

Abstract
Background
Short tandem repeat (STR) expansion disorders are an important cause of human neurological disease. They have an established role in more than 40 different phenotypes including the myotonic dystrophies, Fragile X syndrome, Huntington’s disease, the hereditary cerebellar ataxias, amyotrophic lateral sclerosis and frontotemporal dementia.
Main body
STR expansions are difficult to detect and may explain unsolved diseases, as highlighted by recent findings including: the discovery of a biallelic intronic ‘AAGGG’ repeat in RFC1 as the cause of cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS); and the finding of ‘CGG’ repeat expansions in NOTCH2NLC as the cause of neuronal intranuclear inclusion disease and a range of clinical phenotypes. However, established laboratory techniques for diagnosis of repeat expansions (repeat-primed PCR and Southern blot) are cumbersome, low-throughput and poorly suited to parallel analysis of multiple gene regions. While next generation sequencing (NGS) has been increasingly used, established short-read NGS platforms (e.g., Illumina) are unable to genotype large and/or complex repeat expansions. Long-read sequencing platforms recently developed by Oxford Nanopore Technology and Pacific Biosciences promise to overcome these limitations to deliver enhanced diagnosis of repeat expansion disorders in a rapid and cost-effective fashion.
Conclusion
We anticipate that long-read sequencing will rapidly transform the detection of short tandem repeat expansion disorders for both clinical diagnosis and gene discovery.
Keywords: Tandem, Repeats, Expansion, Neurological, Clinical, Genetics, Disease, Diagnosis, Long-read, Sequencing
Introduction
A large proportion of the human genome is comprised of repetitive DNA sequences known as microsatellites or short tandem repeats (STRs). STRs are small sections of DNA, usually 2–6 nucleotides in length, that are repeated consecutively at a given locus. STRs make up at least 6.77% of the human genome and are highly polymorphic [143]. STR lengths are prone to alteration during DNA replication, due to slippage events on misaligned strands, errors in DNA repair during synthesis and formation of secondary hairpin structures [43]. As a result, STR lengths are relatively unstable, with their frequent mutation providing a source of genetic variation in human populations. STRs have a mutation rate orders of magnitude higher than single nucleotide polymorphisms (SNPs) in non-repetitive contexts [58]. Larger repeats, in general, are more unstable and have an increased propensity to expand during DNA replication.
Large STR expansions may become pathogenic, underpinning various forms of primary neurological disease. There are currently 47 known STR genes that can cause disease when expanded; 37 of these exhibit primary neurological presentations (see Table 1) while 10 present with developmental abnormalities (see Table 2). With increased interest and improving molecular techniques for detecting repeat expansions, the list of known repeat expansion disorders is growing rapidly, with new genes such as RFC1, GIPC1, LRP12, NOTCH2NLC and VWA1 recently implicated. Furthermore, STR expansions have been linked to complex polygenic diseases such as heart disease, bipolar disorder, major depressive disorder and schizophrenia [59]. Some theories also suggest STR variability may account for normal brain and behavioural traits such as anxiety, cognitive function, emotional memory and altruism [41]. Similarly, somatic instability at STR regions is a hallmark of many cancers such as Lynch syndrome-related cancers, gastric cancers, colorectal cancers and endometrial cancers [174]. In this review, we provide an overview of the primary neurological repeat expansion diseases, discuss limitations in current diagnostic methods and developments in long-read sequencing technologies that promise to improve the discovery and diagnosis of STR expansions.
Table 1.
Summary of known neurological diseases caused by short tandem repeat expansions
| Abbreviated phenotype (MIM number) | Gene | Mode of inheritance | Repeat Motif | Location on Gene | Pathogenic repeat numbera | Chromosome | Coordinates (hg38) | Clinical phenotype | References | |
|---|---|---|---|---|---|---|---|---|---|---|
|
C9-FTD C9-ALS (#10550) |
C9orf72 | AD | GGGGCC | 5’ Region | 24–4000 | chr9 | 27573485 | 27573546 | Frontotemporal dementia, amyotrophic lateral sclerosis | [32, 47, 65] |
|
CANVAS (#614575) |
RFC1 | AR |
(AAGGG)400–2000 (ACAGG)exp AAAAG (normal) |
Intron 2 | 400–2000 | chr4 | 39348425 | 39348483 | Cerebellar ataxia, neuropathy, and vestibular areflexia syndrome | [11, 28, 138] |
|
DM1 (#160900) |
DMPK | AD |
CTG (Interruptions: CCG) |
3’ Region | 50–10,000 | chr19 | 45770205 | 45770266 | Myotonic dystrophy 1 | [60, 176] |
|
DM2 (#602668) |
CNBP (ZNF9) | AD | CCTG | Intron 1 | 50–11,000 | chr3 | 129172577 | 129172656 | Myotonic dystrophy 2 | [176] |
|
DRPLA (#125370) |
ATN1 | AD | CAG | Exon 5 | 49–93 | chr12 | 6936717 | 6936775 | Dentatorubral-pallidoluysian atrophy | [78] |
|
EIEE1/XLID (#308350) (#300419) (#300215) |
ARX | XL | GCC | Exon 2 | 17–27 | chrX | 25013654 | 25013697 | Clinical spectrum of disorders including developmental and epileptic encephalopathy 1, hydranencephaly with abnormal genitalia, X-linked lissencephaly 2 and X-linked mental retardation 29 | [73, 150] |
| FAME1 (#601068) | SAMD12 | AD |
TTTCA within TTTTA repeat region |
Intron 4 | 105–3680 | chr8 | 118366813 | 118366918 | Familial adult myoclonic epilepsy 1 | [22, 68] |
|
FAME2 (#607876) |
STARD7 | AD |
ATTTC within ATTTT repeat region |
Intron 1 | 150–460 | chr2 | 96197067 | 96197124 | Familial adult myoclonic epilepsy 2 | [27] |
|
FAME3 (#613608) |
MARCHF6 | AD |
TTTCA within TTTTA repeat region |
Intron 1 | 700–1035 | chr5 | 10356339 | 10356411 | Familial adult myoclonic epilepsy 3 | [40] |
|
FAME6 (#618074) |
TNRC6A | AD |
TTTCA within TTTTA repeat region |
Intron 1 |
? (only 1 family) |
chr16 | 24613439 | 24613532 | Familial adult myoclonic epilepsy 6 | [68] |
|
FAME7 (#618075) |
RAPGEF2 | AD |
TTTCA within TTTTA repeat region |
Intron 14 |
? (only 1 family) |
chr4 | 159342527 | 159342618 | Familial adult myoclonic epilepsy 7 | [68] |
|
FRAXE (#309548) |
FMR2 (AFF2) | XLR | CCG | 5’ Region | > 200 | chrX | 148500605 | 148500753 | Mental retardation, X-linked, FRAXE type | [53] |
|
FRDA (#229300) |
FXN | AR | GAA | Intron 1 | 66–1300 | chr9 | 69037275 | 69037314 | Friedreich ataxia | [5, 19, 162] |
|
FXS (#300624) FXTAS (#300623) |
FMR1 | XL | CGG | 5’ Region |
200–3000 55–200 |
chrX | 147911979 | 147912111 |
Fragile X syndrome Fragile X tremor/ataxia syndrome, premature ovarian failure 1 |
[162] [56] |
|
HD (#143100) |
HTT | AD |
CAG (Interruptions: CAA) |
Exon 1 | 36–250 | chr4 | 3074876 | 3074941 | Huntington disease | [96, 101] |
|
HDL1 (#603218) |
PRNP | AD |
24-base octapeptide PHGGGWGQ |
Exon 2 | 8–14 | chr20 | 4699379 | 4699380 | Huntington disease-like 1 | [108] |
|
HDL2 (#606438) |
JPH3 | AD | CTG | Exon 2A | 40–59 | chr16 | 87604283 | 87604329 | Huntington disease-like 2 | [62] |
| HMN | VWA1 | AR | GGCGCGGAGC | Exon 1 | 3 | chr1 | 1435799 | 1435820 | Hereditary axonal motor neuropathy | [121] |
|
NIID (#603472) |
NOTCH2NLC | AD | CGG | 5' Region | 66–517 | chr1 | 149390803 | 149390842 | Neuronal intranuclear inclusion disease | [55, 118, 146] |
|
OPDM1 (#164310) |
LRP12 | AD | CGG | 5' Region | 90–130 | chr8 | 104588965 | 104588999 | Oculopharyngodistal myopathy | [69] |
|
OPDM2 (#618940) |
GIPC1 | AD | CGG | 5’ Region | 70–164 | chr19 | 14496029 | 14496104 | Oculopharyngodistal myopathy | [172] |
|
OPMD (#164300) |
PABPN1 | AD | GCG | Exon 1 | 7–18 | chr14 | 23321472 | 23321511 | Oculopharyngeal muscular dystrophy | [15, 129] |
|
OPML1 (#618637) |
NUTM2B-AS1 | AD | CGG | 5' Region | 16–160 | chr10 | 79826364 | 79826403 | Oculopharyngeal myopathy with leukoencephalopathy 1 | [69] |
|
SBMA (#313200) |
AR | XLR | CAG | Exon 1 | 38–68 | chrX | 67545317 | 67545419 | Spinal and bulbar muscular atrophy of Kennedy (Kennedy's disease) | [44, 82, 147] |
|
SCA1 (#164400) |
ATXN1 | AD |
CAG (Interruptions: CAT) |
Exon 8 | 39–91 | chr6 | 16327636 | 16327723 | Spinocerebellar ataxia 1 | [120, 141] |
|
SCA2 (#183090) |
ATXN2 | AD |
CAG (Interruptions: CAA, CGG, CGC) |
Exon 1 |
33–200 (29–32 increased ALS risk) |
chr12 | 111598950 | 111599019 | Spinocerebellar ataxia 2 | [18, 133, 141, 148] |
|
SCA3 (#109150) |
ATXN3 | AD | CAG | Exon 10 | 53–87 | chr14 | 92071011 | 92071052 | Spinocerebellar ataxia 3 | [74] |
|
SCA6 (183086) |
CACNA1A | AD | CAG | Exon 47 | 19–33 | chr19 | 13207858 | 13207897 | Spinocerebellar ataxia 6 | [141, 181] |
|
SCA7 (#164500) |
ATXN7 | AD | CAG | Exon 1 | 34–460 | chr3 | 63912685 | 63912716 | Spinocerebellar ataxia 7 | [18, 30] |
|
SCA8 (#608768) |
ATXN8 | AD | CAG/TAG | 3’ UTR | 74–1300 | chr13 | 70139383 | 70139428 | Spinocerebellar ataxia 8 | [79, 141, 155] |
|
SCA10 (#603516) |
ATXN10 | AD |
ATTCT (Interruptions: ATCCT) |
Intron 9 | 280–4500 | chr22 | 45795355 | 45795424 | Spinocerebellar ataxia 10 | [88, 100, 141] |
|
SCA12 (#604326) |
PPP2R2B | AD | CAG | 5’ Region | 51–78 | chr5 | 146878729 | 146878758 | Spinocerebellar ataxia 12 | [63, 94, 141] |
|
SCA17 (#607136) |
TBP | AD |
CAG (Interruptions: CAT, CAA) |
Exon 3 | 43–66 | chr6 | 170561907 | 170562017 | Spinocerebellar ataxia 17, Huntington disease-like 4 | [97, 115, 141] |
|
SCA31 (#117210) |
BEAN1 | AD |
TGGAA within TAAAA and TAGAA repeat region |
Intron/ Intergenic region |
500–760 (> 110 TGGAA repeats) |
chr16 | 66495475 | 66495509 | Spinocerebellar ataxia 31 | [134] |
|
SCA36 (#614153) |
NOP56 | AD | GGCCTG | Intron 1 | 650–2500 | chr20 | 2652733 | 2652775 | Spinocerebellar ataxia 36 | [77] |
|
SCA37 (#615945) |
DAB1 | AD |
ATTTC within (ATTTT)7–400 repeat region |
5’ Region | 31–75 | chr1 | 57367044 | 57367125 | Spinocerebellar ataxia 37 | [139] |
|
ULD (#254800) |
CSTB | AR | CCCCGCCCCGCG |
Upstream 5’ UTR |
30–125 | chr21 | 43776444 | 43776479 | Progressive myoclonic epilepsy 1A (Unverricht and Lundborg disease) | [87, 91] |
ALS, amyotrophic lateral sclerosis; AS, antisense RNA; CANVAS, cerebellar ataxia neuropathy and vestibular areflexia syndrome; DM1; myotonic dystrophy 1; DM2; myotonic dystrophy 2; DRPLA, dentatorubral-pallidoluysian atrophy; EIEE1, early infantile epileptic encephalopathy 1; FAME, familial adult myoclonic epilepsy; FRAXE, fragile-XE syndrome; FRDA, Friedreich’s ataxia; FTD, frontotemporal dementia; FXS, fragile-X syndrome; FXTAS, fragile-x tremor/ataxia syndrome; HMN, hereditary motor neuropathy; HD, Huntington’s disease; HDL2, Huntington disease-like 2; HDL1, Huntington disease-like 1; LMN, lower motor neuron; NIID, neuronal intranuclear inclusion disease; OPDM, oculopharyngodistal myopathy; OPMD, oculopharyngeal muscular dystrophy; OPML, oculopharyngeal myopathy with leukoencephalopathy; SBMA, spinal and bulbar muscular atrophy; SCA, spinocerebellar ataxia; ULD, Unverricht-Lundborg disease; UMN, upper motor neuron; XLID, x-linked intellectual disability;
aThese ranges vary between studies and often the upper limit is unknown. It is important to note that these are only potentially pathogenic. There is a small (< 1%) subsection of the healthy control population who have expanded alleles with no clinical manifestations. Similarly, there are alleles lower than the given range who may have intermediate alleles and premutation syndromes
Table 2.
