C 72.18, H 4.43, N 10.07. MALDI-MS m/z (%): 277.04 ([M − H]−), 279.03 ([M + H]+). FTIR data (KBr, cm−1): 1626 (νC Caromatic), 1579 (C Ost,β-diketone), 1509 (νC N) 1476 (νC Caromatic), 1383 (δCCH), 1295, 1145 (opp CCHaromatic and CCHskeletal), 973 ( CHδ,opp). Elemental analysis (%) calculated for C17H14N2O2·0.15 H2O (281.01 g mol−1) calculated: C 72.66, H 5.13, N 9.97; found: C 72.18, H 4.43, N 10.07.
Synthesis of 135
3pyCCMoid (5 mg, 0.0180 mmol) and Zn(NO3)2·6H2O (16 mg, 0.054 mmol) were added to a mixed solvent involving DMA (0.5 mL) and methanol (1.5 mL). After blending well by sonication at RT, the solution was heated in a closed vessel at 80 °C for 3 days to obtain orange crystals with a size of about 1 mm. The crystals were washed and kept in MeOH. FTIR data (KBr, cm−1): 1633 (νC Caromatic), 1548 (C Ost,β-diketone), 1509 (νC N) 1462 (νC Caromatic), 1383 (δCCH), 1298, 1160 (opp CCHaromatic and CCHskeletal), 970 ( CHδ opp). Elemental analysis (%) calculated for [Zn(3py-CCM)2]n·n/2ZnO or C34H28N4O5.5Zn0.5 (659.6 g mol−1) calculated C:61.8, H:3.96, N:8.48; found: C:61.56, H:4.055, N:8.6.
Synthesis of 235
The amount of the ligand and metal salt used to synthesize 2 was the same as that for 1. The reaction was performed in a mixed solvent involving DMA (0.5 mL) and absolute ethanol (1.5 mL), following the procedure previously described for 1. The obtained orange crystals were washed and kept in EtOH. FTIR data (KBr, cm−1): 1641 (νC Caromatic), 1550 (C Ost,β-diketone), 1512 (νC N) 1463 (νC Caromatic), 1384 (δCCH), 1295, 1160 (opp CCHaromatic and CCHskeletal), 970 ( CHδ,opp). EA (wt%) data for [Zn(3py-CCM)]n·1/2nH2O or C17H14N3O5.5Zn (413.68 g mol−1) calculated C: 49.35, H: 3.41, N:10.15; found C:49.33, H:3.33, N:9.8.
Synthesis of 4
3pyCCMoid (30 mg, 0.108 mmol) was dissolved in 1.5 mL of DMF and placed at the bottom of an elongated vial. Then 1 mL of EtOH was placed on the top. Finally, 64 mg (0.215 mmol) of Zn(NO3)2·6H2O, dissolved in 2.5 mL of EtOH, were added. After the system set-up, it can be observed that the clean EtOH was forming a clearly visible boundary between the solvents keeping the two reagents. After 2–3 weeks, the formation of crystals was observed on the vial walls. These crystals were cleaned several times with fresh EtOH and kept in this solvent. The characterization of 4 by IR spectroscopy and elemental analysis was not possible because only a few crystals are obtained.
Exfoliation of 2
A suspension of 1 mg of the crystalline sample of 2 was prepared in 1 mL of Milli-Q H2O and added to 2 mL vials, which were sonicated for 1 h. Then, the vials were left undisturbed for 6 h. During this time, the deposition of the largest multi-layer nanosheets is expected, while those with only a few layers remain dispersed in the solvent. The presence of residual nanosheets in the solvent is proved by the Tyndall effect using a conventional laser. In the experiment where the effect of H2O on the degradation of the nanosheets is tested, 10 μL of the solvent was placed on the top of a vial containing 50 μL of fresh Milli-Q H2O. The liquids were mixed using a vortex. For analysis, 10 μL of the solvent on the top of the vial was placed on a TEM grid or on a SiO2/Si surface.
