

Abstract
Overexpression of breast cancer resistance protein (BCRP) has been shown to produce multidrug resistance (MDR) in colon cancer, leading to major obstacles for chemotherapy. In this study, we evaluated the effect of regorafenib, an oral multi-kinase inhibitor, in inhibiting BCRP-mediated MDR in silico, in vitro and in vivo. We found that regorafenib significantly sensitized MDR colon cancer cells to BCRP substrates by increasing their intracellular accumulation. There are no significant changes in the expression level or the subcellular distribution of BCRP in the cells exposed to regorafenib. Investigation of the mechanism revealed that regorafenib stimulated BCRP ATPase activity. Our induced-fit docking and molecular dynamics simulations suggested the existence of a strong and stable interaction between regorafenib and the transmembrane domain of human crystalized BCRP. In vivo tumor xenograft study revealed that the combination of regorafenib and topotecan exhibited synergistic effects on mitoxantrone-resistant S1–M1–80 xenograft tumors. In conclusion, our studies indicate that regorafenib would be beneficial in combating MDR in colon cancer.
Keywords: Reversal of multidrug resistance, Combination chemotherapy with regorafenib, Synergy
1. Introduction
Among different types of cancer, colorectal cancers are the third leading cancer cases in both male and female [1]. Because patients with early stages of the disease usually do not experience any symptoms, colorectal cancers are often diagnosed at late stages [1]. As a result, the five-year survival rate of colorectal cancers is less than 40%, with over 20% of the patients seeking medical attention when the disease has already advanced, and up to 25% of patients in this group will already have isolated liver metastasis [2]. Radiation treatments are not often used for colorectal cancers due to the sensitivity of the bowels to radiation [3]. Clinically, the most common treatment options for colorectal cancers are surgery and chemotherapy. However, chemotherapy eventually induces colorectal cancer cells to evade cell death by a phenomenon known as drug resistance [4]. Resistance to chemotherapy remains one of the biggest challenges in the long-term management of colorectal cancer [5–7].
When cancer cells develop resistance to one chemotherapeutic drug, it may simultaneously confer resistance to other drugs, which may have different structures and mechanisms of action. This phenomenon is known as multidrug resistance (MDR) [8]. Defined as the insensitivity of cells to cytotoxic actions of a number of structurally and functionally unrelated drugs, MDR in cancer cells poses a major challenge to effective cancer treatment [9]. In the past few decades, many research efforts were devoted to understanding the mechanisms by which cancer cells develop MDR. Some of these mechanisms have since been identified, such as reduced uptake of drugs, cell cycle arrest, altered drug targets, increased efflux of drugs, apoptosis regulation, autophagy regulation, DNA repair, and sequestration of drugs in lysosomes [10]. Among those factors, the most prominent cause of MDR is the increased efflux of anticancer drugs by ATP-binding cassette (ABC) transporters [11]. ABC transporters are a group of active membrane transporter proteins that have various functions and ubiquitous distribution in both prokaryotes and eukaryotes, and they utilize the energy from ATP hydrolysis to transport endogenous ligands or exogenous drugs across the membranes. In human, genetic studies have divided the ABC transporter superfamily into seven subfamilies based on sequence similarities [12]. Breast cancer resistance protein (BCRP, also known as ABCG2) is a 72 kDa protein and is also the second member of the G subfamily of ABC transporter superfamily [13]. The crystal structure of human BCRP was recently resolved by Taylor et al. via cryo-electron microscopy [14]. The newly-revealed multidrug-binding pocket between the transmembrane domain facilitates our molecular simulation studies. BCRP has been reported to be a mediator of MDR colon cancer, inducing resistance to mitoxantrone, 9-aminocamptothecin, topotecan, irinotecan and SN-38 [15]. In this work, a mitoxantrone-selected MDR colon cancer cell line S1–M1–80 will be utilized, in which BCRP is overexpressed as compared with the parental colon cancer cell line S1.
Receptor tyrosine kinases play crucial roles in tumor vasculature [16]. Regorafenib, a novel oral multikinase inhibitor, targets VEGFR2-TIE2 (Vascular endothelial growth factor receptor 2 and tyrosine kinase with immunoglobulin and epidermal growth factor homology domain 2) and produces anti-angiogenic effects [17]. Furthermore, regorafenib has also been demonstrated to potently inhibit other angiogenic kinase (VEGFR1/3, PDGFR-β and fibroblast growth factor receptor 1) [17]. Because of its potent anti-angiogenic effects, regorafenib has been shown to increase the overall survival of patients with metastatic colorectal cancer, and was approved by the United States Food and Drug Administration (US FDA) on September 27, 2012. Moreover, the US FDA expanded the approved use of regorafenib to treat patients with advanced gastrointestinal stromal tumors (GIST) on February 25, 2013. Recently, regorafenib was approved as a second-line treatment for hepatocellular carcinoma patients who previously received sorafenib. In this work, we hypothesized that regorafenib could reverse the BCRP-related MDR in colorectal cancer by inhibiting the efflux function of BCRP. The combination of regorafenib with conventional chemotherapeutic drugs could exert synergistic effect in colorectal cancer especially when MDR develops. This combination could potentially benefit patients struggling with MDR colorectal cancer or metastatic liver cancer.
