

Abstract
Aim:
Alcohol intake alters DNA methylation profiles and methylation might mediate the association between alcohol and disease, but limited number of positive CpG sites repeatedly replicated.
Materials & methods:
In total, 57 monozygotic (MZ) twin pairs discordant for alcohol drinking from the Chinese National Twin Registry and 158 MZ and dizygotic twin pairs in the Swedish Adoption/Twin Study of Aging were evaluated. DNA methylation was detected using the Infinium HumanMethylation450 BeadChip.
Results:
Among candidate CpG sites, cg07326074 was significantly correlated with drinking after adjusting for covariates in MZ twins in both datasets but not in the entire sample or dizygotic twins.
Conclusion:
The hypermethylation of cg07326074, located in the tumor-promoting gene C16orf59, was associated with alcohol consumption.
Keywords: : alcohol, C16orf59, CARD10, CpG sites, discordant twin design, DNA methylation, hypermethylation
Alcohol consumption affects human health, and there is evidence that alcohol intake results in DNA methylation changes associated with chronic diseases [1–8]. For example, cardiovascular disease-related genes (volume-regulating peptide gene vasopressin precursor [1] and atrial natriuretic peptide gene [1]), metabolism-related genes (SSTR4, ALDH1L2 and GAD1 [3]) and 70 CpG sites in liver function, BMI, and lipid metabolism pathways [8] are influenced by alcohol consumption. Several important malignant tumor-related genes (dehydrogenase family ALDH1L2 and ALDH1A2 [4,5], HRAS proto-oncogene [6], TP53 tumor suppressor [6] and other tumor-associated abnormal expression genes, including ER-α, E-cadherin, p15, MGMT, p14ARF, SFRP1, fusel 18, Septin9 and p16INKa [7]) affected to varying degrees by alcohol consumption have also been identified. Those genes mentioned above were all reported the possibility in involving in the genesis of tumors previously. For example, genes SSTR4, ALDH1L2, GAD1, DBH and GABRP may participate in the biological process of alcohol dependence [3]. SSTR4 activates GPCRs as a regulatory peptide and SSTR4 was markedly high expressed in hepatocellular carcinoma [9], while the ALDH gene code for the alcohol metabolizing enzyme ALDH [10]. However, most research has focused on relatively small populations or lacks verification. Alcohol consumption and DNA methylation are both influenced by age and genetic factors. It is hard to control for the effects of confounders, including age and genetic background, on methylation in the general population [11,12].
Discordant monozygotic (MZ) samples have a natural advantage for controlling potential confounders, like genetic background, and for exploring the relationship between environmental factors and differential epigenetic regulation [13,14]. However, few studies have evaluated alcohol-related DNA methylation in twins; in particular, studies of Asian twins are lacking [15].
In this study, we examined genome-wide CpG sites associated with alcohol consumption in Chinese discordant MZ twin pairs and validated in Swedish twins.
Materials & methods
Study design
A two-stage design was used, including discovery and validation stages, with Chinese National Twin Registry (CNTR) and Swedish Adoption/Twin Study of Aging (SATSA) populations, respectively. In the discovery stage, a genome-wide methylation association study was used to identify top CpG sites related to alcohol consumption, followed by the validation of these signals in the second stage.
For both analyses, ethics approval was obtained. The CNTR analysis was approved by the Peking University Biomedical Ethics Committee and the SATSA analysis was approved by the Regional Ethics Board at Karolinska Institute, Stockholm. Informed consent was obtained from all participants in both studies.
Discovery population
Study overview
In total, 57 twin pairs discordant for drinking surveyed in CNTR were included in the discovery stage. CNTR is the largest twin registry in China and in Asia, as described in detail elsewhere [16]. Drinking status was obtained by a self-reported questionnaire. Twins discordant for alcohol drinking were defined as twins with a difference in daily alcohol consumption of ≥5 g. Twins aged ≤18 years, pregnant, without a blood sample and unpaired were excluded from our analysis. Finally, 57 discordant MZ twin pairs met the inclusion criteria.
Measurements of alcohol consumption & other covariates
The CNTR standardized questionnaire was used to collect twin information in the discovery population, including the following variables: age, gender and lifestyle factors (including smoking, alcohol drinking, vegetable intake and physical activity). Anthropometric data (height and weight) was obtained by a physical examination. BMI was calculated as weight (kg) per height (m2). Drinking behavior was self-reported, including drinking habits in the past year, current drinking frequency, types of drinks consumed and capacity. Alcohol consumption was calculated based on the amount and frequency of drinking of different types of alcohol.
DNA methylation measurements
Whole blood samples from CNTR twins were stored in -80°C refrigerators. After thawing in a water bath, DNA was extracted using the Whole Blood DNA Extraction Kit (Bioteke Co., Beijing, China). Unmethylated cytosine was converted to thymine by using a bisulphite conversion kit named EZ DNA Methylation™ Kit (Zymo Research, CA, USA). Methylated cytosine could not be converted to thymine and could be distinguished. Whole genome DNA methylation levels were detected using the Infinium HumanMethylation450 (Infinium Methylation 450 K; Illumina, Inc. CA, USA). DNA methylation quality control and processing were performed using R package minfi, and more details has been described elsewhere before [17].
Validation population
Study overview
The SATSA study was initiated in 1984 as a sub-study of the Swedish Twin Registry [18]. The project collected twin pairs in Sweden for a longitudinal assessment of aging-related phenotypes with up to ten repeated waves of assessments until 2014. The components of the assessment included questionnaires concerning health-related behaviors and health status, and in-person testing (IPT) for physical and cognitive examination and the collection of blood sample. The questionnaires and IPTs were collected in parallel longitudinal collections, resulting in different years for the waves.
Status of alcohol drinking was repeatedly assessed in four waves denoted as the first (1984), second (1987) and third (1990) questionnaire assessments as well as the survey during the second IPT (1989). Genome-wide DNA methylation was repeatedly measured in some participants, leading to DNA methylation data distributed in the waves of the third (1992–1994), fifth (1999–2001), sixth (2002–2004), eighth (2008–2010), ninth (2010–2012) and tenth (2012–2014) IPT. For consistency with the definition applied in the CNTR study, the SATSA sample used for the validation analysis included MZ or dizygotic (DZ) twins discordant for alcohol consumption, defined as a difference in daily alcohol intake within twin pairs ≥5 g in at least one wave. Subjects had at least one methylation measurement.
There were 59 singletons and 238 twin pairs (in total, 535 participants) with at least one measurement of DNA methylation, with 1399 total measurements. The following cases were removed: singletons (59 participants), pairs in which both individuals did not drink in any waves of the questionnaire investigation (13 pairs) or only one of two had drinking data (31 pairs), and twins whose alcohol drinking differences were less than 5 g/d (36 pairs). In total, 158 twin pairs were involved in the validation analyses, including 56 MZ twin pairs and 102 DZ twin pairs.
