

Abstract
Objectives:
Three Phase 3 trials have demonstrated the efficacy and safety of SPN-812 in pediatric subjects with ADHD. Here, we report the results of a fourth trial.
Methods:
Eligible adolescent subjects (N = 297) were randomized to SPN-812 (400- or 600-mg/day) or placebo. The primary efficacy endpoint was change from baseline (CFB) at end of study (EOS) in the ADHD Rating Scale-5 (ADHD-RS-5) Total score. Statistical analyses included sequential testing for multiple treatment comparisons. Key secondary endpoints included: Clinical Global Impression-Improvement (CGI-I) score at EOS and CFB at EOS in the Conners 3–Parent Short Form (Conners 3–PS) Composite T-score and Weiss Functional Impairment Rating Scale–Parent (WFIRS–P) Total average score.
Results:
The CFB at EOS ADHD-RS-5 Total score (least square [LS] means ± SE) for 400-mg/day, 600-mg/day SPN-812, and placebo was −18.3 ± 1.36, −16.7 ± 1.39, and –13.2 ± 1.38, respectively. The difference vs. placebo was statistically significant only for the 400-mg/day SPN-812 treatment group (600 mg/day: p = 0.0712; 400 mg/day: p = 0.0082). Neither dose could be considered superior to placebo due to the use of statistical method of sequential testing. Significant improvements were observed on a number of secondary endpoints. SPN-812 was well tolerated at both doses, with <5% discontinuation rate due to adverse events.
Conclusions:
Treatment with 400- but not 600-mg/day SPN-812 resulted in statistically significant improvement in the primary endpoint. The negative result seen in the 600-mg/day SPN-812 group was likely due to an unusually high placebo response. Safety data were consistent across all doses in the SPN-812 trials.
Keywords: viloxazine, attention-deficit/hyperactivity disorder, adolescents
Introduction
In the United States, it is estimated that approximately 10.2% of children and adolescents1 and 4.4% of adults2 have been diagnosed with attention-deficit/hyperactivity disorder (ADHD). Although multiple stimulant (various formulations of methylphenidate and amphetamine) and nonstimulant (atomoxetine, guanfacine, and clonidine) medications have demonstrated efficacy in the treatment of ADHD,3,4 it is estimated that approximately 20% to 40% of patients with ADHD do not achieve treatment response or symptomatic remission.5–7
In addition, many of these medications are either contraindicated or should be used with caution in certain patient populations; for instance, in patients with cardiac abnormalities or cardiovascular problems,8–12 agitation, Tourette syndrome, tics, sleep disturbances, bipolar disorder,8,9 sedation, or somnolence.11,12 Common safety and tolerability issues that have been associated with stimulant treatment include insomnia, irritability, nausea, decreased appetite, and weight decrease.8,9 Issues that have been associated with nonstimulant treatment include somnolence, sedation, fatigue, hypotension, and bradycardia.10–12 Further, stimulants are controlled substances (schedule II drugs [CII]), which are also associated with a risk of abuse, misuse, and diversion13–16 and have certain restrictions for initial prescriptions and refills.17
All ADHD medications currently approved by the United States Food and Drug Administration (FDA) require periods of time for dose and/or regimen optimization.18,19 For instance, the duration of action of stimulants often does not meet the all-day demands of ADHD patients,4,20 while current FDA-approved nonstimulant medications have been described as having a slow onset of action and/or requiring long titration periods.11,12,19,21,22
Management of ADHD symptoms in adolescents presents additional challenges. As young children age into adolescence, cognitive tasks become more complex and require more independence. In addition, there is a growing importance of peer interactions that can further place higher demands on these patients, which may lead to difficulties in academic performance, self-perception, family, and peer relations.23–26 In adolescents, ADHD is associated with higher rates of risky driving behavior, obesity, suicidal thoughts, and illicit drug use.23,27–29 In addition, adolescents and young adults with ADHD are likely to be diagnosed with other psychiatric comorbidities, including major depression, bipolar disorder, anxiety, antisocial disorders, tics/Tourette’s syndrome, and substance use disorders.29,30 Considering the limitations of current FDA-approved ADHD medications and the challenges faced in effectively managing ADHD symptoms in pediatric and adolescent populations, new effective and well-tolerated treatment options are needed.
SPN-812 (viloxazine extended-release) is a nonstimulant therapy that has demonstrated activity at the norepinephrine transporter and has been shown to increase serotonin.31 SPN-812 is under investigation as a novel therapy for ADHD in pediatric and adult patients. Two Phase 3 placebo-controlled trials (NCT03247530, NCT03247543) investigated the efficacy and safety of 100-, 200-, and 400-mg/day of SPN-812 in children (6–11 years) with ADHD.32 Another Phase 3 placebo-controlled trial (NCT03247517) investigated the efficacy and safety of 200- and 400-mg/day doses of SPN-812 in adolescents (12–17 years) with ADHD. In all three trials, SPN-812 significantly reduced ADHD symptoms as measured by the ADHD-RS-5, an improvement that was determined to be clinically meaningful as measured by the CGI-I scale. Further, SPN-812 was well tolerated at the dose levels tested in these trials.32–34 Here, we report the results of a fourth pediatric Phase 3 placebo-controlled trial evaluating the efficacy and safety of 400- and 600-mg/day doses of SPN-812 in adolescents (12–17 years of age) with ADHD.
Materials and Methods
Study Design
A randomized, double-blind, placebo-controlled, three-arm, parallel-group trial was conducted at 27 clinical sites in the United States (NCT03247556). Following a screening phase (up to 28 days), 297 subjects were randomized in a 1:1:1 ratio to placebo, 400-mg/day SPN-812, or 600-mg/day SPN-812. All subjects, regardless of the treatment group assignment, were administered three oral capsules daily in the morning with or without food throughout the treatment, with an option to sprinkle over soft food (e.g., apple sauce) to facilitate swallowing if needed.32 All capsules were identical in appearance. Subjects assigned to the placebo group took three placebo capsules daily for 7 weeks; subjects in the 400-mg/day SPN-812 treatment group took one 200-mg capsule of SPN-812 and two placebo capsules daily during Week 1 followed by two 200-mg capsules of SPN-812 and one placebo capsule daily for the remaining 6 weeks; subjects in the 600-mg/day SPN-812 treatment group took one 200-mg SPN-812 capsule and two placebo capsules daily during Week 1, two 200-mg SPN-812 capsules and one placebo capsule daily during Week 2, followed by three 200-mg capsules daily for the remaining 5 weeks (Figure 1). The parent(s)/guardian(s) was asked to accommodate dosing into the family’s morning routine as consistently as possible, though some flexibility in the timing of the daily dose was permitted if an adverse event (AE) precluded or delayed study medication (SM) administration.32
Figure 1.

Study Design
n represents intent-to-treat (ITT) population. ADHD-RS-5, ADHD Rating Scale-5; CFB, change from baseline; CGI-I, Clinical Global Impression-Improvement; CGI-S, Clinical Global Impression-Severity of Illness; Conners 3-PS, Conners 3-Parent Short Form; Conners 3-SRS, Conners 3-Self-Report Short Form; EOS, end of study; SIPA, Stress Index for Parents of Adolescents; WFIRS-P, Weiss Functional Impairment Rating Scale-Parent.
