

Abstract
Background:
Although divalent zinc (Zn2+) is known to bind FXII and affect its sensitivity to autoactivation, little is known about the role of Zn2+ in the binding of FXII to platelets, where FXII activation is thought to occur in vivo, and the function of Zn2+ during thrombus formation following vascular injury remains poorly understood.
Objectives:
To evaluate the role of Zn2+ in platelet-dependent FXIIa generation.
Methods:
FXII binding to platelets and FXII activation by stimulated platelets were assessed using flow cytometry and a platelet-dependent thrombin generation assay. The mouse cremaster laser injury model was used to evaluate the impact of Zn2+ chelation on thrombus formation in vivo.
Results:
Our data demonstrate that stimulated platelets support FXII-dependent thrombin generation and that FXII activation by platelets requires the presence of Zn2+. By contrast, thrombin generation by stimulated endothelial cells occurred independently of FXII and Zn2+. Using flow cytometry, we found that FXII-FITC binds to the surfaces of stimulated platelets in a specific and Zn2+-dependent manner, whereas resting platelets demonstrated minimal binding. Other physiologically-relevant divalent cations are unable to support this interaction. Consistent with these findings, the Zn2+-specific chelator CaEDTA confers thromboprotection in the mouse cremaster laser injury model without causing increased bleeding. We observed an identical phenotype in FXII null mice tested in the same system.
Conclusions:
Our results suggest a novel role for Zn2+ in the binding and activation of FXII at the platelet surface, an interaction that appears crucial to FXII-dependent thrombin generation but dispensable for hemostasis.
Keywords: Factor XII, Contact Pathway, Zinc, Platelet-dependent thrombin generation, FXII knockout mice
Introduction
Platelet-dependent thrombin generation is a key contributor to thrombosis and is the target of many currently available antithrombotic therapies. It is well-established that platelet procoagulant activity is due in part to the ability of stimulated platelet surfaces to ligate and co-localize humoral coagulation factors in a way that greatly accelerates thrombin generation. In addition to this function, it has been known since the 1970s that platelets can directly initiate the coagulation cascade via activation of factor XII (FXII), a circulating 80,000 kDa serine protease [1, 2]. Once FXII undergoes cleavage activation to factor XIIa (FXIIa), it is able to trigger the contact pathway of coagulation and downstream thrombin generation as well as activation of the kinin-kallikrein pathway [3, 4]. Interestingly, severe congenital deficiency of FXII in humans is not associated with a bleeding diathesis, and the physiologic relevance of FXII activation by platelets remained unclear for decades. More recently however, FXII blockade or deficiency has been shown in several animal models to protect against thrombosis [5–8], highlighting the potential therapeutic value of targeting FXII to prevent thrombosis without adversely affecting hemostasis. These observations suggest that the process of platelet-dependent FXIIa generation may be linked to pathologic processes in vivo and has intensified interest in understanding the interaction of platelets and FXII. Nevertheless, the mechanism by which FXII is recruited and activated by platelets remains poorly understood.
Cationic zinc (Zn2+) is a cofactor that plays an important role in several aspects of the contact system, including the binding of FXII and high molecular weight kininogen (HK) to endothelial cells [9, 10] and potentiation of FXII autoactivation upon binding to artificial surfaces [11–15]. FXII has been shown to bind weakly to stimulated washed platelets [16, 17], but the interaction of platelets and FXII in the presence of physiologic concentrations of Zn2+ has not been previously investigated, even though Zn2+ is likely required for FXII-dependent processes in vivo. Likewise, the function of Zn2+ in platelet-dependent FXIIa generation has not been studied. Here we show that FXII binding to the surfaces of stimulated platelets is greatly enhanced by concentrations of Zn2+ similar to those found in plasma. Further, we demonstrate that the Zn2+ typically available in normal platelet-rich plasma (PRP) is sufficient to support platelet-dependent FXIIa generation and that depletion of Zn2+ impairs thrombus formation in vivo. Taken together, our data indicate that Zn2+ plays a key role in the regulation of FXII recruitment and activation at the platelet surface during thrombus formation.
Materials and Methods
Materials
The PAR-1 agonist peptide (SFLLRN), ethylenediaminetetraacetic acid calcium disodium salt (CaEDTA), diethylenetriaminepentaacetic acid calcium salt (CaDTPA), fluorescein-5-isothiocyanate (FITC), soybean trypsin inhibitor (SBTI), and 2-deoxy-D-glucose (2DDG) were purchased from Sigma Aldrich. Anti-FXIIa antibody 3F7 was custom manufactured by Viva Biotech (Shanghai, China) based on complementarity-determining region (CDR) sequences obtained from the published international patent (WO2014207199A1), and its performance was confirmed by aPTT assay (Figure S1). Polyclonal anti-FXII western blot antibody was purchased from Affinity Biosciences. Purified human plasma-derived FXII was purchased from Enzyme Research Labs. C-terminal amide Gly-Pro-Arg-Pro (GPRP) was manufactured by the custom peptide division at Thermo Fisher Scientific. Anti-human tissue factor (clone IIID8) and anti-human factor VIIa (clone IIH2) antibodies were purchased from Biomedica Diagnostics. SN-20 fluorogenic thrombin substrate (BOC-L-FPR-ANSNH-C2H5) was purchased from Haematologic Technologies. Phycoerythrin-conjugated anti-human p-selectin (CD62P) was purchased from Thermo. S-2302 chromogenic FXIIa/kallikrein substrate was purchased from Diapharma. TNF-α was purchased from Millopore Sigma. The source of Zn2+ in all experiments was ZnCl2.
Preparation of Human Platelets
Whole blood was drawn into 0.4% sodium citrate (final concentration w/v) and centrifuged at 160 rcf for 10 minutes to generate PRP. To generate washed platelets, acid citrate dextrose (ACD) solution was added to PRP (final concentration 20% v/v), and platelets were washed twice by centrifugation at 650 rcf in Hepes-buffered Tyrode’s solution with glucose (HTG, 134 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12 mM NaHCO3, 20mM HEPES, and 1mM MgCl2). All procedures were performed with the approval of the Institutional Review Board at Beth Israel Deaconess Medical Center.
