

Supplemental Digital Content is available in the text.
Keywords: coronary artery disease, cytokines, flow cytometry, furin, pulmonary artery
Objective:
Patients with coronary artery disease (CAD) are at increased risk for cardiac death and respiratory failure following severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Platelets are crucially involved in pathogenesis of CAD and might also contribute to pathophysiology of SARS-CoV-2 infection.
Approach and Results:
We enrolled a cohort of 122 participants from February 2020 to July 2020 including 55 patients with preexisting CAD and acute SARS-CoV-2 infection (CAD-SARS-CoV-2positive), 28 patients with CAD and without SARS-CoV-2 (CAD-SARS-CoV-2negative), and 39 healthy controls. Clinical and cardiac examination of the CAD-SARS-CoV-2positive group included blood sampling, echocardiography, and electrocardiography within 24 hours after hospital admission. Phenotyping of platelets was performed by flow cytometry; plasma levels of chemokines were analyzed by ELISA. Respiratory failure of patients was stratified by the Horovitz index as moderately/severely impaired when Horovitz index <200 mm Hg. The clinical end point was defined as Horovitz index <200 mm Hg with subsequent mechanical ventilation within a follow-up of 60 days. CAD-SARS-CoV-2positive patients display a significant enhanced platelet activation and hyper-inflammation early at time of hospital admission. Circulating platelet/leukocyte co-aggregates correlate with plasma levels of cytokines/chemokines like IL (interleukin)-6, CCL2, and CXCL10 (chemokine [C-X-C motif] ligand) as well as activation of platelets is associated with CCL5 and elevation of pulmonary artery pressure. Furthermore, furin is stored and released from activated platelets. High furin plasma levels are associated with poor clinical prognosis in CAD-SARS-CoV-2positive patients.
Conclusions:
Patients with CAD and SARS-CoV-2 infection exhibit elevated systemic platelet activation and enhanced plasma levels of the subtilisin-like proprotein convertase furin, which may contribute to an unfavorable clinical prognosis.
Highlights.
Significant changes in platelet activation, platelet/leukocyte co-aggregation, and plasma levels of cytokines and chemokines can already be detected in the very early phase of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) associated infection of patients with coronary artery disease.
Plasma levels of the subtilisin-like proprotein convertase furin released from activated platelets serve as independent prognostic factor for progression of respiratory failure in SARS-CoV-2 associated infection of patients with coronary artery disease.
Platelets, platelet activation, and platelet-derived mediators play an important role in the immuno-defense of SARS-CoV-2 infection in patients with coronary artery disease.
Early phenotyping of platelets and platelet/leucocytes co-aggregates using simple flow cytometry along with simple analysis of furin plasma levels might predict worsening of respiratory failure at an early stage of SARS-CoV-2 infection in high-risk coronary artery disease patients.
Targeting the release of platelet-derived furin may be a potential treatment strategy to interfere in the early course of disease progression of SARS-CoV-2 associated infection.
Platelet activation and release of inflammatory mediators play a critical role in inflammation, atherosclerosis, and in infectious diseases.1,2 Platelets interact with a variety of pathogens including bacteria and viruses.2 Virus infection is often associated with systemic inflammation and changes in hemostasis (eg, platelet activation, thrombocytopenia) and coagulation (triggered by thrombin activation among others).3 Virus-induced respiratory tract infection like an acute severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is associated with platelet activation and characteristic coagulopathy resulting in thrombotic complications including deep vein thrombosis and pulmonary embolism.4–6 In the case of RNA virus infections such as influenza, platelet activation and formation of platelet/leukocyte co-aggregates may occur early in the course of disease progression7,8 followed by enhanced activation of coagulation and thrombin generation.9 Enhanced platelet activation and formation of circulating platelet/leukocyte co-aggregates was additionally observed in SARS-CoV-2 infected patients10 which is associated with a worse clinical outcome of patients with coronavirus disease 2019 (COVID-19).11 The modulation of the renin-angiotensin-aldosterone system plays also an important role in this context.12 Renin-angiotensin-aldosterone system is a cascade of peptides, which coordinate important processes in human physiology metabolizing angiotensin II (a potent vasoconstrictor) to generate angiotensin (1–7; a vasodilator). Renin-angiotensin-aldosterone system can interface with SARS-CoV-2 via ACE-2 (angiotensin-converting enzyme 2).13,14 Therefore, ACE-2 has been described as key component during SARS-CoV-2 infections. ACE-2 is expressed on the surface of multiple immune cells like monocytes and macrophages as well as in respiratory and vascular endothelial cells and can enable viral entry into cells by binding to SARS-CoV-2 spike protein domains.12,15,16 Recently, it was demonstrated, that ACE-2 is also expressed on the surface of platelets and thereby trigger thrombotic events in patients infected with SARS-CoV-2.17 Enhanced ACE-2 expression may explain the predisposition to severe disease in certain patient groups, for example, patients with cardiovascular disease. Furthermore, viral attachment of SARS-CoV-2 is facilitated by various membrane and circulating proteases and convertases, such as the subtilisin-like proprotein convertase furin, minting severe clinical manifestations during infection.18–21 Furin is expressed ubiquitously and regulates a variety of physiological functions and thus is involved in the blood-clotting and complement system as well as in the cleavage of membrane receptors, viral-envelope glycoproteins, and bacterial exotoxins.22 Furin has been described to play a pivotal role in cardiovascular disease like coronary artery disease (CAD) and cardiovascular risk factors like diabetes. A very recent publication also shows that furin plasma levels are associated with all-cause mortality and cardiovascular events in patients with acute myocardial infarction.23 Additionally, furin was linked to atheroprogression. In advanced atherosclerotic plaques, furin is expressed primarily in macrophages. CAD-associated variants of furin were identified, and these variants altered furin expression are affecting monocyte/macrophage behavior.24–29 But the role of platelet-derived furin has yet not been investigated thoroughly. A preexisting cardiovascular disease is linked to worse outcomes and increased risk of death in patients with COVID-19, whereas COVID-19 itself can also induce myocardial injury, arrhythmia, acute coronary syndrome, perimyocarditis, and venous thromboembolism. CAD, along with attributable other comorbidities like diabetes, has been identified as high-risk condition in patients with COVID-19 and is associated with all-cause mortality, a high risk for severe respiratory failure and organ failure following SARS-CoV-2 infection.30–35 Concomitant CAD is present in ≈1 out of 10 patients hospitalized for COVID-19 but age and gender also are important risk factors.36 Mechanisms underlying COVID-19-induced acute coronary syndrome might involve plaque rupture, coronary spasm or microthrombi owing to systemic inflammation or cytokine storm.33,37 Potential drug–disease interactions affecting patients with COVID-19 and comorbid cardiovascular diseases are also becoming a serious concern and should be accounted for.35 Finally, impaired cardiac function is associated with poor prognosis in COVID-19 positive patients. Consequently, treatment of these patients should include careful guideline-conform cardiovascular evaluation and treatment.30 There is accumulating evidence that SARS-CoV-2 associated respiratory distress is associated with a hyper-thrombotic state and hyper-inflammation.38,39 Further, platelets exhibit local and systemic thrombo-inflammatory activities and are able to alter immune cell functions inducing plaque formation in CAD. They function as connection between local inflammatory responses at vascular wall and development of an atherogenetic milieu.40,41 Further, it could be shown that platelet survival is shortened in patients with CAD.42
We hypothesized that changes in platelet activity and systemic thrombo-inflammation at an early stage of SARS-CoV-2 infection is of prognostic relevance for the course of the disease. We found that significant changes in platelet activation, platelet/leukocyte co-aggregation, and plasma levels of cytokines and chemokines occur initial in the early phase of SARS-CoV-2 associated infection. We identified the plasma levels of furin released from activated platelets as critical prognostic factor for progression of respiratory failure. Targeting release of platelet-derived furin may be a potential target to interfere in the course of the disease progression.
Materials and Methods
Because of the sensitive nature of the data collected for this study, requests to access the data set from qualified researchers trained in human subject confidentiality protocols may be sent to Carolin Langnau at University Hospital Tuebingen, Department of Cardiology and Angiology, Eberhard Karls University Tuebingen, Germany.
Study Design, Study Populations, and Inclusion Criteria
In this study, 122 participants were consecutively enrolled at the Department of Cardiology and Angiology of the University Hospital Tübingen, Germany from February 2020 until July 2020. Fifty-five patients with preexisting CAD and an acute SARS-CoV-2 infection were included within 24 hours after hospital admission (CAD-SARS-CoV-2positive). Twenty-eight patients with CAD but without SARS-CoV-2 infection (CAD-SARS-CoV-2negative) were included in the study as well as 39 healthy controls. All patients received a clinical and cardiac examination including echocardiography, electrocardiography, concomitant medication, comorbidities and blood sampling for routine laboratory parameters, marker expression on platelets, and chemokine profiling. SARS-CoV-2 infection was confirmed by real-time reverse transcriptase-polymerase chain reaction from nasopharyngeal secretions. CAD was determined by coronary angiography and was characterized as >25% to 50% stenosis of one or more coronary vessels.
Inclusion criteria of our study were age older than 18 years, confirmed CAD and subsequent SARS-CoV-2 infection. Exclusion criteria were other microbial infections. The study was approved by the local ethics committee (240/2018B02) and complies with the declaration of Helsinki and the good clinical practice guidelines on the approximation of the laws, regulations, and administrative provisions of the member states relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. Written informed consent was obtained from every patient.
We determined NT-proBNP (N-terminal pro-B-type natriuretic peptide, >300 ng/L), hs TNI (high sensitive troponin I, >37 ng/L), and CRP (C-reactive protein, >0.5 mg/dL) as elevated laboratory markers of myocardial and inflammatory distress. As echocardiographic parameters, the left and right ventricular function, right ventricular dilatation, presence of tricuspid valve regurgitation, and pericardial effusion were included according to current guidelines.43,44
Clinical Follow-Up
The clinical study end point was characterized as rapidly progressive respiratory failure with indication to mechanical ventilation. Respiratory failure was defined by a moderately to severely impaired Horovitz index (moderately impaired HI defined as 101–200 mm Hg, severely impaired HI defined as ≤100 mm Hg). The occurrence of the clinical study end point HI <200 mm Hg was obtained during a follow-up period of 60 days.
