

Abstract
Endothelial dysfunction, characterised by impaired nitric oxide (NO) bioavailability, arises in response to a variety of cardiovascular risk factors and precedes atherosclerosis. NO is produced by tight regulation of endothelial nitric oxide synthase (eNOS) activity in response to vasodilatory stimuli. This regulation of eNOS is mediated in part by store-operated calcium entry (SOCE). We hypothesised that both ATP- and flow-induced eNOS activation are regulated by SOCE derived from Orai1 channels and members of the transient receptor potential canonical (TRPC) channel family. Bovine aortic endothelial cells (BAECs) were pre-treated with pharmacological inhibitors of TRPC channels and Orai1 to examine their effect on calcium signaling and eNOS activation in response to flow and ATP. The peak and sustained ATP-induced calcium signal and the resulting eNOS activation were attenuated by inhibition of TRPC3, which we found to be store operated. TRPC4 blockade reduced the transient peak in calcium concentration following ATP stimulation, but did not significantly reduce eNOS activity. Simultaneous TRPC3 & 4 inhibition reduced flow-induced NO production via alterations in phosphorylation-mediated eNOS activity. Inhibition of TRPC1/6 or Orai1 failed to lower ATP-induced calcium entry or eNOS activation. Our results suggest that TRPC3 is a store-operated channel in BAECs and is the key regulator of ATP-induced eNOS activation, whereas flow stimulation also recruits TRPC4 into the pathway for the synthesis of NO.
Keywords: Store-operated, Calcium, TRPC, Orai1, Nitric oxide
1. Introduction
Nitric Oxide (NO) is released in response to hemodynamic forces and agonists in endothelial cells. It regulates vascular tone and angiogenesis, suppresses smooth muscle growth, and inhibits platelet adhesion and aggregation. Impaired NO production is the hallmark of endothelial dysfunction. It is a precursor to, and a complication of several cardiovascular comorbidities such as atherosclerosis, hypertension, and diabetes [1]. Our efforts to elucidate the key molecular players in flow-induced NO production are motivated by the need for specific druggable targets for the management of endothelial dysfunction.
We have previously reported that shear stress-induced NO production is dependent on ATP autocrine signaling and store-operated calcium entry (SOCE) [2, 3]. Figure 1A outlines the mechanism underlying the ATP-SOCE pathway of flow-induced NO synthesis and the hypothesized role of transient receptor potential canonical (TRPC) channels and Orai1; however, this pathway has not been fully elucidated. It is well known that endothelial nitric oxide synthase (eNOS) activity is a function of several modulating influences (Figure 1B) [4]. In addition to its substrate l-arginine, its cofactors and the inhibitory caveolin1 (cav1) association, both calcium/calmodulin (Ca2+/CaM) binding and eNOS phosphorylation at the ser1179 residue promote eNOS activation. The calcium responses to acute hemodynamic stimuli are transient so that sustained eNOS activation in response to ongoing flow is thought to be generated by changes in phosphorylation [5]. Phosphorylation at eNOS-ser1179 is sensitive to Ca2+/CaM as is phosphorylation at eNOS-threonine497 (thr497) which lies in the calmodulin binding domain[6], where it imposes an inhibitory influence on eNOS activity.
Figure 1.
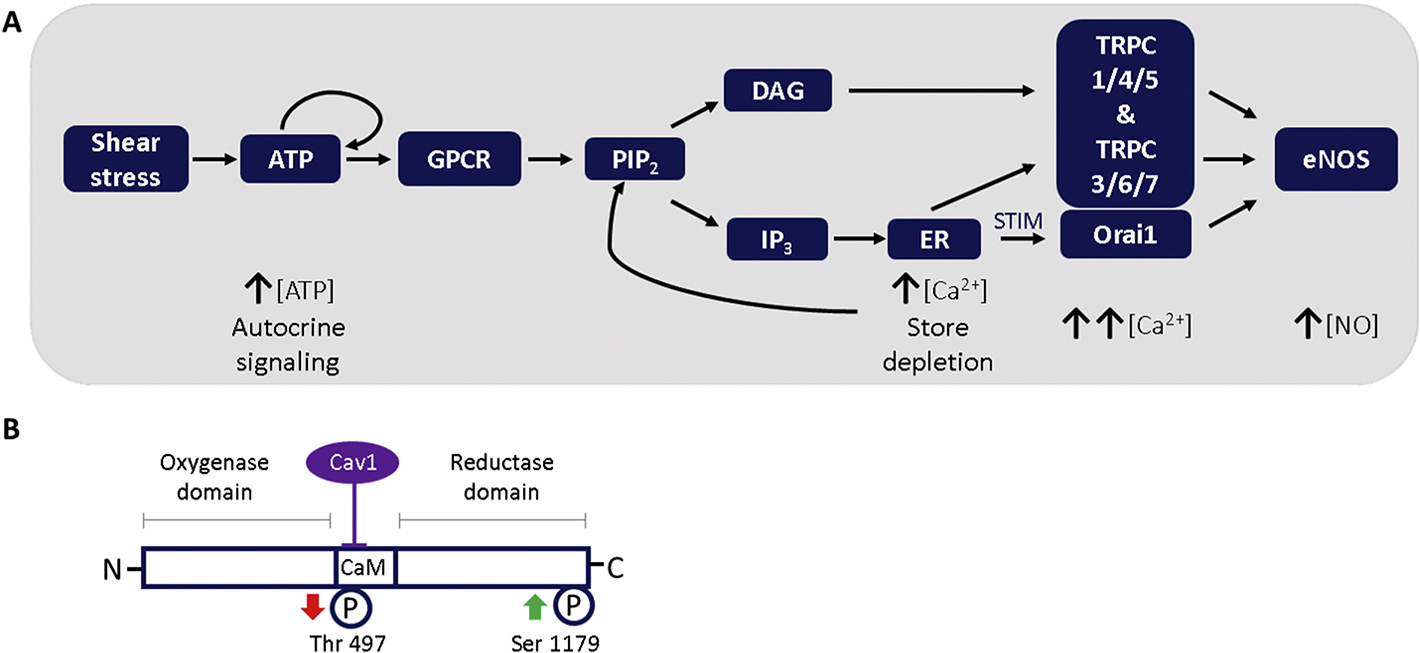
A: Schematic of hypothesized flow-induced eNOS activation regulated by TRPC- and ORAI - mediated SOCE. Shear stress induces autocrine ATP release. ATP activates PLC via G-protein receptors allowing PLC to hydrolyze PIP2 to form DAG and IP3. IP3 diffuses and binds to receptors on the endoplasmic reticulum (ER) causing store depletion which activates SOCs. DAG promotes opening of TRPCs. The increase in intracellular calcium allows for Ca2+/Calmodulin-mediated eNOS activation which leads to NO production. B: Key regulatory sites of endothelial nitric oxide synthase. The inhibitory Cav1 binding site in the calcium/calmodulin binding domain is shown. Phosphorylation sites are numbered according to the bovine eNOS sequence. The arrows indicate the effect of phosphorylation on eNOS activity. Phosphorylation of Ser1179 increases eNOS activity, whereas phosphorylation of Thr497 is inhibitory. Cav1-caveolin1, Ser—serine, Thr—threonine, CaM—calmodulin.
Of the 7 members of the TRPC family of non-selective, receptor-activated cation channels, TRPC1 (the predominant isotype in human endothelial cells)[7, 8], and TRPC 4 are the main candidates for endothelial SOCs [9, 10]. Some reports indicate that only TRPC1 & 4 are expressed in bovine aortic endothelial cells (BAECs)[11], while in HUVECS, TRPC3 is important for SOCE [11, 12]. Some TRP channels have recently been reported to localise to raft domains (TRPC 1,3,4,5) [13, 14]. All TRPCs have a putative caveolin binding motif [9], and plasma membrane (PM) localisation of TRPC4 has been shown to depend on cav1 [15]. The TRPC 1/4/5 subfamily is reported to hetero-tetramerize to form SOCs, while TRPC 3/6/7 form diacylglycerol (DAG)-activated channels[16]. The lack of consensus regarding the role of these channels in the regulation of eNOS is a natural consequence of the diversity of this class of channels.
STIM1, a calcium-sensing protein localized in the endoplasmic reticulum (ER) and caveolae partners with Orai1 channels on the PM and has been shown to activate TRPC1, although this process is not well understood [17]. There is increasing support for Orai1, a calcium release-activated ion channel being a SOC [18]. Nascent theories suggest a positive interaction between TRPC channels and the STIM/Orai machinery [19, 20]. For the purposes of understanding calcium regulation of NO production, we examined which of these channels are recruited by vasodilatory stimuli.
In the present study we endeavoured to quantify the relative contributions of STIM1/Orai1 and TRPC channels to eNOS-regulating calcium signals evoked by flow and ATP. We thus evaluate ATP-stimulated calcium signaling to determine which channels are involved in this pathway and then proceeded to examine how interfering with these channels affected ATP-induced eNOS activation. Given that ATP proportionally, temporally and spatially links shear stress to calcium signaling and NO production, we compared ATP-induced and flow-induced eNOS activation and NO production. The goal of this study was to determine which channels are involved in NO regulation and whether they are store operated. We hypothesized that SOCE would be more important for NO production, based on our previous work.
