

Abstract
Many proteins have one or more surface-exposed patches of non-polar residues; our observations here suggest that PEGylation near such locations might be a useful strategy for increasing protein conformational stability. Specifically, we show that conjugating a PEG-azide to a propargyloxyphenylalanine via the copper (I)-catalyzed azide-alkyne cycloaddition can increase the conformational stability of the WW domain due to a favorable synergistic effect that depends on the hydrophobicity of a nearby patch of non-polar surface residues.
Graphical Abstract
Modification of protein side chains with polyethylene glycol (PEG) has been used for more than thirty years to enhance the stability and pharmacokinetic properties of protein drugs.1–8 Early efforts employed non-specific conjugation strategies that resulted in a heterogeneous mixture of PEGylated protein isoforms, which differed in the number and location of PEGylation sites; such strategies were easy to use, but frequently resulted in diminished stability and biological activity.5 Powerful chemoselective side-chain modification chemistry now allows researchers to avoid PEGylation near enzyme active sites and protein binding interfaces,9–16 but beyond this, there are few structure- or sequence-based guidelines for selecting PEGylation sites that optimally balance stability, pharmacokinetic benefits, and biological activity. We previously showed that the ability of a small PEGylated model protein to evade degradation by a cocktail of proteases depends strongly on the location of the PEGylation site, with the best sites characterized by the ability of PEG to increase protein conformational stability. Motivated by this observation, we seek to understand the molecular basis for PEG-based protein stabilization and to develop structure-based predictive guidelines for identifying optimal PEGylation sites.
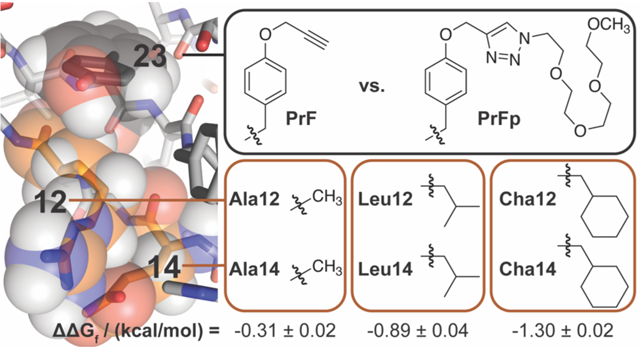