Flow Cytometry Staining
One hundred twenty-two blood samples of patients with CAD (with or without SARS-CoV-2 infection) and healthy controls were collected in citrat-phosphat-dextrose-adenin monovette for flow cytometry to characterize platelets and platelet/leukocyte co-aggregates. Platelets were analyzed in 5 µL whole blood, diluted 1:10 in HEPES Tyrode buffer (pH 7.4) in a 96 well plate. To avoid unspecific binding, the Fc part of cellular receptors was blocked using human IgG (0.01 mg/mL; Sigma Aldrich Co St Luis, MO) and incubated for 20 minutes at 4 °C. Extracellular staining of platelets was performed for 20 minutes at 4 °C using following fluorochrome-conjugated antibodies: CD41 PaBl (clone HIP8), CD42b PerCP-Cy5.5 (clone HIP1), CD62P PE-Cy7 (clone AK4), CD61 FITC (clone VI-PL2), and CD31 BV711 (clone WM59; BioLegend, San Diego, California). For life/dead staining Zombie NIR (BioLegend, San Diego, California) was used and performed together with the extracellular antibody staining in one master mix tube. Zombie NIR from BioLegend is an amine-reactive fluorescent dye which is permeant to the cells with compromised membranes in contrast to nonpermeant live cells. Low-temperature incubations did not influence platelet activation, tested by measurement of CD62P surface expression (data not shown). Cells were fixed overnight with FoxP3/transcription factor staining buffer set containing formaldehyde (Thermo Fisher Scientific, Waltham, MA) according to manufacturer’s instructions. After each staining, cells were washed with PBS containing 1% FCS, 2 mmol/L EDTA, and 1% sodium azide. At least 100 000 cells were acquired using a LSR Fortessa flow cytometer with the DIVA software and were further analyzed using FlowJo 10.6.2 software (all from BD Biosciences, Franklin Lakes, New Jersey). Platelets and platelet/leukocyte co-aggregates were characterized according their size and granularity. All samples were gated as follows: time/singlets/platelets/LiveDead FSC-low or FSC-high. Platelets (blue plots) were gated as follows: FSC-low/CD41+CD42b+. Platelet/leukocyte co-aggregates (green plots) were gated as follows: FSC-high/CD42b+CD61+ and for highly activated CD42b+ platelet/leukocyte co-aggregates expression of CD41+CD61+CD62P+ was measured (Figure IA in the Data Supplement). Compensation was measured and calculated by FACS DIVA software. Surface marker expression was quantified as median fluorescence intensity and frequency of living cells. Fluorescence minus one controls were used for gating on marker positive platelets.
Activation of Platelets and Flow Cytometry Staining
Blood samples of healthy controls were collected in citrat-phosphat-dextrose-adenin monovette to quantify furin surface expression upon activation. Platelet-rich plasma was prepared by centrifugation for 20 minutes at 210g without a break. For each sample, 1×104 platelets were incubated with either 5 µmol/L ADP, 10 µmol/L TRAP (thrombin receptor activating peptide; TRAP6, F. HOFFMANN LA-ROCHE AG, BASEL, SWITZERLAND), 1 µg/mL CRP (CRP-XL, CambCol, Cambridge, United Kingdom), or 1 U/mL Thrombin (F. Hoffmann La-Roche AG, Basel, Switzerland). Platelet surface staining was performed with the following fluorochrome-conjugated antibodies: CD62P PE (clone CLBThromb/6, Beckman-Coulter, Brea, CA) and furin AF488 (clone 222722, R&D Systems, Minneapolis, MN) for 30 minutes at room temperature in combination with the agonists. Platelets were fixed with 0.5% formaldehyde for 30 minutes in the dark. To measure the effects of the PAR-1 (protease-activated receptor-1) inhibitor ML161, samples were preincubated for 5 minutes with 50 µmol/L ML161 at RT before activation and staining for 30 minutes as described above. Measurements were performed using a Calibur (BD Biosciences, Franklin Lakes, NJ) and DIVA software (BD Biosciences, Franklin Lakes, NJ) followed by data analysis with FlowJo software V.10.6.2 (BD Biosciences, Franklin Lakes, NJ).
Imaging Flow Cytometry
Imaging flow cytometry was performed of CAD-SARS-CoV-2negative patients in the same way as described above for the flow cytometry staining of platelets. Following fluorochrome-conjugated antibodies were used: CD42b PerCP-Cy5.5 (clone HIP1) and CD14 FITC (clone M5E2) from BioLegend, San Diego, CA. For live/dead staining, Zombie Aqua (BioLegend, San Diego, California) was included. For gating of platelet/leukocyte co-aggregates CD14 and CD42b surface expression was performed. Platelet/leukocyte co-aggregates were gated as follows: focus/cells/lymphocytes/live/CD14+/CD42b+/platelet-monocyte-complex (Figure IB in the Data Supplement). Measurements were performed with an Amnis Image Stream MK II (Luminex, Austin, TX), and data analysis was done with the software IDAS Version 6.2.
Determination of Plasma Levels of Cytokines/Chemokines (LEGENDPlex)
To quantify the concentrations of several chemokines and cytokines in human plasma, a LEGENDPlex inflammation panel 1 (BioLegend, San Diego, CA) and LEGENDPlex proinflammatory chemokine panel (BioLegend, San Diego, CA) were performed. Only 79 frozen plasma samples were available and could be analyzed, consisting of 19 CAD-SARS-CoV-2negative, 31 CAD-SARS-CoV-2positive patients, and 29 healthy controls. The assays were performed according to the manufacturers’ manual. FACS Lyric (BD Biosciences, Franklin Lakes, NJ) was used for the measurement and data analysis was performed with the LEGENDPlex Data Analysis Software (BioLegend, San Diego, CA).
Isolation of Human Platelets
Human washed platelets were isolated as previously described.45 Washed platelets were resuspended in HEPES Tyrode buffer (pH 7.4, supplemented with 1 mmol/L CaCl2) or lysed in RIPA lysis buffer for further experiments. For activation, isolated platelets were stimulated with CRP (CRP-XL, CambCol, Cambridge, United Kingdom), TRAP (TRAP6, F. Hoffmann La-Roche AG, Basel, Switzerland), or thrombin (F. Hoffmann La-Roche AG, Basel, Switzerland). Individual concentrations and exposure times are indicated within figures and legends.
Generation of Supernatant From Activated Platelets
To gain activated platelet supernatant, washed platelets were activated by addition of 1 U/mL thrombin (F. Hoffmann La-Roche AG, Basel, Switzerland) and incubated at room temperature for 30 minutes each. Afterwards, samples were centrifuged at 340g for 5 minutes, and the supernatants were used for further experiments. The protein concentration of the activated platelet supernatant was determined by a standard Bradford Assay.46
Determination of Plasma Levels of Furin
To quantify furin concentration in human plasma, a furin human ELISA Kit (R&D Systems, Minneapolis, MN) was performed. Frozen plasma samples of CAD-SARS-CoV-2negative (n=20), CAD-SARS-CoV-2positive patients (n=35), and healthy controls (n=28) were measured.
To quantify platelet furin levels, we used lysates from resting human platelets and activated platelet supernatant from platelets activated with 1 U/mL thrombin (F. Hoffmann La-Roche AG, Basel, Switzerland). Each sample was prepared from a suspension containing 2×109 washed platelets/mL. The assays were performed according to the manufacturer manuals.
Immunofluorescence Microscopy
Isolated human platelets in PBS (4×108 platelets/mL) were supplemented with 1 mmol/L CaCl2, activated with 1 µg/mL CRP (CRP-XL CambCol, Cambridge, United Kingdom), and incubated on poly-L-Lysin coated coverslips for 30 minutes at room temperature. The platelets were fixated with 2% formaldehyde and blocked with BSA. The samples were stained with anti-mouse furin AF488 conjugated antibody (Mouse monoclonal IgG2B, clone 222722 R&D Systems, Minneapolis, MN). An IgG2B antibody (Mouse monoclonal IgG2B Isotype control, clone 20116, R&D Systems, Minneapolis, MN) with an AF568 secondary antibody (A1161, invitrogen) was used as isotype control. The coverslips were mounted onto slides and several images from randomly selected areas were taken (Nikon Eclipse Ti2-A, 100×DIC [differential interference contrast] objective). The images were analyzed with the NIS-Elements AR software (Nikon, Japan).
Furin Activation Assay
To analyze the furin enzyme activity of platelets, the fluorogenic peptide substrate pERTKR-AMC Substrate (100 µmol/L; R&D Systems, Minneapolis, MN) was combined with 200 ng furin or isolated platelets (5×107/mL) stimulated with CRP (CRP-XL CambCol, Cambridge, United Kingdom), TRAP (TRAP6, F. HOFFMANN LA-ROCHE AG, BASEL, SWITZERLAND) or thrombin (F. Hoffmann La-Roche AG, Basel, Switzerland).47,48 Additionally, experiments were performed in the same manner with isolated platelets (1×109/mL) diluted in Tyrode buffer (pH 7.4) or platelet-poor plasma derived from the donor of the sample by 10 minutes centrifugation of PRP at 2500g. The resulting fluorescent signal of the digested peptide was immediately detected over 60 minutes using a plate reader (380/460 nm; Glomax, Promega, Madison, WI).
Statistical Analysis
Statistical analysis of participants’ clinical and laboratory baseline characteristics in relation to measured platelet phenotypes, and marker expression was performed. Non-normally distributed continuous data are represented as median with interquartile range (IQR) and normally distributed continuous data are represented as mean with SD. Two group comparisons for non-normally distributed continuous variables were performed using a Mann-Whitney U test while normally distributed continuous variables were compared using Student t test. One-Way ANOVA was performed for >2 group comparison and for post hoc analysis and multiple comparison Dunnett test was used. Categorical variables are represented as total numbers, and proportions of participants and comparison was performed using χ2 test. Survival curves of patients were calculated by Kaplan-Meier analyses and compared using the log-rank test. Correlation analysis was calculated by Spearman rank correlation coefficient. Cox proportional-hazards regression analysis was performed for multivariable analysis to assess furin plasma level association with progressive respiratory failure (HI <200 mm Hg). Volcano plots were performed with JMP Version 15.0. Each data point was tested using Mann-Whitney U test. Comparisons were considered statistically significant if 2-sided P value was ≤0.050. All statistical analysis was performed with IBM SPSS Statistics software version 26 (SPSS, Inc) and GraphPad Prism Version 8.4.0 (GraphPad Software).