The study protocol was approved by Advarra Institutional Review Board (IRB) and conducted in accordance with the Helsinki Declaration and the International Council for Harmonisation (ICH) Good Clinical Practice (GCP) Guidelines. Each subject and parent(s)/legally authorized guardian(s) provided written informed consent/assent prior to screening. The subject and the parent/guardian were informed about the nature and purpose of the study, as well as of its risks and benefits. It was explained that the subject could withdraw from the study at any time for any reason and that this decision would not affect any future medical care. Subjects who completed the 7-week treatment phase and continued to meet eligibility criteria were offered the opportunity to participate in a long-term, open-label extension trial (NCT02736656).
Subjects
Male and female subjects (12 to 17 years of age, weighing ≥ 35 kg) were eligible to participate if they had a primary diagnosis of ADHD per the Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5),35 confirmed via the Mini International Neuropsychiatric Interview for Children and Adolescents (MINI-KID),36 and had an ADHD-RS-5 Total score ≥ 28, and a Clinical Global Impression-Severity of Illness (CGI-S) score ≥ 4 (i.e., overall illness severity of moderate or greater) at screening.32 Further, in order to be eligible to participate in this study, subjects had to be considered medically healthy by the study investigator based on a physical examination, medical history, clinical laboratory tests, vital signs, and electrocardiogram (ECG). Subjects were also required to refrain from taking other ADHD medications for a minimum of 1 week prior to randomization and for the study duration.
Subjects were not eligible to participate in the trial if they had a current diagnosis of a major psychiatric disorder, a major neurological disorder (including seizures or a history of seizure disorder within the immediate family, or a history of seizure-like events), a significant systemic disease, evidence of suicidality (defined as active suicidal plan or thoughts, or more than one lifetime suicide attempt) within 6 months of screening or at screening, and/or a body mass index (BMI) > 95th percentile for the appropriate age and gender.32 Subjects with major depressive disorder who were free of episodes within the 6 months prior to screening were eligible to participate. In addition, subjects were not eligible to participate if they had a history of intolerance or allergic reaction to viloxazine or its excipients, received any investigational drug within 30 days or 5 half-lives prior to first dosing of SM, or in the opinion of the site investigator, had any reason that contraindicated participation.
Other exclusion criteria included positive urine drug screen at the screening visit (a positive test for amphetamines was allowed for subjects receiving a prescription stimulant ADHD medication and not responding to treatment—the subject was required to discontinue the stimulant at least one week prior to the baseline visit), pregnancy, breastfeeding, or refusal to practice abstinence or use an acceptable birth control beginning 30 days prior to the first dose and throughout the study.32
The safety population included all randomized subjects who received at least 1 dose of SM; the intent-to-treat (ITT) population for efficacy included all subjects who were randomized, took at least one dose of SM, and had a baseline and at least one post-randomization ADHD-RS-5 assessment.
Assessments
The primary efficacy assessment, the investigator-rated ADHD-RS-5, was conducted at each outpatient study visit from Visit 1 (screening) to Visit 9 (EOS) (Figure 1). The investigator-rated CGI-S scale was conducted at Visit 1 and Visit 2 (baseline), and the investigator-rated CGI-I scale was conducted at weekly, post-baseline study visits (Visit 3 to Visit 9). Safety assessments (such as vital signs, the Columbia Suicide Severity Rating Scale [C-SSRS], review of AEs, and concomitant medications) were performed at all study visits. ECGs, laboratory tests, and physical examinations were performed at Visits 1 and 9. Parent/guardian- or self-administered ratings (including the Conners 3–PS, WFIRS–P, Stress Index for Parents of Adolescents [SIPA], and Conners 3–Self-Report Short Form [Conners 3–SRS]) were performed at Visit 2 (baseline) and Visit 9 (EOS).
Statistical Analysis
Sample size calculations indicated that 72 subjects per treatment group in the ITT population would yield 90% power across treatment groups at a significance level of 0.05 (two-sided) using a two-sample t-test, based on an effect size of 0.547 obtained in a previous Phase 2b trial for ADHD-RS-5 Total score at SPN-812 dose of 200 mg vs. placebo.37 Based on this and accounting for an anticipated early discontinuation rate of 27.9%, a total of 300 subjects (100 per treatment arm) were projected for the randomized population.
The primary efficacy endpoint was CFB at EOS in the ADHD-RS-5 Total score compared to placebo (Figure 1). The ADHD-RS-5 is an ADHD-specific scale that consists of 18 items (corresponding to 18 DSM-5 symptoms that are further subdivided into two subscales: Inattention [9 items] and Hyperactivity/Impulsivity [9 items]).38 The key secondary efficacy endpoints included: CGI-I39 score at EOS, CFB in the Conners 3–PS40 Composite T-score at EOS, and CFB in the WFIRS–P41,42 Total average score at EOS.32 More detailed descriptions of these scales are provided in another report (Nasser et al., 2020, submitted).
Additional secondary endpoints included: the proportion of ADHD-RS-5 responders (where “responders” are defined as subjects who had a • 50≥ reduction in the CFB ADHD-RS-5 Total score at EOS); the CFB in the SIPA Total score at EOS (this scale identifies areas of stress in parent-adolescent interactions across three domains [adolescent, parent, and adolescent-parent] and Life Stressors Scale); the CFB in each ADHD-RS-5 subscale (Inattention and Hyperactivity/Impulsivity) score at EOS; the CFB in the Conners 3–SRS Composite T-score at EOS; and the proportion of CGI-I responders (where “responder” is defined as a subject who has a CGI-I score of 1 or 2). Exploratory endpoints included CFB in the Conners 3–PS/SRS T-score for each content scale at EOS, CFB in WFIRS–P average score for each domain at EOS and CFB in the SIPA score for each domain and the Life Stressors Scale at EOS.41,42
The primary efficacy endpoint, which has been previously described,32 was analyzed using a mixed model for repeated measures (MMRM). MMRM assumes missing data are missing at random (MAR). The key secondary measures were analyzed using analysis of covariance (ANCOVA).32 The LS means of treatment groups, differences between the LS means of treatment groups and placebo, and p values were determined for all measures. Due to multiple treatment comparisons (400-mg/day SPN-812 vs. placebo and 600-mg/day SPN-812 vs. placebo), the statistical analysis of the primary endpoint included a sequential testing procedure43 with a fixed testing method of the null hypotheses to control the overall type I error rate at 0.05. This procedure involves testing of 600-mg/day SPN-812 vs. placebo first, followed by 400-mg/day SPN-812 vs. placebo only if the first null hypothesis was rejected. If the first null hypothesis was not rejected, the second null hypothesis would not be tested. If both null hypotheses are rejected, then the results of the study would be considered positive. Statistical analyses were performed using SAS® version 9.2 or higher.
Safety and tolerability were assessed by monitoring the incidence of AEs and evaluating clinical laboratory tests, vital signs, physical examinations, ECGs, and suicidal ideation and behavior (C-SSRS44).32 All AEs in this study were recorded following the first administration of SM and were, thus, considered treatment-emergent (TEAEs, henceforth referred to as AEs).32 The relationship to study treatment, seriousness, and severity of AEs were evaluated by the site investigator and considered mild if the subject easily tolerated the symptom(s), moderate if discomfort was enough to interfere with daily activity and may have warranted intervention, and severe if the symptom/event significantly affected the subject’s daily activity or clinical status and warranted intervention.32
Results
Demographics and Baseline Characteristics
A total of 417 subjects were screened (Figure 2); 28.8% failed screening and were not randomized. A total of 297 subjects (12–17 years of age) were randomized (600-mg/day SPN-812, n = 100; 400-mg/day SPN-812, n = 100; placebo, n = 97), with the safety population consisting of 296 subjects (600-mg/day, n = 99; 400-mg/day, n = 100; placebo, n = 97) and ITT population consisting of 292 subjects (600-mg/day, n = 97; 400-mg/day, n = 99; placebo, n = 96). The majority of subjects were male (67.8%), and either White (66.1%) or African American (29.1%). Demographics and baseline characteristics in the placebo and either SPN-812 treatment group were similar (Table 1).
