

Abstract
The secretin receptor (SCTR) is a prototypic Class B1 G protein-coupled receptor (GPCR) that represents a key target for the development of therapeutics for the treatment of cardiovascular, gastrointestinal, and metabolic disorders. However, no non-peptidic molecules targeting this receptor have yet been disclosed. Using a high-throughput screening campaign directed at SCTR to identify small molecule modulators, we have identified three structurally related scaffolds positively modulating SCTRs. Here we outline a comprehensive study comprising a structure-activity series based on commercially available analogs of the three hit scaffold sets A (2-sulfonyl pyrimidines), B (2-mercapto pyrimidines) and C (2-amino pyrimidines), which revealed determinants of activity, cooperativity and specificity. Structural optimization of original hits resulted in analog B2, which substantially enhances signaling of truncated secretin peptides and prolongs residence time of labeled secretin up to 13-fold in a dose-dependent manner. Furthermore, we found that investigated compounds display structural similarity to positive allosteric modulators (PAMs) active at the glucagon-like peptide-1 receptor (GLP-1R), and we were able to confirm cross-recognition of that receptor by a subset of analogs. Studies using SCTR and GLP-1R mutants revealed that scaffold A, but not B and C, likely acts via two distinct mechanisms, one of which constitutes covalent modification of Cys-347GLP−1R known from GLP-1R-selective modulators. The scaffolds identified in this study might not only serve as novel pharmacologic tools to decipher SCTR- or GLP-1R-specific signaling pathways, but also as structural leads to elucidate allosteric binding sites facilitating the future development of orally available therapeutic approaches targeting these receptors.
Keywords: Secretin receptor, positive allosteric modulator, G protein-coupled receptor
1. Introduction
The secretin receptor (SCTR), the founding member of the secretin-like class B1 G protein-coupled receptor (GPCR) family, belongs to a small group of 18 receptors naturally activated by moderate-length peptide hormones incorporating between 27 and 44 amino acid residues [1–3]. Members of this peptide receptor family have well established physiologic functions and potential therapeutic roles for psychiatric disorders, pain, bone disease, cancer, type 2 diabetes (T2D), obesity and cardiovascular disease [1, 4, 5]. Despite their widely recognized clinical relevance, the potential of class B GPCRs has been minimally exploited since only agonists based on natural peptide ligands have been advanced as therapeutic or diagnostic agents so far [6, 7]. One prominent example are parenterally administered glucagon-like peptide-1 (GLP-1) mimetics that act by stimulating GLP-1 receptors (GLP-1Rs) and are currently the most efficient non-surgical treatment for T2D with positive effects on weight reduction and cardiovascular health [8]. Key mechanisms of GLP-1R signaling are thereby Gαs-induced elevation of cyclic adenosine monophosphate (cAMP) and increased glucose-sensitive secretion of insulin by pancreatic beta cells. Although peptide analogs are generally restricted by short half-lives and poor oral bioavailability [1, 9], the development of potent small molecule agonists for this receptor family has remained elusive, likely due to complex binding mechanisms of natural orthosteric ligands and the highly open conformation of the extracellular helical bundle in the active state [1, 10, 11].
Recently, the concept of allosteric modulation, which exploits the interplay between spatially distinct, but conformationally linked, receptor binding pockets, has emerged as an opportunity to develop small molecule compounds targeting class B GPCRs [1]. To overcome the inherent limitations of peptide-based GLP-1R agonists, T2D drug discovery campaigns have pursued the search for GLP-1R positive allosteric modulators (PAMs), which led to the identification of several chemical scaffolds supporting the presence of an allosteric binding site in GLP-1Rs [7, 12–16]. Compound 2 [17] and BETP [12] are two of the most extensively studied allosteric modulators potentiating GLP-1R-mediated cAMP accumulation by partial agonists, comprising endogenous stimulators, such as oxyntomodulin or GLP-1(9–36), as well as synthetic peptide analogs and peptidomimetics [7]. Despite their structurally distinct chemotypes, both small molecule modulators share an electrophilic reactivity resulting in covalent modification of a free cysteine Cys-347 (C347) in the third intracellular loop at the interface between helices 5 and 6 of the receptor [7, 18–21]. Furthermore, this irreversible stabilization of the receptor in an active state conformation has been determined to be the driving force of their PAM activity, since replacing Cys-347 with an alanine at GLP-1Rs did not hamper the signaling of peptide agonists but eradicated the potentiating effects of both modulators [7, 20, 21]. Beyond that, a range of structurally diverse pharmacophores has been shown to act in a Cys-347-dependent way, revealing the irreversible mechanism-of-action (MOA) of a majority of GLP-1R PAMs [7, 22]. Probably due to their electrophilic function, identified GLP-1R PAMs demonstrated poor pharmacokinetic properties that terminated their clinical development [7, 22]. Attempts to generate potent reversible analogs were challenging, as the creation of the non-reactive BETP analog th-BETP, in which a thioether replaced the sulfoxide moiety, resulted in a complete loss of activity. Moreover, the existence of a free cysteine at position 347 in the interface of transmembrane (TM) 5 and 6 is unique to GLP-1Rs within the class B receptor family [7]. Thus, aiming to develop small molecule compounds targeting this binding site at other class B GPCRs, such as SCTRs, was not a promising strategy, even though the quest for allosteric modulators induced a breakthrough in class B GPCR drug discovery overall.
Despite the success of GLP-1 mimetics, bariatric surgery like Roux-en-Y bypass (RYGB) represents the most effective way to resolve obesity-induced T2D [23]. In addition to increased GLP-1 levels, a recent study determined that glucose-sensitive S cells in the distal small intestine may contribute to more than double postprandial secretin plasma concentrations after RYGB [24]. Secretin (Sec-FL) is released by S cells reacting to acidic content in the duodenum and exerts its physiological effects by activating SCTRs [3, 25]. Typical for class B GPCRs, SCTR signaling is predominantly mediated by Gαs proteins leading to enhanced adenylate cyclase activity resulting in an increase of cAMP production. Primarily known to stimulate biliary and pancreatic secretion, SCTRs have also been implicated to promote beneficial effects on insulin secretion, cardiac output and gastric accommodation/emptying [3, 9, 25, 26]. Beyond that, SCTR activation is crucial for meal-induced brown fat thermogenesis (BAT) resulting in satiation and short-term food intake reduction in mice [27]. However, despite the broad implications and potential benefits of SCTR activation, no ligands other than closely related secretin peptide analogs had been developed at all [11, 25, 28–30]. In a previous report [30], we described a testing funnel directed to identify small molecule compounds acting as positive allosteric modulators at SCTRs upon binding of secretin peptides. By comparing three different cAMP detection methods, and evaluating the effects of individual or mixtures of full and partial peptide agonists utilized as orthosteric stimulator probes in primary PAM screening efforts, we discovered three related scaffolds (A: 2-sulfonyl pyrimidines, B: 2-mercapto pyrimidines and C: 2-amino pyrimidines) with substantial SCTR PAM activity, but with no to minor intrinsic activity and devoid of significant off-target effects on type 2 arginine vasopressin receptor (AVP2R)-overexpressing or parental CHO-K1 cell lines.
Here we outline intensive structure-activity relationship (SAR) and allosteric activity studies of the first SCTR PAMs, which enabled the elucidation of structural components contributing to distinct pharmacological profiles in SCTR-overexpressing and endogenously expressing cell lines. We identified analog B2, which exerted improved positive cooperativity to partial secretin peptide agonist Sec(3–27) and substantially prolonged secretin residence time on SCTRs. Being aware of the electrophilic moiety incorporated in 2-sulfonyl pyrimidines (scaffold A), we discovered significant structural similarities to established GLP-1R small molecule modulators such as BETP. By also screening with GLP-1Rs, we were able to determine that a subset of SCTR PAMs potentiate not only SCTR activation, but also GLP-1R signaling. Subsequent evaluation and comparison of cooperativity factors elucidated distinct receptor and ligand selectivity profiles for each scaffold. We further confirmed that particularly scaffold A augmented GLP-1R activity in cAMP accumulation and insulin secretion studies on endogenously receptor expressing INS-1 832/3 cells. To distinguish between irreversible and reversible-acting PAMs, we deployed glutathione reactivity and cAMP-washout experiments and additionally generated SCTR and GLP-1R mutants, which were applied to explore the potential binding sites of newly discovered PAMs.
To our knowledge, this is the first report disclosing the discovery and comprehensive functional characterization of a novel class of SCTR PAMs, with some exerting structure-related activity on GLP-1Rs. These newly found agents constitute not only useful tool compounds to elucidate GLP-1R or SCTR-specific signaling processes and allosteric binding sites, but also structural leads to advance the development of orally available therapeutics targeting these important receptors to eventually treat metabolic disorders, such as obesity and T2D.
2. Materials and Methods
2.1. Materials
2.1.1. Peptides and compounds:
Sec-FL (full length human secretin (1–27), #4031250), GLP-1 (glucagon-like peptide-1 trifluoroacetate salt, #4030663) and AVP ((Arg⁸)-vasopressin trifluoroacetate salt, #4012215) were received from Bachem AG (Bubendorf, Switzerland). Secretin 1–23 (Sec(1–23), HSDGTFTSELSRLREGARLQRLL-OH) and Secretin 3–27 (Sec(3–27), DGTFTSELSRLREGARLQRLLQGLV-NH2) were synthesized by Biopeptide (San Diego, CA, USA). GLP-1(9–36) (GLP-1(9–36) amide, #AS-65070) was purchased from Anaspec (Fremont, CA, USA). BETP (4-(3-(Benzyloxy)phenyl)-2-(ethylsulfinyl)-6-(trifluoromethyl)pyrimidine) was obtained from Sigma Aldrich (St. Louis, MO, USA, #SML0558, purity ≥98% (HPLC)). Na125Iodine used for secretin peptide radioiodination was from PerkinElmer (Waltham, MA, USA). Fluorescently tagged full length secretin (Fluo-Sec) was synthesized as described previously [30]. Dry powders of compounds were ordered via MolPort (Beacon, NY, USA) supplied by ChemBridge Corporation (San Diego, CA, USA), ChemDiv, Inc. (San Diego, CA, USA) or Vitas-M Laboratory (Champaign, IL, USA) with purity ≥ 90%. Compound IDs correspond to the following MolPort IDs: A1 (MolPort-007–583-364), A2 (MolPort-001–603-825), A3 (MolPort-001–603-762), A4 (MolPort-001–603-814), A5 (MolPort-001–603-819), A6 (MolPort-001–603-826), A7 (MolPort-007–583-366), A8 (MolPort-007–583-368), A9 (MolPort-001–603-798), A10 (MolPort-000–162-762), A11 (MolPort-000–160-830), A12 (MolPort-002–769-455), B1 (MolPort-001–603-579), B2 (MolPort-001–986-676), B3 (MolPort-002–122-137), B4 (MolPort-001–986-686), B5 (MolPort-001–603-618), B6 (MolPort-001–628-009), B7 (MolPort-001–985-074), B8 (MolPort-002–283-488), C1 (MolPort-001–604-034), C2 (MolPort-001–603-977), C3 (MolPort-001–603-980), C4 (MolPort-001–604-037), C5 (MolPort-007–589-843), C6 (MolPort-002–772-185), C7 (MolPort-002–775-111), C8 (MolPort-001–604-001), C9 (MolPort-001–604-035), C10 (MolPort-001–604-036), C11 (MolPort-002–775-047) and C13 (MolPort-000–433-551). Compound C12 was obtained by Sundia (SB6308, Shanghai, China). For experiments, compounds were dissolved in 100% DMSO and stored at room temperature in Echo Qualified 384-well low dead volume (384LDV) microplates (Labcyte, San Jose, CA, USA) in a desiccator as 16-point 2-fold dilutions. Compound stock concentrations ranged from 0 to 10 mM.
2.1.2. Cells and culture reagents:
Human Embryonic Kidney (HEK)-293(T), Chinese Hamster Ovary (CHO-K1) and NG108–15 cells were obtained from ATCC (Manassas, VA, USA). INS-1 832/13 rat insulinoma cell line was obtained from Sigma Aldrich (SCC207). CHO-K1 cells were maintained in CHO cell growth media (Ham’s F-12K (Kaighn’s modification), Corning Life Sciences, Tewksbury, MA, USA, Cellgro #10–025-CV), 5% Fetal Bovine Serum (FBS) Clone II (GE Healthcare Life Sciences, Marlborough, MA, USA, Hyclone #SH30066.03), 1% penicillin (10,000 units)/ streptomycin (10 mg) (Pen/Strep, Gibco from Thermo Fisher Scientific, Waltham, MA, USA, #15140122), 1% L-glutamine (200 mM) (Gibco #25030081). HEK-293(T) cells were maintained in HEK cell growth media (Dulbecco’s Modified Eagles Medium (DMEM) Corning Life Sciences, Cellgro #10–013-CV), 10% FBS (Omega Scientific, Tarzana, CA, USA, #FB-12), 1% Pen/Strep, 1% L-glutamine). Cells were detached using TrypLE Express (Gibco #12605036). NG108–15 cells were maintained in NG108 growth media consisting of DMEM with 4.5g/L glucose without L-glutamine and sodium pyruvate, (Corning, #15–017-CV), 10% FBS, 1% L-glutamine, 0.1mM hypoxanthine, 400 nM aminopterin, 0.016mM thymidine (H.A.T., ATCC, #69-X-2mL), 1.5 g/L sodium bicarbonate (Sigma Aldrich, #S5761–1KG). INS-1 832/13 cells were maintained INS-1 growth media containing RPMI 1640 without L-glutamine (Corning, #15–040-CV), 10% FBS, 1% Pen/Strep, 1% L-glutamine, 1 mM sodium pyruvate (Gibco, #11360–070), 10 mM HEPES (Gibco, #15630–080), 0.05 mM 2-mercapto ethanol (Gibco, #21985–023) and 0.3mg/mL G418 (Omega Scientific, Tarzana, CA, USA, #GN-04).
2.1.3. Receptor constructs:
GLP-1R C347A construct was prepared using Q5 Site-Directed Mutagenesis Kit (New England Biolabs (NEB), Ipswich, MA) following manufacturer’s protocol. The final product was verified by DNA sequencing. Secretin receptor constructs, SCTR K317A and SCTR K317C (SCTR numbering is without the signal sequence (21 amino acid)) were prepared by oligonucleotide-directed approach using QuikChange site-directed mutagenesis (Agilent Technologies, Santa Clara, CA), using manufacturer’s instructions, and the products were verified by DNA sequencing.
2.2. Methods
2.2.1. cAMP (cyclic adenosine monophosphate) accumulation assays
cAMP assays were performed as described previously [30] with minor modifications. In brief, selectivity screens and allosteric activity studies were performed on frozen stocks of SCTR- or AVP2R-overexpressing CHO-K1 cells or GLP-1R-overexpressing HEK-293T cells, all derived from a single cell clone at a low passage number. After reaching 80–90% confluency, cells were detached using TrypLE Express, centrifuged and re-suspended in freeze media (10% DMSO in growth media) to dilute to a final concentration of cell stocks of 20 million cells/mL. Respective ligand standard curves were measured for each batch of cells to determine adequate EC20 and EC95 concentrations.