Results
Demographic and Clinical Characteristics of Patients With CAD and SARS-CoV-2-Associated Respiratory Failure
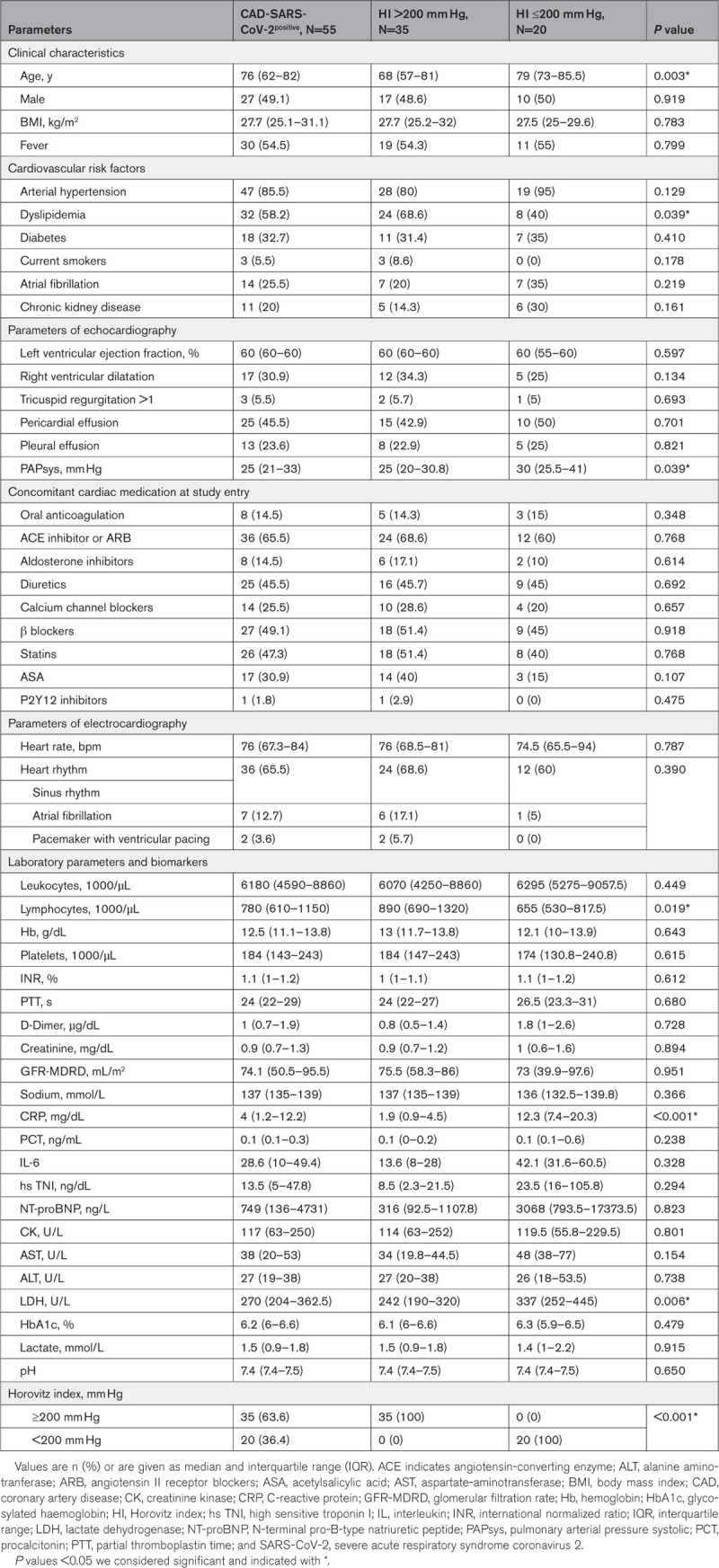
We prospectively studied a consecutive cohort of 55 patients in spring 2020 during the first wave of symptomatic patients with CAD that were admitted to our hospital for suspected respiratory infection and found to be positive for SARS-CoV-2 (CAD-SARS-CoV-2positive; Table 1). Twenty-eight patients with symptomatic CAD without any signs of respiratory infection and with a negative reverse transcriptase-polymerase chain reaction test for SARS-CoV-2 (CAD-SARS-CoV-2negative) were matched by age, gender, and presence of stable CAD on admission to the CAD-SARS-CoV-2positive group (Table 1). The median age of the population was 63.5 (IQR, 47–78) years; 57 (46.7%) patients were men (Table 1). About one-fourth (21.8%) of CAD-SARS-CoV-2positive patients showed a BMI ≥30 compared with 10.7% of CAD-SARS-CoV-2negative patients and to 10.3% of healthy control group (P=0.045). Pericardial effusion was significantly more often present in the CAD-SARS-CoV-2positive group, possible mirroring concomitant perimyocarditis (P<0.001; Table 1). Interestingly, there were no significant differences of D-dimer levels between groups upon admission (P=0.882). Progressive respiratory failure as indicated by an HI of ≥200 mm Hg was found in 35 (63.6%) of CAD-SARS-CoV-2positive patients. Out of 55 patients, 22 (36.4%) required mechanical ventilation in the time course during their hospital stay (Table 2).
Table 1.
Baseline Characteristics of Patient Population
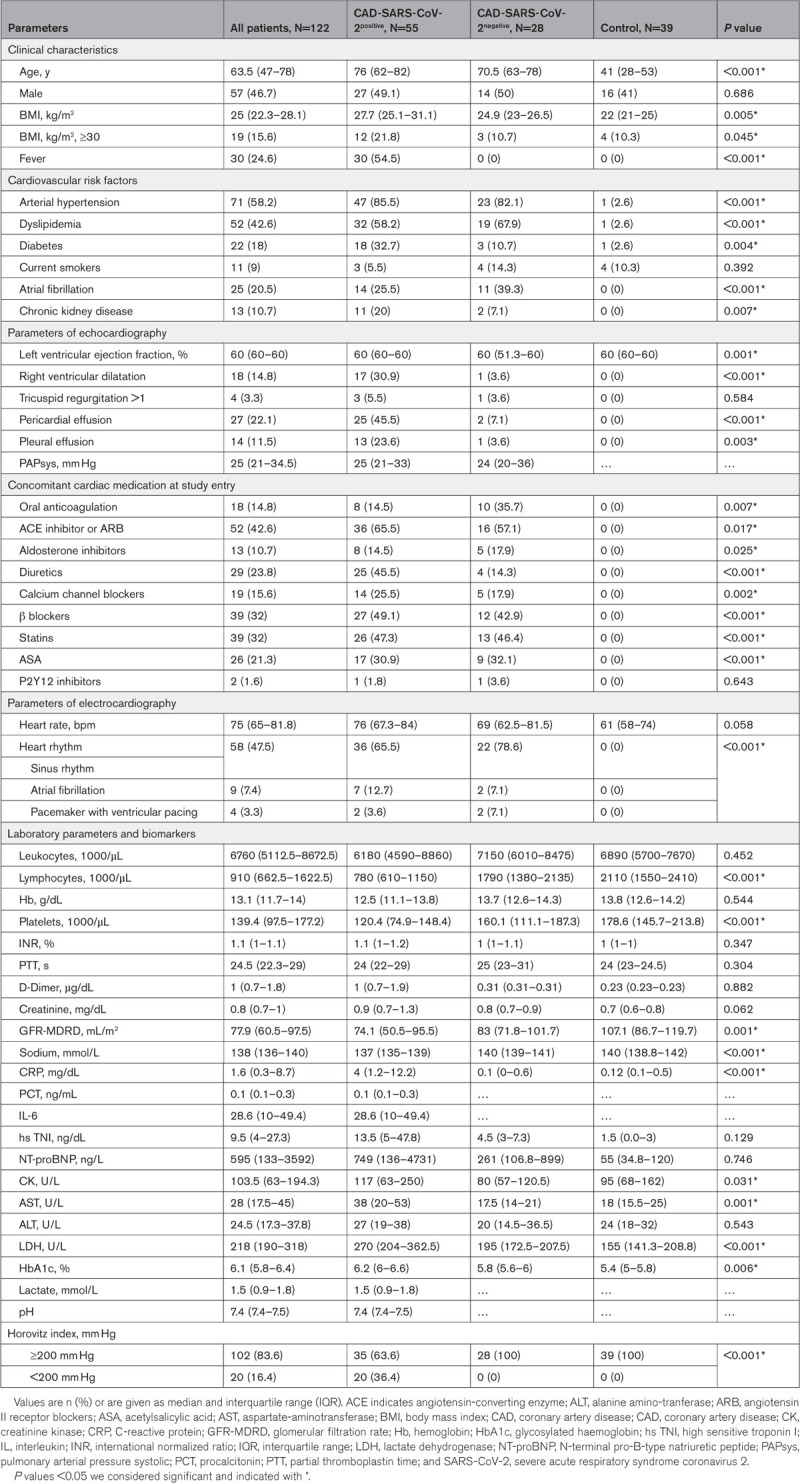
Table 2.
Baseline Characteristics of Patient Population Stratified by the Occurrence of the Clinical End Point (Progressive Respiratory Failure Defined by HI <200 mm Hg and Mechanical Ventilation)
CAD-SARS-CoV-2positive Patients Show Significantly Higher Systemic Platelet Activation and Hyper-Inflammation
At time of hospital admission all patients received a prespecified complete clinical assessment and evaluation of ECG, echocardiography, and extensive routine laboratory parameters including troponin I, NT-proBNP, and D-dimers. Blood samples were characterized by flow cytometric platelet activation parameters and cytokine/chemokine plasma profiling.
CAD-SARS-CoV-2positive patients showed a significantly reduced peripheral platelet count compared with CAD-SARS-CoV-2negative patients and healthy controls (healthy control versus CAD-SARS-CoV-2positive versus –negative; median, IQR; 1.79×108/mL (1.46×108×/mL–2.14×108/mL) versus 1.20×108/mL (0.75×108/mL–1.48×108/mL) versus 1.60×108/mL (1.11×108/mL–1.87×108/mL); healthy control versus CAD-SARS-CoV-2positive P<0.0001, healthy control versus CAD-SARS-CoV-2negative P=0.4610, CAD-SARS-CoV-2positive versus –negative P=0.0365; Figure 1A). Further, activation of circulating platelets of CAD-SARS-CoV-2positive patients, determined by surface expression of CD62P, was significantly enhanced in the infected group compared with uninfected CAD patients and the healthy control group (%CD62P+ platelets; healthy control versus CAD-SARS-CoV-2positive versus –negative; median (IQR); 13.5% (9.7%–19.3%) versus 18.7% (14.6%–25.2%) versus 10.8% (7.4%–15.5%); healthy control versus CAD-SARS-CoV-2positive P=0.0139, healthy control versus CAD-SARS-CoV-2negative P=0.4994, CAD-SARS-CoV-2positive versus –negative P=0.0002; Figure 1B). Further significantly enhanced CD42b+CD61+ platelet/leukocyte co-aggregates were observed in the infection group compared with CAD-SARS-CoV-2negative patients and the healthy control group (healthy control versus CAD-SARS-CoV-2positive versus –negative; median (IQR); 4.6% (3.4%–7.1%), 8.4% (5.6%–11.2%) versus 4.4% (3.3%–5.0%); healthy control versus CAD-SARS-CoV-2positive P=0.0002, healthy control versus CAD-SARS-CoV-2negative P=0.7580, CAD-SARS-CoV-2positive versus –negative P<0.0001; Figure 1C). Moreover, enhanced platelet activation in CAD-SARS-CoV-2positive patients was reflected by an increase of CD41+CD61+CD62P+ surface expression of CD42b+ platelet/leukocyte co-aggregates in comparison to healthy and CAD-SARS-CoV-2negative patients (Figure 1D). Platelet/leukocyte co-aggregates were gated first for double-positive CD41+CD61+ and further for CD62P+ representing highly activated platelet/leukocyte co-aggregates. For visualization of the platelet/leukocyte co-aggregates, we performed imaging flow cytometry analysis and showed that CD42b+ platelets adhere to CD14+ monocytes, represented by red and green fluorescence, respectively (Figure 1E).
