

Abstract
Objectives:
Although clozapine exhibited high efficacy for treating the symptoms of patients with treatment-resistant schizophrenia (TRS), its precise action mechanisms have not been fully understood. Recently, accumulating evidence has suggested the presence of abnormalities in the gamma-aminobutyric acid (GABA) systems in patients with schizophrenia, and the potential effects of clozapine on GABA receptors have gained a great deal of attention.
Experimental Designs:
In the present study, the cortical silent period (CSP), an electrophysiological parameter of GABA function via GABAB receptors, was measured using with the transcranial magnetic stimulation in patients with schizophrenia and healthy control subjects. Then the CSP of patients treated with clozapine (N = 12) was compared with that of patients treated with other antipsychotics (N = 25) and with that of healthy controls (N = 27).
Principal Observations:
The CSP of the patients treated with clozapine was significantly longer compared to those of the other two groups. The CSP of patients treated with other antipsychotics was similar to that of healthy subjects. There was a positive correlation between CSP and global assessment of function (GAF) in patients with TRS.
Conclusions:
The present study indicated that CSP was prolonged in patients receiving clozapine, and suggested that clozapine enhances the transmission signal via GABAB receptors.
Keywords: antipsychotic, cortical inhibition, GABA, transcranial magnetic stimulation, treatment-resistant
Introduction
Clozapine is the last resort in pharmacological medications for patients with schizophrenia, and it is the only antipsychotic agent that has been established to be effective for patients with treatment-resistant schizophrenia (TRS).1 It has been demonstrated that clozapine did not only exhibit a high efficacy against positive symptoms, but also against aggressive behaviors and suicidal-related symptoms, although its beneficial effects on primary negative symptoms and cognitive functions seemed to be restricted.2–4
To date, however, the action mechanism accounting for its superior efficacy in comparison to all other antipsychotics has yet to be clarified. Although several mechanical actions have been proposed, such as fast dissociation from dopamine D2 receptors,5 the effect of norclozapine, its main active metabolite,6 and a higher affinity to dopamine D4 receptors,7,8 none of these hypotheses has been decisively confirmed. The glutamate hypothesis and gamma-aminobutyric acid (GABA) hypothesis emerged over 10 years ago,9 but it is difficult to assess these hypotheses in an individual subject.
The possibility that clozapine has an action mechanism that affects the GABA system has been explored mainly in animal models and electrophysiological studies. Accumulating evidence suggests that clozapine works in any of the neuronal circuits in the mesocortical, mesolimbic, and nigrostriatal pathways, through the GABA and glutamine neurotransmissions.10,11 However, it is still difficult to examine this mechanism at the individual patient level.
The cortical silent period (CSP) is a temporal inhibition of neuronal activity which is induced by a single pulse of transcranial magnetic stimulation (TMS).12 In this silent period, transmission in the pyramidal tract from the central nervous system (CNS) to the peripheral nerves is inhibited. It was established that the latter half of the CSP is especially characterized by the inhibition of CNS via GABAB receptors.13
Although some studies have suggested that this measure detects possible alterations of GABA functions over disease stage in schizophrenia patients,14–17 most studies have examined the potential effects of various antipsychotics on CSP in patients taking these agents.18,19 However, none of these studies have reached a firm conclusion on this issue due to a small sample size or to their inclusion of patients receiving heterogeneous treatments.20 Among these studies, some commonly reported that clozapine prolonged CSP,21–23 but these did not attempt to clarify the relationship of CSP with clinical parameters.
Thus the significance of CSP has been uncertain in both contexts of pharmacology and the disease etiology. In the present study, we aimed to examine CSP in TRS patients under treatment with clozapine, and then compare them with the CSP of TRS or non-TRS patients under other antipsychotic. This design would contribute to reach the firm conclusion on the effect of clozapine on CSP value. Furthermore, this would provide important evidence on the pharmacological mechanism of clozapine.
