

Abstract
Molecular dimers have been frequently found to play an important role in room temperature phosphorescence (RTP), but its inherent working mechanism has remained unclear. Herein a series of unique characteristics, including singlet excimer emission and thermally activated delayed fluorescence, were successfully integrated into a new RTP luminogen of CS-2COOCH3 to clearly reveal the excited-state process of RTP and the special role of molecular dimers in persistent RTP emission.
The first purely organic room temperature phosphorescence (RTP) luminogen, with singlet excimer emission and thermally activated delayed fluorescence (TADF) effect, was successfully developed. 
Introduction
Persistent room temperature phosphorescence (RTP), an afterglow from an excited triplet state, has drawn considerable attention for its wide applications in displays, anti-fake, information storage, bio-imaging and so on.1 Until now, persistent RTP phenomena have been mainly limited to inorganic compounds or organometallic complexes, but relatively scarce in purely organic luminogens because of their weak spin–orbit coupling and inferior stability of triplet excitons, accompanied by unclear understanding of the exact inherent mechanism.2
Because of the enthusiasm of scientists, a series of persistent organic RTP systems have been developed and the internal mechanism of RTP is gradually being revealed accordingly. Since the persistent RTP effect just exists in the solid state, the relationship between molecular packing and the effect has attracted much attention.3 Particularly, the molecular dimer, the most important intermolecular interaction form, has been explored frequently.4 As one of the most impressive examples, Huang and coworkers have successfully developed a series of carbazole derivatives with an ultralong RTP effect, in which molecular dimers with H-aggregation could largely prolong the lifetimes of triplet excitons.4a Furthermore, dynamic RTP color tuning from violet to green was achieved in a single-component molecular crystal by utilizing the different energy levels between monomer and dimers in H-aggregation.4b Besides, Chi and coworkers proposed the key role of the intermolecular n–π* electronic coupling in molecular dimers for achieving efficient persistent RTP.4c Also, our group identified that strong π–π stacking within molecular dimers could alter the lifetimes of triplet excitons through decreasing their radiative and non-radiative rates in a series of phenothiazine 5,5-dioxide derivatives.4d,e But what about the exact role of molecular dimers in crystals? Where does the persistent RTP exactly come from, monomer-dominated emission (P1) or a triplet excimer (P2) (Fig. 1)?
Fig. 1. (A) The query about the role of dimers in the persistent RTP effect and the proposed solution, in which F1 indicates monomer-dominated fluorescence, P1 indicates monomer-dominated RTP, F2 indicates singlet excimer fluorescence and P2 indicates possible triplet excimer RTP. (B) The molecular structure and design strategy of CS-2COOCH3.

After an overview of previous reports concerning purely organic RTP, it was found that the RTP was more likely to be monomer-dominated emission (P1), as just the corresponding monomer-dominated fluorescence (F1) occurred and no excimer fluorescence (F2) could be observed.1–4 However, there has been no direct and powerful proof to confirm this point, for the lack of appropriate RTP examples. As mentioned above, the π–π stacking in molecular dimers could lead to the persistent RTP effect in phenothiazine 5,5-dioxide derivatives, and also a singlet excimer could be formed as a result of π–π interactions in some cases.5 Thus, persistent RTP and singlet excimer emission (F2) could be integrated together, with suitable intermolecular π–π interactions. Then, through a careful comparison of emission peaks between RTP and singlet excimer, the above questions might be answered, as the triplet state is always lower in energy than the corresponding singlet one in theory. Besides, to well distinguish RTP from monomer-dominated emission or a triplet excimer, a small energy gap (ΔEST) between excited singlet (S1) and triplet (T1) states should be necessary. With this, the fluorescence and phosphorescence could closely appear in pairs (F1/P1 and F2/P2). Thus, thermally activated delayed fluorescence (TADF) luminogens with minimized energy gap between S1 and T1 states would be a good choice.6
In order to realize integrated RTP, with singlet excimer and TADF effects in one luminogen, two RTP building blocks of phenothiazine 5,5-dioxide and dimethyl isophthalate were combined together through the twisted linkage mode of a C–N bond to yield CS-2COOCH3.4d,e,7 With this structure, three main purposes could be achieved. Firstly, the persistent RTP effect could be maintained with the integration of two simple RTP building blocks. Secondly, the small ΔEST, as well as the TADF effect, would be more likely to be realized with this twisted D–A structure, in which the phenothiazine 5,5-dioxide group acted as electron donor (D) and dimethyl isophthalate as acceptor (A). Thirdly, the planar conformations of donor and acceptor units could promote multiple intermolecular π–π interactions, thus allowing realization of the formation of an excimer. Fortunately, persistent RTP and singlet excimer emissions, as well as the TADF effect, were all integrated into the target compound CS-2COOCH3, just as expected. With this, in-depth analyses of the excited state process in RTP became possible. And it was found that the persistent RTP was monomer dominated (P1), rather than from a triplet excimer (P2).