2.2.1.1. General procedure:
(using Cisbio cAMP Gs Dynamic HTRF detection kit (Cisbio GsD, Cisbio US, Inc., Bedford, MA, USA) Orthosteric stimulator dilutions and titrations were prepared freshly in DMSO and transferred to 384LDV microplates. Ligands and compounds were dispensed onto dry 1536-well plate (Corning #3725) with an Echo liquid handler (Labcyte). Frozen cell stocks were thawed in a water bath at 37°C and immediately diluted in stimulation buffer (HBSS (Hank’s Balanced Salt Solution with Ca2+ and Mg2+, Gibco #24020117), 5 mM HEPES (hydroxyethyl piperazineethanesulfonic acid), 0.5 mM IBMX (3-isobutyl-1-methylxanthine, Sigma-Aldrich) and 0.075% BSA (7.5% DTPA-purified Bovine Serum Albumin, PerkinElmer). 5 μL of cell suspension were added per well utilizing a Multidrop Combi dispenser (Thermo Fisher Scientific). The microplate was centrifuged at 1000 rpm for 1 min, covered with a lid and kept at room temperature (RT) for 30 min. cAMP standard dilutions were prepared in stimulation buffer and added to designated wells. Detection reagents, cAMP-d2 and anti-cAMP cryptate, were diluted in cAMP detection buffer and 4 μL were added per well. The microplate was centrifuged at 1000 rpm for 1 min, covered with a lid and, after a 30 min incubation at RT, read on a PHERAstar FSX microplate reader (BMG Labtech, Ortenberg, Germany) using the HTRF (homogeneous time resolved fluorescence) module.
2.2.1.2. Selectivity screens:
In all formats, DMSO was added to obtain the same final DMSO content in each well. For selectivity studies of compounds, wells contained 0 – 50 μM compound and peptide ligand or DMSO. Selectivity screening data were uploaded and analyzed on CBIS (Chemical and Biology Information System software, ChemInnovation Software, Inc., San Diego, CA, USA). For PAM assays, EC20 of ligand and for agonist assays, DMSO was set as the negative control. EC95 served as the positive control in both cases. Further characterization was conducted using the TIBCO Spotfire software (PerkinElmer). Experiments were performed in duplicate in three to five independent experiments. In GLP-1R agonist and PAM formats A2, B1 and C10 have been tested in duplicate in two independent experiments due to limited compound availability.
2.2.1.3. Allosteric activity studies:
For allosteric activity studies, wells contained 0 – 25 μM of compound and Sec-FL (0–100 pM), Sec(1–23) (0–100 nM), Sec(3–27) (0–2.5 μM), GLP-1 (0–250 pM) or GLP-1(9–36) (0–1.25 μM) as 16-point 2-fold dilutions. DMSO was added to obtain the same final DMSO content in each well. All experiments were performed in duplicate in at least three independent experiments. TR-FRET ratio values were converted into cAMP concentrations (nM) employing the cAMP standard curves recorded with each experiment and the “interpolate the standard curve” function in GraphPad Prism 8.4.0. For determination of allosteric activity parameters, the “operational model of allosterism” (equation (1)) was used as described by Leach, K., et al. [31],
| (1) |
which is based on the allosteric ternary complex model presented by Ehlert [32] and the Black/Leff operational model of agonism [33] (equation (2)):
| (2) |
The operational model of allosterism comprises the pharmacological response of allosteric modulator by describing its concentration [B], equilibrium dissociation constant KB, intrinsic activity τB, which incorporates total receptor density and efficiency of stimulus-response coupling, as well as cooperativity factors with respect to binding affinity (α) and efficacy (β). The allosteric modulator effect is dependent on the orthosteric ligand that is described by concentration [A], equilibrium dissociation constant KA and intrinsic activity τA. Em stands for the maximal possible system response and n denotes the slope factor of the dose-response curves. By applying the Black/Leff operational model, KA and τA were determined for each peptide ligand dose-response curve without allosteric modulator present. To evaluate KB, α and β for allosteric modulators, KA and τA, as well as basal and n were held constant. In some instances, the “Allosteric EC50 shift” equation in GraphPad Prism (San Diego, CA, USA) had to be employed to get initial KB and α values for determining cooperativity factor β. αβ values between 0 and 1 describe negative, αβ values of 1 neutral, and αβ values higher than 1 denote positive cooperativity toward the specific receptor-ligand complex investigated.
2.2.1.4. Assays with endogenously expressing cell lines:
Following the general protocol, cAMP assays were conducted in cultured NG108–15 or INS-1 832/13 cells with a passage count between 8–17 or 18–29, respectively. After reaching 80–90% confluency, cells were detached, re-suspended in growth media, centrifuged at 300xg for 3 min and diluted in stimulation buffer. For the detection of cAMP accumulation in INS-1 832/13 cells, LANCE Ultra cAMP detection kit (PerkinElmer) was employed as follows. After incubation of compound-ligand mixtures with cells at RT for 30 min, 4 μL of 100-fold-diluted Eu-cAMP stock and 300-fold-diluted Ulight Ab stock in detection buffer was added. TR-FRET signal was recorded after incubation at RT for 60 min using HTRF module. Assay wells contained 12.5 or 25 μM of test compound and peptide ligand as 16-point 2-fold dilution, i.e. Sec-FL (0– 50 nM), Sec(1–23) (0–5 μM) or Sec(3–27) (0–50 μM) for studies in NG108–15 and GLP-1 (0–500 nM) or GLP-1(9–36) (0–5 μM) for studies in INS-1 832/13 cells. DMSO was added to obtain the same final DMSO content in each well. All experiments were performed in duplicate in at least three independent experiments. Curves were fitted using nonlinear regression equation “[Agonist] vs. response -- Variable slope (four parameters)” in GraphPad Prism 8.4.0.
2.2.1.5. Co-incubation versus pre-incubation:
In general, compounds and ligand were dispensed onto dry microplates and co-incubated with cells for 30 min at RT before addition of detection reagent. In case of mutational studies, allosteric activity studies employing HEK-293T-GLP-1R cells and GLP-1(9–36) as well as for studies in INS-1 832/12 cells, group A compounds and BETP were added after a 10–20 min pre-incubation of ligand at RT to achieve maximal compound response.
2.2.1.6. Wash-out experiments:
Wash-out experiments were performed according to general procedure with the following modifications: A frozen vial of SCTR-overexpressing CHO-K1 cells or GLP-1R-overexpressing HEK-293T cells was thawed quickly at 37 °C and diluted in growth media to obtain desired cell densities. Cells were dispensed 5 μL per well into TC-treated 384-well microplates (Greiner Bio-One small volume 784080). For HEK-293-GLP-1R cells, Poly-D-Lysine coated microplates were employed. Plates were centrifuged at 500 rpm for 15 sec, covered with a lid and incubated overnight at 37 °C and 5% CO2. 25 μM of compounds or DMSO were added to one half of the plate, followed by 20–30 min incubation at RT. Subsequently, compound-containing media was removed and cells were washed three times with PBS (CHO-SCTR) or HBSS (HEK-293T-GLP-1R) using BlueWasher (Blue Cat Bio, Concord, MA, USA). 5 μL of stimulation buffer was added per well, plates were centrifuged 500 rpm for 1 min and 25 μM of compounds or DMSO were added to the second half of the plate. On top, peptide ligands (Sec-FL (0–100 pM) or GLP-1 (0–250 pM)) as 16-point 2-fold dilutions were dispensed across the microplate. After centrifugation at 1000 rpm at 1 min, plates were incubated at RT for 30 min. Detection of cAMP accumulation was conducted following the general protocol but dispensing 5 μL of detection mixture per well. All experiments were performed in duplicate in at least three independent experiments. Curves were fitted using nonlinear regression equation “[Agonist] vs. response -- Variable slope (four parameters)” in GraphPad Prism 8.4.0.
2.2.1.7. Mutational studies:
Receptor mutant constructs were characterized in cAMP assays following the general procedure with few minor modifications: HEK-293 cells in HEK cell growth media were seeded in 6-well TC-plates and incubated overnight at 37 °C and 5% CO2. SCTR WT (wildtype), SCTR K317A, SCTR K317C, GLP-1R WT or GLP-1R C347A were transfected using TransIT®-LT1 transfection reagent following manufacturer’s manual (Mirus Bio, Madison, WI, USA) delivering 1 μg DNA of SCTR construct or 0.5 μg DNA of GLP-1R construct per well. After 24 h at 37 °C and 5% CO2, cells expressing receptor constructs were harvested using TrypLE Express, centrifuged at 300 g for 3 min and diluted in stimulation buffer. 5 μL of cells were added per well to a 1536-well microplate (Corning #3725), which already contained DMSO or 12.5–25 μM of test compound. Plates were centrifuged for 1 min at 1000 rpm. After a 10–20 min incubation at RT, peptide ligand (Sec-FL 0–100 pM, Sec(1–23) 0–100 nM, Sec(3–27) 0–2.5 μM, GLP-1 0–250 pM, GLP-1(9–36) 0–1.25 μM) as 16-point 2-fold dilutions were dispensed on top. After centrifugation at 1000 rpm at 1 min, plates were incubated at RT for 30 min. Detection of cAMP accumulation was conducted following the general protocol. All experiments were performed in duplicate in at least three independent experiments. Curves were fitted using nonlinear regression equation “[Agonist] vs. response -- Variable slope (four parameters)” in GraphPad Prism 8.4.0.
2.2.2. TR-FRET SNAP-SCTR binding
Binding experiments were performed as previously described [30], with the following modifications:
2.2.2.1. Competition binding:
Equilibrium dissociation constant KD of Sec-Fluo was determined by saturation binding experiments described in our previous report [30]. For evaluation of competition binding, Fluo-Sec and Sec-FL were diluted in DMSO and dispensed into a 384LDV plate. Ligand titrations were prepared in DMSO in adjacent wells. Fluo-Sec was added (6 nM final) to all test wells of a 1536-well plate (Corning #3725). DMSO (in positive control wells), Sec-FL (5 μM final, negative control wells) or ligand/compound titrations (Sec-FL (0–500 nM), test compounds (0–50 μM), 16-point 2-fold dilutions) were dispensed on top. Using a dounce homogenizer HEK-293 SNAP-SCTR membranes, thawed at RT and pre-labeled with Lumi-4 Terbium cryptate (Cisbio Tag-lite), were diluted in binding buffer (10 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 1 mM ascorbic acid, 0.2% BSA). Membrane solution was added at 5 μL per well. The microplate was centrifuged 1000 rpm for 1 min and incubated for 2 h at RT. Fluo-Sec bound to SNAP-SCTRs was recorded by PHERAstar FSX (LanthaScreen module). Data was uploaded and analyzed via CBIS to report EC50 and efficacy values with respect to in- or decrease of Fluo-Sec receptor binding for compounds or Sec-FL, respectively. Experiments were performed in duplicate in three to five independent experiments.
2.2.2.2. Dissociation binding
Fluo-Sec (10 nM final) was dispensed into a 1536-well plate (Corning #3725). For negative control wells Sec-FL (5 μM final) and for positive control wells and sample wells DMSO were added on top. With a dounce homogenizer thawed HEK-293 SNAP-SCTR membranes were diluted in binding buffer (10 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 1 mM ascorbic acid, 0.2% BSA). Membrane suspension was added at 5 μL/well and the microplate was centrifuged for 1 min at 1000 rpm. After 2 h incubation at RT, the plate was read for baseline fluorescence using PHERAstar FSX. DMSO (negative control) or test compounds (3.1, 6.25 or 12.5 μM) were dispensed into sample wells and after centrifugation at 1000 rpm for 1 min, the microplate was read with PHERAstar FSX. Immediately, dissociation buffer (binding buffer, 5 μM Sec-FL, 100 μM GTPγS) was dispensed into sample wells and binding buffer was added to control wells. Dissociation binding was detected using PHERAstar FSX (LanthaScreen, kinetic mode, 10 cycles, 45 min). Dissociation half-lives (t1/2 [min]) were determined using GraphPad Prism 8.4.0 applying equation “Dissociation – One phase exponential decay”. Experiments were performed in triplicates in at least three independent experiments.
2.2.3. Glucose stimulated insulin secretion (GSIS) assay
GSIS was performed according to Merglen, A., et al. [34] with the following modifications: INS-1 832/13 were maintained in INS-1 growth media. After reaching 80–90% confluency, cells were harvested using TrypLE Express and diluted in growth media. 25 μL of cells were seeded per well into a 384-well TC-treated microplate (Greiner 781098). After 2 days at 37 °C and 5% CO2, media was displaced with 21 μL of glucose-free INS-1 growth media using Blue Cat Bio Blue Washer. Microplate was centrifuged at 500 rpm for 15 sec and incubated at 37 °C and 5% CO2 for 2 h. Media was displaced with 21 μL of KRSHB buffer (Krebs-Ringer Solution, HEPES-buffered (Alfa Aesar, Haverhill, MA, USA) containing 120 mM sodium chloride, 5 mM potassium chloride, 2 mM calcium chloride, 1 mM magnesium chloride, 25 mM sodium bicarbonate, 5.5 mM HEPES and 1.1 mM D-glucose with addition of 0.1% BSA). Microplate was centrifuged at 500 rpm for 15 sec, incubated at 37 °C and 5% CO2 for 1 h and the BlueWasher step was repeated leaving 21 μL of KRSHB buffer. Test compounds (6.25x for 20 μM final) or DMSO and GLP-1(9–36) (6.25x for 1 μM final) or DMSO had been dispensed into a Corning NBS (non-binding surface) 384 well microplate and were diluted with KRSHB buffer containing additional glucose (6.25x for 15 mM final). 4 μL of compound-ligand mixtures were transferred to the assay plate, which was subsequently centrifuged at 500 rpm for 15 sec and incubated for 2 h at 37 °C and 5% CO2 followed by 15 min at RT. The Insulin High Range Kit (Cisbio) was employed for quantification of secreted insulin according to manufacturer’s instructions. Insulin standard, diluent and detection reagents were prepared on ice according to manufacturer’s instructions. 5 μL of conditioned media were transferred per well into a cooled 384 well NBS microplate (Corning). 45 μL of diluent (Cisbio) were added on top of each well, followed by centrifugation at 1000 rpm for 1 min. 1 μL of the 10-fold diluted media was transferred per well into a cooled white 384 well small volume microplate (Greiner), which contained 1 μL of the insulin standard titration in adjacent wells. After addition of 15 μL ice-cold detection mixture (Cisbio), the small volume plate was spun at 1000 rpm for 1 min, sealed with foil and incubated in the dark overnight. The plate was read using PHERAstar FSX (HTRF). Experiments were performed as quintuplicates in three independent experiments. Insulin concentrations were calculated in Excel using TR-FRET ratio values and the insulin standard curve equation: Insulin concentration (ng/mL) = ((0.0000003)*(TR-FRET ratio)2)+(0.0006*TR-FRET ratio)+0.6336; R2 = 0.9988. Values were multiplied by 10 to determine insulin secretion as ng/mL per source well and plotted as bar graphs using GraphPad Prism 8.4.0. Statistical significance of compound and ligand treatment as well as of interactions between compound and ligand treatment were determined using ordinary two-way ANOVA and Tukey’s multiple comparisons test with individual variances computed for each comparison (99% confidence interval). P values were illustrated according to the following classification: (*) adjusted P values between 0.01 and 0.05, (**) adjusted P values between 0.01 and 0.001, (***) adjusted P values between 0.001 and 0.0001, (****) adjusted P values <0.0001.
2.2.4. Cell culture and transfections for receptor mutant-binding assays
SCTR constructs were transiently expressed in African green monkey kidney (COS-1) cells. Cells were grown in sterile 10 cm tissue culture plates in DMEM media supplemented with 5% Fetal Clone II, 1% penicillin and streptomycin mixture in a 37 °C incubator in a humidified environment containing 5% carbon dioxide. At an approximate confluence of 80 percent, the cells were transfected with 1.0 μg of DNA per dish using the diethylaminoethyl (DEAE)-dextran method [35].