The desolvation and burial of non-polar residues in the interior of protein tertiary and quaternary structures provides a major driving force for protein folding, via enthalpic and entropic contributions known collectively as the hydrophobic effect.17–21 This process allows the ordered water molecules that surrounded the non-polar residues in the unfolded conformation to be released to bulk solvent upon folding; it also maximizes the contact surface area between non-polar side chains in the folded conformation. Previous observations suggest that side-chain PEGylation similarly releases surface bound water molecules in the immediate vicinity of the PEGylation site, thereby increasing protein conformational stability via an entropic effect.22,23 We recently found that the strength of a salt bridge between Glu12 and Arg14 in the WW domain (hereafter called WW) increases when a propargyloxyphenylalanine residue at position 23 (PrF23) is modified with a PEG-azide via the copper-catalyzed azide-alkyne cycloaddition (compare ER vs pER in Table 1; Figure 1).23 Presumably the resulting PEGylated PrF residue (PrFp23) strengthens the Glu12-Arg14 salt bridge by shielding it from interfering water molecules. Based on this observation, we wondered whether PrFp23 might be similarly able to shield or partially desolvate non-polar residues at positions 12 and 14, thereby increasing WW conformationally stability via the hydrophobic effect.
Table 1.
Sequences, melting temperatures and folding free energy changes of PEGylated WW variants relative to their non-PEGylated counterparts.a
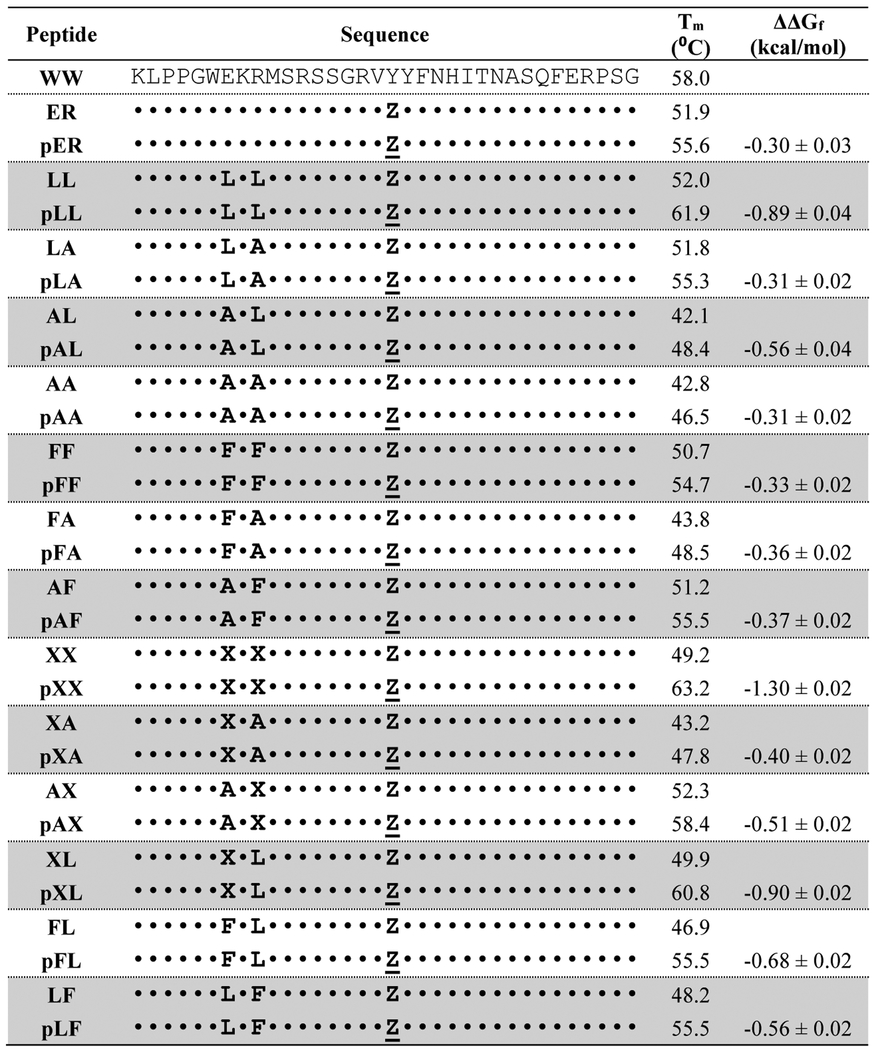
|
X = cyclohexylalanine (Cha), Z = propargyloxyphenylalanine (PrF); Z = PrFPEG. ΔΔGf value for each PEGylated variant is given ± standard error relative to its non-PEGylated counterpart in kcal/mol at 50 μM protein concentration, in 20 mM sodium phosphate buffer (pH 7), at the melting temperature of the non-PEGylated protein. Data for WW are from reference 22. Data for variants ER and pER are from reference 23.
Figure 1.
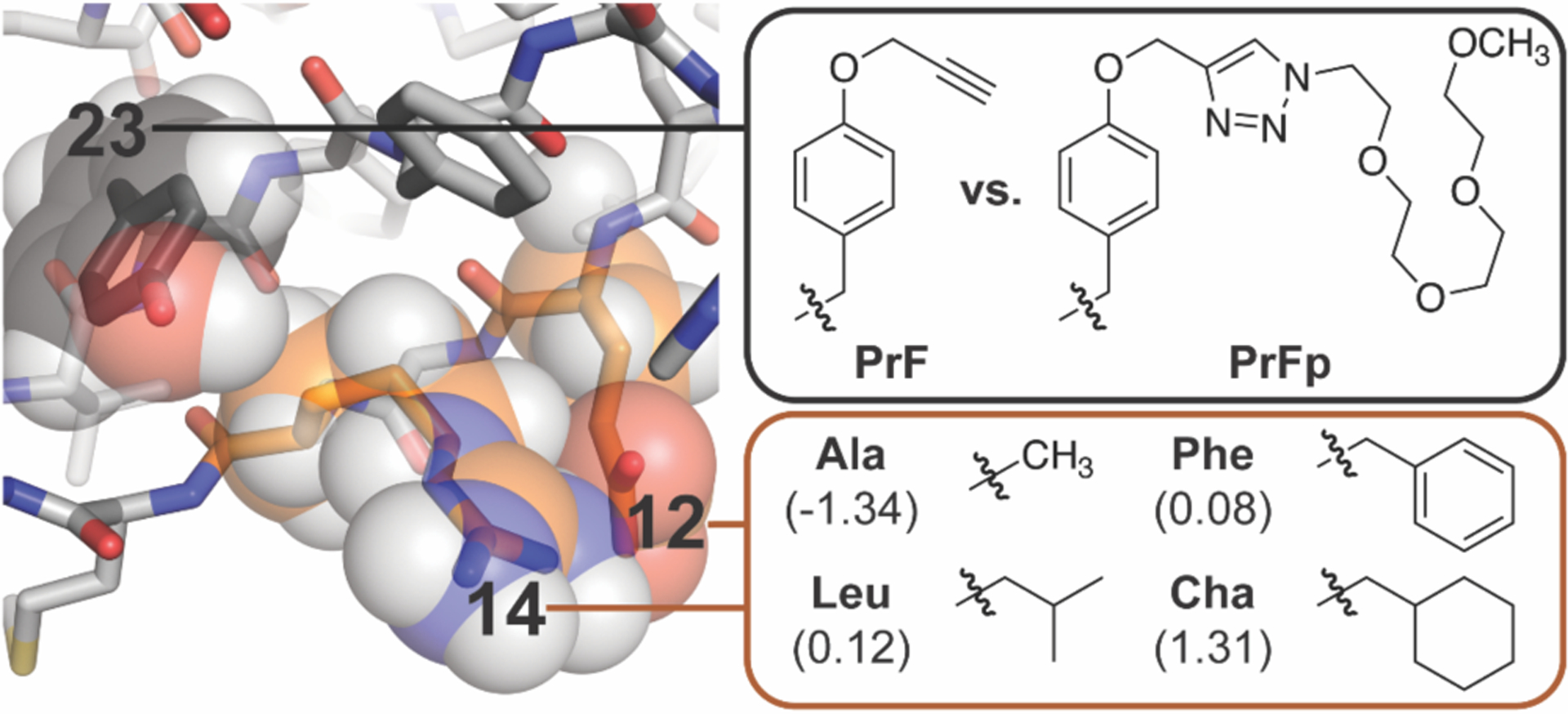
Ribbon diagram of the parent WW domain (PDB ID: 1PIN) from which the variants described here were derived. The orange outlined box shows the non-polar residues that we incorporated at positions 12 and 14, which are highlighted in orange space-filling spheres. Shown in parentheses are the c log P values for the corresponding N-acetyl N-methyl amino acid amide. The black outlined box shows the PrF and PrFp residues that we incorporated at position 23, which is highlighted in black space-filling spheres.