Methods
Subjects
The schizophrenia group (SCH) included patients with schizophrenia and schizoaffective disorder as a main diagnosis according to the Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-5). The TRS patients were selected based on the following non-responder criteria: no response of positive symptoms to at least two different classes of antipsychotics with a sufficient dose (chlorpromazine equivalent dose [CP-eq.] of 600 mg or greater) for a sufficient duration (4 weeks or longer) in each trial, and no excess of 41 points in global assessment of functioning (GAF) during the previous year. The present study’s TRS patients included both those under treatment with clozapine and those under treatment with other antipsychotics. The healthy control (HC) group included the subjects without a present or past history of any psychiatric disorders.
The other major inclusion criteria in the SCH and HC groups were as follows: 20–65 years old when giving informed consent for the study and, right-handedness evaluated using the Edinburgh Handedness Inventory.24 The exclusion criteria were as follows: pregnancy, including suspected pregnancy; having had a past physical illness including any CNS disorder such as cerebrovascular diseases, epilepsy, and so on; and having a substance abuse/dependent disorder other than nicotine dependence.
The present study was conducted after approval by the ethics committee at Chiba University Graduate School of Medicine. All the subjects provided their informed consent after receiving a full and detailed explanation of the study.
Measurements
Throughout the measurement of CSP, each subject sat reluctantly on the sofa. For each subject, CSP was measured by giving a single stimulation pulse to the left-side motor cortex corresponding to the right-side first dorsal interosseous muscle, using the TMS device [Magstim Rapid 2: the Magstim Company Ltd]. First, the resting motor threshold (RMT) was established by the most common method. That is, the RMT was defined at the minimum output level of the TMS device exhibiting a magnitude of 50 μV or greater on an electroencephalogram (EEG) monitor at a ratio of over 50% of 10 stimulations.
Then each subject was asked to maintain 20% of the maximum strength level of his or her right hand, which was confirmed on the EEG monitor. A single pulse with an intensity 1.2 times that of the RMT was delivered, and the silent period appearing on the EEG monitor was defined as the CSP. The CSP was measured a total of five times, and the results were averaged. The CSP on the EEG monitor was defined as the period from the time point of the TMS pulse to the time point at which the electromyogram reemerged.
The psychopathology and daily function of patients in the SCH group were evaluated with Positive and Negative Syndrome Scale (PANSS)25 and the GAF, respectively.
Statistical Procedures
All statistical procedures were performed with SPSS ver. 23.0 software (IBM, New York, NY). Regarding the comparisons among the three groups, an analysis of variance (ANOVA) was performed on the continuous variables, and Fisher’s exact test was applied to the categorical values. Regarding the two-group comparisons, Student’s t test was applied to the continuous variables, and Fisher’s exact test was applied to the categorical values. The threshold of statistical significance was set at P = 0.05 (two-tailed).
Results
Thirty-seven schizophrenia patients (CLZ group, N = 12; Non-CLZ group, N = 25) and 27 HC participated in the present study (Table 1). Regarding the demographic and treatment variables, the ANOVA for age revealed a significant difference, and a post-hoc comparison revealed that the average age of the subject in Non-CLZ group was higher than that of subjects in the HC group. There was no significant difference in age between the CLZ group and the Non-CLZ group. The psychopathology as determined by the PANSS and daily functioning as determined by the GAF showed poorer results for the CLZ group compared to the Non-CLZ group, but these differences did not reach significant levels. The medication data showed that the CLZ group received a higher antipsychotic dose than the Non-CLZ group. The patients in the CLZ group took a CP-eq. dose of 125 ∼ 1000 mg (clozapine-dose 12.5 mg ∼ 425 mg; 2 patients took other antipsychotics, namely risperidone at 1.5 mg and perospirone at 8 mg, respectively). The medications used by the patients in the Non-CLZ group were as follows: olanzapine, N = 5, CP-eq. 200 mg ∼ 500 mg; quetiapine, N = 2, CP-eq. 56.8 mg ∼ 340 mg; aripiprazole, N = 12, CP-eq. 150 mg ∼ 933 mg; risperidone/paliperidone, N = 2, CP-eq. 200 mg ∼ 700 mg, blonanserin, N = 2, CP-eq. 300 mg ∼ 625 mg; haloperidol, N = 1, CP-eq. 150 mg: zotepine, N = 1, CP-eq. 75.8 mg.