2. Materials and methods
2.1. Reagents
Regorafenib was obtained from Bayer HealthCare Pharmaceuticals Inc. (Whippany, NJ). Dulbecco’s modified Eagle’s medium (DMEM), penicillin/streptomycin and trypsin 0.25% were purchased from Corning Life Sciences (Tewksbury, MA). Fetal bovine serum (FBS) and bovine calf serum were purchased from Hyclone, GE Healthcare Life Science (Pittsburgh, PA). Mitonxantrone, SN-38, and cisplatin were products from Medkoo Biosciences, Inc. (Chapel Hill, NC). Bovine serum albumin (BSA), monoclonal antibody BA3R (against beta-actin), Alexa Fluor 488 conjugated goat anti-mouse IgG secondary antibody, and RNase A were purchased from Thermo Fisher Scientific Inc. (Rockford, IL). Monoclonal antibody BXP-21 (against BCRP) was purchased from GeneTex (Irvine, CA). HRP-labeled rabbit anti-mouse secondary IgG was purchased from Cell Signaling Technology (Danvers, MA). 3-(4,5-dimethylthiazol-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethylsulfoxide (DMSO), Triton X-100, propidium iodide and paraformaldehyde were purchased from Sigma-Aldrich (St. Louis, MO). [3H]-mitoxantrone (2.5 Ci/mmol) were purchased from Moravek Biochemicals, Inc. (Brea, CA). Fumitremorgin C (FTC) was a gift from Dr. Susan Bates (NIH, Bethsda, MD).
2.2. Cell lines and cell culture
Human colon cancer cell line S1 and its mitoxantrone-selected BCRP-overexpressing S1–M1–80 cells were kindly provided by Drs. Susan E. Bates and Robert Robey (NIH, Bethesda, MD) [18]. All the cell lines were grown as adherent monolayers in poly-D-lysine coated flasks with DMEM supplemented with 10% FBS or bovine calf serum and 1% penicillin/streptomycin in a humidified incubator containing 5% CO2 at 37 °C. All cells were tested every 12 weeks for detection of mycoplasma contamination.
2.3. Animals
Male athymic NCR (nu/nu) nude mice (13–15 g, age 4 weeks) were purchased from the Taconic Farms (NCRNU-M, Homozygous, Albany, NY) and were used for tumor xenograft. All the animals were maintained on an alternating 12 h light/dark cycle with free access to water and rodent chow ad libitum. The mice were maintained at St. John’s University Animal Facility and were monitored closely for tumor growth by palpation and visual examination. Institutional Animal Care & Use Committee (IACUC) of St. John’s University approved this project, and the research was conducted in compliance with Animal Welfare Act and other federal statutes.
2.4. Cytotoxicity assay
Modified MTT colorimetric assay [19] was used to measure the changes of cytotoxicities of anticancer drugs with or without inhibitors. The protocol used in this study is identical to that of a previously described study [19].
2.5. Live cell Hoechst 33342 accumulation and fluorescence microscopic analysis
Hoechst 33342 accumulation assays were performed in the absence or presence of regorafenib (3 μM) as previously described [20].
2.6. [3H]-labeled substrates intracellular accumulation and efflux assay
[3H]-labeled mitoxantrone accumulation and efflux assay were performed on S1 and S1–M1–80 cells in the presence or absence of regorafenib (3 μM) or FTC (3 μM) as followed by our previously established protocols [19,20].
2.7. Preparation of total cell lysates and Western blotting
S1–M1–80 cells were treated with regorafenib (3 μM) for up to 72 h. Samples at different times were collected and blotted using our previously established protocols [20].
2.8. Immunofluorescence of BCRP
S1–M1–80 cells were seeded on sterile poly-D-lysine coated coverslips and were allowed to grow overnight, followed by treatment with either PBS or 3 μM regorafenib for 72 h. Fluorescence microscopy was performed as previously described [20].
2.9. BCRP ATPase assay
BCRP containing crude membrane were prepared from High-Five insect cells as previously described [21]. Regorafenib-induced BCRP ATPase activities were determined as previously described [20].
2.10. Induced-fit docking and molecular dynamic simulations
Human BCRP crystal structure (PDB ID: 5NJ3) was prepared and the docking grid was confined at cavity 1 as described by Taylor et al. [14]. The default Glide extensive sampling induce-fit docking (IFD) protocol was followed and the docking score (kcal/mol) was calculated [22,23]. The regorafenib-BCRP complex was then subjected to another 10 ns molecular dynamic (MD) simulation [19]. All calculations mentioned above were performed on a 6-core Xeon processor except MD jobs which were performed on a Nvidia GPU.
2.11. MDR colon cancer xenograft tumor model
S1 and S1–M1–80 cells (1.0 × 107) were injected subcutaneously under the armpits of nude mice. When the subcutaneous tumors reached approximately 5 × 5 mm in size, the mice were randomized into 4 treatment groups, with 8 mice in each group. Group 1 animals received the vehicle A (polypropylene glycol/PEG400/Pluronic F68/Saline, 34%/34%/12%/20%) orally every 3rd days, 1 h prior to intraperitoneal administration of saline. Group 2 animals received 60 mg/kg regorafenib orally (prepared in vehicle A) administered every 3rd day, 1 h prior to intraperitoneal administration of saline. Group 3 animals received vehicle A orally every 3rd day, 1 h prior to 5 mg/kg intraperitoneal topotecan administration. Group 4 animals received a combination of regorafenib, administered every 3rd day orally, 1 h prior to intraperitoneal topotecan administration. The body weights of the mice were monitored and the two perpendicular diameters of tumors (A and B) were recorded every 3rd day to calculate the tumor volume. The animals were euthanized using carbon dioxide after 18 days of treatments. The tumor tissues were excised, weighed and stored at −80 °C.
The ratio of growth inhibition (IR) for tumor weight (IRW) and tumor volume (IRV, at the end of 18-days treatments) were estimated according to the formula given below,
To analyze the additive or synergistic effects of different treatment groups, the Bliss definition was applied. The additive effect was defined as the fractional response of a combination of two treatments equaling the sum of the two fractional responses minus their product, assuming two treatments are independent of each other. The effect was defined as synergistic when IRcombination > IRthreshold. The threshold was defined as the total response to a mixture of two additive treatments (A and B) and was calculated according to the formula given below.
2.12. Statistics
All experiments were performed at least three times and the differences were determined by one-way ANOVA following Newman-Keuls post hoc analysis. Differences were considered significant when p < 0.05.