To test this hypothesis, we prepared WW variants LL and pLL, in which Leu occupies positions 12 and 14, with either PrF or PrFp, respectively at position 23 (one letter code: Z for PrF, Z for PrFp). Variable temperature circular dichroism (CD) experiments reveal that pLL is −0.89 ± 0.04 kcal/mol more stable than non-PEGylated LL (Table 1). We used triple mutant box analysis23–31 to explore the origins of this stabilizing effect by replacing Leu12 and/or Leu14 with Ala to generate variants LA, AL, and AA, along with their PEGylated counterparts pLA, pAL, and pAA. Comparing the folding free energies of these variants reveals that Leu12 and Leu14 engage in a favorable interaction in pLL (ΔΔΔGf = −0.44 ± 0.04 kcal/mol, Table 2), but not in non-PEGylated LL (ΔΔΔGf = −0.06 ± 0.05 kcal/mol, Table 2). PEGylation enhances the Leu12-Leu14 interaction by −0.38 ± 0.06 kcal/mol (ΔΔΔΔΔGf in Table 2), a synergistic effect that represents a substantial fraction of the overall difference in stability between pLL and LL (see above). Based on this result, we wondered whether PEGylation of PrF23 would be similarly stabilizing for WW variants with other non-polar residues at positions 12 and 14, with more non-polar WW variants experiencing greater PEG-based stabilization.
Table 2.
Triple mutant box analysis of the impact of PEGylation the interaction between residues 12 and 14 within variants pLL, pFF, pXX, pXL, pFL and pLF.a
| Peptide | ΔGf (kcal/mol) | ΔΔΔGfb (kcal/mol) | ΔΔΔΔGfc(kcal/mol) |
|---|---|---|---|
| AA | 0.60 ± 0.02 | ||
| LA | −0.12 ± 0.02 | ||
| AL | 0.61 ± 0.04 | ||
| LL | −0.17 ± 0.02 | −0.06 ± 0.05 | |
| pAA | 0.30 ± 0.01 | ||
| pLA | −0.46 ± 0.01 | ||
| pAL | 0.15 ± 0.02 | ||
| pLL | −1.05 ± 0.02 | −0.44 ± 0.03 | −0.38 ± 0.06 |
| AA | 0.53 ± 0.02 | ||
| FA | 0.44 ± 0.02 | ||
| AF | −0.16 ± 0.01 | ||
| FF | −0.19 ± 0.02 | 0.06 ± 0.03 | |
| pAA | 0.22 ± 0.01 | ||
| pFA | 0.05 ±0.01 | ||
| pAF | −0.53 ±0.01 | ||
| pFF | −0.46 ±0.01 | 0.25 ± 0.02 | 0.19 ± 0.04 |
| AA | 0.63 ± 0.02 | ||
| XA | 0.42 ±0.01 | ||
| AX | −0.16 ±0.02 | ||
| XX | 0.19 ±0.02 | 0.56 ± 0.03 | |
| pAA | 0.33 ± 0.01 | ||
| pXA | 0.23 ± 0.03 | ||
| pAX | −0.67 ± 0.02 | ||
| pXX | −1.18 ± 0.01 | −0.41 ± 0.04 | −0.97 ± 0.05 |
| AA | 0.40 ± 0.02 | ||
| XA | 0.26 ± 0.01 | ||
| AL | 0.43 ± 0.03 | ||
| XL | −0.24 ±0.01 | −0.53 ± 0.04 | |
| pAA | 0.10 ± 0.01 | ||
| pXA | −0.01 ±0.03 | ||
| pAL | −0.07 ± 0.02 | ||
| pXL | −1.09 ±0.02 | −0.91 ± 0.04 | −0.39 ± 0.06 |
| AA | 0.33 ±0.02 | ||
| FA | 0.25 ± 0.02 | ||
| AL | 0.43 ± 0.03 | ||
| FL | −0.01 ±0.02 | −0.28 ± 0.04 | |
| pAA | 0.02 ± 0.01 | ||
| pFA | −0.13 ± 0.01 | ||
| pAL | −0.14 ± 0.02 | ||
| pFL | −0.69 ± 0.01 | −0.39 ± 0.03 | −0.11 ± 0.05 |
| AA | 0.66 ± 0.02 | ||
| LA | −0.06 ± 0.02 | ||
| AF | −0.03 ± 0.02 | ||
| LF | 0.23 ± 0.01 | 0.99 ± 0.03 | |
| pAA | 0.36 ± 0.01 | ||
| pLA | −0.39 ± 0.01 | ||
| pAF | −0.40 ± 0.01 | ||
| pLF | −0.36 ± 0.01 | 0.79 ± 0.03 | −0.20 ± 0.04 |
ΔGf, ΔΔΔGf, and ΔΔΔΔGf values are given ± standard error. ΔGf values for each of the eight peptides within the same triple mutant box were calculated at their average melting temperature: 323.3 K for pLL and its derivatives; 322.3 K for pFF and its derivatives; 323.5 K for pXX and its derivatives; 320.8 K for pXL and its derivatives; 320.0 K for pFL and its derivatives; 324.0 K for pLF and its derivatives. Because variants AA, AL, LA, AF, FA, and XA and their PEGylated counterparts appear in more than one triple mutant box, their ΔGf values are necessarily presented at more than one temperature.