2.2.5. Receptor mutant-binding assays
Receptor expressing COS-1 cells were incubated with ~5 pM 125I-Tyr10-secretin (prepared and purified in our laboratory to yield approximate specific radioactivity of 2,000 Ci/mmol) in the absence or presence of increasing concentrations (0 to 0.1 μM) of unlabeled secretin peptide for 1 h at room temperature in KRH medium (25 mM HEPES, pH 7.4, 104 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM KH2PO4, 1.2 mM MgSO4) pH 7.4, containing 0.01% soybean trypsin inhibitor and 0.2% bovine serum albumin. The receptor-bound fraction was washed with ice-cold KRH medium containing 0.01% soybean trypsin inhibitor and 0.2% bovine serum albumin to separate free from receptor bound ligand, and were subsequently lysed with 0.5 M NaOH. Receptor bound radioactivity was quantified in a γ–counter with approximate 70% counting efficiency. Competition-binding studies were performed in duplicate and repeated in at least three independent experiments. Non-specific binding was determined in the presence of 0.1 μM unlabeled secretin and represented less than 15% of total binding. Saturable binding data was analyzed using the non-linear least-squares curve-fitting routine in Prism 6.0 (GraphPad, San Diego, CA).
2.2.6. Covalent reactivity with Glutathione (GSH)
In 1.5 mL Eppendorf tubes 100 μL of GSH (Sigma Aldrich, 10 mM stock in PBS, 4 mM final, 2 eq.), 50 μL of compound A1 or B2 (10 mM stock in DMSO, 2 mM final) and 100 μL of DMSO were mixed and incubated at 37 °C. Control reactions in PBS/DMSO (1:1.5, 250 μL) containing compounds or GSH only were prepared in parallel. After 5 min and after 4 h, samples (20 μL) of the reaction mixtures were taken, diluted in 1 mL of DMSO and presence of GS-adduct formation was monitored using Waters Acquity UPLC/MS (Phenomenex Kinetex C18 2.6 μM, 20 × 2.1 mm; flow 0.8 mL/min; linear gradient in water (0.05% formic acid): 10% to 99% methanol (0.05% formic acid) in 2 min, 99% methanol (0.05% formic acid) for 0.8 min, 99% to 10% methanol (0.05% formic acid) in 0.7 min; tR: 3.5 min) by comparing molecular weights of compounds and putative compound-glutathione adducts.
3. Results
3.1. Structure-activity relationships of SCTR PAM scaffolds elucidate key structural components for compound activity
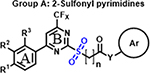
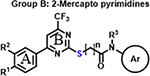
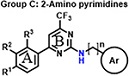
We previously established and validated a SCTR screening platform and demonstrated its ability to identify PAMs, although specific structures have not previously been disclosed [27]. We got particularly interested in three scaffold sets, which shared a structural motif and were described as A: 2-sulfonyl pyrimidines, B: 2-mercapto pyrimidines and C: 2-amino pyrimidines. The three compound groups differ mainly in their substituents at ring A, their substituent and corresponding reactivity at position 2, as well as the length and nature of the linker connecting to a third aromatic moiety (Ar) (Table 1). The original screening hits showed significant SCTR PAM activity, but no to minor intrinsic activity and negligible off-target effects on non-receptor bearing parental cell lines or on type 2 arginine vasopressin receptor (AVP2R)-overexpressing cells used as a class A GPCR counterpart that is also coupled with Gs proteins.
Table 1: Structure-activity studies of 33 analogs in functional selectivity screens and SNAP-SCTR binding assays.
Average potencies (EC50 [μM] ± standard deviation (SD)) and average compound efficacy (Emax [%] ± SD) of compound dose response studies in screening mode. Compounds were tested in Cisbio GsD cAMP accumulation assays and SNAP-SCTR binding experiments; NT: not tested; n.d.: not detected (response less than mean ± 3xSD of negative control); data analysis via CBIS and Microsoft Excel; experiments were performed in duplicates in three to five independent experiments.
   | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Structural specifications | SCTR PAM (3-pep mix) | SCTR Sec-FL (PAM) | SCTR agonist | SNAP-SCTR Binding | AVP PAM (AVP) | GLP-1R PAM (GLP-1) | GLP-1R agonist | ||||||||||||||
| CMPD ID | R1 | R2 | R3 | X | n | Y | Ar | EC50 [μM] | Emax [%] | EC50 [μM] | Emax [%] | EC50 [μM] | Emax [%] | EC50 [μM] | Emax [%] | EC50 [μM] | Emax [%] | EC50 [μM] | Emax [%] | EC50 [μM] | Emax [%] |
| A1 | OCH3 | OCH3 | H | 3 | 3 | NH |  |
7.7 ± 4.0 | 60 ± 16 | 8.5 ± 6.8 | 45 ± 14 | 34 ± 11 | 32 ± 5.4 | 6.3 ± 3.8 | 137 ± 11 | 19 ± 17 | 26 ± 11 | 12 ± 1.0 | 49 ± 21 | 22 ± 19 | 77 ± 51 |
| A2 | OCH3 | OCH3 | H | 3 | 3 | NH | 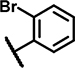 |
8.0 ± 4.4 | 72 ± 12 | 12 ± 13 | 55 ± 12 | 29 ± 16 | 26 ± 12 | 5.2 ± 1.2 | 144 ± 11 | 26 ± 20 | 26 ± 7.2 | 11 ± 5.3 | 57 ± 13 | 20 ± 14 | 70 ± 29 |
| A3 | H | H | OCH3 | 3 | 3 | NH | 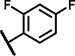 |
7.0 ± 3.9 | 40 ± 11 | 18 ± 22 | 21 ± 4.5 | n.d. | n.d. | 11 ± 8.8 | 149 ± 23 | n.d. | n.d. | 6.7 ± 3.5 | 62 ± 43 | 7.3 ± 4.7 | 48 ± 19 |
| A4 | OCH3 | OCH3 | H | 3 | 3 | NH | 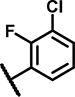 |
7.7 ± 5.4 | 41 ± 1.3 | 12 ± 13 | 21 ± 3.3 | 26 ± 18 | 13 ± 2.9 | 6.8 ± 2.9 | 142 ± 5.9 | n.d. | n.d. | 7.3 ± 4.7 | 58 ± 34 | 4.4 ± 1.9 | 46 ± 15 |
| A5 | OCH3 | OCH3 | H | 3 | 3 | NH | 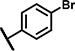 |
8.4 ± 4.4 | 43 ± 15 | 15 ± 18 | 23 ± 3.2 | 26 ± 9.5 | 22 ± 6.1 | 3.4 ± 1.7 | 138 ± 10 | 38 ± 14 | 25 ± 6.5 | 5.4 ± 1.0 | 42 ± 18 | 6.2 ± 3.6 | 49 ± 15 |
| A6 | OCH3 | OCH3 | H | 3 | 3 | NH | 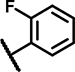 |
12 ± 5.3 | 77 ± 24 | 19 ± 11 | 61 ± 29 | 24 ± 7.9 | 25 ± 8.5 | 7.8 ± 3.0 | 148 ± 13 | 13 ± 13 | 26 ± 8.2 | 12 ± 1.6 | 61 ± 27 | 15 ± 2.3 | 61 ± 26 |
| A7 | OCH3 | OCH3 | H | 3 | 3 | NH | 8.4 ± 2.9 | 31 ± 5.0 | 12 ± 10 | 18 ± 3.9 | 45 ± 7.7 | 20 ± n.d. | 3.2 ± 2.2 | 128 ± 15 | 46 ± 6.3 | 29 ± n.d. | 28 ± 18 | 64 ± 30 | 9.5 ± 3.5 | 36 ± 14 | |
| A8 | OCH3 | OCH3 | H | 3 | 3 | NH | 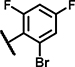 |
13 ± 5.5 | 63 ± 22 | 15 ± 12 | 30 ± 1.3 | 18 ± 0.6 | 27 ± 1.7 | 3.9 ± 2.7 | 128 ± 15 | 44 ± 4.0 | 52 ± 3.7 | 26 ± 13 | 57 ± 31 | 18 ± 13 | 56 ± 37 |
| A9 | H | H | OCH3 | 3 | 3 | NH | 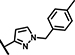 |
3.4 ± 1.7 | 42 ± 12 | 11 ± 19 | 38 ± 15 | n.d. | n.d. | 6.8 ± 10 | 147 ± 30 | n.d. | n.d. | 18 ± 22 | 48 ± 3.9 | 3.7 ± 1.9 | 32 ± 13 |
| A10 | H | H | OCH3 | 3 | 3 | OH | - | 28 ± 16 | 20 ± 4.8 | 40 ± 14 | 6.6 ± n.d. | n.d. | n.d. | n.d. | n.d. | 29 ± 21 | 14 ± n.d. | n.d. | n.d. | 39 ± 8.3 | 26 ± 0.1 |
| A11 | F | H | H | 2 | ethyl | - | - | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 38 ± 8.8 | 76 ± 1.6 | 42 ± 5.8 | 118 ± 15 |
| A12 | H | H | OCH3 | 3 | methyl | - | - | 18 ± 8.5 | 49 ± 18 | 28 ± 21 | 29 ± 3.3 | n.d. | n.d. | 33 ± 24 | 104 ± n.d. | 18 ± 13 | 16 ± 2.9 | 15 ± 11 | 71 ± 15 | 11 ± 6.1 | 75 ± 21 |
| B1 | OCH3 | OCH3 | CH3 | - | 1 | - | 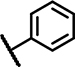 |
3.6 ± 0.6 | 47 ± 16 | 1.3 ± 0.3 | 33 ± 12 | 22 ± 19 | 16 ± 6.5 | 4.2 ± 2.8 | 160 ± 24 | 44 ± 8.6 | 31 ± n.d. | 3.9 ± 1.0 | 32 ± 1.3 | 36 ± 14 | 20 ± n.d. |
| B2 | OCH3 | H | CH3 | - | 1 | - | 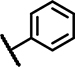 |
2.5 ± 0.5 | 93 ± 18 | 2.1 ± 0.9 | 34 ± 21 | 43 ± 14 | 15 ± n.d. | 2.4 ± 0.3 | 172 ± 26 | n.d. | n.d. | 23 ± 20 | 28 ± 4.3 | n.d. | n.d. |
| B3 | H | H | H | - | 1 | - | 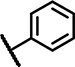 |
n.d. | n.d. | n.d. ± n.d. | n.d. ± n.d. | 37 ± 19 | 7.2 ± n.d. | 0.7 ± 0.7 | 116 ±7.4 | n.d. | n.d. | n.d. | n.d. | 42 ± 11 | 5.1 ± n.d. |
| B4 | OCH3 | H | H | - | 1 | - | 19 ± 12 | 23 ± 8.6 | 41 ± 13 | 16 ± n.d. | 40 ± 15 | 20 ± n.d. | 29 ± 8.1 | 144 ± 12 | n.d. | n.d. | n.d. | n.d. | 42 ± 12 | 20 ± n.d. | |
| B5 | OCH3 | H | H | - | 2 | - | 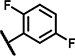 |
18 ± 23 | 26 ± 3.4 | 17 ± 23 | 31 ± 1.2 | n.d. | n.d. | 0.4 ± 0.2 | 128 ± 11 | n.d. | n.d. | 17 ± 23 | 30 ± 13 | n.d. | n.d. |
| B6 | H | H | H | - | 1 | - | 18 ± 23 | 20 ± 3.4 | 35 ± 21 | 29 ± n.d. | n.d. | n.d. | 33 ± 24 | 130 ± n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | |
| B7 | H | H | H | - | 1 | - | 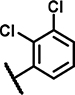 |
19 ± 22 | 32 ± 1.6 | 34 ± 22 | 29 ± n.d. | n.d. | n.d. | 6.4 ± 3.3 | 140 ± 24 | n.d. | n.d. | 33 ± 24 | 29 ± n.d. | n.d. | n.d. |
| B8 | Cl | H | H | - | 1 | - | 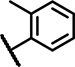 |
18 ± 23 | 14 ± 1.6 | 35 ± 22 | 19 ± n.d. | n.d. | n.d. | 1.0 ± 0.8 | 121 ± 9.2 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| C1 | OCH3 | OCH3 | H | - | 1 | - | 6.9 ± 1.8 | 57 ± 11 | 8.3 ± 11 | 34 ± 13 | 42 ± 12 | 27 ± 1.1 | 2.8 ± 2.6 | 132 ± 12 | n.d. | n.d. | 19 ± 22 | 54 ± 4.3 | n.d. | n.d. | |
| C2 | OCH3 | H | H | - | 1 | - | 7.4 ± 0.8 | 37 ± 5.4 | 34 ± 23 | 20 ± n.d. | 27 ± 14 | 17 ± 1.0 | 2.7 ± 2.2 | 131 ± 9.8 | n.d. | n.d. | 19 ± 22 | 31 ± 9.2 | n.d. | n.d. | |
| C3 | OCH3 | H | H | - | 1 | - | 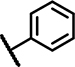 |
7.9 ± 1.6 | 56 ± 11 | 13 ± 19 | 37 ± 14 | 39 ± 16 | 19 ± 0.9 | 5.5 ± 3.4 | 151 ± 11 | n.d. | n.d. | 20 ± 21 | 29 ± 8.6 | n.d. | n.d. |
| C4 | OCH3 | OCH3 | H | - | 1 | - | 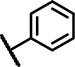 |
6.2 ± 1.0 | 76 ± 10 | 12 ± 19 | 38 ± 13 | 27 ± 19 | 20 ± 1.2 | 4.4 ± 1.8 | 147 ± 11 | n.d. | n.d. | 20 ± 21 | 52 ± 2.7 | 46 ± 5.2 | 19 ± n.d. |
| C5 | OCH3 | OCH3 | H | - | 1 | - | 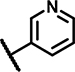 |
20 ± 0.3 | 71 ± 15 | 20 ± 17 | 27 ± 7.4 | 36 ± 11 | 27 ± 7.0 | 23 ± 11 | 165 ± 12 | n.d. | n.d. | 22 ± 20 | 35 ± 0.8 | 41 ± 13 | 15 ± n.d. |
| C6 | H | OCH3 | H | - | 1 | - | 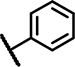 |
7.4 ± 2.0 | 62 ± 11 | 21 ± 23 | 36 ± 13 | 45 ± 8.6 | 17 ± 2.3 | 11 ± 15 | 152 ± 20 | n.d. | n.d. | 19 ± 22 | 32 ± 10 | n.d. | n.d. |
| C7 | H | OCH3 | H | - | 1 | - | 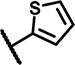 |
8.2 ± 2.3 | 45 ± 1.6 | 34 ± 23 | 16 ± n.d. | 36 ± 20 | 7.2 ± n.d. | 5.0 ± 2.8 | 141 ± 9.0 | n.d. | n.d. | 19 ± 22 | 37 ± 8.3 | n.d. | n.d. |
| C8 | H | H | OCH3 | - | 1 | - | 17 ± 8.7 | 39 ± 3.4 | 34 ± 23 | 14 ± n.d. | 44 ± 8.3 | 15 ± n.d. | 2.5 ± 2.1 | 135 ± 12 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | |
| C9 | OCH3 | OCH3 | H | - | 1 | - | 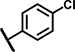 |
7.3 ± 3.2 | 67 ± 8.6 | 11 ± 19 | 37 ± 12 | 37 ± 17 | 23 ± 3.1 | 3.1 ± 2.4 | 140 ± 9.7 | n.d. | n.d. | 2.4 ± 0.9 | 51 ± 7.4 | 39 ± 15 | 15 ± n.d. |
| C10 | OCH3 | OCH3 | H | - | 1 | - | 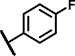 |
5.4 ± 0.6 | 84 ± 9.1 | 5.6 ± 4.8 | 48 ± 17 | 28 ± 22 | 15 ± 3.0 | 3.7 ± 1.0 | 155 ± 11 | n.d. | n.d. | 2.6 ± 0.2 | 53 ± 16 | n.d. | n.d. |
| C11 | H | OCH3 | H | - | 1 | - | 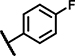 |
5.4 ± 2.0 | 70 ± 4.0 | 33 ± 23 | 25 ± n.d. | 19 ± 22 | 9.9 ± 2.1 | 5.2 ± 2.9 | 159 ± 12 | n.d. | n.d. | 33 ± 17 | 46 ± n.d. | 39 ± 15 | 6.8 ± n.d. |
| C12 | H | H | H | - | 1 | - | 21 ± 5.0 | 47 ± 12 | 34 ± 23 | 20 ± n.d. | 37 ± 19 | 6.9 ± n.d. | 22 ± 17 | 167 ± 13 | n.d. | n.d. | 35 ± 21 | 34 ± n.d. | n.d. | n.d. | |
| C13 | H | H | H | - | 2 | - | 5.7 ± 0.6 | 45 ± 19 | 19 ± 22 | 42 ± 3.3 | n.d. | n.d. | 18 ± 23 | 113 ± 1.5 | n.d. | n.d. | 18 ± 23 | 37 ± 15 | n.d. | n.d. | |
| BETP | - | - | - | - | - | - | - | 24 ± 12 | 51 ± 25 | 31 ± 19 | 42 ± 2.0 | n.d. | n.d. | 17 ± 14 | 152 ± 11 | n.d. | n.d. | 0.8 ± 0.2 | 76 ± 22 | 0.8 ± 0.6 | 91 ± 16 |
| Sec-FL [pM] | - | - | - | - | - | - | - | 0.5 ± 0.2 | 117 ± 11 | 0.4 ± 0.3 | 104 ± 4.4 | 0.5 ± 0.2 | 113 ± 8.3 | 3766 ±2300 | 104 ± 9.0 | NT | NT | NT | NT | NT | NT |
| GLP-1 [pM] | - | - | - | - | - | - | - | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT | 0.6 ± 0.1 | 121 ± 5.3 | 0.6 ± 0.1 | 119 ± 6.8 |
| AVP [pM] | - | - | - | - | - | - | - | NT | NT | NT | NT | NT | NT | NT | NT | 1.3 ± 0.3 | 109 ± 2.5 | NT | NT | NT | NT |
Encouraged by the promising profiles of original hits, we expanded structural diversity within scaffolds by obtaining commercially available analogs (analog-by-catalog, ABC) for each scaffold to further validate and optimize putative leads. In total, we acquired 33 compounds, 12 of which are structural analogs of 2-sulfonyl pyrimidines, 8 analogs for 2-mercapto pyrimidines and 13 compounds incorporating 2-aminopyrimdine scaffold. All molecules were subjected to SCTR-specific cAMP and binding assays and, in parallel, to a screen against AVP2Rs to exclude undesirable cross-selectivity. For PAM screening purposes and high-throughput detection of structure-activity relationships (SARs), we established and performed cAMP accumulation assays using a fixed concentration (EC10-EC20) of peptide ligand(s) to achieve a basal orthosteric stimulator response necessary to visualize PAM activity [30]. We deployed the natural full-length ligands secretin and AVP (arginine vasopressin) for basal activation of SCTR and AVP2Rs, respectively. In addition, we tested the set of structural analogs in SCTR cAMP assays stimulated with 3-peptide mix (3-pep mix), which comprises full-length secretin (Sec-FL) in combination with its C- terminally truncated, low-potent but fully efficacious analog secretin 1–23 (Sec(1–23)) and N-terminally cleaved, partially active secretin 3–27 (Sec(3–27)). In a previous study [30], the application of 3-pep mix resulted not only in substantially increased assay sensitivity, but also allowed the detection of probe-dependent hits. Since PAMs may exert intrinsic activity, we additionally conducted SCTR cAMP agonist assays in the absence of peptide ligands (SCTR agonist). To further profile and validate structural analogs with respect to target engagement and MOA, we performed TR-FRET based receptor binding assays by monitoring the effect of compounds on the interaction of N-terminal SNAP-tagged and Lumi-4 terbium cryptate-labeled SNAP-SCTRs with Fluo-Sec, a version of secretin linked to a C-terminal fluorescein molecule. Table 1 (Column 1–6) summarizes potencies and efficacies of 33 structural analogs determined by dose-response studies in the five described assay formats. 28 of 33 compounds demonstrated significant activity in SCTR PAM (3-pep mix) assay with negligible intrinsic agonist activity nor showing substantial off-target effects on AVP2Rs. Since all compounds, especially those with scaffold B and C, showed similar overall profiles but lower responses in full-length secretin (Sec-FL) stimulated SCTR PAM assays, we decided to concentrate on 3-pep mix assays for characterizing PAM activities described below. Consistent with the functional data, all but 6 analogs were able to significantly increase Fluo-Sec binding to SNAP-SCTRs.