Table 1. Demographic and Treatment Information and CSP Results of the Three Groups.
| Variables | CLZ Group [N = 12] |
Non-CLZ Group [N = 25] |
HC Group [N = 27] |
Statistical Values | ||||
| Age [y] | 39.1 [9.5] | 47.4 [10.3] | 37.4 [13.1] |
F(2,62) = 5.061, P = 0.009 Non-CLZ > HC |
||||
| Sex: Male/Female | 7/5 | 16/9 | 11/16 | Chi-sq = 2.511, P = 0.285 | ||||
| Handedness [R/L] | 12/0 | 25/1 | 11/16 | — | ||||
| Illness duration [y] | 18.3 [9.00] | 22.3 [10.3] | – | t(36) = −1.170, P = 0.250 | ||||
| Psychopathology | ||||||||
| PANSS-total | 77.1 [30.6] | 65.8 [23.1] | – | t(36) = 1.260, P = 0.216 | ||||
| PANSS-positive | 15.8 [7.0] | 13.1 [5.4] | – | t(36) = 1.289, P = 0.206 | ||||
| PANSS-negative | 20.4 [10.7] | 18.5 [8.7] | – | t(36) = 0.598, P = 0.554 | ||||
| PANSS-general | 40.9 [14.9] | 34.3 [11.4] | – | t(36) = 1.519, P = 0.137 | ||||
| GAF | 45.3 [10.5] | 50.0 [16.9] | – | t(36) = −1.054, P = 0.300 | ||||
| RMT [%] | 65.0 [13.0] | 69.0 [13.0] | 64.6 [10.7] | F(2,62) = 0.979, P = 0.382 | ||||
| CSP [msec] | 168.1 [33.6] | 104.3 [36.2] | 106.4 [35.9] |
F(2,62) = 15.157, P < 0.01 CLZ > Non-CLZ/HC |
||||
| Antipsychotic dose [CP-eq. mg] | 539.6 [270.4] | 325.2 [205.9] | – |
t(36) = 2.699, P = 0.011 CLZ > Non-CLZ |
Each cell indicates the mean value ± [standard deviation]. CP-eq: chlorpromazine-equivalent; CSP: cortical silent period; GAF: global assessment of functioning; L: Left handedness; R: Right handedness; RMT: resting motor threshold.
Regarding the results of the measurements with TMS, there was no significant difference in RMT among the three groups. As shown in Fig. 1, the CSP results showed a significant difference among the three groups, and a post-hoc analysis revealed that the CSPs of the CLZ group were significantly longer than those of the Non-CLZ group or the HC group. Since this prolongation in the CLZ group might have been induced by the greater severity of their disease psychopathology, but not by the treatment with CLZ, an additional analysis was performed by comparing 12 patients in the CLZ group with 6 patients in the Non-CLZ group who met the TRS criteria. The latter group’s CSP was 109.0 ± 33.7 msec, which was still significantly different from the CSP value of the CLZ group (t16 = −3.509, P = 0.003). Furthermore, when patients treated with olanzapine/quetiapine were selected (N = 7), their CSP was shorter than that of the CLZ group, and was similar to that of the HC group.
Figure 1.
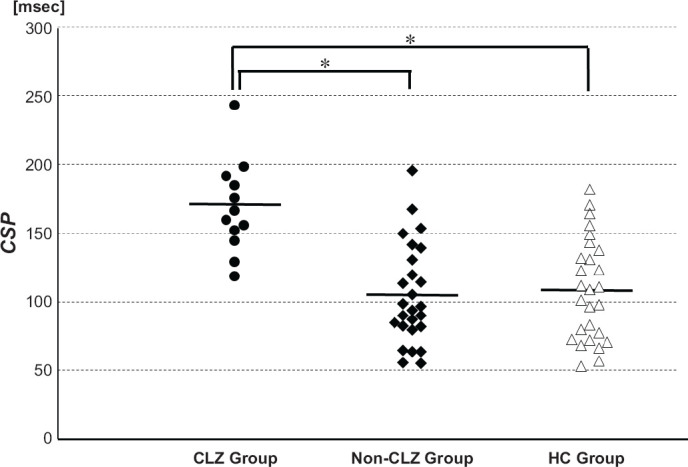
The CSP Values of the CLZ, Non-CLZ, and HC Groups
Horizontal lines indicates the mean values of the respective groups. *P < 0.05.