Alcohol consumption measurement
Alcohol consumption was investigated by the questionnaire survey. The items related to alcohol drinking in the questionnaire were whether subjects ever drank alcoholic beverages (yes or no) and questions about the frequency and amount of drinking for different types of alcohol separately, including beer, wine (red wine or white wine) and hard liquor. There were nine options for drinking frequency: never, once a year or less, two to six-times a year, once a month, twice a month, once a week, twice a week, every other day and every day. There were seven options for volumes of beer from one glass or less to one to six bottles, six options for wine (i.e., 10 cl, 20 cl, 37 cl, 60 cl, 75 cl, and numbers of bottles if drinking more than one bottle), and nine options for hard liquor (i.e., 4 cl, 6 cl, 8 cl, 12 cl, 18 cl, 37 cl, 60 cl, 75 cl, or the exact amount of bottles if drinking more than one bottle).
Referring to the alcohol standards for different types of alcohol [19], one bottle of beer (12 ounces, 5%) contains 14 g alcohol, one bottle of wine (75 cl = 25.3 ounces, 12%) contains approximately 70.8 g alcohol, and one bottle of liquor (75 cl = 25.3 ounces, 40%) contains 236.1 g of alcohol. Volumes of beer, wine, and hard liquor were obtained separately for each wave and then summed and divided by 365.25 to generate the daily alcohol consumption (units: g/day) at each wave. The absolute differences in daily alcohol assumption within each twin pair in each wave were calculated to identify twins discordant for drinking in SATSA.
DNA methylation measurements
Genome-wide DNA methylation was measured using the 450K array and the Illumina EPIC Human Methylation Microarray (EPIC Array). CpG sites in both of the arrays were included in the analysis, and DNA methylation levels were processed with background correction, normalization and adjusted for cell counts and batch effects. Details are described in a previous study [20]. We used methylation β-value (the methylated probe intensity divided by the sum of the methylated and unmethylated probe intensities) for the data analysis. The datasets generated and analyzed in SATSA are available in Array Express database of EMBL-EBL (www.ebi.ac.uk/arrayexpress, accession number E-MTAB-7309).
Covariate measurements
Height, weight and smoking status were collected in each IPT. BMI was calculated as weight in kg divided by the square of height (m2). Smoking status was distinguished as nonsmoker, ex-smoker and current smoker. Moreover, type of bead-chip for DNA methylation measurements was also included as a covariate in the data analysis.
Statistical analysis
In the discovery stage, in the primary epigenome-wide association study (EWAS), the DNA methylation β-value was used as the outcome variable. The average daily alcohol consumption (g/day) was natural log-transformed and used as the predictor variable. The mixed effect model (ME) was used to analyze differentially methylated CpG sites (DMCs) related to drinking and to control for familial aggregation. In MZ twin pairs discordant for drinking, the moderated t-test with empirical Bayes priors was used to compare methylation levels and to identify drinking-related differentially methylated CpGs. An empirical Bayesian pairwise analysis can provide higher sensitivity for the detection of internal differential expression and can naturally match shared information for twin pairs [21]. Considering that MZ co-twins shared the same age and sex, multivariate models were adjusted for smoking, BMI and surrogate variable (SVA). A false discovery rate (FDR) of less than 0.05 was used to correct for multiple testing. Considering the correlation between smoking and drinking behavior [22,23], the sensitivity analysis was adjusted for cigarette smoking.
In the validation stage, significant CpG sites identified in the first stage were included in the analysis. A linear ME model was established for each CpG site to evaluate the association between DNA methylation and alcohol consumption, with DNA methylation level as the dependent variable and average daily alcohol consumption of the four waves as the independent variable. Alcohol drinking and DNA methylation were measured at different time points, and all measurements of alcohol drinking occurred before measurements of DNA methylation; therefore, the average alcohol consumption of the four waves, but not the single measurement of alcohol consumption, was taken as the analysis variable. Individual and twin identity numbers were entered as a random effect (i.e., every study wave nested in person and person nested in twin). The model was adjusted for age, sex, Beadchip type, BMI and smoking. The analysis was performed using the entire population, MZ twins only and DZ twins only. Two sensitivity analyses were performed: one analysis only included twins within whom two individuals had DNA methylation measurements at the same time point, and the other analysis used a cross-sectional design by using the last measurement of alcohol consumption and first measurement of DNA methylation. In the latter sensitivity analysis, all covariates were measured at the same time point as alcohol drinking and the definitions of independent and dependent variables and covariates were the same as those in the main analyses, except that only the twin identity number was used as a random effect in the mixed model. The significance level was set to 0.0125 for the Bonferroni correction (two-sided).
The final validation analysis was to validate the DNA methylation prediction model of alcohol consumption in both CNTR and SATSA populations. The alcohol predictor was established using DNA methylation at 144 CpG sites based on a large population sample [24] and was validated in another independent population [25].
In the remaining CpG sites after the quality control, 92 of the 144 CpG sites in the alcohol predictor could be found in SATSA population (Supplementary Table 5). A total of 144 CpG sites in the alcohol predictor were found in CNTR population (Supplementary Table 6). We first calculated alcohol predictor for each participant using the formula below,
Where i represents the serial number of CpG site, n represents the total number of CpG sites in the alcohol predictor, β represents the CpG site coefficients from Liu et al. study, and CpG represents the DNA methylation β-value at the corresponding CpG sites from either CNTR or SATSA populations. We fitted exactly the same ME model as in the main analysis except that alcohol predictor was used as the dependent variable. The significance level of p-value was set to 0.05 (two-sided).
All analyses were performed using Stata 16.0 (StataCorp LLC, TX, USA).
Results
General characteristics of study samples
The discovery population (CNTR) consisted of 57 twin pairs discordant for drinking. A total of 86% of participants were male and aged 19–74 years. The average daily alcohol consumption was 21.5 g, and 67.5% of subjects reported current drinking (Table 1).
Table 1. . General characteristics of the two study samples.
| CNTR (discovery) | SATSA (validation) | |
|---|---|---|
| n of participants (MZ twin pairs, DZ twin pairs) | 114 (57, 0) | 316 (56, 102) |
| Age (years), mean (SD) | 46.4 (11.5) | IPT3: 66.1 (8.8) IPT5: 66.8 (9.8) IPT6: 69.3 (8.6) IPT8: 73.6 (7.8) IPT9: 75.0 (7.3) IPT10: 76.8 (6.8) |
| Male (%) | 98 (86.0) | 160 (50.6) |
| Average daily alcohol consumption (g), median (IQR) | 21.5 (0–33.6) | 16.8 (4.7–42.0) |
| Current smoking (%) | 39 (34.2) | IPT3: 47 (26.9) IPT5: 50 (24.4) IPT6: 35 (20.8) IPT8: 26 (15.6) IPT9: 20 (14.9) IPT10: 4 (12.9) |
| BMI (kg/m2), mean (SD) | 25.6 (3.7) | IPT3: 25.8 (3.7) IPT5: 26.1 (3.9) IPT6: 26.4 (3.8) IPT8: 26.7 (3.8) IPT9: 26.7 (4.0) IPT10: 26.5 (3.4) |
| WC (cm), mean (SD) | 89.8 (9.9) | - |
| Current drinking (%) | 77 (67.5) | 306 (96.8) |
CNTR: Chinese National Twin Registry; DZ: Dizygotic twin; IPT: In-person testing; IQR: Interquartile range; MZ: Monozygotic twin; SATSA: Swedish Adoption/Twin Study of Aging; SD: Standard deviation; WC: Waist circumference.