Platelet-dependent Thrombin and FXIIa Generation Assays
Platelet-dependent thrombin and FXIIa generation assays were performed using a protocol modified from Jurk, et al. [18]. In the first step, either 50 μM SFLLRN or vehicle was added to 200 ul single-donor PRP (platelet count 250 × 103/μl) anticoagulated with 10 mM GPRP to prevent fibrin polymerization and containing increasing amounts of CaCl2 (4, 6, 8, 10, and 12 mM). A calcium titration was necessary due to donor-dependent differences in plasma calcium levels as well as subtle variability in blood draw techniques. The PRP mixture was incubated at room temperature for one hour, after which it was diluted 1:5 in PBS assay buffer containing 0.5 mM of the FXIIa-specific substrate S-2302, 12 mM EDTA (Sigma), 30 μM SBTI, and 2 units/ml hirudin, and absorbance was measured at 405 nm for 20 minutes. The concentration of Ca2+ that provided maximum signal separation between resting and SFLLRN-stimulated platelets was then chosen for use in the second step. In this phase, the inhibitor being tested was added to PRP containing a fixed concentration of Ca2+ and anticoagulated with GPRP, which was then stimulated with SFLLRN, incubated, and read in a manner identical to that used in Step 1. The platelet-dependent thrombin generation assay was performed in a similar fashion, except PBS assay buffer (1.05 mM KH2PO4, 155 mM NaCl, 3 mM, Na2HPO4-7H2O, pH 7.4, Gibco) containing the thrombin-specific substrate (SN-20) at 50 μM and 12 mM EDTA was substituted for S-2302 containing buffer, and fluorescence was read by excitation at 352 nm and emission at 470 nm.
For mouse platelet-dependent FXIIa generation assays, we obtained pooled citrated PRP from two groups of five animals each that were treated 30 minutes previously with either vehicle or 250 mg/kg CaEDTA (platelet count 225 × 103/μl). GPRP (final concentration 10 mM) was added to the PRP as well as 0, 2, 4, 6, or 8 mM of CaCl2 (total volume per condition: 100 μl), and the sample was then stimulated with 500 μM of the PAR-4 agonist AYPGKF or vehicle (2 conditions per calcium concentration, 10 conditions total). After incubation for two hours at room temperature, each condition was diluted 1:5 in PBS assay buffer containing 0.5 mM S-2302, 12 mM EDTA, 30 μM SBTI, 300 μg/ml apixaban, and 2 units/ml hirudin, and absorbance was measured at 405 nm for 30 minutes. For each calcium concentration, the level of FXIIa generation in the AYPGKF-stimulated condition was corrected against the corresponding resting platelet condition (background), and results are reported as the peak FXIIa generation across the five calcium concentrations.
Activated Partial Thromboplastin (aPTT) Assay
The PTT assay was performed using the PTT Automate assay (Stago) on the Start4 coagulometer (Stago) per the manufacturer’s instructions.
HUVEC Thrombin Generation Assay
HUVEC (Lonza, P1-P5) monolayers cultured in Endothelial Growth Medium (EGM)-2 (Lonza) on gelatin coated culture vessels were sub-cultured on 96-well plates. Confluent HUVEC monolayers were stimulated with vehicle or TNF-α (10 ng/mL) in endothelial basal medium (EBM)-2 (Lonza) for 4 hours at 37°C. For thrombin generation experiments, stimulation medium was removed from wells and replaced with a 25 μL of 80% platelet poor plasma (PPP) mixture composed of pooled PPP from 4–5 donors in 5 mM GPRP and HEPES Buffered Saline (20 mM HEPES, 150 mM NaCl, pH 7.4, HBS) containing 10 mM CaCl2, with or without inhibitors for 15 minutes. Following incubation, thrombin levels were measured with the addition of SN-20 fluorogenic substrate in PBS with 2 mM CaCl2. Fluorescence was measured every minute for 60 minutes using the Synergy4 plate reader (Biotek). Thrombin levels were determined by converting reaction rate to units/ml.
Generation of Recombinant FXII
Triple mutant FXII (R334A, R343A, and R353A, which we designated “FXII-3T”) [19] was cloned into a pcDNA™ 3.4 backbone using Gibson Assembly® and was transiently transfected and expressed in Expi293F™ cells for 96 hours. Following harvesting, conditioned media was treated with Sodium Acetate pH 5.2 to a final concentration of 50mM and applied to a 5mL HiTrap Heparin HP column (GE Lifesciences). Protein was eluted off the column using a linear gradient of 2M NaCl. Peak fractions were pooled and applied to a 5mL Strep-Tactin®XT Superflow® high capacity cartridge (IBA Lifesciences) followed by elution by buffer containing 50mM biotin. Peaks were pooled and concentrated for injection on a Superdex™ S200 Increase 10/300 GL. The peak corresponding to soluble protein was combined and concentrated, and dialyzed into TBS storage buffer (20 mM Tris, 150 mM NaCl, pH 8.3) to be used as the final product.
Platelet Flow Cytometry
Washed platelets (400–500K/μl) were stimulated with 50 μM SFLLRN for 10 minutes. Platelets were then diluted in HTG at 10,000 per μl together with 2 mM CaCl2, Zn2+ at 0, 3, or 10 μM, and 100 nM FITC-labeled FXII, FXIIa (Enzyme Research Labs), or FXII-3T. The mixture was incubated at room temperature for 20 minutes and then diluted 1:10 in HTG containing the condition-dependent Zn2+ concentration and analyzed by flow cytometry on a Gallios instrument (Beckman Coulter). For experiments with 2-deoxy-D-glucose (2DDG) and sodium azide, platelets were pre-incubated with these two agents for 30 minutes prior to activation with SFLLRN.
Mouse Laser Injury Model
The intravital mouse cremaster arteriole laser injury model was performed as described previously [20, 21]. The operator was blinded to the genotype and treatment assignment (vehicle vs. CaEDTA) of each subject animal. Digital images were captured with an Orca Flash 4.0v2 sCMOS camera (Hamamatsu Photonics K.K., Shizuoka Pref., Japan). Representative images are presented, and the median curves include the full data. The kinetics of platelet thrombus formation were analyzed using SlideBook 6.0 (Intelligent Imaging Innovations, Denver, CO).
Male wild-type C57/BL6J mice were purchased from Jackson Laboratory. Male FXII null mice were the kind gift of Dr. David Gailani. All protocols involving the use of animals were in compliance with guidelines from the National Institutes of Health and the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
Size Exclusion Chromatography
Size exclusion chromatography was performed on a Superdex 200 column, and HBS with varying concentrations of Zn2+ was used as a running buffer. Prior to loading on the column, purified human FXII was dissolved at 0.66 mg/ml in HBS with the indicated concentrations of Zn2+ and incubated for 10 minutes at room temperature.