3. Results
3.1. Regorafenib significantly sensitized S1–M1–80 cells to mitoxantrone and SN-38, but not to cisplatin
As compared with parental S1 cells, the drug-selected resistant S1–M1–80 cells exhibited significant multidrug resistance to BCRP substrates mitoxantrone and SN-38 due to the overexpression of BCRP. In order to determine the reversal effect of regorafenib, non-toxic concentrations (1 μM and 3 μM) of regorafenib were applied to cells as chemosensitizers prior to chemotherapeutical agents. As shown in Fig. 1A, regorafenib at 1 μM and 3 μM significantly sensitized S1–M1–80 cells to mitoxantrone. Regorafenib at 3 μM decreased the mitoxantrone IC50 from 30.21 μM to 0.31 μM. Compared with parental S1 cells, the resistant-fold of mitoxantrone in S1–M1–80 cells was decreased from 86.2-fold to 1.1-fold by 3 μM of regorafenib. Moreover, regorafenib did not sensitize parental S1 cells. A known BCRP inhibitor, FTC at 3 μM, was used as a positive control chemosensitizer. As shown in Fig. 1B, we calculated the percentage of concentration remaining to reach IC50 after BCRP inhibition followed our previous methods [19]. BCRP inhibition by both regorafenib and FTC did not significantly alter the sensitivities to mitoxantrone in S1 cells. However, only 9.14% of mitoxantrone concentration was needed to reach IC50 when co-administered with regorafenib at 1 μM. Similarly, 3 μM treatment of regorafenib led to 98.97% mitoxantrone dose reduction in S1–M1–80 cells. The inhibitory effects of regorafenib on BCRP followed a concentration-dependent pattern.
Fig. 1.
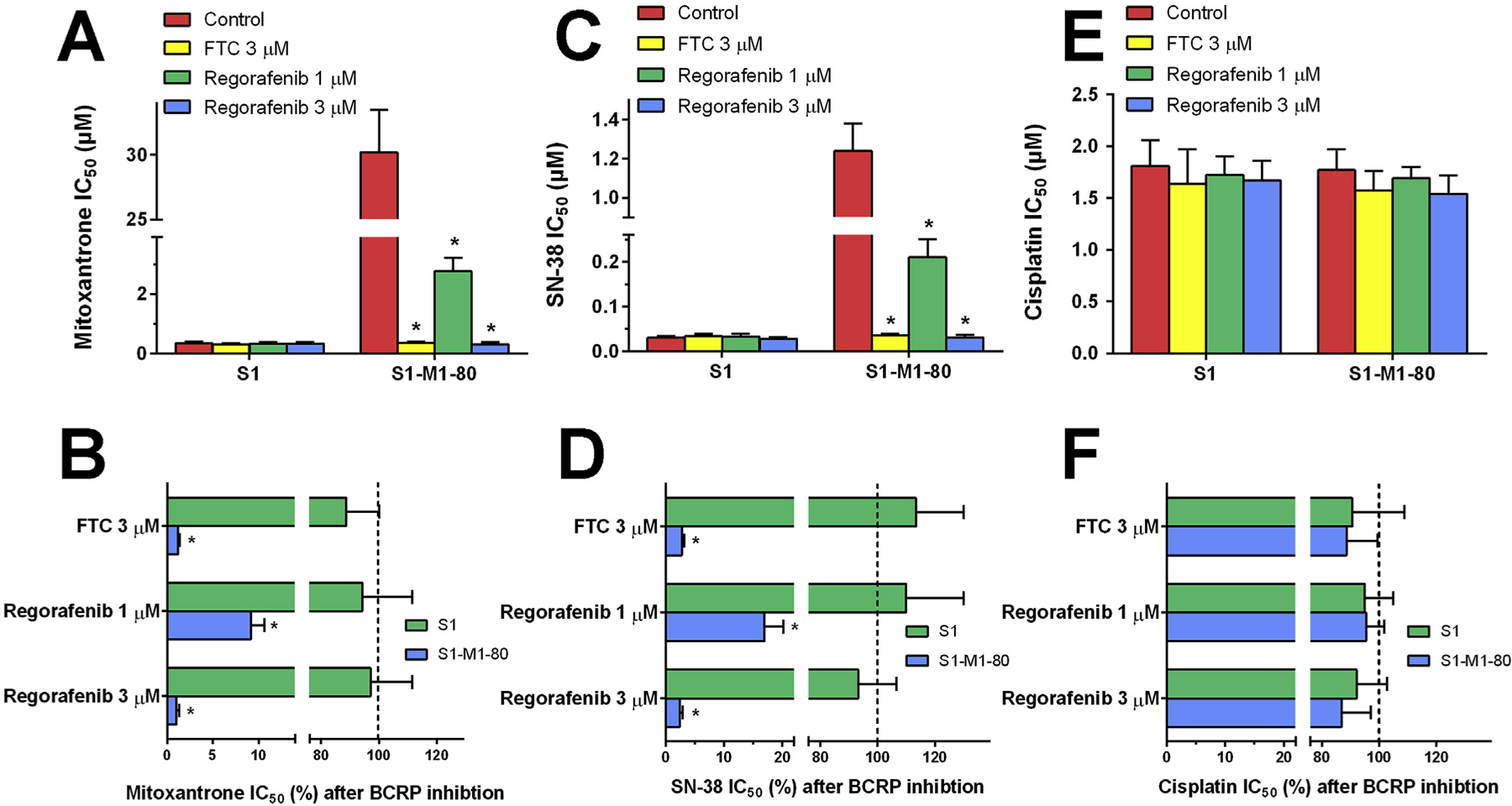
Effects of regorafenib on the IC50 values of (A) mitoxantrone, (C) SN-38 and (E) cisplatin in parental S1 and drug-selected BCRP-overexpressing multidrug resistant S1–M1–80 cells. Error bars represent SD. FTC 3 μM is used as a positive control. The percentage of dose remaining for (B) mitoxantrone, (D) SN-38 and (F) cisplatin after BCRP inhibition to reach IC50 in parental and MDR cells. Dashed lines show the mitoxantrone, SN-38 or cisplatin dose prior to BCRP inhibition (100%). Error bars represents the SD. FTC 3 μM is used as a positive control.