In the SATSA sample, there were 56 MZ and 102 DZ twin pairs with discordant alcohol consumption. 50.6% of the participants were male and the mean age ranged from 66.1 to 76.8 years in different waves of IPTs. The average alcohol consumption was 16.8 g/d, and 96.8% of the participants reported current drinking during the investigation (Table 1).
EWAS results for CNTR in the discovery stage
Mixed effect model
Taking the average daily alcohol consumption (g/day) as the independent variable, three CpG sites (cg21716444, cg07326074 and cg04163847) were significant (FDR = 1.71×10-2,1.71×10-2 and 2.48×10-2, respectively) in model 1 after adjusting for age, sex, BMI, project area and SVA agent variables as the fixed effects and twin ID as the random effects. A Manhattan plot was generated to visualize the -log10 p-values for all CpG sites and their locations (the p threshold of 1.00×10-6 is shown as a red line) (Figure 1).
Figure 1. . Genome-wide CpG sites associated with alcohol intake.
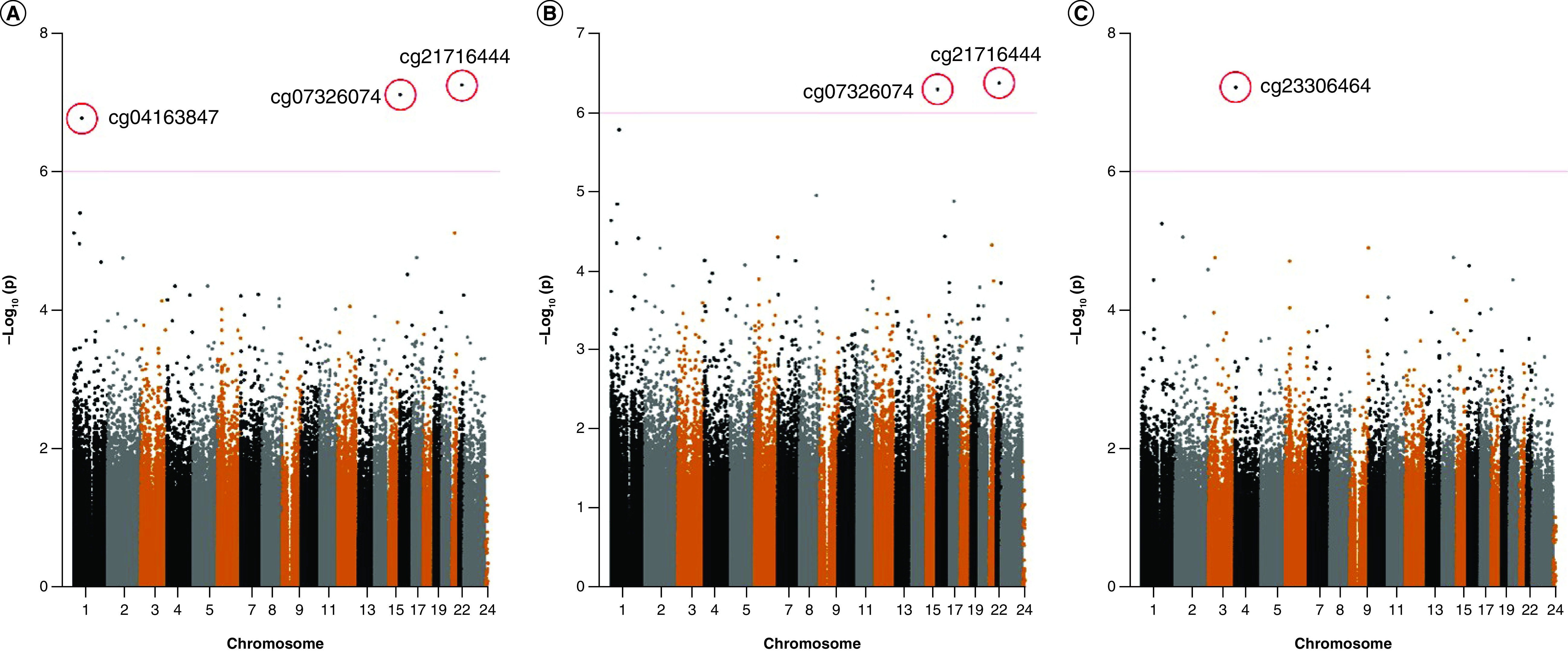
(A) Discovery population, model 1, adjusting age, sex, BMI and surrogate variable as mixed effect, twin identity number as random effect; (B) discovery population, model 2, additional adjusted with smoking based on model 1; (C) difference ≥40 g/day group, model 1,adjusting age, sex, BMI and surrogate variable as mixed effect, twin identity number as random effect.
The lowest p-value was obtained for cg21716444 located in the caspase recruitment domain 10 (CARD10) gene on chromosome 22 (FDR=1.71×10-2). The top three CpG sites with the lowest p-values were cg07326074, cg21716444, and cg04163847 located in C16orf59, CARD10, and an unknown gene, respectively. The top ten sites with the lowest p-values and their locations are shown in Table 2.
Table 2. . Top ten CpG sites associated with alcohol drinking (discovery population, model 1).
| Site | Chromosome | Location on the chromosome | Gene | Relationship with gene | Regression coefficients | Standard error | Original p-value | FDR |
|---|---|---|---|---|---|---|---|---|
| cg21716444* | 22 | 36245196 | CARD10 | TSS200 | 1.02E-03 | 1.29E-04 | 5.56E-08 | 1.71E-02 |
| cg07326074* | 16 | 2450389 | C16orf59 | Body | 3.39E-04 | 4.39E-05 | 7.71E-08 | 1.71E-02 |
| cg04163847* | 1 | 59395182 | - | - | -9.20E-04 | 1.25E-04 | 1.68E-07 | 2.48E-02 |
| cg21268223 | 1 | 45761497 | PRDX1 | TSS1500 | -1.04E-03 | 1.73E-04 | 3.93E-06 | 4.35E-01 |
| cg20256117 | 1 | 1083198 | - | - | 7.67E-04 | 1.34E-04 | 7.66E-06 | 5.66E-01 |
| cg06464111 | 21 | 31853603 | TIAM1 | TSS1500 | 2.14E-04 | 3.73E-05 | 7.67E-06 | 5.66E-01 |
| cg13946806 | 1 | 41742149 | - | - | -3.26E-04 | 5.83E-05 | 1.10E-05 | 6.93E-01 |
| cg24613923 | 17 | 31115636 | C17orf50 | Body | 1.14E-04 | 2.11E-05 | 1.74E-05 | 8.67E-01 |
| cg09071112 | 2 | 112529645 | TMEM87B | 1stExon | 1.10E-04 | 2.05E-05 | 1.76E-05 | 8.67E-01 |
| cg18384097 | 1 | 200396189 | PTPN7; PTPN7 | TSS1500; 5’UTR | 2.19E-04 | 4.11E-05 | 2.01E-05 | 8.90E-01 |
- The gene is unknown.