2. Materials & Methods
2.1. Experiments
To determine the participation of TRPC1/3/4&6 and Orai1 in direct flow stimulation and surrogate agonist stimulation, cells were probed for expression of these channels then treated with drugs inhibiting these channels and their responses to stimuli compared to controls. To identify channels involved in ATP-mediated endothelial calcium handling, cells were stimulated with ATP, an NO agonist, and calcium signaling was examined microscopically. Secondarily, cells were exposed to store-depleting drug and assessed for SOCE to determine which channels were store-dependent in this system. ATP-evoked eNOS activation was assessed by evaluating the eNOS phosphorylation response of bovine aortic endothelial cells (BAECs) stimulated with ATP under the influence of channel inhibitors. To compare with agonist-stimulated experiments, cells were mechanically stimulated, and flow-induced NO production was evaluated by real time measurement of NO in cells perfused for 3 min. Finally, BAECs from flow experiments were assessed for eNOS phosphorylation to determine flow-induced eNOS phosphorylation.
In order to identify which channels participate in calcium handling, we examined the effect of pretreatment with 3μM of TRPC3 inhibitor Pyr3 [21, 22], 1μM TRPC4 inhibitor ML204 [23], combined TRPC3&4 inhibition, TRPC1/6 inhibition (100nM GsMTx4 ) and Orai1 inhibition (10μM AnCoA4 [24]).We used a second set of calcium channels blockers to verify the participation of the TRPC subtypes. Pyr10, a novel TRPC3-selective inhibitor was applied at 3μM [25], as well as TRPC4 antagonist HC-070 (46nM)[26] and 5μM of TRPC6 channel blocker larixyl acetate[27]. All drugs were applied at lowest effective concentration which was greater than or equal to the cell free IC50 from literature. Control group cells were held in PBS with calcium for the same duration as drug treatment. PBS pre-incubation did not affect calcium, eNOS or NO responses.
2.2. Cell culture
Primary bovine aortic endothelial cells (BAEC) were obtained from Dr. Peter Davies laboratory (University of Pennsylvania). Batches of freshly harvested BAECs were maintained at 37°C and 5% CO2. Experiments were performed on 90– 100% confluent cells in passage 7–14. Cells were assessed for viability microscopically and for ideal morphology. BAECs were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Sigma Aldrich), 2mM/L L-glutamine (Mediatech) and penicillin-streptomycin (Sigma Aldrich).
2.3. Chemicals and reagents
ATP, 1-[4-[(2,3,3-Trichloro-1-oxo-2-propen-1-yl)amino]phenyl]-5-(trifluoromethyl)-1H-pyrazole-4-carboxylic acid (Pyr3), 4-Methyl-2-(1-piperidinyl)quinoline (ML204), ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), L-arginine, Dulbecco’s phosphate buffered saline with calcium and magnesium (PBS) and Thapsigargin (Tg) were sourced from Sigma Aldrich, while the calcium dye Fluo8 was from Abcam. GsMTx4, a tarantula venom toxin, was purchased from Tocris, and 3-(6-Methoxy-1,3-benzodioxol-5-yl)-8,8-dimethyl-4H,8H-benzo[1,2-b:3,4-b]dipyran-4-one (AnCoA4) also known as Jamaicin was obtained from EMD Millipore.
2.4. Calcium Imaging
BAECs were seeded 48 hrs prior to experiments. Cells were loaded with the calcium-sensitive dye Fluo-8 by incubation with 5μM of the acetoxymethyl ester form of the dye (Fluo-8AM) in the dark for 30 minutes at 37°C. Following dye loading, cells were incubated a further 30 min in PBS or inhibitor in the dark and then stimulated with agonist or flow. Changes in fluorescence intensity were viewed with an inverted microscope (Nikon TE300 Eclipse & Leica DMI3000) at 20x magnification using a Leica HCX PL Fluotar L 20x/0.4 n.a. objective. Microscopy was performed in an enclosure held at 37°C, in which cells were placed for ~1min before imaging commenced. A field of up to 100 −200 cells was imaged and all cells with distinguishable boundary were manually selected as ROIs for analysis in ImageJ. Four images were acquired 3s apart to establish baseline fluorescence immediately before stimulation. We found the baseline readings to be stable (0.1 – 5% deviation from mean). An image was acquired every 3 seconds for a duration of 10 minutes.
Fluorescence measurements were analysed using Image J software (National Institutes of Health) and expressed as a ratio of fluorescence measurements to the average basal fluorescence intensity.
2.4.1. Agonist-stimulated Calcium Imaging
Confluent BAECS in 35mm2 dishes were stimulated by the addition of 100μM ATP to the side of the dish following acquisition of baseline images. Store-operated calcium entry was initiated by stimulating dye-loaded cells with 1μM Thapsigargin (Tg) in calcium-free PBS for 5 min before removing the Tg and continuing to image the cells in PBS with calcium for 5 min.
2.4.2. Flow-stimulated Calcium Imaging
Confluent BAECs were grown in Ibidi μ-slide I luer 0.4mm polymer channel slides according to the manufacturer’s protocol. Calcium imaging was set up as described in §2.4. The channel slide was secured to the stage and connected to a syringe pump at the inlet and waste bottle at outlet with tubing. After baseline imaging, cells were perfused with PBS with calcium at 10 dyn/cm2 laminar shear stress at 37°C for 3 min.
2.5. Western Blotting
BAECs were grown to confluence in 60 mm dishes and pretreated for 30 min in PBS or inhibitor. Cells were stimulated with 100 μM ATP for 0 (no ATP),1,3,5 or 10 min in each treatment condition at 37°C. For channel expression assessment, cells were not pretreated. BAECs were then washed in ice-cold PBS, lysed on ice, and collected with scraping in 1x RIPA lysis buffer supplemented with 0.5nM dithiothreitol (DTT) (Amresco), HALT protease inhibitor cocktail and HALT phosphatase inhibitor cocktails (Thermo Fisher Scientific). Samples were agitated at 4°C for 30min and then centrifuged for 20 min and 16,000 x g at 4°C to remove insoluble material. Supernatant protein content was assayed using the bicinchoninic protein assay kit (Thermo Scientific). Cell lysates were then normalized for protein content and heated at 70°C for 10 minutes in laemmli buffer and centrifuged for 1 min. Proteins were resolved by SDS-PAGE on NuPAGE 4%−20% Bis-Tris Gel (Invitrogen) and transferred to nitrocellulose membrane using the iBlot2 (Invitrogen).
Membranes were briefly stained in Ponceau S stain and evaluated for even transfer before destaining in TBS/0.1% Tween. For phospho-proteins, blots were preincubated in Supersignal western blot enhancer (Thermo Fisher) for 10 min before blocking non-specific sites in 5% BSA/TBST. For remaining proteins, the blocking step was performed in 5% milk/TBST. Membranes were probed with primary antibodies p-eNOS ser1179 (Thermo Fisher), p-eNOS thr495 (Cell Signaling), eNOS (BD biosciences), GAPDH (Novus Biologicals), orai1,TRPC1, TRPC3 (Santa Cruz Biotech.), TRPC4 and TRPC6 (Abcam) for 30 min at room temperature then overnight at 4°C. Membranes were then rinsed and incubated for 2 hours at room temperature with secondary antibodies, anti-Rabbit (Thermo Fisher Scientific) and anti-Mouse (KPL Inc.) conjugated to horse radish peroxidase and visualized with enhanced chemiluminescence kit (Thermo Fisher Scientific) using a chemiluminescence detector. Densitometry was performed in ImageJ and results were reported as the fold change of phospho-eNOS/total eNOS at each time point normalised to the ‘zero’ time point (no ATP treatment condition) where applicable.
2.6. Shear stress stimulation and NO electrode measurements
Cells were prepared for NO readings as previously described [2]. Real-time, direct measurements of NO production were acquired using a parallel-plate flow device developed in our lab with an NO sensitive electrode that produces a polarographic current which is converted to NO concentration by calibration according to manufacturer’s protocol [2]. Briefly, BAECs were grown to confluence on the underside of polyester transwell™ membrane. Cells were washed in experimental solution (PBS supplemented with 70μM L-arginine) and preincubated in either L-arginine supplemented PBS or inhibitor solution for 30min at 37°C. The transwell was then transferred to the flow chamber which sits in a 37°C water bath and the internal chamber temperature allowed to equilibrate for 10 min. When the cells were perfused in the chamber, measurements were acquired from the upper compartment containing stagnant experimental fluid where a NO-sensitive electrode tip abuts the transwell membrane. BAECs were exposed to 10 dyn/cm2 shear for 3 minutes. Baseline readings were acquired in the 10 min before shear onset. Results are reported as the change in NO concentration.
2.6.1. Western Blotting of sheared cells
Directly following the flow period, the transwell was extracted from the chamber, placed in cold PBS and rinsed. The transwell was inverted on ice and cells collected by scraping in lysis buffer. Western blotting was performed as described in section 2.5.
2.7. Statistical analysis
Results are expressed as mean ± SEM. Data sets from control and treatment conditions were compared using the Student’s or Dunnett’s t-test or ANOVA with Tukey’s HSD post hoc analysis. For calcium imaging, n is reported as number of different dishes with ~ 150 cells analysed per dish. Differences were considered statistically significant when p < 0.05. In the figures, notations indicate the following: ns- non significant difference, *p < 0.05, **p < 0.01, ***p < 0.001. Effect size was calculated with a correction for small and unequal sample sizes and reported as Hedge’s g where g ≥ 0.8 is large, 0.8 > g ≥ 0.5 is medium and 0.5 > g ≥ 0.2 is considered to be a small effect. Statistical testing was performed with SPSS software (SPSS Inc).