Within the group of 2-sulfonyl pyrimidines, compounds A1, A2, A4, A5, A6, A7 and A8 share 3, 4-dimethoxy substituents at the phenyl (ring A) connected to the pyrimidine, but carry different substitution patterns on aryl function (Ar) that is connected to the sulfonyl pyrimidine (ring B) by a butanamide. Although having a similar activity profile overall, the substituents at the aniline (Ar) had an impact on PAM activity at SCTRs. A1, possessing a 2, 4-fluoro substitution at ring C, exerted PAM activity with a maximal efficacy of around 60% on SCTRs. Interestingly, the 3-chloro-4-fluoro analog A4 as well as the 2-methoxy-4-chloro variant A7 lost SCTR PAM activity (Emax ~30–40%), whereas A6 (2-fluoro analog) and A8 (2-bromo-4,6-fluoro, substitution) gained PAM efficacy for SCTRs (EC50 8–13 μM, Emax 63–77%). Compound A3, like A1 holding 2,4-fluoro substitutions at the aniline (Ar) but a 2-methoxy group at ring A, demonstrated moderate PAM activity on SCTRs (EC50 7.0 μM, Emax 40%). To evaluate the relevance of the aryl (Ar) function, we tested A9, A10 and A12, which share the 2-methoxy substitution at ring A but possess either no third aromatic moiety (A10, A12) or a fourth aromatic group (A9). A10 whose sulfonyl group is attached to butyric acid, displayed no significant activity in all assays, while 2-methylsulfonyl pyrimidine A12 showed a low-potent effect on SCTRs (EC50 18 μM, Emax 49%). Strikingly, despite showing substantial functional responses, A12 did not enhance Fluo-Sec binding to SNAP-SCTRs. A9, which contains a 1-[(4-methylphenyl)methyl]3-amino-1H-pyrazole instead of a halogen-substituted aniline, suffered from a loss of efficacy (Emax 42%) but was the most potent compound in this series (EC50 ~3 μM) in SCTR PAM mode. We also tested a 4-fluorophenyl-difluoromethyl pyrimidine analog (A11) that had no effect on SCTR binding or activation.
2-Mercapto pyrimidines (Table 1, Column 1–6) appear as reduced analogs of 2-sulfonyl pyrimidines due to the substitution of the sulfonyl into a thioether function. Other characteristics of group B analogs include a shorter linker missing one to two carbon atoms and a methylated amide function for select compounds. Compound B1, the original hit, possesses as A1 3, 4-dimethoxy substituents at ring A but a methylated acetamide linker connecting the 2-mercapto pyrimidine core to an unsubstituted phenyl (Ar). In SCTR PAM assay, B1 demonstrated moderate activity (EC50 3.6 μM, Emax 47%) and was devoid of significant intrinsic activity or effects on AVP2Rs. In addition, B1 substantially increased Fluo-Sec binding at SNAP-SCTRs (EC50 4.2 μM, 60% increase of binding) [30]. All 7 structural analogs of the original HTS hit B1, demonstrate clean and selective PAM activity, devoid of significant effects in AVP2R PAM as well as in SCTR agonist assays. Intriguingly, B2, which differs from B1 by lacking the 3-methoxy group at ring A, showed the strongest PAM activity toward SCTRs amongst all 33 analogs with 2.4 μM potency and 93% efficacy in 3-pep mix cAMP assays and a 76 percent increase of Fluo-Sec/SNAP-SCTR complexes (EC50 2.5 μM). In contrast, analog B3 was inactive in all assays, probably due to the removal of all substituents at ring A and/or the missing methyl group at the amide function. Similarly, compound B4 whose amide is mono-substituted with a 4-methylpyridine lacked significant activity in any formats. We wondered if the elongation of the structure led to attenuated PAM activity. Therefore, we tested additionally a version of B2 incorporating a propanamide linker, which is attached to a 2, 5-difluorophenyl group (B5). Even though this modification resulted in an increase of potency (EC50 0.3–1.4 μM) B5 seemed inferior to B2 with respect to PAM efficacy (Emax ~30%). To explore the influence of substituents on the third aromatic head (Ar), we evaluated the responses of two analogs of B3, which bear either an indanyl (B6) or a 2, 3-chlorophenyl (B7) function instead of the plain phenyl group present at B3. Both structural analogs were able to slightly increase PAM activity at SCTRs (EC50 1.6–2.9 μM, Emax 20–32%) compared to B3, however, the indanyl analog B6 was not able to increase Fluo-Sec binding to SNAP-SCTRs. Compound B8, carrying a 4-chloro substituent at ring A and a 2-methyl substitution at aniline (Ar), also lacked the ability to modulate SCTRs.
The third set of compounds (Table 1, Column 1–6) consisted of structural analogs incorporating the 4-phenyl-6-trifluoromethyl pyrimidine core with a benzylamine (C1-12) or phenethylamine (C13) substitution at position 2. The original HTS hit C1, contained a 4-methoxybenzylamine function and, like A1 and B1, 3, 4-dimethoxy groups at ring A. Similar to all structural analogs within group C, C1 elevated Fluo-Sec binding to SNAP-SCTRs (EC50 2.5 μM, 36% increase of binding), was devoid of off-target effects at AVP2Rs and hardly displayed intrinsic agonist activity at SCTRs. However, C1 demonstrated significant PAM activity with single-digit micromolar potency and maximal efficacy around 60 percent [30]. We divided 2-amino pyrimidine analogs into five distinct sets according to their substitution pattern at ring A. The first set comprising C1, C4, C5, C9 and C10 bears 3, 4-dimethoxy groups at cycle A but differ from their substitutions at the benzylamine function. Comparing to the original hit C1, the unsubstituted benzylamine analog C4 gains PAM effects with respect to SCTR cAMP accumulation (EC50 6.2 μM, Emax 76%) and binding (EC50 4.4 μM, 4.7% increase of binding). Replacement of the phenyl by a 3-pyridine ring (C5) led to a three-fold loss of potency toward SCTRs. The 4-chloro (C9) and 4-fluoro (C10) analogs demonstrated significant PAM activity similar to C4. The second set of compounds comprising C2 and C3, that are structural analogs of C1 and C4, respectively, lacking the 3-methoxy group at ring A. Interestingly, while maintaining a similar overall profile, C2 and C3 showed slightly lower PAM effects across receptor functional assays (SCTR PAM Emax 37–56%). Compounds C6, C7 and C11 constituted the third set bearing a methoxy substitution at position 3 of ring A and displayed equally significant PAM effects as their direct analogs C3, C6 and C10, respectively. Strikingly, C8 (set 4), structurally related to C1 and C2 due to the 4-methoxybenzylamine function but containing a 2-methoxy substitution at ring A, suffered from a loss of potency in SCTR PAM assays. The fifth set within scaffold C comprised analogs C12 and C13, which lack any substitution at cycle A. To explore the impact of the aromatic benzylamine moiety, we examined the effects of C12 whose 2-amino pyrimidine is connected to an ethyl acetate group. Even though it suffers from lower potencies (EC50 21–34 μM) in all formats, C12 was able to enhance SCTR activation and binding. We additionally investigated the influence of an elongation of the linker by testing the 3,4-dimethoxy phenethylamine analog C13, which was demonstrating substantial PAM activity at SCTRs in functional (EC50 5.7 μM, Emax 45%) but not in binding assays (EC50 18 μM, 13% increase of binding).
For more detailed allosteric activities and MOA studies, we selected two analogs for each scaffold group (Fig. 1). To facilitate further SAR conclusions, we focused on structural analogs displaying moderate to strong PAM effects that differed by a single structural feature in each scaffold group. According to these selection criteria, we characterized A1 and A2 with distinct substitution patterns on ring C, B1 and B2, which only differentiate from each other by a methoxy group at position 3 of ring A, as well as C4 and C9 with a plain or 4-chloro substituted benzylamine function (ring C), respectively.
Figure 1: Modulator titration reveals allosteric activity profiles dependent on distinct orthosteric ligands:

cAMP dose-response curves of (A) Sec-FL, (B) Sec(1–23), (C) Sec(3–27) on SCTRs treated with DMSO or six increasing concentrations of compounds (from left to right: A1, A2, B1, B2, C4 and C9) ranging from 0.8 to 25 μM; TR-FRET ratios converted to cAMP concentrations and normalized to corresponding orthosteric ligand; allosteric activity parameters KB, α and β were determined using the complete operational model of allosterism in GraphPad Prism; experiments were performed in duplicate in three independent experiments and data points are shown as mean ± SEM. For select compound concentrations only below error bars are shown for illustrative purposes.
3.2. Cooperativity factors reveal structure-related probe dependencies
To evaluate allosteric activity parameters of putative PAMs, we performed allosteric modulator titrations on orthosteric peptide ligand dose-response curves in cAMP assays. Being aware of allosteric modulator probe dependency [10, 31], the phenomenon that allosteric modulators exert distinct effects depending on the orthosteric stimulator probe present, we evaluated individually the allosteric activities toward full agonists Sec-FL and Sec(1–23) or the partial agonist Sec(3–27), the N-terminally truncated secretin analog. Allosteric activity parameters, which characterize the nature and power of an allosteric modulator, consist of the equilibrium dissociation constant KB and cooperativity factors on ligand-receptor complexes α and β, whereby α describes the allosteric modulation on ligand potency/binding affinity and β delineates cooperative effects on ligand efficacy [31]. Dose-response curves of peptide ligands were recorded with five increasing doses of test compound (Fig. 1, 800 nM to 25 μM) and allosteric activity parameters were determined using the complete operational model of allosterism [31] on the concentrations of cAMP formed during simultaneous compound and ligand treatment. The investigations on 2-sulfonyl pyrimidines A1 and A2 resulted in a very similar profile. Both test molecules demonstrated greater cooperativity toward secretin peptide efficacies than ligand potencies, while their affinities ranged between 0.4 and 6.4 μM. Overall, they appeared to have similar effects on Sec-FL and its truncated analogs. Strikingly, the titrations of structural analogs B1 and B2, whose key distinction is the presence or absence of a 3-methoxy group at ring A, respectively, revealed substantially different allosteric activity profiles. On SCTR-overexpressing cells, B1 demonstrated positive cooperativity in combination with Sec-FL and Sec(1–23) but negative cooperativity with Sec(3–27) with respect to ligand efficacy (β 0.3). B2 discriminated even more, displaying strong positive modulation of potency but not efficacy of Sec(1–23) (α 36; β 0.8), while also demonstrating strong potentiation of Sec(3–27) efficacy (α 6; β 30). In addition, we studied allosteric effects of 2-amino pyrimidine analogs C4 (-H) and C9 (-Cl). In general, we determined relatively similar PAM profiles for both compounds. With dissociation constants KB in the single to double-digit micro molar range, 2-amino pyrimidines exerted significant cooperativities toward secretin peptides with maximal positive modulation for Sec(3–27) (α 5–7; β 8–9).