Subsequently, we determined the reversal effects of regorafenib on SN-38. The resistant-fold of SN-38 in S1–M1–80 was decreased from 41.3-fold to 1.02-fold by 3 μM of regorafenib, which is a 97.6% dose reduction as compared with the SN-38 dose prior to BCRP inhibition (Fig. 1C and D). Moreover, we further studied the reversal effects of regorafenib on cisplatin. Unlike mitoxantrone or SN-38, there was no significant resistance to cisplatin in S1–M1–80 cells as compared with S1 cells. The treatment of regorafenib or FTC did not lead to significant changes in IC50 values or cisplatin dose reductions in both cells (Fig. 1E and F).
3.2. Effects of regorafenib on Hoechst 33342 accumulation
It has been previously reported that BCRP is an efficient Hoechst 33342 efflux pump, therefore Hoechst 33342 could be used as a probe for the evaluation of BCRP function. Fluorescence microscopy revealed that fluorescence intensity was significantly greater in parental S1 cells compared with MDR S1–M1–80 cells after exposure to Hoechst 33342 cells for the same time period. A membrane-bound fluorescence pattern, instead of a traditional nucleus-enriched pattern, was observed in S1–M1–80 cells, suggesting the active efflux role of BCRP (Fig. 2A, third panel). The treatment with regorafenib did not change the intensity or the intracellular pattern of Hoechst 33342 fluorescence in parental S1 cells; however, regorafenib treatment significantly increased the fluorescence intensity in S1–M1–80 cells.
Fig. 2.

(A) Fluorescence microscopic analysis of Hoechst 33342 staining of S1 and S1–M1–80 cells. Cells were treated as described in Materials and Methods. Zoomedin area shows membrane-enriched fluorescence pattern. (B) Effects of regorafenib on intracellular accumulation of [3H]-mitoxantrone in S1 and S1–M1–80 cells. (C) Efflux of [3H]-mitoxantrone in the absence or presence of inhibitors in S1–M1–80 cells. (D) The effects of regorafenib at 3 μM on the expression level of BCRP in S1–M1–80 cells. (E) Quantitative analysis of effects of regorafenib on BCRP expression. (F) The effects of regorafenib at 3 μM on the subcellular localization of BCRP in S1–M1–80 cells.
3.3. Effects of regorafenib on [3H]-mitoxantrone accumulation and efflux
In order to quantify the intracellular accumulation of BCRP substrate, [3H]-mitoxantrone accumulation was measured with or without inhibitors. As shown in Fig. 2B, lower intracellular [3H]-mitoxantrone was observed in drug-resistant S1–M1–80 cells as compared with parental S1 cells. Pretreatment with 1 μM regorafenib increased the intracellular [3H]-mitoxantrone in S1–M1–80 cells from 12.1% to 51.4% of that in parental cells. Pretreatment with 3 μM regorafenib increased the intracellular [3H]-mitoxantrone in S1–M1–80 cells from 12.1% to 79.8% of that in parental cells. FTC at 3 μM was used as a positive control and increased the intracellular [3H]-mitoxantrone in S1–M1–80 cells from 12.1% to 83.2% of that in parental cells. Regorafenib and FTC did not significantly alter the [3H]-mitoxantrone accumulation level in S1 cells.
In order to evaluate the reason for drug accumulation and to quantify the time-course of drug efflux in parental and MDR cells, a tritium-labeled efflux assay was performed (Fig. 2C). Untreated S1–M1–80 cells effluxed 47% of the normalized intracellular [3H]-mitoxantrone in 120 min of incubation in mitoxantrone-free medium. Treatment of cells with 1 and 3 μM regorafenib led to significant intracellular [3H]-mitoxantrone retention (70% and 83%, respectively). [3H]-mitoxantrone by 3 μM FTC was used as a positive control.
3.4. Effects of regorafenib on the expression level and subcellular localization of BCRP
Western blot was used to determine the effects of regorafenib on the expression level of BCRP. As shown in Fig. 2D–E, treatment of 3 μM regorafenib in S1–M1–80 cells for up to 72 h did not significantly alter the expression level of BCRP. We further visualized the subcellular localization of BCRP inside S1–M1–80 cells using immunofluorescence assay. As shown in Fig. 2F, membrane-enriched fluorescence was observed, consistent with the role of BCRP as a membrane-bound efflux pump. Regorafenib treatment for up to 72 h had no significant effects on the subcellular localization of BCRP.
3.5. Regorafenib-associated BCRP ATPase activities
The effects of regorafenib on BCRP ATPase were measured by ATP hydrolysis in the presence of drugs at different concentrations ranging from 0 to 40 μM (Fig. 3A). Regorafenib stimulated BCRP ATPase with a maximal 3.29-fold of the basal activity. Regorafenib had a bell-shaped curve instead of a Michaelis-Menten type curve in stimulating ATPase activity of BCRP. The concentration of regorafenib required to obtain 50% stimulation is 3.59 ± 0.42 μM. At concentrations (1 μM and 3 μM) used in this study, regorafenib stimulated BCRP ATPase at 2.48- and 2.79-fold, respectively.
Fig. 3.