*CpGs with FDR <0.05.
5’UTR: 5’ non-coding region; Body: Gene body; FDR: False discovery rate; TSS: Transcription start site unit base pair range.
After adjusting for age, sex, BMI, project area, smoking, vegetable consumption, fruit consumption, red meat intake, physical activity and SVA agent variables as fixed effects and twin ID as a random effect in model 2, the FDR for all CpG sites exceeded 0.05. A Manhattan plot showing each CpG locus across the genome is presented in Figure 1B. The two lowest p-values were obtained for cg21716444 and cg07326074, as shown in Figure 2 above the red reference line.
Figure 2. . Volcanic plot of monozygotic discordant for drinking.
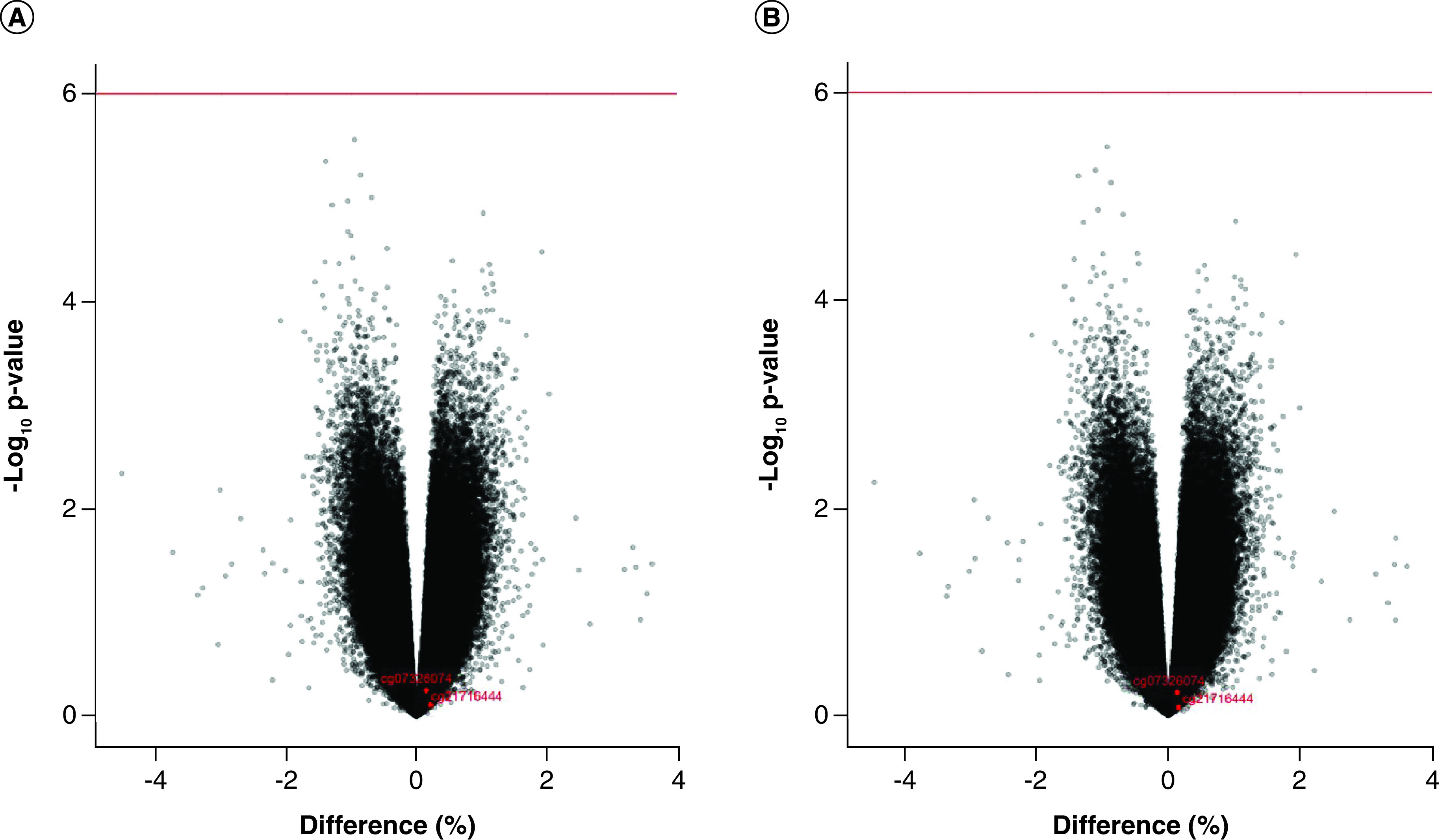
(A) Discovery population, model 1; (B) Discovery population, model 2. (A) Volcanic plot in discovery population in model 1 adjusting age, sex, BMI and SVA as mixed effect, twin identity number as random effect. (B) model 2 additional adjusted with smoking based on model 1. The abscissa indicated the ratio of difference of the average β-value between the high drinking group and low drinking group for any CpG site to the low drinking group. The ordinate represented -log10 p-value calculated by Empirical Bayes paired moderated t-test.
SVA: Surrogate variable.
We further filtered twin pairs into four groups in which the differences in average daily alcohol consumption were ≥10, ≥20, ≥30 and ≥40 g/day for the ME model to improve the detection power. For a difference of ≥40 g/day using model 1 adjusting for age, sex, BMI, project area and SVA agent variables as fixed effects and twin ID as a random effect, the FDR was 0.02 for cg23306464 (located in an unknown gene). A Manhattan plot is displayed in Figure 1C and the top CpG sites with the highest p-values are shown in Table 3. The information of significant CpG sites-associated with alcohol consumption using mixed effect model in model 1 and model 2 were presented in Supplementary Table 1.