Figure 1.
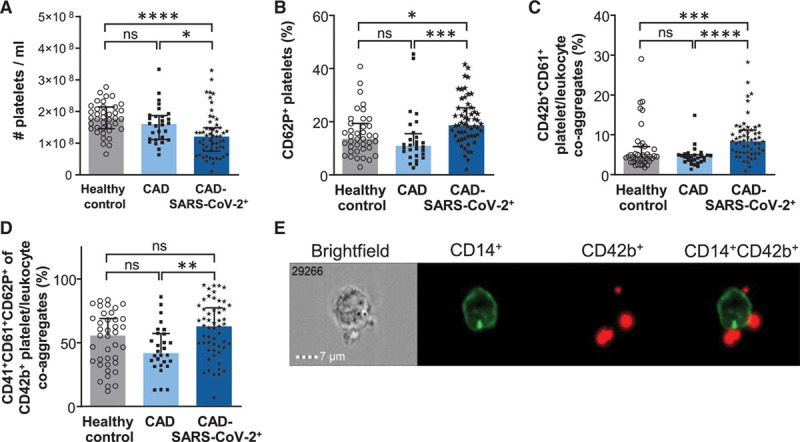
Coronary artery disease severe acute respiratory syndrome coronavirus 2 (CAD-SARS-CoV-2)positive patients present with reduced platelet count but show increased platelet activation and platelet/leukocyte co-aggregates in peripheral blood. Human platelets were analyzed by flow cytometry in whole blood of healthy controls (n=39), CAD-SARS-CoV-2negative patients (CAD; n=28), and patients with CAD with subsequent SARS-CoV-2 infection (CAD-SARS-CoV-2positive; n=55). Graphs show (A) cell count of platelets/mL, (B) frequency of CD62P+ platelets (%), (C) frequency of CD42b+CD61+ platelet/leukocyte co-aggregates (%), and (D) frequency of CD41+CD61+CD62P+ of CD42b+ platelet/leukocyte co-aggregates (%). E, Imaging flow cytometry was performed to analyze platelet/leukocyte co-aggregates by staining with CD42b-PerCP-Cy5.5 and CD14-FITC antibodies. Representative brightfield and fluorescence images are shown. A–D, Plotted: Median with interquartile range (IQR); Statistics: Mann-Whitney U test. ns indicates not significant; *P≤0.050, **P≤0.010, ***P≤0.001, and ****P≤0.0001.
Thus, in contrast to patients with CAD with a negative test for SARS-CoV-2, the virus infection was associated with enhanced platelet activation.
We next measured plasma levels of hyper-inflammation markers in our patient cohort. Among the 17 tested inflammation markers 10 cytokines/chemokines were significantly different expressed at time of hospital admission in CAD-SARS-CoV-2positive in contrast to CAD-SARS-CoV-2negative patients and healthy controls. IFN (interferon)-γ, IL (interleukin) 18, IL-1β, IL-33, CCL2, CXCL8 (chemokine [C-X-C motif] ligand), CXCL9, CXCL10, CXCL11, and furin were significantly increased. IL-6, IL-10, IFN-α, TNF (tumor necrosis factor)-α IL-12p70, CCL5, and CXCL5 were not differently expressed (Figure 2A through 2Q).
Figure 2.
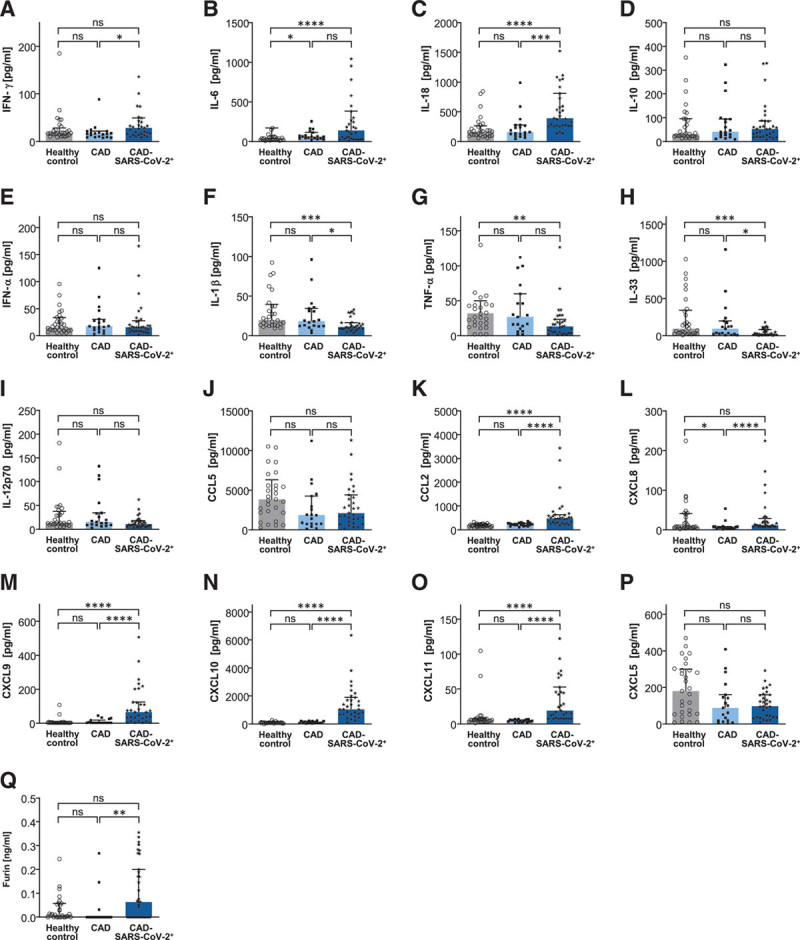
Coronary artery disease severe acute respiratory syndrome coronavirus 2 (CAD-SARS-CoV-2)positive patients showed highly increased plasma concentration of furin and several inflammatory cytokines. LEGENDPlex assays were performed for measuring plasma concentrations of healthy controls (n=29), CAD-SARS-CoV-2negative (n=19), and CAD-SARS-CoV-2positive patients (n=31). Graphs display concentration (pg/mL) of (A) IFN (interferon)-γ, (B) IL (interleukin)-6, (C) IL-18, (D) IL-10, (E) IFN-α, (F) IL-1ß, (G) TNF (tumor necrosis factor)-α, (H) IL-33, (I) IL-12p70, (J) CCL5, (K) CCL2, (L) CXCL8 (chemokine [C-X-C motif] ligand), (M) CXCL9, (N) CXCL10, (O) CXCL11, and (P) CXCL5. Q, Plasma concentration (ng/mL) of Furin in healthy controls (n=28), CAD-SARS-CoV-2negative (n=20) and CAD-SARS-CoV-2positive patients (n=35) was analyzed by ELISA. A–Q, Plotted: Median with interquartile range (IQR); Statistics: Mann-Whitney U test, ns indicates not significant; *P≤0.050, **P≤0.010, ***P≤0.001, and ****P≤0.0001.
Further we found that platelet activation, as indicated by surface expression of the degranulation marker CD62 (P-selectin), correlates with plasma levels of CCL5 (RANTES; r=0.454, P=0.010), which is a prominent platelet chemokine released upon activation (Figure IIIA in the Data Supplement). Further, circulating CD42b+CD61+ platelet/leukocyte co-aggregates correlated with IL-6 (r=0.398, P=0.027) as well as chemokines CCL2 (r=0.481, P=0.006) and CXCL10 (r=0.537, P=0.002) indicating that platelet activation and interaction with leukocytes is a trigger of hyper-inflammation (Figure 3B through 3D in the Data Supplement). Further, in CAD-SARS-CoV-2positive patients, platelet activation and platelet/leukocyte co-aggregation are associated with elevation of pulmonary artery pressure (r=0.428, P=0.007) and plasma levels of TNI (troponin I; r=0.302, P=0.033; Figure 3E and 3F in the Data Supplement).
Plasma Levels of Furin Are Associated With Poor Clinical Prognosis in SARS-CoV-2-Positive CAD Patients, and Furin Is Released From Activated Platelets
We found that plasma levels of furin that has been postulated to activate the spike protein of the SARS-CoV-2 virus and facilitates cell fusion is enhanced in plasma of CAD-SARS-CoV-2positive versus –negative patients (Figure 2Q).
Further, we observed increased furin plasma levels in CAD-SARS-CoV-2positive patients requiring intensive care unit (ICU) treatment. Thus, furin levels on hospital admission are an early marker for progressive respiratory failure and requirement of ICU treatment (Figure 3A). Moreover, plasma levels of furin significantly correlate with numbers of circulating CD42b+CD61+ platelet/leukocyte co-aggregates (r=0.460, P=0.009; Figure 3B). Thus, in CAD-SARS-CoV-2positive patients, platelet activation is associated with enhanced plasma levels of furin. Next, we performed Kaplan-Meier analyses to identify whether plasma furin levels can be used as prognostic marker for patients with CAD infected with SARS-CoV-2. We divided CAD-SARS-CoV-2positive patients according to the calculated median concentration of furin (0.064 ng/mL) into 2 groups. We found that increased plasma levels of the serine protease furin is associated with adverse clinical outcome (ventilation therapy and overall mortality) in CAD-SARS-CoV-2positive patients (log-rank=3.68, P=0.05; Figure 3C; Table 3). In our cohort, 11/55 CAD-SARS-CoV-2positive patients reached the primary end point after 60 days defined as the occurrence of a HI <200 mm Hg and mechanical ventilation. Results showed that significantly more often CAD-SARS-CoV-2positive patients with a furin concentration above the median reached the clinical end point (Figure 3C). Cox regression analysis adjusted for age, gender, pulmonary artery pressure, platelet count, and platelet activation showed that early determination of plasma levels of furin is predictive for progression of respiratory failure in CAD-SARS-CoV-2positive patients (Table 3). Interestingly, 6 events of venous thromboembolism occurred during the 60 days of follow-up within the CAD-SARS-CoV-2positive group of 55 patients. Four patients experienced a deep vein thrombosis and 2 patients a combination of deep vein thrombosis and pulmonary embolism. All events occurred in the CAD-SARS-CoV-2positive patients admitted to ICU.