Characterization
Single crystal X-ray diffraction data for 1 and 4 were collected at the XALOC beamline 13 of Alba-CELLS Synchrotron (Spain) (T = 100 K, λ = 0.82654 Å for 1 and 0.729 Å for 4), data for 3pyCCMoid at the ALS Synchrotron (Berkeley, USA) (T = 100 K, λ = 0.7749 Å) and data for 2 using a Bruker D8 Venture diffractometer at 293(2) K (λ = 0.71073 Å). The structures were solved by intrinsic phasing methods (SHELXT using the XIA package for the data collected on the beamline) and refined on F2.36 Hydrogen atoms were included at the calculated positions, riding on their carrier atoms. Powder X-ray diffraction (PXRD) patterns were collected on a Panalytical X'PERT PRO MPD diffractometer using Cu Kα radiation (λ = 1.5418 Å) at 295 K. The simulated powder patterns were calculated from the single crystal crystallographic data using Mercury 3.7 programme. Fourier transform infrared (FTIR) spectra were obtained using an FT-IR JASCO 4700LE using the ATR (attenuated total reflectance) accessory in the range between 600 and 4000 cm−1. Elemental analyses were carried out using a PerkinElmer 2400 series II analyser. 1H-NMR spectra were obtained on a Bruker Advanced at 300 MHz and 298 K. Thermogravimetric analysis was performed under a N2 atmosphere from room temperature to 1000 °C at a heating rate of 10 °C min−1, using a simultaneous thermogravimetric analysis (TG)-differential scanning calorimetry/differential thermal analysis (heat flow DSC /DTA) system NETZSCH-STA 449 F1 Jupiter. Images of the nanosheets for the morphological analysis were obtained with a TEM 120 kV JEOL 1210. The composition of the nanosheets was roughly estimated using a SEM Magellan 400L working in STEM mode and equipped with an X-Max Ultim Extreme EDX (Oxford Instruments). The determination of the nanosheet thickness was performed by analysing the topographic images obtained with a Keysight 5100 using the software WSxM 5.0.37
Conclusions
This work presents the ligand 3pyCCMoid as a promising heteroditopic ligand in the field of CPs. The synthesis of this molecule together with three new CPs, 1–3, containing exclusively 3pyCCMoid as a linker among the ZnII centres, provides structures that extend from 1D to 3D. This work illustrates the versatile architectures that are possible thanks to this ligand and presents CCMoids as reliable connectors, emphasizing that their use in coordination chemistry should not be limited to curcumin. Our work describes the structures and basic physicochemical properties of the three new CPs, as well as pays great attention to the study of compound 2, due to its 2D nature. Here, stability studies in organic solvents and exfoliation in H2O have been introduced finding a way to achieve extended few-layered materials and analysing the factors that could degrade the system. We would like to extend our studies to other metals as well as heterometallic systems. In addition, extended biocompatibility studies of 3pyCCMoid and CPs will be analysed toward their possible use as active ingredients in therapeutic uses and/or nano-/microdevices.
Author contributions
Conceptualization and writing, N. A.-A, C. D and M.S.; experimental part CPs, L. R.-C., W. Q., J. I.-A. and E. M.-O.; L. R.-C. and W. Q. made equal contributions to this work; experimental part 3-pyCCMoid, A. E.-B.; supervision of experimental work, A.G.-C and A. L.-P.; acquisition and/or resolution of crystallographic data, D. C.-L., E. C. S., O. R. and J. G. P. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 R&D programme (ERC-724981). We also acknowledge the Spanish Government, Ministerio de Ciencia e Innovación (projects CTQ2017-83632, CTQ2015-68370-P, PGC2018-098630-B-I00, MAT2016-77852-C2-1-R, CTQ2016-75150-R, PGC2018-098630-B-I00, PGC2018-102047-B-I00 and Ramon y Cajal grant RYC-2017-22910) and the Generalitat de Catalunya for the grant 2017SGR1277. The work at the University of Chile was supported by ANID, Fondecyt Regular Project 1201962 and 1161775 (M. S.). C. D., N. A.-A., A. L.-P., A. G.-C., and L. R.-C. acknowledge the financial support through the “Severo Ochoa” program for Centres of Excellence in R&D (SEV-2015-0496) under the FUNMAT-FIP-2016 fellowship. Some of the experiments were performed at the XALOC46 and NCD-SWEET beamlines of the ALBA synchrotron with the support of ALBA staff and at the Advanced Light Source (ALS) synchrotron. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. This work (L. R.-C.) has been done in the framework of the doctoral program “Chemistry” of the Universitat Autònoma de Barcelona.