Results and discussion
CS-2COOCH3 could be easily synthesized through carbon-nitrogen coupling reaction, followed by oxidation with hydrogen peroxide (Scheme S1†). The molecular structure and purity have been well determined by 1H and 13C NMR spectra, high-resolution mass spectrum, elemental analysis and high-performance liquid chromatography. A crystal of CS-2COOCH3 showed the expected persistent RTP effect, and its afterglow could last for more than 1 s after turning off 365 nm UV radiation. However, it was strange that the afterglow was much blue-shifted compared to the one under UV irradiation (Fig. 2). This was totally different from the common RTP phenomena in previous reports, as the excited triplet state should be lower in energy than the corresponding singlet one in theory.1 Then photoluminescence (PL) spectra of a crystal, including fluorescence and phosphorescence, were measured at room temperature under air or N2 atmosphere (Fig. 2 and S1†). As shown in Fig. 2A and C, two very different emission peaks at 405 and 505 nm appeared in the fluorescence spectrum, while the main peak of RTP was located at 430 nm, accompanied by two shoulder peaks at 460 and 490 nm. The same went for those under N2 atmosphere (Fig. S1†). All of the three phosphorescence peaks showed similar excitation spectra, indicating the shoulder peaks came from the vibration energy levels of the main phosphorescence one (Fig. S2†). Thus, the occurrence of long-wavelength fluorescence around 505 nm should be the main reason for the visual blue-shifted afterglow. Then why did the CS-2COOCH3 crystal show double fluorescence emissions?
Fig. 2. (A) The normalized steady-state PL spectra of CS-2COOCH3 in different states—crystal, ground state and doped in poly(methyl methacrylate) (PMMA) film in air. (B) The fluorescence decay curves of CS-2COOCH3 crystal at 405 and 505 nm in air. (C) The RTP spectra, acquired after 10 ms delay, of CS-2COOCH3 in different states—crystal, ground state and doped in PMMA film under air atmosphere. (D) The RTP decay curves of CS-2COOCH3 crystal at 430, 460 and 490 nm. Insets: photos of CS-2COOCH3 crystal before and after turning off 365 nm UV radiation in air.

In order to clarify this, CS-2COOCH3 was doped in poly(methyl methacrylate) (PMMA) with a mass fraction of about 5% or dissolved in tetrahydrofuran (THF) to eliminate the influence of intermolecular interactions. As shown in Fig. 2A, C and in S3,† there was just one fluorescence peak at around 420 nm and no phosphorescence peak for the doped film and THF solution, in which the intermolecular interactions could be diminished and the emission was only from the monomer. Thus, the corresponding short-wavelength fluorescence peak at 405 nm for the crystal should be also monomer-dominated emission (F1), while the one at 505 nm was from aggregates, such as singlet excimer (F2).