Measurement of Plasma Labile Zinc Levels (Heparinized PPP)
Labile Zn2+ levels in heparinized human plasma were measured using the chromogenic Zinc Assay Kit from Abcam (ab102507) per the manufacturer’s instructions.
FITC-labeling of FXII
Plasma-derived purified FXII (Enzyme Research Laboratories) was dialyzed into TBS overnight at 4° C, resulting in a FXII solution of approximately 1 mg/ml. FITC-Celite® (Sigma) was dissolved at 50 mg/mL in anhydrous DMSO (Thermo Fisher, D12345) to form a working solution. This working solution was added to the dialyzed FXII to a final concentration of 1 mg/mL of FITC. The reaction mixture was vortexed briefly and incubated in the dark at ambient temperature for 20 minutes. After incubation, the excess Celite was removed by centrifugation at 10,000 g for 5 minutes. The labeled FXII was then dialyzed against PBS overnight at 4°C to remove excess FITC.
Quantification of Platelet FXII Binding Sites
In order to quantify the number of platelet FXII binding sites, a calibration curve was created using standardized Quantum™ FITC-5 Molecules of Equivalent Soluble Fluorochrome (MESF) beads (Bangs Laboratories). The standard curve was generated by plotting the fluorescent intensities of beads with known MESF values, resulting in the following equation:
where MFI is the mean fluorescence intensity reading at any given MESF level, and m and b are derived from best-fit linear regression.
In the second step, SFLLRN-stimulated platelets were added at 103/uL to a series of tubes containing 300 μM Zn2+ in HTG and either 0, 100, 300, 1000, 3000, or 5000 nM FXII-FITC and incubated for 20 minutes before collecting fluorescent reads (Gallios, Beckman Coulter). The value recorded as the curve leveled off was used as the saturation point for FXII binding. The fluorescent read at this saturation point was used to interpolate the MESF value from the standard curve that was previously derived. Subsequently, the number of binding sites could be determined by dividing the MESF value by the fluorophore per protein molecule ratio (F:P) of FXII-FITC:
Results
Platelet-dependent Thrombin Generation Requires FXIIa and Zinc
The mechanism by which stimulated platelets directly activate the coagulation cascade to generate thrombin is not understood. In order to evaluate the role of FXII in platelet-dependent thrombin generation, we stimulated PRP with the PAR-1 agonist peptide SFLLRN (TRAP-6) in the presence of inhibitory antibodies directed against FXIIa (3F7), tissue factor (TF), or factor VIIa (FVIIa) and measured thrombin generation by use of a fluorogenic substrate. Blockade of FXIIa resulted in nearly complete abrogation of platelet-dependent thrombin generation, while inhibition of FVIIa and TF had no effect (Figure 1A).
Figure 1: Thrombin generation by stimulated platelets is FXII and zinc dependent.

(A) Thrombin generation was measured after platelet rich plasma (PRP) was stimulated with 50 μM of the PAR-1 receptor agonist peptide SFLLRN in the presence of either non-immune control antibody (SFLLRN) or inhibitory antibodies against FXIIa (3F7, left, N=12 replicates), factor VIIa (middle, N=12 replicates), or tissue factor (right, N=15 replicates). Each experiment was performed independently two times with 9 replicates per condition. (B) PRP was stimulated with 50 μM SFLLRN in the presence of increasing concentrations of the Zn2+-specific chelator CaEDTA and FXIIa generation was recorded (left, N=9 replicates per condition). P-selectin (CD62P) expression was measured in resting and SFLLRN-stimulated platelets in the presence or absence of 40 mM CaEDTA (right, N=3 replicates per condition). (C) By comparison, the activated partial thromboplastin time (aPTT) was recorded in the presence of increasing concentrations of CaEDTA, N=3 replicates per condition. **P<0.05 compared to activated platelet control by Kruskal-Wallis test with Dunn’s post-test for multiple comparisons.
In light of previous observations that cationic zinc (Zn2+) significantly enhances the activation of purified FXII in buffer and PPP [11–14], we next determined whether Zn2+ is required to support platelet-dependent FXIIa generation ex vivo. We found that at baseline, heparinized pooled human PPP contained labile Zn2+ at a mean ± SEM concentration of 12.16 ± 0.22 μM. PRP was incubated with increasing concentrations of the Zn2+-specific extracellular chelator ethylenediaminetetraacetic acid calcium disodium salt (CaEDTA) [22–24], followed by measurement of FXIIa generation. CaEDTA inhibited platelet-based FXIIa generation in a dose-dependent manner without affecting the activation state of platelets as reflected by P-selectin (CD62P) expression (Figure 1B). Correspondingly, plasma labile Zn2+ declined to undetectable levels in the presence CaEDTA (Figure S2A). We sought to confirm these results using a different Zn2+-specific chelator, calcium diethylenetriaminepentaacetic acid (CaDTPA) [25] and found that FXIIa generation was likewise inhibited (Figure S2B). The presence of CaEDTA did not appear to influence kaolin-mediated FXII autoactivation or the cleavage of FXII to FXIIa by kallikrein in vitro (Figure S3). By contrast, Zn2+ chelation did not prolong coagulation times as measured by the activated partial thromboplastin time (aPTT) performed on PPP (Figure 1C). These data demonstrate that stimulated platelets can directly activate coagulation by generating FXIIa through a process requiring the Zn2+ available in PRP.
Stimulated Endothelial Cells Do Not Require FXII to Generate Thrombin
Endothelial cells stimulated in vitro are known to support thrombin generation by expressing procoagulant molecules such as TF and phosophatidylserine [26]. FXII has been shown to bind endothelial cells via a trimolecular membrane receptor complex in the presence of Zn2+ [9], but the effect of the FXII-endothelial cell interaction on thrombin generation has not been described. We sought to understand whether, like platelets, endothelium supports thrombin generation via a FXIIa-dependent mechanism. We therefore interrogated the roles of zinc and FXIIa in thrombin generation by stimulated human umbilical vein endothelial cells (HUVECs) (Figure 2). HUVECs were overlaid with citrated human PPP and stimulated with tumor necrosis factor alpha (TNF-α) in the presence of CaEDTA, 3F7, or function-blocking antibodies against FVIIa or TF. Inhibition of either FVIIa or TF significantly impaired thrombin generation by TNF-α stimulated HUVECs, whereas neither CaEDTA nor 3F7 was able to inhibit HUVEC procoagulant activity, suggesting that stimulated endothelial cells and platelets utilize different arms of the coagulation system to initiate thrombin generation.