Figure 2.

Disposition of Subjects
aNumber of subjects in the safety population is used as denominator for this section.
Table 1. Demographics and Baseline Characteristics—ITT Population.
| Demographics and Baseline Characteristics | Placebo | SPN-812 | Total | |||||
| 400-mg/day | 600-mg/day | |||||||
| n | 96 | 99 | 97 | 292 | ||||
| Age, years | ||||||||
| Mean (SD) | 13.8 (1.53) | 14.0 (1.74) | 13.7 (1.52) | 13.8 (1.60) | ||||
| Median (min, max) | 14.0 (12, 17) | 14.0 (12, 17) | 13.0 (12, 17) | 14.0 (12, 17) | ||||
| Age group, n (%) | ||||||||
| 12–14 years | 63 (65.6) | 61 (61.6) | 72 (74.2) | 196 (67.1) | ||||
| 15–17 years | 33 (34.4) | 38 (38.4) | 25 (25.8) | 96 (32.9) | ||||
| Gender, n (%) | ||||||||
| Male | 61 (63.5) | 66 (66.7) | 71 (73.2) | 198 (67.8) | ||||
| Female | 35 (36.5) | 33 (33.3) | 26 (26.8) | 94 (32.2) | ||||
| Ethnicity, n (%) | ||||||||
| Not Hispanic and not Latino | 63 (65.6) | 65 (65.7) | 66 (68.0) | 194 (66.4) | ||||
| Hispanic or Latino | 32 (33.3) | 34 (34.3) | 31 (32.0) | 97 (33.2) | ||||
| Not allowed to ask as per local regulations | 1 (1.0) | 0 | 0 | 1 (0.3) | ||||
| Race, n (%) | ||||||||
| White | 64 (66.7) | 63 (63.6) | 66 (68.0) | 193 (66.1) | ||||
| Black or African American | 25 (26.0) | 33 (33.3) | 27 (27.8) | 85 (29.1) | ||||
| Multiple | 6 (6.3) | 3 (3.0) | 2 (2.1) | 11 (3.8) | ||||
| American Indian or Alaska Native | 1 (1.0) | 0 | 1 (1.0) | 2 (0.7) | ||||
| Native Hawaiian or other Pacific Islander | 0 | 0 | 1 (1.0) | 1 (0.3) | ||||
| Weight (kg), mean (SD) | 56.39 (12.897) | 57.19 (11.883) | 55.67 (13.253) | 56.42 (12.657) | ||||
|
Body mass index
(kg/m2), mean (SD) |
21.07 (3.582) | 21.14 (3.512) | 20.99 (3.310) | 21.07 (3.458) | ||||
| ADHD-RS-5, mean (SD) | ||||||||
| Total score | 38.8 (8.06) | 41.2 (7.80) | 39.8 (8.34) | 39.9 (8.10) | ||||
| Inattention | 22.4 (3.59) | 22.5 (3.70) | 22.3 (3.82) | 22.4 (3.69) | ||||
| Hyperactivity/Impulsivity | 16.4 (6.36) | 18.7 (5.59) | 17.5 (6.49) | 17.6 (6.21) | ||||
| CGI-S, mean (SD) | 4.5 (0.66) | 4.8 (0.69) | 4.6 (0.69) | ND | ||||
ADHD-RS-5, ADHD Rating Scale-5; CGI-S, Clinical Global Impression-Severity of Illness; ITT, intent-to-treat; ND, not determined; SD, standard deviation.
ADHD-RS-5
The ADHD-RS-5 Total score at EOS was reduced (improved) from baseline in all three arms; the CFB in ADHD-RS-5 Total score at EOS (LS mean ± SE) was −16.7 ± 1.39 in the 600-mg/day SPN-812 group, −18.3 ± 1.36 in the 400-mg/day SPN-812 group, and −13.2 ± 1.38 in the placebo group (Table 2). The placebo-adjusted LS means difference in CFBs at EOS in the ADHD-RS-5 Total scores were −3.5 ± 1.93 (600-mg/day, p = 0.0712) and −5.1 ± 1.93 (400-mg/day, p = 0.0082). The null hypothesis for 600-mg/day vs placebo comparison (H01) was not rejected. Thus, due to the sequential gatekeeping testing procedure for multiplicity adjustment to control the type I error rate at a significance level of 5%, neither dose group was considered superior to placebo in this study. Hence, all efficacy outcomes reported below are only considered informative.
Table 2. ADHD-RS-5 Results at EOS by Treatment Group.
| ADHD-RS-5 Measure | Placebo | SPN-812 | ||||
| (n = 96) | 400-mg/day (n = 99) |
600-mg/day (n = 97) |
||||
| CFB, LS mean ± SEa | ||||||
| Total score | –13.2 ± 1.38 | –18.3 ± 1.36* | –16.7 ± 1.39 | |||
| Inattention subscale score | –7.1 ± 0.76 | –10.1 ± 0.75* | –8.7 ± 0.78 | |||
| Hyperactivity/Impulsivity subscale score |
–6.4 ± 0.69 | –8.3 ± 0.68* | –7.6 ± 0.71 | |||
| ADHD-RS-5 Responders, n (%)b,c | 32 (32.9%) | 48 (48.2%)* | 45 (46.0%) | |||
aADHD-RS-5 total score analyzed using MMRM ANCOVA model with fixed effect terms for baseline ADHD-RS-5 Total Score, age group, treatment, visit, and treatment-by-visit interaction, as fixed independent variables and Subscales are analyzed using ANCOVA model with baseline, age group, and treatment as fixed independent variables from which the LS Means and P-values are obtained.
bp-values for ADHD-RS-5 Responder is derived from Pearson’s Chi-squared Test or Fisher’s Exact Test; Fisher’s exact test is used when there are expected cell counts less than 5. Otherwise Pearson’s Chi-squared test is used.
cProportion of responders, defined as subjects who achieved a ≥ 50% reduction in ADHD-RS-5 Total score, at EOS.
*p < 0.05 vs placebo.
ADHD-RS-5, ADHD Rating Scale-5; ANCOVA, analysis of covariance; CFB, change from baseline; EOS, end of study; LS, least squares; SE, standard error.
A greater reduction (improvement) in ADHD-RS-5 Total score was observed at every week during treatment for both the 600- and 400-mg/day SPN-812 group compared to placebo (Figure 3). The difference was statistically significant between the 600-mg/day and placebo at Week 2 (p = 0.0456) and Week 3 (p = 0.0238), but not at EOS. The difference was also statistically significant between 400-mg/day SPN-812 and placebo at Week 2 (p = 0.0063) and was sustained through EOS.
Figure 3.