3.3. B2 substantially decelerates secretin dissociation on SNAP-SCTRs
To further decipher the MOA of test compounds on SCTRs, we performed dissociation binding experiments [36]. One characteristic of class B GPCR activation is the two-domain binding model of its natural peptide ligands [2], i.e. the C-terminal portion of full-length secretin binds with moderate to high affinity to the extracellular N-domain (ECD) of the receptor while the N-terminal end of secretin interacts with the helical core bundle (J-domain) with much lower affinity, inducing conformational arrangements that result in the activation of intracellular signaling pathways predominantly through Gαs proteins. Hence, peptide concentration and affinity to the N-domain define receptor occupancy whereas binding to both domains is dependent on receptor conformations mainly regulated by G-protein coupling as reported for class A GPCRs [37], which leads to higher agonist affinities in the G protein-bound state. To facilitate interpretation of data, we added GTPγS to the dissociation buffer, which is described to shift the equilibrium of receptors into the G protein-unbound state and thereby attenuates J-domain binding [38]. Thus, after inducing dissociation, the majority of peptide ligand binds to only one site, the N-domain of receptors, which in addition leads to an elevated off-rate for Fluo-Sec (Fig. 2).
Figure 2: Analog B2 substantially prolongs Fluo-Sec residence time on SNAP-SCTRs:

Effects of DMSO, 12.5 μM of (A) 2-sulfonyl pyrimidines A1 (dark green), A2 (light green), (B) 2-mercapto pyrimidines B1 (light blue), B2 (dark blue), (C) 2-amino pyrimidines C4 (light red), C9 (dark red) or (B) 3.1 or 6.3 μM B2 (dotted lines in blue shades) on dissociation half-lives t1/2 of Fluo-Sec from SNAP-SCTR HEK-293 cell membranes induced by addition of an excess Sec-FL. LanthaScreen ratios normalized to Fluo-Sec bound at time 0; dissociation half-lives (t1/2) determined applying Dissociation – One phase exponential decay-equation in GraphPad Prism 8.4.0; experiments performed in triplicate in at least three independent experiments; data points shown as mean ± SD. Statistical significance of rate constant K changes compared to DMSO determined using GraphPad Prism’s unpaired t-test corrected with Holm-Sidak method, α = 0.01; (A) not significant; (B, C) significant (p < 0.01).
We investigated the impact of putative PAMs on Fluo-Sec dissociation induced by excess concentration of Sec-FL from SNAP-SCTR expressing HEK-293 cell membranes by adding 12.5 μM of test compound or DMSO to the dissociation buffer and recording dissociation binding curves to determine residence time half-lives (t1/2 [min]). 2-Sulfonyl pyrimidines A1 [30] and A2 slightly decelerated Fluo-Sec dissociation compared to control wells (t1/2 14 min) with half-lives between 15 and 16 min (Fig. 2A). The changes in rate constants K were determined to be not statistically significant. Addition of B1 [30], C4 or C9 resulted in a more significant effect, reducing dissociation rates of Fluo-Sec to half-lives between 25 and 30 min (Fig. 2B, C). Even more pronounced was the impact of structural analog B2 prolonging receptor residence time by 13-fold with a dissociation half-life of 177 min (Fig. 2B). To explore the strength of this interaction in more detail, we determined half-lives at 3.1 μM (58 min) and 6.3 μM (108 min) of B2, which confirmed that B2 significantly slows down Fluo-Sec dissociation in a dose-dependent manner.
3.4. SCTR PAMs potentiate signaling in endogenously receptor-bearing NG108 cells
Since receptor overexpressing cell lines and membranes applied in selectivity and allosteric activity studies, may display overly amplified or distorted signaling, we investigated the response of test compounds in the neuroglioma hybrid cell line NG108(−15), which was created by fusion of mouse neuroblastoma with rat glioma cells (ATCC) and has been described to endogenously express SCTRs [39]. To determine PAM activity, we incubated 25 μM of test compound with increasing concentrations of peptide ligands and recorded corresponding changes in cAMP accumulation. Figure 3A illustrates the modulation of 2-sulfonyl pyrimidines A1 and A2 on Sec-FL, Sec(1–23) and Sec(3–27). In this format, the effects of A1 and A2 appeared to be mainly based on intrinsic agonist activity elevating basal activity by 30 to 40 percent. Cooperative effects of group A analogs toward Sec-FL and Sec(1–23) were minor, detected as around two-fold improved potencies and a 20 percent gain of efficacies. The effects of 2-mercapto pyrimidines in NG108 cells are depicted in Figures 3B. Analog B1 displayed an around 20% elevation of basal activity and showed positive cooperativity toward Sec-FL and Sec(1–23) potencies, whereas the 4-methoxy analog B2, even though exerting similar positive effects on full agonists (3–7-fold leftward shift), did not show signs of intrinsic agonist activity. In addition, B2 was able to convert the inactive Sec(3–27) into a partial agonist with 20% efficacy in NG108 cells. In contrast, 2-amino pyrimidines C4 and C9 (Fig. 3C) exhibited negligible effects in this cell model, displaying 2–3-fold improved potencies in combination with Sec-FL and Sec(1–23) and having no effect on Sec(3–27).
Figure 3: SCTR modulators exert PAM activity in NG108–15 cell line:

cAMP dose-response of (A, B, C, left to right) Sec-FL, Sec(1–23) and Sec(3–27) in NG108 cells treated with DMSO (black), 25 μM of (A) 2-sulfonyl pyrimidines A1 (dark green), A2 (light green), (B) 2-mercapto pyrimidines B1 (light blue), B2 (dark blue) or (C) 2-amino pyrimidines C4 (light red), C9 (dark red); TR-FRET ratios resulting from cAMP accumulation normalized to Sec-FL; graphs plotted using GraphPad Prism; experiments performed in duplicates in at least three independent experiments; data points shown as mean ± SEM. Statistical significance determined using GraphPad Prism’s unpaired t-test corrected with Holm-Sidak method.
3.5. Structural similarity to GLP-1R small molecule agonists discloses GLP-1R-modulation by SCTR PAMs
During our SAR studies on SCTR PAMs, we discovered that 2-sulfonyl pyrimidine moieties had been described to exert electrophilic reactivity and possess mild alkylating properties [40, 41]. Beyond that, we found clear structural similarity to described GLP-1R agonists/PAMs Compound A [12] and BETP [7] (Fig. 4A 1.), referring to the 4-phenyl-6-trifluoromethyl pyrimidine core (ring A and B) incorporated in Compound A/BETP structures that is consistently present across all three SCTR PAM scaffolds (Fig. 4A 2.−4.). To investigate whether our newly identified SCTR PAMs also exhibit effects at GLP-1Rs, we performed cAMP assays using GLP-1R-overexpressing HEK-293T cells in agonist and PAM mode, performed in the absence or in the presence of EC20 concentration of GLP-1, respectively (Table 1, Column 7–8). For comparison of our 33 analogs with a thoroughly characterized GLP-1R ago-PAM, BETP was included as reference to all performed assays (Table 1, Column 1–8). As expected, BETP exerted strong PAM and agonist activity toward GLP-1Rs (EC50 0.8 μM, Emax 76–91%). Even though PAM activity at SCTRs has never been reported for BETP, it was also able to positively modulate SCTR binding and activation (SCTR PAM Emax 51 %). Nevertheless, BETP potency toward SCTRs was significantly lower (SCTR PAM EC50 24 μM) resulting in a 30-fold selectivity toward GLP-1Rs. Within our group of SCTR PAMs, only scaffold A analogs exerted significant intrinsic activities (Table 1, Column 8, GLP-1R agonist). However, in GLP-1R PAM format not only 2-sulfonyl pyrimidines but also 2-amino pyrimidines and select analogs of 2-mercapto pyrimidines were able to substantially increase GLP-1 stimulated cAMP accumulation in HEK-293T-GLP-1R cells (Table 1, Column 7, GLP-1R PAM). To assess overall receptor preferences, we depicted the maximal efficacy (Emax [%]) of test compounds with respect to SCTR PAM (3-pep mix) activity in correlation to their PAM effects on GLP-1Rs in a scatterplot (Fig. 4B). Within the group of 2-sulfonyl pyrimidines, A3 (2-methoxy analog of A1), the 3-chloro-4-fluoro analog A4, 2-methoxy-4-chloro variant A7, A11 (4-fluorophenyl-difluoromethyl pyrimidine analog of GLP-1R ago-PAM Compound A) and the 2-methylsulfonyl pyrimidine A12 demonstrated an overall selectivity trend toward GLP-1Rs. Interestingly, group A analogs lacking the third aromatic moiety (Ar) seemed to generally favor GLP-1Rs, whereas both, substitution patterns at ring A and at the aniline Ar, influenced receptor selectivity profiles.
Figure 4: Three related scaffolds demonstrate structural and functional similarity to GLP-1R ago-PAMs Compound A and BETP:

(A) Molecular structures of 1. established GLP-1R PAMs Compound A and BETP, 2. Group A 2-sulfonyl pyrimidines A1, A2 and A3, 3. Group B 2-mercapto pyrimidines B1 and B2, 4. Group C 2-amino pyrimidines C4 and C9. (B) Scatterplot depicting % maximal efficacy (Emax) in SCTR PAM (3-pep mix) cAMP assays (x-axis) in correlation to % Emax in GLP-1R PAM mode (y-axis), color code according to scaffold sets A (blue), B (green), C (red) and positive controls Sec-FL, GLP-1 and BETP (orange); scatterplot created using TIBCO Spotfire.
In contrast, 2-mercapto pyrimidines (group B) as well as 2-amino pyrimidines (group C) appeared to favor SCTRs, even though select analogs showed substantial effects in cAMP GLP-1R PAM assays. Of the six analogs we had investigated in more detail, we found five (A1, A2, B1, C4 and C9) displaying a balanced profile. However, compound B2, which differs from B1 by missing the 3-methoxy group at ring A, strongly preferred modulating SCTRs by displaying only moderate PAM activity at GLP-1Rs.
3.6. SCTR - GLP-1R selectivity profiles considering probe dependencies and GLP-1R cooperativity factors
Although initial screens against SCTRs and GLP-1Rs indicated certain compound selectivity profiles, we were concerned that the use of different orthosteric stimulator probes could have had impacted receptor preferences. To investigate GLP-1R effects in more detail, we performed MOA studies with the same six analogs studied in SCTR experiments. Moreover, to determine binding affinities and power of allosteric modulators, the evaluation of allosteric activity parameters against GLP-1Rs is obligatory. Since BETP was shown to have a much greater effect on GLP-1(9–36) [12, 20], the DPP4 (dipeptidyl-peptidase-4) cleavage product of GLP-1 [42], than on the full-length peptide GLP-1, we additionally included GLP-1(9–36) in all further studies on GLP-1Rs. As illustrated in Figure 5A–B, BETP showed mild allosteric activity on GLP-1-treated GLP-1Rs with a dissociation constant KB of 3 μM and α/β values between 1 and 3, while in combination with GLP-1(9–36) BETP significantly enhanced both potency (α 137) and efficacy (β 5.5), as illustrated in Figure 5A–B, which is in alignment with previous studies and thereby validated our approach [20]. Of note, the highest concentration of BETP equal 25 μM was excluded from analysis as it led to an inversion of its PAM activity. 2-sulfonyl pyrimidines A1 and A2 demonstrated similar profiles with slightly weaker responses. We observed for A1 15–200-fold reduced affinities, compared to its SCTR profile, but substantially increased cooperative effects on GLP-1(9–36) ligand potency (α 247). We recorded similar overall PAM effects for A2, even though its KB and α values remained in a 10-fold lower range (KB 1.5–8 μM; α (GLP-1(9–36)) 21). These discrepancies might be due to the electrophilic nature of 2-sulfonyl pyrimidines, which could interfere with the calculation of equilibrium dissociation constants and associated factors as allosteric pharmacology models do not account for covalent binding principles [19, 43]. We further studied the effects of structural analogs B1 and B2, whose key distinction is the presence or absence of a 3-methoxy group at ring A. Both analogs showed weak PAM effects on GLP-1 against GLP-1Rs. Interestingly, 3, 4-methoxy analog B1 was able to substantially increase potency and efficacy of the N-terminally truncated analog GLP-1(9–36) (α 3.5; β 4.5) whereas 4-methoxy variant B2 only displayed minor PAM effects (α 2.9; β 1). In contrast, 2-amino pyrimidine analogs C4 (-H) and C9 (-Cl) displayed negligible cooperativity on GLP-1(9–36)-treated GLP-1R-overexpressing cells while significantly modulating the full-length peptide GLP-1 in a positive way (α 3–5; β 2).
Figure 5: Cooperativity factors reveal distinct selectivity profiles toward SCTRs/GLP-1R and full-length/truncated peptides:

cAMP dose-response curves of (A) GLP-1 or (B) GLP-1(9–36) on GLP-1Rs treated with DMSO or six increasing concentrations of compounds (from left to right: BETP, A1, A2, B1, B2, C4 and C9) ranging from 0.8 to 25 μM; (C) Modulator titration of BETP on SCTR-stimulated cAMP accumulation by (left to right) Sec-FL, Sec(1–23) or Sec(−27); TR-FRET ratios converted to cAMP concentrations and normalized to corresponding orthosteric ligand; allosteric activity parameters KB, α and β were determined using the complete operational model of allosterism in GraphPad Prism; experiments were performed in duplicate in three independent experiments and data points are shown as mean ± SEM. For select compound concentrations only below error bars are shown for illustrative purposes. (D) Compound selectivity toward GLP-1Rs vs SCTRs determined as Δ log (αβ) (SCTR log (αβ) – GLP-1R log (αβ)) stimulated with either full-length (solid black) or truncated (white/black stripes) peptide agonists; (E) probe dependency toward full-length (FL) vs truncated (TR) peptide ligands determined as Δ log (αβ) (TR log (αβ) – FL log (αβ)) stimulating either SCTRs (black, white dots) or GLP-1Rs (white, vertical stripes).
We further assessed allosteric activity data of BETP against SCTRs (Fig. 5C). The GLP-1R ago-PAM BETP was able to slightly shift the responses of secretin peptides displaying the lowest effects in combination with C-terminally truncated analog Sec(1–23). For better comparison of cooperative strengths and selectivity profiles, we determined the difference between logarithmic cooperativity factors Δ log (αβ) (Fig. 5D). To match results for both receptors, we concentrated on full-length peptides Sec-FL and GLP-1 as well as N-terminally truncated (TR) analogs Sec(3–27) and GLP-1(9–36). For evaluation of receptor selectivity (Fig. 5D), we depicted Δ log (αβ) of full-length peptides (Sec-FL minus GLP-1) or their truncated analogs (Sec(3–27) minus GLP-1(9–36)). To describe probe dependencies within SCTRs or GLP-1Rs (Fig. 5E), we subtracted log (αβ) of full-length peptides from truncated analogs (Sec(3–27) minus Sec-FL; GLP-1(9–36) minus GLP-1). BETP, A2 and B1 were GLP-1R-selective by comparing truncated analogs but displayed slight SCTR preference when treated with full-length peptides. 2-Amino pyrimidines C4 and C9 preferred partial agonist Sec(3–27) stimulation on SCTRs but full agonist GLP-1 treatment on GLP-1Rs. A1 and B2 were solely selective for GLP-1Rs or SCTRs, respectively, independent of the nature of the orthosteric ligand. Overall, the preferences for a distinct receptor were more pronounced while stimulated with a truncated peptide version (Fig. 5E).