Figure 3.
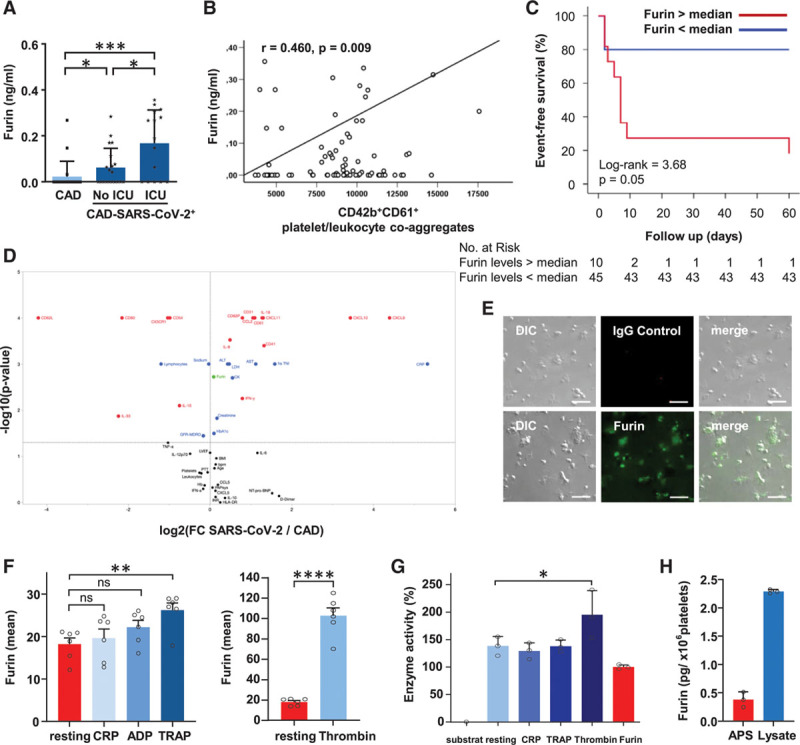
Furin is stored in platelets and furin plasma levels are associated with clinical outcome of coronary artery disease severe acute respiratory syndrome coronavirus 2 (CAD-SARS-CoV-2)positive patients, platelet activation, and presence of platelet/leucocyte co-aggregates. A, Furin plasma levels (ng/mL) were measured by ELISA and stratified by CAD (CAD-SARS-CoV-2negative; n=20) and CAD-SARS-CoV-2positive status (subgroup of CAD-SARS-CoV-2positive admitted to the intensive care unit [ICU; n=15] compared with no ICU [n=20] admission). Plotted: Mean±SD; statistics: 1-way ANOVA (Dunnett), *P≤0.050 and ***P≤0.001. B, Pearson correlation analysis was performed to evaluate associations between CD42b+CD61+ platelet/leukocyte co-aggregates (median) and furin (ng/mL; r=0.460, P=0.009). Statistics: Pearson correlation coefficient r, **P≤0.010. C, Plasma concentration of furin of CAD-SARS-CoV-2positive infected patients was divided into 2 groups based on the calculated median of furin concentration (median 0.064 ng/mL). Kaplan-Meier curve represents the occurrence of the clinical study end point stratified according to furin plasma concentration of all CAD-SARS-CoV-2positive patients within a follow-up time of 60 days. During these 60 days, 11/55 (20%) reached the end point. The clinical study end point was defined as rapidly progressive respiratory failure with a Horovitz index <200 mm Hg and required mechanical ventilation. Nine out of 11 (81.8%) patients had a furin plasma concentration above the calculated median (log-rank 3.68, P=0.05). D, Volcano plot displays analysis of clinical data and flow cytometry and LEGENDPlex measurements. y axis displays P (log10) with cut-off 1.3=−log10(0.05) and x axis fold change between the median of CAD (CAD-SARS-CoV-2negative) and CAD-SARS-CoV-2positive. Test was performed by JMP Version 15.0 Statistics: Mann-Whitney U test. E, Immunofluorescence microscopy was performed to analyze whether platelets store furin. Graph shows images of representative immunofluorescence microscopy pictures of spreaded human platelets stained with furin antibody (AF488) or an IgG2B control antibody (AF568). Responding differential interference contrast (DIC) and merge images are supplied (scale=10 µm). F, For comparison of furin surface expression between differently activated platelets (n=6), graphs display mean fluorescence intensity of furin. Plotted: mean±SD; statistics: Student t test, ns=not significant; **P≤0.010 and **** P≤0.0001. G, For analysis of enzyme activity assay, a pERTKR-AMC fluorogenic peptide substrate was used. Graph shows the statistical end point analysis of the enzyme activity after 60 min incubation with of the fluorogenic peptide substrate with 5×107 platelets and activators (n=3). Plotted: mean±SD; Statistics: 1-way ANOVA (Dunnett), *P≤0.050. H, To quantify platelet furin levels we used lysates from resting human platelets and APS from platelets activated with 1 U/mL thrombin. Each sample was prepared from a platelet suspension of 2×109 cells/mL. For comparison of furin amount in platelets, supernatant of activated platelets (APS) and resting platelet lysate was performed and furin concentration (pg/1×106 platelets) was measured by ELISA (n=3). CRP indicates C-reactive protein; and TRAP, thrombin receptor activating peptide.
Table 3.
Results of Unadjusted and Adjusted Cox Regression Analysis of Progressive Respiratory Failure (HI <200 mm Hg)

Volcano plot analysis confirms that furin plasma levels (green spot) are significantly elevated in CAD-SARS-CoV-2positive patients compared with CAD-SARS-CoV-2negative patients (Figure 3D).
Previously, others and we described platelets as a major source of inflammatory mediators, which play a critical role in atheroprogression and thrombo-inflammation.1,49 Since platelet activation is associated with enhanced plasma levels of furin in CAD-SARS-CoV-2positive patients, we asked whether furin is present in platelets and released upon activation. As shown by immunofluorescence studies, furin is stored in significant amounts in platelets (Figure 3E). Upon activation with TRAP6 or most prominently thrombin, furin is significantly expressed on the platelet surface (flow cytometry; Figure 3F). Furthermore, the enhanced furin expression by TRAP6 can be significantly suppressed with the PAR-1 inhibitor ML161 (10 µmol/L). This result was expected; as TRAP6 mediated activation is solely depended on the PAR-1 receptor (Figure IV in the Data Supplement). In contrast, the thrombin-induced furin expression is only partially inhibited in samples treated with 0.1 U/mL thrombin and 40 µmol/L ML161. When we used 1 U/mL of thrombin activation failed to be inhibited by 40 μmol/L ML161 (Figure IV in the Data Supplement). In both cases, ML161 was used to the maximum applicable concentration, due to the solubility limit of ML161. These findings indicate, that thrombin-induced furin expression is dependent on the PAR-1 as well as the PAR-4 signaling pathway and that pathway involvement may be dependent on the thrombin concentration. CRP activation was used as control for the ML161 specificity. No inhibitory effect of ML161 on CRP activation could be observed (Figure IV in the Data Supplement). The sample activation was confirmed by CD62P expression, which was significantly increased in all samples measured (Figures II and IV in the Data Supplement). Further, comparison of washed platelets and platelet poor plasma regarding enzyme activity upon platelet activation display increased furin activity in the presence of plasma (Figure V in the Data Supplement). Additionally, platelets have the potential ability to degrade the SARS-CoV-2 spike protein indicated by the furin activity assay performed (Figure 3G). To quantify platelet furin levels, we used lysates from resting human platelets and activated platelet supernatant from platelets activated with 1 U/mL thrombin. Each sample was prepared from a platelet suspension of 2×109 cells/mL. Quantification by ELISA revealed that resting platelets contain 2.291±0.0348 (pg/1×106 platelets) furin and release approximately 20% furin upon activation (0.382±0.1225 pg/1×106 platelets; Figure 3H). In summary, platelets are a major source of furin that is released upon activation and may be critical for progression of respiratory failure in SARS-CoV-2 positive patients with CAD.
Discussion
The major findings of our present study are (1) patients with CAD and SARS-CoV-2 infection already exhibit a significantly enhanced platelet activation (Figure 1B) and hyper-inflammation at time of hospital admission (Figure 2) and (2) plasma levels of furin are associated with poor clinical outcome in CAD-SARS-CoV-2positive patients (Figure 3C), in addition, furin is stored and released from activated platelets (Figure 3E and 3F). Our data imply that a prothrombotic state of circulating platelets is critically involved in development of systemic thrombo-inflammation (hyper-inflammation) early in the development of SARS-CoV-2 infection. Moreover, release of the SARS-CoV-2 activated serin protease furin from platelets may promote infection of the RNA virus.