Table 3. . Top ten CpG sites associated with alcohol drinking (difference ≥40 g/day, model 1).
| Site | Chromosome | Location on the chromosome | Gene | Relationship with gene | Regression coefficients | Standard error | Original p-value | FDR |
|---|---|---|---|---|---|---|---|---|
| cg23306464* | 4 | 4919257 | - | - | -8.96E-05 | 2.79E-06 | 6.04E-08 | 2.67E-02 |
| cg07015494 | 1 | 148123673 | HIST2H2BE; HIST2H2AC; HIST2H2BE | 1stExon; TSS1500; 3’UTR | -7.38E-05 | 4.93E-06 | 5.63E-06 | 1.00E+00 |
| cg08701028 | 2 | 54734435 | SPTBN1 | Body | -8.57E-05 | 6.18E-06 | 8.78E-06 | 1.00E+00 |
| cg09420761 | 9 | 135272397 | ADAMTS13; REXO4 | Body; Body | -1.50E-04 | 1.15E-05 | 1.26E-05 | 1.00E+00 |
| cg23335576 | 14 | 103079480 | - | - | 1.73E-04 | 1.40E-05 | 1.73E-05 | 1.00E+00 |
| cg00244462 | 3 | 49921277 | - | - | -1.81E-04 | 1.47E-05 | 1.74E-05 | 1.00E+00 |
| cg13069441 | 6 | 32047301 | STK19 | TSS1500 | 2.27E-04 | 1.88E-05 | 1.95E-05 | 1.00E+00 |
| cg10276272 | 16 | 10179323 | GRIN2A | Body | -1.45E-04 | 1.23E-05 | 2.27E-05 | 1.00E+00 |
| cg07644939 | 2 | 241625331 | SNED1 | Body | 1.82E-04 | 1.58E-05 | 2.60E-05 | 1.00E+00 |
| cg05320259 | 20 | 25318897 | ABHD12 | Body | 8.22E-05 | 7.58E-06 | 3.64E-05 | 1.00E+00 |
- The gene is unknown.
*CpGs with FDR <0.05.
5’UTR: 5’ noncoding region; Body: Gene body; FDR: False discovery rate; TSS: Transcription start site unit base pair range.
The moderated t-test with empirical Bayes priors
The individual with the highest average daily alcohol consumption (g/day) in each twin set was included in the high-drinking group. The other twin in the pair was included in the low-drinking group. In the moderated t-test with empirical Bayes priors, alcohol consumption group was treated as a two-category independent variable, and the DNA methylation level was the dependent variable.
We also filtered twin pairs with respect to average daily alcohol consumption differences (≥10, ≥20, ≥30 and ≥40 g/day) for an empirical Bayes paired moderated t-test. The FDR values for all CpG sites after correction exceeded 0.05, and no significant drinking-related CpG site was found. Volcano plots were generated to show differences in methylation levels between high-drinking and low-drinking groups at each CpG sites genome-wide (model 1 in Figure 2A, model 2 in Figure 2B). The information of significant CpG sites-associated with alcohol consumption using the moderated t-test with empirical Bayes priors in model 1 and model 2 were presented in Supplementary Table 2.
Sensitivity analysis
In the discovery stage, the average daily alcohol consumption was included as a continuous independent variable, and DNA methylation was included as a dependent variable. We adjusted for previous cigarette consumption (years of smoking × average annual cigarette packs) in ME model 1 (adjusting for age, sex, BMI, project area and SVA agent variables as fixed effects and twin ID as a random effect) in the whole discovery population. Similarly, a sensitivity analysis was conducted for ME model 1 adjusted for the same covariates among 17 twin pairs with an average daily alcohol consumption difference of ≥40 g/d.
We found that the correlation between drinking and cg21716444 or cg07326074 was still robust after adjusting for previous cigarette consumption (Table 4).
Table 4. . Sensitivity analyses for the CpG sites after adjusting the previous cigarettes consumption.
| CpG sites | Gene | Original p-value | FDR |
|---|---|---|---|
| cg21716444* | CARD10 | 8.36E-08 | 2.71E-02 |
| cg07326074* | C16orf59 | 1.23E-07 | 2.71E-02 |
| cg04163847* | - | 3.73E-07 | 5.51E-02 |
| cg23306464 | - | 8.83E-06 | 1.00E-00 |
*CpGs with FDR <0.05.
FDR: False discovery rate.
Validation using the SATSA population
Four CpG sites were included in validation phase in the SATSA population: cg21716444, cg07326074, cg04163847 and cg23306464. We did not observe significant associations between each CpG site and alcohol consumption in the entire population, MZ twins only or DZ twins only, irrespective of covariates (Table 5). In the first sensitivity analysis including only twins with methylation data for both individuals in the same wave, no statistically significant associations were observed between CpG sites and alcohol consumption (Supplementary Table 3).
Table 5. . Validation analysis between significant CpG sites found in Chinese National Twin Registry and alcohol consumption in The Swedish Adoption/Twin Study of Aging.
| model 1 | model 2 | |||||
|---|---|---|---|---|---|---|
| β | 95% CI | p-value | β | 95% CI | p-value | |
| Overall samples | ||||||
| cg21716444 | -1.58E-06 | -8.35E-06, 5.19E-06 | 0.648 | -2.20E-06 | -8.98E-06, 4.59E-06 | 0.526 |
| cg07326074 | 1.10E-05 | -1.37E-05, 3.57E-05 | 0.383 | 1.18E-05 | -1.30E-05, 3.65E-05 | 0.351 |
| cg04163847 | 1.14E-06 | -9.94E-05, 1.02E-04 | 0.982 | 8.29E-07 | -9.98E-05, 1.01E-04 | 0.987 |
| cg23306464 | 1.68E-06 | -1.55E-05, 1.88E-05 | 0.848 | 2.59E-06 | -1.45E-05, 1.97E-05 | 0.767 |
| Only monozygotic twin | ||||||
| cg21716444 | -2.45E-06 | -1.21E-05, 7.22E-06 | 0.619 | -3.09E-06 | -1.28E-05, 6.57E-06 | 0.531 |
| cg07326074 | 1.98E-05 | -1.51E-05, 5.48E-05 | 0.266 | 2.03E-05 | -1.48E-05, 5.53E-05 | 0.257 |
| cg04163847 | 4.38E-05 | -7.84E-05, 1.66E-04 | 0.482 | 4.30E-05 | -7.93E-05, 1.65E-04 | 0.491 |
| cg23306464 | 8.35E-06 | -1.24E-05, 2.91E-05 | 0.430 | 8.13E-06 | -1.26E-05, 2.89E-05 | 0.443 |
| Only dizygotic twin | ||||||
| cg21716444 | 1.04E-06 | -8.66E-06, 1.07E-05 | 0.833 | 8.23E-08 | -9.68E-06, 9.84E-06 | 0.987 |
| cg07326074 | -4.73E-06 | -3.94E-05, 2.99E-05 | 0.789 | -3.55E-06 | -3.84E-05, 3.13E-05 | 0.842 |
| cg04163847 | -6.16E-05 | -2.13E-04, 9.00E-05 | 0.426 | -6.39E-05 | -2.16E-04, 8.80E-05 | 0.410 |
| cg23306464 | -4.30E-06 | -3.07E-05, 2.21E-05 | 0.750 | -1.73E-06 | -2.81E-05, 2.47E-05 | 0.898 |
model 1: mixed effect model with DNA methylation level as dependent variable, and average daily alcohol consumption as independent variable as fixed effect. Person’s identity number and twin identity number was entered as random effect. Age, gender, type of chip and BMI were adjusted as covariates in the model as fixed effects. model 2: additional adjusted with smoking based on model 1. β means the change of methylation level for every 1 g/d amount of change in alcohol drinking.