(A) Effects of regorafenib (0–40 μM) on the BCRP ATPase activities. Concentrations of regorafenib are plotted at linear or log scale. (B) The polar interactions between regorafenib and nearby residues inside the BCRP binding cavity. Regorafenib and interacting residues are depicted as sticks. BCRP backbones are depicted in cartoon style. All atoms are colored with CPK coloring scheme, whereas the carbon atoms in regorafenib are presented in orange and the carbon atoms in BCRP are presented in green. (C) The superimposition of induced-fit docked pose and MD pose of regorafenib within the binding cavity of BCRP. Ligands are depicted as sticks with CPK coloring, whereas the carbon atoms in induced-fit docked regorafenib are presented in orange and carbon atoms in MD pose of regorafenib are presented in green. The protein cartoon for BCRP are presented in red or blue for pre- and post-MD, respectively. (D) RMSD trajectory of BCRP and regorafenib in regorafenib-BCRP complex over the 10 ns simulation run.
3.6. Regorafenib-BCRP molecular docking and molecular dynamics
We first equilibrated the BCRP crystal structure by 10 ns of molecular dynamics simulations. There was no significant difference between the pre- and post-equilibration protein structure (RMSD: 2.7 Å). We then performed induced-fit docking to study the interaction between regorafenib and BCRP. As shown in Fig. 4B, the best-scored docked pose of regorafenib were predicted within the transmembrane domain of BCRP. The regorafenib ligand was stabilized into the hydrophobic cavity formed by nearby residues. Three hydrogen bonds were observed between regorafenib and Asn 436 and Thr542 (Fig. 4B). We further performed molecular dynamics simulations for the regorafenib-BCRP complex to validate the docked pose. The superimposition of regorafenib in pre- and post-MD complex is depicted in Fig. 4C. The binding pocket did not have significant steric changes and there was no turn-over in the regorafenib binding pose. The predicted hydrogen bonds between regorafenib and BCRP within the binding pockets persisted for more than 80% of the simulation time. Also, by monitoring the root-mean-square deviation (RMSD) shown in in Fig. 4D, we concluded that stable conformations of regorafenib persisted until the end of the simulation.
Fig. 4.
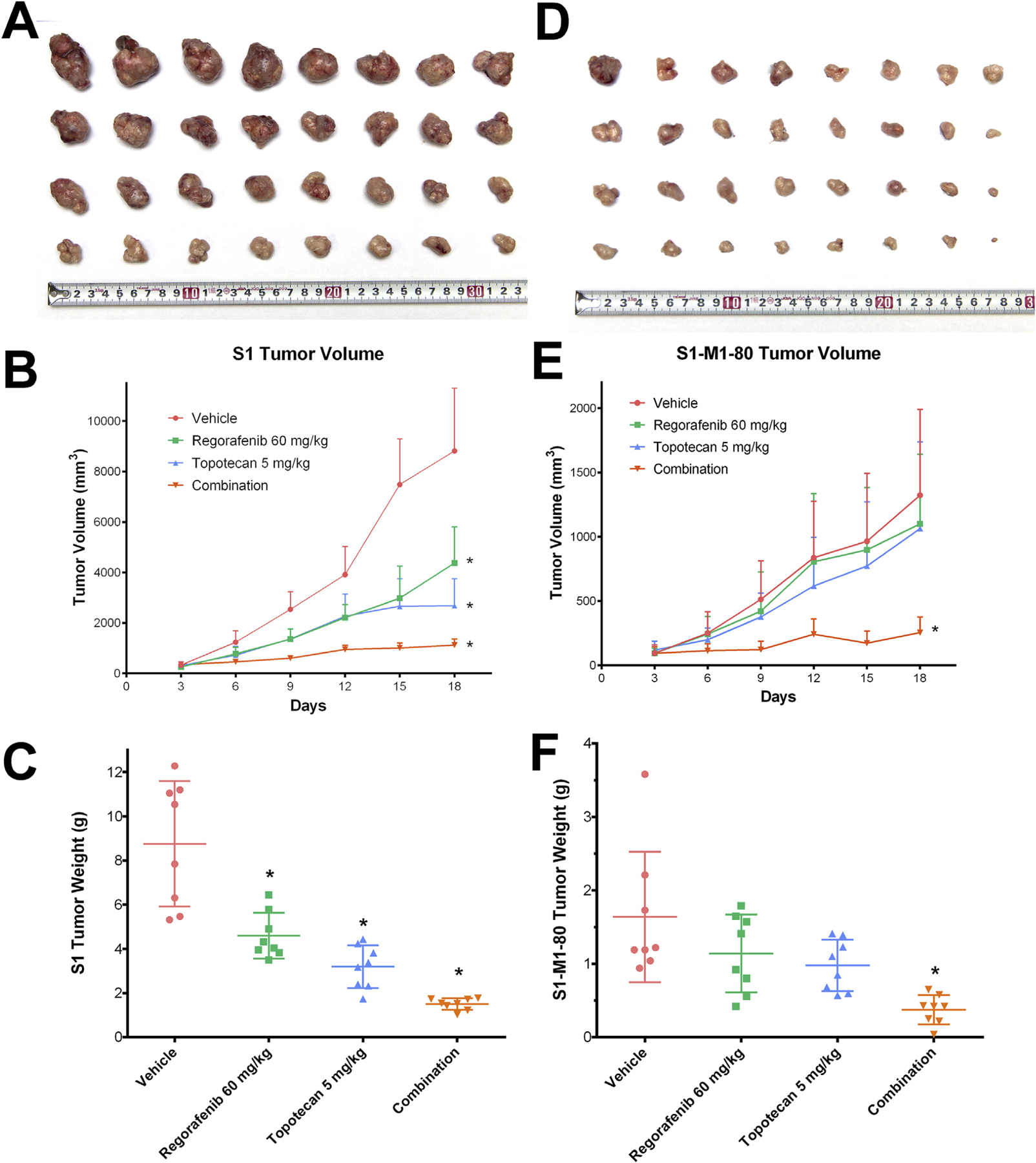
Effect of regorafenib and topotecan on the growth of S1 tumors in nude athymic mice. (A) The images of excised S1 tumors implanted subcutaneously in athymic NCR nude mice (n = 8) that were treated with vehicle, regorafenib, topotecan and the combination of regorafenib and topotecan. (B) Changes in tumor volume over time following the implantation. Data points represent the mean tumor volumes for each treatment group. (C) The mean weight of the excised S1 tumors from the mice treated with vehicle, regorafenib, topotecan and the combination of regorafenib and topotecan, at the end of the 18-day treatment period. Effect of regorafenib and topotecan on the growth of S1–M1–80 tumors in nude athymic mice. (D) The images of excised S1–M1–80 tumors implanted subcutaneously in athymic NCR nude mice (n = 8) that were treated with vehicle, regorafenib, topotecan and the combination of regorafenib and topotecan. (E) The changes in tumor volume over time following the implantation. (F) The mean weight of the excised S1–M1–80 tumors from the mice treated with vehicle, regorafenib, topotecan and the combination of regorafenib and topotecan, at the end of the 18-day treatment period. Error bars, SD. *p < 0.05 versus the vehicle group.