Targeting platelet activation and attenuating release of inflammation markers at an early state of the infection may help to control progression of COVID-19. Former studies showed that acute SARS-CoV-2 infection is associated with platelet activation, increased amount of platelet/leucocyte co-aggregates and characteristic coagulopathy resulting in thrombotic complications including deep vein thrombosis and pulmonary embolism.4–6,10 Platelet activation and platelet/leucocyte co-aggregates can predict an unfavorable clinical outcome of patients with COVID-19.11 This might be in particular be relevant for patients with CAD due to their increased risk of mortality, respiratory, and organ failure. Mechanisms underlying COVID-19-induced acute coronary syndrome might involve plaque rupture, coronary spasm, or microthrombi owing to systemic inflammation or cytokine storm. Potential drug–disease interactions affecting patients with COVID-19 and comorbid cardiovascular diseases are also becoming a serious concern and should be accounted for. Therefore, addressing platelet activation and release of mediators like furin as therapeutic target might be of clinical relevance to improve outcome of these patients.32–35,50 Furthermore, it has been described that platelet activation and the amount of circulating platelet/leucocyte co-aggregates are associated with the stability of CAD and can serve as reliable biomarkers during atheroprogression in cardiovascular disease.51–54 In our cohort, we could not observe statistically significant differences in platelet activation and the amount of platelet/leukocyte co-aggregates between healthy controls and CAD-SARS-CoV-2negative patients, while CAD-SARS-CoV-2positive patients were characterized by significantly increased platelet activation and elevated numbers of platelet/leukocyte co-aggregates. As we analyzed a relatively small patient cohort this might explain why we did not detect statistically significant differences in platelet activation and amount of platelet/leukocyte co-aggregates between healthy controls and CAD-SARS-CoV-2negative patients as described before.51–54 On the contrary, we included patients with stable CAD that were on long-term antiplatelet therapy during their course of the disease. This aspect might also explain why there was no statistically significant difference between healthy controls and CAD-SARS-CoV-2negative patients reflecting efficient antiplatelet treatment in this subgroup. Patients with acute coronary syndrome and myocardial infarction were explicitly excluded, while this subgroup has been investigated thoroughly in former studies and was also characterized by enhanced platelet activation and increased amount of platelet/leukocyte co-aggregates.51–56
An underlying cardiac disease is a major risk factor for unfavorable clinical outcome in COVID-19.30,57,58 Platelet hyper-reactivity promotes development of acute coronary syndromes,59 and COVID-19 has been described to be associated with myocardial injury and mortality.60,61
In line with previous findings, we found that 21.8% in CAD-SARS-CoV-2positive patients showed a BMI ≥30 compared with 10.7% in the CAD-SARS-CoV-2negative group and 10.3 % in the healthy control group (Table 1; P=0.045). With this significant difference between the groups, BMI might be another confounding factor regarding outcome, in particular of the end point defined as respiratory failure. Recent studies have shown that obesity is a risk factor of unfavorable outcome and progressive respiratory failure. Obesity increases risk for hospitalization, ICU admission, requirement of invasive mechanical ventilation, and death among patients with COVID-19.62–64
A prothrombotic disease state has been recognized as critical factor in COVID-19.5 Patients with SARS-CoV-2 infection are at increased risk for thrombosis (deep vein thrombosis and pulmonary embolism) and for multiple organ failure most likely due to microcirculatory arrest.65,66 Recently, it has been shown that development of thrombocytopenia, elevated D-dimers and markers of systemic inflammation are of prognostic relevance in SARS-CoV-2-positive patients.67,68 In our cohort, there were no significant differences between groups in their D-dimer levels upon admission (Table 1). This might be explained by the low patient number and by the time point when samples for D-dimer measurements were obtained. All blood samples were taken within 24 hours of hospital admission and, therefore, also correlated our findings and presented baseline characteristics including laboratory parameters upon admission. There were no associations of D-dimer levels on admission and outcome. However, we did not correlate repeated D-dimer levels during the course of the disease and correlated these with the occurrence of the clinical outcome, which might explain the observed difference. The impact of D-dimer has been discussed controversially and studies showed miscellaneous aspects and impacts of D-dimer. D-dimer was independently associated with incidence of critical illness, thrombosis, acute kidney injury, and all-cause mortality.69 Another study showed that D-dimer levels could not predict thrombotic events.70 Thus, levels of D-dimer may help to estimate the individual risk for an unfavorable course of the disease and to identify patients at risk. In regards to our findings, we have to take into account comorbidities and other clinical features in addition to D-dimer levels to estimate patients’ prognosis. Therefore, the additive analysis of furin levels and platelet-derived furin can improve an intensified risk assessment of patients with COVID-19 regarding an unfavorable course of the disease depending on their furin levels as additional biomarker. Our findings implicate, that several biomarkers and clinical parameters should be taken into account, to identify patients at risk of an unfavorable course of the disease, for example, furin.69,70
Patients with uncontrolled hyper-inflammation are at high risk to develop progressive respiratory failure, multiple organ failure, or of death.4,71 In the present study, we evaluated whether changes of platelet activation occur in an early state of SARS-CoV-2 infection defined as time of hospital admission. Beyond thrombosis, platelets are key mediators of inflammation and thrombo-immunity.2 During pathogen infections, platelets release a variety of potent cytokines or chemokines72,73 and interact with leukocytes as well as endothelial cells and propagate vascular and tissue inflammation.8,74 It is well known that virus infections are associated with thrombocytopenia, platelet hyper-reactivity, and formation of platelet/leukocyte co-aggregates leading to enhanced sequestration within the microcirculation.75 In critically affected patients, nonsurvivors showed a trend towards a drop in platelet count and leukocytosis.76 Recently, in critical COVID-19 ICU patients an enhanced platelet apoptosis has been described77 and excessive platelet and neutrophil activation.38,39 Our data confirm and extent the published findings and show that platelet activation not only is evident in progressive SARS-CoV-2 associated infection but substantially is present at an early disease stage as soon as SARS-CoV-2 infection was verified by reverse transcriptase polymerase chain reaction. Most strikingly, our study shows that platelet activation and CD42b+CD61+ platelet/leukocyte co-aggregates correlate with enhanced plasma levels of cytokine IL-6 and chemokines CCL2, CCL5, and CXCL10 which represent prominent markers of hyper-inflammation (Figure III in the Data Supplement). Platelet activation was also associated with enhanced pulmonary artery pressure and elevation of the cardiac injury marker troponin I indicating its linkage to myocardial stretch and subsequent myocardial necrosis in our patient cohort. Further, CD42b+CD61+ platelet/leukocyte co-aggregates correlate with furin plasma levels, which in turn is associated with progression of respiratory failure and poor clinical prognosis (Figure 3B). Furin has been suggested to cleave the SARS-CoV-2 spike protein, a putative critical mechanism for syncytium formation and virus replication.18,19,78 Platelets contain significant amounts of furin.50 We further showed that furin is released from activated platelets implying that platelets are a major source of furin in the microenvironment of platelet activation (Figure 3F). Although we do not provide direct evidence that platelet-derived furin is involved in SARS-CoV-2 activation, it is tempting to speculate that activated platelets promote vascular and tissue inflammation and might contribute to virus-induced cell fusion and replication. Limiting platelet release of furin by antithrombotic therapy early in the disease state may be a feasible and effective prevention for worse clinical outcome and thromboembolic events. This might be most relevant in patients with underlying cardiovascular diseases to prevent progression of CAD and threatening organ failure.
Furthermore, the thrombin-specific phenotype observed must be transmitted via the thrombin receptors PAR1 or PAR4 (Figure IV in the Data Supplement). The TRAP-6 did also induce a significant increase in furin, even though the effect is not as prominent as the Thrombin activation. This effect is, as expected, fully inhibited by the PAR-1 receptor inhibitor ML161, therefore, PAR1 is probably involved in furin activation. But this does not exclude PAR4 as additional pathway and the involvement of both receptors may explain the strong activation. By activation with thrombin or TRAP, more furin will be available especially free furin, which is responsible for the measured increase in enzyme activity. Some studies suggest that the dual receptor system PAR1 and PAR4 offers an intriguing possibility of pharmacologically fine-tuning thrombin signaling in platelets by taking advantage of the individual contributions of PAR1 and PAR4, but with distinct kinetics. PAR1 is described to respond to much lower concentrations of thrombin than PAR4 suggesting that PAR4 might only be a backup thrombin receptor. Indeed, our results do match with this hypothesis. We did observe, that low concentration of thrombin (0.1 U/mL) stimulation could be partially inhibited by the PAR-1 inhibitor ML161 but not high concentration like 1 U/mL thrombin, which were unaffected by the inhibitor even at maximum concentrations. These results imply the involvement of the PAR4 signaling pathway upon strong thrombin activation. Blocking the sustained signaling from PAR4 may limit thrombosis, while leaving the transient PAR1 signaling mechanism available to initiate hemostasis and limit bleeding. The first in-class antiplatelet therapy targeting PAR1, vorapaxar, was approved by the Food and Drug Administration in 2014 for secondary prevention of thrombotic events in stable patients. Importantly, vorapaxar was approved for use in addition to the standard antiplatelet therapy, but not as a stand-alone therapy or as a substitute for aspirin or clopidogrel. Recent data support the hypothesis that blocking PAR4-mediated sustained signaling for stable thrombus formation, while preserving PAR1 signaling for initial thrombus formation, may be a safe and effective antithrombotic strategy. Vorapaxar has not gained widespread clinically use. One issue is a difficult clinical management due to its pharmacokinetics and pharmacodynamics. The reversibility and elimination characteristics of the PAR4 inhibitor BMS-986120 may prove to be advantageous in this regard. PAR1 and PAR4 inhibition might regulate furin signaling and, therefore, serve as therapeutic targets among others. But larger studies focusing on underlying mechanisms and clinical relevance are needed to clarify these questions.79–87
Therefore, we investigated the prognostic impact of platelet activation, platelet-derived furin and furin plasma levels along with other established risk factors for unfavorable outcome of COVID-19. Blocking the activity of furin might reduce severity of viral infections and cardiovascular damage and could be beneficial for the treatment of both, SARS-CoV-2 infection and cardiovascular diseases.
Sources of Funding
This work was supported by the German Research Foundation (DFG)—Project number 374031971–TRR 240 and by the Ministry of Science, Research and the Arts of the State of Baden-Württemberg (COVID-19 funding). The funder had no role in study design, data collection, data analysis, data interpretation, or writing of the article.
Disclosures
None.
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- ACE-2
- angiotensin-converting enzyme 2
- CAD
- coronary artery disease
- COVID-19
- coronavirus disease 2019
- CRP
- C-reactive protein
- CXCL
- chemokine (C-X-C motif) ligand
- HI
- Horovitz index
- ICU
- intensive care unit
- IFN
- interferon
- IL
- interleukin
- IQR
- interquartile range
- NT-proBNP
- N-terminal pro-B-type natriuretic peptide
- SARS-CoV-2
- severe acute respiratory syndrome coronavirus 2
- TNF
- tumor necrosis factor
- TRAP
- thrombin receptor activating peptide
C. Langnau and A.-K. Rohlfing contributed equally.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.315698.
For Sources of Funding and Disclosures, see page 2094.
Contributor Information
Carolin Langnau, Email: Carolin.Langnau@med.uni-tuebingen.de.
Anne-Katrin Rohlfing, Email: Anne-Katrin.Rohlfing@med.uni-tuebingen.de.
Sarah Gekeler, Email: Sarah.Gekeler@med.uni-tuebingen.de.
Manina Günter, Email: Manina.Guenter@dkfz.de.
Simone Pöschel, Email: Simone.Poeschel@med.uni-tuebingen.de.
Álvaro Petersen-Uribe, Email: Alvaro.Petersen@med.uni-tuebingen.de.
Philippa Jaeger, Email: Philippa.Jaeger@med.uni-tuebingen.de.
Alban Avdiu, Email: Albanavdiu93@gmail.com.
Tobias Harm, Email: Tobias.Harm@med.uni-tuebingen.de.
Klaus-Peter Kreisselmeier, Email: Klaus-Peter.Kreisselmeier@med.uni-tuebingen.de.
Tatsiana Castor, Email: Tatsiana.Castor@med.uni-tuebingen.de.
Tamam Bakchoul, Email: tamam.bakchoul@med.uni-tuebingen.de.
Dominik Rath, Email: dominik.rath@med.uni-tuebingen.de.
Meinrad Paul Gawaz, Email: meinrad.gawaz@med.uni-tuebingen.de.
Stella E. Autenrieth, Email: stella.autenrieth@dkfz-heidelberg.de.