To further confirm this point, the UV-visible absorption spectra of CS-2COOCH3 in different states were measured. As shown in Fig. S4,† one more shoulder peak at long wavelength of about 365 nm could be observed for CS-2COOCH3 crystal, while this peak disappeared in the solution state. This indicated the strong intermolecular coupling in the crystal. After it was ground heavily, the shoulder peak at 365 nm weakened largely, accompanied by nearly disappearing excimer emission at 505 nm, as a result of the destruction of molecular packing (Fig. S4† and 2A). Also, the excitation spectra of the two fluorescence peaks in the crystal were similar, which were both red-shifted in comparison with that in the solution state (Fig. S5†). These could well demonstrate the formation of a singlet excimer in the crystal with a particular molecular packing mode.8 Also, powder X-ray diffraction (PXRD) was carried out to monitor the molecular packing in the crystal and ground states of CS-2COOCH3. As shown in Fig. S6,† the PXRD pattern of the CS-2COOCH3 crystal was similar to the computed XRD pattern (as derived from X-ray crystallography), which could rule out the possible presence of two crystalline morphologies. After it was ground heavily, the PXRD peaks nearly disappeared as a result of the destroyed molecular packing. At the same time, the PL spectrum and corresponding excitation spectrum in the ground state were similar to those for the doped film (Fig. S7†), while their PL lifetimes were much different (Fig. S8†). It was believed that the rigid environment from PMMA could decrease the non-radiative motion of CS-2COOCH3 and restrict the diffusion of oxygen, thus leading to the much longer emission lifetime in PMMA film. Besides, the crystal and doped PMMA film of CS-2COOCH3 showed a similar TADF effect for their short-wavelength emissions at 405 or 420 nm with average lifetimes (τFs) of about 1 μs, while the one at 505 nm was much longer (10.6 μs) (Table 1). This could be well consistent with the character of a singlet excimer.8 On the other hand, an increasing component was observed in the time-resolved curve of 505 nm (Fig. 2B), which might come from its special PL process in the excited state. According to these experimental results, it was confirmed that the 505 nm emission in the fluorescence spectrum was indeed from the singlet excimer (F2), while the one at 405 nm was monomer-dominated emission (F1).
The lifetimes of CS-2COOCH3 in different states (τF = A1τ1 + A2τ2; A1: fraction of the shorter component; τ1: lifetime of the shorter component; A2: fraction of longer-lived component; τ2: lifetime of the longer component).
| State | λ F (nm) | τ 1 (ns) | A 1 (%) | τ 2 | A 2 (%) | τ F | Φ F (%) | λ P (nm) | τ P (ms) |
|---|---|---|---|---|---|---|---|---|---|
| Solution | 430 | 18.0 | 100 | — | — | 18.0 ns | 21.82 | — | — |
| Crystal | 405 | 55.2 | 13.14 | 1.45 μs | 86.86 | 1.27 μs | 7.96 | 430/460/490 | 91/87/86 |
| 505 | 10.0 | 3.74 | 11.0 μs | 96.26 | 10.6 μs | ||||
| Ground | 410 | 19.3 | 73.39 | 61.47 ns | 26.61 | 33.6 ns | 15.58 | — | — |
| Doped | 420 | 30.0 | 56.36 | 2.20 μs | 43.64 | 0.97 μs | 10.40 | — | — |
Interestingly, a unique double TADF effect was observed for the CS-2COOCH3 crystal in both monomer-dominated emission (F1) and excimer emission (F2) (Fig. 2B). To the best of our knowledge, this is the first example of double TADF effects in both monomer-dominated emission and excimer emission.9 Besides, the ΔEST of the crystal was measured to be only about 0.17 eV according to the low-temperature (77 K) PL spectra (Fig. S9†), which should be small enough to allow the intersystem crossing between S1 and T1 states, and thus to lead to the TADF effect. Then the temperature-dependent PL spectra of the CS-2COOCH3 crystal were measured from 77 K to room temperature. As shown in Fig. 3, the fluorescence peaks at 386 and 495 nm were much lower than those of phosphorescence (407 and 430 nm) at 77 K. With increasing temperature, the ratios of fluorescence peaks both increased, while those of phosphorescence decreased. At last, just two fluorescence peaks at about 405 and 505 nm could be observed at room temperature, and those of phosphorescence were much weakened. Since the TADF effect should be promoted with the increased temperature, these experimental results could well confirm the emission peak at 505 nm as the thermally activated delayed singlet excimer emission, rather than phosphorescence. Also, because of this unique TADF effect, the monomer-dominated fluorescence (F1) (405 nm) and phosphorescence (P1) (430 nm) could closely appear in pairs, just as we expected. Thus, the ideal PL spectra were achieved to identify the role of molecular dimer in the persistent RTP effect. With this, it was easy to determine that the persistent RTP should be monomer-dominated emission (P1), rather than triplet excimer emission (P2).