Summary of known congenital and developmental disorders caused by short tandem repeat expansions.
Adapted from Khristich and Mirkin [76]
| Phenotype (OMIM #) | Gene | Motif | Pathogenic repeat number | Location | (hg38) | References | ||
|---|---|---|---|---|---|---|---|---|
|
BPES (#110100) |
FOXL2 | GCG | 22–24 | Exon | chr3 | 138946022 | 138946062 | [116] |
|
CCHS (#209880) |
PHOX2B | GCG | 24–33 | Exon | chr4 | 41745976 | 41746022 | [7] |
|
DBQD2 (#615777) |
XYLT1 | GGC | 100–800 | 5’ Region | chr16 | 17470869 | 17470967 | [86] |
|
FECD3 (#613267) |
TCF4 | TGC | > 50 | Intron | chr18a | 55222184a | 55635956a | [167] |
|
GDPAG (#618412) |
GLS | GCA | > 300 | 5’ Region | chr2 | 190880873 | 190880920 | [159] |
|
HFG (#140000) |
HOXA13 | GCG | 24–26 | Exon | chr7 | 27199827 | 27199967 | [50] |
|
HPE5 (#609637) |
ZIC2 | GCG | 25 | Exon | chr13 | 99985449 | 99985494 | [17] |
|
HSAN8 (#616488) |
PRDM12 | GCG | 18–19 | Exon | chr9 | 130681606 | 130681641 | [23] |
|
SPD1 (#186000) |
HOXD13 | GCG | 22–29 | Exon | chr2 | 176093058 | 176093099 | [2] |
|
XLMR (#300123) |
SOX3 | GCG | 15–26 | Exon | chr3 | 181712415 | 181712456 | [89] |
BPES, blepharophimosis, epicanthus inversus, and ptosis; CCHS, congenital central hypoventilation syndrome; DBQD2, Desbuquois dysplasia 2; FECD3, Fuchs endothelial corneal dystrophy 3; GDPAG, global developmental delay, progressive ataxia, and elevated glutamine; HFG, hand-foot-genital syndrome; HPE5, holoprosencephaly 5; SPD1, synpolydactyly 1; XLMR, x-linked mental retardation
aLocation of entire gene listed
General characteristics of repeat expansion disorders
Molecular mechanisms
Repeat expansion diseases have a wide range of pathogenic mechanisms, which depend on the location of the expanded STR within a gene loci, and the nature and function of the gene. It is often hard to determine the specific mechanism as multiple may occur simultaneously and all may contribute to the disease form. The mechanisms may be broadly categorised as loss-of-function (LOF) or toxic gain-of-function (GOF).
LOF mechanisms include hypermethylation and gene silencing [43, 132], defective transcription, and increased messenger RNA (mRNA) degradation [154]; all effects that can be elicited by an STR expansion within a gene locus. DNA methylation is an epigenetic process that contributes to genome stability and maintenance, and regulation of gene expression during development, with aberrant methylation profiles often implicated in disease [2]. Large expanded STRs may induce local hypermethylation, thereby silencing gene expression. One such classic example is an expanded STR in the promoter region of FMR1, seen in Fragile X syndrome (FXS). The expansion causes hypermethylation of the FMR1 promoter region leading to silencing of transcription and LOF in the FMR1 gene. Therefore, the methylation state of relevant genes, in addition to STR length, may be informative for diagnosis of repeat expansion diseases.
Toxic GOF mechanisms include RNA toxicity, aberrant alternative splicing, repeat-associated non-AUG (RAN) translation, increased promoter activity, coding tract expansions and polyglutamine aggregation [85, 154, 180]. Repeat expansions in coding and non-coding regions may disrupt RNA function in many ways, with multiple coexisting mechanisms potentially contributing to pathogenicity. For example, post-mortem examination of brain tissue in patients with an expanded ‘GGGGCC’ repeat in the 5’ region of C9orf72 ALS/FTD, revealed multiple potential pathogenic RNA species: RNA that had been stalled at repeat locations, RAN proteins, antisense transcription of repeat regions and alternative splicing of intron 1 containing the repeat [48]. These species are considered “toxic” as they accumulate as RNA foci within the neurons, astrocytes, microglia and oligodendrocytes and form complexes with RNA-binding proteins to dysregulate translation and modify transcription [48, 49].
The other common toxic GOF mechanism is expansion of homopolymer amino acid tracts resulting in misfolding and proteinopathy. In neurological repeat expansion diseases, exonic ‘CAG’ repeat expansions code for the amino acid glutamine; when expanded, they create polyglutamine tract expansions which can reach hundreds of amino acids long. This is thought to alter and expand the transcribed protein creating insoluble protein aggregates within neuronal cells (primarily in the cerebellum), leading to perturbations of intracellular homeostasis and cell death [81]. This mechanism is commonly seen in the hereditary spinocerebellar ataxias. In congenital and developmental repeat expansion diseases, exonic ‘GCG’ coding tracts expand to create polyalanine tract expansions (Table 2). However, they are quite different to polyglutamine tract expansions seen in neurological repeat expansion disorders; they are smaller and generally meiotically stable when transmitted between generations, thus they do not exhibit the same large pathogenic range seen in neurological repeat expansion disorders. For example, a normal allele in HOXA13 contains 15–18 alanine residues while a pathogenic allele only contains between 7 and 15 extra residues [50]. Thus, the mechanism of mutation in polyalanine disorders is thought to be different and hypothesised to be due to unequal crossing between mispaired alleles and duplication during replication rather than dynamic trinucleotide expansions [164]. This would explain the relative stability of transmission and small pathogenic ranges. Furthermore, these polyalanine tract repeat expansion disorders are more commonly caused by other mutations such as missense and frameshift mutations. Interestingly, several studies show that an expansion of polyalanine tracts results in low levels of the protein found in the nucleus thereby exhibiting LOF, rather than increased protein levels and proteinopathy seen in polyglutamine tract expansions [23, 64].
Repeat length and disease severity
The size of STR expansions has been shown to quantitively affect disease severity, with larger expansions often associated with earlier onset of disease and more severe symptoms. For example, the repeat size in myotonic dystrophy type 1 (DM1) has a very broad pathogenic range (Fig. 1). Typically, 50–150 repeats cause a late-onset (20–70 years) mild phenotype with cataracts and myotonia, 100–1000 repeats cause onset in adolescence/early adulthood (10–30 years) with a classical phenotype of weakness, myotonia, cataracts, balding and arrhythmias, while even larger expansions cause early-onset (birth to 10 years) disease with infantile hypotonia, respiratory involvement and intellectual disability [13, 176].
Fig. 1.

Healthy and pathogenic ranges in neurological short tandem repeat expansion disorders. Box plot indicates the range of observed sizes for the pathogenic STR in known neurological STR expansion disorders (see Table 1). For each disorder, the range of STR sizes observed among unaffected individuals is shown in black, and the sizes observed in affected individuals is shown in pink
Slightly expanded STR regions, known as premutation alleles, may be associated with mild or variable phenotypes. For example, in Huntington’s disease (HD), there is full penetrance in all individuals with greater than 39 repeats of ‘CAG’ within exon 1 of the HTT gene, and partial penetrance in individuals with 36–39 repeats [101]. Approximately 50–70% of the variability in age of onset in Huntington’s disease is directly correlated to repeat length variability [54, 170]. Another classical example is FXS. In 1991, it was found that a ‘CGG’ repeat in the 5’ promoter region of the FMR1 gene normally contains an unmethylated STR of up to 45 ‘CGG’ repeats [55]. In individuals with expansions greater than 200 repeats, the FMR1 promoter region undergoes hypermethylation and transcriptional silencing of Fragile X mental retardation protein (FMRP) [109]. Loss of the FMRP protein, which is vital for synaptic plasticity in the CNS, leads to FXS [10]. However, the premutation allele (55–200 repeats) is known to cause late-onset Fragile X-associated tremor/ataxia syndrome (FXTAS) in men [90]. While in women, a 55–200 repeat-allele may present with a primary ovarian insufficiency due to absent menarche or premature follicular depletion [109]. This premutation allele does not exhibit hypermethylation, and in fact increases promoter region activity and transcription, resulting in production of toxic RNA species [59]. Thus, two allele sizes in the same STR region may exhibit opposing molecular mechanisms corresponding with distinct clinical phenotypes. This highlights the importance of accurate repeat sizing for these genes.
It is important to note that the exact point at which STR pathogenicity occurs is still the subject of ongoing investigation and debate. For example, there is some uncertainty over the pathogenic cut-off for SCA8 and SCA17, since expanded alleles have been detected in a healthy control population [142, 178]. Moreover, the pathogenic link between the STR expansion in ATXN8 and SCA8 has been questioned [136, 149, 169]. Rates of expanded repeats in healthy populations exist in other STR regions, such as C9orf72 and FMR1, where 0.1–0.4% of the healthy population have a repeat expansion [69]. Hence, in these cases it is difficult to determine the significance of an expanded or slightly expanded allele. Furthermore, due to intrinsic limitations in current clinical diagnostic methods, the upper range of STR expansions is often difficult to accurately define, with large expansions exceeding the capabilities of established molecular diagnostic techniques (see below). For example, the sizing of SCA31 repeats has been imprecise or absent, with no accurate literature defining the upper end of pathological repeat sizes [67]. Generally, genetic reports for C9orf72 indicate three size ranges: normal, intermediate and pathogenic [16]. The pathogenic range is generally reported as “ > 30” repeats [16].
Clinical anticipation
As mentioned earlier, STRs have an intrinsic tendency to expand during replication. This means that, while most repeat expansion diseases are inherited, there may be sporadic cases with no previous family history. STR instability also explains a phenomenon known as clinical anticipation. Anticipation is the seemingly increasing severity of disease and/or symptoms appearing at an earlier age as generations continue. Because of this phenomenon, the premutation allele in FXS is commonly seen in maternal carriers and maternal grandfathers of affected individuals. Over generations, the unstable premutation allele favours continual expansion and may sporadically present as full FXS in male children. Anticipation is also commonly seen in HD, with larger repeats being more unstable [130]. Intermediate alleles of 34–35 ‘CAG’ repeats in HTT have a high risk of expanding and causing new mutations [140]. Interestingly, anticipation in HD is much more commonly seen in paternal transmission, with larger expansion juvenile-onset HD often inherited from the father; although, there are some cases of maternal transmission [113, 127]. This is thought to be due to large STR instability and variation in spermatogenesis seen in fathers [166]. This paternal transmission pattern of anticipation is also seen in SCA1, SCA2, SCA7 and DRPLA [6, 51, 66, 99], while in SCA8 there is a pattern of maternal transmission thought to be due to en masse STR contractions in paternal sperm [110]. ATN1 (DRPLA) and ATXN7 (SCA7) are especially unstable [125]; anticipation in SCA7 may be so severe that young children develop symptoms before an affected parent or grandparent.