Strength of interaction between residues 12 and 14.
Impact of PEGylation on the strength of the interaction between residues 12 and 14.
To explore this possibility, we prepared and characterized additional derivatives of LL and pLL, in which Leu12 and/or Leu14 were replaced by Phe, Ala, or cyclohexylalanine (Cha; one-letter abbreviation is X) to give variants FF, FA, AF, XX, XA, AX, XL, LX, FL, and LF, along with their PEGylated counterparts pFF, pFA, pAF, pXX, pXA, pAX, pXL, pLX, pFL, and pLF. We estimated the hydrophobicity of each side chain by calculating the logarithm of the water-octanol partition coefficient (c log P) of the corresponding N-acetyl amino acid N-methyl amide in ChemDraw; we calculated the overall hydrophobicity of residues 12 and 14 by averaging their individual c log P values. A plot of the impact of PEGylation on the folding free energy of each variant (ΔΔGf) vs. this composite c log P value is shown in Figure 2A. The data in these plots fit reasonably to a linear equation with a slope of −0.37 ± 0.07 kcal/mol per log unit (R2 = 0.70), indicating that PEGylation at position 23 is more stabilizing when the residues that occupy positions 12 and 14 are more hydrophobic.
Figure 2.
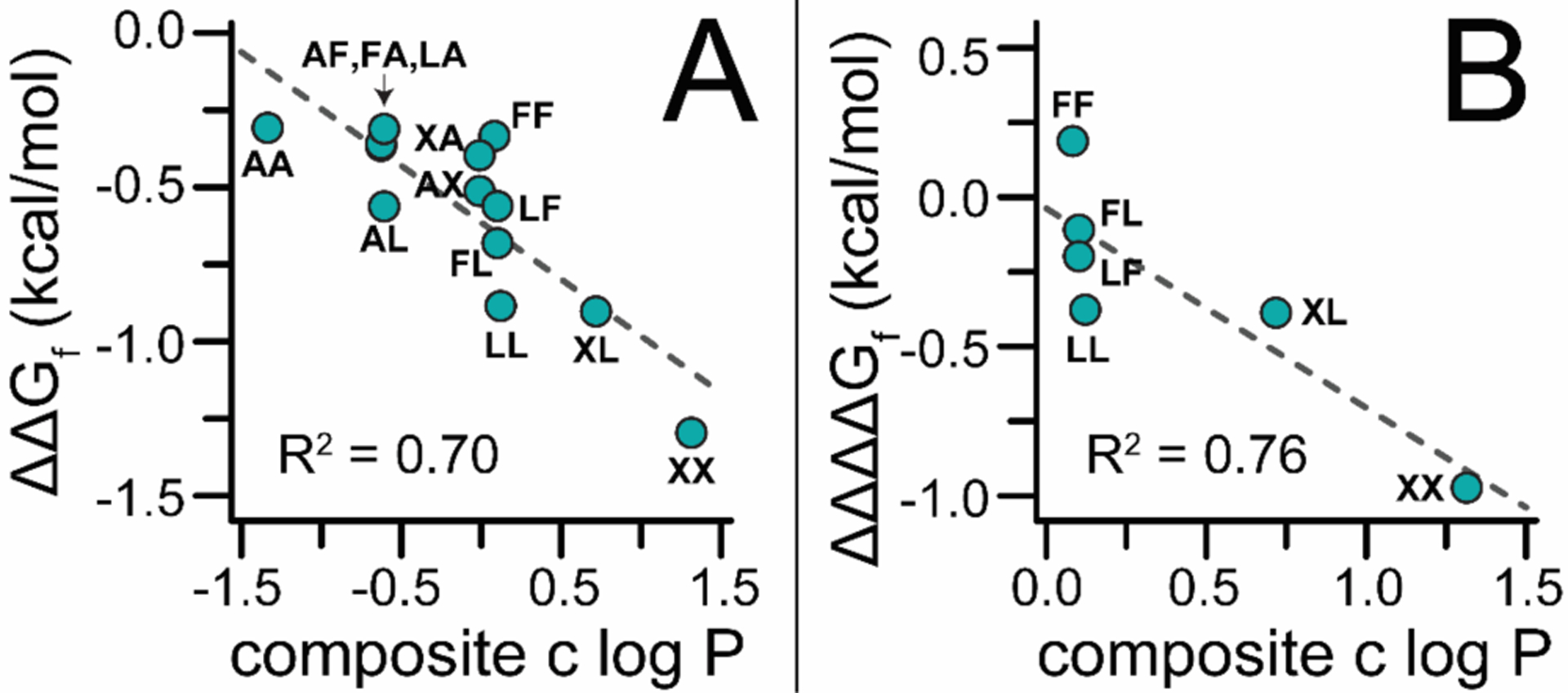
(A) Plot of the impact of PrF-PEGylation at position 23 the conformational stability of WW variants pAA, pLA, pAL, pLL, pFA, pAF, pFF, pXA, pAX, pXX, pXL, pFL, and pLF relative to their non-PEGylated counterparts (ΔΔGf) vs. the composite c log P for residues at positions 12 and 14. Dotted line is a fit of the data to a linear equation with the indicated R2 value. (B) Plot of the impact of PrF-PEGylation on the synergistic interaction between residues 12 and 14 (ΔΔΔΔGf) in variants pLL, pFF, pXX, pXL, pFL, and pLF vs. the composite c log P values for residues at these positions. Dotted line is a fit of the date to a linear equation with the indicated R2 value.