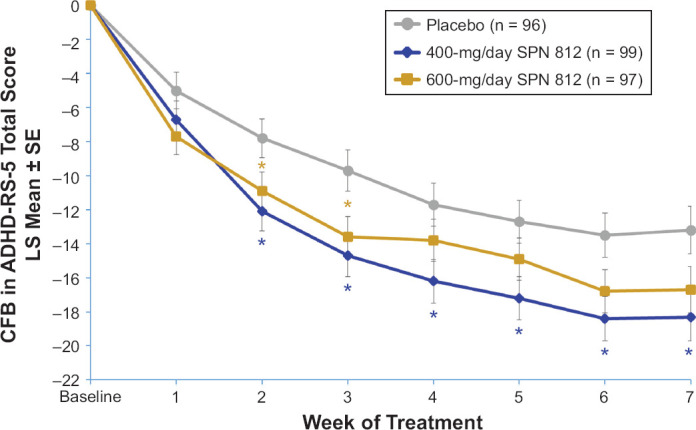
Profile of the Change from Baseline in ADHD-RS-5 Total Score by Treatment Group and Week of Treatment
*p < 0.05. ADHD-RS-5, ADHD Rating Scale-5; CFB, change from baseline; LS, least squares.
The CFB in the ADHD-RS-5 Inattention and Hyperactivity/Impulsivity subscale scores were not significantly different from placebo in the 600-mg/day SPN-812 group (p = 0.1392 and p = 0.2084, respectively). However, differences between the 400-mg/day SPN-812 group and the placebo group in Inattention and Hyperactivity/Impulsivity subscale scores were statistically significant (p = 0.0042 and p = 0.0484, respectively) (Table 2).
There were 46.0%, 48.2%, and 32.9% ADHD-RS-5 responders in the 600-mg/day SPN-812 (p = 0.0688), 400-mg/day SPN-812 (p = 0.0340), and placebo group, respectively. The difference in the proportion of ADHD-RS-5 responders between 600-mg/day SPN-812 group and the placebo group was not statistically significant. A significantly higher percentage of ADHD-RS-5 responders was observed in the 400-mg/day SPN-812 group compared to the placebo group.
CGI-I
The mean CGI-S score at baseline was similar among treatment groups (Table 1). The mean CGI-I score at EOS was lower for the 600- and 400-mg/day SPN-812 treatment group compared to the placebo group; however, a statistically significant difference was only noted in the 400-mg/day SPN-812 treatment group compared to placebo (p = 0.0051).
There was a higher proportion of CGI-I responders for each week of treatment in both the 600- and 400-mg/day SPN-812 treatment group compared to the placebo group. This difference was statistically significant in the 600-mg/day SPN-812 group at Week 3 and in the 400-mg/day SPN-812 group at Weeks 2–4 and 6–7 (Figure 4).
Figure 4.
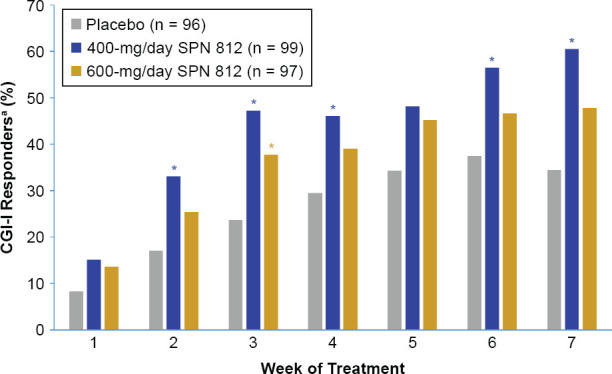
Proportion of CGI-I Responders by Treatment Group and Week of Treatment
aResponder is defined as a subject who has a CGI-I score of 1 or 2. *p < 0.05. CGI-I, Clinical Global Impression-lmprovement scale.
Conners 3
The CFB in Conners 3-PS Composite T-score at EOS was numerically lower in both SPN-812 arms compared to placebo, but this difference was not statistically significant (600 mg/day, p = 0.3130; 400-mg/day, p = 0.1377). However, there was a statistically significant reduction, compared to placebo, in the CFB T-score at EOS for the Conners 3-PS executive functioning content scale in the 400-mg/day (p = 0.0434) (Table 3). There was also a reduction in the CFB in Conners 3-SRS Composite T-score at EOS in both SPN-812 arms compared to placebo, but the difference was only statistically significant in the 600-mg/day SPN-812 group (p = 0.0432).
Table 3. Conners 3–PS and WFIRS–P Results by Treatment Group and Domain.
| Conners 3–PS | Placebo | SPN-812 | ||||
| n = 96 | 400-mg/day n = 99 |
600-mg/day n = 97 |
||||
| Composite T-score | ||||||
| Baseline, Mean ± SD (absolute value) | 73.1 ± 8.91 | 73.2 ± 8.87 | 73.9 ± 8.82 | |||
| CFB at EOS | ||||||
| LS mean ± SE | –5.6 ± 0.89 | –7.5 ± 0.87 | –6.9 ± 0.88 | |||
| Difference of LS mean (± SE) (vs. placebo) |
– | –1.9 ± 1.26 | –1.3 ± 1.25 | |||
| 95% CI of difference | – | (–4.3, 0.6) | (–3.7, 1.2) | |||
| p-value (vs. placebo) | – | 0.1377 | 0.3130 | |||
| Content Scale T-score | ||||||
| CFB at EOS, LS mean ± SE | ||||||
| Inattention | –6.1 ± 1.27 | –9.5 ± 1.24 | –9.6 ± 1.28 | |||
| Hyperactivity | –7.1 ± 1.39 | –8.9 ± 1.36 | –8.5 ± 1.38 | |||
| Learning Problems | –4.4 ± 1.08 | –5.8 ± 1.05 | –7.0 ± 1.07 | |||
| Executive Functioning | –6.2 ± 1.10 | –9.3 ± 1.08* | –5.8 ± 1.11 | |||
| Defiance/Aggression | –4.9 ± 1.39 | –5.4 ± 1.35 | –5.7 ± 1.38 | |||
| Peer Relations | –5.7 ± 1.45 | –6.0 ± 1.42 | –4.8 ± 1.45 | |||
| WFIRS–P | Placebo | SPN-812 | ||||
| n = 96 | 400-mg/day n = 99 |
600-mg/day n = 97 |
||||
| Total Average Score | ||||||
| Baseline, Mean ± SD (absolute value) | 0.97 ± 0.480 | 0.96 ± 0.442 | 0.99 ± 0.450 | |||
| CFB at EOS | ||||||
| LS mean ± SE | –0.23 ± 0.035 | –0.32 ± 0.034 | –0.23 ± 0.035 | |||
| Difference of LS mean (± SE) (vs. placebo) |
– | –0.09 ± 0.049 | –0.00 ± 0.050 | |||
| 95% CI of difference | – | (–0.19, 0.01) | (–0.10, 0.10) | |||
| P value (vs. placebo) | – | 0.0698 | 0.9756 | |||
| Domain Average Score | ||||||
| CFB at EOS, LS mean ± SE | ||||||
| Family | –0.30 ± 0.053 | –0.42 ± 0.052 | –0.26 ± 0.053 | |||
| Self-concept | –0.26 ± 0.065 | –0.33 ± 0.064 | –0.13 ± 0.064 | |||
| School | –0.32 ± 0.059 | –0.44 ± 0.057 | –0.36 ± 0.059 | |||
| Life Skills | –0.23 ± 0.052 | –0.31 ± 0.052 | –0.24 ± 0.052 | |||
| Social Activities | –0.22 ± 0.049 | –0.29 ± 0.048 | –0.21 ± 0.048 | |||
| Risky Activities | –0.08 ± 0.025 | –0.15 ± 0.025 | –0.09 ± 0.025 | |||
LS means, 95% CIs and p-values are from ANCOVA model with baseline and treatment as fixed independent variables. *p < 0.05.