The phenomenon of genetic anticipation may not be true for all repeat expansion diseases, for example, clinical anticipation is not seen in families with OPMD or FRDA [52, 71], and while studies show evidence of clinical anticipation in C9orf72 expanded alleles [160], carrier alleles may variably contract or expand over generations [42]. Furthermore, the repeat length has been found to differ within the same patient, indicating cells in brain tissue and cells in blood have different repeat sizes (similar patterns of somatic mutation are seen in other repeat expansion disorders such as HD and DM1) [123]. Thus, further accurate genotyping of C9orf72 affected families is required to better understand the correlation between repeat size and phenotype.
Common clinical features
Repeat expansion diseases tend to cluster around shared phenotypes. It would be difficult to find a repeat expansion disorder that did not exhibit of one or more of the following phenotypes: cerebellar ataxia, chorea or HD phenocopies, tremor, cognitive impairment, muscular dystrophies, myoclonic seizures, amyotrophic lateral sclerosis and peripheral neuropathies.
Hereditary cerebellar ataxias
Patients with hereditary cerebellar ataxia exhibit abnormal eye movements, dysarthria, limb and gait ataxia. These may be due to a plethora of different STR expansions including the spinocerebellar ataxias (SCA), dentatorubral-pallidoluysian atrophy (DRPLA), Friedreich’s Ataxia (FRDA) and the cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS, see section below) [12], and may also be due to point mutations, duplications, and deletions [71].
The most common STR expansions in patients with hereditary cerebellar ataxia is an expanded ‘CAG’ repeat within polyglutamine tracts found in SCA1, SCA2, SCA3, SCA6, SCA7, SCA12 and SCA17 [131]. For these disorders, there are efficient cost-effective repeat-primed polymerase chain reaction (RP-PCR) methods for diagnostic testing, however a majority of patients referred for these panels return with negative test results [72]. Testing other STR regions is not as straight forward, and requires time-consuming methods of individual gene sequencing [8]. In a German cohort of 440 of people who returned negative for SCA1, 2, 3, 6 and 7, there were five patients with expanded SCA8 repeats, one patient with an FXTAS expanded allele and four with possible FXTAS alleles, and one C9orf72 expansion [8]. This study shows that, while they are uncommon, other STR expansions may cause undiagnosed late-onset progressive ataxia. Recently, SCA37 was linked to a novel expansion of ‘ATTTC’ within a ‘ATTTT’ polymorphism in DAB1 [139]. The repeat length and conformation of the repeat expansion could only be accurately assessed with long-read sequencing [139]. It has a similar phenotype to other spinocerebellar ataxias, suggesting there are more novel expansions which may explain cases of undiagnosed ataxia.
Myoclonus epilepsies
Unverricht-Lundborg disease (ULD) is one of the most common single causes of progressive myoclonus epilepsy worldwide; it is characterised by childhood-onset stimulus-sensitive myoclonus epilepsy, ataxia and cognitive and behavioural abnormalities [91]. Other repeat expansion diseases may also present with myoclonus epilepsies, usually with large repeat sizes and severe phenotypes; these include SCA7, SCA10 and DRPLA [92, 103, 161, 177]. Furthermore, a group of familial adult myoclonus epilepsies (FAME1, 2, 3 and 6) have recently been linked to STR expansions, discussed further below.
Huntington’s disease and Huntington’s disease phenocopies
HD is caused by a ‘CAG’ repeat in the HTT gene and is characterised by chorea with psychiatric symptoms and cognitive decline, with mean age of symptom onset between 35 to 44 years old [20]. The most common HD phenocopies or HD-like syndromes are seen in STR expansions within C9orf72 [111] (discussed below), however, others include PRNP (Huntington disease-like 1, HDL1), JPH3 (HDL2), TBP (SCA17 or HDL4), ATXN8 (SCA8), FXN (Friedreich’s ataxia) and ATN1 (DRPLA), in addition to sequencing variants/deletions in VPS13A, TITF1, ADCY5, RNF216 and FRRS1L [135]. HDL2 shares molecular characteristics with HD: they are both due to polyglutamine tract expansion caused by a ‘CAG’ repeat in exon 1 of their respective genes, and there is evidence to suggest that similar CREB-binding protein (CBP) sequestration in nuclear bodies drives both pathological processes [62, 168]. Given numerous examples of HD phenocopies and the overlap between several repeat expansion diseases, one may suspect that further phenocopies of HD might have an undiscovered genetic basis in STR regions.
C9orf72-related disorders
Since its discovery in 2011, the ‘GGGGCC’ hexanucleotide repeat in C9orf72 has been studied extensively. It is the most common cause of familial frontotemporal dementia (FTD) and familial amyotrophic lateral sclerosis (ALS) [32]. Interestingly, the C9orf72 repeat expansion has also been linked to a range of clinical phenotypes including typical Parkinson’s disease, atypical parkinsonian syndromes, schizophrenia and bipolar disorder [14, 49]. In a recent retrospective study, movement disorders were the second most common initial presentation of C9orf72-related diseases, following cognitive signs in FTD [37]. These patients frequently present with one or several of the following: parkinsonism, myoclonus, dystonia, chorea and ataxia [37]. The phenotypic heterogeneity is difficult to explain, consistent with the concept that the mechanisms of disease caused by STR expansions are poorly understood [59].
Interruptions
Some STR expansions contain internal sequence interruptions that may directly affect the phenotype or lead to overestimation of repeat sizes. These interruptions have long been found in Fragile X, Huntington’s disease, hereditary cerebellar ataxias and myotonic dystrophies, however their origins and effect are poorly understood. There has been more research in this area due to new methods of long-read sequencing, combined with specific RP-PCR and Southern blot primers to establish a stronger consensus on repeat motifs [156]. This has allowed new discoveries in the role of interruptions. For example, three groups have shown that a loss of a ‘CAA’ interruption within expanded ‘CAG’ tracts in HTT leads to earlier onset Huntington’s disease [170]. It is estimated that this variant is associated with 9.5 years earlier onset in Huntington’s disease [39], particularly in those with reduced penetrance alleles of 36–39 ‘CAG’ repeats. The ‘CAA’ interruption is also a genetic modifier of other polyglutamine repeat expansions, such as SCA2 and SCA17 [25, 45]. These ‘CAA’ interruptions fall within ‘CAG’ coding tracts and therefore still translate to glutamine, however the interrupted alleles preferentially form shorter branching hairpin structures which reduce strand slippage and increase stability of the repeat [145, 173]. Thus, it is proposed that the pathogenic mechanism of this interruption may be due to increased instability during somatic expansion of the repeat, and longer polyglutamine tracts leading to increased toxic GOF [170]. Interestingly, in SCA2, ‘CAA’, ‘CGG’ and ‘CGC’ interruptions are linked to autosomal dominant levodopa-responsive Parkinson’s disease, demonstrating interruptions may modify phenotype as well as age of onset [122].
Similarly, a DM1 family was found to have ‘CCG’ interruptions within the ‘CTG’ STR expansion in DMPK resulting in atypical traits such as severe axial and proximal weakness and late onset of symptoms [9].
Pentanucleotide STR regions are very unstable and dynamic in nature, often containing large amounts of heterogeneity in controls as well as patients. For example, pathogenic ‘ATTCT’ repeats in ATXN10 (SCA10) likely exist within a dynamic structure of pentanucleotide, hexanucleotide and heptanucleotide motifs [102]. Interruptions with the specific ‘ATCCT’ motif is strongly associated with epilepsy [88, 103], while pure ‘ATTCT’ tracts are associated with parkinsonism [137]. The mechanism of disease caused by these interruptions is difficult to discern; further genotyping of these regions is first required. This complex motif structure is commonly seen in several newly discovered pentanucleotide repeat expansions such as RFC1 or SAMD12, which show that pathogenic sequences are often extremely dynamic in nature [3, 107, 138].
Recent discoveries for neurological repeat expansion disorders
Most of the repeat expansion disorders listed in Table 1 have been discussed extensively in literature, however, in the last three years, 12 novel neurological repeat expansion disorders have been classified – these include SCA37, CANVAS, neuronal intranuclear inclusion disease (NIID), OPML, OPDM, OPDM2, FAME1, FAME2, FAME3, FAME6, FAME7 and recessive hereditary motor neuropathy (HMN) (Table 1).
In 2019, a heterozygous ‘CGG’ expansion in the Notch homolog 2N-terminal-like C (NOTCH2NLC) gene was found to be the cause of NIID by numerous independent groups [34, 69, 146]. Of note, the expansion was detected or confirmed using long-read sequencing. Some patients have been identified to have ‘AGG’ interruptions, with evidence in a small East–Asian cohort showing interruptions may be linked to earlier age of onset [24]. NIID is a neurodegenerative condition characterized by eosinophilic intranuclear inclusions in neuronal and glial cells, which have characteristic findings on brain MRI, including high diffusion-weighted imaging signals along the corticomedullary junction [4, 95, 152]. The NOTCH2NLC expansion has also been found in a rapidly growing number of phenotypes, including leukoencephalopathy, essential tremor, Parkinson’s disease, multiple system atrophy (MSA) and amyotrophic lateral sclerosis [38, 69, 95, 117, 119, 175]. Further long-read sequencing studies have found noncoding CGG repeat expansions in LOC642361/NUTM2B-AS1, LRP12 and GIPC1 [69, 172]. These STR expansions correspond to similar phenotypes: oculopharyngeal myopathy with leukoencephalopathy (OPML), and oculopharyngodistal myopathy 1 and 2 (OPDM1 and OPDM2), emphasising the need for screening multiple genetic causes in patients presenting with these clinical features. For example, a recent study screened a cohort of 211 patients clinically diagnosed with OPDM and found seven patients with ‘CGG’ expansions in NOTCH2NLC [118]. Similarly, in a cohort of 189 patients clinically diagnosed with MSA, five were found to have ‘GCC’ repeats in NOTCH2NLC [38].
In 2019, an intronic biallelic ‘AAGGG’ repeat in the RFC1 gene was linked to patients presenting with cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS) [28, 126]. CANVAS is characterised by a collection of clinical features which often present later in life [21]. Previously determined idiopathic [171], the newly discovered repeat expansion was found in 22% of all patients (n = 150) with undiagnosed late-onset ataxia. This percentage increased to 63% if they also had sensory neuronopathy and up to 92% of patients with full CANVAS syndrome features [28], however these numbers seem to be an overestimation in non-European populations [3]. RFC1 expansions can also mimic other disorders such as Sjogren’s syndrome, hereditary sensory neuropathy with cough or paraneoplastic syndrome [29, 83]. Interestingly, in this case, the pathogenic repeat ‘AAGGG’ is a conformational variation on the normal ‘AAAAG’ motif, suggesting a disease mechanism associated with the expansion of variant motifs. Many studies have shown the dynamic nature of the repeats within RFC1. A study of 608 healthy controls used flanking and RP-PCR, Southern blot analysis and Sanger sequencing to demonstrate an allelic distribution of 75.5% for the ‘(AAAAG)11’ allele, 13.0% for the ‘(AAAAG)exp’ allele, 7.9% for the ‘(AAAGG)exp’ allele and 0.7% for the ‘(AAGGG)exp’ allele [28]. The average size of normally expanded alleles ‘AAAAG’ and ‘AAAGG’ was 15–200 repeats and 40–1000 repeats respectively. Another study reports two other heterozygous conformations, ‘AAGAG’ and ‘AGAGG’, which have an average size of 160 repeats and a frequency of approximately 2% in healthy populations and 7% in CANVAS cases [3].