Figure 2: Thrombin generation by stimulated endothelial cells is not FXII or zinc dependent.

Human umbilical vein endothelial cells were stimulated with 10 ng/ml TNF-α, and thrombin generation was recorded in the presence of inhibitory antibodies (10 μg/ml) or calcium EDTA (40 mM) as indicated. Antibody 3F7 was used for FXIIa inhibition. N=4 replicates per condition, **P<0.05 for comparison to TNF-α condition.
FXII Binding to the Platelet Surface is Zinc-dependent and Specific
Given the finding that stimulated platelets can support FXII activation, we sought to assess FXII binding to the platelet surface. Resting or stimulated washed platelets were incubated with FITC-labeled FXII (FXII-FITC) and increasing concentrations of Zn2+ prior to analysis by flow cytometry (Figure 3A). Consistent with previous literature [16], in the absence of Zn2+ washed platelets stimulated with SFLLRN displayed increased binding of FXII-FITC relative to resting platelets. However, physiologic concentrations of Zn2+ greatly enhanced FXII-FITC binding to stimulated and, to a lesser degree, resting platelets. The binding of FXII-FITC was readily reversible with unlabeled FXII (Figure 3B), indicating the specific nature of the FXII-FITC/platelet interaction. We next recombinantly generated a single-chain form of FXII (FXII-3T) in which the major cleavage sites at R334, R343, and R353 have each been replaced with alanine [19]; this form of FXII cannot be converted to FXIIa. When we assayed the ability of FITC-labeled FXIIa and FXII-3T to bind the platelet surface in the presence of 10 μM ZnCl2, we found that these two forms of FXII performed similarly to FXII-FITC zymogen (Figure S4). We next sought to assess the possibility that Zn2+ was inducing the formation of FXII aggregates that were non-specifically adhering to platelets in our assay. We found that the presence of Zn2+ even at supraphysiologic concentrations did not lead to the formation of detectable FXII aggregates in solution (Figure S5). Previous data have suggested that FXII binds to the GPIbα receptor on the platelet surface [27], though this finding has never been corroborated. We therefore evaluated whether platelets supported FXII-FITC binding in an energy-independent manner as would be expected with GPIbα, a constitutively active receptor. FXII-FITC binding was largely prevented when platelets were pre-incubated with 2-deoxy-D-glucose (2DDG) and sodium azide in order to deplete available energy stores (Figure 3C), suggesting that the Zn2+-mediated binding of FXII-FITC to stimulated platelets is regulated by an energy-dependent process. Because Zn2+ is itself a platelet activator [28], we evaluated the level of CD62P expression on resting platelets after exposure to Zn2+ (Figure S6). At 10 μM Zn2+, we noted a small but measurable increase in platelet CD62P expression, suggesting that platelet activation may be responsible in part for the increased binding of FXII-FITC observed with resting platelets in the presence of Zn2+.
Figure 3: FXII binding to the platelet surface is specific and zinc-dependent.
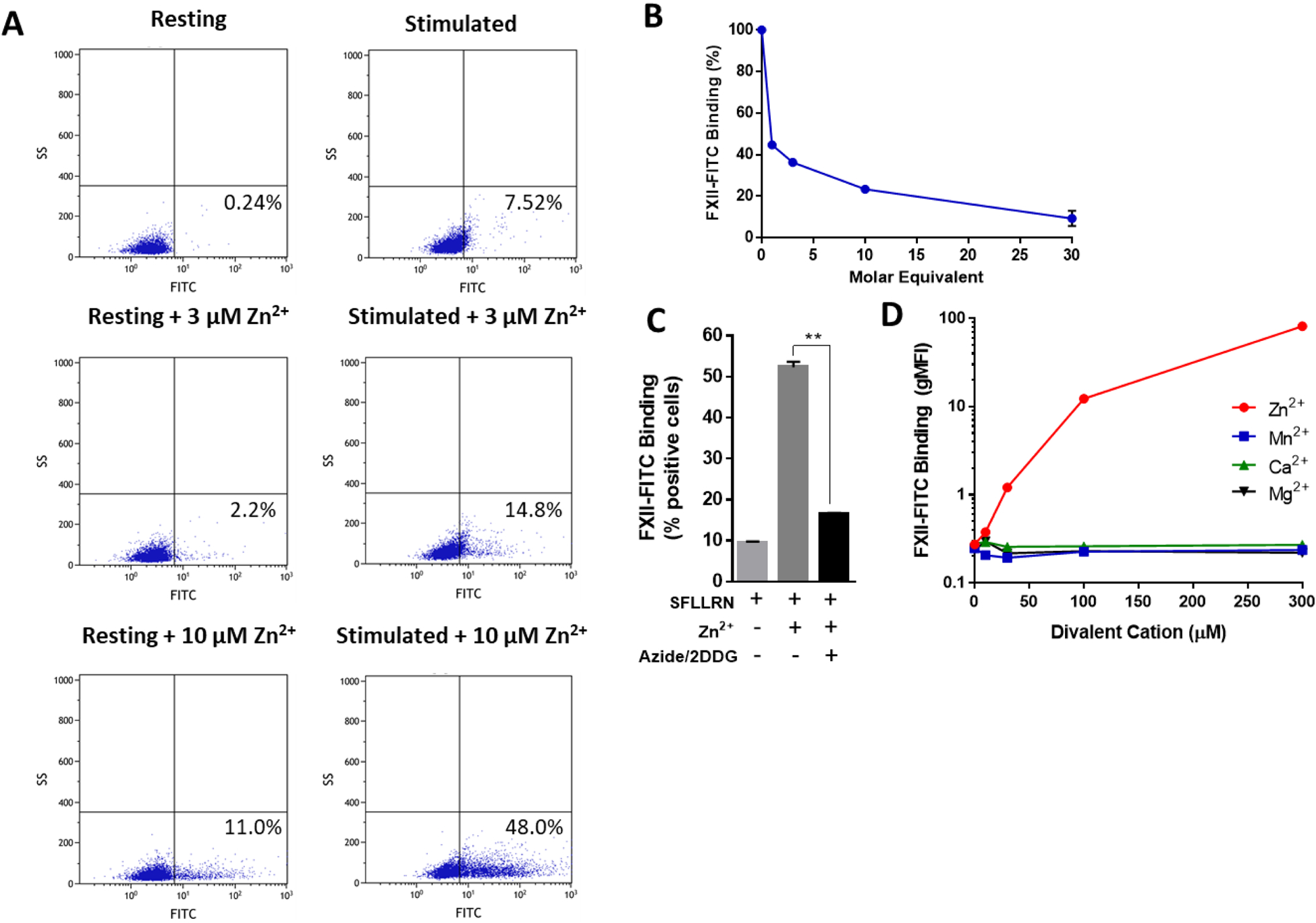
(A) The binding of FXII-FITC (100 nM) to resting platelets and platelets stimulated with 50 μM SFLLRN was measured by flow cytometry in the absence or presence of Zn2+. Each experiment was performed with 6 replicates, and representative scatter plots are shown here. (B) Binding of FXII-FITC (100 nM) to stimulated platelets in the presence of 10 μM Zn2+ and increasing molar equivalents of unlabeled FXII was measured by flow cytometry (representative plot from 3 independent experiments with 3 replicates for each condition). (C) Binding of FXII-FITC to stimulated platelets was measured in the presence or absence of 10 μM Zn2+ and with or without 30-minute preincubation with 0.2% sodium azide and 4 mg/ml 2-deoxy-D-glucose (2DDG), N=6 replicates, **P<0.0001 (D) FXII-FITC (100 nM) binding to stimulated washed platelets was recorded by flow cytometry at increasing concentrations of the indicated divalent metal cations (N=3 replicates per condition).