ANCOVA, analysis of covariance; CFB, change from baseline; CI, confidence interval; Conners 3–PS, Conners 3–Parent Short Form; LS, least squares; SD, standard deviation; SE, standard error; WFIRS–P, Weiss Functional Impairment Rating Scale–Parent.
WFIRS–P
The CFB in WFIRS–P Total average score at EOS was also numerically lower in both SPN-812 groups compared to the placebo group, but this difference was not statistically significant for either the 600-mg/day or 400-mg/day SPN-812 treatment arms (p = 0.9756 and p = 0.0698, respectively).
SIPA
The CFB in SIPA Total score at EOS was numerically lower only in the 400-mg SPN-812 arm compared to placebo; however, the difference was not statistically significant (p = 0.1259).
Safety and Tolerability
Overall, SPN 812 was well-tolerated in this trial with a low AE-related discontinuation rate of 4.5% (i.e., 9 subjects) and no deaths. The most common treatment-related AEs that occurred in • 5% of subjects in any SPN-812 treatment group were somnolence (15.1%), fatigue (10.6%), headache (8.0%), nausea (6.5%), and decreased appetite (6.0%) (Table 4).
Table 4. Summary of Adverse Events.
| Safety measure, n (%) | Placebo | SPN-812 | ||||||
| (n = 97) | 400-mg/day (n = 100) |
600-mg/day (n = 99) |
Overall (n = 199) |
|||||
| At least one AE | 39 (40.2) | 58 (58.0) | 55 (55.6) | 113 (56.8) | ||||
|
Treatment-Related AEs ≥ 5% Somnolence Fatigue Headache Nausea Decreased appetite |
3 (3.1) 4 (4.1) 3 (3.1) 2 (2.1) 2 (2.1) |
13 (13.0) 11 (11.0) 9 (9.0) 5 (5.0) 6 (6.0) |
17 (17.2) 10 (10.1) 7 (7.1) 8 (8.1) 6 (6.1) |
30 (15.1) 21 (10.6) 16 (8.0) 13 (6.5) 12 (6.0) |
||||
|
AEs leading to discontinuation, n (%) Totala Tachycardia Abdominal pain upper Nausea Vomiting Fatigue Dizziness postural Headache Somnolence Depressed mood Suicidal ideation Suicide attempt Vitiligo |
1 (1.0) 0 0 0 0 0 0 0 0 0 1 (1.0) 0 0 |
4 (4.0) 0 1 (1.0) 1 (1.0) 1 (1.0) 0 0 1 (1.0) 0 0 0 1 (1.0) 0 |
5 (5.1) 1 (1.0) 0 0 0 1 (1.0) 1 (1.0) 0 2 (2.0) 1 (1.0) 0 0 1 (1.0) |
9 (4.5) 1 (0.5) 1 (0.5) 1 (0.5) 1 (0.5) 1 (0.5) 1 (0.5) 1 (0.5) 2 (1.0) 1 (0.5) 0 1 (0.5) 1 (0.5) |
||||
aSubjects who discontinued due to 1 or more AE incidents. AE, adverse event.
The majority of AEs reported in the SPN-812 treatment groups were mild or moderate: ≥ 1 mild AEs occurred in 29.1% of subjects, ≥ 1 moderate AEs occurred in 26.1% of subjects, and ≥ 1 severe AEs occurred in 1.5% of subjects. One subject (400-mg/day SPN-812) experienced a suicide attempt, which was reported by the Site Investigator as a serious AE and possibly related to SM and/or the patient’s psychiatric history. The subject was discontinued from the study and subsequently admitted to the hospital for observation and treatment. Another subject (400-mg/day SPN-812) experienced severe abnormal skin odor that was considered possibly related to treatment but did not require SM interruption. A third subject (600-mg/day SPN-812) experienced two severe AEs, postural dizziness (possibly related to treatment) and orthostatic hypotension (not related to treatment), as well as tachycardia, somnolence, and depressed mood. This subject was discontinued from the study due to the AEs of postural dizziness, depressed mood, and tachycardia.
Overall, AEs leading to study discontinuation in the subjects receiving SPN-812 (n = 9, 4.5 %) included tachycardia, upper abdominal pain, nausea, vomiting, fatigue, postural dizziness, headache, depressed mood, suicide attempt, and vitiligo, each occurring in 1 subject; and somnolence, which occurred in 2 subjects (Table 4). In the placebo group, one subject experienced post-baseline suicidal ideation, which led to study discontinuation.
One subject (400-mg/day) experienced mild syncope possibly related to treatment, which was reported as serious AE; the subject fully recovered on the same day; however, the SM was discontinued at a later time point due to an AE of vomiting.
No discontinuations due to AEs reported for abnormal values in laboratory tests occurred during SPN-812 treatment. The most common laboratory test abnormalities observed in this trial were: alanine aminotransferase (ALT) above normal in 0 (0.0%), 9 (10.5%), and 9 (10.2%) subjects; glucose below normal in 2 (2.3%), 11 (12.8%), and 6 (6.8%) subjects; and neutrophils below normal in 9 (10.5%), 9 (10.2%), and 4 (4.5%) subjects in the placebo, 400-mg/day SPN-812, and 600-mg/day SPN-812 treatment group, respectively.
Increased ALT and aspartate aminotransferase (AST) were reported as AEs in one subject (400-mg/day SPN-812) and considered mild in severity and unrelated to SM. In another subject (400-mg/day SPN-812), mild increase in ALT was reported as an AE and considered possibly related to treatment. In both subjects with elevated ALT, bilirubin values were normal. An AE of elevated values of hematocrit, red blood cell count, and hemoglobin was reported in one subject (400-mg/day SPN-812), while an AE of decreased white blood cell count was reported in another subject (600-mg/day SPN-812); both AEs were considered mild in severity and unrelated to the SM.
Changes in the vital signs were generally small and infrequent, with the exception of BP below normal (> 10% of subjects) observed at one time point in each of the SPN-812 arms. The mean ± SD CFB at EOS in systolic and diastolic BP were 1.5 ± 9.92 mmHg and 1.2 ± 7.77 mmHg in the placebo group, 0.5 ± 9.48 mmHg and 2.9 ± 10.47 mmHg in the 400-mg/day group, and 2.5 ± 9.78 mmHg and 3.4 ± 9.17 in the 600-mg/day group, respectively. The mean ± SD CFBs at EOS in HR (beats per min, bpm) were 0.5 ± 11.05 (placebo), 4.9 ± 12.07 bpm (400-mg/day), and 6.3 ± 14.41 bpm (600-mg/day). Mild vital signs abnormalities were reported as AEs in 6 subjects receiving SPN-812, including increased diastolic and systolic BP in 1 subject (400-mg/day), increased HR in 1 subject (400-mg/day), and diastolic hypertension in 1 subject (600-mg/day), which occurred during the titration period; increased orthostatic hypotension in 2 subjects (600-mg/day) and increased HR in 1 subject (600mg/day), which occurred during maintenance period.