DZ: Dizygotic twin; MZ: Monozygotic twin.
In a cross-sectional sensitivity analysis, 175 (55.4%) participants were evaluated for DNA methylation for the first time in the third wave of IPT, 106 (33.5%) participants were first tested in the fifth wave, 26 (8.2%) were first tested in the sixth wave, and 7 (2.2%) and 2 (0.6%) participants were first tested in the eighth and ninth waves of IPT, respectively. We found marginally significant associations in MZ twins between cg07326074 and alcohol consumption in model 1 with adjustment for age, sex and BMI (β = 2.91×10-5, 95% CI [-5.46×10-6, 6.37×10-5], p = 0.099), and similar results for model 2 with additional adjustment for smoking based on model 1; however, we did not find significant results for the entire sample or DZ twins (Supplementary Table 4).
Validation using the alcohol predictor
We established DNA methylation based alcohol predictor using 92 CpG sites referred to Liu et al. study, and found that alcohol predictor was significantly associated with alcohol consumption in the entire population, as well as in MZ twins and DZ twins in SATSA sample (Supplementary Table 5). However, the association was not found between alcohol predictor and alcohol consumption in CNTR either in ME or twin paired analysis (Supplementary Table 6). And the CpGs used in the alcohol predictor were listed in Supplementary Table 7.
Discussion
In the CNTR study, after adjusting for genetic factors and common environmental factors, four CpG sites (cg21716444, cg07326074, cg04163847 and cg23306464) were associated with alcohol drinking. Both cg21716444 and cg07326074 were hypermethylated in the high drinking group, while cg04163847 and cg23306464 were hypomethylated in the high drinking group. The correlations between drinking and cg21716444 or cg07326074 were still robust after adjusting for previous cigarette consumption. The association of cg07326074 with alcohol drinking was validated in the SATSA study, although the association was only marginally significant. Both studies were based on twins from the general population and both yielded statistically significant results for MZ twins. Although CNTR used a cross-sectional design, SATSA was a longitudinal study in which all measurements of alcohol consumption were obtained before measurements of methylation levels; therefore, the current results indicated that alcohol drinking may alter DNA methylation, especially in the short term.
Most recent studies use a case–control design and only compare patients with alcoholism to normal controls. Relatively fewer studies have focused on the relationship between quantitative alcohol consumption and DNA methylation modifications [24,26,27].
Correlations between alcohol drinking and the sites identified in this study have not been reported to date. Cg21716444 is located in CARD10, which is involved in tumor invasion and metastasis [28,29]. Cg07326074 is located in C16orf59 (i.e.,TEDC2), which has a significantly different methylation patterns in the male schizophrenia population than in controls [30].
The CARMA3 gene, in which the significant CpG site cg21716444 was located, is involved in NF-κB signaling [31]. NF-κB is an important downstream target of redox signaling. Alcohol can activate the NF-κB signaling pathway in triple-negative breast cancer cells [28]. Studies have shown that the activation of the NF-κB pathway is associated with the alcohol-induced progression and metastasis of human hepatocellular carcinoma [32]. Previous studies have suggested that alcohol can activate NF-κB-related signaling pathways and further promote the progression, invasion and metastasis of human hepatoma cells. In this study, we found that the cg21716444 site in the CARMA3 gene is differentially methylated in the drinking population. Accordingly, the CARMA3 gene may act as an intermediator in alcohol-related progression, invasion and metastasis of tumor cells. The significant CpG site cg07326074 is located in the TEDC2 gene and the observed correlation between this site and alcohol consumption was validated in SATSA. TEDC2 exhibits significant differences in methylation profiles between the male schizophrenia population and the controls [30]. TEDC2 genes/transcripts/exonic regions are candidates for the tumorigenesis of lung adenocarcinoma and for the construction of accurate classification systems [33]. The human protein atlas reported that TEDC2 is the prognostic marker in liver cancer (unfavorable) and lung cancer (unfavorable), though the mechanism behind is still unclear. However, further studies are needed to clarify the function of TEDC2.
Liang et al. separately identified 83 phosphatidylethanol (an objective measure of alcohol consumption)-associated CpGs, but no CpG significantly associated with self-reported alcohol consumption in people who reported hazardous alcohol drinking [34]. Unfortunately, there was no overlap of CpG sites in our study. The difference of ethnicity and the definition of alcohol consumption, and the selection criteria of participants might account for the heterogeneity of results. And phosphatidylethanol only provided reliably detects ethanol levels up to 21 days after the last drink [35], which presented less information on long-term drinking status than self-reported alcohol consumption. Similarly, the CpG sites we found in our study were not identified by Lohoff et al., though it was the largest and most comprehensive EWAS on alcohol use disorder to date [36]. Our study is a population-based twin study and the alcohol consumption were self-reported while the participants in Lohoff’s study were recruited at the National Institute on Alcohol Abuse and Alcoholism. The differences in cumulative alcohol consumption possibly contributed to the heterogeneity of the results.
Furthermore, the association between alcohol predictor and alcohol consumption was successfully validated in SATSA population but not in CNTR. The alcohol predictor was built based on the European population [24] and it may be due to the ethnicity that the alcohol predictor could be applied and validated in SATSA but failed in CNTR. The relatively small sample size in CNTR was also a possible reason for the null finding in the validation phase.
Although important results were obtained, our study had the following limitations. The sample size of this study was relatively small. The sample size has been estimated for the EWAS study design of discordant twins with given epigenetic risk effect sizes (EWAS genome-wide significance threshold p = 1.00×10-6) [37,38]. If a genome-wide methylation difference was ⋝15%, 50 pairs of MZ samples would have 80% power to detect a difference in t-test. Despite the limited difference in genome-wide methylation levels in our research, we found four significant CpG sites. To our knowledge, this is the largest MZ twin study of alcohol-related methylation CpG sites, providing new insights into the pathophysiological effects of alcohol consumption. In addition, owing to the cross-sectional design of this study, our research may not be generalizable beyond blood markers and causality could not be inferred. Alcohol affects tumor invasion and metastasis via certain key genes [39–42]. Future studies are warranted to investigate whether drinking-related methylation in the blood and other tissues affects CARMA3 and TEDC2 gene function, and to investigate how drinking-related epigenetic modifications influence the progression of diseases, such as cancer. In addition, we lack the wet lab data to validate the biological pathway of our result. However, existing evidence has already demonstrated the role of CARD10 and C16orf59 genes in promoting liver diseases [43–45].