3. Results
Expression of the orai1 and TRPC channels of interest was established by western blotting whole lysate of BAECs with isotype-targeting antibodies against known channel-expressing positive control lysates. All target channel subtypes (orai1, TRPC3, TRPC4, TRPC6) are expressed in our cells (Figure 2). We then investigated the participation of these channels in ATP-induced NO production and flow-stimulated NO production in parallel.
Fig. 2.

Expression of orai1 and TRPC variants in BAECs. Untreated BAECs were lysed and probed by western blotting for orai1 and TRPC 1,3,4,& 6 alongside positive controls MCF-7 whole cell lysate, and Jurkat whole cell lysate. All proteins appeared at or near predicated molecular weights: orai1 ~ 38/50 kDa, TRPC1 ~88 kDa, TRPC3 ~97/106/100 kDa, TRPC4 ~110 kDa, and TRPC6 ~106 kDa.
3.1. TRPC & Orai1 involvement in endothelial ATP-induced calcium influx
To identify which channels participate in calcium handling, we assessed the effect of TRPC3 inhibitor Pyr3, TRPC4 inhibitor ML204, combined TRPC3&4 inhibition, TRPC1/6 inhibitor GsMTx4 and Orai1 inhibitor AnCoA4 on ATP-evoked calcium signaling in BAECs. When stimulated with ATP, dye-loaded cells showed a rapid increase in calcium fluorescence followed by a slower decline to a steady state (Figure 3).
Figure 3:

Effect of TRPC3 and TRPC4 inhibition on ATP-induced calcium entry. BAECs were preincubated with PBS for 30min (control)-black, TRPC3 inhibitor Pyr3 (A,B), TRPC4 inhibitor ML204 (C,D), or a combination of Pyr3 & ML204 (E,F) –grey then stimulated with 100μM ATP. A,C,E: Averaged traces of ATP-induced calcium fluorescence (F) normalized to baseline fluorescence (F0) B,D,F: Bar graph depicting relative calcium fluorescence at the peak of the calcium transient as well as at 1min and 3min of ATP stimulation. Results are reported as mean ± S.E.M., * p<0.05, ** p<0.01, *** p<0.001, Control & Pyr3 n= 14,17 Control & ML204 n=14, 20, Control & Pyr3+ML204 n=6, 10.
3.1.1. TRPC 3&4 participate in endothelial ATP-induced calcium influx
TRPC3 inhibition caused a decline in both the peak and steady-state calcium signal as evinced by significant reductions in the fluorescence ratio at the peak as well as the 1 and 3 minute mark (22%, 36% & 23% reduction respectively) (Figure 3A & B). TRPC4 inhibitor, ML204 (Figure 3C & D), reduced the peak of the calcium transient and the signal at 1min but did not affect the fluorescence ratio at 3min of ATP stimulation (22%, 19% and 5% reduction in fluorescence). Combined inhibition of TRPC3 and TRPC4 produced a 22% decrease in peak calcium and significant suppression of the calcium fluorescence ratio at 1 and 3 min but did not alter the sustained response (Figure 3E & F).
3.1.2. TRPC 1/6 do not participate in endothelial ATP-induced calcium influx
Application of 100nM GsMTx4, a TRPC1/6 inhibitor, did not affect calcium entry following ATP-stimulation. We found the fluorescence ratio at the peak of the calcium transient to be 2.16 ± 0.18 and 2.07 ± 0.13 for the GsMTX4 and control groups respectively. Calcium fluorescence was also similar between groups at later time points (Figure 4A & B).
Figure 4:

Effect of TRPC1/6 and ORAI1 inhibition on ATP-induced calcium entry. BAECs were preincubated with PBS for 30min (control)-black, TRPC1/6 inhibitor GsMTx4 (A,B) or Orai1 inhibitor AnCoA4 (C,D) –grey then stimulated with 100μM ATP. A,C: Averaged traces of ATP-induced calcium fluorescence (F) normalized to baseline fluorescence (F0). B,D: Bar graph representing relative calcium fluorescence at the peak of the calcium transient as well as at 1 min and 3 min of ATP stimulation. There is no significant difference between the calcium signals. Results are shown as mean ± S.E.M., Control & GsMTx4 n=9, 8, Control & AnCoA4 n=5, 7.
3.1.3. ATP-induced calcium influx is not Orai1-dependent
Inhibition of Orai1 channels by incubating BAECs for 30min in 10μM AnCoA4 produced no significant change in ATP-stimulated calcium entry at the peak of the calcium transient or at later time points compared to the PBS control. The fluorescence ratio at the peak of the calcium transient was 2.15 ± 0.1 and 1.83 ± 0.16 for the Orai1 inhibition and control groups respectively (Figure 4C & D).
3.1.4. Inhibition of TRPC3 has a robust effect on ATP-induced calcium signaling
The second set of TRPC subtype-specific drugs we employed had a similar effect on ATP-induced calcium handling. TRPC3 inhibition with Pyr10 produced a significant reduction in calcium entry at 1 min of ATP stimulation Figure 5. As with the first set of drugs, TRPC3 and TRPC4 targeting had larger effects on the peak of the calcium transient (~12% and ~12%) while TRPC6 had less effect (~9% reduction). TRPC3 inhibition had the greatest impact (hedges g> 1.17) compared to TRPC4 targeting with HC-070 and TRPC6 inhibition with larixyl acetate (hedge’s g = 0.34 and 0.5) at 1 min.
Figure 5.
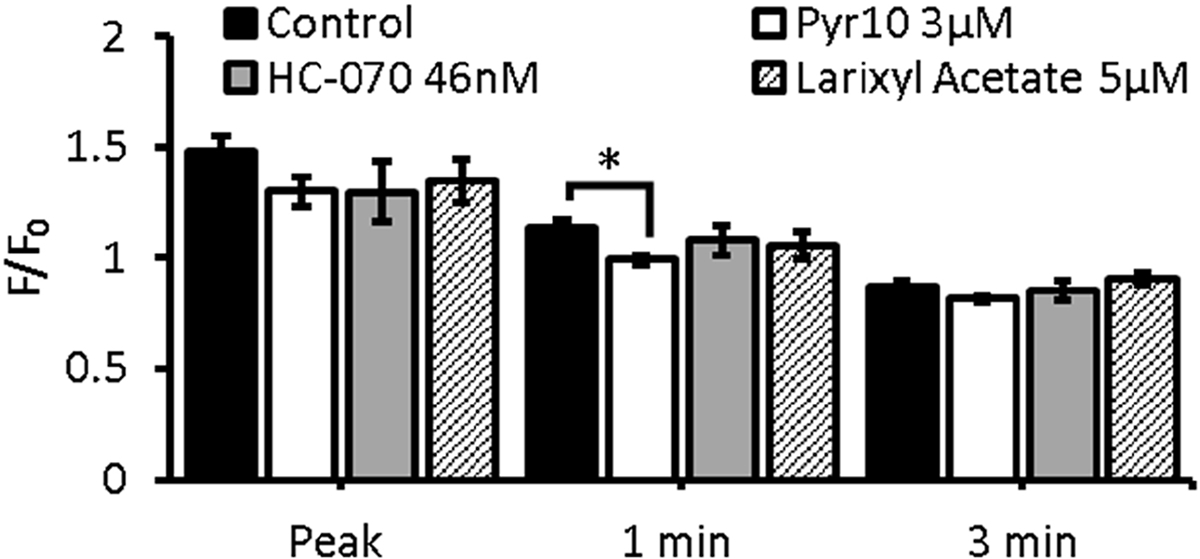
Effect of TRPC3,TRPC4 and TRPC6 inhibition on ATP-induced calcium entry. BAECs were preincubated with PBS for 30min (control)-black, TRPC3 inhibitor Pyr10 (white), TRPC4 inhibitor HC-070 (grey), or TRPC6 inhibitor, larixyl acetate(hatched) then stimulated with 100μM ATP. Bar graph depicts calcium fluorescence (F) normalized to baseline fluorescence (F0) at the peak of the calcium transient as well as at 1min and 3min of ATP stimulation. Results are reported as mean ± S.E.M., * p<0.05, Control n=17, PYr10 n=5, HC-070 n=, Larixyl Acetate n=4.
3.1.5. The kinetics of calcium handling are disrupted by TPRC channel inhibition
To further characterize the effect of channel inhibition on the intracellular calcium concentration, we evaluated the time to peak of the ATP-evoked calcium transient following pre-treatment with the channel-perturbing drugs. We found the mean time to peak to be 16.3s (Figure 6A). TRPC3 inhibition as well as TRPC1/6 inhibition significantly shortened the period between ATP stimulation and the peak of the calcium transient while TRPC4 inhibition significantly increased the time to peak (Figure 6B). Neither combined pretreatment with Pyr3 & ML204 nor application of Orai1 inhibitor, AnCoA4, altered the mean time to maximal calcium fluorescence.