We next tested the ability of other physiologic divalent cations to support binding of FXII-FITC to the platelet surface. SFLLRN-stimulated washed platelets and FXII-FITC (100 nM) were incubated with increasing concentrations of divalent calcium, magnesium, manganese, or zinc, and FXII-FITC platelet binding was measured by flow cytometry (Figure 3D). Enhanced FXII-FITC binding could be readily detected when Zn2+ was included, but binding remained unchanged in the presence of Ca2+ (CaCl2), Mg2+ (MgCl2), and Mn2+ (MnCl2). Using reagent beads containing known amounts of fluorochrome (Quantum™ MESF), we estimated that each stimulated platelet maximally bound on average 135,922 ± 4117 molecules of FXII-FITC at 300 μM Zn2+. These data are consistent with the unique role that Zn2+ is known to play in the biology of the contact system.
Zinc Chelation Prevents Thrombus Formation in vivo Without Increasing Bleeding
In order to interrogate the role of Zn2+ in thrombus formation in vivo, we used CaEDTA to deplete Zn2+ in the mouse cremaster arteriole laser injury model of thrombosis. Mice received intravenous infusion of CaEDTA (250 mg/kg) or vehicle control immediately prior to laser injury. Treatment with CaEDTA led to significantly decreased platelet accumulation and fibrin formation at sites of vascular injury (Figure 4A–E). When Zn2+ levels were assessed in mouse plasma ex vivo, we found that CaEDTA completely eliminated detectable levels of labile Zn2+ from mouse plasma (Figure 4F) in a manner similar to that seen with human plasma (Figure S2A). Despite preventing thrombus formation in this model, CaEDTA did not lead to an increased tendency for bleeding as gauged by a tail transection assay (Figure 4G and S7). In a separate experiment, we assayed FXIIa generation ex vivo in stimulated pooled PRP taken from mice treated with either vehicle or 250 mg/kg CaEDTA (N=5 animals per group). Our results confirm that platelet-dependent FXIIa generation was significantly reduced in mice treated with CaEDTA (Figure S8).
Figure 4:

Zinc chelation prevents thrombus formation in vivo. (A) Mice infused with 250 mg/kg of CaEDTA or vehicle control underwent quantitative assessment of thrombus formation (platelet accumulation in red, fibrin formation in green) in the cremaster arteriole laser injury model, and representative intravital images are shown, along with median curves and AUC distributions for platelet accumulation (B-C) and fibrin formation (D-E). A total of 56 thrombi were evaluated in each group. (F) Mice were treated with either CaEDTA (250 mg/kg) or vehicle control (N=4 in each group), and plasma Zn2+ concentrations were measured at 1 hour. (G) Tail bleeding times were compared between CaEDTA and vehicle treated animals (N=6 per group). **P<0.05 compared to vehicle control.
Using homozygous FXII knockout (FXII−/−) mice, we compared the in vivo effects of Zn2+ chelation with those of FXII deficiency in this model. FXII−/− mice significantly protected from thrombosis following laser-induced injury to the cremaster arterioles (Figure 5A–E), despite these mice having no demonstrable bleeding diathesis [7]. These data suggest that Zn2+ plays a role in thrombosis but not hemostasis and support the concept that Zn2+ may be necessary to facilitate FXIIa generation in vivo.
Figure 5:

Factor XII deficiency inhibits thrombus formation in vivo. (A) Wild-type (WT) or FXII knockout mice (FXII KO) mice underwent quantitative assessment of thrombus formation in the cremaster arteriole laser injury model (platelet accumulation in red, fibrin formation in green). Representative intravital images are shown, together with median curves and AUC distributions for platelet accumulation (B-C) and fibrin formation (D-E). **P<0.05 compared to WT.
Discussion
Given the longstanding observation that platelet function is critical to initiating and propagating arterial thromboses, platelets are likely a major contributor of FXIIa at sites of vascular injury [29]. Nevertheless, the interaction between platelets and FXII remains uncharacterized. Cationic zinc lies at the intersection of the twin processes of FXII surface binding and FXIIa generation; therefore, understanding the function of Zn2+ in this setting is key to elucidating the broader mechanism of FXII in thrombus formation. Our data provide direct evidence that Zn2+ plays a crucial role in the process of FXII ligation and activation at the stimulated platelet surface and demonstrate the importance of Zn2+ in thrombus formation.
An inhibitory anti-FXIIa antibody completely abrogated platelet-dependent thrombin generation in plasma, whereas blockade of TF and FVIIa appeared to have no effect, indicating that direct activation of coagulation by stimulated platelets occurs via the contact pathway exclusively. By contrast, endothelial cells stimulated with TNF-α mediate thrombin generation by initiating the extrinsic arm of the coagulation cascade, with little need for FXII. These findings provide a previously unrecognized basis for thrombin generation assays performed in PRP [18, 30, 31] during which addition of thrombin to PRP leads to significant downstream thrombin generation. Our use of SFLLRN as a platelet agonist rules out an enzymatic contribution to thrombin generation from the small amounts of thrombin used as the initiator in some versions of this assay, which is thought to mimic the physiologic process of thrombus propagation driven by feedback activation from the “thrombin burst.” During this phase, coagulation factors V and VIII undergo activation and greatly increase the reaction rates of the tenase and prothrombinase complexes at the platelet surface [32]. While this model assumes that the initial source of thrombin is the TF-FVII pathway, our data indicate that FXIIa generation quickly becomes an important, perhaps the dominant, contributor once stimulated platelets are present in large numbers at sites of vascular injury. Platelet-dependent FXIIa generation therefore provides a plausible explanation for the observation that FXII blockade or deficiency prevents arterial thrombosis in preclinical models [7, 33, 34].