The ECG-related AEs during the study were tachycardia reported in 0 (0.0%), 3 (3.0%), and 1 (1.0%) subjects in the placebo, 400-mg/day SPN-812, and 600-mg/day SPN-812 groups, respectively. Additionally, two subjects had a QTcF (QT interval corrected for HR using Fridericia’s method) of > 450 ms; one subject in the placebo group had a QTcF of 454 ms, while one subject in the 400-mg/day SPN-812 group had a QTcF of 453 ms. Neither of these events were considered clinically significant. No CFB in QTcF of > 60 ms were observed. Small ECG changes included a CFB in QTcF of 30 ms to 60 ms occurring in 3 subjects (3.4%) in the placebo group, 5 subjects (5.5%) in the 400-mg/day group, and 1 subject (1.1%) in the 600-mg/day group.
Weight fluctuation considered unrelated to SM occurred in 1 subject in the 600-mg/day SPN-812 group (0.1 kg decrease was observed at EOS). Weight decrease was observed in 1 subject in the 400-mg/day SPN-812 group (1.5 kg), 1 subject in the 400-mg/day SPN-812 group (5.9 kg), and 1 subject in 600-mg/day SPN-812 group (4.3 kg). Poor weight gain was reported in 2 subjects in the 400-mg/day SPN-812 group. All these events were mild or moderate and considered related to the SM. No BMI or height changes were reported as AEs.
Discussion
The primary efficacy endpoint (CFB at EOS ADHD-RS-5 Total score) was not met in this study. Due to multiple treatment comparisons (400-mg/day SPN-812 vs. placebo and 600-mg/day SPN-812 vs. placebo), the statistical analysis of the primary endpoint included a sequential testing procedure43 with a fixed testing method of the null hypotheses to control the overall type I error rate at 0.05. In this type of analysis, two separate null hypotheses (each SPN-812 dose vs. placebo) are tested sequentially. If the first null hypothesis (in this study: no treatment mean difference between 600-mg/day SPN-812 and placebo) is not rejected, then the second null hypothesis (in this study: no treatment mean difference between 400-mg/day SPN-812 and placebo) is not tested, and no conclusion can be reached about the efficacy of either dose.
Based on this pre-specified approach, even though the CFB in ADHD-RS-5 Total score did separate from placebo following treatment with 400-mg/day SPN-812 – starting at Week 2 after the initiation of treatment and through EOS – it could not be considered statistically different from placebo in this study.
In three of four Phase 3 clinical trials of SPN-812, the primary endpoint (which was the same in all four trials) was met: 100- to 400-mg/day doses have demonstrated statistically significant improvements vs. placebo in the CFB at EOS in ADHD-RS-5 Total score in children and adolescents with ADHD. Although CFB at EOS in ADHD-RS-5 Total score was similar across doses 100–600 mg, it was rather surprising that in this fourth trial (presented here), the highest tested dose of SPN-812 (600-mg/day) did not separate from placebo on this measure. One possible explanation could be a high placebo response in this study, which was >44% higher than in the other three trials.45 In fact, it has been demonstrated earlier that a number of non-specific factors unrelated to the actual study medication can influence the success rate of placebo-controlled randomized clinical trials, such as flaws in the study design, changes in the patient referral patterns, or early drop-out rates.46 High placebo response is one of most common non-specific factors that contributes to clinical trial failure, which prevent detection of the statistically significant effects of psychiatric medications.47–50 Other explanations (e.g., flaws in the study design, small sample size, high drop-out rates) are rather unlikely to explain the negative finding of this Phase 3 trial of SPN-812 considering that all four of the Phase 3 trials of SPN-812 had, functionally, an identical study design (barring changes in dose, age of study participants, and drug tapering procedures), were sufficiently powered, and had similar drop-out rates across the studies.
If high placebo response was a contributing factor to this negative trial, it could have impacted the ability to detect statistically significant differences on the primary endpoint (CFB at EOS for ADHD-RS-5), as well as the various secondary endpoints, for both doses of SPN-812 versus placebo. Although no statistically significant differences were found between 400-mg/day or 600-mg/day SPN-812 and placebo in the Conners 3–PS Composite T-score or the WFIRS–P Total average score in the present trial, this hypothesis is supported by the fact that differences were reported with lower doses of SPN-812 in the other Phase 3 studies.32–34
Despite the lack of statistically significant difference vs. placebo in the 600-mg/day dose group in the primary efficacy analysis in the present study, statistically significant improvements vs. placebo were observed for a number of other secondary endpoints: CFBs at EOS in the ADHD-RS-5 Inattention and Hyperactivity/Impulsivity subscale scores (400-mg/day SPN-812), proportion of ADHD-RS-5 responders (400-mg/day SPN-812), the CGI-I score at EOS (400-mg/day SPN-812), proportion of CGI-I responders at various time points during the study (400-mg/day and 600-mg/day SPN-812), CFB at EOS Conners 3-PS executive functioning content scale score (400-mg/day SPN-812), and CFB in Conners 3-SRS Composite T-score at EOS (600-mg/day SPN-812). These results are consistent with the data reported in the other Phase 3 trials of SPN-812.32–34
Low incidence of AEs and no clinically relevant trends observed in the clinical laboratory tests, vital signs, or ECG in this study are also consistent with previously reported Phase 3 data.32–34
A high inter-individual variability of treatment response in ADHD18,51,52 suggests that a proportion of individuals with ADHD may benefit from a higher dose of a nonstimulant medication, such as SPN-812, with a favorable safety and efficacy profile. Future exploratory analyses may help identify those patients and guide a personalized treatment approach to the management of ADHD.
Conclusion
In this study, one of the two doses of SPN-812 (600-mg/day) did not separate from placebo. This was possibly due to high placebo response, which may have confounded the treatment effect. The 400-mg/day dose of SPN-812 did separate from placebo, however, due to the nature of the statistical analysis (600-mg/day dose was tested first), these study results can only be interpreted as informative. Although this Phase 3 study of SPN-812 did not reach statistical significance for the primary endpoint, interpreting these results within the context of all four Phase 3 clinical trials of SPN-812 is important. That is, three previous Phase 3 trials of SPN-812 have demonstrated that SPN-812 was effective in reducing the symptoms of ADHD in a pediatric population (6 to17 years of age).32–34 Further, all doses of SPN-812 tested in the Phase 3 program were well tolerated as evidenced by the low AE-related discontinuation rates in these trials.32–34
Acknowledgments
This work was funded by Supernus Pharmaceuticals, Inc. Supernus Pharmaceuticals, Inc. employees, AN, TL, TA, NF, JTH, FC, GDB, ZM, and SS, participated in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Footnotes
Disclosures
AN, TL, TA, NF, JTH, FC, GDB, ZM, and SS are employees of Supernus Pharmaceuticals, Inc.
AC is a consultant for Adlon Therapeutics, Aevi Genomics, AiCure, Akili Interactive, Arbor Pharmaceuticals, Atentive, Ironshore, KemPharm, Lundbeck, MedAvante-ProPhase, Neos Therapeutics, NLS Pharma, Otsuka, Purdue, Shire, Sunovion, Supernus, Takeda, and Tris Pharma. He has received speaker/promotional honoraria from Arbor Pharmaceuticals, Ironshore, Lundbeck, Neos Therapeutics, Otsuka, Shire, Sunovion, Supernus, Takeda, and Tris Pharma. Dr. Cutler has received research grants from Aevi Genomics, Akili Interactive, Arbor Pharmaceuticals, Ironshore, KemPharm, Lundbeck, Neos Therapeutics, Otsuka, Purdue, Shire, Sunovion, Supernus, Takeda, and Tris Pharma. He is an employee and board member of the Neuroscience Education Institute.