Electronic supplementary information (ESI) available. CCDC 2044029 (3pyCCMoid), 2044078 (1), 2044079 (2) and 2043842 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt00708d
Notes and references
- (a) Ariga K. Nishikawa M. Mori T. Takeya J. Shrestha L. K. Hill J. P. Sci. Technol. Adv. Mater. 2019;20:51–95. doi: 10.1080/14686996.2018.1553108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kitagawa S. Kitaura R. Noro S. Angew. Chem., Int. Ed. 2004;43:2334–2375. doi: 10.1002/anie.200300610. [DOI] [PubMed] [Google Scholar]; (c) Moulton B. Zaworotko M. J. Chem. Rev. 2001;101(6):1629–1658. doi: 10.1021/cr9900432. [DOI] [PubMed] [Google Scholar]
- (a) Wang J. L. Wang X. Y. Wang Y. Hu X. Y. Lian J. R. Guan Y. L. Chen H. He Y. Wang H. S. Coord. Chem. Rev. 2020;411(213256):1–21. [Google Scholar]; (b) Leong W. L. Vittal J. J. Chem. Rev. 2011;111:688–764. doi: 10.1021/cr100160e. [DOI] [PubMed] [Google Scholar]
- Janiak C. Dalton Trans. 2003:2781–2804. [Google Scholar]
- (a) Haga M.-A. Kobayashi K. Terada K. Coord. Chem. Rev. 2007;251:2688–2701. [Google Scholar]; (b) Novio F. Simmchen J. Vázquez-Mera N. Amorín-Ferré L. Ruiz-Molina D. Coord. Chem. Rev. 2013;257:2839–2847. [Google Scholar]
- (a) Uemura K. Matsuda R. Kitagawa S. J. Solid State Chem. 2005;178:2420–2429. [Google Scholar]; (b) Murdock C. R. Hughes B. C. Lu Z. Jenkins D. M. Coord. Chem. Rev. 2014;258:119–136. [Google Scholar]
- (a) Robin A. Y. Fromm K. M. Coord. Chem. Rev. 2006;250:2127–2157. [Google Scholar]; (b) Ye B. H. Tong M. L. Chen X. M. Coord. Chem. Rev. 2005;249:545–565. [Google Scholar]
- (a) Basu Baul T. S. Chaurasiya A. Duthie A. Montes-Tolentino P. Hopfl H. Cryst. Growth Des. 2019;19:6656–6671. [Google Scholar]; (b) Wang F. Zhang J. Ding X. Dong S. Liu M. Zheng B. Li S. Wu L. Yu Y. Gibson H. W. Huang F. Angew. Chem., Int. Ed. 2010;49:1090–1094. doi: 10.1002/anie.200906389. [DOI] [PubMed] [Google Scholar]
- Maxim C. Branzea D. Allain M. Andruh M. Clerac R. Iorga B. Avarvari N. CrystEngComm. 2012;14:3096–3102. [Google Scholar]
- (a).Rojas S. Devic T. Horcajada P. J. Mater. Chem. B. 2017;5:2560–2573. doi: 10.1039/c6tb03217f. [DOI] [PubMed] [Google Scholar]
- (a) Su H. Sun F. Jia J. He H. Wang A. Zhu G. Chem. Commun. 2015;51:5774–5777. doi: 10.1039/c4cc10159f. [DOI] [PubMed] [Google Scholar]; (b) Feng X. Wang Y. Muhammad F. Sun F. Tian Y. Zhu G. Cryst. Growth Des. 2019;19:889–895. [Google Scholar]
- Portoles-Gil N. Lanza A. Aliaga-Alcalde N. Ayllon J. A. Gemmi M. Mugnaioli E. Lopez-Periago A. M. Domingo C. ACS Sustainable Chem. Eng. 2018;6:12309–12319. [Google Scholar]
- Rodríguez-Cid L. Sañudo E. C. López-Periago A. M. González-Campo A. Aliaga-Alcalde N. Domingo C. Cryst. Growth Des. 2020;20(10):6555–6564. [Google Scholar]
- Philip E. A., Progress in Hetercyclic chemistry, ed. G. Gribble and J. A. Joule, Elsevier Ltd, 2011, vol. 22, pp. 349–391 [Google Scholar]
- Hamada Y., Pyridine, ed. P. P. Pandey, IntechOpen, 2018, vol. 9, pp. 9–25 [Google Scholar]
- Guan A. Y. Liu C. L. Sun X. F. Xie Y. Wang M. A. Bioorg. Med. Chem. 2016;24:342–353. doi: 10.1016/j.bmc.2015.09.031. [DOI] [PubMed] [Google Scholar]