Given the importance of FXIIa generation for platelet-dependent thrombin generation, we further explored the FXII-platelet interaction. Platelet FXIIa generation in plasma was abolished in the presence of Zn2+-specific chelators, indicating that Zn2+ is required for the binding and/or activation of FXII at the platelet surface. Similarly, recent work by Wang and colleagues has demonstrated that FXII autoactivation by polyphosphate as well as FXII activation by kallikrein in buffer are Zn2+-dependent processes, with peak Zn2+ cofactor activity occurring between 5 and 10 μM of the cation [15]. Resting platelets demonstrated minimal binding to FXII, whereas stimulated platelets were able to bind FXII in a manner that was greatly enhanced by the presence of Zn2+ and appeared to be specific. Moreover, because FXII binding to stimulated platelets required metabolic energy, it is intriguing to speculate that an activatable platelet surface protein and/or phospholipid exposure may serve as the principal platelet binding partner for FXII. At variance with a previous report demonstrating an in vitro interaction between FXII and platelet glycoprotein 1bα (GP1bα) [27], it appears unlikely based on our data that this role is played by a constitutively active surface molecule such as the GP1b-V-IX complex.
It has been proposed that FXII has between 4 and 8 discrete binding sites for Zn2+ localized to the fibronectin type II domain of its heavy chain [11, 12, 35, 36], and Zn2+ potentiates contact-driven FXIIa generation, likely by inducing FXII to assume a conformational state that is amenable to autoactivation [11, 12]. FXII almost certainly circulates bound to one or more Zn2+ atoms. Further studies will be needed to determine whether Zn2+ acts by facilitating FXII autoactivation, by enabling FXII binding to the platelet surface, or via both of these mechanisms. By contrast, zinc is not required for FXII activation in standard clinical coagulation assays such as the activated partial thromboplastin time (aPTT), suggesting that platelet-mediated FXIIa generation may represent a physiologic process that is distinct from that triggered by artificial surfaces. In interpreting our results, it is important to note that while CaEDTA is considered a highly specific Zn2+ chelating agent under most physiologic circumstances [22, 23, 37], it does have activity as a chelator for several non-physiologic and/or trace metal cations (e.g. thorium, palladium, uranium, cobalt, etc.), and we cannot rule out the possibility that chelation of one or more of these other elements could be contributing to our findings.
An important consideration is whether endogenous Zn2+ is sufficient under physiologic conditions to support the binding and activation of FXII by platelets. The availability of adequate Zn2+ in platelets and plasma for these reactions is strongly corroborated by our observation that Zn2+ chelation in PRP abrogates platelet-dependent FXIIa generation. It is also noteworthy that the concentration of Zn2+ in platelets is approximately 25- to 60-fold higher than plasma and that this zinc resides largely in alpha granules [9, 38–40]. Upon stimulation at sites of vascular injury, platelets are thought to secrete Zn2+ into the local microenvironment [9, 38], which allows them to serve as an ancillary source of Zn2+ and perhaps also provide an extra layer of regulatory control for FXIIa generation. Consistent with this idea, mice treated with CaEDTA were protected from thrombosis without experiencing impaired hemostasis, a phenotype identical to that observed with FXIIa blockade in the same model.
Although total plasma Zn2+ levels are in the range of 10–20 μM [41, 42], a remarkable degree of disagreement persists regarding the level of protein unbound or “free” Zn2+, which has proven difficult to measure precisely. Estimates range between 1 nM and 2 μM depending on the method used [38, 43–45]. However, free Zn2+ does not accurately reflect the true exchangeable or “labile” pool of zinc that is available to participate in biological reactions; it has been estimated that plasma in fact contains on average 8.1 ± 4.8 μM of labile zinc [46], similar to our own readings of ~12 μM. This is because approximately 80% of plasma Zn2+ is bound loosely to albumin [41] and serves as a ready source of Zn2+ in the body. Albumin contains two Zn2+ binding sites, including a low-affinity site with a Kd of ~65 μM [47]. By comparison, the high-affinity Zn2+ binding site on FXII is associated with a Kd of approximately 0.1 μM [12], suggesting that FXII is able to draw on the pool of labile plasma Zn2+. Therefore, our use of 10 μM Zn2+ in these experiments is likely similar to the plasma concentration of Zn2+ available in vivo. Taken together, these results indicate that Zn2+ is central to the process of platelet-dependent FXIIa generation and form the basis for further experiments aimed at elucidating the interaction between FXII and potential platelet surface binding partners.
Supplementary Material
Essentials.
The mechanism of FXII recruitment and activation and the platelet surface remains unclear
We find that cationic zinc (Zn2+) is necessary for platelet-dependent FXII activation
Stimulated endothelium generates thrombin via a FXII and Zn2+-independent mechanism
Zn2+ chelation blocks thrombus formation and phenocopies FXII deletion in vivo
Acknowledgements
This work was funded in part by the National Institutes of Health (1K08 HL136840-;02 to P.K.B., R35-HL135775, R01-HL125275, and T32-HL007917 to R.F.).
Footnotes
Conflicts of Interest
Dr. Bendapudi reports receiving consulting fees from Anthos Pharmaceuticals. S.A.C., M.S., L.T., S.H., J.R., K.E., and R.B. report no conflicts of interest. Dr. Flaumenhaft has financial interests in and is a founder of PlateletDiagnostics. His interests are reviewed and managed by Beth Israel Deaconess Medical Center in accordance with their conflict-of-interest policies.