Figure 6:

Time to peak calcium fluorescence. Graphs indicate the length of the interval between ATP stimulation and peak calcium fluorescence. Cells were stimulated with 100μM ATP following pretreatment. A: Mean time to peak (t) for ATP-induced calcium entry in control group. B: Bar graph indicates normalized time to peak for PBS (control), TRPC3 inhibitor Pyr3, TRPC4 inhibitor ML204, a combination of Pyr3 + ML204, TRPC1/6 inhibitor GsMTx4 or Orai1 inhibitor AnCoA4. TRPC3 inhibition significantly shortened the time to peak as did TRPC1/6 inhibition. Inhibition of TRPC4 prolonged the time to peak significantly while combined TRPC3 & TRPC4 inhibition did not affect the mean time to peak. TRPC1/6 inhibition produced a significant reduction in the time to peak while Orai1 inhibition had no effect. Results are shown as mean ± S.E.M., * p<0.05, ** p<0.01. Control & Pyr3 n=14,13, Control & ML204 n=14,19, Control & Pyr3 + ML204 n=6,10, Control & GxMTx4 n=9,8, Control & AnCoA4 n=4,4.
3.1.6. TRPC3 calcium channels are store-operated
Following the finding that TRPC3 and 4, but not TRPC1/6 or Orai1 participate in ATP-stimulated calcium handling in BAECs, we sought to determine which of these TRPC channels are store operated. We treated cells with Tg in the absence of extracellular calcium to induce store depletion. Calcium influx due to subsequent addition of extracellular calcium is indicative of SOCE (Figure 7A). Pre-treatment with TRPC3 inhibitor, Pyr3, caused a 24% diminution in the mean peak calcium fluorescence (p = 0.008) (Figure 7B). In contrast, pre-treatment with the TPRC4 inhibitor, ML204, led to a 18% reduction in peak calcium fluorescence, which was not statistically significant (Figure 7C).
Figure 7.

Store-dependence of TRPC channels. Dye-loaded BAECs were preincubated with PBS for 30min (control), TRPC3 inhibitor Pyr3 (A,B) or TRPC4 inhibitor ML204 (A,C), then stimulated with 1μM Tg without calcium, followed by a calcium add-back phase (A-C). A: Representative traces of Tg-induced store-operated calcium fluorescence (F) normalized to baseline fluorescence (F0). B,C: Graph depicting peak calcium fluorescence. TRPC3 inhibition significantly diminished the peak calcium while TRPC4 did not produce a significant effect. Results are reported as mean ± S.E.M., ** p<0.01, Control & Pyr3 n= 13,8, Control & ML204 n=7,5.
3.2. Regulation of ATP-stimulated eNOS activation
We assessed the effect of pretreatment with the calcium channel inhibitors on the level of basal eNOS phosphorylation (Figure 8). Concurrent Pyr3 and ML204 treatment had a large effect (g = 1.45) on peNOS ser1179 compared to Pyr3 alone. Although ML204 and GsMTx4 pretreatment evoked pronounced changes, producing a 74% & 92% increase in peNOS-thr497 phosphorylation respectively compared to the control, these responses were not statistically significant (ML204 p = 0.16, GsMTx4 p = 0.09). These two drugs did, however, produce the largest effect on eNOS-thr497 phosphorylation (ML204 g = 0.93, GsMTx4 g = 1.13).
Figure 8.

Basal eNOS-ser1179 phosphorylation following channel inhibition. BAECs were treated for 30 min with PBS with calcium (control) or Pyr3, ML204, Pyr3 + ML204, GsMTX4 and AnCoA4 (TRPC3, TRPC4, TRPC3&4, TRPC1,6 and Orai1 inhibitors respectively) and eNOS-ser1179 and eNOS-th497 phosphorylation was assessed by western blotting. (A) Western blot of basal eNOS phosphorylation following channel inhibition. (B) Graph shows relative eNOS phosphorylation. There were no significant differences in eNOS phosphorylation produced by drug pretreatment compared to the PBS control. Results are shown as mean peNOS/eNOS ± S.E.M., * p<0.05. peNOS ser1179 & peNOS thr497 n=9,5.
3.2.1. TRPC3 inhibition attenuates ATP-induced eNOS activation
We then investigated how the interference with calcium entry would be propagated to eNOS responses. ATP treatment for 0 (no ATP),1,3,5 or 10 min induced a transient increase in eNOS ser1179 and eNOS thr497 phosphorylation in the absence of pharmacological inhibition (Figure 9). Pretreatment with Pyr3 reduced the 1 min peNOS ser1179/eNOS ratio to 2.63 ± 0.27 compared 3.36 ± 0.28 under control conditions (g = 0.96) and produced concurrent non-significant increases in peNOS thr497/eNOS (g = 0.82, Figure 9A & B). The largest effects as quantified by Hedge’s g occurred at 1 min for eNOS-ser1179 phosphorylation and 10 m in for eNOS-thr497. TRPC4 inhibitor, ML204, did not cause a significant perturbation of the eNOS ser1179 or thr497 phosphorylation (Figure 9C & D). At 1 min post-stimulation under ML204 treatment, peNOS thr497/eNOS was reduced by 28% (n.s., p = 0.057) compared to untreated control (g = 0.93). Combined TRPC3 and TRPC4 inhibition did not produce a significant attenuation of eNOS activation. The effect of combined TRPC3 & TRPC4 inhibition on peNOS-thr497/eNOS (Figure 9F) was most pronounced at 10 min after ATP stimulation (Hedge’s g = 0.84, n.s., p = 0.062).
Fig. 9.
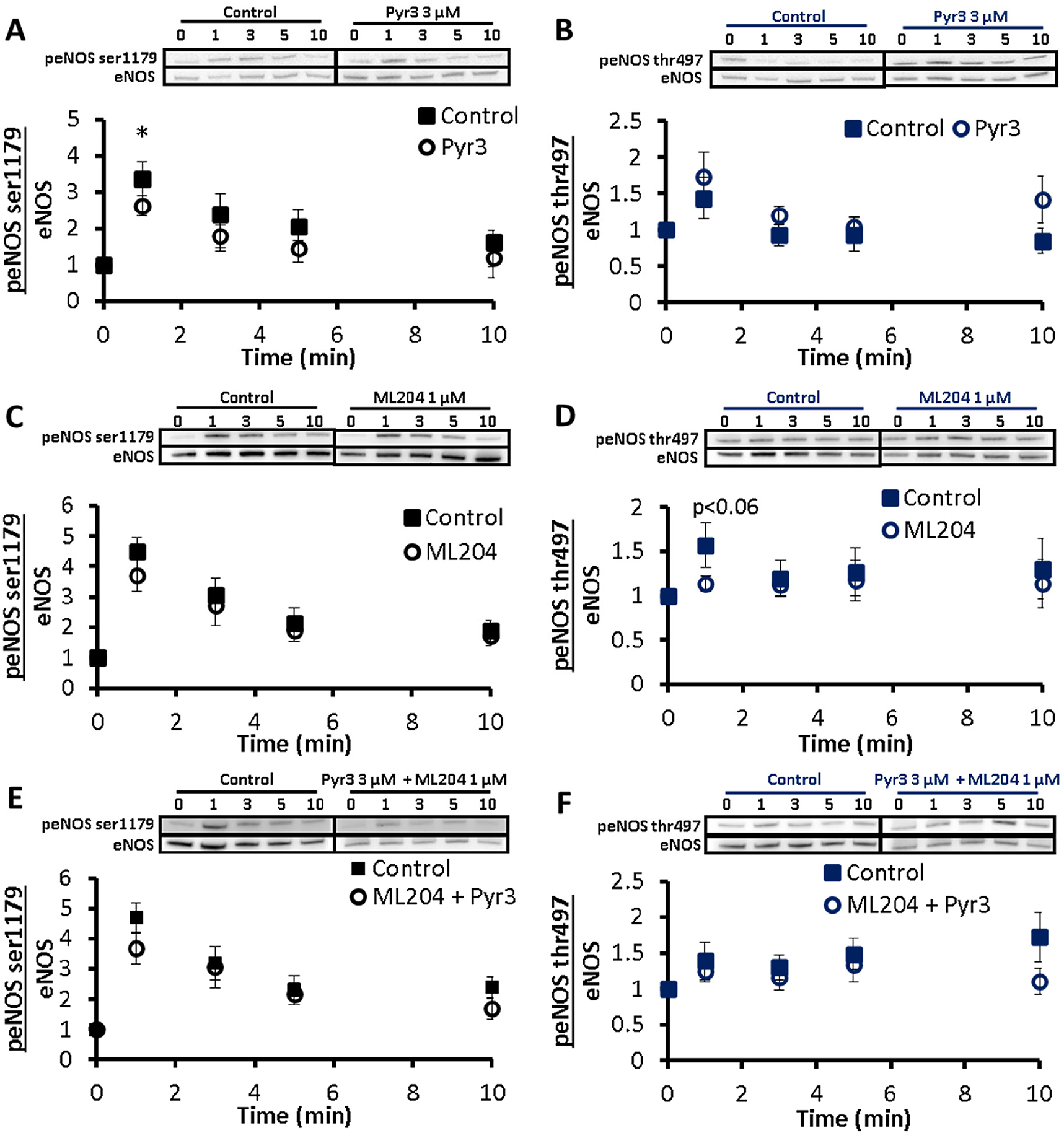
Effect of TRPC3 & TRPC4 inhibition on ATP-induced eNOS activation. BAECs were preincubated with PBS for 30 min (control)-solid square, TRPC3 inhibitor Pyr3 (A,B), TRPC4 inhibitor ML204 (C,D), or a combination of Pyr3 + ML204 (E,F) – open circle. Cells were stimulated with 100 μM ATP for 0 (no ATP),1,3,5 and 10 min and assessed via western blotting. A,C,E: Exemplar western blot and p-eNOS ser1179 normalized to eNOS. B,D,F: Exemplar western blot and p-eNOS thr497 normalized to eNOS. All peNOS/eNOS ratios are normalized to t = 0. Results are reported as mean ± S.E.M., *p < 0.05, Control & Pyr3 n = 6, 7, Control & ML204 n = 6, 7, Control & Pyr3 + ML204 n = 6, 7.