3.7. Select analogs enhance cAMP formation and insulin secretion in INS-1 (832/3) rat insulinoma cells
To evaluate the response of test compounds in a naturally GLP-1R-bearing environment, we used INS-1 (832/3) rat insulinoma cells as cell model with natural GLP-1R expression, which have been used for studies on insulin secretion and pancreatic islet beta-cell function [34]. Since the response in INS-1 cells upon GLP-1R activation was hard to detect with the Gs dynamic cAMP kit (Cisbio) that had been used in all other cAMP assays described in this study, we switched to the LANCE Ultra kit (PerkinElmer) with higher sensitivity for the detection of cAMP accumulation in INS-1 cells. We applied otherwise the same conditions as in our studies in NG108–15 cells (25 μM of test compound, peptide ligand titration), except for BETP, which had displayed inverse effects at 25 μM in GLP-1R overexpressing cells. Hence, we reduced its test concentration to 12.5 μM. Figure 6A illustrates the effects on cAMP accumulation of GLP-1 or GLP-1(9–36) stimulated INS-1 cells in combination with DMSO, BETP or 2-sulfonyl pyrimidines (A1, A2). BETP did not show a significant positive effect on GLP-1 treatment but was able to convert GLP-1(9–36) signaling from initially inverse agonism to around 50 percent receptor activation (106% increase of efficacy). 2-sulfonyl pyrimidines A1 and A2 displayed comparable PAM activity on partial agonist GLP-1(9–36), showing 84% elevation of efficacy, while demonstrating a 2-fold leftward shift of the dose-response curve together with a 20% increase of basal activity toward full agonist GLP-1. Similarly, compound A3, a structural 2-sulfonyl pyrimidines analog with higher GLP-1R selectivity, exerted strong PAM activity on GLP-1(9–36) in INS-1 cells comparable to BETP leading to a 105% gain of efficacy and demonstrated positive cooperativity toward GLP-1 like its close structural analogs A1 and A2. By contrast, both 2-mercapto pyrimidine analogs, B1 and B2, demonstrated rather negative effects on basal receptor activation with only negligible effects on peptide ligand potencies (Fig. 6B). 2-Amino pyrimidine C4 and C9 analogs addition resulted in a similar profile but very minor increase of basal activation (Fig. 6C).
Figure 6: Select SCTR PAMs potentiate stimulatory and insulinotropic effects in pancreatic rat insulinoma cell line:

cAMP dose-response of (A, B, C, left to right) GLP-1 and GLP-1(9–36) in INS-1 cells treated with DMSO (black), 12.5–25 μM of (A, B) BETP (purple) or 2-sulfonyl pyrimidines A1 (dark green), A2 (light green), A3 (light blue-green), (C, D) 2-mercapto pyrimidines B1 (light blue), B2 (dark blue) or (E, F) 2-amino pyrimidines C4 (light red), C9 (dark red); TR-FRET ratios resulting from cAMP accumulation normalized to GLP-1; graphs plotted using GraphPad Prism; experiments performed in duplicate in at least three independent experiments; data points shown as mean ± SEM. Statistical significance determined using GraphPad Prism’s unpaired t-test corrected with Holm-Sidak method. (D) Increase of insulin secretion compared to DMSO (black) in DMSO or GLP-1(9–36) co-treated INS-1 cells with 20 μM of BETP (purple), A1 (dark green), A2 (light green), A3 (light blue-green), B1 (light blue), B2 (dark blue), C9 (red) or negative control compound (NEG CTRL, beige); TR-FRET ratio resulting from insulin secretion converted to insulin concentration (ng/mL) per source well and plotted as bar graphs using GraphPad Prism 8.4.0.; experiments were performed as quintuplicates in three independent experiments and data is presented as mean ± SD; statistical significance was determined using ordinary two-way ANOVA and Tukey’s multiple comparisons test (99% confidence interval). P values were illustrated according to the following classification: (*p = 0.01–0.05), (**p = 0.01–0.001), (***p = 0.001–0.0001), (****p < 0.0001).
To further explore biologically relevant activities, we utilized the INS-1 cell line in a glucose-stimulated insulin secretion (GSIS) experiment (Fig. 6D). Since a previous study investigating BETP effects on insulin secretion in isolated mouse islets revealed BETP was most effective together with GLP-1(9–36) in a high glucose environment [7], we focused on evaluating test compounds (BETP, A1, A2, A3, B1, B2 and C9) in these conditions, studying synergistic compound effects in combination with high glucose concentrations (15 mM) and with or without GLP-1(9–36) stimulation. As negative control, we utilized a compound that was inactive in cAMP assays on GLP-1R-expressing cell lines (data not shown). We determined statistical significance of compound or ligand treatment as well as of interactions between compound and ligand treatment in GraphPad Prism applying ordinary two-way ANOVA and Tukey’s multiple comparisons test with individual variances computed for each comparison. In the absence of GLP-1(9–36), insulin secretion was significantly enhanced by BETP, C9 (****p < 0.0001), A1, A2, B1 (**p < 0.01) and B2 (*p < 0.05). Co-stimulation with GLP-1(9–36) elevated insulin secretion for all tested analogs, which was more significant for BETP, A1, A2 and C9 (****p < 0.0001) than B2 (***p < 0.001) or B1 (**p < 0.01). Surprisingly, 2-sulfonyl pyrimidine A3 did not enhance insulin secretion in either condition. Further analysis accredited solely BETP (****p < 0.0001) and A1 (***p = 0.0005) significant interactions with GLP-1(9–36). The negative control did not display any significant effects.
3.8. Scaffold A demonstrates electrophilic reactivity and potential covalent mechanism of action
As reported in previous studies [7, 20], Compound A and BETP belong to a group of GLP-1R PAMs that act via covalent modification of cysteine 347 (Cys-347, C347) in GLP1-Rs. Furthermore, 2-sulfonyl pyrimidines constituting the core structure shared between Compound A (Fig. 4A, 1.) and group A analogs, have been shown to alkylate cysteines via a nucleophilic aromatic substitution reaction due to electron deficiency at position 2. To determine the potential electrophilic reactivity of analogs incorporating scaffold A, we incubated A1 with glutathione (GSH) for up to four hours and monitored putative A1-GS adduct formation via HPLC/MS. Indeed, we observed a complete conversion of A1 into A1-GS adduct and A1-sulfinic acid (Fig. 7A) already after 5 minutes reaction time. In parallel, we performed the same experiment using 2-mercapto pyrimidine B2. In accordance with its lower electrophilicity, we did not observe GS-adduct formation in up to 4 hours of incubation with B2 at 37 °C. Having confirmed that 2-sulfonyl pyrimidines are capable of covalently modifying cysteines, we investigated PAM scaffolds in a cell-based setting with respect to a potential irreversible MOA. Inspired by a previously reported compound-washout cAMP assay [20], we recorded cAMP accumulation in SCTR-overexpressing CHO-K1 cells (Fig. 7B) or GLP-1R-overexpressing HEK-293T cells (Fig. 7C, pretreated with test compounds prior to a triple wash step. The corresponding peptide ligands Sec-FL and GLP-1 were added after the washout of PAMs. Intriguingly, 2-sulfonyl pyrimidines A1 and A2 maintained the ability to augment the cAMP response of Sec-FL after washout supporting a potential covalent attachment to SCTRs as the basis for PAM function. In contrast, 2-mercapto pyrimidine B2 and 2-amino pyrimidine C9 lost their positive cooperativity if added prior to the washout confirming the reversible nature of these two scaffolds. We observed similar responses with GLP-1Rs (Fig. 7C). BETP and 2-sulfonyl pyrimidines A1 and A2 exerted substantial intrinsic activity manifested as an elevation of basal response for both conditions, added before or after the wash step. In this case, a pretreatment of the electrophilic compounds seemed to further enhance their effects on GLP-1Rs.
Figure 7: Scaffold A but not B and C demonstrate electrophilic reactivity and potential covalent mechanism of action:

(A) Covalent reaction of glutathione (GSH) and A1 leading to the formation CMPD A1- GS adduct and CMPD A1 sulfinic acid confirmed by HPLC-MS analysis. cAMP dose-response curves of (B) Sec-FL stimulating SCTRs and (C) GLP-1 stimulating GLP-1Rs; both pre-treated (dotted lines) prior a triple wash step with DMSO, irreversible scaffold A (A1 wash, A2 wash, BETP wash, left panels) or reversible scaffolds (B2 wash, C9 wash, right panels) as well as co-treated (solid lines) with DMSO, irreversible scaffold A (A1, A2, BETP, left panels) or reversible scaffolds (B2, C9, right panels) added after wash; TR-FRET ratios resulting from cAMP accumulation normalized to corresponding agonist; graphs plotted using GraphPad Prism; experiments performed in duplicate in at least three independent experiments; data points shown as mean ± SEM. Statistical significance determined using GraphPad Prism’s unpaired t-test corrected with Holm-Sidak method, α = 0.01; (B) all potency shifts statistically significant (p < 0.01) if not indicated otherwise (ns = not significant); (C) EC50 shifts for all conditions not statistically significant except BETP wash (p < 0.01) and C9 (p < 0.05). Intrinsic activity (% basal activity) statistically significant (p < 0.01) for BETP, BETP wash, A1, A1 wash, A2 and A2 wash.
3.9. Mutational studies reveal a putative secondary mechanism for 2-sulfonyl pyrimidines
Encouraged by these strong indications for covalent MOAs of 2-sulfonyl pyrimidines, we explored their utility as tools to investigate their potential site of interaction with both receptors. Since Cys-3476.36 was determined to be the site of action for BETP and related compounds [20], we performed a sequence alignment (GPCRdb) of SCTR and GLP-1R. As expected, we found no cysteine, but a lysine (Lys-3176.36) at the analogous position in SCTRs (Fig. 8A). Depending on the structural environment, lysine residues have been shown to be covalently modified by electrophilic chemotypes [44], however, we considered it less likely, since there was no evidence for this conjugation with 2-sulfonyl pyrimidines. Nevertheless, we continued with site-directed mutagenesis yielding GLP-1R C347A (cysteine-to-alanine), SCTR K317A (lysine-to-alanine) and SCTR K317C (lysine-to-cysteine) constructs to investigate a possible common site of interaction on both receptors. Wild type (WT) and mutant constructs were transiently expressed in HEK-293 cells and their PAM responses on corresponding peptide ligands in cAMP accumulation assays were compared. Intrinsic agonist and cooperative effects of BETP on GLP-1(9–36) were eradicated in the GLP-1R C347A mutant (Fig. 8B), which validated the results of a previous study [20]. We detected a substantial loss of activities for 2-sulfonyl pyrimidines A1, A2 and A3 toward GLP-1(9–36) stimulating GLP-1R C347A compared to WT receptor, however, the PAM effect was not completely forfeited and the potentiating activity for GLP-1 even remained in the same range. Of note, structural analog A3 lost its intrinsic activity at the GLP-1R C347A mutant whereas the increased basal activation of A1 and A2 was consistent in both, WT and mutant receptors. To ensure that lack of PAM activities of putative irreversible scaffolds was caused by the missing cysteine and not by potential receptor conformational arrangements induced by the mutation, we additionally tested 2-mercapto pyrimidines B1 and B2 as reversible control compounds on GLP-1 C347A (Fig. 8C). As expected, cooperative activities of both compounds remained unchanged in cells expressing the GLP-1R C347A mutant receptor.
Figure 8: Irreversible scaffold A acts likely via two distinct mechanisms:

(A) Sequence alignment of SCTR and GLP-1R, specifically TM5, ICL3 and TM6 and highlighting homolog residues C347GLP−1R and K317SCTR; generated by GPCRdb. cAMP dose-response of (B, C, top panels) GLP-1, (B, C, bottom panels) GLP-1(9–36), Sec-FL (D, E, top panels) or Sec(3–27) (D, E, bottom panels) stimulating (B, C, left panels) GLP-1R-WT, (B, C, right panels) GLP-1R C347A, (D, E, left panels) SCTR WT, (D, E, middle panels) SCTR K317A or (D, E, right panels) SCTR K317C treated with DMSO (black), 12.5–25 μM of (B, D) BETP (purple) or 2-sulfonyl pyrimidines A1 (dark green), A2 (light green), A3 (light blue-green) or (C, E) 2-mercapto pyrimidines B1 (light blue), B2 (dark blue); TR-FRET ratios resulting from cAMP accumulation normalized to corresponding ligands; graphs plotted using GraphPad Prism; experiments performed in duplicate in at least three independent experiments; data points shown as mean ± SEM. Statistical significance for EC50 shifts determined using GraphPad Prism’s unpaired t-test corrected with Holm-Sidak method, α = 0.01; all results statistically significant (p < 0.01) if not indicated otherwise (ns = not significant).
Similarly, we generated and characterized SCTR K317A and SCTR K317C to investigate whether the MOA of our compounds is covalent attachment to Lys-317 and to determine whether incorporation of a cysteine residue at the homologous position at SCTRs would further increase sensitivity toward BETP as it was reported for glucagon receptors (GCGRs) [20], respectively. To ensure functional integrity of receptor constructs, we subjected transiently expressing COS-1 cells to radioligand binding experiments using 125I-Tyr10-secretin and Sec-FL. Both, SCTR K317A and SCTR K317C, were able to maintain the ability to bind secretin peptides similarly to WT SCTRs (Table 2).
Table 2: Binding and biological activity parameters for SCTR mutant constructs expressed in COS-1 cells.
Values are expressed in means ± SEM from 4 independent experiments performed in duplicate.
| Constructs | pKi | pEC50 |
|---|---|---|
| SCTR WT | 9.2±0.2 | 11.6±0.4 |
| SCTR (K317C) | 8.7±0.1 | 10.9±0.4 |
| SCTR (K317A) | 8.9±0.1 | 10.5±0.2 |
In addition, we transiently transfected the SCTR constructs in HEK-293 cells and tested effect of the mutations on responses of A and B group compounds in cAMP assays (Fig. 8D and E). Compared to WT receptors, secretin peptides demonstrated a similar activation profile on both receptor mutants even though potencies against SCTRs K317C were slightly reduced by two to three-fold. Strikingly, neither mutating Lys-317 to alanine nor to cysteine had an impact on the potentiating effects of irreversible analogs BETP, A1 or A2. Surprisingly, reversible control B2 seemed less effective on SCTR K317A receptors. Based on these results, we concluded that covalent modulators target very likely an allosteric binding site at SCTRs distinct from the reported site of interaction at GLP-1Rs [21].
4. Discussion
Here we report the discovery and detailed characterization of three structurally related scaffolds that originated from a HTS screening campaign against SCTRs. Expansion of structural analogs within scaffold sets and subsequent SAR studies led to the selection of two promising derivatives from each group for detailed allosteric activity studies and analysis of effects in naturally SCTR-expressing NG108 cells. Intriguingly, we recognized that our newly discovered SCTR PAMs share their core scaffold with reported GLP-1R activators Compound A and BETP. Based on this structural similarity, we screened novel SCTR PAMs against GLP-1Rs and determined probe-dependent activities for scaffold groups in GLP-1R-overexpressing and endogenously expressing cell lines. Moreover, we distinguished between irreversibly and reversibly acting analogs based on their electrophilicity at the 2-position of the pyrimidine ring, further confirmed by GSH-reactivity and wash-out experiments. Mutational studies at position 6.36 at both, GLP-1Rs and SCTRs, not only confirmed a common covalent MOA for scaffold A with other GLP-1R PAMs such as BETP, but also discerned reversible interactions with compounds B1 and B2 and indicated a secondary mechanism for 2-sulfonyl pyrimidine analogs.