When we examined the possible correlation between the CSP and PANSS/GAF, there were no significant relationships between CSP and any parameters in the CLZ and Non-CLZ groups. However, there was a significant positive correlation between the CSP and GAF in the TRS patients (r = 0.688, P = 0.003; derived from N = 12 in the CLZ group and N = 5 in the Non-CLZ group; Fig. 2). This positive correlation was significant when age and antipsychotic dose (which were significant different among the three groups and might affect the value of CSP) were dealt as covariates (i.e., partial correlation: r = 0.613, P = 0.011). There was no any significant relationship between the CSP and PANSS/GAF in any of the patients (i.e., the SCH group) or in the patients without TRS. Lastly, there was no relationship between the CSP and the clozapine dose in the CLZ group. These results were the same when age and antipsychotic dose were included as covariates.
Figure 2.
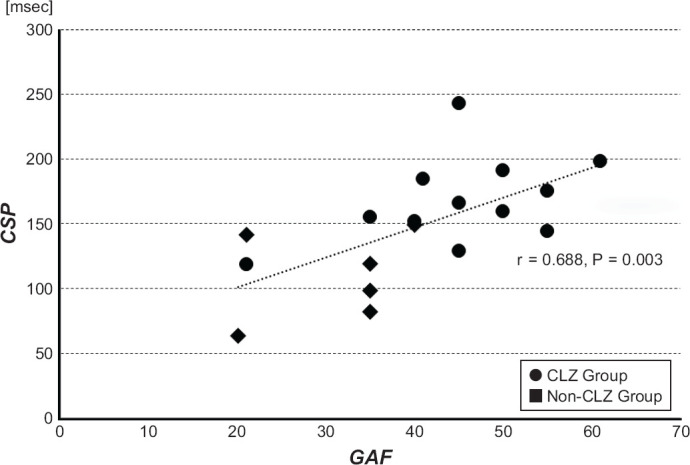
The Correlation Between CSP and GAF in the TRS Patients
TRS patients included 12 patients in the CLZ group and 5 patients in the Non-CLZ group. This correlation was also significant when their age and antipsychotic dose were included in covariates.
Discussion
The present study revealed that almost all of the patients receiving clozapine exhibited CSP prolongation compared to the HCs. This finding was not found in patients with a TRS pathology who were treated with other agents, strongly suggesting that clozapine itself prolonged the CSP. This finding also supported similar results reported in several studies.21–23 Recently, the action mechanism of clozapine on GABAB agonism has gained attention as a possible explanation for why clozapine exhibits high efficacy to patients with severe psychopathology.26 Our findings supported this possible mechanism of clozapine. On the other hand, olanzapine and quetiapine were developed from clozapine as a prototype,27 and these agents were suggested to have actions on GABA receptors.28–30 However, our results did not support this.
When we looked at the patients with TRS (i.e., all of the patients in the CLZ group and 5 patients in the Non-CLZ group), their GAF was significantly correlated with CSP, whereas there were no significant correlations in either patients treated with clozapine (the CLZ group) or all of the patients (the SCH group). These negative findings regarding the absence of a relationship in the latter subgroups might be due to the small size of the samples and the small variances of CSP and GAF in the subgroups (particularly in the CLZ group). However, the functional outcomes that the patients with TRS who were treated with clozapine showed a significantly higher GAF, and those treated with non-clozapine agents showed a significantly lower GAF, might reflect that the improvement observed with clozapine and the increase in GAF were related to the prolongation of CSP.
Overall, the present study may suggest that extreme prolongation of CSP could be related to the occurrence of symptomatic improvement through the GABAB receptors. A GABAB agonist, baclofen, which also showed prolongation of the CSP, did not show an efficacy as an antipsychotic even close to that of clozapine in several earlier studies.31–35 Therefore, clozapine’s high efficacy cannot be explained by GABAB agonism alone. It is possible that clozapine works efficiently in combination with actions on glutamatergic and 5-THnergic neurotransmissions, resulting in its high efficacy.11
One limitation of the present study was the small size of the sample, although the findings strongly confirmed previous similar results. Further, the relationship between the CSP and the treatment factors like the clozapine dose and degree of improvement brought about by clozapine treatment was uncertain. Thus, further clinical study with a higher number of patients is warranted.