RF receives or has received research support, acted as a consultant and/or has received honoraria from Acadia, Adamas, Aevi, Afecta, Akili, Alcobra, Alkermes, Allergan, Amerex, American Academy of Child & Adolescent Psychiatry, American Psychiatric Press, Arbor, Axsome, Daiichi-Sankyo, Gedeon Richter, Genentech, Idorsia, Intra-Cellular Therapies, KemPharm, Luminopia, Lundbeck, MedAvante-ProPhase, Merck, NIH, Neurim, Noven, Nuvelution, Otsuka, PCORI, PaxMedica, Pfizer, Physicians Postgraduate Press, Q BioMed, Receptor Life Sciences, Roche, Sage, Signant Health, Sunovion, Supernus Pharmaceuticals, Syneos, Syneurx, Takeda, Teva, Tris, TouchPoint, and Validus.
References
- 1.Xu G, Strathearn L, Liu B, Yang B, Bao W. Twenty-Year Trends in Diagnosed Attention-Deficit/Hyperactivity Disorder Among US Children and Adolescents, 1997–2016. JAMA Netw Open. 2018;1(4):e181471. doi: 10.1001/jamanetworkopen.2018.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klassen AF, Miller A, Fine S. Health-related quality of life in children and adolescents who have a diagnosis of attention-deficit/hyperactivity disorder. Pediatrics. 2004;114(5):e541–547. doi: 10.1542/peds.2004-0844. [DOI] [PubMed] [Google Scholar]
- 3.Childress AC. Methylphenidate HCL for the treatment of ADHD in children and adolescents. Expert Opin Pharmacother. 2016;17(8):1171–1178. doi: 10.1080/14656566.2016.1182986. [DOI] [PubMed] [Google Scholar]
- 4.Childress A, Tran C. Current Investigational Drugs for the Treatment of Attention-Deficit/Hyperactivity Disorder. Expert Opin Investig Drugs. 2016;25(4):463–474. doi: 10.1517/13543784.2016.1147558. [DOI] [PubMed] [Google Scholar]
- 5.Biederman J, Mick E, Faraone SV. Age-dependent decline of symptoms of attention deficit hyperactivity disorder: impact of remission definition and symptom type. Am J Psychiatry. 2000;157(5):816–818. doi: 10.1176/appi.ajp.157.5.816. [DOI] [PubMed] [Google Scholar]
- 6.Mattingly GW, Weisler RH, Young J et al. Clinical response and symptomatic remission in short- and long-term trials of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. BMC Psychiatry. 2013;13:39. doi: 10.1186/1471-244X-13-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen MH, Huang KL, Hsu JW, Tsai SJ. Treatment-resistant Attention-deficit Hyperactivity Disorder: Clinical Significance, Concept, and Management. Taiwanese Journal of Psychiatry (Taipei) 2019;33:66–75. [Google Scholar]
- 8.Titusville, NJ: Janssen Pharmaceuticals, Inc; 2017. CONCERTA®. Prescribing information. [Google Scholar]
- 9.Lexington, MA: Shire US Inc.; 2018. VYVANSE®. Prescribing information. [Google Scholar]
- 10.Indianapolis, IN: Lilly USA, LLC; 2017. STRATTERA®. Prescribing information. [Google Scholar]
- 11.Lexington, MA: Shire US Inc; 2019. INTUNIV®. Prescribing information. [Google Scholar]
- 12.St. Michael, Barbados: Concordia Pharmaceuticals Inc; 2016. KAPVAY®. Prescribing information. [Google Scholar]
- 13.Wilens TE, Gignac M, Swezey A, Monuteaux MC, Biederman J. Characteristics of adolescents and young adults with ADHD who divert or misuse their prescribed medications. J Am Acad Child Adolesc Psychiatry. 2006;45(4):408–414. doi: 10.1097/01.chi.0000199027.68828.b3. [DOI] [PubMed] [Google Scholar]
- 14.Weyandt LL, Oster DR, Marraccini ME et al. Pharmacological interventions for adolescents and adults with ADHD: stimulant and nonstimulant medications and misuse of prescription stimulants. Psychol Res Behav Manag. 2014;7:223–249. doi: 10.2147/PRBM.S47013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kollins SH. Abuse liability of medications used to treat attention-deficit/hyperactivity disorder (ADHD) Am J Addict. 2007;(16 Suppl 1:35–42) doi: 10.1080/10550490601082775. quiz 43–34. [DOI] [PubMed] [Google Scholar]
- 16.Kroutil LA, Van Brunt DL, Herman-Stahl MA, Heller DC, Bray RM, Penne MA. Nonmedical use of prescription stimulants in the United States. Drug Alcohol Depend. 2006;84(2):135–143. doi: 10.1016/j.drugalcdep.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 17. Code of Federal Regulations Title 21, Vol 9, Part 1305 Orders for Schedule I and II Controlled Sustance. https://www.govinfo.gov/app/details/CFR-2017-title21-vol9/CFR-2017-title21-vol9-part1305/context. [Google Scholar]
- 18.Huss M, Duhan P, Gandhi P, Chen CW, Spannhuth C, Kumar V. Methylphenidate dose optimization for ADHD treatment: review of safety, efficacy, and clinical necessity. Neuropsychiatr Dis Treat. 2017;13:1741–1751. doi: 10.2147/NDT.S130444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asherson P, Bushe C, Saylor K, Tanaka Y, Deberdt W, Upadhyaya H. Efficacy of atomoxetine in adults with attention deficit hyperactivity disorder: an integrated analysis of the complete database of multicenter placebo-controlled trials. J Psychopharmacol. 2014;28(9):837–846. doi: 10.1177/0269881114542453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faraone SV, Schachar RJ, Barkley RA, Nullmeier R, Sallee FR. Early Morning Functional Impairments in Stimulant-Treated Children with Attention-Deficit/Hyperactivity Disorder Versus Controls: Impact on the Family. J Child Adolesc Psychopharmacol. 2017;27(8):715–722. doi: 10.1089/cap.2016.0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montoya A, Hervas A, Cardo E et al. Evaluation of atomoxetine for first-line treatment of newly diagnosed, treatment-naive children and adolescents with attention deficit/hyperactivity disorder. Curr Med Res Opin. 2009;25(11):2745–2754. doi: 10.1185/03007990903316152. [DOI] [PubMed] [Google Scholar]
- 22.Kolar D, Keller A, Golfinopoulos M, Cumyn L, Syer C, Hechtman L. Treatment of adults with attention-deficit/hyperactivity disorder. Neuropsychiatr Dis Treat. 2008;4(2):389–403. doi: 10.2147/ndt.s6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brahmbhatt K, Hilty DM, Hah M, Han J, Angkustsiri K, Schweitzer JB. Diagnosis and Treatment of Attention Deficit Hyperactivity Disorder During Adolescence in the Primary Care Setting: A Concise Review. J Adolesc Health. 2016;59(2):135–143. doi: 10.1016/j.jadohealth.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolraich ML, Wibbelsman CJ, Brown TE et al. Attention-deficit/hyperactivity disorder among adolescents: a review of the diagnosis, treatment, and clinical implications. Pediatrics. 2005;115(6):1734–1746. doi: 10.1542/peds.2004-1959. [DOI] [PubMed] [Google Scholar]
- 25.Baweja R, Mattison RE, Waxmonsky JG. Impact of Attention-Deficit Hyperactivity Disorder on School Performance: What are the Effects of Medication. Paediatr Drugs. 2015;17(6):459–477. doi: 10.1007/s40272-015-0144-2. [DOI] [PubMed] [Google Scholar]