Next, we wondered whether these observations depended similarly on PEG-based strengthening of favorable synergistic interactions between residues at positions 12 and 14 as we observed above for pLL. Triple mutant box analysis of variant pXX (Table 2) reveals that PEGylation shifts the Cha12-Cha14 interaction from unfavorable (ΔΔΔGf = 0.56 ± 0.03 kcal/mol) to favorable (ΔΔΔGf = −0.41 ± 0.04 kcal/mol), a synergistic effect worth ΔΔΔΔGf = −0.97 ± 0.05 kcal/mol. Similarly, PEG increases the strength of already favorable interactions between Cha12 and Leu14 in pXL and between Phe12 and Leu14 in pFL (ΔΔΔΔGf = −0.39 ± 0.06 and −0.11 ± 0.05 kcal/mol, respectively). In contrast, PEGylation makes an unfavorable interaction in pLF between Leu12 and Phe14 less unfavorable (ΔΔΔΔGf = −0.20 ± 0.04 kcal/mol) and has a destabilizing effect on the interaction between Phe12 and Phe14 in pFF (ΔΔΔΔGf = 0.19 ± 0.04 kcal/mol). The variability among these ΔΔΔΔGf values correlates inversely with the composite c log P values for residues 12 and 14 (R2 = 0.77; slope = −0.67 ± 0.18 kcal/mol per log unit), indicating such that PEGylation better enhances the interaction between more hydrophobic residues (Figure 2B).
Many proteins have one or more surface-exposed patches of non-polar residues;32 our observations here suggest that PEGylation near such locations might be a useful strategy for increasing protein conformational stability. Moreover, it is interesting that variants pLL, pXX and pXL (Tm = 61.9, 63.2, and 60.8 °C, respectively) are each more stable than the parent WW domain from which they were derived (Tm = 58.0 °C), highlighting the possibility of simultaneously engineering a PEGylation site and a nearby surface-exposed hydrophobic patch in place of native residues to achieve superior conformational stability relative to the native sequence.
Experimental Section
Peptide Synthesis.
WW variants AA and pAA were synthesized previously.23 WW variants AL, LA, LL, AF, FA, FF, XA, AX, XX, XL, FL, and LF were prepared as the C-terminal acids via Fmoc-based solid-phase peptide synthesis on Fmoc-Gly-Wang resin (EMD Biosciences) as described previously.22,23,33 Fmoc-protected amino acids with acid-labile side-chain protecting groups were purchased from Advanced ChemTech and used without further purification; we also used previously synthesized N-[(9H-Fluoren-9-ylmethoxy)-O-2-propyn-1yl-L-tyrosine.33,34 Coupling reactions were carried out using 5 eq of the appropriate Fmoc-protected amino acid, 5 eq of 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), 5 eq of N-hydroxybenzotriazole hydrate (HOBt), and 10 eq of N,N-diisopropylethylamine in N-methyl-2-pyrrolidinone (NMP). Fmoc deprotection reactions were carried out using 20% piperidine in N,N-dimethylformamide (DMF). Following the final deprotection step, WW variants pAL, pLA, pLL, pAF, pFA, pFF, pXA, pAX, pXX, pXL, pFL, and pLF were prepared from AL, LA, LL, AF, FA, FF, XA, AX, XX, XL, FL, and LF via on-resin copper (I) catalyzed azide-alkyne cycloaddition using previously synthesized PEG-azide 13-azido-2,5,8,11-tetraoxatridecane.33,34
WW variants were cleaved from resin and side-chain protecting groups were globally removed by stirring the resin for 2–4 hr in a solution of phenol (0.0625 g), water (62.5 μL), thioanisole (62.5 μL), ethanedithiol (31 μL) and triisopropylsilane (12.5 μL) in trifluoroacetic acid (TFA, 1 mL). The TFA solution was drained from the resin and proteins were precipitated by addition of diethyl ether (~40 mL). Following centrifugation, the ether was decanted and the pellet was dissolved in ~40 mL 1:1 water/acetonitrile, then was flash frozen over dry ice in acetone and lyophilized to remove volatile impurities. The resulting powder was stored at −20°C until purification.