References
- 1.Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115:3378–3384. doi: 10.1172/JCI27196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. 2018;122:337–351. doi: 10.1161/CIRCRESAHA.117.310795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, Yao C, Rade J, Levy D, Wang JP, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. 2019;10:1780. doi: 10.1038/s41467-019-09607-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guzik TJ, Mohiddin SA, Dimarco A, Patel V, Savvatis K, Marelli-Berg FM, Madhur MS, Tomaszewski M, Maffia P, D’Acquisto F, et al. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020;116:1666–1687. doi: 10.1093/cvr/cvaa106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135:2033–2040. doi: 10.1182/blood.2020006000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, Limami Y, Zaid N, Sadki K, El Haj RB, et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in covid-19. Circ Res. 2020;127:1404–1418. doi: 10.1161/CIRCRESAHA.120.317703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lê VB, Schneider JG, Boergeling Y, Berri F, Ducatez M, Guerin JL, Adrian I, Errazuriz-Cerda E, Frasquilho S, Antunes L, et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med. 2015;191:804–819. doi: 10.1164/rccm.201406-1031OC [DOI] [PubMed] [Google Scholar]
- 8.Dib PRB, Quirino-Teixeira AC, Merij LB, Pinheiro MBM, Rozini SV, Andrade FB, Hottz ED. Innate immune receptors in platelets and platelet-leukocyte interactions. J Leukoc Biol. 2020;108:1157–1182. doi: 10.1002/JLB.4MR0620-701R [DOI] [PubMed] [Google Scholar]
- 9.Boilard E, Paré G, Rousseau M, Cloutier N, Dubuc I, Lévesque T, Borgeat P, Flamand L. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood. 2014;123:2854–2863. doi: 10.1182/blood-2013-07-515536 [DOI] [PubMed] [Google Scholar]
- 10.Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, Petrey AC, Tolley ND, Guo L, Cody M, et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136:1317–1329. doi: 10.1182/blood.2020007214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, Righy C, Franco S, Souza TML, Kurtz P, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. 2020;136:1330–1341. doi: 10.1182/blood.2020007252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clarke NE, Turner AJ. Angiotensin-converting enzyme 2: the first decade. Int J Hypertens. 2012;2012:307315. doi: 10.1155/2012/307315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jarcho JA, Ingelfinger JR, Hamel MB, D’Agostino RB, Sr, Harrington DP. Inhibitors of the renin-angiotensin-aldosterone system and covid-19. N Engl J Med. 2020;382:2462–2464. doi: 10.1056/NEJMe2012924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-angiotensin-aldosterone system inhibitors in patients with covid-19. N Engl J Med. 2020;382:1653–1659. doi: 10.1056/NEJMsr2005760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Datta PK, Liu F, Fischer T, Rappaport J, Qin X. SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics. 2020;10:7448–7464. doi: 10.7150/thno.48076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abassi Z, Knaney Y, Karram T, Heyman SN. The lung macrophage in SARS-CoV-2 infection: a friend or a foe? Front Immunol. 2020;11:1312. doi: 10.3389/fimmu.2020.01312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, Liu M, Zhao X, Xie Y, Yang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. 2020;13:120. doi: 10.1186/s13045-020-00954-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176:104742. doi: 10.1016/j.antiviral.2020.104742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, Li F. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA. 2020;117:11727–11734. doi: 10.1073/pnas.2003138117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakayama K. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem J. 1997;327(pt 3):625–635. doi: 10.1042/bj3270625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang YK, Tang JN, Han L, Liu XD, Shen YL, Zhang CY, Liu XB. Elevated FURIN levels in predicting mortality and cardiovascular events in patients with acute myocardial infarction. Metabolism. 2020;111:154323. doi: 10.1016/j.metabol.2020.154323 [DOI] [PubMed] [Google Scholar]
- 24.Adu-Agyeiwaah Y, Grant MB, Obukhov AG. The potential role of osteopontin and furin in worsening disease outcomes in COVID-19 patients with pre-existing diabetes. Cells. 2020;9:E2528. doi: 10.3390/cells9112528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yakala GK, Cabrera-Fuentes HA, Crespo-Avilan GE, Rattanasopa C, Burlacu A, George BL, Anand K, Mayan DC, Corlianò M, Hernández-Reséndiz S, et al. FURIN inhibition reduces vascular remodeling and atherosclerotic lesion progression in mice. Arterioscler Thromb Vasc Biol. 2019;39:387–401. doi: 10.1161/ATVBAHA.118.311903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao G, Yang W, Wu J, Chen B, Yang X, Chen J, McVey DG, Andreadi C, Gong P, Webb TR, et al. Influence of a coronary artery disease-associated genetic variant on FURIN expression and effect of furin on macrophage behavior. Arterioscler Thromb Vasc Biol. 2018;38:1837–1844. doi: 10.1161/ATVBAHA.118.311030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang X, Yang W, McVey DG, Zhao G, Hu J, Poston RN, Ren M, Willeit K, Coassin S, Willeit J, et al. FURIN expression in vascular endothelial cells is modulated by a coronary artery disease-associated genetic variant and influences monocyte transendothelial migration. J Am Heart Assoc. 2020;9:e014333. doi: 10.1161/JAHA.119.014333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stawowy P, Kallisch H, Borges Pereira Stawowy N, Stibenz D, Veinot JP, Gräfe M, Seidah NG, Chrétien M, Fleck E, Graf K. Immunohistochemical localization of subtilisin/kexin-like proprotein convertases in human atherosclerosis. Virchows Arch. 2005;446:351–359. doi: 10.1007/s00428-004-1198-7 [DOI] [PubMed] [Google Scholar]
- 29.Fernandez C, Rysä J, Almgren P, Nilsson J, Engström G, Orho-Melander M, Ruskoaho H, Melander O. Plasma levels of the proprotein convertase furin and incidence of diabetes and mortality. J Intern Med. 2018;284:377–387. doi: 10.1111/joim.12783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rath D, Petersen-Uribe A, Avdiu A, Witzel K, Jaeger P, Zdanyte M, Heinzmann D, Tavlaki E, Muller K, Gawaz MP. Impaired cardiac function is associated with mortality in patients with acute covid-19 infection. Clin Res Cardiol. 2020;109:1491–1499. doi: 10.1007/s00392-020-01683-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. 2020;17:259–260. doi: 10.1038/s41569-020-0360-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie J, Tong Z, Guan X, Du B, Qiu H. Clinical characteristics of patients who died of coronavirus disease 2019 in China. JAMA Netw Open. 2020;3:e205619. doi: 10.1001/jamanetworkopen.2020.5619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong TY, Redwood S, Prendergast B, Chen M. Coronaviruses and the cardiovascular system: acute and long-term implications. Eur Heart J. 2020;41:1798–1800. doi: 10.1093/eurheartj/ehaa231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishiga M, Wang DW, Han Y, Lewis DB, Wu JC. COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol. 2020;17:543–558. doi: 10.1038/s41569-020-0413-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.European Society of Cardiology. Esc guidance for the Diagnosis and Management of CV Disease during the Covid-19 Pandemic. 2020. https://www.escardio.org/Education/COVID-19-and-Cardiology/ESCCOVID-19-Guidance. Accessed February 13, 2021
- 36.Li B, Yang J, Zhao F, Zhi L, Wang X, Liu L, Bi Z, Zhao Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin Res Cardiol. 2020;109:531–538. doi: 10.1007/s00392-020-01626-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18:1094–1099. doi: 10.1111/jth.14817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicolai L, Leunig A, Brambs S, Kaiser R, Weinberger T, Weigand M, Muenchhoff M, Hellmuth JC, Ledderose S, Schulz H, et al. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation. 2020;142:1176–1189. doi: 10.1161/CIRCULATIONAHA.120.048488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mueller KAL, Langnau C, Günter M, Pöschel S, Gekeler S, Petersen-Uribe Á, Kreisselmeier KP, Klingel K, Bösmüller H, Li B, et al. Numbers and phenotype of non-classical CD14dimCD16+ monocytes are predictors of adverse clinical outcome in patients with coronary artery disease and severe SARS-CoV-2 infection. Cardiovasc Res. 2021;117:224–239. doi: 10.1093/cvr/cvaa328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wirtz TH, Tillmann S, Strüßmann T, Kraemer S, Heemskerk JW, Grottke O, Gawaz M, von Hundelshausen P, Bernhagen J. Platelet-derived MIF: a novel platelet chemokine with distinct recruitment properties. Atherosclerosis. 2015;239:1–10. doi: 10.1016/j.atherosclerosis.2014.12.039 [DOI] [PubMed] [Google Scholar]
- 41.Reinthaler M, Braune S, Lendlein A, Landmesser U, Jung F. Platelets and coronary artery disease: Interactions with the blood vessel wall and cardiovascular devices. Biointerphases. 2016;11:029702. doi: 10.1116/1.4953246 [DOI] [PubMed] [Google Scholar]
- 42.Steele P, Battock D, Genton E. Effects of clofibrate and sulfinpyrazone on platelet survival time in coronary artery disease. Circulation. 1975;52:473–476. doi: 10.1161/01.cir.52.3.473 [DOI] [PubMed] [Google Scholar]
- 43.Parasuraman S, Walker S, Loudon BL, Gollop ND, Wilson AM, Lowery C, Frenneaux MP. Assessment of pulmonary artery pressure by echocardiography-A comprehensive review. Int J Cardiol Heart Vasc. 2016;12:45–51. doi: 10.1016/j.ijcha.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2015;16:233–270. doi: 10.1093/ehjci/jev014 [DOI] [PubMed] [Google Scholar]
- 45.Borst O, Schmidt EM, Münzer P, Schönberger T, Towhid ST, Elvers M, Leibrock C, Schmid E, Eylenstein A, Kuhl D, et al. The serum- and glucocorticoid-inducible kinase 1 (SGK1) influences platelet calcium signaling and function by regulation of Orai1 expression in megakaryocytes. Blood. 2012;119:251–261. doi: 10.1182/blood-2011-06-359976 [DOI] [PubMed] [Google Scholar]
- 46.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999 [DOI] [PubMed] [Google Scholar]