Recently, more novel pathogenic RFC1 conformations have been implicated with CANVAS. ‘ACAGG’ was found to have expanded in two Asia–Pacific families [138] who demonstrated additional clinical features, namely fasciculations and elevated serum kinase. Another study showed a ‘(AAAGG)10–25(AAGGG)exp’ allele was the predominant pathogenic allele found in Māori populations, with no apparent phenotypic differences when compared to the European populations [11]. Accurately genotyping the conformation of the expanded allele in RFC1 is vital for diagnosing CANVAS and discovering novel pathogenic conformations. Long-read sequencing has been used to read entire lengths of repeat regions and overcomes traditional problems of mapping novel conformations with short-reads or creating repeat-primed probes with RP-PCR and Southern blot. This is also seen in SCA37 and the five FAME subtypes, whereby a variant conformation is expanded within the patient cohort [68, 139].
In 2019, five subtypes of familial adult myoclonus-epilepsies (FAME) were linked to ‘TTTCA’ intronic repeats in their respective genes [68]. Using PacBio long-read sequencing, the 2.2–18.4 kb expanded alleles in SAMD12 (FAME1) could be accurately and efficiently sized [68, 107] and were found to have expanded ‘TTTCA’ segments rather than the ‘TTTTA’ motif found in control patients. FAME6 and FAME7 only have genotype–phenotype linkage in one family each, thus evidence regarding these two diseases is still limited [68].
It is possible a shared motif/repeat location may cause similar clinical syndromes. The ‘TTTCA’ intronic repeats in SAMD12, MARCHF6, TNRC6A and RAPGEF2 are all responsible for FAME [68]. Similarly, the ‘CGG’ non-coding repeat in NIID, OPML and OPDM also have overlapping phenotypes with some common typical MRI findings.
Very recently, a 10 base pair expansion in the gene VWA1 was identified as a cause of recessive distal hereditary motor neuropathy (HMN), further underscoring that repeat expansions can be linked with neuropathy phenotypes and highlighting the rapid rate of new STR expansions [121].
Current clinical testing approaches for repeat expansion diseases are time-consuming to develop, and often cannot accurately assess larger STR regions with high ‘GC’ content. We must establish a new robust clinical pipeline for STR genotyping, that can be developed at a rapid pace, to match the rate of discovery of novel repeat expansion diseases as seen in Fig. 2.
Fig. 2.
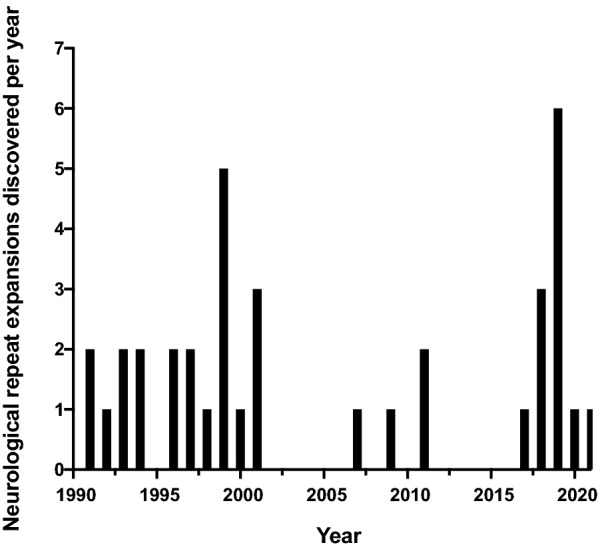
Rate of discovery of neurological short tandem repeat expansions. Bar plot indicates the number of new pathogenic STR expansion discoveries published each year during the period 1990–2021 (see Table 1 for references to original publications for each gene)
Molecular diagnostics
The established approach for molecular diagnosis of repeat expansion diseases involves genotyping STRs by repeat-primed precise PCR (RP-PCR) and/or Southern blot assays for sizing larger expansions (Fig. 3). The clinician must decide which STRs warrant testing, which can be difficult due to phenotypic heterogeneity and overlap between various repeat expansion disorders. Moreover, since both methods require separate primers/probes for each STR, parallel analysis of multiple candidates in a single assay is not possible.
Fig. 3.

Current molecular diagnostic methods. Flow chart shows an example of two current diagnostic methods for diagnosing STR expansions: Southern blot and repeat-primed PCR. The sample analysis shown in both diagnostic methods was taken from a patient with Friedrich’s ataxia with a heterozygous ‘GAA’ expansion in the FXN gene (approximately 90 and 900 repeats). The RP-PCR graph shows the characteristic tailing/stuttering pattern of expanded alleles caused by the repeat-primed probes binding to more sites within the STR expansion. For sizing, Southern blot is performed. The larger 900-repeat ‘GAA’ allele cannot be seen using the Southern blot sizing ladder shown above
Southern blot assays are regarded as the gold-standard for detecting large polynucleotide repeat expansions, but this method is time-consuming, inefficient, costly and requires large quantities (up to 10 μg) of high-quality DNA for a single analysis [4]. In certain STR expansions, Southern blotting has been replaced by RP-PCR, which is cheaper and more efficient [151]. However, because the highly repetitive region is amplified and then fragmented into shorter reads, PCR stutter errors make it difficult to accurately determine the length of an expanded repeat. Furthermore, in large repeats with high ‘GC’ content, repetitive flanking regions or flanking variants, it can be highly challenging to establish an effective diagnostic PCR assay. This is evident in testing regimes for C9orf72, which have not been standardised across labs [4]. Currently, optimised PCR methods can detect expanded repeat sizes up to 900 hexanucleotide repeats, However, accurate quantitative sizing may only be reported up to 140 repeats [26, 151].
Furthermore, while interruptions may be detected within a repeat, their exact motif may be challenging to determine [61]. Due to the high concentration of guanine-cystine (GC) content in some of these repeat and interruption motifs, there is a high chance of secondary structure formation and allelic dropout of PCR amplification leading to further sequencing errors [61, 75].
Next generation sequencing
Next-generation sequencing (NGS) provides an alternative approach for genotyping STRs. STR expansions can be detected across the entire genome, using established short-read NGS platforms (e.g., Illumina), and a growing number of bioinformatics tools have been developed for this purpose (e.g., ExpansionHunter, LobSTR, RepeatSeq, HipSTR and GangSTR) [35, 57, 84, 112]. These tools also allow researchers to link STR regions in affected family members, making them good methods for identifying novel expansions, thereby leading to a recent wave of discoveries (as described earlier). The major advantage of whole-genome sequencing is that, in theory, all STRs in the genome are profiled simultaneously, as well as STR contraction and non-STR mutations, which may also be implicated in disease. While NGS remains relatively expensive, avoiding the need for repeated molecular testing on multiple targets means this can be cost effective, and will be increasingly competitive as sequencing prices continue to fall.
However, the utility of short-read NGS for repeat expansion diagnosis is hampered by several limitations. Firstly, highly repetitive and/or ‘GC’ rich genome regions are refractory to NGS library preparation, PCR amplification and sequencing, making it difficult to obtain sufficient coverage in many STR regions. PCR amplification during the library preparation can also introduce stutter errors, although this can be alleviated through the use of PCR-free library preparations [104]. Secondly, the repetitive nature of STR regions can cause ambiguous alignment or misalignment of short NGS reads to the reference genome. More fundamentally, the short-read length (~ 100–150 bp) of established NGS technologies is insufficient to span large STR expansions, making it impossible to precisely determine their length (see Fig. 4). Lastly, standard NGS does not detect epigenetic modifications, such as 5-methylcytosine, which are diagnostically important in some cases [132, 144]. Although NGS has proven useful for the discovery of new disease-related repeat expansions, these limitations have so far prevented widespread adoption of NGS for clinical diagnosis and replacement of low-throughout molecular tests like Southern blotting.
Fig. 4.

NGS and Long-read sequencing for diagnosing short tandem repeat expansions. Flow chart shows the use of short-read NGS and two long-read sequencing methods for genotyping STR expansions: PacBio single-molecule real-time (SMRT) sequencing and Oxford Nanopore Technology (ONT) long-read sequencing. The alignment of reads to the genome can be seen for all three methods; short-reads are ‘tiled’ together to estimate the repeat size and sequence, while long reads easily span repeat and flanking regions. Nanopore sequencing high error rates can be overcome via sufficient coverage
Outlook: efficient and accurate diagnosis of repeat expansion disorders with long-read sequencing
For thorough evaluation of a suspected repeat expansion disorder, clinicians must be able to: (1) screen for all the relevant genes (including any newly discovered candidates); (2) accurately assess the size of any detected expansion and; (3) look for additional diagnostic or prognostic markers such as repeat interruptions and DNA methylation state. Emerging long-read sequencing platforms from Oxford Nanopore Technology (ONT) and Pacific Biosciences (PacBio) have the potential to address these requirements, while overcoming the limitations of conventional Illumina short-read sequencing platforms [84].
ONT devices measure the displacement of ionic current as a DNA strand passes through a biological nanopore and subsequently translate this data into DNA sequence information (see Fig. 4). ONT sequencing has no theoretical upper limit on read length, with > 10 kb average read length considered standard for genomic DNA sequencing and some examples achieving maximum read lengths in excess of 1 Mb [98]. Therefore, unlike for short-read NGS, individual ONT reads may span the entire length of large pathogenic repeat expansions (see Fig. 4 below). In one study, between 80 and 99.5% of reads successfully spanned expanded ‘GGCCTG’ repeats in NOP56 (median 37 repeats) and ‘CCCCGG’ repeats in C9orf72 (median 406 repeats), allowing direct measurement of STR lengths [36]. Nanopore reads currently exhibit relatively high sequencing error rates when compared to NGS, due to inaccuracies in the base-calling process, however, accurate consensus sequence determination is possible with sufficient coverage [70] and several studies have demonstrated accurate genotyping of repeat expansions with ONT [36, 46, 146]. Additionally, analysis of ONT signal data allows the methylation status of a given loci to be determined in parallel, providing an additional marker for the diagnosis of relevant repeat expansion disorders, such as FXS [46].
PacBio Single Molecule, Real-Time (SMRT) sequencing technology detects, in real-time, fluorescent signals from nucleotides as they are being incorporated to a single DNA template-polymerase [128]. SMRT sequencing achieves greater than 99% accuracy via circular consensus sequencing (CCS), whereby large DNA strands are ligated on either end to form a circular DNA molecule such that the DNA polymerase completes multiple passes of the same DNA fragment in a single read to achieve high coverage (average read-length 13.5 kb) [165]. An advantage of the long and highly accurate reads generated by PacBio SMRT sequencing, is the ability to resolve the STR length and sequence, as well as detecting and phasing possible variants in the surrounding regions. For example, a recent study developed a haplotype phasing protocol for the HTT gene using PacBio SMRT sequencing, enabling detection of relevant SNPs and ‘CAG’ expansions in HTT on the same amplicon [153]. Several new bioinformatics tools, such as IsoPhase [163], SHAPEIT4 [33] and NanoCaller [1], use long reads to accurately phase SNV, insertions and deletions. Thus, both ONT and PacBio SMRT technologies have the potential to replace current clinical molecular diagnostics by accurately generating reads spanning the length of large pathogenic repeat expansions.
Despite these promising recent developments, the computational analysis of long-read sequencing data to accurately genotype repeats is an active area of development, with several important hurdles yet to be overcome. Multiple software packages have been recently created for this purpose, including tandem-genotypes [106], NanoSatellite [31], STRique [46], RepeatHMM [93] and PacmonSTR [158], with each demonstrating the capability to measure the size of expanded STRs. However, discordant results between some tools [106] highlight the need for more rigorous benchmarking on a broad selection of different repeat types and sizes. Furthermore, the ability to resolve challenging cases such as STR interruptions, mixed conformations (e.g., the Māori-specific RFC1 conformation [11]) and allelic differences in conformations, has yet to be demonstrated. Furthermore, the detection of novel pathogenic STR expansions remains another major unsolved challenge given the polymorphic nature of STRs and the vast STR diversity encountered in human populations [93, 106].