3.2.2. TRPC1/6 and Orai1 do not mediate ATP-induced eNOS activation
TRPC1/6 inhibitor, GsMTx4 did not affect ser1179 eNOS phosphorylation (Figure 10A) whereas eNOS-thr497 phosphorylation was significantly suppressed at every measured time point compared to the respective controls Figure 10B). The largest effect on eNOS-thr497 phosphorylation was observed at 3 min with Hedge’s g = 2.18 (p = 0.04). Inhibition of Orai1 did not affect eNOS activation (Figure 10C & D). At the peak of the ser1179 phosphorylation response to ATP (1 min), PBS-treated cells showed peNOS-ser1179/eNOS of 3.78 ± 0.67 in the AnCoA4 treated group compared to 3.45 ± 0.06 under control conditions.
Figure 10.

Effect of TRPC1/6 & Orai1 inhibition on ATP-induced eNOS activation. BAECs were preincubated with PBS for 30min (control), TRPC1/6 inhibitor GsMTx4 (A,B) or Orai1 inhibitor AnCoA4 (C,D). Cells were stimulated with 100μM ATP for 0 (no ATP),1,3,5 and 10 min and assessed via western blotting. A,C: Exemplar western blot and p-eNOS ser1179 normalized to eNOS B,D: Exemplar western blot and p-eNOS thr497 normalized to eNOS. All peNOS/eNOS ratios are normalised to t=0. There was a significant suppression in ATP-induced eNOS-thr497 phosphorylation at all time points under TRPC1/6 inhibition. Results are shown as mean ± S.E.M., * p<0.05, ** p<0.01. Control & GsMTx4 n=5, 4, Control & AnCoA4 n=4, 4.
3.2.3. Inhibition of TRPC3 has a robust effect on ATP-induced eNOS phosphorylation
For the purposes of confirming these findings we employed an alternative set of TRPC subtype-specific inhibitors to confirm the roles of TRPC3 (Pyr10), TRPC4 (HC-070) and TRPC6 (larixyl acetate) in eNOS activation following ATP stimulation. As shown in Figure 11, the phosphorylation response to 1 min of ATP with calcium was significantly impacted by TRPC3 inhibition while the effect of TRPC4 and TRPC6 targeting was smaller. Pyr10 pretreatment altered relative eNOS phosphorylation by ~50%.
Figure 11.

Effect of TRPC3, TRPC4 & TRPC6 inhibition on ATP-induced eNOS activation. BAECs were preincubated with PBS for 30min (control), TRPC3 inhibitor Pyr10, TRPC4 inhibitor HC-070, or TRPC6 inhibitor, larixyl acetate. Cells were stimulated with ATP for 0 (no ATP) or 1min and assessed via western blotting (A). peNOS ser1179 and peNOS thr497 are normalized to eNOS. All peNOS/eNOS ratios are normalised to t=0 (B). Results are reported as mean ± S.E.M.,*** p<0.001, Control n= 13,12, Pyr10 n= 6,6, HC-070 n=10,9, larixyl acetate n=10,7.
3.3. Flow-stimulated calcium signaling
We compared the BAEC NO response to agonist stimulation with the mechanically-evoked NO response by shearing cells at 10dyn/cm2 for 3 min. Although we observed a reduction in the peak of the calcium transient under channel inhibition there was no significant effect associated with any TRPC channel subtype or orai1 (Figure 12A).
Figure 12.

Flow-induced calcium and nitric oxide readings. BAECs were treated for 30 min with PBS with calcium (control)-black (B-F), (B) Pyr3, (C) ML204, (D) Pyr3 + ML204, (E) GsMTX4 or (F) AnCoA4 -grey then exposed to 3 min of 10dyn/cm2 shear stress. A: Graph depicts relative calcium fluorescence (F/F0) at the peak of the calcium transient. B-F: Traces represent readings from single experiment, with 10s of baseline readings before onset of flow. Bars represent mean ± S.E.M., ns., n=4.
3.4. Regulation of flow-induced eNOS activation by TRPCs and Orai1
BAECs were exposed to 10 dyn/cm2 of shear stress for 3 min and the effect of channel inhibition on eNOS examined. Representative flow-induced NO readings are shown in Figure 12. Cells were treated for 30 min with PBS (control), 3μM Pyr3 (Figure 12A), 1μM ML204 (Figure 12B), Pyr3 + ML204 (Figure 12C), 100nM GsMTX4 (Figure 12D), or 10μM AnCoA4 (Figure 12E). NO response curves depict the initial decrease in NO concentration following the onset of flow followed by an increase in NO production, a phenomenon arising from transport effects as described by our group [2]. It is important to note from Figure 12 that, temporally, the peak occurs between 1 min and 3 min with the NO signal decreasing slightly at 3 min as it approaches a plateau under continued flow.
3.4.1. Flow-stimulated eNOS activation is mediated by TRPC3- & TRPC4 – derived calcium
Following TRPC3 and TRPC4 inhibition (Figure 13A), the mean change in peak NO concentration was reduced by 16% and 30% respectively. Similarly, TRPC3 inhibitor Pyr3 produced a less than 20% reduction in NO concentration at 1 and 3 min of flow (n.s.) and pretreatment with TRPC4 inhibitor produced a halving of the NO signal at both 1 and 3 min (n.s., p= 0.0727 at 3 min). When TRPC3 and TRPC4 inhibitors were applied concurrently, a marked reduction (p< 0.05) in Δ[NO] in comparison to the control group was observed at the peak and at 3 min of flow. This dual inhibition produced a larger and statistically significant impairment in the flow-evoked NO signal compared to TRPC4 inhibition alone at the 1 min mark. The effect of combined inhibition was large at all time points (Hedge’s g> 0.9). The largest effects of dual Pyr3 & ML204 combination on Δ[NO] were compared to Pyr3 at 3 min (g= 1.29) after flow-onset and with ML204 at 1 min (g= 1.79).
Figure 13.

Flow-induced NO production and eNOS phosphorylation. BAECs were treated for 30 min with PBS (control), Pyr3, ML204, Pyr3 + ML204, GsMTX4 or AnCoA4 before temperature equilibration in the flow chamber and onset of 3min of 10 dyn/cm2 shear. Cells were lysed and eNOS phosphorylation assessed by western blotting. A: Bar graph showing peak change in NO concentration and Δ[NO] following 1 min and 3 min of flow. Combined Pyr3 + ML204 inhibition significantly suppressed NO production and produced further reduction in Δ[NO] than application of TRPC4 inhibitor alone. B: Western blots of eNOS phosphorylation following calcium inhibition under static and flow conditions. C: peNOS ser1179/eNOS in static and sheared cells. GsMTx4 pretreatment significantly reduced flow induced eNOS-ser1179 phosphorylation compared to static group. D: peNOS thr497/eNOS in static and sheared cells. Pyr3 and combined Pyr3 + ML204 pretreatment significantly increased eNOS-thr497 phosphorylation in sheared cells compared to static group. Results are shown as mean ± S.E.M., * p<0.05, ** p<0.01, Flow: Control n=9, Pyr3 n=9, ML204 n=7, Pyr3 & ML204 n=7, GxMTx4 n=6, AnCoA4 n=7, phosphorylation: static & flow n=4,5.
3.4.2. TRPC1/6 and Orai1 do not mediate flow-evoked NO production
Pre-treatment with GsMTx4 produced a 22% decrease in peak NO concentration along with a 24% increase in both the 1 and 3 min mean Δ[NO] readings. Application of AnCoA4 produced its largest effect on the NO signal at 1 min of flow, producing a 31% lower NO level compared to the control group. Neither treatment produced a significant effect on the flow-induced NO production (Figure 13A).
3.4.3. TRPC and Orai1 modulation of flow-induced eNOS phosphorylation
We proceeded to examine the phosphorylation of eNOS in the sheared cells compared to their static counterparts by western blotting. Under static conditions, there was no significant difference in eNOS-ser1179 or eNOS-thr497 phosphorylation following drug treatment compared to the control group. We found significant suppression of eNOS-ser1179 phosphorylation in the TRPC1/6 inhibitor treated cells in response to flow and a marked, but non-significant difference between the PBS and AnCoA4 pre-treated groups under static conditions (Figure 13C). When we probed for eNOS-thr497 phosphorylation, we observed a significant increase in the peNOS-thr497/eNOS signal following flow as compared to the static condition in the cases of TRPC3 inhibition and simultaneous TRPC3 & 4 inhibition (Figure 13D). On the other hand, GsMTx4 preincubation reduced the flow-evoked peNOS thr497/eNOS signal compared to the static condition (n.s., p=0.0886). This drug also produced a 20% reduction in flow-induced eNOS-thr497 phosphorylation compared to the PBS pre-treated control flow condition (n.s., p=0.066).