Proteins were purified by preparative reverse-phase high performance liquid chromatography (HPLC) on a C18 column using a linear gradient of acetonitrile in water with 0.1% v/v TFA. Fractions containing the desired protein product were pooled, frozen, and lyophilized. Proteins were identified by electrospray ionization time of flight mass spectrometry (ESI-TOF); expected and observed exact masses appear below. Protein purity was assessed by analytical HPLC on a C18 column using a 10–60% gradient of acetonitrile in water with 0.1% v/v TFA over 50 min at a flow rate of 1 mL/min. Mass spectra and HPLC chromatograms for all peptides are available in the Supporting Information.
Variant LL. Molecular formula C181H273N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 991.2633; observed [M+4H+]/4 = 991.2594. Retention time on analytical HPLC: 31.7 min.
Variant LA. Molecular formula C178H267N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 980.7516; observed [M+4H+]/4 = 980.7500. Retention time on analytical HPLC: 36.2 min.
Variant AL. Molecular formula C178H267N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 980.7516; observed [M+4H+]/4 = 980.7511. Retention time on analytical HPLC: 36.3 min.
Variant pLL. Molecular formula C190H292N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1049.5477; observed [M+4H+]/4 = 1049.5450. Retention time on analytical HPLC: 28.2 min.
Variant pLA. Molecular formula C187H286N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1039.0360; observed [M+4H+]/4 = 1039.0326. Retention time on analytical HPLC: 35.7 min.
Variant pAL. Molecular formula C187H286N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1039.0360; observed [M+4H+]/4 = 1039.0357. Retention time on analytical HPLC: 35.5 min.
Variant FF. Molecular formula C187H269N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 1008.2555; observed [M+4H+]/4 = 1008.2572. Retention time on analytical HPLC: 46.0 min.
Variant FA. Molecular formula C181H265N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 989.2476; observed [M+4H+]/4 = 989.2472. Retention time on analytical HPLC: 40.3 min.
Variant AF. Molecular formula C181H265N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 989.2476; observed [M+4H+]/4 = 989.2480. Retention time on analytical HPLC: 42.3 min.
Variant pFF. Molecular formula C196H288N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1066.5399; observed [M+4H+]/4 = 1066.5338. Retention time on analytical HPLC: 30.4 min.
Variant pFA. Molecular formula C190H284N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1047.5321; observed [M+4H+]/4 = 1047.5375. Retention time on analytical HPLC: 33.2 min.
Variant pAF. Molecular formula C190H284N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1047.5321; observed [M+4H+]/4 = 1047.5337. Retention time on analytical HPLC: 24.4 min.
Variant XX. Molecular formula C187H281N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 1011.2790; observed [M+4H+]/4 = 1011.2825. Retention time on analytical HPLC: 36.2 min.
Variant XA. Molecular formula C181H271N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 990.7594; observed [M+4H+]/4 = 990.7580. Retention time on analytical HPLC: 37.7 min.
Variant AX. Molecular formula C181H271N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 990.7594; observed [M+4H+]/4 = 990.7629. Retention time on analytical HPLC: 37.7 min.
Variant pXX. Molecular formula C196H300N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1069.5634; observed [M+4H+]/4 = 1069.5557. Retention time on analytical HPLC: 32.8 min.
Variant pXA. Molecular formula C190H290N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1049.0438; observed [M+4H+]/4 = 1049.0425. Retention time on analytical HPLC: 36.8 min.
Variant pAX. Molecular formula C190H290N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1049.0438; observed [M+4H+]/4 = 1049.0381. Retention time on analytical HPLC: 37.4 min.
Variant XL. Molecular formula C184H277N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 1001.2711; observed [M+4H+]/4 = 1001.2705. Retention time on analytical HPLC: 38.9 min.
Variant FL. Molecular formula C184H271N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 999.7594; observed [M+4H+]/4 = 999.7586. Retention time on analytical HPLC: 37.7 min.
Variant LF. Molecular formula C184H271N51O48S; ESI-TOF MS: expected [M+4H+]/4 = 999.7594; observed [M+4H+]/4 = 999.7577. Retention time on analytical HPLC: 37.9 min.
Variant pXL. Molecular formula C193H296N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1059.5555; observed [M+4H+]/4 = 1059.5534. Retention time on analytical HPLC: 38.3 min.
Variant pFL. Molecular formula C193H290N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1058.0438; observed [M+4H+]/4 = 1058.0419. Retention time on analytical HPLC: 36.5 min.
Variant pLF. Molecular formula C193H290N54O52S; ESI-TOF MS: expected [M+4H+]/4 = 1058.0438; observed [M+4H+]/4 = 1058.0412. Retention time on analytical HPLC: 37.1 min.
CD Measurements.