Fig. 3. Normalized PL spectra of CS-2COOCH3 crystal from 77 K to 298 K.

To explore the internal mechanism, the single-crystal structure of CS-2COOCH3 was carefully analyzed (Table S1†). As shown in Fig. 4, CS-2COOCH3 presented a twisted D–A conformation for the C–N linkage mode, which would be much beneficial for its TADF effect. Because of the planar conformations of phenothiazine 5,5-dioxide and dimethyl isophthalate units, they both presented parallel stacks facing each other in the crystal. Besides, the π–π vertical distance for adjacent phenothiazine 5,5-dioxide units was as short as 3.76 Å, while that of adjacent dimethyl isophthalate units was 3.59 Å, indicating the existence of multiple π–π interactions. With these, the unique molecular dimers were formed, thus allowing realization of singlet excimer emission. Furthermore, all of the molecular dimers were perpendicular or parallel to each other, which could well ensure the relatively strong singlet excimer emission at 505 nm. Also, with the effective π–π interactions, the persistent RTP effect could be achieved for the decreased phosphorescent radiative and non-radiative rates, just as in our previous reports.4d,e Then why was the RTP a monomer-dominated emission, rather than from a triplet excimer?
Fig. 4. (A) The single-crystal structure and molecular dimer (side view and top view) of CS-2COOCH3. (B) The natural transition orbitals (NTOs) of the T1 state for the monomer and dimer of CS-2COOCH3.

To answer this question, the natural transition orbitals (NTOs) of the T1 state for the monomer and dimer (derived from its single-crystal structure) of CS-2COOCH3 were evaluated by time-dependent density functional theory calculation.10 As shown in Fig. 4B, both monomer and dimer gave a charge transfer (CT)-dominated T1 state with hole located at phenothiazine 5,5-dioxide unit and particle mainly at dimethyl isophthalate unit. Particularly, only one molecule was involved in the NTO of the T1 state for the molecular dimer, which should be the main reason for the monomer-dominated RTP emission of the CS-2COOCH3 crystal. Besides, owing to the twisted D–A structure of CS-2COOCH3, the well-separated HOMOs and LUMOs could be achieved in both the monomer and dimer of CS-2COOCH3 (Fig. S10†). With this, a minimized ΔEST could be achieved, resulting in the TADF effect and close fluorescence/phosphorescence peaks. Thus, although molecular dimers existed in the crystal, the RTP from the excited triplet state was still monomer-dominated emission, rather than triplet excimer emission.
Then how did the monomer (involved in π–π stacking) generate persistent RTP? Actually, we could get some clues from the corresponding fluorescence properties. The fluorescent radiative rate (kr) and non-radiative rate (knr) for CS-2COOCH3 in different states were estimated based on the corresponding fluorescence lifetimes (τ) and quantum yields (ΦF), including the doped PMMA film (@420 nm), monomer-dominated emission in the crystal (@405 nm) and excimer emission in the crystal (@505 nm) (Table S2†). Their changing tendency is summarized in Fig. 5. Among them, the molecules doped in PMMA gave the highest kr and knr for the eliminated intermolecular interaction. As for the monomer involved in π–π stacking in the crystal, its kr and knr (@405 nm) decreased, thus benefiting a prolonged fluorescence lifetime according to the equation of τ = 1/(kr + knr).4d,7a Similarly, the lifetimes of triplet excitons could also be prolonged for the monomer involved in π–π interactions with much decreased phosphorescent radiative and non-radiative rates, thus resulting in the persistent RTP effect. As for the triplet state of the excimer, its phosphorescence radiative rate would be too slow to compete with the corresponding singlet one, and thus no emission could be realized for it.