Conclusion
In conclusion, the present study demonstrated that clozapine prolonged the CSP in patients taking clozapine, and suggested that the CSP could be a promising tool for explaining the difference in antipsychotic effects between clozapine and other agents.
Footnotes
Author Contributions
Conceptualization: N.K, Data curation: A.M, Formal analysis: A.M, Funding acquisition: N.K, Investigation: A.M., Y.N., A.K., Y.O, Methodology: A.M., N.K., S.K, Project administration: N.K, Resource: N.K, Software: M.A., N.K, Supervision: S.K., H. K., H.W., M.I, Validation: N.K., Y.N, Visualization: N.K, Writing-original draft: A.M., N.K, Writing-review and editing: H.K., Y.O.
Disclosures and Acknowledgments:
Funding Source
The present study was supported by a grant from the Watanabe Foundation.
Conflicts of Interest
N.K. reports honoraria from Otsuka Pharmaceutical Co., Ltd., Sumitomo Dainippon Pharma Co., Ltd., Janssen Pharmaceutical K.K., Meiji Seika Pharma Co., Ltd., and Mochida Pharmaceutical Co., Ltd. H.K. reports honoraria from Otsuka Pharmaceutical Co., Ltd., and Janssen Pharmaceutical K.K. Y.O. reports honoraria from Otsuka Pharmaceutical Co., Ltd., Sumitomo Dainippon Pharma Co., Ltd., and Meiji Seika Pharma Co., Ltd. H.W. reports lecture fees from MSD K.K. from Mochida Pharmaceutical Co., Ltd. M.I. received consultant fees from Eli Lilly Japan K.K., Sumitomo Dainippon Pharma Co., Ltd., Pfizer Japan Inc., Abbott Japan Co., Ltd. and Janssen Pharmaceutical K.K., and reports honoraria from Janssen Pharmaceutical K.K., Eli Lilly Japan K.K., Otsuka Pharmaceutical Co., Ltd., Meiji Seika Pharma Co., Ltd., Astellas Pharma Inc., Sumitomo Dainippon Pharma Co., Ltd., Ono Pharmaceutical Co., Ltd., GlaxoSmithKline K.K., Takeda Pharmaceutical Co., Ltd., Mochida Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., MSD K.K., Eisai Co. Ltd., Daiichi-Sankyo Co. Ltd., Novartis Pharma K.K., Teijin Ltd., Shionogi & Co., Ltd., Hisamitsu Pharmaceutical Co., Inc. and Asahi Kasei Corporation. A.M., Y.N., S.K., and A.K. have no conflict of interest to declare.
Location of work
Department of Psychiatry, Graduate School of Medicine, Chiba University Hospital, the Center for Forensic Mental Health, Chiba University, Chiba, Japan.
References
- 1.Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45:789–796. doi: 10.1001/archpsyc.1988.01800330013001. [DOI] [PubMed] [Google Scholar]
- 2.Huhn M, Nikolakopoulou A, Schneider-Thoma J, Krause M, Samara M, Peter N et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: a systematic review and network meta-analysis. Lancet. 2019;394:939–951. doi: 10.1016/S0140-6736(19)31135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krakowski MI, Czobor P, Citrome L, Bark N, Cooper TB. Atypical antipsychotic agents in the treatment of violent patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2006;63:622–629. doi: 10.1001/archpsyc.63.6.622. [DOI] [PubMed] [Google Scholar]
- 4.Meltzer HY, Alphs L, Green AI, Altamura AC, Anand R, Bertoldi A et al. Clozapine treatment for suicidality in schizophrenia: International Suicide Prevention Trial (InterSePT) Arch Gen Psychiatry. 2003;60:82–91. doi: 10.1001/archpsyc.60.1.82. [DOI] [PubMed] [Google Scholar]
- 5.Kapur S, Seeman P. Does fast dissociation from the dopamine d(2) receptor explain the action of atypical antipsychotics?: A new hypothesis. Am J Psychiatry. 2001;158:360–369. doi: 10.1176/appi.ajp.158.3.360. [DOI] [PubMed] [Google Scholar]
- 6.Mendoza MC, Lindenmayer JP. N-desmethylclozapine: is there evidence for its antipsychotic potential. Clin Neuropharmacol. 2009;32:154–157. doi: 10.1097/WNF.0b013e31818d46f5. [DOI] [PubMed] [Google Scholar]