Measurements were made with an Aviv 420 Circular Dichroism Spectropolarimeter, using quartz cuvettes with a path length of 0.1 cm. Solutions of each WW variant were prepared in 20 mM sodium phosphate buffer, pH 7; solution concentrations were determined spectroscopically based on tyrosine and tryptophan absorbance at 280 nm in 6 M guanidine hydrochloride + 20 mM sodium phosphate (εTrp = 5690 M−1cm−1, εTyr = 1280 M−1cm−1).35 CD spectra of 50 μM solutions were obtained from 260 to 200 nm at 25°C.
Variable temperature CD data were obtained at least in triplicate (one sample was made and then aliquoted into three different cuvettes) by monitoring the molar ellipticity [θ] at 227 nm of 50 μM solutions of each WW variant in 20 mM sodium phosphate (pH 7) from 1 to 95 °C, at 2 °C intervals, with 120 s equilibration time between data points and 30 s averaging time. Triplicate variable temperature CD data for each peptide were fit globally to a two-state model for thermally induced unfolding. We used nonlinear least-squares regression to fit the three data sets globally to equations derived from the two-state folding model of folding for each variant to obtain a melting temperature Tm and temperature-dependent folding free energy ΔGf for each variant, as described in detail in the Supporting Information, which also contains statistics for the fits, including R2 and sum of the square residuals.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health Grant R15 GM116055-01.
Footnotes
Supporting Information
Mass spectra, HPLC chromatograms, CD spectra, global fits of variable temperature CD data, and statistics for all fits.
References
- (1).Abuchowski A; Vanes T; Palczuk NC; Davis FF “Alteration of Immunological Properties of Bovine Serum-Albumin by Covalent Attachment of Polyethylene-Glycol.”J. Biol. Chem 1977, 252, 3578. [PubMed] [Google Scholar]
- (2).Abuchowski A; Mccoy JR; Palczuk NC; Vanes T; Davis FF “Effect of Covalent Attachment of Polyethylene-Glycol on Immunogenicity and Circulating Life of Bovine Liver Catalase.”J. Biol. Chem 1977, 252, 3582. [PubMed] [Google Scholar]
- (3).Harris JM; Chess RB “Effect of pegylation on pharmaceuticals.”Nat. Rev. Drug Discov 2003, 2, 214. [DOI] [PubMed] [Google Scholar]
- (4).Frokjaer S; Otzen DE “Protein drug stability: A formulation challenge.”Nat Rev Drug Discov 2005, 4, 298. [DOI] [PubMed] [Google Scholar]
- (5).Fishburn CS “The pharmacology of PEGylation: Balancing PD with PK to generate novel therapeutics.”J. Pharm. Sci 2008, 97, 4167. [DOI] [PubMed] [Google Scholar]
- (6).Veronese FM; Mero A “The Impact of PEGylation on Biological Therapies.”Biodrugs 2008, 22, 315. [DOI] [PubMed] [Google Scholar]
- (7).PEGylated Protein Drugs: Basic Sience and Clinical Applications; Veronese FM, Ed.; Birkhauser Verlag: Basel, 2009. [Google Scholar]
- (8).Jevsevar S; Kunstelj M; Porekar VG “PEGylation of therapeutic proteins.”Biotechnol. J 2010, 5, 113. [DOI] [PubMed] [Google Scholar]
- (9).Dawson PE; Kent SBH “SYNTHESIS OF NATIVE PROTEINS BY CHEMICAL LIGATION 1.”Annu. Rev. Biochem 2000, 69, 923. [DOI] [PubMed] [Google Scholar]
- (10).Rosendahl MS; Doherty DH; Smith DJ; Carlson SJ; Chlipala EA; Cox GN “A Long-Acting, Highly Potent Interferon α-2 Conjugate Created Using Site-Specific PEGylation.”Bioconjugate Chem. 2005, 16, 200. [DOI] [PubMed] [Google Scholar]
- (11).Dirksen A; Dawson PE “Expanding the scope of chemoselective peptide ligations in chemical biology.”Curr. Opin. Chem. Biol 2008, 12, 760—766. [DOI] [PubMed] [Google Scholar]
- (12).Brocchini S; Godwin A; Balan S; Choi J.-w.; Zloh M; Shaunak S “Disulfide bridge based PEGylation of proteins.”Adv. Drug Deliv. Rev 2008, 60, 3. [DOI] [PubMed] [Google Scholar]
- (13).Van de Vijver P; Suylen D; Dirksen A; Dawson PE; Hackeng TM “N-epsilon-(Thiaprolyl)-lysine as a Handle for Site-Specific Protein Conjugation.” Biopolymers 2010, 94, 465. [DOI] [PubMed] [Google Scholar]
- (14).Cho H; Daniel T; Buechler YJ; Litzinger DC; Maio Z; Putnam AM; Kraynov VS; Sim BC; Bussell S; Javahishvili T; Kaphle S; Viramontes G; Ong M; Chu S; Becky GC; Lieu R; Knudsen N; Castiglioni P; Norman TC; Axelrod DW; Hoffman AR; Schultz PG; DiMarchi RD; Kimmel BE “Optimized clinical performance of growth hormone with an expanded genetic code.”Proc. Natl. Acad. Sci. USA 2011, 108, 9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tada S; Andou T; Suzuki T; Dohmae N; Kobatake E; Ito Y “Genetic PEGylation.”Plos One 2012, 7, e49235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Levine PM; Craven TW; Bonneau R; Kirshenbaum K “Semisynthesis of Peptoid-Protein Hybrids by Chemical Ligation at Serine.”Org. Lett 2014, 16, 512. [DOI] [PubMed] [Google Scholar]