3.7. Regorafenib potentiated the anticancer activity of topotecan in BCRP-overexpressed colon cancer xenograft model
After evaluating the in vitro reversal effects of regorafenib on BCRP-overexpressing colon cancer cells, we further translated these findings into the in vivo colon cancer xenograft model. As shown in Fig. 4, after 18-days of treatment, regorafenib alone demonstrated strong tumor inhibitory effects on S1 tumors, with 46% inhibition in tumor weights and 50% inhibition in tumor volumes. Regorafenib alone also exhibited moderate but non-significant inhibitory effects on S1–M1–80 tumors, with 33% inhibition in tumor weights and 17% inhibition in tumor volume. Treatment of topotecan demonstrated significant growth retardation in S1 tumors; however, its inhibitory effects in S1–M1–80 cells were much weaker when compared with that in S1 tumors (64% IRW in S1 tumors and 42% IRW in S1–M1–80 tumors; 70% IRV in S1 tumors and 20% IRV in S1–M1–80 tumors). As shown in Fig. 4E, the increase in tumor volume of S1–M1–80 cells was not successfully controlled by either regorafenib or topotecan treatment. The growth of xenograft tumors was significantly inhibited in the regorafenib-topotecan combination group as compared with the vehicle group. The combination treatment exhibited 83% IRW and 87% IRV in parental S1 tumors. IRW and IRV in S1–M1–80 tumors for the combination group were 78% and 81% respectively. The threshold for synergistic effects were calculated as 81% IRW, 85% IRV for S1 tumors and 61% IWR, 33% IRV for S1–M1–80 tumors based on the IRW and IRV values of regorafenib-alone and topotecan-alone groups (Table 1). Therefore, we concluded that the combination of regorafenib and topotecan exhibited additive effects in S1 xenograft tumors and synergistic effects in S1–M1–80 xenograft tumors.
Table 1.
The ratios of growth inhibition (IR) for tumor weight (IRW) and tumor volume (IRV) of S1 and S1–M1–80 xenograft tumors at the end of 18-days treatments.
| S1 tumors | S1–M1–80 tumors | |||
|---|---|---|---|---|
| IRWa (%) | IRVb (%) | IRWa (%) | IRVb (%) | |
| Regorafenib | 46* | 50* | 33 | 17 |
| Topotecan | 64* | 70* | 42 | 20 |
| Calculated threshold of synergy c | 81 | 85 | 61 | 33 |
| Combination of regorafenib and topotecan | 83* | 87* | 78* | 81* |
IRW and IRV are calculated as compared with the vehicle-only control group as described in the Materials and methods section.
Threshold of synergy are calculated based on the average growth inhibition values of regorafenib and topotecan treatment using the Bliss method as describe in the Materials and Methods section.
Significant (p < 0.05) as compared with vehicle-only group.
4. Discussion
In this study, we validated the sensitizing effects of regorafenib in MDR colorectal cancers, which could be beneficial in developing more potent targeted combination chemotherapy for colorectal cancer patients. Regorafenib is currently used for colorectal cancer as an FDA-approved target therapy; therefore, there are clinically translational advantages to develop novel combinational therapies using regorafenib.
The sensitizing effects of regorafenib on S1–M1–80 cells in our cytotoxicity assays were clearly related to BCRP as demonstrated by our experimental design for the following reasons. First, regorafenib did not significantly sensitize isogenic parental S1 cells, in which BCRP is not overexpressed (Supplemental Figure S1). Second, cisplatin is not a substrate of BCRP, as demonstrated by the similarity of cisplatin IC50 values in both parental S1 and MDR S1–M1–80 cells. Regorafenib did not sensitize both these cells towards cisplatin, suggesting that the sensitizing effects of regorafenib is limited to BCRP substrates in those cells. Lastly, the BCRP-transfected HEK/ABCG2 cells exhibited resistance to BCRP substrate, and this resistance can be reversed by regorafenib (Supplemental Figure S2). Therefore, we could conclude that regorafenib sensitizes MDR in S1–M1–80 cells by targeting the BCRP transporter.
Inhibition of active efflux pumps such as BCRP would lead to decreased substrates efflux, and therefore increased intracellular substrate accumulation, and finally result in increased drug efficacy. Our drug efflux assay showed rapid [3H]-mitoxantrone efflux in S1–M1–80 cells, suggesting active BCRP function in these cells. The inhibition of the BCRP pump by regorafenib resulted in significant decreases in drug efflux. Reduced drug efflux is not a guarantee for intracellular drug accumulation especially in cancer cells, since the intracellular drugs may be deactivated due to altered metabolic pathways [24]. Therefore, Hoechst dye and [3H]-mitoxantrone were used to directly show intracellular drug accumulation qualitatively and quantitatively. These results indicated that the mechanism of regorafenib in sensitizing MDR is by regorafenib inhibiting the efflux of BCRP and elevating intracellular drug accumulation which ultimately leads to cell death.