Conclusion
In this study, a two-stage design was used to explore genome-wide drinking-related DNA methylation sites in twins. Four significant CpG sites were found to be associated with drinking, and cg07326074 was validated in SATSA. This locus should be further validated in other populations or using larger sample sizes. DNA methylation might mediate the relationship between alcohol drinking and diseases.
Future perspective
In this study, we explored the genome-wide association between alcohol consumption and DNA methylation in CNTR and validated the results in SATSA. In this discordant twin study, the hypermethylation of cg07326074, located in the tumor-promoting gene C16orf59, was associated with alcohol consumption. Those results demonstrated that alcohol consumption might have significant impact on the prognosis of tumor. Due to the potential reversibility of DNA methylation, the alcohol consumption-related CpG sites provide potential targets with practical value for new drug development and new intervention methods for tumor. In the future, the integrated analysis of transcription, translation and other omics studies might contribute to elucidate the mechanism of alcohol-related genes and disease. And advanced evidence from other population or larger sample sizes will improve the credibility of this study and find more specific alcohol consumption-related methylation sites.
Summary points.
DNA methylation might mediate alcohol consumption and health status, but most research has focused on relatively small populations or lacks verification.
Discordant monozygotic samples have a natural advantage for controlling potential confounders, like genetic background and early environment factors.
There is limited evidence from twin on the association of alcohol consumption and DNA methylation.
In this study, a two-stage design was used to explore genome-wide drinking-related DNA methylation sites in twins.
Four significant CpG sites were found to be associated with drinking, and cg07326074 was validated in Swedish Adoption/Twin Study of Aging.
In this discordant twin study, the hypermethylation of cg07326074, located in the tumor-promoting gene C16orf59, was associated with alcohol consumption.
Due to the potential reversibility of DNA methylation, the alcohol consumption-related CpG sites provide potential targets with practical value for new drug development and new intervention methods.
More multi-omics analyses or studies conducted in other populations or using larger sample sizes are needed to improve the credibility of the conclusion in the future.
Acknowledgments
We gratefully appreciate the support from the Centers of Disease Control and Prevention in Qingdao, Zhejiang, Jiangsu and Sichuan for their dedication that made this research possible. We acknowledge The Swedish Twin Registry for access to data. Methylation profiling on the Infinium MethylationEPIC BeadChip was performed by the SNP&SEQ Technology Platform in Uppsala (www.genotyping.se). The facility is part of the National Genomics Infrastructure (NGI) Sweden and Science for Life Laboratory.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/suppl/10.2217/epi-2021-0039
Financial & competing interests disclosure
This study was supported by National Natural Science Foundation of China (82073633, 81973126, 81573223) and special fund for Health scientific research in the public welfare (201502006, 201002007). SATSA was funded by NIH (grants AG04563 and AG10175), the MacArthur Foundation Research Network on Successful Aging, the Swedish Research Council for Working Life and Social Research (FAS; grants 97:0147:1B, 2009-0795) and the Swedish Research Council (825-2007-7460 and 825-2009-6141). This work was supported by the Swedish Research Council (2015-03255), The Foundation for Gamla Tjänarinnor (2018-00688), the KI Foundation, the Strategic Research Area in Epidemiology at Karolinska Institutet, Erik Rönnbergs donation for scientific studies in aging and age-related diseases. X Qin was supported by King Gustaf V’s and Queen Victorias Freemason Foundation, The Foundation for Gamla Tjänarinnor (2018-00688), and the China Scholarship Council for one year study at Karolinska Institutet between 2020 and 2021. The SNP&SEQ Platform is also supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation. The Swedish Twin Registry is managed by Karolinska Institutet and receives funding through the Swedish Research Council under the grant no 2017-00641. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing assistance was utilized in the production of this manuscript by Editage using funding from National Natural Science Foundation of China (82073633, 81973126, 81573223).
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Hillemacher T, Frieling H, Luber K et al. Epigenetic regulation and gene expression of vasopressin and atrial natriuretic peptide in alcohol withdrawal. Psychoneuroendocrinology 34(4), 555–560 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Lee BH, Choi MR, Chai YG, Lee JS. P.6.b.009 Changes of DNA methylation of the HERP gene promoter and its mRNA expression during alcohol detoxification. Eur. Neuropsychopharmacol. 23(1), S562–S563 (2013). [Google Scholar]
- 3.Zhao R, Zhang R, Li W et al. Genome-wide DNA methylation patterns in discordant sib pairs with alcohol dependence. Asia Pac. Psychiatry 5(1), 39–50 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Zhang R, Miao Q, Wang C et al. Genome-wide DNA methylation analysis in alcohol dependence. Addict. Biol. 18(2), 392–403 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Nicole H, Bryan AD, Thayer RE, Karoly HC, Niles O, Hutchison KE. Methylation of a CpG site near the ALDH1A2 gene is associated with loss of control over drinking and related phenotypes. Alcohol. Clin. Exp. Res. 38(3), 713–721 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hill SY, Rompala G, Homanics GE, Zezza N. Cross-generational effects of alcohol dependence in humans on HRAS and TP53 methylation in offspring. Epigenomics 9(9), 1189–1203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varela-Rey M, Woodhoo A, Martinez-Chantar ML, Mato JM, Lu SC. Alcohol, DNA methylation, and cancer. Alcohol Res. 35(1), 25–35 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu K, Montalvo-Ortiz JL, Zhang X et al. Epigenome-wide dna methylation association analysis identified novel loci in peripheral cells for alcohol consumption among European American male veterans. Alcohol. Clin. Exp. Res. 43(10), 2111–2121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie Y-M, Yan L-N, Wei B, Guo M-M, Tang C-W. [Correlation of somatostatin receptor expression in human hepatocellular carcinoma tissue to serum alpha-fetoprotein concentration]. Ai Zheng 26(7), 688–692 (2007). [PubMed] [Google Scholar]
- 10.Higuchi S, Matsushita S, Masaki T et al. Influence of genetic variations of ethanol-metabolizing enzymes on phenotypes of alcohol-related disorders. Ann. NY Acad. Sci. 1025, 472–480 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Kaminsky ZA, Thomas T, Sun-Chong W et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 41(2), 240–245 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Hasler R, Begun A, Freitag-Wolf S et al. Genetic control of global gene expression levels in the intestinal mucosa: a human twin study. Physiol. Genomics 38(1), 73–79 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Tan Q, Christiansen L, Von Bornemann Hjelmborg J, Christensen K. Twin methodology in epigenetic studies. J. Exp. Biol. 218(Pt 1), 134–139 (2015). [DOI] [PubMed] [Google Scholar]; • Provides methodology in epigenetic studies of twin study.
- 14.Castillo-Fernandez JE, Spector TD, Bell JT. Epigenetics of discordant monozygotic twins: implications for disease. Genome Med. 6(7), 60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Describes natural advantage of discordant monozygotic twins in epigenetic studies.