- (17).Nicholls A; Sharp KA; Honig B “Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons.”Proteins 1991, 11, 281. [DOI] [PubMed] [Google Scholar]
- (18).Lins L; Brasseur R “The hydrophobic effect in protein folding.”FASEB J. 1995, 9, 535. [DOI] [PubMed] [Google Scholar]
- (19).Dill KA “The Meaning of Hydrophobicity.”Science 1990, 250, 297. [DOI] [PubMed] [Google Scholar]
- (20).Dill KA “Dominant Forces in Protein Folding.”Biochemistry 1990, 29, 7133. [DOI] [PubMed] [Google Scholar]
- (21).Dill KA; Bromberg S; Yue KZ; Fiebig KM; Yee DP; Thomas PD; Chan HS “Principles of Protein-Folding - a Perspective from Simple Exact Models.”Protein Sci. 1995, 4, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lawrence PB; Gavrilov Y; Matthews SS; Langlois MI; Shental-Bechor D; Greenblatt HM; Pandey BK; Smith MS; Paxman R; Torgerson CD; Merrell JP; Ritz CC; Prigozhin MB; Levy Y; Price JL “Criteria for Selecting PEGylation Sites on Proteins for Higher Thermodynamic and Proteolytic Stability.”J. Am. Chem. Soc 2014, 136, 17547. [DOI] [PubMed] [Google Scholar]
- (23).Xiao Q; Draper SRE; Smith MS; Brown N; Pugmire NAB; Ashton DS; Carter AJ; Lawrence EEK; Price JL “Influence of PEGylation on the Strength of Protein Surface Salt Bridges.”ACS Chem. Biol 2019, 14, 1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Horovitz A; Fersht AR “Strategy for Analyzing the Cooperativity of Intramolecular Interactions in Peptides and Proteins.”J. Mol. Biol 1990, 214, 613. [DOI] [PubMed] [Google Scholar]
- (25).Luisi DL; Snow CD; Lin JJ; Hendsch ZS; Tidor B; Raleigh DP “Surface salt bridges, double-mutant cycles, and protein stability: an experimental and computational analysis of the interaction of the Asp 23 side chain with the N-terminus of the N-terminal domain of the ribosomal protein L9.”Biochemistry 2003, 42, 7050. [DOI] [PubMed] [Google Scholar]
- (26).Price JL; Powers DL; Powers ET; Kelly JW “Glycosylation of the Enhanced Aromatic Sequon is Similarly Stabilizing in Three Distinct Reverse Turn Contexts.”Proc. Natl. Acad. Sci. USA 2011, 108, 14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Price JL; Powers ET; Kelly JW “N-PEGylation of a Reverse Turn is Stabilizing in Multiple Sequence Contexts unlike N-GlcNAcylation.”ACS Chem. Biol 2011, 6, 1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Smith MS; Billings WM; Whitby FG; Miller MB; Price JL “Enhancing a long-range salt bridge with intermediate aromatic and nonpolar amino acids.”Org. Biomol. Chem 2017, 15, 5882. [DOI] [PubMed] [Google Scholar]
- (29).Chao SH; Matthews SS; Paxman R; Aksimentiev A; Gruebele M; Price JL “Two Structural Scenarios for Protein Stabilization by PEG.”J. Phys. Chem. B 2014, 118, 8388. [DOI] [PubMed] [Google Scholar]
- (30).Pandey BK; Enck S; Price JL “Stabilizing Impact of N-Glycosylation on the WW Domain Depends Strongly on the Asn-GlcNAc Linkage.”ACS Chem. Biol 2013, 8, 2140. [DOI] [PubMed] [Google Scholar]
- (31).Chen WT; Enck S; Price JL; Powers DL; Powers ET; Wong CH; Dyson HJ; Kelly JW “Structural and Energetic Basis of Carbohydrate-Aromatic Packing Interactions in Proteins.”J Am Chem Soc 2013, 135, 9877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lijnzaad P; Berendsen HJ; Argos P “Hydrophobic patches on the surfaces of protein structures.” Proteins 1996, 25, 389. [DOI] [PubMed] [Google Scholar]
- (33).Draper SRE; Lawrence PB; Billings WM; Xiao Q; Brown NP; Becar NA; Matheson DJ; Stephens AR; Price JL “Polyethylene Glycol Based Changes to beta-Sheet Protein Conformational and Proteolytic Stability Depend on Conjugation Strategy and Location.”Bioconjugate Chem. 2017, 28, 2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lawrence PB; Billings WM; Miller MB; Pandey BK; Stephens AR; Langlois MI; Price JL “Conjugation Strategy Strongly Impacts the Conformational Stability of a PEG-Protein Conjugate.”ACS Chem. Biol 2016, 11, 1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Edelhoch H “Spectroscopic Determination of Tryptophan and Tyrosine in Proteins.”Biochemistry 1967, 6, 1948. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.