- Jha N. N. Ghosh D. Das S. Anoop A. Jacob R. S. Singh P. K. Maji S. K. Sci. Rep. 2016;6:1–15. doi: 10.1038/srep28511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anto R. J. Kuttan G. Babu K. D. Rajasekharan K. N. Kuttan R. Pharm. Pharmacol. Commun. 1998;4:103–106. [Google Scholar]
- Zhao M. Huang Y. Peng Y. Huang Z. Ma Q. Zhang H. Chem. Soc. Rev. 2018;47:6267–6295. doi: 10.1039/c8cs00268a. [DOI] [PubMed] [Google Scholar]
- Pabon H. J. J. Recl. Trav. Chim. Pays-Bas. 1964;83:379. [Google Scholar]
- Chen Y. Huang P. Chen H. Wang S. Wang H. Guo J. Zhang X. Zhang S. Yan J. Xia J. Xu Z. ACS Biomater. Sci. Eng. 2018;4:4095–4103. doi: 10.1021/acsbiomaterials.8b00957. [DOI] [PubMed] [Google Scholar]
- Tamames-Tabar C. Imbuluzqueta E. Guillou N. Serre C. Miller S. R. Elkaïm E. Horcajada P. Blanco-Prieto M. J. CrystEngComm. 2015;17:456–462. [Google Scholar]
- Aliaga-Alcalde N. Rodríguez L. Ferbinteanu M. Höfer P. Weyhermüller T. Inorg. Chem. 2012;51:864–873. doi: 10.1021/ic201420d. [DOI] [PubMed] [Google Scholar]
- The two planes used to measure planarity were made using the meta-positions of the aromatic rings and the central carbon of the studied 3pyCCMoid, hence N′–C8′′C1 and N–C8–C1 atoms from Fig. 1
- Seetharaj R. Vandana P. V. Arya P. Mathew S. Arabian J. Chem. 2019;12:295–315. [Google Scholar]
- Lan Y. Q. Jiang H. L. Li S. L. Xu Q. Inorg. Chem. 2012;51:7484–7491. doi: 10.1021/ic202635a. [DOI] [PubMed] [Google Scholar]
- Díaz-Torres R. Alvarez S. Dalton Trans. 2011;40:10742–10750. doi: 10.1039/c1dt11000d. [DOI] [PubMed] [Google Scholar]
- (a) Kręzel A. Maret W. Arch. Biochem. Biophys. 2016;611:3–19. doi: 10.1016/j.abb.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vahrenkamp H. Dalton Trans. 2007:4751–4759. doi: 10.1039/b712138e. [DOI] [PubMed] [Google Scholar]; (c) Erxleben A. Coord. Chem. Rev. 2003;246:203–228. [Google Scholar]
- Song Y. Yang J. Wang L. Xie Z. ChemMedChem. 2020;15:416–419. doi: 10.1002/cmdc.201900664. [DOI] [PubMed] [Google Scholar]
- Zhao M. Huang Y. Peng Y. Huang Z. Ma Q. Zhang H. Chem. Soc. Rev. 2018;47:6267–6295. doi: 10.1039/c8cs00268a. [DOI] [PubMed] [Google Scholar]
- Amo-Ochoa P. Zamora F. Coord. Chem. Rev. 2014;276:34–58. [Google Scholar]
- Bazargan M. Mirzaei M. Akbari M. J. Mol. Struct. 2019;1188:129–141. [Google Scholar]
- Tan C. L. Cao X. H. Wu X.-J. He Q. Y. Yang J. Zhang X. Chen J. Z. Zhao W. Han S. K. Nam G.-H. Sindoro M. Zhang H. Chem. Rev. 2017;117:6225. doi: 10.1021/acs.chemrev.6b00558. [DOI] [PubMed] [Google Scholar]
- Qian W. González-Campo A. Pérez-Rodríguez A. Rodríguez-Hermida S. Imaz I. Wurst K. Maspoch D. Ruiz E. Ocal C. Barrena E. Amabilino D. B. Aliaga-Alcalde N. Chem. – Eur. J. 2018;24:12950–12960. doi: 10.1002/chem.201802031. [DOI] [PubMed] [Google Scholar]
- Antina E. V. Bumagina N. A. V'yugin A. I. Solomonov A. V. Dyes Pigm. 2017;136:368–381. [Google Scholar]
- Qian W., PhD thesis, Universitat Autónoma de Barcelona, Spain, 2018, https://ddd.uab.cat/pub/tesis/2018/hdl_10803_664220/weqi1de1.pdf [Google Scholar]
- Sheldrick G. Acta Crystallogr., Sect. C: Struct. Chem. 2015;71:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horcas I. Fernández R. Gomez-Rodriguez J. M. Colchero J. W. Gómez-Herrero J. W. Baro A. M. Rev. Sci. Instrum. 2007;78:013705. doi: 10.1063/1.2432410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.