As a membrane-bound transporter, BCRP may also have lost its function by being down-regulated or translocated into the cytoplasm. Since pretreatment with regorafenib for 1 h exhibited sufficient inhibitory effects in our efflux/accumulation assays, it is not likely that the inhibitory effects were mainly related to down-regulation. This hypothesis was further examined by Western blotting analysis. No obvious down-regulation of BCRP was observed after the cells were treated with regorafenib for up to 72 h. Therefore, regorafenib did not function as a BCRP down-regulator. Another potential factor is translocation of BCRP. Unlike down-regulation, translocation of membrane transporters could happen in milliseconds [25], however our immunofluorescence assay did not shown BCRP translocation. Taking all these results into consideration, we conclude that regorafenib may directly target BCRP and inhibit its substrates-pumping function.
BCRP uses ATP as energy source to transport substrates across cell membranes. Given the fact that regorafenib belongs to a class of tyrosine kinase inhibitors, it is rational to hypothesize that regorafenib may inhibit ATP binding and therefore inhibit BCRP activity. Interestingly, our result indicated that regorafenib stimulated BCRP ATPase activities, while our previous results indicated that regorafenib inhibited P-gp ATPase activities [26]. Regorafenib may serve as a competitive inhibitor of BCRP. Our molecular simulation studies provided a further look into the molecular interactions between regorafenib and the BCRP drug-binding pocket. The crystal structure of BCRP is very stable during our initial MD simulation (RMSD: 2.4 Å). Our induced-fit docking algorithm then predicted high binding affinity of regorafenib at the drug-binding cavity at transmembrane domain of BCRP. Moreover, the docked pose of regorafenib matched the shape of the previously illustrated cavity by Taylor et al. [14]. However, several studies have revealed the problem that some well-docked ligand might ‘fly’ away from the binding pocket in MD simulation, suggesting unreliable docked posse [27]. We further performed 10 ns MD for the regorafenib-BCRP complex. Hydrogen bond interactions between regorafenib and residue Thr542 persisted through more than 80% of the simulation time. The maximal RMSD of regorafenib was recorded at 1.6 Å. Therefore, we concluded that the regorafenib binding pose is very stable and reliable in the cavity within BCRP through 10 ns of simulation. These in silico results suggested that regorafenib might have high affinity to the substrate binding cavity and therefore block the pumping function of BCRP in a competitive way.
In vitro experiments are regarded as non-physiological and have great limitations. Unlike formulated culture medium, living creatures are biologically complex, especially for higher order animals. The in vivo tumor environment is dependent on several sophisticated systems including the circulatory system and the immune system. Therefore, while the results discovered from experiments carried out in cultured cancer cells is important, in vivo approaches are necessary to investigate how drugs can produce therapeutic effects in a whole living organism. Many in vivo interactions are complex and cannot be obtained from in vitro experiments, such as the anti-angiogenesis effects of regorafenib. Thus, we performed in vivo evaluations on xenograft tumors using in vitro data as guidance. It is interesting to note that S1–M1–80 tumors proliferated much slower than S1 tumors, however S1–M1–80 tumors were resistant to either topotecan or regorafenib therapy. No significant benefits were observed in the topotecan-treated group in S1–M1–80 xenograft tumors. Regorafenib alone exhibited 50% IRV in S1 tumors; however, it only exhibited 17% (non-significant) IRV in S1–M1–80 tumors. The poorer response of regorafenib in S1–M1–80 tumors is consistent with our previous hypothesis that regorafenib may be a substrate of BCRP. Although S1–M1–80 tumors failed to respond to topotecan- or regorafenib-treatment, the development of S1–M1–80 tumors were successfully controlled by the combination of topotecan and regorafenib. Moreover, in order to evaluate whether topotecan and regorafenib exhibit synergistic or additive effects in combination, we used the Bliss’ synergy algorithm to calculate the threshold inhibition rate. Additive effects of regorafenib and topotecan was observed in parental S1 tumors. The combination of regorafenib and topotecan exhibited strong synergistic effects in S1–M1–80 tumors, with a tumor inhibition rate significantly lower than threshold values based on the effects of regorafenib and topotecan alone. This synergistic effect may be explained by the in vivo inhibitory effect of BCRP by regorafenib, leading to higher topotecan efficacy in S1–M1–80 tumors.
In conclusion, we report that regorafenib reversed BCRP-mediated MDR in colorectal cancer cell lines by binding at the transmembrane cavity of BCRP, inhibiting the efflux function and increasing intracellular concentrations of BCRP substrates. In vivo studies revealed that a combination therapy of topotecan and regorafenib exerts synergistic effects on inhibiting the development of MDR human colorectal xenograft tumors. These results suggest that the usage of regorafenib would have the great potential to be expanded as a combination therapy with conventional chemotherapeutic agents. Patients bearing colorectal cancers or metastatic liver cancers, especially in those with BCRP overexpressed cancers, may benefit from regorafenib combination chemotherapy.
Supplementary Material
Acknowledgements
We are thankful for Drs. Susan E. Bates and Robert W. Robey (NIH, MD) for providing FTC, S1 cells and S1–M1–80 cells. We thank Dr. Tanaji T. Talele (St. John’s University, New York) for providing the computational resources of induced-fit dockings. We thank Dr. Yangmin Chen (MediMedia Managed Markets, ICON plc, Yardley, PA) for editorial support.
Fundings
This work was supported by funds from NIH (No. 1R15CA143701 and No. 1R15GM116043–01) and St. John’s University Research Seed Grant (No. 579–1110–7002) to Z. Chen. S.S and S.V.A were supported by the Intramural Research Program of National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.canlet.2018.10.032.