4. Discussion
4.1. Role of TRPC3 and TRPC4 channels in ATP-initiated eNOS activation
The importance of ATP and SOCE in transducing shear-induced eNOS activation has been confirmed by studies showing functional dependence of shear-induced NO production on ATP production [3] and by imaging studies which show colocalisation of shear-induced ATP-sparks with calcium waves [28]. To identify the molecular participants in the shear-ATP-SOCE eNOS activation pathway, we first probed the effect of isotype-wise interference with TRPCs on ATP-induced calcium entry.
We found that both TRPC3 and TRPC4 were involved in ATP-induced calcium handling in BAECs. However, inhibition of TRPC3 produces a more pronounced effect in the plateau phase of the calcium transient compared to TRPC4 inhibition. Combined TRPC3 & 4 inhibition afforded no additional suppression of calcium signaling, although the suppression of the sustained calcium response seen with Pyr3 was partially preserved under combined inhibition. These data are consistent with literature reports that TRPC3 is important for the sustained calcium response in endothelial cells [12, 29]. This role for TRPC3 in ATP-evoked calcium handling was confirmed using Pyr10, another TRPC3-specific inhibitor. As with the Pyr3 and ML204, inhibition of TRPC4 using HC-070 reduced peak calcium (non-significantly) to the same extent as TRPC3 inhibition with Pyr10 but had a lesser effect at 1 and 3min.
Considering our previous finding that ATP-induced eNOS activation is strongly store-dependent, we proceeded to identify which of these TRPC channels may be store-operated in this system. The store-dependence of TRPC channels is controversial. Some studies suggest that TPRC1/4/5 are store operated and TRPC3/6/7 are DAG-dependent [16, 29]. However, recent evidence indicates that TRPC4 can be DAG-activated [30] which would make TRPC4 responsive to agonists. The suppression of ATP-induced calcium entry we observed following TRPC4 inhibition is likely due to TRPC4 behaving in a receptor-activated mode in BAECs since TRPC4 inhibition failed to inhibit marked Tg-stimulated SOCE. Disagreement also exists regarding the activation of TRPC3 channels. Orai1 has been shown to modulate TRPC3/6/7 by direct binding which confers calcium store-status dependence [20]. We observed no effect on calcium signaling following Orai1 inhibition which suggests that the store-operated behaviour of TRPC3 we report is not Orai1-mediated, although this does not discount the possibility that STIM affords store-operated behaviour on TRPC3 directly, as seen elsewhere with SOCE mediated by STIM1/TRPC1/4 complexes [31]. Work by Trebak and colleagues pointed to TRPC3 acting in both store-operated, and receptor-activated modes[32]. The variety of discrepant findings regarding TRPC regulation is likely due to differential cell-type and species-specific expression of the channels and their differential sensitivity to inhibitors and stimuli as well as the large number of permutations of heteromultimeric channels. Our data strongly supports TRPC3 being store operated in our cells. Inhibition of TRPC3 markedly inhibited the sustained phase of the calcium response to ATP stimulation. Furthermore, the calcium response to the stimulation of SOCE by Tg treatment was blocked by TRPC3 inhibition.
Given that TRPC3 is store-operated in this system, there is a loss of the store-operated phase of the calcium response which occurs when TRPC3-inhibited BAECs are stimulated with ATP. The calcium fluorescence curve thus shifts to the left. This shift is reflected in the reduction in the time to peak fluorescence that occurs following TRPC3 inhibition of ATP-induced calcium entry. The shorter time to peak is indicative of an ER store release dominated signal in the absence of the accompanying SOCE.
Next, we wanted to determine if the effect of perturbed TRPC3 and TRPC4 calcium fluxes was propagated to eNOS phosphorylation. Neither the TRPC3 nor the TRPC4 inhibitors altered the level of constitutive eNOS phosphorylation at the two residues we probed indicating that all the fluctuations in eNOS phosphorylation observed with the varying drugs are not due to changes in baseline phosphorylation. TRPC3 inhibition, which produced a sustained diminution of the ATP-induced calcium signal also significantly reduced ATP-stimulated eNOS-ser1179 phosphorylation at 1 min (Hedge’s g> 0.5) while it resulted in non-significant, but large (g> 0.8) impairment of ser1179 phosphorylation at 3,5 and 10 min of ATP stimulation. This effect was even more pronounced with TRPC3 inhibitor Pyr10. We observed concomitant increase in eNOS-thr497 phosphorylation. In ML204 treated cells, there was a large effect (g=0.93) on eNOS-thr497 after 1 min ATP stimulation. This response would indicate an activating influence on eNOS, but it was not statistically significant at the α=0.05 level.
It is interesting to consider the relationship between the effects of TRPC3 and TRPC4 inhibition (individually or in combination) on the characteristics of the calcium response to ATP stimulation and the resultant eNOS phosphorylation. While inhibition of TRPC3 and TRPC4 both reduced the calcium response, the temporal dynamics of the responses were different. Furthermore, combined inhibition did not have an additive effect but rather produced a response that shares features of the individual responses during different phases of the response. Importantly, the profound inhibition of the sustained calcium influx by TRPC3 inhibition was not seen in the case of combined inhibition. It is noteworthy, then, that only singly inhibiting TRPC3 produced a significant decrease in eNOS activation via ser1179 phosphorylation. Taken together, these data indicate that TRPC3 is a key molecular player in ATP-initiated eNOS activation as summarised in Figure 14. We postulate that eNOS discriminates between calcium entering from TRPC3 & 4 channels, possibly as a result of TRPC3 being store-operated, a characteristic which we have previously shown to promote eNOS activity [3].
Figure 14.

Summary of the flow- and ATP-induced eNOS activation pathway. Shear stress induces autocrine ATP release. ATP activates PLC via G-protein receptors allowing PLC to hydrolyze PIP2 to form DAG and IP3. IP3 diffuses and binds to receptors on the endoplasmic reticulum (ER) causing store depletion which activates the SOC TRPC3 and (putatively) Orai1. DAG promotes opening of store-independent TRPC4s. There is a combined effect of TRPC3 and TRPC4 activation. The increase in intracellular calcium allows for Ca2+/Calmodulin-mediated eNOS activation which leads to NO production regulated by TRPC3- and TRPC4- mediated calcium entry. We deduced that ATP-independent recruitment of TRPC4 occurs in response to flow. The participation of Orai1 in this pathway is unclear (grey dotted line).
4.2. Role of TRPC3 and TRPC4 channels in flow-initiated eNOS activation
TRPC3 inhibition produced a slight reduction in NO synthesis (g = 1.27) which was accompanied by a concomitant increase in eNOS-thr497 phosphorylation, while TRPC4 inhibition produced a larger suppression of NO synthesis (g = 1.79). Freichel and colleagues [33] observed a similar impairment of agonist-induced relaxation when they silenced TRPC4 in Mouse aortic cells. We observed that combined TRPC3 and TRPC4 inhibition produced a significant reduction in Δ[NO] at 3 min of flow. There was also a marked increase in flow-induced eNOS-thr497 phosphorylation under combined TRPC 3&4 inhibition compared to the control at this time point. This dual inhibition resulted in significantly lower NO production than TRPC4 inhibition at 1 min of flow indicating a compounding of NO suppression. On the backdrop of reports proposing different mechanisms of TRPC3/6/7 vs TRPC1/4/5 regulation, we can thus postulate that the contributions of TRPC3 and 4 to intracellular calcium both regulate eNOS activity. There is a combined effect produced by inhibiting both TRPC subfamilies, however, it is not necessarily additive. Dual inhibition did not increase inhibition of ATP-induced eNOS-ser1179 phosphorylation, but it did increase inhibition of NO production, which suggest that additional pathways are evoked by shear in addition to the ATP-induced signalling cascade.
Given that TRPC4 is not widely reported to be mechanosensitive, it is apparent that another significant pathway is initiated by mechanical stimulation of endothelial cells that acts outside the ATP-SOCE pathway to activate eNOS via TRPC3 & 4-derived calcium fluxes (Figure 14). This idea is supported by the limited perturbation of flow-invoked calcium signaling by inhibition of all the target channels. Wang and colleagues have shown that flow activates P2Y 2 via piezo1 and produces eNOS-ser1179 phosphorylation via Gq-mediated initiation of the src-Pi3K -AKT cascade [34]. This finding, taken with reports of the activation of TRPC4 via Gq signaling [30], may reconcile the evidence shown here for differential TRPC-mediated activation of eNOS by ATP and flow. We demonstrate that the contribution of TRPC4-mediated calcium to flow-instigated eNOS activation is not solely dependent on the ATP-SOCE pathway and is consistent with the engagement of TRPC4 via additional pathways.
The previously reported pattern of eNOS activation by ser1179 phosphorylation / thr497 dephosphorylation in tandem [6, 35] occurred in response to flow. Despite the marked disruption in NO production under combined TRPC3 & TRPC4 activation, we observed only a slight alteration in eNOS phosphorylation at the ser1179 residue but a marked increase in thr497 phosphorylation that could explain the reduction in NO production. Similarly, in a recent study in preconstricted mouse arteries, flow did not produce changes in eNOS-thr495 or -ser1179 phosphorylation [36]. It is therefore evident that the pattern of eNOS phosphorylation is dependent on the particular stimulation conditions.