Whole-genome analysis with both ONT and PacBio long-read sequencing platforms is now feasible and will likely aid in the discovery of many novel disease-related STR expansions in the near future. For example, Sone and colleagues recently discovered a ‘GGC’ repeat in the NOTCH2NLC gene in 13 patients affected with NIID using long-read whole-genome sequencing combined with bioinformatics tool tandem-genotypes [146]. They then confirmed their findings with RP-PCR on positive and healthy controls. Similarly, a ‘TTTCA’ repeat expansion was discovered in SAMD12 and linked to FAME1; the study used low-coverage (~ 10×) PacBio long-read sequencing with STR detection tools RepeatHMM and inScan to target the locus identified by linkage analysis [179]. It should also be noted that the ‘TTTCA’ expansion in the SAMD12 gene was also discovered independently by Ishiura and colleagues, who used linkage analysis followed by repeat-primed PCR and Southern blotting to detect the expansion, then used PacBio to elucidate the motif structure [68].
Given the high cost and large data volumes generated using whole-genome, targeted sequencing of candidate genes represents a more viable and cost-effective pathway to clinical adoption. This requires the establishment of reliable methods for amplification-free enrichment and sequencing of long DNA fragments spanning STR regions.
One promising strategy involves the use of CRISPR-Cas9 guide-ribonucleoproteins (RNPs) for selective cleavage of target loci, followed by ligation of a magnetic adaptor that allows isolation of target molecules prior to PacBio SMRT sequencing [157]. To date, this method has been applied for genotyping STR expansions in HTT, C9orf72, ATXN10 and NOTCH2NLC [146, 157]. ONT sequencing is amenable to an analogous strategy, where ONT sequencing adapters are directly ligated to Cas9 cleavage sites to enable their selective sequencing [46, 48]. In establishing this approach, Giesselmann et al. found a single ONT MinION flow-cell could generate greater than 40-fold coverage over the expanded ‘GGGGCC’ region in C9orf72 [46], sufficient for accurate determination of repeat length. Furthermore, using their own raw signal algorithm termed STRique, they were able to profile ‘CpG’ methylation of the STR and its flanking regions, with hypermethylation observed at the C9orf72 promoter in mutated alleles. In the study by Sone et al. mentioned above, they also used Cas9-mediated enrichment to achieve high sequencing depth (100–1795×) following their initial low-coverage whole-genome sequencing [146]. Furthermore, this method aided in identifying a ‘AAGGG’ repeat in a Japanese family in the RFC1 gene as well as benign ‘TAAAA’ and ‘TAGAA’ expansions in BEAN1 [114]. Cas9-mediated target enrichment is amenable to multiplexing, making it feasible to target multiple disease alleles in parallel, for more efficient and cost-effective diagnosis. For example, Tsai et al. demonstrated parallel enrichment of C9orf72, HTT, FMR1 and ATXN10, achieving 150–2000-fold coverage depth with SMRT sequencing on all targets in a single assay [157]. This capability is advantageous from a diagnostic perspective, avoiding the need to order multiple tests, as is the case with standard molecular diagnostics.
Another recent innovation in ONT sequencing is programmable target selection, using ONT’s Read Until API. Via real-time identification and rejection of off-target DNA fragments, Read Until affords enriched sequencing depth across target regions of the user’s choice without requiring any upstream molecular target enrichment [80, 124]. One unpublished study has already applied this new approach to the detection of repeat expansions, simultaneously determining repeat size and methylation status in patients with pathogenic expansions in FMR1, FXN, ATXN3, ATXN8, or XYLT1 [105]. Besides the obvious advantage in avoiding cumbersome molecular methods of target enrichment, the Read Until method allows hundreds or even thousands of candidate loci to be targeted in parallel, and the specific set of targets can be easily customised for a given patient depending on their phenotype and family history. These advantages could see programmable ONT sequencing become the preferred method for both diagnosis and discovery of repeat expansion disorders in the near future.
Conclusions
Short tandem repeat expansion disorders are highly important in human disease, particularly in the field of neurology. The list of repeat expansion disorders is currently over 40 and growing rapidly. This is highlighted by the recent findings that several important disorders in neurology (such as CANVAS and NIID) have been found to be caused by short tandem repeat expansions. The established methods for diagnosing these disorders are cumbersome and time consuming. However, long-read sequencing offers the opportunity to transform the detection of repeat expansion disorders, allowing for rapid and accurate genotyping. This would provide a more in-depth understanding of healthy and pathogenic repeat ranges, transmission and clinical anticipation, and the role of interruptions. Further research is required to overcome the technical hurdles and fully exploit the potential of long-read sequencing. Additionally, cost-effectiveness studies are required to compare the cost associated with long-read sequencing approaches to traditional methods of detecting repeat expansion disorders prior to widespread use in clinical practice.
Acknowledgements
Not applicable.
Authors' contributions
S.R.C. was responsible for cataloguing known repeat expansion disorders, creating figures and diagrams and writing a majority of the main body of the article. K.R.K., S.S.P. and I.W.D. all contributed to writing the main body of the article as well as creating the figures. All authors contributed equally to editing and preparing the final manuscript. All authors read and approved the final manuscript.
Funding
There is no specific funding for this paper. Dr Deveson is supported by the following funding sources: MRFF Investigator Grant MRF1173594 and philanthropic support from The Kinghorn Foundation (to I.W.D.). Dr Kumar is supported by a philanthropic grant from the Paul Ainsworth Family Foundation, a research award from the Michael J. Fox Foundation, Aligning Science Against Parkinson’s disease initiative, and honorarium from Seqirus.
Availability of data and material
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sanjog R. Chintalaphani, Email: s.chintalaphani@student.unsw.edu.au
Sandy S. Pineda, Email: s.pineda-gonzalez@garvan.org.au
Ira W. Deveson, Email: i.deveson@garvan.org.au
Kishore R. Kumar, Email: k.kumar@garvan.org.au
References
- 1.Ahsan U, Liu Q, Fang L, Wang K (2020) NanoCaller for accurate detection of SNPs and indels in difficult-to-map regions from long-read sequencing by haplotype-aware deep neural networks. bioRxiv. 10.1101/2019.12.29.890418 [DOI] [PMC free article] [PubMed]
- 2.Akarsu A, Stoilov I, Yilmaz E, Sayil B, Sarfarazi M. Genomic structure of HOXD13 gene: a nine polyalanine duplication causes synpolydactyly in two unrelated families. Hum Mol Genet. 1996;5:945–952. doi: 10.1093/hmg/5.7.945. [DOI] [PubMed] [Google Scholar]
- 3.Akçimen F, Ross JP, Bourassa CV, Liao C, Rochefort D, Gama MTD, et al. Investigation of the RFC1 repeat expansion in a Canadian and a Brazilian ataxia cohort: identification of novel conformations. Front Genet. 2019;10:1219. doi: 10.3389/fgene.2019.01219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akimoto C, Volk AE, Van Blitterswijk M, Van Den Broeck M, Leblond CS, Lumbroso S, et al. A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J Med Genet. 2014;51:419–424. doi: 10.1136/jmedgenet-2014-102360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Mahdawi S, Ging H, Bayot A, Cavalcanti F, La Cognata V, Cavallaro S, et al. Large interruptions of GAA repeat expansion mutations in Friedreich ataxia are very rare. Front Cell Neurosci. 2018;12:443–443. doi: 10.3389/fncel.2018.00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Almaguer-Mederos LE, Mesa JML, González-Zaldívar Y, Almaguer-Gotay D, Cuello-Almarales D, Aguilera-Rodríguez R, et al. Factors associated with ATXN2 CAG/CAA repeat intergenerational instability in spinocerebellar ataxia type 2. Clin Genet. 2018;94:346–350. doi: 10.1111/cge.13380. [DOI] [PubMed] [Google Scholar]
- 7.Amiel J, Laudier B, Attié-Bitach T, Trang H, de Pontual L, Gener B, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. 2003;33:459–461. doi: 10.1038/ng1130. [DOI] [PubMed] [Google Scholar]
- 8.Aydin G, Dekomien G, Hoffjan S, Gerding WM, Epplen JT, Arning L. Frequency of SCA8, SCA10, SCA12, SCA36, FXTAS and C9orf72 repeat expansions in SCA patients negative for the most common SCA subtypes. BMC Neurol. 2018;18:3. doi: 10.1186/s12883-017-1009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ballester-Lopez A, Koehorst E, Almendrote M, Martínez-Piñeiro A, Lucente G, Linares-Pardo I, et al. A DM1 family with interruptions associated with atypical symptoms and late onset but not with a milder phenotype. Hum Mutat. 2020;41:420–431. doi: 10.1002/humu.23932. [DOI] [PubMed] [Google Scholar]
- 10.Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beecroft SJ, Cortese A, Sullivan R, Yau WY, Dyer Z, Wu TY, et al. A Māori specific RFC1 pathogenic repeat configuration in CANVAS, likely due to a founder allele. Brain. 2020;143:2673–2680. doi: 10.1093/brain/awaa203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bird TD, et al. Hereditary ataxia overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews. Seattle: University of Washington; 2019. [Google Scholar]
- 13.Bird TD, et al. Myotonic dystrophy type 1. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews. Seattle: University of Washington; 1993. [Google Scholar]
- 14.Bourinaris T, Houlden H. C9orf72 and its relevance in parkinsonism and movement disorders: a comprehensive review of the literature. Mov Disord Clin Pract. 2018;5:575–585. doi: 10.1002/mdc3.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brais B, Bouchard J-P, Xie Y-G, Rochefort DL, Chrétien N, Tomé FM, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- 16.Bram E, Javanmardi K, Nicholson K, Culp K, Thibert JR, Kemppainen J, et al. Comprehensive genotyping of the C9orf72 hexanucleotide repeat region in 2095 ALS samples from the NINDS collection using a two-mode, long-read PCR assay. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:107–114. doi: 10.1080/21678421.2018.1522353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown LY, Odent S, David V, Blayau M, Dubourg C, Apacik C, et al. Holoprosencephaly due to mutations in ZIC2: alanine tract expansion mutations may be caused by parental somatic recombination. Hum Mol Genet. 2001;10:791–796. doi: 10.1093/hmg/10.8.791. [DOI] [PubMed] [Google Scholar]
- 18.Cagnoli C, Stevanin G, Michielotto C, Gerbino Promis G, Brussino A, Pappi P, et al. Large pathogenic expansions in the SCA2 and SCA7 genes can be detected by fluorescent repeat-primed polymerase chain reaction assay. J Mol Diagn. 2006;8:128–132. doi: 10.2353/jmoldx.2006.050043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 20.Caron NS, Wright GEB, Hayden MR, et al. Huntington disease. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews. Seattle: University of Washington; 1993. [Google Scholar]
- 21.Cazzato D, Bella ED, Dacci P, Mariotti C, Lauria G. Cerebellar ataxia, neuropathy, and vestibular areflexia syndrome: a slowly progressive disorder with stereotypical presentation. J Neurol. 2016;263:245–249. doi: 10.1007/s00415-015-7951-9. [DOI] [PubMed] [Google Scholar]