References
- [1].Fleming M, Ravula S, Tatishchev SF, Wang HL, Colorectal carcinoma: pathologic aspects, J. Gastrointest. Oncol 3 (2012) 153–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Simmonds PC, Primrose JN, Colquitt JL, Garden OJ, Poston GJ, Rees M, Surgical resection of hepatic metastases from colorectal cancer: a systematic review of published studies, Br. J. Canc 94 (2006) 982–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N, Colorectal cancer, Lancet 375 (2010) 1030–1047. [DOI] [PubMed] [Google Scholar]
- [4].Gottesman MM, Ludwig J, Xia D, Szakacs G, Defeating drug resistance in cancer, Discov. Med 6 (2006) 18–23. [PubMed] [Google Scholar]
- [5].Van Cutsem E, Borras JM, Castells A, Ciardiello F, Ducreux M, Haq A, Schmoll HJ, Tabernero J, Improving outcomes in colorectal cancer: where do we go from here? Eur. J. Canc 49 (2013) 2476–2485. [DOI] [PubMed] [Google Scholar]
- [6].Zahreddine H, Borden KL, Mechanisms and insights into drug resistance in cancer, Front. Pharmacol 4 (2013) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hammond WA, Swaika A, Mody K, Pharmacologic resistance in colorectal cancer: a review, Ther Adv Med Oncol 8 (2016) 57–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wu Q, Yang Z, Nie Y, Shi Y, Fan D, Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches, Cancer Lett 347 (2014) 159–166. [DOI] [PubMed] [Google Scholar]
- [9].Amiri-Kordestani L, Basseville A, Kurdziel K, Fojo AT, Bates SE, Targeting MDR in breast and lung cancer: discriminating its potential importance from the failure of drug resistance reversal studies, Drug Resist. Updates 15 (2012) 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li W, Zhang H, Assaraf YG, Zhao K, Xu X, Xie J, Yang DH, Chen ZS, Overcoming ABC transporter-mediated multidrug resistance: molecular mechanisms and novel therapeutic drug strategies, Drug Resist. Updates 27 (2016) 14–29. [DOI] [PubMed] [Google Scholar]
- [11].Zhang YK, Wang YJ, Gupta P, Chen ZS, Multidrug resistance proteins (MRPs) and cancer therapy, AAPS J 17 (2015) 802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dean M, Allikmets R, Complete characterization of the human ABC gene family, J. Bioenerg. Biomembr 33 (2001) 475–479. [DOI] [PubMed] [Google Scholar]
- [13].Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD, A multidrug resistance transporter from human MCF-7 breast cancer cells, Proc. Natl. Acad. Sci. U. S. A 95 (1998) 15665–15670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Taylor NMI, Manolaridis I, Jackson SM, Kowal J, Stahlberg H, Locher KP, Structure of the human multidrug transporter ABCG2, Nature 546 (2017) 504–509. [DOI] [PubMed] [Google Scholar]
- [15].Robey RW, Polgar O, Deeken J, To KW, Bates SE, ABCG2: determining its relevance in clinical drug resistance, Cancer Metastasis Rev 26 (2007) 39–57. [DOI] [PubMed] [Google Scholar]
- [16].Jeltsch M, Leppanen VM, Saharinen P, Alitalo K, Receptor tyrosine kinasemediated angiogenesis, Cold Spring Harb Perspect Biol 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schutz G, Thierauch KH, Zopf D, Regorafenib (BAY 73–4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity, Int. J. Canc 129 (2011) 245–255. [DOI] [PubMed] [Google Scholar]
- [18].Miyake K, Mickley L, Litman T, Zhan Z, Robey R, Cristensen B, Brangi M, Greenberger L, Dean M, Fojo T, Bates SE, Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes, Cancer Res 59 (1999) 8–13. [PubMed] [Google Scholar]
- [19].Zhang YK, Zhang XY, Zhang GN, Wang YJ, Xu H, Zhang D, Shukla S, Liu L, Yang DH, Ambudkar SV, Chen ZS, Selective reversal of BCRP-mediated MDR by VEGFR-2 inhibitor ZM323881, Biochem. Pharmacol 132 (2017) 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhang YK, Zhang GN, Wang YJ, Patel BA, Talele TT, Yang DH, Chen ZS, Bafetinib (INNO-406) reverses multidrug resistance by inhibiting the efflux function of ABCB1 and ABCG2 transporters, Sci. Rep 6 (2016) 25694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang YK, Zhang H, Zhang GN, Wang YJ, Kathawala RJ, Si R, Patel BA, Xu J, Chen ZS, Semi-synthetic ocotillol analogues as selective ABCB1-mediated drug resistance reversal agents, Oncotarget 6 (2015) 24277–24290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes, J. Med. Chem 49 (2006) 6177–6196. [DOI] [PubMed] [Google Scholar]
- [23].Farid R, Day T, Friesner RA, Pearlstein RA, New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies, Bioorg. Med. Chem 14 (2006) 3160–3173. [DOI] [PubMed] [Google Scholar]
- [24].Herling A, Konig M, Bulik S, Holzhutter HG, Enzymatic features of the glucose metabolism in tumor cells, FEBS J 278 (2011) 2436–2459. [DOI] [PubMed] [Google Scholar]
- [25].Winterfeld S, Ernst S, Borsch M, Gerken U, Kuhn A, Real time observation of single membrane protein insertion events by the Escherichia coli insertase YidC, PloS One 8 (2013) e59023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang YJ, Zhang YK, Zhang GN, Al Rihani SB, Wei MN, Gupta P, Zhang XY, Shukla S, Ambudkar SV, Kaddoumi A, Shi Z, Chen ZS, Regorafenib overcomes chemotherapeutic multidrug resistance mediated by ABCB1 transporter in colorectal cancer: in vitro and in vivo study, Cancer Lett 396 (2017) 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen YC, Beware of docking!, Trends Pharmacol. Sci 36 (2015) 78–95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.