References
- 1.Walsh PN, Griffin JH. Platelet-coagulant protein interactions in contact activation. Ann N Y Acad Sci. 1981; 370: 241–52. 10.1111/j.1749-6632.1981.tb29737.x. [DOI] [PubMed] [Google Scholar]
- 2.Walsh PN. The role of platelets in the contact phase of blood coagulation. British journal of haematology. 1972; 22: 237–54. [DOI] [PubMed] [Google Scholar]
- 3.Schmaier AH. The elusive physiologic role of Factor XII. J Clin Invest. 2008; 118: 3006–9. 10.1172/jci36617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nickel KF, Long AT, Fuchs TA, Butler LM, Renne T. Factor XII as a Therapeutic Target in Thromboembolic and Inflammatory Diseases. Arteriosclerosis, thrombosis, and vascular biology. 2017; 37: 13–20. 10.1161/atvbaha.116.308595. [DOI] [PubMed] [Google Scholar]
- 5.Cheng Q, Tucker EI, Pine MS, Sisler I, Matafonov A, Sun MF, White-Adams TC, Smith SA, Hanson SR, McCarty OJ, Renne T, Gruber A, Gailani D. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010; 116: 3981–9. 10.1182/blood-2010-02-270918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matafonov A, Leung PY, Gailani AE, Grach SL, Puy C, Cheng Q, Sun MF, McCarty OJ, Tucker EI, Kataoka H, Renne T, Morrissey JH, Gruber A, Gailani D. Factor XII inhibition reduces thrombus formation in a primate thrombosis model. Blood. 2014; 123: 1739–46. 10.1182/blood-2013-04-499111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Renne T, Pozgajova M, Gruner S, Schuh K, Pauer HU, Burfeind P, Gailani D, Nieswandt B. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005; 202: 271–81. jem.20050664 [pii] 10.1084/jem.20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larsson M, Rayzman V, Nolte MW, Nickel KF, Bjorkqvist J, Jamsa A, Hardy MP, Fries M, Schmidbauer S, Hedenqvist P, Broome M, Pragst I, Dickneite G, Wilson MJ, Nash AD, Panousis C, Renne T. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Science translational medicine. 2014; 6: 222ra17. 10.1126/scitranslmed.3006804. [DOI] [PubMed] [Google Scholar]
- 9.Mahdi F, Madar ZS, Figueroa CD, Schmaier AH. Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, gC1qR, and cytokeratin 1 on endothelial cell membranes. Blood. 2002; 99: 3585–96. 10.1182/blood.v99.10.3585. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Qiu Q, Mahdi F, Shariat-Madar Z, Rojkjaer R, Schmaier AH. Assembly and activation of HK-PK complex on endothelial cells results in bradykinin liberation and NO formation. American journal of physiology Heart and circulatory physiology. 2001; 280: H1821–9. 10.1152/ajpheart.2001.280.4.H1821. [DOI] [PubMed] [Google Scholar]
- 11.Bernardo MM, Day DE, Olson ST, Shore JD. Surface-independent acceleration of factor XII activation by zinc ions. I. Kinetic characterization of the metal ion rate enhancement. J Biol Chem. 1993; 268: 12468–76. [PubMed] [Google Scholar]
- 12.Rojkjaer R, Schousboe I. The surface-dependent autoactivation mechanism of factor XII. European journal of biochemistry / FEBS. 1997; 243: 160–6. 10.1111/j.1432-1033.1997.0160a.x. [DOI] [PubMed] [Google Scholar]
- 13.Shimada T, Kato H, Iwanaga S. Accelerating effect of zinc ions on the surface-mediated activation of factor XII and prekallikrein. Journal of biochemistry. 1987; 102: 913–21. 10.1093/oxfordjournals.jbchem.a122132. [DOI] [PubMed] [Google Scholar]
- 14.Schousboe I Contact activation in human plasma is triggered by zinc ion modulation of factor XII (Hageman factor). Blood coagulation & fibrinolysis : an international journal in haemostasis and thrombosis. 1993; 4: 671–8. [PubMed] [Google Scholar]
- 15.Wang Y, Ivanov I, Smith SA, Gailani D, Morrissey JH. Polyphosphate, Zn(2+) and high-molecular-weight kininogen modulate individual reactions of the contact pathway of blood clotting. J Thromb Haemost. 2019. 10.1111/jth.14612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Back J, Sanchez J, Elgue G, Ekdahl KN, Nilsson B. Activated human platelets induce factor XIIa-mediated contact activation. Biochem Biophys Res Commun. 2010; 391: 11–7. 10.1016/j.bbrc.2009.10.123. [DOI] [PubMed] [Google Scholar]
- 17.Zakharova NV, Artemenko EO, Podoplelova NA, Sveshnikova AN, Demina IA, Ataullakhanov FI, Panteleev MA. Platelet surface-associated activation and secretion-mediated inhibition of coagulation factor XII. PloS one. 2015; 10: e0116665. 10.1371/journal.pone.0116665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jurk K, Lahav J, H VANA, Brodde MF, Nofer JR, Kehrel BE. Extracellular protein disulfide isomerase regulates feedback activation of platelet thrombin generation via modulation of coagulation factor binding. J Thromb Haemost. 2011; 9: 2278–90. 10.1111/j.1538-7836.2011.04509.x. [DOI] [PubMed] [Google Scholar]
- 19.Ivanov I, Matafonov A, Sun MF, Cheng Q, Dickeson SK, Verhamme IM, Emsley J, Gailani D. Proteolytic properties of single-chain factor XII: a mechanism for triggering contact activation. Blood. 2017; 129: 1527–37. 10.1182/blood-2016-10-744110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jasuja R, Passam FH, Kennedy DR, Kim SH, Hessem LV, Lin L, Bowley SR, Joshi SS, Dilks JR, Furie B, Furie BC, Flaumenhaft R. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest. 2012. 61228 [pii] 10.1172/JCI61228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nature medicine. 2002; 8: 1175–81. 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 22.Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996; 272: 1013–6. 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- 23.Radford RJ, Lippard SJ. Chelators for investigating zinc metalloneurochemistry. Current opinion in chemical biology. 2013; 17: 129–36. 10.1016/j.cbpa.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nyborg JK, Peersen OB. That zincing feeling: the effects of EDTA on the behaviour of zinc-binding transcriptional regulators. The Biochemical journal. 2004; 381: e3–4. 10.1042/bj20041096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arts J, Bade S, Badrinas M, Ball N, Hindle S. Should DTPA, an Aminocarboxylic acid (ethylenediamine-based) chelating agent, be considered a developmental toxicant? Regulatory toxicology and pharmacology : RTP. 2018; 97: 197–208. 10.1016/j.yrtph.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 26.Rao LV, Pendurthi UR. Regulation of tissue factor coagulant activity on cell surfaces. J Thromb Haemost. 2012; 10: 2242–53. 10.1111/jth.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradford HN, Pixley RA, Colman RW. Human factor XII binding to the glycoprotein Ib-IX-V complex inhibits thrombin-induced platelet aggregation. J Biol Chem. 2000; 275: 22756–63. 10.1074/jbc.M002591200. [DOI] [PubMed] [Google Scholar]