ATP-stimulation produced large changes in eNOS-ser1179 phosphorylation with a lesser change in eNOS-thr497 phosphorylation in most of our study conditions. On the other hand, in response to flow, we observed large changes in eNOS-thr497 phosphorylation compared to small changes in ser1179. We previously reported that ATP autocrine stimulation is a component of the flow response[3], but in this study the pattern of phosphorylation changes produced by flow do not match those from ATP-stimulation alone. These data are supported by literature reports of differential ser1179/thr497 phosphorylation and dephosphorylation occurring with chemical vs mechanical stimulation [37, 38] of endothelial cells and in the case of basal and stimulated NO production and based on duration of the stimulus as evinced by phospho-proteomic analysis [39]. Jagnadan and colleagues [40] hypothesised that the dysfunctional eNOS produced by the molecule’s mislocalization within the cell was a result of its inability to access calcium calmodulin. Others have described a framework in which different agonists [6, 41, 42] and the intracellular localisation of eNOS [35, 43, 44] are tied to differential phosphorylation of eNOS at various residues (in addition to other post-translational modifications). These reports, along with the data we report here on eNOS activation, support the idea that eNOS discriminates between calcium fluxes of equal amplitude from different channels and encodes these differences in the pattern of phosphorylation.
4.3. Role of TRPC1/6 in eNOS activation
Interestingly, although application of GsMTx4, a TRPC1/6 gating modifier [45, 46] to our cells did not affect the size of the calcium response, it did reduce the time to peak. We do not, however, believe that this perturbation in the calcium response affected eNOS phosphorylation as there was no change in eNOS-ser1179 phosphorylation (which is calcium-dependent) at the earliest time point we assessed phosphorylation (1 min). There was marked dysregulation of the canonical ser1179 and thr497 reciprocal phosphorylation/dephosphorylation in response to ATP-stimulation following pretreatment with the TRPC1/6 inhibitor. We found a consistent and striking reduction in eNOS-thr497 phosphorylation compared to controls in response to agonist at all time points, without a discernible effect on eNOS-ser1179. This response suggests that application of GsMTx4 increased eNOS activity; however, in isolation, without an accompanying alteration in calcium flux, this seems unlikely.
The observed disruption of ser1179/thr497 dual phosphorylation/dephosphorylation suggests disruption of kinase/phosphatase signaling targeting the thr497 eNOS residue [47, 48]. TRPC1 has been shown to bind the cav1 scaffolding domain which binds eNOS [49]. Application of GsMTx4 may interfere with eNOS thr497 phosphorylation by interrupting the caveolin scaffolding domain. It is therefore possible that GsMTx4, a tarantula venom toxin, has secondary effects on eNOS in this system, independent of its effect on calcium entry.
The GsMTx4 peptide acts by partitioning into the membrane and interacting with lipids[50]. Since eNOS activity is regulated by membrane localisation [44] and membrane lipids [51], we therefore surmise that in this system the effect of GsMTx4 on membrane lipids, outweighs the limited influence of TRPC1&6 on ATP-induced calcium entry. Furthermore, our confirmatory investigation into the role of TRPC6 in this pathway using larixyl acetate indicated little to no participation of this isotype in calcium handling.
Although GsMTx4 did not produce a large effect on calcium signaling or NO production under flow, it did significantly decrease eNOS-ser1179 phosphorylation while also lowering thr497 phosphorylation non-significantly. These results point to concurrent deactivation and activation of eNOS. This dichotomy is another instance of the atypical ser/thr phosphorylation observed under GsMTx4 treatment and supports our hypothesis of secondary effects of this drug beyond the scope of this study.
Suppression of eNOS-ser1179 phosphorylation occurred in cells exposed to flow under GsMTx4 treatment. This suggests that the introduction of the mechano-transductive component of eNOS activation which was brought on by shearing the cells and absent in ATP experiments, may recruit ser1179 to the regulation of eNOS activity. Indeed, GsMTx4 has been found to block both mechanosensitive and agonist-evoked TRPC6 activity [52]. It is therefore evident, as in the case of TRPC4 inhibition under flow, that shearing cells initiates signaling pathways which may recruit the TRPC1/6 channel but do not act entirely through the ATP-SOCE pathway. This differential involvement of TRPC channels by agonist stimulation as compared to flow is supported by the almost uniform inhibition of flow-induced calcium entry by inhibition of TRPC1,3,4, and 6, as opposed to varying participation of these channels in ATP-induced calcium entry when inhibited with same drugs.
4.4. Role of Orai1 in flow and agonist-initiated eNOS activation
Orai1 inhibition did not produce an observable difference in the agonist-operated calcium influx, which is at odds with studies reporting that Orai1 is a key SOC [12, 53]. Our data is, however, consistent with the influence of STIM1/Orai1 on signaling being exerted, not through Orai1 calcium fluxes per se, but via its modulation of TRPCs in our system. It is also possible that orai2 and/or orai3 are the main SOC in this pathway, however others have shown that they usually operate as SOCs in heteromultimeirc channels with orai1 [54].
Although Orai1 inhibition did not reduce ATP-induced calcium entry, it precipitated a small reduction in flow-induced calcium signaling and 31% reduction in flow-induced NO production at 1 min which was a medium effect (g=0.66). This discrepancy affirms the participation of a mechano-transductive component to eNOS activation which is not mediated by ATP (Figure 14).
Conclusions
This body of work identifies some of the key molecular players in calcium-mediated flow and ATP-evoked eNOS activation and subsequent NO production. TRPC3 exhibited store-dependant characteristics which is important for flow-stimulated, ATP-mediated eNOS activation. TRPC4 was not explicitly store-operated in this system. TRPC3 inhibition significantly attenuated ATP-induced eNOS activation. Although both TRPC3 and TRPC4 inhibition individually lowered NO synthesis under flow, it was the effect of dual TRPC3 and TRPC4 disruption that resulted in a marked and statistically significant suppression of NO production. The influence of TRPC4 on flow-stimulated eNOS activation was absent in response to agonist challenge. This suggests that TRPC4-mediated calcium entry is initiated by mechanisms beyond the ATP-SOCE pathway which is the focus of this work.
An interesting observation in this study was the limited effect on endothelial calcium signaling and eNOS activity produced by interference with Orai1 compared to some reports which implicate it as the main SOC in endothelial cells. These data suggest a framework for the differential activation of eNOS by the TRPC1,4,5 and TRPC3,6,7 subfamilies with a limited role for Orai1 in response to vasodilatory inputs. Specifically, flow recruited channels from both TRPC subfamilies while ATP-stimulation regulates eNOS mainly via TRPC3-mediated calcium. Further inquiry is required to unravel the intricacies of calcium-handling in the regulation of eNOS activity. Notwithstanding, our findings highlight the need for channel subtype-specific drug design to ameliorate the reduction of bio-available NO, which occurs in the early stages of cardiovascular disease.
Acknowledgements
We thank Dr Peter Davies at University of Pennsylvania for supplying the bovine aortic endothelial cells.