- 22.Cen Z, Jiang Z, Chen Y, Zheng X, Xie F, Yang X, et al. Intronic pentanucleotide TTTCA repeat insertion in the SAMD12 gene causes familial cortical myoclonic tremor with epilepsy type 1. Brain. 2018;141:2280–2288. doi: 10.1093/brain/awy160. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y-C, Auer-Grumbach M, Matsukawa S, Zitzelsberger M, Themistocleous AC, Strom TM, et al. Transcriptional regulator PRDM12 is essential for human pain perception. Nat Genet. 2015;47:803–808. doi: 10.1038/ng.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Z, Xu Z, Cheng Q, Tan YJ, Ong HL, Zhao Y, et al. Phenotypic bases of NOTCH2NLC GGC expansion positive neuronal intranuclear inclusion disease in a Southeast Asian cohort. Clin Genet. 2020;98:274–281. doi: 10.1111/cge.13802. [DOI] [PubMed] [Google Scholar]
- 25.Choudhry S, Mukerji M, Srivastava AK, Jain S, Brahmachari SK. CAG repeat instability at SCA2 locus: anchoring CAA interruptions and linked single nucleotide polymorphisms. Hum Mol Genet. 2001;10:2437–2446. doi: 10.1093/hmg/10.21.2437. [DOI] [PubMed] [Google Scholar]
- 26.Cleary EM, Pal S, Azam T, Moore DJ, Swingler R, Gorrie G, et al. Improved PCR based methods for detecting C9orf72 hexanucleotide repeat expansions. Mol Cell Probes. 2016;30:218–224. doi: 10.1016/j.mcp.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, et al. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat Commun. 2019;10:4920. doi: 10.1038/s41467-019-12671-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yau WY, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet. 2019;51:649–658. doi: 10.1038/s41588-019-0372-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cortese A, Tozza S, Yau WY, Rossi S, Beecroft SJ, Jaunmuktane Z, et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome due to RFC1 repeat expansion. Brain. 2020;143:480–490. doi: 10.1093/brain/awz418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.David G, Abbas N, Stevanin G, Dürr A, Yvert G, Cancel G, et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65–70. doi: 10.1038/ng0997-65. [DOI] [PubMed] [Google Scholar]
- 31.De Roeck A, De Coster W, Bossaerts L, Cacace R, De Pooter T, Van Dongen J, et al. NanoSatellite: accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. Genome Biol. 2019 doi: 10.1186/s13059-019-1856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dejesus-Hernandez M, Bradley I, Baker M, Nicola AM, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9orf72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delaneau O, Zagury J-F, Robinson MR, Marchini JL, Dermitzakis ET. Accurate, scalable and integrative haplotype estimation. Nat Commun. 2019;10:5436. doi: 10.1038/s41467-019-13225-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng J, Gu M, Miao Y, Yao S, Zhu M, Fang P, et al. Long-read sequencing identified repeat expansions in the 5'UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. J Med Genet. 2019;56:758–764. doi: 10.1136/jmedgenet-2019-106268. [DOI] [PubMed] [Google Scholar]
- 35.Dolzhenko E, van Vugt JJFA, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 2017;27:1895–1903. doi: 10.1101/gr.225672.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ebbert MTW, Farrugia SL, Sens JP, Jansen-West K, Gendron TF, Prudencio M, et al. Long-read sequencing across the C9orf72 ‘GGGGCC’ repeat expansion: implications for clinical use and genetic discovery efforts in human disease. Mol Neurodegener. 2018;13:46. doi: 10.1186/s13024-018-0274-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Estevez-Fraga C, Magrinelli F, Hensman Moss D, Mulroy E, Di Lazzaro G, Latorre A, et al. Expanding the spectrum of movement disorders associated With C9orf72 hexanucleotide expansions. Neurol Genet. 2021;7:e575. doi: 10.1212/nxg.0000000000000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang P, Yu Y, Yao S, Chen S, Zhu M, Chen Y, et al. Repeat expansion scanning of the NOTCH2NLC gene in patients with multiple system atrophy. Ann Clin Transl Neurol. 2020;7:517–526. doi: 10.1002/acn3.51021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Findlay Black H, Wright GEB, Collins JA, Caron N, Kay C, Xia Q, et al. Frequency of the loss of CAA interruption in the HTT CAG tract and implications for Huntington disease in the reduced penetrance range. Genet Med. 2020;22:2108–2113. doi: 10.1038/s41436-020-0917-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Florian RT, Kraft F, Leitão E, Kaya S, Klebe S, Magnin E, et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with familial adult myoclonic epilepsy type 3. Nat Commun. 2019;10:4919. doi: 10.1038/s41467-019-12763-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fondon JW, Hammock EAD, Hannan AJ, King DG. Simple sequence repeats: genetic modulators of brain function and behavior. Trends Neurosci. 2008;31:328–334. doi: 10.1016/j.tins.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 42.Fournier C, Barbier M, Camuzat A, Anquetil V, Lattante S, Clot F, et al. Relations between C9orf72 expansion size in blood, age at onset, age at collection and transmission across generations in patients and presymptomatic carriers. Neurobiol Aging. 2019;74:234.e231–234.e238. doi: 10.1016/j.neurobiolaging.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 43.Francastel C, Magdinier F. DNA methylation in satellite repeats disorders. Essays Biochem. 2019;63:757–771. doi: 10.1042/ebc20190028. [DOI] [PubMed] [Google Scholar]
- 44.Fratta P, Collins T, Pemble S, Nethisinghe S, Devoy A, Giunti P, et al. Sequencing analysis of the spinal bulbar muscular atrophy CAG expansion reveals absence of repeat interruptions. Neurobiol Aging. 2014;35:443.e441–443.e443. doi: 10.1016/j.neurobiolaging.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao R, Matsuura T, Coolbaugh M, Zühlke C, Nakamura K, Rasmussen A, et al. Instability of expanded CAG/CAA repeats in spinocerebellar ataxia type 17. Eur J Med Genet. 2008;16:215–222. doi: 10.1038/sj.ejhg.5201954. [DOI] [PubMed] [Google Scholar]
- 46.Giesselmann P, Brändl B, Raimondeau E, Bowen R, Rohrandt C, Tandon R, et al. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat Biotechnol. 2019;37:1478–1481. doi: 10.1038/s41587-019-0293-x. [DOI] [PubMed] [Google Scholar]
- 47.Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Engelborghs S, De Bleecker J, et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol Psychiatry. 2016;21:1112–1124. doi: 10.1038/mp.2015.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilpatrick T, Lee I, Graham JE, Raimondeau E, Bowen R, Heron A, et al. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat Biotechnol. 2020;38:433–438. doi: 10.1038/s41587-020-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glasmacher SA, Wong C, Pearson IE, Pal S. Survival and prognostic factors in C9orf72 repeat expansion carriers. JAMA Neurol. 2020;77:367. doi: 10.1001/jamaneurol.2019.3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goodman FR, Bacchelli C, Brady AF, Brueton LA, Fryns JP, Mortlock DP, et al. Novel HOXA13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am J Hum Genet. 2000;67:197–202. doi: 10.1086/302961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gouw LG, Castañeda MA, McKenna CK, Digre KB, Pulst SM, Perlman S, et al. Analysis of the dynamic mutation in the SCA7 gene shows marked parental effects on CAG repeat transmission. Hum Mol Genet. 1998;7:525–532. doi: 10.1093/hmg/7.3.525. [DOI] [PubMed] [Google Scholar]
- 52.Grewal RP, Karkera JD, Grewal RK, Detera-Wadleigh SD. Mutation analysis of oculopharyngeal muscular dystrophy in hispanic American families. Arch Neurol. 1999;56:1378. doi: 10.1001/archneur.56.11.1378. [DOI] [PubMed] [Google Scholar]
- 53.Gu Y, Shen Y, Gibbs RA, Nelson DL. Identification of FMR2, a novel gene associated with the FRAXE CCG repeat and CpG island. Nat Genet. 1996;13:109–113. doi: 10.1038/ng0596-109. [DOI] [PubMed] [Google Scholar]
- 54.Gusella JF, MacDonald ME, Lee JM. Genetic modifiers of Huntington's disease. Mov Disord. 2014;29:1359–1365. doi: 10.1002/mds.26001. [DOI] [PubMed] [Google Scholar]
- 55.Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB, Moine H, Kooy RF, et al. Fragile X syndrome. Nat Rev Dis Primers. 2017;3:17065. doi: 10.1038/nrdp.2017.65. [DOI] [PubMed] [Google Scholar]
- 56.Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/WNL.57.1.127. [DOI] [PubMed] [Google Scholar]
- 57.Halman A, Oshlack A. Accuracy of short tandem repeats genotyping tools in whole exome sequencing data. F1000Research. 2020;9:200. doi: 10.1101/2020.02.03.933002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hannan AJ. Tandem repeat polymorphisms. New York: Springer; 2012. [Google Scholar]
- 59.Hannan AJ. Tandem repeats mediating genetic plasticity in health and disease. Nat Rev Genet. 2018;19:286–298. doi: 10.1038/nrg.2017.115. [DOI] [PubMed] [Google Scholar]
- 60.He F, Todd P. Epigenetics in nucleotide repeat expansion disorders. Semin Neurol. 2011;31:470–483. doi: 10.1055/s-0031-1299786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Höijer I, Tsai Y-C, Clark TA, Kotturi P, Dahl N, Stattin E-L, et al. Detailed analysis of HTT repeat elements in human blood using targeted amplification-free long-read sequencing. Hum Mutat. 2018;39:1262–1272. doi: 10.1002/humu.23580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Holmes SE, O'Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, et al. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease–like 2. Nat Genet. 2001;29:377–378. doi: 10.1038/ng760. [DOI] [PubMed] [Google Scholar]
- 63.Holmes SE, O'Hearn EE, McInnis MG, Gorelick-Feldman DA, Kleiderlein JJ, Callahan C, et al. Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12. Nat Genet. 1999;23:391–392. doi: 10.1038/70493. [DOI] [PubMed] [Google Scholar]
- 64.Hughes J, Piltz S, Rogers N, McAninch D, Rowley L, Thomas P. Mechanistic insight into the pathology of polyalanine expansion disorders revealed by a mouse model for X-linked hypopituitarism. PLoS Genet. 2013;9:e1003290. doi: 10.1371/journal.pgen.1003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iacoangeli A, Al Khleifat A, Jones AR, Sproviero W, Shatunov A, Opie-Martin S, et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol Commun. 2019;7:115. doi: 10.1186/s40478-019-0724-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ikeuchi T, Koide R, Tanaka H, Onodera O, Igarashi S, Takahashi H, et al. Dentatorubral-pallidoluysian atrophy: clinical features are closely related to unstable expansions of trinucleotide (CAG) repeat. Ann Neurol. 1995;37:769–775. doi: 10.1002/ana.410370610. [DOI] [PubMed] [Google Scholar]
- 67.Ishige T, Sawai S, Itoga S, Sato K, Utsuno E, Beppu M, et al. Pentanucleotide repeat-primed PCR for genetic diagnosis of spinocerebellar ataxia type 31. J Hum Genet. 2012;57:807–808. doi: 10.1038/jhg.2012.112. [DOI] [PubMed] [Google Scholar]
- 68.Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet. 2018;50:581–590. doi: 10.1038/s41588-018-0067-2. [DOI] [PubMed] [Google Scholar]
- 69.Ishiura H, Shibata S, Yoshimura J, Suzuki Y, Qu W, Doi K, et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet. 2019;51:1222–1232. doi: 10.1038/s41588-019-0458-z. [DOI] [PubMed] [Google Scholar]
- 70.Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36:338–345. doi: 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jayadev S, Bird TD. Hereditary ataxias: overview. Genet Med. 2013;15:673–683. doi: 10.1038/gim.2013.28. [DOI] [PubMed] [Google Scholar]
- 72.Kang C, Liang C, Ahmad KE, Gu Y, Siow S-F, Colebatch JG, et al. High degree of genetic heterogeneity for hereditary cerebellar ataxias in Australia. Cerebellum. 2019;18:137–146. doi: 10.1007/s12311-018-0969-7. [DOI] [PubMed] [Google Scholar]