4.1. Irreversible 2-sulfonyl pyrimidine scaffold likely acts via an alternative mechanism in addition to Cys347GLP−1R alkylation
We identified the 4-phenyl-6-trifluoromethyl pyrimidine core as a shared structural motif amongst 2-sulfonyl pyrimidines (group A), 2-mercapto pyrimidines (group B), 2-amino pyrimidines (groupC) and BETP, which belongs to a group of characterized GLP-1R ago-PAMs [7, 12, 19, 20]. Beyond that, similarly to BETP 2-sulfonyl pyrimidines incorporate an electrophilic moiety, which is crucial for the potentiating effects of many GLP-1R allosteric modulators by covalently modifying Cys-347 located at TM6 close to intracellular loop (ICL) 3, which leads to stabilization of GLP-1R active conformation by promoting engagement with G proteins via formation of a 90° kink in TM6. Willard, et. al. [19] developed a putative binding model of BETP in G-protein bound GLP-1Rs (Fig. 9A). Besides an irreversible linkage to Cys-347, they proposed additional halogen-bonding interactions between the trifluoromethyl group and the positively charged ε-amino function of K346 as well as attractions of the aromatic moieties by hydrophobic amino acid residues (F324, I328, V332, L354) through van-der-Waals forces. Mutagenesis studies with C347 mutant receptors supported that the activity of 2-sulfonyl pyrimidines on GLP-1Rs was predominantly dependent on a putative covalent interaction with Cys-347. Thus, structural modifications targeting the linker or the aniline ring (ring C) should play a minor role in GLP-1R modulation with the 4-phenyl-6-trifluoromethyl pyrimidine moiety residing in the proposed allosteric binding pocket (Fig. 9B, C). Therefore, 2-sulfonyl pyrimidines can be distinguished between A1-like compounds incorporating a 3, 4-methoxy substitution, and A3-like analogs which bear a 2-methoxy group instead. Both types of 2-sulfonyl pyrimidines are lacking the 3-benzyloxy function that is part of the BETP molecule, resulting in a dramatic loss of potency (Table 1) in both cases, putatively due to missing hydrophobic interactions with F324 and I328. Since A3 displayed significantly higher responses in GLP-1R related cAMP assays and was more affected by the cysteine-to-alanine mutation than its direct analog A1, we hypothesized that the hydroxy function of threonine T353 might be involved in potential hydrogen-bonding interactions with the 2- or 3-methoxy substituents in scaffold A analogs. In addition, A3-like analog A12 demonstrated similar superior activity in GLP-1R cAMP assays, which supports the relevance of the 2-methoxy group and negligibility of the butanamide linker and third aromatic moiety in order to interact with GLP-1Rs. Interestingly, A11 suffered from a loss of potency likely due to the difluoromethyl substitution and/or the lack of the 2-methoxy function supporting receptor-ligand interactions proposed in the binding model. Otherwise, the number and location of methoxy substituents as well as the nature of the tail attached to sulfonyl groups could influence the on-rate of compounds, their positioning for Cys-347 engagement and the ability to pass through the cell membrane, which is required to act via Cys-347 and the surrounding binding pocket. This prerequisite could also explain the inactivity of the butyric acid analog A10. In addition, our mutational studies revealed that the potentiating effects of scaffold A analogs could not be completely diminished by replacing the nucleophilic cysteine residue. Hence, we assume the existence of a second allosteric mechanism, either through reversible effects or interactions with a second binding site that is not targeted by BETP and could further explain SAR of analogs.
Figure 9: Comparison of BETP and 2-sulfonyl pyrimidines covalently bound to active-state GLP-1Rs:

Model of BETP’s (purple sticks, left panel) binding pocket, adapted from ref. [19], illustrating covalent modification of Cys347 in TM6 at the GLP-1R (cyan) in active conformation and additional putative influences of surrounding residues, such as van-der Waals interactions with hydrophobic residues or halogen attraction of Lys346; model was modified for A1-like (green sticks, middle panel) and A3-like (pale blue sticks, right panel) 2-sulfonyl pyrimidine analogs; illustrations generated using Chimera 1.13.1.
Beyond targeting GLP-1Rs, 2-sulfonyl pyrimidines show PAM activities toward SCTRs superior to BETP, which are also likely due to a covalent mechanism as supported by washout experiments. Our mutagenesis studies suggested that newly discovered 2-sulfonyl pyrimidines do not discriminate between WT, K317A or K317C SCTRs and therefore do not target an allosteric binding site at SCTRs equivalent to the pocket in GLP-1Rs. In contrast to the phenylalanine-to-cysteine substitution in GCGRs, the lysine-to-cysteine mutation could not engender sensitivity to BETP in SCTRs. Even though alignment of SCTR and GLP-1R sequence by GPCRdb suggested Lys-317SCTR as the homolog residue, a multiple sequence alignment of class B GPCRs proposed Cys-347GLP−1R might be the result of a single amino acid insertion that had occurred within a glucagon receptor sub-family (GLP-1R, GLP-2R, GCGR and GIPR) [19]. Thus, our results might lack direct comparability to the GCGR study. Nevertheless, the indifference toward SCTR K317A lacking the potential halogen interaction partner, supports the hypothesis of an alternative site of interaction. Another indicator that 2-sulfonyl pyrimidines may target a distinct location at SCTRs is the different probe dependency profile in allosteric activity studies on SCTR or GLP-1R-overexpressing cell lines comparing potentiating effects on full-length or truncated peptides. While highly selective for the N-terminally truncated analog GLP-1(9–36) over full-length peptide GLP-1, analogs A1 and A2 were less discriminating in case of secretin and truncated analogs Sec(1–23) and Sec(3–27). Furthermore, in an endogenous signaling environment scaffold A compounds converted GLP-1(9–36) from an inverse agonist to a partial agonist in INS-1 cells whereas they were unable to rescue inactive Sec(3–27) in the NG108 cell line. Beyond that, we would expect that a PAM or allosteric agonist targeting an allosteric hub at TM6 that is crucially involved in stabilizing the active ligand-GPCR-G-protein ternary complex conformation, would lead to a substantial increase in orthosteric agonist residence time as it has been shown for BETP at GLP-1Rs [45–47]. However, we observed only minor increase of Fluo-Sec binding to SNAP-SCTRs overall and negligible effects on Fluo-Sec dissociation for analogs incorporating the 2-sulfonyl pyrimidine scaffold A compared to reversible PAM scaffolds B and C. Altogether, although our data strongly supports an alternative and possibly shared allosteric mechanism for 2-sulfonyl pyrimidines on both, SCTRs and GLP-1Rs, in addition to the thoroughly characterized modification of Cys-347GLP−1R, further investigations and mutational studies are necessary to confirm and decipher the potential novel allosteric binding site as well as its impact on SARs of structural analogs.
4.2. Reversible analogs exert PAM activity with distinct selectivity profiles
Since highly reactive compounds like 2-sulfonyl pyrimidines may be impractical for drug development purposes, we were excited to have identified two related scaffolds devoid of electrophilic reactivity. Glutathione reactivity, cAMP washout and mutant receptor studies confirmed the absence of an apparent covalent MOA for 2-mercapto pyrimidines, which validated that the sulfur atom had not been targeted by air oxidation. Selectivity screens around scaffold B revealed that a mono or di-methoxy substituted phenyl ring and/or a methylated acetamide linker are crucial for significant PAM activity in both SCTRs and GLP-1Rs. Since our SAR studies were solely based on commercially available structural analogs, a synthetic approach would be necessary to decipher the impact of individual functional groups, which was not part of the current study. Focusing on the two most potent analogs B1 and B2 differing from each other by one methoxy group at the core phenyl ring, we determined surprisingly distinct selectivity profiles and probe dependencies for the individual molecules. B1 augmented not only GLP-1(9–36)–triggered effects on GLP-1Rs to a much greater extent than B2 but also appeared to preferentially enhance Sec-FL and Sec(1–23) over Sec(3–27) signaling. In contrast, beyond significant cooperative activity toward potencies of full agonists Sec-FL and Sec(1–23), B2 drastically potentiated both, potency and efficacy, of SCTR signaling upon binding of partial agonist Sec(3–27). In NG108 cells expressing endogenous SCTR, this powerful enhancement was also evident with B2 being the only analog tested inducing a seven-fold leftward shift of Sec(1–23) dose-response and converting the inactive Sec(3–27) into a partial agonist. In addition, B2 had by far the greatest effect on prolonging residence time of Fluo-Sec at SNAP-SCTRs, which was comparable to similar studies with BETP in SNAP-GLP-1Rs [45–47]. We hypothesized the formidable deceleration of Fluo-Sec dissociation in conditions attenuating G protein coupling could be due to either trapping the peptide ligand at the N-domain of the receptor or stabilizing the receptor in an active-state conformation by binding to an allosteric hub in the J-domain. Considering both the binding and functional data of B2, we believe the latter explanation to be more likely. The loss of activity in SCTR K317A might be another hint pointing to a potential B2 binding site but further studies, e.g. investigations on ligand association kinetics or cooperative effects between individual PAM scaffolds, are needed to support this hypothesis. Even though B1 and B2 significantly enhanced GLP-1(9–36)-stimulated GLP-1R responses in overexpressing cell lines, both compounds decreased receptor basal activity in naturally expressing INS-1 cells rather than having potentiating effects on the partial agonist. Interestingly, while th-BETP, the 2-mercapto pyrimidine analog of BETP, was inactive in GLP-1R overexpressing conditions [7], another study reported the discovery of a structural similar 3-(trifluoromethyl)quinoxaline, sharing the aniline substituted thioacetamide with scaffold B analogs, by screening compounds against a constitutively active GLP-1R C347QQYR mutant [48]. As an aside, unlike covalently acting 2-sulfonyl pyrimidines, reversible scaffolds B and C displayed stronger potentiating effects at GLP-1Rs if added simultaneously with the orthosteric agonist, supporting their preference to bind and act via receptors in active confirmation (comparison of data not shown). Parental INS-1 cells displayed a very low sensitivity to GLP-1R stimulation with partial agonist GLP-1(9–36) even decreasing basal receptor activity implying an overall low number of active receptor populations. Hence, we argued that the augmenting activity of current 2-mercapto pyrimidines might be too weak to intrinsically stabilize active receptor conformations and would need further structural optimization to modulate responses in that setting. Interestingly, a recent study [49] identified a low molecular weight, reversibly acting GLP-1R PAM via in-silico screening against a predicted allosteric binding site in the TM domain based on the recent cryo-EM structure of the GLP-1R-G protein complex (PDB ID: 5VAI) [50]. Subsequent site-specific mutagenesis studies supported the presence of a novel allosteric binding pocket, that is close to but distinct from the BETP-binding site, located between TM6 and helix 8, at the interaction site of the alpha-helical C-term of the G protein alpha subunit to GLP-1R [49]. We concluded that in future studies it might be worthwhile to investigate potential interactions of our novel reversible PAMs at this new allosteric binding pocket, which might elucidate their preference for co-stimulated receptors.
For more detailed analysis of 2-amino pyrimidines, we concentrated on two analogs C4 and C9 incorporating 3,4-dimethoxy groups at the core phenyl ring, which appeared to be beneficial for modulation of both, SCTRs and GLP-1Rs. Strikingly, both analogs elevated efficacies of agonist peptides on SCTRs without pronounced discrimination between Sec-FL, Sec(1–23) and Sec(3–27), while 2-amino pyrimidines strongly favored the full-length peptide GLP-1 over partial agonist GLP-1(9–36) on GLP-1Rs, strongly suggesting different MOAs for modulating each receptor. Unfortunately, C4 and C9 only showed marginal potentiation of both Sec-FL and Sec(1–23) signaling in NG108 and insignificant activities in INS-1 cells, which reinforces that further optimization might be necessary to achieve agents suitable to study the receptors in an endogenous environment. As 2-amino pyrimidines were able to double Sec-Fluo residence time at SNAP-SCTRs similarly to B1, we assume both reversible scaffolds could target the same allosteric binding site at SCTRs. In contrast, scaffold C exerted probe dependencies reverse to scaffold B at GLP1-Rs implying different mechanisms. We hypothesize that while 2-mercapto pyrimidines might act on or close to the established allosteric pocket of BETP, 2-amino pyrimidines could target the alternative binding site of 2-sulfonyl pyrimidines. However, further studies are needed to support this hypothesis.
4.3. Select analogs of irreversible and reversible scaffolds elevate insulin secretion in INS-1 cells
GLP-1R activation by synthetic GLP-1 mimetics has become a crucial treatment option for T2D improving pancreatic beta cell function and sensitivity to glucose-stimulated insulin secretion (GSIS) [46, 51]. To explore whether newly discovered PAMs have a physiologically relevant effect on GLP-1R signaling, we investigated the effect on GSIS of select analogs in INS-1 cells with and without GLP-1(9–36) co-stimulation and compared with BETP. Strikingly, all but one analog significantly enhanced secreted insulin concentrations in conditioned media. Similar to GLP-1(9–36)-mediated cAMP formation in INS-1 cells, BETP demonstrated superior response in GSIS assays compared to our PAMs. At the same time, substantial discrepancies became apparent when comparing compound activities in modulating INS-1 cAMP accumulation and effects on insulin secretion. For example, A3, demonstrating stronger potentiating effects on GLP-1(9–36) cAMP signaling in INS-1 cells than its structural analogs A1 and A2, was paradoxically inactive in GSIS, while A1 and A2 had pronounced effects on GLP-1(9–36)-mediated insulin secretion. Similarly contradictory was the substantial gain of insulin secretion upon C9 treatment that lacked significant activity in INS-1 cAMP assays. These results were unexpected since several studies reported a tight correlation between rising cAMP levels and promotion of insulin secretion in pancreatic beta cell lines [46, 52]. It is likely that the mechanism might be more complex and several signaling processes influence GLP-1R-mediated GSIS, such as functional selectivity, receptor internalization, nanodomain clustering, endosomal and compartmentalized cAMP elevation and receptor-ligand binding kinetics [46, 47, 52]. Unique signaling profiles of identified PAMs and their analogs would help to elucidate underlying pharmacological signaling pathways; however, investigations on these processes were outside the scope of our study.
4.4. Novel SCTR PAMs provide unique opportunities for developing tool compounds and lead scaffolds
To our knowledge, we discovered and characterized the first small molecule PAMs targeting SCTRs. By pursuing an ABC-driven approach, we were able to optimize original hits leading to analog B2, which demonstrated impressive cooperativity toward agonist-stimulated SCTR signaling. The disclosed SCTR hit scaffolds have structural and functional similarity to some GLP-1R agonists/PAMs. Both class B GPCRs are highly involved in mechanisms of metabolic disorders. Even though newly discovered scaffolds were inferior to BETP in GLP-1R stimulation, they suggest that the sulfone moiety responsible for electrophilic reactivity and covalent modification of Cys-347 at GLP-1Rs, could be replaced with a thioether or amino function without eradicating PAM activity at GLP-1Rs. Both features have never been reported for previous modulators of GLP-1Rs [7, 19]. Moreover, for the first time we now disclose the structures of small molecules able to interact and modulate SCTRs, since only peptide ligands have been described so far [25, 29, 30]. One example is the SCTR/GLP-1R co-agonist GUB06–046, a structural hybrid of Sec-FL and GLP-1, whose administration has been shown to be beneficial for appetite regulation and glucose homeostasis devoid of proliferative effects on exocrine or total pancreas mass [11]. The preservation of beta cell mass by dual stimulation of SCTRs and GLP-1Rs indicates a potential advantage compared with selective GLP-1 agonists. Hence, the discovery of orally available small molecules targeting both of these receptors appears particularly attractive. Our SAR studies of commercially available analogs provide useful starting points for the development of highly efficient tool compounds through further chemical optimization to decipher GLP-1R or SCTR-specific signaling processes and drug development options for metabolic diseases, such as obesity and diabetes. Beyond that, intensive allosteric activity analysis and mutational studies led to the hypothesis of an alternative allosteric mechanism at both, SCTRs and GLP-1Rs, building the foundation for future investigations to elucidate a potential novel as well as distinct binding pocket applicable for structure-based design of further low molecular weight and reversible agents on these class B GPCRs.
Acknowledgements
This work was supported by a grant from the National Institutes of Health, HL133501.