- 47.Ferguson TE, Reihill JA, Walker B, Hamilton RA, Martin SL. A selective irreversible inhibitor of furin does not prevent pseudomonas aeruginosa exotoxin A-induced airway epithelial cytotoxicity. PLoS One. 2016;11:e0159868. doi: 10.1371/journal.pone.0159868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huygens C, Liénart S, Dedobbeleer O, Stockis J, Gauthy E, Coulie PG, Lucas S. Lysosomal-associated transmembrane protein 4B (LAPTM4B) decreases transforming growth factor β1 (TGF-β1) production in human regulatory T cells. J Biol Chem. 2015;290:20105–20116. doi: 10.1074/jbc.M115.655340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Massberg S, Brand K, Grüner S, Page S, Müller E, Müller I, Bergmeier W, Richter T, Lorenz M, Konrad I, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196:887–896. doi: 10.1084/jem.20012044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leblond J, Laprise MH, Gaudreau S, Grondin F, Kisiel W, Dubois CM. The serpin proteinase inhibitor 8: an endogenous furin inhibitor released from human platelets. Thromb Haemost. 2006;95:243–252. doi: 10.1160/TH05-08-0561 [DOI] [PubMed] [Google Scholar]
- 51.Linden MD, Furman MI, Frelinger AL, 3rd, Fox ML, Barnard MR, Li Y, Przyklenk K, Michelson AD. Indices of platelet activation and the stability of coronary artery disease. J Thromb Haemost. 2007;5:761–765. doi: 10.1111/j.1538-7836.2007.02462.x [DOI] [PubMed] [Google Scholar]
- 52.Allen N, Barrett TJ, Guo Y, Nardi M, Ramkhelawon B, Rockman CB, Hochman JS, Berger JS. Circulating monocyte-platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis. 2019;282:11–18. doi: 10.1016/j.atherosclerosis.2018.12.029 [DOI] [PubMed] [Google Scholar]
- 53.Furman MI, Benoit SE, Barnard MR, Valeri CR, Borbone ML, Becker RC, Hechtman HB, Michelson AD. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J Am Coll Cardiol. 1998;31:352–358. doi: 10.1016/s0735-1097(97)00510-x [DOI] [PubMed] [Google Scholar]
- 54.Brambilla M, Camera M, Colnago D, Marenzi G, De Metrio M, Giesen PL, Balduini A, Veglia F, Gertow K, Biglioli P, et al. Tissue factor in patients with acute coronary syndromes: expression in platelets, leukocytes, and platelet-leukocyte aggregates. Arterioscler Thromb Vasc Biol. 2008;28:947–953. doi: 10.1161/ATVBAHA.107.161471 [DOI] [PubMed] [Google Scholar]
- 55.Ibrahim H, Kleiman NS. Platelet pathophysiology, pharmacology, and function in coronary artery disease. Coron Artery Dis. 2017;28:614–623. doi: 10.1097/MCA.0000000000000519 [DOI] [PubMed] [Google Scholar]
- 56.Malakar AK, Choudhury D, Halder B, Paul P, Uddin A, Chakraborty S. A review on coronary artery disease, its risk factors, and therapeutics. J Cell Physiol. 2019;234:16812–16823. doi: 10.1002/jcp.28350 [DOI] [PubMed] [Google Scholar]
- 57.Clerkin KJ, Fried JA, Raikhelkar J, Sayer G, Griffin JM, Masoumi A, Jain SS, Burkhoff D, Kumaraiah D, Rabbani L, et al. COVID-19 and cardiovascular disease. Circulation. 2020;141:1648–1655. doi: 10.1161/CIRCULATIONAHA.120.046941 [DOI] [PubMed] [Google Scholar]
- 58.Inciardi RM, Adamo M, Lupi L, Metra M. Atrial fibrillation in the COVID-19 era: simple bystander or marker of increased risk? Eur Heart J. 2020;41:3094. doi: 10.1093/eurheartj/ehaa576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin JF, Kristensen SD, Mathur A, Grove EL, Choudry FA. The causal role of megakaryocyte–platelet hyperactivity in acute coronary syndromes. Nat Rev Cardiol. 2012;9:658–670. doi: 10.1038/nrcardio.2012.131 [DOI] [PubMed] [Google Scholar]
- 60.Bonow RO, O’Gara PT, Yancy CW. Cardiology and COVID-19. JAMA. 2020;324:1131–1132. doi: 10.1001/jama.2020.15088 [DOI] [PubMed] [Google Scholar]
- 61.Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, Wang H, Wan J, Wang X, Lu Z. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5:811–818. doi: 10.1001/jamacardio.2020.1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang Y, Lu Y, Huang YM, Wang M, Ling W, Sui Y, Zhao HL. Obesity in patients with COVID-19: a systematic review and meta-analysis. Metabolism. 2020;113:154378. doi: 10.1016/j.metabol.2020.154378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soeroto AY, Soetedjo NN, Purwiga A, Santoso P, Kulsum ID, Suryadinata H, Ferdian F. Effect of increased BMI and obesity on the outcome of COVID-19 adult patients: a systematic review and meta-analysis. Diabetes Metab Syndr. 2020;14:1897–1904. doi: 10.1016/j.dsx.2020.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tamara A, Tahapary DL. Obesity as a predictor for a poor prognosis of COVID-19: a systematic review. Diabetes Metab Syndr. 2020;14:655–659. doi: 10.1016/j.dsx.2020.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lax SF, Skok K, Zechner P, Kessler HH, Kaufmann N, Koelblinger C, Vander K, Bargfrieder U, Trauner M. Pulmonary arterial thrombosis in COVID-19 with fatal outcome: results from a prospective, single-center, clinicopathologic case series. Ann Intern Med. 2020;173:350–361. doi: 10.7326/M20-2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Poissy J, Goutay J, Caplan M, Parmentier E, Duburcq T, Lassalle F, Jeanpierre E, Rauch A, Labreuche J, Susen SLille ICU Haemostasis COVID-19 Group. Pulmonary embolism in patients with COVID-19: awareness of an increased prevalence. Circulation. 2020;142:184–186. doi: 10.1161/CIRCULATIONAHA.120.047430 [DOI] [PubMed] [Google Scholar]
- 67.Gu SX, Tyagi T, Jain K, Gu VW, Lee SH, Hwa JM, Kwan JM, Krause DS, Lee AI, Halene S, et al. Thrombocytopathy and endotheliopathy: crucial contributors to COVID-19 thromboinflammation. Nat Rev Cardiol. 2021;18:194–209. doi: 10.1038/s41569-020-00469-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Levi M, Thachil J, Iba T, Levy JH. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020;7:e438–e440. doi: 10.1016/S2352-3026(20)30145-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berger JS, Kunichoff D, Adhikari S, Ahuja T, Amoroso N, Aphinyanaphongs Y, Cao M, Goldenberg R, Hindenburg A, Horowitz J, et al. Prevalence and outcomes of D-dimer elevation in hospitalized patients with COVID-19. Arterioscler Thromb Vasc Biol. 2020;40:2539–2547. doi: 10.1161/ATVBAHA.120.314872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Elbadawi A, Elgendy IY, Sahai A, Bhandari R, McCarthy M, Gomes M, Bishop GJ, Bartholomew JR, Kapadia S, Cameron SJ. Incidence and outcomes of thrombotic events in symptomatic patients with COVID-19. Arterioscler Thromb Vasc Biol. 2021;41:545–547. doi: 10.1161/ATVBAHA.120.315304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jose RJ, Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med. 2020;8:e46–e47. doi: 10.1016/S2213-2600(20)30216-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yeaman MR. Platelets in defense against bacterial pathogens. Cell Mol Life Sci. 2010;67:525–544. doi: 10.1007/s00018-009-0210-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maouia A, Rebetz J, Kapur R, Semple JW. The immune nature of platelets revisited. Transfus Med Rev. 2020;34:209–220. doi: 10.1016/j.tmrv.2020.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li C, Li J, Ni H. Crosstalk between platelets and microbial pathogens. Front Immunol. 2020;11:1962. doi: 10.3389/fimmu.2020.01962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rapkiewicz AV, Mai X, Carsons SE, Pittaluga S, Kleiner DE, Berger JS, Thomas S, Adler NM, Charytan DM, Gasmi B, et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine. 2020;24:100434. doi: 10.1016/j.eclinm.2020.100434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rondina MT, Tatsumi K, Bastarache JA, Mackman N. Microvesicle tissue factor activity and interleukin-8 levels are associated with mortality in patients with influenza A/H1N1 infection. Crit Care Med. 2016;44:e574–e578. doi: 10.1097/CCM.0000000000001584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Althaus K, Marini I, Zlamal J, Pelzl L, Haeberle H, Mehrlaender M, Hammer S, Schulze H, Bitzer M, Malek N, et al. Severe covid-19 infection is associated with increased antibody-1 mediated platelet apoptosis. Blood. 2021. doi: 10.1101/2020.09.03.20187286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheng YW, Chao TL, Li CL, Chiu MF, Kao HC, Wang SH, Pang YH, Lin CH, Tsai YM, Lee WH, et al. Furin inhibitors block SARS-CoV-2 spike protein cleavage to suppress virus production and cytopathic effects. Cell Rep. 2020;33:108254. doi: 10.1016/j.celrep.2020.108254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hasan A, Paray BA, Hussain A, Qadir FA, Attar F, Aziz FM, Sharifi M, Derakhshankhah H, Rasti B, Mehrabi M, et al. A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J Biomol Struct Dyn. 2021;39:3025–3033. doi: 10.1080/07391102.2020.1754293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schoergenhofer C, Schwameis M, Gelbenegger G, Buchtele N, Thaler B, Mussbacher M, Schabbauer G, Wojta J, Jilma-Stohlawetz P, Jilma B. Inhibition of protease-activated receptor (PAR1) reduces activation of the endothelium, coagulation, fibrinolysis and inflammation during human endotoxemia. Thromb Haemost. 2018;118:1176–1184. doi: 10.1055/s-0038-1655767 [DOI] [PubMed] [Google Scholar]
- 82.Cheng JW. Impact of selective platelet inhibition in reducing cardiovascular risk - role of vorapaxar. Vasc Health Risk Manag. 2016;12:263–268. doi: 10.2147/VHRM.S81342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Han X, Nieman MT. PAR4 (Protease-Activated Receptor 4): PARticularly important 4 antiplatelet therapy. Arterioscler Thromb Vasc Biol. 2018;38:287–289. doi: 10.1161/ATVBAHA.117.310550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li S, Tarlac V, Hamilton JR. Using PAR4 inhibition as an anti-thrombotic approach: why, how, and when? Int J Mol Sci. 2019;20:E5629. doi: 10.3390/ijms20225629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rovai ES, Alves T, Holzhausen M. Protease-activated receptor 1 as a potential therapeutic target for covid-19. Exp Biol Med (Maywood). 2021;246:688–694. doi: 10.1177/1535370220978372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hosokawa K, Ohnishi T, Miura N, Sameshima H, Koide T, Tanaka KA, Maruyama I. Antithrombotic effects of PAR1 and PAR4 antagonists evaluated under flow and static conditions. Thromb Res. 2014;133:66–72. doi: 10.1016/j.thromres.2013.10.037 [DOI] [PubMed] [Google Scholar]
- 87.Wu CC, Huang SW, Hwang TL, Kuo SC, Lee FY, Teng CM. YD-3, a novel inhibitor of protease-induced platelet activation. Br J Pharmacol. 2000;130:1289–1296. doi: 10.1038/sj.bjp.0703437 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.