- 73.Kato M, Saitoh S, Kamei A, Shiraishi H, Ueda Y, Akasaka M, et al. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome) Am J Hum Genet. 2007;81:361–366. doi: 10.1086/518903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet. 1994;8:221–228. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- 75.Kebschull JM, Zador AM. Sources of PCR-induced distortions in high-throughput sequencing data sets. Nucl Acids Res. 2015;43:e143–e143. doi: 10.1093/nar/gkv717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khristich AN, Mirkin SM. On the wrong DNA track: molecular mechanisms of repeat-mediated genome instability. J Biol Chem. 2020;295:4134–4170. doi: 10.1074/jbc.REV119.007678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet. 2011;89:121–130. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, et al. Unstable expansion of CAG repeat in hereditary dentatorubral–pallidoluysian atrophy (DRPLA) Nat Genet. 1994;6:9–13. doi: 10.1038/ng0194-9. [DOI] [PubMed] [Google Scholar]
- 79.Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8) Nat Genet. 1999;21:379–384. doi: 10.1038/7710. [DOI] [PubMed] [Google Scholar]
- 80.Kovaka S, Fan Y, Ni B, Timp W, Schatz MC. Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. Nat Biotechnol. 2020;39:431–441. doi: 10.1038/s41587-020-0731-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kratter IH, Finkbeiner S. PolyQ disease: too many Qs, too much function? Neuron. 2010;67:897–899. doi: 10.1016/j.neuron.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kuhlenbäumer G, Kress W, Ringelstein EB, Stögbauer F. Thirty-seven CAG repeats in the androgen receptor gene in two healthy individuals. J Neurol. 2001;248:23–26. doi: 10.1007/s004150170265. [DOI] [PubMed] [Google Scholar]
- 83.Kumar KR, Cortese A, Tomlinson SE, Efthymiou S, Ellis M, Zhu D, et al. RFC1 expansions can mimic hereditary sensory neuropathy with cough and Sjögren syndrome. Brain. 2020;143:e82. doi: 10.1093/brain/awaa244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kumar KR, Cowley MJ, Davis RL. Next-generation sequencing and emerging technologies. Semin Thromb Hemost. 2019;45:661–673. doi: 10.1055/s-0039-1688446. [DOI] [PubMed] [Google Scholar]
- 85.Kuyumcu-Martinez NM, Cooper TA. Misregulation of alternative splicing causes pathogenesis in myotonic dystrophy. Prog Mol Subcell Biol. 2006;44:133–159. doi: 10.1007/978-3-540-34449-0_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.LaCroix AJ, Stabley D, Sahraoui R, Adam MP, Mehaffey M, Kernan K, et al. GGC repeat expansion and exon 1 methylation of XYLT1 is a common pathogenic variant in baratela-scott syndrome. Am J Hum Genet. 2019;104:35–44. doi: 10.1016/j.ajhg.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lalioti MD, Scott HS, Buresi C, Rossier C, Bottani A, Morris MA, et al. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature. 1997;386:847–851. doi: 10.1038/386847a0. [DOI] [PubMed] [Google Scholar]
- 88.Landrian I, McFarland KN, Liu J, Mulligan CJ, Rasmussen A, Ashizawa T. Inheritance patterns of ATCCT repeat interruptions in spinocerebellar ataxia type 10 (SCA10) expansions. PLoS ONE. 2017;12:e0175958–e0175958. doi: 10.1371/journal.pone.0175958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Laumonnier F, Ronce N, Hamel BC, Thomas P, Lespinasse J, Raynaud M, et al. Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet. 2002;71:1450–1455. doi: 10.1086/344661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Leehey MA. Fragile X-associated tremor/ataxia syndrome: clinical phenotype, diagnosis, and treatment. J Investig Med. 2009;57:830–836. doi: 10.2310/JIM.0b013e3181af59c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lehesjoki A, Kälviäinen R, et al. Unverricht-Lundborg disease. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews. Seattle: University of Washington; 2014. [Google Scholar]
- 92.Linhares SDC, Horta WG, Marques Júnior W. Spinocerebellar ataxia type 7 (SCA7): family princeps’ history, genealogy and geographical distribution. Arch Neuropsychiatry. 2006;64:222–227. doi: 10.1590/s0004-282x2006000200010. [DOI] [PubMed] [Google Scholar]
- 93.Liu Q, Tong Y, Wang K. Genome-wide detection of short tandem repeat expansions by long-read sequencing. BMC Bioinform. 2020;21:542. doi: 10.1186/s12859-020-03876-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lone WG, Khan IA, Poornima S, Shaik NA, Meena AK, Rao KP, et al. Exploration of CAG triplet repeat in nontranslated region of SCA12 gene. J Genet. 2016;95:427–432. doi: 10.1007/s12041-016-0624-3. [DOI] [PubMed] [Google Scholar]
- 95.Ma D, Tan YJ, Ng ASL, Ong HL, Sim W, Lim WK, et al. Association of NOTCH2NLC repeat expansions With parkinson disease. JAMA Neurol. 2020;77:1–5. doi: 10.1001/jamaneurol.2020.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 97.Maltecca F, Filla A, Castaldo I, Coppola G, Fragassi NA, Carella M, et al. Intergenerational instability and marked anticipation in SCA-17. Neurology. 2003;61:1441–1443. doi: 10.1212/01.wnl.0000094123.09098.a0. [DOI] [PubMed] [Google Scholar]
- 98.Mantere T, Kersten S, Hoischen A. Long-read sequencing emerging in medical genetics. Front Genet. 2019;10:426. doi: 10.3389/fgene.2019.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matilla T, Volpini V, Genís D, Rosell J, Corral J, Dávalos A, et al. Presymptomatic analysis of spinocerebellar ataxia type 1 (SCA1) via the expansion of the SCA1 CAG-repeat in a large pedigree displaying anticipation and parental male bias. Hum Mol Genet. 1993;2:2123–2128. doi: 10.1093/hmg/2.12.2123. [DOI] [PubMed] [Google Scholar]
- 100.Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26:191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- 101.McColgan P, Tabrizi SJ. Huntington's disease: a clinical review. Eur J Neurol. 2018;25:24–34. doi: 10.1111/ene.13413. [DOI] [PubMed] [Google Scholar]
- 102.McFarland KN, Liu J, Landrian I, Godiska R, Shanker S, Yu F, et al. SMRT sequencing of long tandem nucleotide repeats in SCA10 reveals unique insight of repeat expansion structure. PLoS ONE. 2015;10:e0135906. doi: 10.1371/journal.pone.0135906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McFarland KN, Liu J, Landrian I, Zeng D, Raskin S, Moscovich M, et al. Repeat interruptions in spinocerebellar ataxia type 10 expansions are strongly associated with epileptic seizures. Neurogenetics. 2014;15:59–64. doi: 10.1007/s10048-013-0385-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Meienberg J, Bruggmann R, Oexle K, Matyas G. Clinical sequencing: is WGS the better WES? Hum Genet. 2016;135:359–362. doi: 10.1007/s00439-015-1631-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Miller DE, Sulovari A, Wang T, Loucks H, Hoekzema K, Munson KM et al (2020) Targeted long-read sequencing resolves complex structural variants and identifies missing disease-causing variants. bioRxiv. 10.1101/2020.11.03.365395
- 106.Mitsuhashi S, Frith MC, Mizuguchi T, Miyatake S, Toyota T, Adachi H, et al. Tandem-genotypes: robust detection of tandem repeat expansions from long DNA reads. Genome Biol. 2019 doi: 10.1186/s13059-019-1667-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mizuguchi T, Toyota T, Adachi H, Miyake N, Matsumoto N, Miyatake S. Detecting a long insertion variant in SAMD12 by SMRT sequencing: implications of long-read whole-genome sequencing for repeat expansion diseases. J Hum Genet. 2019;64:191–197. doi: 10.1038/s10038-018-0551-7. [DOI] [PubMed] [Google Scholar]
- 108.Moore RC, Xiang F, Monaghan J, Han D, Zhang Z, Edström L, et al. Huntington disease phenocopy is a familial prion disease. Am J Hum Genet. 2001;69:1385–1388. doi: 10.1086/324414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mor-Shaked H, Eiges R. Reevaluation of FMR1 hypermethylation timing in Fragile X syndrome. Front Mol Neurosci. 2018;11:31. doi: 10.3389/fnmol.2018.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moseley ML, Schut LJ, Bird TD, Koob MD, Day JW, Ranum LP. SCA8 CTG repeat: en masse contractions in sperm and intergenerational sequence changes may play a role in reduced penetrance. Hum Mol Genet. 2000;9:2125–2130. doi: 10.1093/hmg/9.14.2125. [DOI] [PubMed] [Google Scholar]
- 111.Moss DJH, Poulter M, Beck J, Hehir J, Polke JM, Campbell T, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology. 2014;82:292–299. doi: 10.1212/WNL.0000000000000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mousavi N, Shleizer-Burko S, Yanicky R, Gymrek M. Profiling the genome-wide landscape of tandem repeat expansions. Nucl Acids Res. 2019;47:e90–e90. doi: 10.1093/nar/gkz501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Myers RH. Huntington's disease genetics. NeuroRx. 2004;1:255–262. doi: 10.1602/neurorx.1.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nakamura H, Doi H, Mitsuhashi S, Miyatake S, Katoh K, Frith MC, et al. Long-read sequencing identifies the pathogenic nucleotide repeat expansion in RFC1 in a Japanese case of CANVAS. J Hum Genet. 2020;65:475–480. doi: 10.1038/s10038-020-0733-y. [DOI] [PubMed] [Google Scholar]
- 115.Nakamura K, Jeong S-Y, Uchihara T, Anno M, Nagashima K, Nagashima T, et al. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10:1441–1448. doi: 10.1093/hmg/10.14.1441. [DOI] [PubMed] [Google Scholar]
- 116.Nallathambi J, Moumné L, De Baere E, Beysen D, Usha K, Sundaresan P, et al. A novel polyalanine expansion in FOXL2: the first evidence for a recessive form of the blepharophimosis syndrome (BPES) associated with ovarian dysfunction. Hum Genet. 2007;121:107–112. doi: 10.1007/s00439-006-0276-0. [DOI] [PubMed] [Google Scholar]
- 117.Ng ASL, Lim WK, Xu Z, Ong HL, Tan YJ, Sim WY, et al. NOTCH2NLC GGC repeat expansions are associated with sporadic essential tremor: variable disease expressivity on long-term follow-up. Ann Neurol. 2020;88:614–618. doi: 10.1002/ana.25803. [DOI] [PubMed] [Google Scholar]
- 118.Ogasawara M, Iida A, Kumutpongpanich T, Ozaki A, Oya Y, Konishi H, et al. CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations. Acta Neuropathol Commun. 2020;8:204. doi: 10.1186/s40478-020-01084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Okubo M, Doi H, Fukai R, Fujita A, Mitsuhashi S, Hashiguchi S, et al. GGC repeat expansion of NOTCH2NLC in adult patients with leukoencephalopathy. Ann Neurol. 2019;86:962–968. doi: 10.1002/ana.25586. [DOI] [PubMed] [Google Scholar]
- 120.Orr HT, Chung M-y, Banfi S, Kwiatkowski TJ, Servadio A, Beaudet AL, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- 121.Pagnamenta AT, Kaiyrzhanov R, Zou Y, Da'as SI, Maroofian R, Donkervoort S, et al. An ancestral 10-bp repeat expansion in VWA1 causes recessive hereditary motor neuropathy. Brain. 2021 doi: 10.1093/brain/awaa420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Park H, Kim H-J, Jeon BS. Parkinsonism in spinocerebellar ataxia. BioMed Res Int. 2015;2015:125273–125273. doi: 10.1155/2015/125273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Paulson H. Repeat expansion diseases. Handb Clin Neurol. 2018;147:105–123. doi: 10.1016/B978-0-444-63233-3.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Payne A, Holmes N, Clarke T, Munro R, Debebe B, Loose M (2020) Nanopore adaptive sequencing for mixed samples, whole exome capture and targeted panels. bioRxiv. 10.1101/2020.02.03.926956
- 125.La Spada RA. Trinucleotide repeat instability: genetic features and molecular mechanisms. Brain Pathol. 1997;7:943–963. doi: 10.1111/j.1750-3639.1997.tb00895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rafehi H, Szmulewicz DJ, Bennett MF, Sobreira NLM, Pope K, Smith KR, et al. Bioinformatics-based identification of expanded repeats: a non-reference intronic pentamer expansion in RFC1 causes CANVAS. Am J Hum Genet. 2019;105:151–165. doi: 10.1016/j.ajhg.2019.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ranen NG, Stine OC, Abbott MH, Sherr M, Codori AM, Franz ML, et al. Anticipation and instability of IT-15 (CAG)n repeats in parent-offspring pairs with Huntington disease. Am J Hum Genet. 1995;57:593–602. [PMC free article] [PubMed] [Google Scholar]