Footnotes
Declaration of Conflicting Interests
We declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Wootten D, Miller LJ, Structural Basis for Allosteric Modulation of Class B G Protein-Coupled Receptors, Annu Rev Pharmacol Toxicol 60 (2020) 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hoare SRJ, Allosteric Modulators of Class B G-Protein-Coupled Receptors, Current Neuropharmacology 5(3) (2007) 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Afroze S, Meng F, Jensen K, McDaniel K, Rahal K, Onori P, Gaudio E, Alpini G, Glaser SS, The physiological roles of secretin and its receptor, Annals of Translational Medicine 1(3) (2013) 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].de Graaf C, Song G, Cao C, Zhao Q, Wang MW, Wu B, Stevens RC, Extending the Structural View of Class B GPCRs, Trends Biochem Sci 42(12) (2017) 946–960. [DOI] [PubMed] [Google Scholar]
- [5].Dengler DG, Sun Q, Harikumar KG, Beutel J, Holleran J, Pollari S, Ma C-T, Miller LJ, Sergienko E, Identification and Characterization of Dual-Acting Small Molecule Modulators targeting Secretin Receptors, The FASEB Journal 34(S1) (2020) 1–1. [Google Scholar]
- [6].Lagerstrom MC, Schioth HB, Structural diversity of G protein-coupled receptors and significance for drug discovery, Nat Rev Drug Discov 7(4) (2008) 339–57. [DOI] [PubMed] [Google Scholar]
- [7].Bueno AB, Showalter AD, Wainscott DB, Stutsman C, Marin A, Ficorilli J, Cabrera O, Willard FS, Sloop KW, Positive Allosteric Modulation of the Glucagon-like Peptide-1 Receptor by Diverse Electrophiles, J Biol Chem 291(20) (2016) 10700–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sloop KW, Briere DA, Emmerson PJ, Willard FS, Beyond Glucagon-like Peptide-1: Is G-Protein Coupled Receptor Polypharmacology the Path Forward to Treating Metabolic Diseases?, ACS Pharmacology & Translational Science 1(1) (2018) 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brandler J, Miller LJ, Wang XJ, Burton D, Busciglio I, Arndt K, Harmsen WS, Camilleri M, Secretin effects on gastric functions, hormones and symptoms in functional dyspepsia and health: randomized crossover trial, Am J Physiol Gastrointest Liver Physiol 318(4) (2020) G635–G645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Congreve M, Oswald C, Marshall FH, Applying Structure-Based Drug Design Approaches to Allosteric Modulators of GPCRs, Trends Pharmacol Sci 38(9) (2017) 837–847. [DOI] [PubMed] [Google Scholar]
- [11].van Witteloostuijn SB, Dalboge LS, Hansen G, Midtgaard SR, Jensen GV, Jensen KJ, Vrang N, Jelsing J, Pedersen SL, GUB06–046, a novel secretin/glucagon-like peptide 1 co-agonist, decreases food intake, improves glycemic control, and preserves beta cell mass in diabetic mice, J Pept Sci 23(12) (2017) 845–854. [DOI] [PubMed] [Google Scholar]
- [12].Sloop KW, Willard FS, Brenner MB, Ficorilli J, Valasek K, Showalter AD, Farb TB, Cao JX, Cox AL, Michael MD, Gutierrez Sanfeliciano SM, Tebbe MJ, Coghlan MJ, Novel small molecule glucagon-like peptide-1 receptor agonist stimulates insulin secretion in rodents and from human islets, Diabetes 59(12) (2010) 3099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Morris LC, Nance KD, Gentry PR, Days EL, Weaver CD, Niswender CM, Thompson AD, Jones CK, Locuson CW, Morrison RD, Daniels JS, Niswender KD, Lindsley CW, Discovery of (S)-2-cyclopentyl-N-((1-isopropylpyrrolidin2-yl)-9-methyl-1-oxo-2,9-dihydro-1H-py rrido[3,4-b]indole-4-carboxamide (VU0453379): a novel, CNS penetrant glucagon-like peptide 1 receptor (GLP-1R) positive allosteric modulator (PAM), J Med Chem 57(23) (2014) 10192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wootten D, Simms J, Koole C, Woodman OL, Summers RJ, Christopoulos A, Sexton PM, Modulation of the glucagon-like peptide-1 receptor signaling by naturally occurring and synthetic flavonoids, J Pharmacol Exp Ther 336(2) (2011) 540–50. [DOI] [PubMed] [Google Scholar]
- [15].Cheong YH, Kim MK, Son MH, Kaang BK, Two small molecule agonists of glucagon-like peptide-1 receptor modulate the receptor activation response differently, Biochemical and biophysical research communications 417(1) (2012) 558–63. [DOI] [PubMed] [Google Scholar]
- [16].Morris LC, Days EL, Turney M, Mi D, Lindsley CW, Weaver CD, Niswender KD, A Duplexed High-Throughput Screen to Identify Allosteric Modulators of the Glucagon-Like Peptide 1 and Glucagon Receptors, J Biomol Screen 19(6) (2014) 847–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Knudsen LB, Kiel D, Teng M, Behrens C, Bhumralkar D, Kodra JT, Holst JJ, Jeppesen CB, Johnson MD, de Jong JC, Jorgensen AS, Kercher T, Kostrowicki J, Madsen P, Olesen PH, Petersen JS, Poulsen F, Sidelmann UG, Sturis J, Truesdale L, May J, Lau J, Small-molecule agonists for the glucagon-like peptide 1 receptor, Proceedings of the National Academy of Sciences 104(3) (2007) 937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Eng H, Sharma R, McDonald TS, Edmonds DJ, Fortin JP, Li X, Stevens BD, Griffith DA, Limberakis C, Nolte WM, Price DA, Jackson M, Kalgutkar AS, Demonstration of the innate electrophilicity of 4-(3-(benzyloxy)phenyl)-2-(ethylsulfinyl)-6-(trifluoromethyl)pyrimidine (BETP), a small-molecule positive allosteric modulator of the glucagon-like peptide-1 receptor, Drug Metab Dispos 41(8) (2013) 1470–9. [DOI] [PubMed] [Google Scholar]
- [19].Willard FS, Ho JD, Sloop KW, Discovery and pharmacology of the covalent GLP-1 receptor (GLP-1R) allosteric modulator BETP: A novel tool to probe GLP-1R pharmacology, Adv Pharmacol 88 (2020) 173–191. [DOI] [PubMed] [Google Scholar]
- [20].Nolte WM, Fortin JP, Stevens BD, Aspnes GE, Griffith DA, Hoth LR, Ruggeri RB, Mathiowetz AM, Limberakis C, Hepworth D, Carpino PA, A potentiator of orthosteric ligand activity at GLP-1R acts via covalent modification, Nat Chem Biol 10(8) (2014) 629–31. [DOI] [PubMed] [Google Scholar]
- [21].Song G, Yang D, Wang Y, de Graaf C, Zhou Q, Jiang S, Liu K, Cai X, Dai A, Lin G, Liu D, Wu F, Wu Y, Zhao S, Ye L, Han GW, Lau J, Wu B, Hanson MA, Liu ZJ, Wang MW, Stevens RC, Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators, Nature 546(7657) (2017) 312–315. [DOI] [PubMed] [Google Scholar]
- [22].Willard FS, Bueno AB, Sloop KW, Small molecule drug discovery at the glucagon-like peptide-1 receptor, Exp Diabetes Res 2012 (2012) 709893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Buchwald H, Estok R, Fahrbach K, Banel D, Jensen MD, Pories WJ, Bantle JP, Sledge I, Weight and Type 2 Diabetes after Bariatric Surgery: Systematic Review and Meta-analysis, The American Journal of Medicine 122(3) (2009) 248–256.e5. [DOI] [PubMed] [Google Scholar]
- [24].Modvig IM, Andersen DB, Grunddal KV, Kuhre RE, Martinussen C, Christiansen CB, Orskov C, Larraufie P, Kay RG, Reimann F, Gribble FM, Hartmann B, Bojsen-Moller KN, Madsbad S, Wewer Albrechtsen NJ, Holst JJ, Secretin release after Roux-en-Y gastric bypass reveals a population of glucose-sensitive S cells in distal small intestine, Int J Obes (Lond) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dong M, Harikumar KG, Raval SR, Milburn JE, Clark C, Alcala-Torano R, Mobarec JC, Reynolds CA, Ghirlanda G, Christopoulos A, Wootten D, Sexton PM, Miller LJ, Rational development of a high-affinity secretin receptor antagonist, Biochem Pharmacol 177 (2020) 113929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Grossini E, Molinari C, Morsanuto V, Mary DA, Vacca G, Intracoronary secretin increases cardiac perfusion and function in anaesthetized pigs through pathways involving beta-adrenoceptors and nitric oxide, Exp Physiol 98(5) (2013) 973–87. [DOI] [PubMed] [Google Scholar]
- [27].Li Y, Schnabl K, Gabler S-M, Willershäuser M, Reber J, Karlas A, Laurila S, Lahesmaa M, u Din M, Bast-Habersbrunner A, Virtanen KA, Fromme T, Bolze F, O’Farrell LS, Alsina-Fernandez J, Coskun T, Ntziachristos V, Nuutila P, Klingenspor M, Secretin-Activated Brown Fat Mediates Prandial Thermogenesis to Induce Satiation, Cell 175(6) (2018) 1561–1574.e12. [DOI] [PubMed] [Google Scholar]
- [28].Wu N, Meng F, Invernizzi P, Bernuzzi F, Venter J, Standeford H, Onori P, Marzioni M, Alvaro D, Franchitto A, Gaudio E, Glaser S, Alpini G, The secretin/secretin receptor axis modulates liver fibrosis through changes in transforming growth factor-beta1 biliary secretion in mice, Hepatology 64(3) (2016) 865–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Singh K, Senthil V, Arokiaraj AW, Leprince J, Lefranc B, Vaudry D, Allam AA, Ajarem J, Chow BK, Structure-Activity Relationship Studies of N- and C-Terminally Modified Secretin Analogs for the Human Secretin Receptor, PLoS One 11(3) (2016) e0149359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dengler DG, Sun Q, Holleran J, Pollari S, Beutel J, Brown BT, Shinoki Iwaya A, Ardecky R, Harikumar KG, Miller LJ, Sergienko EA, Development of a Testing Funnel for Identification of Small-Molecule Modulators Targeting Secretin Receptors, SLAS DISCOVERY: Advancing the Science of Drug Discovery 26(1) (2021) 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Leach K, Sexton PM, Christopoulos A, Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology, Trends Pharmacol Sci 28(8) (2007) 382–9. [DOI] [PubMed] [Google Scholar]
- [32].Ehlert FJ, Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods, Molecular pharmacology 33(2) (1988) 187–194. [PubMed] [Google Scholar]
- [33].Black JW, Leff P, Operational models of pharmacological agonism, Proceedings of the Royal Society of London. Series B. Biological Sciences 220(1219) (1983) 141–162. [DOI] [PubMed] [Google Scholar]
- [34].Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P, Glucose Sensitivity and Metabolism-Secretion Coupling Studied during Two-Year Continuous Culture in INS-1E Insulinoma Cells, Endocrinology 145(2) (2004) 667–678. [DOI] [PubMed] [Google Scholar]
- [35].Lopata MA, Cleveland DW, Sollner-Webb B, High level transient expression of a chloramphenicol acetyl transferase gene by DEAE-dextran mediated DNA transfection coupled with a dimethyl sulfoxide or glycerol shock treatment, Nucleic Acids Res 12(14) (1984) 5707–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Klein MT, Vinson PN, Niswender CM, Approaches for probing allosteric interactions at 7 transmembrane spanning receptors, Prog Mol Biol Transl Sci 115 (2013) 1–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rodbell M, Birnbaumer L, Pohl SL, Krans HM, The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. V. An obligatory role of guanylnucleotides in glucagon action, J Biol Chem 246(6) (1971) 1877–82. [PubMed] [Google Scholar]
- [38].DeVree BT, Mahoney JP, Velez-Ruiz GA, Rasmussen SG, Kuszak AJ, Edwald E, Fung JJ, Manglik A, Masureel M, Du Y, Matt RA, Pardon E, Steyaert J, Kobilka BK, Sunahara RK, Allosteric coupling from G protein to the agonist-binding pocket in GPCRs, Nature 535(7610) (2016) 182–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gossen D, Tastenoy M, Robberecht P, Christophe J, Secretin receptors in the neuroglioma hybrid cell line NG108–15. Characterization and regulation of their expression, European journal of biochemistry 193(1) (1990) 149–54. [DOI] [PubMed] [Google Scholar]
- [40].Bauer MR, Joerger AC, Fersht AR, 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells, Proc Natl Acad Sci U S A 113(36) (2016) E5271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Förster T, Shang E, Shimizu K, Sanada E, Schölermann B, Huebecker M, Hahne G, López-Alberca MP, Janning P, Watanabe N, Sievers S, Giordanetto F, Shimizu T, Ziegler S, Osada H, Waldmann H, 2-Sulfonylpyrimidines Target the Kinesin HSET via Cysteine Alkylation, European Journal of Organic Chemistry 2019(31–32) (2019) 5486–5496. [Google Scholar]
- [42].Mulvihill EE, Drucker DJ, Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors, Endocr Rev 35(6) (2014) 992–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].May LT, Leach K, Sexton PM, Christopoulos A, Allosteric modulation of G protein-coupled receptors, Annu Rev Pharmacol Toxicol 47 (2007) 1–51. [DOI] [PubMed] [Google Scholar]
- [44].Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S, Crystal structure of the micro-opioid receptor bound to a morphinan antagonist, Nature 485(7398) (2012) 321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jones BJ, Scopelliti R, Tomas A, Bloom SR, Hodson DJ, Broichhagen J, Potent Prearranged Positive Allosteric Modulators of the Glucagon-like Peptide-1 Receptor, ChemistryOpen 6(4) (2017) 501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jones B, Buenaventura T, Kanda N, Chabosseau P, Owen BM, Scott R, Goldin R, Angkathunyakul N, Correa IR Jr., Bosco D, Johnson PR, Piemonti L, Marchetti P, Shapiro AMJ, Cochran BJ, Hanyaloglu AC, Inoue A, Tan T, Rutter GA, Tomas A, Bloom SR, Targeting GLP-1 receptor trafficking to improve agonist efficacy, Nat Commun 9(1) (2018) 1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Buenaventura T, Bitsi S, Laughlin WE, Burgoyne T, Lyu Z, Oqua AI, Norman H, McGlone ER, Klymchenko AS, Correa IR Jr., Walker A, Inoue A, Hanyaloglu A, Grimes J, Koszegi Z, Calebiro D, Rutter GA, Bloom SR, Jones B, Tomas A, Agonist-induced membrane nanodomain clustering drives GLP-1 receptor responses in pancreatic beta cells, PLoS Biol 17(8) (2019) e3000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kopin AB, M., Methods and Compositions for the Treatment of Metabolic Disorders, USA, 2004. [Google Scholar]
- [49].Redij T, Ma J, Li Z, Hua X, Li Z, Discovery of a potential positive allosteric modulator of glucagon-like peptide 1 receptor through virtual screening and experimental study, J Comput Aided Mol Des 33(11) (2019) 973–981. [DOI] [PubMed] [Google Scholar]
- [50].Zhang Y, Sun B, Feng D, Hu H, Chu M, Qu Q, Tarrasch JT, Li S, Sun Kobilka T, Kobilka BK, Skiniotis G, Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein, Nature 546(7657) (2017) 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Holst JJ, The physiology of glucagon-like peptide 1, Physiol Rev 87(4) (2007) 1409–39. [DOI] [PubMed] [Google Scholar]
- [52].Kuna RS, Girada SB, Asalla S, Vallentyne J, Maddika S, Patterson JT, Smiley DL, DiMarchi RD, Mitra P, Glucagon-like peptide-1 receptor-mediated endosomal cAMP generation promotes glucose-stimulated insulin secretion in pancreatic beta-cells, Am J Physiol Endocrinol Metab 305(2) (2013) E161–70. [DOI] [PubMed] [Google Scholar]