- 28.Watson BR, White NA, Taylor KA, Howes JM, Malcor JD, Bihan D, Sage SO, Farndale RW, Pugh N. Zinc is a transmembrane agonist that induces platelet activation in a tyrosine phosphorylation-dependent manner. Metallomics : integrated biometal science. 2016; 8: 91–100. 10.1039/c5mt00064e. [DOI] [PubMed] [Google Scholar]
- 29.Muller F, Renne T. Platelet polyphosphates: the nexus of primary and secondary hemostasis. Scandinavian journal of clinical and laboratory investigation. 2011; 71: 82–6. 10.3109/00365513.2010.550312. [DOI] [PubMed] [Google Scholar]
- 30.Hemker HC, Giesen P, Al Dieri R, Regnault V, de Smedt E, Wagenvoord R, Lecompte T, Beguin S. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiology of haemostasis and thrombosis. 2003; 33: 4–15. 10.1159/000071636. [DOI] [PubMed] [Google Scholar]
- 31.Stopa JD, Neuberg D, Puligandla M, Furie B, Flaumenhaft R, Zwicker JI. Protein disulfide isomerase inhibition blocks thrombin generation in humans by interfering with platelet factor V activation. JCI insight. 2017; 2: e89373. 10.1172/jci.insight.89373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furie B, Furie BC. Mechanisms of thrombus formation. The New England journal of medicine. 2008; 359: 938–49. 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 33.Kleinschnitz C, Stoll G, Bendszus M, Schuh K, Pauer HU, Burfeind P, Renne C, Gailani D, Nieswandt B, Renne T. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006; 203: 513–8. jem.20052458 [pii] 10.1084/jem.20052458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuijpers MJ, van der Meijden PE, Feijge MA, Mattheij NJ, May F, Govers-Riemslag J, Meijers JC, Heemskerk JW, Renne T, Cosemans JM. Factor XII regulates the pathological process of thrombus formation on ruptured plaques. Arteriosclerosis, thrombosis, and vascular biology. 2014; 34: 1674–80. 10.1161/atvbaha.114.303315. [DOI] [PubMed] [Google Scholar]
- 35.Stavrou E, Schmaier AH. Factor XII: what does it contribute to our understanding of the physiology and pathophysiology of hemostasis & thrombosis. Thrombosis research. 2010; 125: 210–5. 10.1016/j.thromres.2009.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rojkaer R, Schousboe I. Partial identification of the Zn2+-binding sites in factor XII and its activation derivatives. European journal of biochemistry / FEBS. 1997; 247: 491–6. 10.1111/j.1432-1033.1997.00491.x. [DOI] [PubMed] [Google Scholar]
- 37.Ceccom J, Bouhsira E, Halley H, Daumas S, Lassalle JM. Differential needs of zinc in the CA3 area of dorsal hippocampus for the consolidation of contextual fear and spatial memories. Learning & memory (Cold Spring Harbor, NY). 2013; 20: 348–51. 10.1101/lm.029017.112. [DOI] [PubMed] [Google Scholar]
- 38.Kiran Gotru S, van Geffen JP, Nagy M, Mammadova-Bach E, Eilenberger J, Volz J, Manukjan G, Schulze H, Wagner L, Eber S, Schambeck C, Deppermann C, Brouns S, Nurden P, Greinacher A, Sachs U, Nieswandt B, Hermanns HM, Heemskerk JWM, Braun A. Defective Zn(2+) homeostasis in mouse and human platelets with alpha- and delta-storage pool diseases. Scientific reports. 2019; 9: 8333. 10.1038/s41598-019-44751-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gorodetsky R, Mou X, Blankenfeld A, Marx G. Platelet multielemental composition, lability, and subcellular localization. American journal of hematology. 1993; 42: 278–83. [DOI] [PubMed] [Google Scholar]
- 40.Marx G, Korner G, Mou X, Gorodetsky R. Packaging zinc, fibrinogen, and factor XIII in platelet alpha-granules. J Cell Physiol. 1993; 156: 437–42. 10.1002/jcp.1041560302. [DOI] [PubMed] [Google Scholar]
- 41.Foote JW, Delves HT. Albumin bound and alpha 2-macroglobulin bound zinc concentrations in the sera of healthy adults. J Clin Pathol. 1984; 37: 1050–4. 10.1136/jcp.37.9.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whitehouse RC, Prasad AS, Rabbani PI, Cossack ZT. Zinc in plasma, neutrophils, lymphocytes, and erythrocytes as determined by flameless atomic absorption spectrophotometry. Clinical chemistry. 1982; 28: 475–80. [PubMed] [Google Scholar]
- 43.Bloxam DL, Tan JC, Parkinson CE. Non-protein bound zinc concentration in human plasma and amniotic fluid measured by ultrafiltration. Clinica chimica acta; international journal of clinical chemistry. 1984; 144: 81–93. [DOI] [PubMed] [Google Scholar]
- 44.Kelly E, Mathew J, Kohler JE, Blass AL, Soybel DI. Redistribution of labile plasma zinc during mild surgical stress in the rat. Translational research : the journal of laboratory and clinical medicine. 2011; 157: 139–49. 10.1016/j.trsl.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cunningham BC, Bass S, Fuh G, Wells JA. Zinc mediation of the binding of human growth hormone to the human prolactin receptor. Science. 1990; 250: 1709–12. 10.1126/science.2270485. [DOI] [PubMed] [Google Scholar]
- 46.Zalewski P, Truong-Tran A, Lincoln S, Ward D, Shankar A, Coyle P, Jayaram L, Copley A, Grosser D, Murgia C, Lang C, Ruffin R. Use of a zinc fluorophore to measure labile pools of zinc in body fluids and cell-conditioned media. BioTechniques. 2006; 40: 509–20. 10.2144/06404rr02. [DOI] [PubMed] [Google Scholar]
- 47.Handing KB, Shabalin IG, Kassaar O, Khazaipoul S, Blindauer CA, Stewart AJ, Chruszcz M, Minor W. Circulatory zinc transport is controlled by distinct interdomain sites on mammalian albumins. Chemical science. 2016; 7: 6635–48. 10.1039/c6sc02267g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.