- 128.Rhoads A, Au KF. PacBio sequencing and its applications. Genom Proteom Bioinform. 2015;13:278–289. doi: 10.1016/j.gpb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Richard P, Trollet C, Stojkovic T, de Becdelievre A, Perie S, Pouget J, et al. Correlation between PABPN1 genotype and disease severity in oculopharyngeal muscular dystrophy. Neurology. 2017;88:359–365. doi: 10.1212/WNL.0000000000003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ridley RM, Frith CD, Farrer LA, Conneally PM. Patterns of inheritance of the symptoms of Huntington's disease suggestive of an effect of genomic imprinting. J Med Genet. 1991;28:224–231. doi: 10.1136/jmg.28.4.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. 2014;42:174–183. doi: 10.1159/000358801. [DOI] [PubMed] [Google Scholar]
- 132.Russ J, Liu EY, Wu K, Neal D, Suh E, Irwin DJ, et al. Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol. 2015;129:39–52. doi: 10.1007/s00401-014-1365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996;14:277–284. doi: 10.1038/ng1196-277. [DOI] [PubMed] [Google Scholar]
- 134.Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, et al. Spinocerebellar ataxia type 31 is associated with "inserted" penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85:544–557. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schneider SA, Bird T. Huntington's disease, Huntington's disease look-alikes, and benign hereditary chorea: what's new? Mov Disord Clin Pract. 2016;3:342–354. doi: 10.1002/mdc3.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schöls L, Bauer I, Zühlke C, Schulte T, Kölmel C, Bürk K, et al. Do CTG expansions at the SCA8 locus cause ataxia? Ann Neurol. 2003;54:110–115. doi: 10.1002/ana.10608. [DOI] [PubMed] [Google Scholar]
- 137.Schüle B, McFarland KN, Lee K, Tsai Y-C, Nguyen K-D, Sun C, et al. Parkinson’s disease associated with pure ATXN10 repeat expansion. NPJ Parkinsons Dis. 2017;3:27. doi: 10.1038/s41531-017-0029-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Scriba CK, Beecroft SJ, Clayton JS, Cortese A, Sullivan R, Yau WY, et al. A novel RFC1 repeat motif (ACAGG) in two Asia-Pacific CANVAS families. Brain. 2020;143:2904–2910. doi: 10.1093/brain/awaa263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Seixas AI, Loureiro JR, Costa C, Ordóñez-Ugalde A, Marcelino H, Oliveira CL, et al. A pentanucleotide ATTTC repeat insertion in the non-coding region of DAB1, mapping to SCA37, causes spinocerebellar ataxia. Am J Hum Genet. 2017;101:87–103. doi: 10.1016/j.ajhg.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Semaka A, Kay C, Doty C, Collins JA, Bijlsma EK, Richards F, et al. CAG size-specific risk estimates for intermediate allele repeat instability in Huntington disease. J Med Genet. 2013;50:696–703. doi: 10.1136/jmedgenet-2013-101796. [DOI] [PubMed] [Google Scholar]
- 141.Sequeiros J, Seneca S, Martindale J. Consensus and controversies in best practices for molecular genetic testing of spinocerebellar ataxias. Eur J Hum Genet. 2010;18:1188–1195. doi: 10.1038/ejhg.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shin JH, Park H, Ehm GH, Lee WW, Yun JY, Kim YE, et al. The pathogenic role of low range repeats in SCA17. PLoS ONE. 2015;10:e0135275. doi: 10.1371/journal.pone.0135275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shortt JA, Ruggiero RP, Cox C, Wacholder AC, Pollock DD. Finding and extending ancient simple sequence repeat-derived regions in the human genome. Mob DNA. 2020;11:11. doi: 10.1186/s13100-020-00206-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Smith SS, Laayoun A, Lingeman RG, Baker DJ, Riley J. Hypermethylation of telomere-like foldbacks at codon 12 of the human c-Ha-ras gene and the trinucleotide repeat of the FMR-1 gene of fragile X. J Mol Biol. 1994;243:143–151. doi: 10.1006/jmbi.1994.1640. [DOI] [PubMed] [Google Scholar]
- 145.Sobczak K, Krzyzosiak WJ. CAG repeats containing CAA interruptions form branched hairpin structures in spinocerebellar ataxia type 2 transcripts. J Biol Chem. 2005;280:3898–3910. doi: 10.1074/jbc.M409984200. [DOI] [PubMed] [Google Scholar]
- 146.Sone J, Mitsuhashi S, Fujita A, Mizuguchi T, Hamanaka K, Mori K, et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet. 2019;51:1215–1221. doi: 10.1038/s41588-019-0459-y. [DOI] [PubMed] [Google Scholar]
- 147.Spada ARL, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 148.Sproviero W, Shatunov A, Stahl D, Shoai M, Van Rheenen W, Jones AR, et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol Aging. 2017;51:178.e171–178.e179. doi: 10.1016/j.neurobiolaging.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stevanin G, Herman A, Dürr A, Jodice C, Frontali M, Agid Y, et al. Are (CTG)n expansions at the SCA8 locus rare polymorphisms? Nat Genet. 2000;24:213–213. doi: 10.1038/73408. [DOI] [PubMed] [Google Scholar]
- 150.Strømme P, Mangelsdorf ME, Shaw MA, Lower KM, Lewis SME, Bruyere H, et al. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet. 2002;30:441–445. doi: 10.1038/ng862. [DOI] [PubMed] [Google Scholar]
- 151.Suh E, Grando K, Van Deerlin VM. Validation of a long-read PCR assay for sensitive detection and sizing of C9orf72 hexanucleotide repeat expansions. J Mol Diagn. 2018;20:871–882. doi: 10.1016/j.jmoldx.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Suthiphosuwan S, Sasikumar S, Munoz DG, Chan DK, Montanera WJ, Bharatha A. MRI diagnosis of neuronal intranuclear inclusion disease leukoencephalopathy. Neurol Clin Pract. 2019;9:497–499. doi: 10.1212/cpj.0000000000000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Svrzikapa N, Longo KA, Prasad N, Boyanapalli R, Brown JM, Dorset D, et al. Investigational assay for haplotype phasing of the Huntingtin gene. Mol Ther Methods Clin Dev. 2020;19:162–173. doi: 10.1016/j.omtm.2020.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Swinnen B, Robberecht W, Van Den Bosch L. RNA toxicity in non-coding repeat expansion disorders. EMBO J. 2019 doi: 10.15252/embj.2018101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Todd PK, Paulson HL. RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol. 2010;67:291–300. doi: 10.1002/ana.21948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tomé S, Gourdon G. Fast assays to detect interruptions in CTG.CAG repeat expansions. Methods Mol Biol. 2020;2056:11–23. doi: 10.1007/978-1-4939-9784-8_2. [DOI] [PubMed] [Google Scholar]
- 157.Tsai YC, Greenberg D, Powell J, Höijer I, Ameur A, Strahl M, et al. Amplification-free, CRISPR-Cas9 targeted enrichment and SMRT sequencing of repeat-expansion disease causative genomic regions. BioRxiv. 2017 doi: 10.1101/203919. [DOI] [Google Scholar]
- 158.Ummat A, Bashir A. Resolving complex tandem repeats with long reads. Bioinformatics. 2014;30:3491–3498. doi: 10.1093/bioinformatics/btu437. [DOI] [PubMed] [Google Scholar]
- 159.Van Kuilenburg ABP, Tarailo-Graovac M, Richmond PA, Drögemöller BI, Pouladi MA, Leen R, et al. Glutaminase deficiency caused by short tandem repeat expansion in GLS. N Engl J Med. 2019;380:1433–1441. doi: 10.1056/nejmoa1806627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Van Mossevelde S, van der Zee J, Gijselinck I, Sleegers K, De Bleecker J, Sieben A, et al. Clinical evidence of disease anticipation in families segregating a C9orf72 repeat expansion. JAMA Neurol. 2017;74:445–452. doi: 10.1001/jamaneurol.2016.4847. [DOI] [PubMed] [Google Scholar]
- 161.Veneziano L, Frontali M, et al. DRPLA. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews. Seattle: University of Washington; 2016. [Google Scholar]
- 162.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu Y-H, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 163.Wang B, Tseng E, Baybayan P, Eng K, Regulski M, Jiao Y, et al. Variant phasing and haplotypic expression from long-read sequencing in maize. Commun Biol. 2020;3:78. doi: 10.1038/s42003-020-0805-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Warren ST, Muragaki Y, Mundlos S, Upton J, Olsen BR. Polyalanine expansion in synpolydactyly might result from unequal crossing-over of HOXD13. Science. 1997;275:408–409. doi: 10.1126/science.275.5298.408. [DOI] [PubMed] [Google Scholar]
- 165.Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol. 2019;37:1155–1162. doi: 10.1038/s41587-019-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wheeler VC, Persichetti F, McNeil SM, Mysore JS, Mysore SS, MacDonald ME, et al. Factors associated with HD CAG repeat instability in Huntington disease. J Med Genet. 2007;44:695–701. doi: 10.1136/jmg.2007.050930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, et al. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2–2) gene predicts Fuchs corneal dystrophy. PLoS ONE. 2012;7:e49083–e49083. doi: 10.1371/journal.pone.0049083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu X, et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron. 2011;70:427–440. doi: 10.1016/j.neuron.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Worth PF, Houlden H, Giunti P, Davis MB, Wood NW. Large, expanded repeats in SCA8 are not confined to patients with cerebellar ataxia. Nat Genet. 2000;24:214–215. doi: 10.1038/73411. [DOI] [PubMed] [Google Scholar]
- 170.Wright GEB, Black HF, Collins JA, Gall-Duncan T, Caron NS, Pearson CE, et al. Interrupting sequence variants and age of onset in Huntington's disease: clinical implications and emerging therapies. Lancet Neurol. 2020;19:930–939. doi: 10.1016/s1474-4422(20)30343-4. [DOI] [PubMed] [Google Scholar]
- 171.Wu TY, Taylor JM, Kilfoyle DH, Smith AD, McGuinness BJ, Simpson MP, et al. Autonomic dysfunction is a major feature of cerebellar ataxia, neuropathy, vestibular areflexia ‘CANVAS’ syndrome. Brain. 2014;137:2649–2656. doi: 10.1093/brain/awu196. [DOI] [PubMed] [Google Scholar]
- 172.Xi J, Wang X, Yue D, Dou T, Wu Q, Lu J, et al. 5' UTR CGG repeat expansion in GIPC1 is associated with oculopharyngodistal myopathy. Brain. 2020;144(2):601–614. doi: 10.1093/brain/awaa426. [DOI] [PubMed] [Google Scholar]
- 173.Xu P, Pan F, Roland C, Sagui C, Weninger K. Dynamics of strand slippage in DNA hairpins formed by CAG repeats: roles of sequence parity and trinucleotide interrupts. Nucl Acids Res. 2020;48:2232–2245. doi: 10.1093/nar/gkaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yamamoto H, Imai K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin Oncol. 2019;46:261–270. doi: 10.1053/j.seminoncol.2019.08.003. [DOI] [PubMed] [Google Scholar]
- 175.Yuan Y, Liu Z, Hou X, Li W, Ni J, Huang L, et al. Identification of GGC repeat expansion in the NOTCH2NLC gene in amyotrophic lateral sclerosis. Neurology. 2020;95(24):e3394–e3405. doi: 10.1212/wnl.0000000000010945. [DOI] [PubMed] [Google Scholar]
- 176.Yum K, Wang ET, Kalsotra A. Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr Opin Genet Dev. 2017;44:30–37. doi: 10.1016/j.gde.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zaheer F, Fee D. Spinocerebellar ataxia 7: a report of unaffected siblings who married into different SCA 7 families. Case Rep Neurol Med. 2014;2014:1–3. doi: 10.1155/2014/514791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zeman A, Stone J, Porteous M, Burns E, Barron L, Warner J. Spinocerebellar ataxia type 8 in Scotland: genetic and clinical features in seven unrelated cases and a review of published reports. J Neurol Neurosurg Psychiatry. 2004;75:459–465. doi: 10.1136/jnnp.2003.018895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zeng S, Zhang M-Y, Wang X-J, Hu Z-M, Li J-C, Li N, et al. Long-read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy. J Med Genet. 2019;56:265–270. doi: 10.1136/jmedgenet-2018-105484. [DOI] [PubMed] [Google Scholar]
- 180.Zhang N, Ashizawa T. RNA toxicity and foci formation in microsatellite expansion diseases. Curr Opin Genet Dev. 2017;44:17–29. doi: 10.1016/j.gde.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.