Fig. 5. Changing tendency of the fluorescent radiative rate (kr) and non-radiative rate (knr) for CS-2COOCH3 in different states.

As revealed by the experimental results, coupled with theoretical calculations, it could be concluded that the persistent RTP was monomer-dominated emission (P1), rather than from a triplet excimer (P2). Thus, the PL process in the excited state of CS-2COOCH3 could be understood clearly (Fig. 6). First, monomers absorbed photons to form singlet excitons. Because of the small energy gap between S1 and T1 states, efficient intersystem crossing (ISC) and reverse intersystem crossing (RISC) between them could happen for some excitons, leading to the TADF emission with a lifetime of about 1 μs. Subsequently, some of the singlet excitons could interact with adjacent molecules in the ground state to form singlet excimers, for which the emission lifetime would further increase to about 10 μs for the decreased fluorescent radiative and non-radiative rates. On the other hand, some of the formed triplet excitons, originating from the ISC transition, would be trapped in 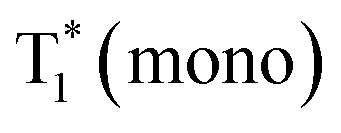 state with much decreased phosphorescent radiative and non-radiative rates for the π–π interactions. Finally, monomer-dominated persistent RTP occurred with lifetime up to about 90 ms. As for the triplet state of the excimer, no emission could be achieved for the ultra-low radiative rate.
state with much decreased phosphorescent radiative and non-radiative rates for the π–π interactions. Finally, monomer-dominated persistent RTP occurred with lifetime up to about 90 ms. As for the triplet state of the excimer, no emission could be achieved for the ultra-low radiative rate.
Fig. 6. The proposed PL process of CS-2COOCH3.

Analyzing the PL process in Fig. 6 carefully, it was not difficult to find that the microsecond-scale component of the singlet excimer (@505 nm) could be promoted through two ways (1 and 2), while that of the nanosecond-scale one was just from one way (3):
(1) S0 → S1(mono) → S1(excimer) → T1(excimer) → S1(excimer) → S0 (microsecond fluorescence);
(2) S0 → S1(mono) → T1(mono) → S1(mono) → S1(excimer) → S0 (microsecond fluorescence);
(3) S0 → S1(mono) → S1(excimer) → S0 (nanosecond fluorescence).
As additional microsecond-scale excitons could be formed through way (2) in the singlet excimer emission, in comparison with traditional TADF luminogens, an increasing component could be observed in the delayed component of 505 nm (Fig. 2B). Also, this could confirm the accuracy of the proposed PL process in Fig. 6 from another perspective.
Conclusions
In summary, the unique singlet excimer emission and TADF effect have been successfully integrated into the RTP luminogen of CS-2COOCH3 for the first time. Furthermore, the excited-state process of RTP was revealed clearly with the aid of these unique effects, especially for the role of molecular dimers in the persistent RTP emission. In previous reports, molecular dimers were frequently found to play an important role in the RTP effect, but their specific working mechanism has not been revealed in detail or just been simulated by theoretical calculation. This work has made the first attempt to reveal the actual role of dimers in the RTP effect based on solid experimental results. Of note, the ideal target RTP luminogen with two other unique emission effects was obtained with both rational molecular design and good luck. Incorporating the excited-state characteristics of singlet excimer emission and TADF, it has been well demonstrated that the persistent RTP was monomer dominated, rather than from a triplet excimer. The information obtained from this study would be of great importance for gaining a clear and deep understanding of the whole RTP process, thus guiding the further development of this research area.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We are grateful to the starting grants of Tianjin University and Tianjin Government, National Natural Science Foundation of China (no. 51903188) and Natural Science Foundation of Tianjin City (No. 19JCQNJC04500) for financial support.