- 7.Seeman P. Therapeutic receptor-blocking concentrations of neuroleptics. Int Clin Psychopharmacol. 1995;10(3):5–13. Suppl. [PubMed] [Google Scholar]
- 8.Reynolds GP. The importance of dopamine D4 receptors in the action and development of antipsychotic agents. Drugs. 1996;51:7–11. doi: 10.2165/00003495-199651010-00002. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt MJ, Mirnics K. Neurodevelopment, GABA system dysfunction, and schizophrenia. Neuropsychopharmacology. 2015;40:190–206. doi: 10.1038/npp.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daskalakis ZJ, George TP. Clozapine, GABA(B), and the treatment of resistant schizophrenia. Clin Pharmacol Ther. 2009;86:442–446. doi: 10.1038/clpt.2009.115. [DOI] [PubMed] [Google Scholar]
- 11.O’Connor WT, O’Shea SD. Clozapine and GABA transmission in schizophrenia disease models: establishing principles to guide treatments. Pharmacol Ther. 2015;150:47–80. doi: 10.1016/j.pharmthera.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Rossini PM, Burke D, Chen R, Cohen LG, Daskalakis Z, Di Iorio R et al. Non-invasive electrical and magnetic stimulation of the brain, spinal cord, roots and peripheral nerves: Basic principles and procedures for routine clinical and research application. An updated report from an I.F.C.N. Committee. Clin Neurophysiol. 2015;126:1071–1107. doi: 10.1016/j.clinph.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woler A, Ziemann U, Benecke R. New York, YN: Oxford University Press Inc; The cortical silent period. In Epstein CM, Wassermann EM, Ziemann U (Eds.), The Oxford Handbook of Transcranial Stimulation; pp. 91–102. [Google Scholar]
- 14.Hasan A, Nitsche MA, Rein B, Schneider-Axmann T, Guse B, Gruber O et al. Dysfunctional long-term potentiation-like plasticity in schizophrenia revealed by transcranial direct current stimulation. Behav Brain Res. 2011;224:15–22. doi: 10.1016/j.bbr.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 15.Hasan A, Wobrock T, Grefkes C, Labusga M, Levold K, Schneider-Axmann T et al. Deficient inhibitory cortical networks in antipsychotic-naive subjects at risk of developing first-episode psychosis and first-episode schizophrenia patients: a cross-sectional study. Biol Psychiatry. 2012;72:744–751. doi: 10.1016/j.biopsych.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Strube W, Wobrock T, Bunse T, Palm U, Padberg F, Malchow B et al. Impairments in motor-cortical inhibitory networks across recent-onset and chronic schizophrenia: a cross-sectional TMS Study. Behav Brain Res. 2014;264:17–25. doi: 10.1016/j.bbr.2014.01.041. [DOI] [PubMed] [Google Scholar]
- 17.Tang Y, Zhang T, Edelman B, Zeng B, Zhao S, Li C et al. Prolonged cortical silent period among drug-naive subjects at ultra-high risk of psychosis. Schizophr Res. 2014;160:124–130. doi: 10.1016/j.schres.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Frank E, Landgrebe M, Poeppl TB, Schecklmann M, Kreuzer PM, Prasser J, Rupprecht R et al. Antipsychotic treatment with quetiapine increases the cortical silent period. Schizophr Res. 2014;156:128–132. doi: 10.1016/j.schres.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 19.Ustohal L, Mayerova M, Hublova V, Prikrylova Kucerova H, Ceskova E, Kasparek T. Risperidone increases the cortical silent period in drug-naive patients with first-episode schizophrenia: A transcranial magnetic stimulation study. J Psychopharmacol. 2017;31:500–504. doi: 10.1177/0269881116662650. [DOI] [PubMed] [Google Scholar]