- 15.Ruggeri B, Nymberg C, Vuoksimaa E et al. Association of protein phosphatase PPM1G with alcohol use disorder and brain activity during behavioral control in a genome-wide methylation analysis. Am. J. Psychiatry 172(6), 543–552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao W, Cao W, Lv J et al. The Chinese National Twin Registry: a ‘gold mine’ for scientific research. J. Intern. Med. 286(3), 299–308 (2019). [DOI] [PubMed] [Google Scholar]; •• Provides description of Chinese National Twin Registry in more details.
- 17.Li C, Gao W, Gao Y et al. Age prediction of children and adolescents aged 6–17 years: an epigenome-wide analysis of DNA methylation. Aging 10(5), 1015–1026 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pedersen NL, Mcclearn GE, Plomin R, Nesselroade JR, Berg S, Defaire U. The Swedish adoption twin study of aging: an update. Acta Genet. Med. Gemellol. 40(1), 7–20 (1991). [DOI] [PubMed] [Google Scholar]
- 19.National Institutes of Health (NIH) National Institute on Alcohol Abuse and Alcoholism. What is a standard drink? (2021). https://www.niaaa.nih.gov/alcohols-effects-health/overview-alcohol-consumption/what-standard-drink/
- 20.Wang Y, Karlsson R, Lampa E et al. Epigenetic influences on aging: a longitudinal genome-wide methylation study in old Swedish twins. Epigenetics 13(9), 975–987 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trejo Banos D, Mccartney DL, Patxot M et al. Bayesian reassessment of the epigenetic architecture of complex traits. Nat. Commun. 11(1), 2865 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Develops methodology that can naturally match shared information for twin pairs named empirical Bayesian pairwise analysis.
- 22.Liu R, Chen L, Zhang F et al. Trends in alcohol intake and the association between socio-demographic factors and volume of alcohol intake amongst adult male drinkers in China. Int. J. Environ. Res. Public Health 16(4), 573 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu RY, Chen L, Zeng H et al. Tobacco and alcohol consumption rates among Chinese women of reproductive age in 2004–2011: rate and sociodemographic influencing factors. Int. J. Environ. Res. Public Health 16(1), 56 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu C, Marioni RE, Hedman AK et al. A DNA methylation biomarker of alcohol consumption. Mol. Psychiatry 23(2), 422–433 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Provides 144 CpG sites in the alcohol predictor. The study has focused on the relationship between quantitative alcohol consumption and DNA methylation modifications.
- 25.Yousefi PD, Richmond R, Langdon R et al. Validation and characterisation of a DNA methylation alcohol biomarker across the life course. Clin. Epigenetics 11(1), 163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Philibert RA, Penaluna B, White T et al. A pilot examination of the genome-wide DNA methylation signatures of subjects entering and exiting short-term alcohol dependence treatment programs. Epigenetics 9(9), 1212–1219 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perrier F, Viallon V, Ambatipudi S et al. Association of leukocyte DNA methylation changes with dietary folate and alcohol intake in the EPIC study. Clin. Epigenetics 11(1), 57 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Guo Y, Huang WJ et al. CARD10 is a novel caspase recruitment domain/membrane-associated guanylate kinase family member that interacts with BCL10 and activates NF-kappa B. J. Biol. Chem. 276(24), 21405–21409 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Wang F, Yang JL, Yu KK et al. Activation of the NF-kappa B pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma. Mol. Cancer 14(1), 10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rukova B, Staneva R, Hadjidekova S, Stamenov G, Milanova V, Toncheva D. Whole genome methylation analyses of schizophrenia patients before and after treatment. Biotechnol. Biotechnol. Equip. 28(3), 518–524 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao M, Howard EW, Parris AB, Guo Z, Zhao Q, Yang X. Alcohol promotes migration and invasion of triple-negative breast cancer cells through activation of p38 MAPK and JNK. Mol. Carcinog. 56(3), 849–862 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Wang F, Yang JL, Yu K et al. Activation of the NF-κB pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma. Mol. Cancer 14(1), 1–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides the evidence of CARD10 gene that interacts with BCL10 and activates NF-⋝B.
- 33.Hsu MK, Wu IC, Cheng CC et al. Triple-layer dissection of the lung adenocarcinoma transcriptome: regulation at the gene, transcript, and exon levels. Oncotarget 6(30), 28755–28773 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang X, Justice AC, So-Armah K, Krystal JH, Sinha R, Xu K. DNA methylation signature on phosphatidylethanol, not on self-reported alcohol consumption, predicts hazardous alcohol consumption in two distinct populations. Mol. Psychiatry (2020) (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides the information of TEDC2 gene that can be potentially important genes/transcripts/exonic regions for the tumorigenesis of lung adenocarcinoma.
- 35.Schröck A, Thierauf-Emberger A, Schürch S, Weinmann W. Phosphatidylethanol (PEth) detected in blood for 3 to 12 days after single consumption of alcohol–a drinking study with 16 volunteers. Int. J. Legal Med. 131(1), 153–160 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Lohoff FW, Roy A, Jung J et al. Epigenome-wide association study and multi-tissue replication of individuals with alcohol use disorder: evidence for abnormal glucocorticoid signaling pathway gene regulation. Mol. Psychiatry (2020) (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graw S, Henn R, Thompson JA, Koestler DC. pwrEWAS: a user-friendly tool for comprehensive power estimation for epigenome wide association studies (EWAS). BMC Bioinformatics 20(1), 218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai PC, Bell JT. Power and sample size estimation for epigenome-wide association scans to detect differential DNA methylation. Int. J. Epidemiol. 44(4), 1429–1441 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang J, Tan W, Ling Z et al. Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nat. Commun. 8, 15290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Floris A, Luo J, Frank J et al. Star-related lipid transfer protein 10 (STARD10): a novel key player in alcohol-induced breast cancer progression. J. Exp. Clin. Cancer Res. 38(1), 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu M, Ren Z, Wang X et al. ErbB2 and p38gamma MAPK mediate alcohol-induced increase in breast cancer stem cells and metastasis. Mol. Cancer 15(1), 52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu M, Wang S, Qi Y et al. Role of MCP-1 in alcohol-induced aggressiveness of colorectal cancer cells. Mol. Carcinog. 55(5), 1002–1011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hou H, Li WX, Cui X, Zhou DC, Zhang B, Geng XP. CARMA3/NF-κB signaling contributes to tumorigenesis of hepatocellular carcinoma and is inhibited by sodium aescinate. World J. Gastroenterol. 25(36), 5483–5493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Beek M, Oravecz-Wilson KI, Delekta PC et al. Bcl10 links saturated fat overnutrition with hepatocellular NF-kB activation and insulin resistance. Cell Rep. 1(5), 444–452 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papatheodorou I, Moreno P, Manning J et al. Expression Atlas update: from tissues to single cells. Nucleic Acids Res. 48(D1), D77–d83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]