Funding
This work was supported by the National Heart, Lung and Blood Institute Grant: U01HL116256
Abbreviations
- NO
Nitric Oxide
- eNOS
Endothelia nitric oxide synthase
- ER
Endoplasmic reticulum
- PBS
Dulbecco’s phosphate buffered saline
- PLC
Phospholipase C
- BAEC
Bovine aortic endothelial cells
- SOCE
Store-operated calcium entry
- DAG
Diacylglycerol
- IP3
Inositol triphosphate
- PIP2
P phosphatidylinositol 4,5-bisphosphate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bredt DS, Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic Res, 2000. 31(6): p. 577–96. [DOI] [PubMed] [Google Scholar]
- 2.Andrews AM, et al. , Direct, real-time measurement of shear stress-induced nitric oxide produced from endothelial cells in vitro. Nitric Oxide, 2010. 23(4): p. 335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews AM, et al. , Shear stress-induced NO production is dependent on ATP autocrine signaling and capacitative calcium entry. Cell Mol Bioeng, 2014. 7(4): p. 510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dudzinski DM and Michel T, Life history of eNOS: Partners and pathways. Cardiovascular Research, 2007. 75(2): p. 247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fleming I, Molecular mechanisms underlying the activation of eNOS. Pflugers Arch, 2010. 459(6): p. 793–806. [DOI] [PubMed] [Google Scholar]
- 6.Fleming I, et al. , Phosphorylation of Thr495 Regulates Ca2+/Calmodulin-Dependent Endothelial Nitric Oxide Synthase Activity. Circ Res, 2001. 88(11): p. e68–e75. [DOI] [PubMed] [Google Scholar]
- 7.Tiruppathi C, et al. , Role of Ca2+ signaling in the regulation of endothelial permeability. Vascular pharmacology, 2002. 39(4–5): p. 173–185. [DOI] [PubMed] [Google Scholar]
- 8.Groschner K, et al. , Trp proteins form store-operated cation channels in human vascular endothelial cells. FEBS Letters, 1998. 437(1): p. 101–106. [DOI] [PubMed] [Google Scholar]
- 9.Ambudkar IS, et al. , Functional organization of TRPC-Ca2+ channels and regulation of calcium microdomains. Cell Calcium, 2006. 40(5–6): p. 495–504. [DOI] [PubMed] [Google Scholar]
- 10.Ahmmed GU, et al. , Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem, 2004. 279(20): p. 20941–9. [DOI] [PubMed] [Google Scholar]
- 11.Antoniotti S, et al. , Interaction between TRPC channel subunits in endothelial cells. J Recept Signal Transduct Res, 2006. 26(4): p. 225–40. [DOI] [PubMed] [Google Scholar]
- 12.Antigny F, et al. , Thapsigargin activates Ca2+ entry both by store-dependent, STIM1/Orai1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium, 2011. 49(2): p. 115–127. [DOI] [PubMed] [Google Scholar]
- 13.Brownlow SL and Sage SO, Transient receptor potential protein subunit assembly and membrane distribution in human platelets. Thromb Haemost, 2005. 94(4): p. 839–45. [DOI] [PubMed] [Google Scholar]
- 14.Adebiyi A, Narayanan D, and Jaggar JH, Caveolin-1 assembles type 1 inositol 1,4,5-trisphosphate receptors and canonical transient receptor potential 3 channels into a functional signaling complex in arterial smooth muscle cells. J Biol Chem, 2011. 286(6): p. 4341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murata T, et al. , Genetic evidence supporting caveolae microdomain regulation of calcium entry in endothelial cells. J Biol Chem, 2007. 282(22): p. 16631–43. [DOI] [PubMed] [Google Scholar]
- 16.Hofmann T, et al. , Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature, 1999. 397(6716): p. 259–63. [DOI] [PubMed] [Google Scholar]
- 17.Pani B and Singh BB, Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium, 2009. 45(6): p. 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clapham DE, Calcium signaling. Cell, 2007. 131(6): p. 1047–58. [DOI] [PubMed] [Google Scholar]
- 19.Saul S, et al. , How ORAI and TRP channels interfere with each other: interaction models and examples from the immune system and the skin. Eur J Pharmacol, 2014. 739: p. 49–59. [DOI] [PubMed] [Google Scholar]
- 20.Liao Y, et al. , Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci U S A, 2007. 104(11): p. 4682–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiyonaka S, et al. , Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proceedings of the National Academy of Sciences, 2009. 106(13): p. 5400–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang J-H, et al. , TRPC3 channel contributes to nitric oxide release: significance during normoxia and hypoxia–reoxygenation. Cardiovascular Research, 2011. 91(3): p. 472–482. [DOI] [PubMed] [Google Scholar]
- 23.Miller M, et al. , Identification of ML204, a Novel Potent Antagonist That Selectively Modulates Native TRPC4/C5 Ion Channels. The Journal of biological chemistry, 2011. 286: p. 33436–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadaghiani Amir M., et al. , Identification of Orai1 Channel Inhibitors by Using Minimal Functional Domains to Screen Small Molecule Microarrays. Chemistry & Biology, 2014. 21(10): p. 1278–1292. [DOI] [PubMed] [Google Scholar]
- 25.Schleifer H, et al. , Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca(2+) entry pathways. Br J Pharmacol, 2012. 167(8): p. 1712–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Just S, et al. , Treatment with HC-070, a potent inhibitor of TRPC4 and TRPC5, leads to anxiolytic and antidepressant effects in mice. PloS one, 2018. 13(1): p. e0191225–e0191225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urban N, et al. , Identification and Validation of Larixyl Acetate as a Potent TRPC6 Inhibitor. Molecular Pharmacology, 2016. 89(1): p. 197. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto K, et al. , Visualization of flow-induced ATP release and triggering of Ca2+ waves at caveolae in vascular endothelial cells. J Cell Sci, 2011. 124(Pt 20): p. 3477–83. [DOI] [PubMed] [Google Scholar]
- 29.Nilius B, Droogmans G, and Wondergem R, Transient receptor potential channels in endothelium: solving the calcium entry puzzle? Endothelium, 2003. 10(1): p. 5–15. [DOI] [PubMed] [Google Scholar]
- 30.Storch U, et al. , Dynamic NHERF interaction with TRPC4/5 proteins is required for channel gating by diacylglycerol. Proc Natl Acad Sci U S A, 2017. 114(1): p. E37–e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sundivakkam PC, et al. , The Ca(2+) sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca(2+) entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol Pharmacol, 2012. 81(4): p. 510–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trebak M, et al. , Comparison of human TRPC3 channels in receptor-activated and store-operated modes. Differential sensitivity to channel blockers suggests fundamental differences in channel composition. J Biol Chem, 2002. 277(24): p. 21617–23. [DOI] [PubMed] [Google Scholar]
- 33.Freichel M, et al. , Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nature Cell Biology, 2001. 3: p. 121. [DOI] [PubMed] [Google Scholar]
- 34.Wang S, et al. , P2Y2 and Gq/G11 control blood pressure by mediating endothelial mechanotransduction. The Journal of Clinical Investigation, 2015. 125(8): p. 3077–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin MI, et al. , Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J Biol Chem, 2003. 278(45): p. 44719–26. [DOI] [PubMed] [Google Scholar]
- 36.Looft-Wilson RC, et al. , Flow does not alter eNOS phosphoryation at Ser1179 or Thr495 in preconstricted mouse mesenteric arteries. Physiol Rep, 2018. 6(17): p. e13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallis B, et al. , Identification of Flow-dependent Endothelial Nitric-oxide Synthase Phosphorylation Sites by Mass Spectrometry and Regulation of Phosphorylation and Nitric Oxide Production by the Phosphatidylinositol 3-Kinase Inhibitor LY294002. Journal of Biological Chemistry, 1999. 274(42): p. 30101–30108. [DOI] [PubMed] [Google Scholar]
- 38.Sessa WC, eNOS at a glance. Journal of Cell Science, 2004. 117(12): p. 2427–2429. [DOI] [PubMed] [Google Scholar]
- 39.Iring A, et al. , Shear stress–induced endothelial adrenomedullin signaling regulates vascular tone and blood pressure. The Journal of Clinical Investigation, 2019. 129(7): p. 2775–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jagnandan D, Sessa WC, and Fulton D, Intracellular location regulates calcium-calmodulin-dependent activation of organelle-restricted eNOS. Am J Physiol Cell Physiol, 2005. 289(4): p. C1024–33. [DOI] [PubMed] [Google Scholar]
- 41.Michell BJ, et al. , Identification of regulatory sites of phosphorylation of the bovine endothelial nitric-oxide synthase at serine 617 and serine 635. J Biol Chem, 2002. 277(44): p. 42344–51. [DOI] [PubMed] [Google Scholar]
- 42.Harris MB, et al. , Reciprocal Phosphorylation and Regulation of Endothelial Nitric-oxide Synthase in Response to Bradykinin Stimulation. Journal of Biological Chemistry, 2001. 276(19): p. 16587–16591. [DOI] [PubMed] [Google Scholar]
- 43.Garcia-Cardena G, et al. , Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem, 1997. 272(41): p. 25437–40. [DOI] [PubMed] [Google Scholar]
- 44.Fulton D, et al. , Localization of Endothelial Nitric-oxide Synthase Phosphorylated on Serine 1179 and Nitric Oxide in Golgi and Plasma Membrane Defines the Existence of Two Pools of Active Enzyme. Journal of Biological Chemistry, 2002. 277(6): p. 4277–4284. [DOI] [PubMed] [Google Scholar]
- 45.Bowman CL, et al. , Mechanosensitive ion channels and the peptide inhibitor GsMTx-4: history, properties, mechanisms and pharmacology. Toxicon : official journal of the International Society on Toxinology, 2007. 49(2): p. 249–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gnanasambandam R, et al. , GsMTx4: Mechanism of Inhibiting Mechanosensitive Ion Channels. Biophys J, 2017. 112(1): p. 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michell BJ, et al. , Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J Biol Chem, 2001. 276(21): p. 17625–8. [DOI] [PubMed] [Google Scholar]
- 48.Chen ZP, et al. , AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett, 1999. 443(3): p. 285–9. [DOI] [PubMed] [Google Scholar]
- 49.Sundivakkam PC, et al. , Caveolin-1 scaffold domain interacts with TRPC1 and IP3R3 to regulate Ca2+ store release-induced Ca2+ entry in endothelial cells. Am J Physiol Cell Physiol, 2008. 296(3): p. C403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jung HJ, et al. , Lipid membrane interaction and antimicrobial activity of GsMTx-4, an inhibitor of mechanosensitive channel. Biochem Biophys Res Commun, 2006. 340(2): p. 633–8. [DOI] [PubMed] [Google Scholar]
- 51.Andrews AM, et al. , Cholesterol Enrichment Impairs Capacitative Calcium Entry, eNOS Phosphorylation & Shear Stress-Induced NO Production. Cellular and molecular bioengineering, 2017. 10(1): p. 30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spassova MA, et al. , A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proceedings of the National Academy of Sciences of the United States of America, 2006. 103(44): p. 16586–16591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abdullaev IF, et al. , Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res, 2008. 103(11): p. 1289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanwar J, Trebak M, and Motiani RK, Cardiovascular and Hemostatic Disorders: Role of STIM and Orai Proteins in Vascular Disorders. Adv Exp Med Biol, 2017. 993: p. 425–452. [DOI] [PubMed] [Google Scholar]