Electronic supplementary information (ESI) available. CCDC 1944189 and 1963173. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04632a
Notes and references
- (a) Kabe R. Adachi C. Nature. 2017;550:384–387. doi: 10.1038/nature24010. [DOI] [PubMed] [Google Scholar]; (b) Fateminia S. Mao Z. Xu S. Yang Z. Chi Z. Liu B. Angew. Chem., Int. Ed. 2017;56:12160–12164. doi: 10.1002/anie.201705945. [DOI] [PubMed] [Google Scholar]; (c) Fang M. Yang J. Xiang X. Xie Y. Dong Y. Peng Q. Li Q. Li Z. Mater. Chem. Front. 2018;2:2124–2129. doi: 10.1039/C8QM00396C. [DOI] [Google Scholar]; (d) Huang L. Chen B. Zhang X. Trindle C. Liao F. Wang Y. Miao H. Luo Y. Zhang G. Angew. Chem., Int. Ed. 2018;57:16046–16050. doi: 10.1002/anie.201808861. [DOI] [PubMed] [Google Scholar]; (e) Salla C. A. M. Farias G. Rouzières M. Dechambenoit P. Durola F. Bock H. Souza B. d. Bechtold I. H. Angew. Chem., Int. Ed. 2019;58:6982–6986. doi: 10.1002/anie.201901672. [DOI] [PubMed] [Google Scholar]; (f) Hirata S. Adv. Sci. 2019;6:1900410. doi: 10.1002/advs.201900410. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Jinnai K. Kabe R. Adachi C. Adv. Mater. 2018:1800365. doi: 10.1002/adma.201800365. [DOI] [PubMed] [Google Scholar]; (h) Ogoshi T. Tsuchida H. Kakuta T. Yamagishi T. Taema A. Ono T. Sugimoto M. Mizuno M. Adv. Funct. Mater. 2018:1707369. doi: 10.1002/adfm.201707369. [DOI] [Google Scholar]; (i) Shoji Y. Ikabata Y. Wang Q. Nemoto D. Sakamoto A. Tanaka N. Seino J. Nakai H. Fukushima T. J. Am. Chem. Soc. 2017;1397:2728–2733. doi: 10.1021/jacs.6b11984. [DOI] [PubMed] [Google Scholar]; (j) Ward J. S. Nobuyasu R. S. Batsanov A. S. Data P. Monkman A. P. Diasb B. Bryce M. R. Chem. Commun. 2016;52:2612–2615. doi: 10.1039/C5CC09645F. [DOI] [PubMed] [Google Scholar]
- (a) Kenry Chen C. Liu B. Nat. Commun. 2019;10:2111. doi: 10.1038/s41467-019-10033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ma X. Wang J. Tian H. Acc. Chem. Res. 2019;52:738–748. doi: 10.1021/acs.accounts.8b00620. [DOI] [PubMed] [Google Scholar]; (c) Hirata S. Adv. Opt. Mater. 2017:1700116. doi: 10.1002/adom.201700116. [DOI] [Google Scholar]
- (a) Yang J. Ren Z. Xie Z. Liu Y. Wang C. Xie Y. Peng Q. Xu B. Tian W. Zhang F. Chi Z. Li Q. Li Z. Angew. Chem., Int. Ed. 2017;56:880–884. doi: 10.1002/anie.201610453. [DOI] [PubMed] [Google Scholar]; (b) Yang J. Gao X. Xie Z. Gong Y. Fang M. Peng Q. Chi Z. Li Z. Angew. Chem., Int. Ed. 2017;56:15299–15303. doi: 10.1002/anie.201708119. [DOI] [PubMed] [Google Scholar]; (c) Cai S. Shi H. Tian D. Ma H. Cheng Z. Wu Q. Gu M. Huang L. An Z. Peng Q. Huang W. Adv. Funct. Mater. 