- 20.Radhu N, de Jesus DR, Ravindran LN, Zanjani A, Fitzgerald PB, Daskalakis ZJ. A meta-analysis of cortical inhibition and excitability using transcranial magnetic stimulation in psychiatric disorders. Clin Neurophysiol. 2013;124:1309–1320. doi: 10.1016/j.clinph.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 21.Daskalakis ZJ, Christensen BK, Fitzgerald PB, Moller B, Fountain SI, Chen R. Increased cortical inhibition in persons with schizophrenia treated with clozapine. J Psychopharmacol. 2008;22:203–209. doi: 10.1177/0269881107084002. [DOI] [PubMed] [Google Scholar]
- 22.Liu SK, Fitzgerald PB, Daigle M, Chen R, Daskalakis ZJ. The relationship between cortical inhibition, antipsychotic treatment, and the symptoms of schizophrenia. Biol Psychiatry. 2009;65:503–509. doi: 10.1016/j.biopsych.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 23.Kaster TS, de Jesus D, Radhu N, Farzan F, Blumberger DM, Rajji TK et al. Clozapine potentiation of GABA mediated cortical inhibition in treatment resistant schizophrenia. Schizophr Res. 2015;165:157–162. doi: 10.1016/j.schres.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Oldfield RC. The assessment and analysis of handedness. Neuropsychologia. 1971;9:97–113. doi: 10.1016/0028-3932(71)90067-4. The Edinburgh inventory. [DOI] [PubMed] [Google Scholar]
- 25.Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13:261–276. doi: 10.1093/schbul/13.2.261. [DOI] [PubMed] [Google Scholar]
- 26.Nair PC, McKinnon RA, Miners JO, Bastiampillai T. Binding of clozapine to the GABA(B) receptor: clinical and structural insights. Mol Psychiatry. 2020;25:1910–1919. doi: 10.1038/s41380-020-0709-5. [DOI] [PubMed] [Google Scholar]
- 27.Kuroki T, Nagao N, Nakahara T. Neuropharmacology of second-generation antipsychotic drugs: a validity of the serotonin-dopamine hypothesis. Prog Brain Res. 2008;172:199–212. doi: 10.1016/S0079-6123(08)00910-2. [DOI] [PubMed] [Google Scholar]
- 28.Lian J, Deng C. Early antipsychotic exposure affects NMDA and GABAA receptor binding in the brains of juvenile rats. Psychiatry Res. 2019;273:739–745. doi: 10.1016/j.psychres.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Xu S, Gullapalli RP, Frost DO. Olanzapine antipsychotic treatment of adolescent rats causes long term changes in glutamate and GABA levels in the nucleus accumbens. Schizophr Res. 2015;161:452–457. doi: 10.1016/j.schres.2014.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Locchi F, Dall’olio R, Gandolfi O, Rimondini R. Olanzapine counteracts stress-induced anxiety-like behavior in rats. Neurosci Lett. 2008;438:146–149. doi: 10.1016/j.neulet.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 31.Beckmann H, Frische M, Rüther E, Zimmer R. Baclofen (para-chlorphenyl-GABA) in schizophrenia. Pharmakopsychiatr Neuropsychopharmakol. 1977;10:26–31. doi: 10.1055/s-0028-1094515. [DOI] [PubMed] [Google Scholar]
- 32.Davis KL, Hollister LE, Berger PA. Letter: Bacloffen in schizophrenia. Lancet. 1976;1:1245. doi: 10.1016/s0140-6736(76)92198-x. [DOI] [PubMed] [Google Scholar]
- 33.Simpson GM, Branchey MH, Shrivastava RK. Letter: Baclofen in schizophrenia. Lancet. 1976;1:966–967. doi: 10.1016/s0140-6736(76)92749-5. [DOI] [PubMed] [Google Scholar]
- 34.Gulmann NC, Bahr B, Andersen B, Eliassen HM. A double-blind trial of baclofen against placebo in the treatment of schizophrenia. Acta Psychiatr Scand. 1976;54:287–293. doi: 10.1111/j.1600-0447.1976.tb00122.x. [DOI] [PubMed] [Google Scholar]
- 35.Bigelow LB, Nasrallah H, Carman J, Gillin JC, Wyatt RJ. Baclofen treatment in chronic schizophrenia: a clinical trial. Am J Psychiatry. 1977;134:318–320. doi: 10.1176/ajp.134.3.318. [DOI] [PubMed] [Google Scholar]