- 26.Scholtens S, Rydell AM, Yang-Wallentin F. ADHD symptoms, academic achievement, self-perception of academic competence and future orientation: a longitudinal study. Scand J Psychol. 2013;54(3):205–212. doi: 10.1111/sjop.12042. [DOI] [PubMed] [Google Scholar]
- 27.Barkley RA, Cox D. A review of driving risks and impairments associated with attention-deficit/hyperactivity disorder and the effects of stimulant medication on driving performance. J Safety Res. 2007;38(1):113–128. doi: 10.1016/j.jsr.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 28.Shaw M, Hodgkins P, Caci H et al. A systematic review and analysis of long-term outcomes in attention deficit hyperactivity disorder: effects of treatment and non-treatment. BMC Med. 2012;10:99. doi: 10.1186/1741-7015-10-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Childress AC, Berry SA. Pharmacotherapy of attention-deficit hyperactivity disorder in adolescents. Drugs. 2012;72(3):309–325. doi: 10.2165/11599580-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 30.Biederman J, Monuteaux MC, Mick E et al. Young adult outcome of attention deficit hyperactivity disorder: a controlled 10-year follow-up study. Psychol Med. 2006;36(2):167–179. doi: 10.1017/S0033291705006410. [DOI] [PubMed] [Google Scholar]
- 31.Yu C, Garcia-Olivares J, Candler S, Schwabe S, Maletic V. New Insights into the Mechanism of Action of Viloxazine: Serotonin and Norepinephrine Modulating Properties. J Exp Pharmacol. 2020;12:285–300. doi: 10.2147/JEP.S256586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nasser A, Liranso T, Adewole T et al. A Phase III, Randomized, Placebo-controlled Trial to Assess the Efficacy and Safety of Once-daily SPN-812 (Viloxazine Extended-release) in the Treatment of Attention-deficit/Hyperactivity Disorder in School-age Children. Clin Ther. 2020;42(8):1452–1466. doi: 10.1016/j.clinthera.2020.05.021. [DOI] [PubMed] [Google Scholar]
- 33.Nasser A, Hull JT, Chowdhry FA, Adewole T, Liranso T, Schwabe S. 112 A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study (P302): Efficacy and Safety of Extended-Release Viloxazine in Adolescents with ADHD. CNS Spectr. 2020;25(2):272–273. [Google Scholar]
- 34.Nasser A, Hull JT, Chowdhry FA, Adewole T, Liranso T, Schwabe S. 113 Phase 3, Randomized, Double-Blind, Placebo-Controlled Study (P303) Assessing Efficacy and Safety of Extended-Release Viloxazine in Children with ADHD. CNS Spectr. 2020;25(2):273–274. [Google Scholar]
- 35.5th ed. Washington, DC: American Psychiatric Publishing; 2013. Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association. [Google Scholar]
- 36.Sheehan DV, Sheehan KH, Shytle RD et al. Reliability and validity of the Mini International Neuropsychiatric Interview for Children and Adolescents (MINI-KID) J Clin Psychiatry. 2010;71(3):313–326. doi: 10.4088/JCP.09m05305whi. [DOI] [PubMed] [Google Scholar]
- 37.Johnson JK, Liranso T, Saylor K et al. A Phase II Double-Blind, Placebo-Controlled, Efficacy and Safety Study of SPN-812 (Extended-Release Viloxazine) in Children With ADHD. J Atten Disord. 2020;24(2):348–358. doi: 10.1177/1087054719836159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DuPaul GJ, Power TJ, Anastopoulos AD . New York, NY: The Guilford Press; 2016. ADHD Rating Scale-5 for Children and Adolescents: Checklists, Norms, and Clinical Interpretation. [Google Scholar]
- 39.Guy W. Rockville, MD: U.S. Department of Health, Education, and Welfare; Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration; National Institute of Mental Health; Psychopharmacology Research Branch; Division of Extramural Research Programs; 1976. Clinical Global Impression (CGI). Early Clinical Drug Evaluation Unit (ECDEU) Assessment Manual for Psychopharmacology; pp. 218–222. [Google Scholar]
- 40.Sparrow EP. Hoboken, NJ: John Wiley & Sons, Inc; 2010. Essentials of Conners Behavior Assessments. [Google Scholar]
- 41.Gajria K, Kosinski M, Sikirica V et al. Psychometric validation of the Weiss Functional Impairment Rating Scale-Parent Report Form in children and adolescents with attention-deficit/hyperactivity disorder. Health Qual Life Outcomes. 2015;13:184. doi: 10.1186/s12955-015-0379-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson T, Lloyd A, Joseph A, Weiss M. The Weiss Functional Impairment Rating Scale-Parent Form for assessing ADHD: evaluating diagnostic accuracy and determining optimal thresholds using ROC analysis. Qual Life Res. 2017;26(7):1879–1885. doi: 10.1007/s11136-017-1514-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Westfall PH, Tobias RD, Wolfinger RD. Multiple comparisons and multiple tests using SAS. 2011 SAS Institute. [Google Scholar]
- 44.Posner K, Brown GK, Stanley B et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–1277. doi: 10.1176/appi.ajp.2011.10111704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Supernus Pharmaceuticals, Inc., data on file. [Google Scholar]
- 46.Montgomery SA. The failure of placebo-controlled studies. ECNP Consensus Meeting, September 13, 1997, Vienna. European College of Neuropsychopharmacology. Eur Neuropsychopharmacol. 1999;9(3):271–276. doi: 10.1016/s0924-977x(98)00050-9. [DOI] [PubMed] [Google Scholar]
- 47.Khan A, Detke M, Khan SR, Mallinckrodt C. Placebo response and antidepressant clinical trial outcome. J Nerv Ment Dis. 2003;191(4):211–218. doi: 10.1097/01.NMD.0000061144.16176.38. [DOI] [PubMed] [Google Scholar]
- 48.Papakostas GI, Fava M. Does the probability of receiving placebo influence clinical trial outcome? A meta-regression of double-blind, randomized clinical trials in MDD. Eur Neuropsychopharmacol. 2009;19(1):34–40. doi: 10.1016/j.euroneuro.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 49.Walsh BT, Seidman SN, Sysko R, Gould M. Placebo response in studies of major depression: variable, substantial, and growing. JAMA. 2002;287(14):1840–1847. doi: 10.1001/jama.287.14.1840. [DOI] [PubMed] [Google Scholar]
- 50.Weimer K, Colloca L, Enck P. Placebo effects in psychiatry: mediators and moderators. Lancet Psychiatry. 2015;2(3):246–257. doi: 10.1016/S2215-0366(14)00092-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kratochvil CJ, Michelson D, Newcorn JH et al. High-dose atomoxetine treatment of ADHD in youths with limited response to standard doses. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1128–1137. doi: 10.1097/chi.0b013e318074eeb3. [DOI] [PubMed] [Google Scholar]
- 52.Tsuda Y, Matsuo Y, Matsumoto S, Wajima T. Population pharmacokinetic and exposure-response analyses of guanfacine in Japanese pediatric ADHD patients. Drug Metab Pharmacokinet. 2019;34(6):365–371. doi: 10.1016/j.dmpk.2019.07.001. [DOI] [PubMed] [Google Scholar]