2018:1705045. doi: 10.1002/adfm.201705045. [DOI] [Google Scholar]; (d) Lucenti E. Forni A. Botta C. Carlucci L. Giannini C. Marinotto D. Pavanello A. Previtali A. Righetto S. Cariati E. Angew. Chem., Int. Ed. 2017;56:16302–16307. doi: 10.1002/anie.201710279. [DOI] [PubMed] [Google Scholar]; (e) Li Q. Tang Y. Hu W. Li Z. Small. 2018:1801560. doi: 10.1002/smll.201801560. [DOI] [PubMed] [Google Scholar]; (f) Zhou B. Yan D. Adv. Funct. Mater. 2019;29:1807599. doi: 10.1002/adfm.201807599. [DOI] [Google Scholar]; (g) Yuan J. Wang S. Ji Y. Chen R. Zhu Q. Wang Y. Zheng C. Tao Y. Fan Q. Huang W. Mater. Horiz. 2019;6:1259–1264. doi: 10.1039/C9MH00220K. [DOI] [Google Scholar]; (h) Yang J. Chi Z. Zhu W. Tang B. Li Z. Sci. China: Chem. 2019;62:1090–1098. doi: 10.1007/s11426-019-9512-x. [DOI] [Google Scholar]; (i) Fang M. Yang J. Li Z. Chin. J. Polym. Sci. 2019;37:383–393. doi: 10.1007/s10118-019-2218-z. [DOI] [Google Scholar]; (j) Li Q. Li Z. Sci. China Mater. 2019 doi: 10.1007/s40843-019-1172-2. [DOI] [Google Scholar]; (k) Chen X. Luo W. Ma H. Peng Q. Yuan W. Zhang Y. Sci. China: Chem. 2018;61:351–359. doi: 10.1007/s11426-017-9114-4. [DOI] [Google Scholar]
- (a) An Z. Zheng C. Tao Y. Chen R. Shi H. Chen T. Wang Z. Li H. Deng R. Liu X. Huang W. Nat. Mater. 2015;14:685–690. doi: 10.1038/nmat4259. [DOI] [PubMed] [Google Scholar]; (b) Gu L. Shi H. Bian L. Gu M. Ling K. Wang X. Ma H. Cai S. Ning W. Fu L. Wang H. Wang S. Gao Y. Yao W. Huo F. Tao Y. An Z. Liu X. Huang W. Nat. Photonics. 2019;13:373–375. doi: 10.1038/s41566-019-0447-x. [DOI] [Google Scholar]; (c) Yang Z. Mao Z. Zhang X. Ou D. Mu Y. Zhang Y. Zhao C. Liu S. Chi Z. Xu J. Wu Y. Lu P. Lien A. Bryce M. R. Angew. Chem., Int. Ed. 2016;55:2181–2185. doi: 10.1002/anie.201509224. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yang J. Zhen X. Wang B. Gao X. Ren Z. Wang J. Xie Y. Li J. Peng Q. Pu K. Li Z. Nat. Commun. 2018;9:840. doi: 10.1038/s41467-018-03236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yang J. Gao H. Wang Y. Yu Y. Gong Y. Fang M. Ding D. Hu W. Tang B. Z. Li Z. Mater. Chem. Front. 2019;3:1391–1397. doi: 10.1039/C9QM00108E. [DOI] [Google Scholar]; (f) Wang X. Xiao H. Chen P. Yang Q. Chen B. Tung C. Chen Y. Wu L. J. Am. Chem. Soc. 2019;141:5045–5050. doi: 10.1021/jacs.9b00859. [DOI] [PubMed] [Google Scholar]
- (a) Wei Y. Zhang Z. Chen Y. Wu C. Liu Z. Ho S. Liu J. Lin J. Chou P. Chem. Commun. 2019;2:10. doi: 10.1038/s42004-019-0113-8. [DOI] [Google Scholar]; (b) Yuan M. Wang D. Xue P. Wang W. Wang J. Tu Q. Liu Z. Liu Y. Zhang Y. Wang J. Chem. Mater. 2014;26:2467–2477. doi: 10.1021/cm500441r. [DOI] [Google Scholar]
- (a) Uoyama H. Goushi K. Shizu K. Nomura H. Adachi C. Nature. 2012;492:234–238. doi: 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]; (b) Wada Y. Kubo S. Kaji H. Adv. Mater. 2018:1705641. doi: 10.1002/adma.201705641. [DOI] [PubMed] [Google Scholar]; (c) Freeman D. M. E. Musser A. J. Frost J. M. Stern H. L. Forster A. K. Fallon K. J. Rapidis A. G. Cacialli F. McCulloch I. Clarke T. M. Friend R. H. Bronstein H. J. Am. Chem. Soc. 2017;139:11073–11080. doi: 10.1021/jacs.7b03327. [DOI] [PubMed] [Google Scholar]; (d) Im Y. Kim M. Cho Y. J. Seo J. Yook K. S. Lee J. Y. Chem. Mater. 2017;29:1946–1963. doi: 10.1021/acs.chemmater.6b05324. [DOI] [Google Scholar]; (e) Park I. S. Komiyamaa H. Yasuda T. Chem. Sci. 2017;8:953–960. doi: 10.1039/C6SC03793C. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kondo Y. Yoshiura K. Kitera S. Nishi H. Oda S. Gotoh H. Sasada Y. Yanai M. Hatakeyama T. Nat. Photonics. 2019;13:678–682. doi: 10.1038/s41566-019-0476-5. [DOI] [Google Scholar]; (g) Kotadiya N. B. Blom P. W. M. Wetzelaer G. A. H. Nat. Photonics. 2019;13:765–769. doi: 10.1038/s41566-019-0488-1. [DOI] [Google Scholar]
- (a) Tian S. Ma H. Wang X. Lv A. Shi H. Geng Y. Li J. Liang F. Su Z. An Z. Huang W. Angew. Chem., Int. Ed. 2019;58:6645–6649. doi: 10.1002/anie.201901546. [DOI] [PubMed] [Google Scholar]; (b) Gong Y. Zhao L. Peng Q. Fan D. Yuan W. Zhang Y. Tang B. Z. Chem. Sci. 2015;6:4438–4444. doi: 10.1039/C5SC00253B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y. Zhang P. Gu Y. Wang J. Han M. Chen C. Zhan X. Xie Z. Zou B. Peng Q. Chi Z. Li Z. Adv. Opt. Mater. 2018;6:1800198. doi: 10.1002/adom.201800198. [DOI] [Google Scholar]
- (a) Yang Z. Mao Z. Xie Z. Zhang Y. Liu S. Zhao J. Xu J. Chi Z. Aldred M. P. Chem. Soc. Rev. 2017;46:915–1016. doi: 10.1039/C6CS00368K. [DOI] [PubMed] [Google Scholar]; (b) Huang T. Jiang W. Duan L. J. Mater. Chem. C. 2018;6:5577–5596. doi: 10.1039/C8TC01139G. [DOI] [Google Scholar]; (c) Aydemir M. Polym. Bull. 2019;76:6429–6436. doi: 10.1007/s00289-019-02729-8. [DOI] [Google Scholar]; (d) Wang T. Hua X. Yu Y. Yuan Y. Fung M. Jiang Z. Chin. J. Org. Chem. 2019;39:1436–1443. doi: 10.6023/cjoc201810016. [DOI] [Google Scholar]; (e) Wang Z. Cai J. Zhang M. Zheng C. Ji B. Acta Chim. Sin. 2019;77:263–268. doi: 10.6023/A18100437. [DOI] [Google Scholar]
- Cai S. Shi H. Zhang Z. Wang X. Ma H. Gan N. Wu Q. Cheng Z. Ling K. Gu M. Ma C. Gu L. An Z. Huang W. Angew. Chem., Int. Ed. 2018;57:4005–4009. doi: 10.1002/anie.201800697. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.