

Abstract
Metabolic syndrome is a clustering of metabolic disorder with unclear molecular mechanism. Increasing studies have found that the pathogenesis and progression of metabolic syndrome are closely related to inflammation. Here, we report celastrol, a traditional Chinese medicine, can improve high fat diet-induced metabolic syndrome through suppressing resistin-induced inflammation. Mechanistically, celastrol binds to adenylyl cyclase associated protein 1 (CAP1) and inhibits the interaction between CAP1 and resistin, which restrains the cyclic adenylate monophosphate (cAMP)–protein kinase A (PKA)–nuclear factor kappa-B (NF-κB) signaling pathway and ameliorates high fat diet-induced murine metabolic syndrome. Knockdown of CAP1 in macrophages abrogated the resistin-mediated inflammatory activity. In contrast, overexpression of CAP1 in macrophages aggravated inflammation. Taken together, our study identifies celastrol, which directly targets CAP1 in macrophages, might be a promising drug candidate for the treatment of inflammatory metabolic diseases, such as metabolic syndrome.
KEY WORDS: Metabolic syndrome, Celastrol, CAP1, Resistin, Inflammation
Graphical abstract
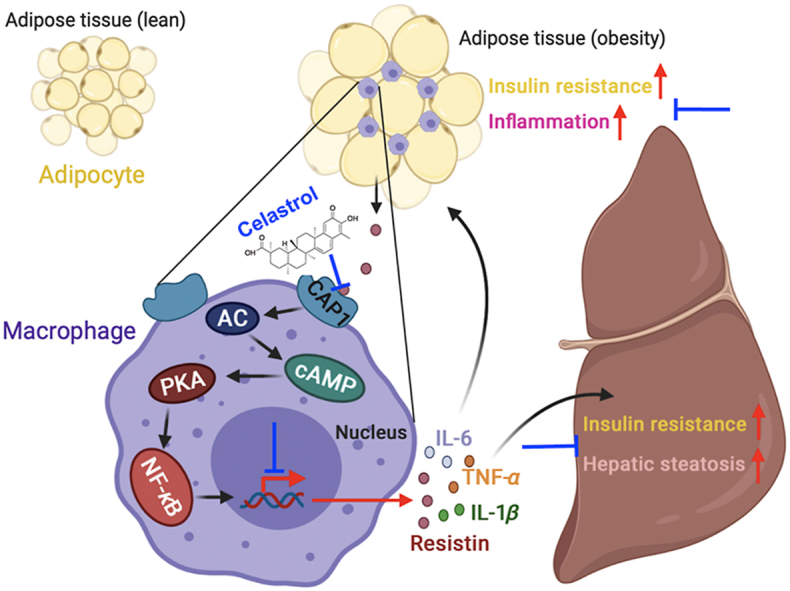
In obesity, adipose tissue generates resistin, which binds to CAP1 on macrophages and activates the cAMP–PKA–NF-κB signaling pathway and promotes proinflammatory cytokines expression. These factors further aggravate inflammation, insulin resistance, and hepatic steatosis. Celastrol ameliorates metabolic syndrome via binding to CAP1 and inhibiting its interaction with resistin.
1. Introduction
Metabolic syndrome (MetS) is a clustering of clinical findings made up of abdominal obesity, high glucose, high triglyceride, low high-density lipoprotein cholesterol levels, and hypertension1, 2, 3, 4, 5. In recent years, the incidence rate of MetS has risen sharply worldwide. This increase is related to the global epidemic of obesity and diabetes6. Because MetS not only increases the risk of diabetes, but also increases the risk of cardiovascular disease7, 8, 9, 10, 11, there is an urgent need to develop strategies to prevent this emerging global epidemic. Increasing studies have found that the pathogenesis and progression of MetS are closely related to inflammation12, and is usually called chronic low-grade inflammation, which interacts with insulin resistance and obesity13, and eventually worsens the MetS.
Resistin was found to cause insulin resistance14, 15, 16, 17. It can completely reverse insulin resistance by normalizing plasma resistin level. Subsequently, a large number of studies show that resistin plays an important role in the occurrence and development of a variety of inflammatory diseases15,16. It has been suggested that resistin may be a molecular link between inflammation, metabolism, and vascular dysfunction18, leading to the risk of MetS, type 2 diabetes, and cardiovascular disease. Adenylyl cyclase associated protein 1 (CAP1), a functional receptor for human resistin19, could be a target in the treatment of inflammatory diseases.
Celastrol is a pentacyclic triterpene compound derived from the roots of the Tripterygium wilfordii. Celastrol is widely used for a number of indications, including chronic inflammatory and immunity disorders20, 21, 22. Recently, celastrol has emerged as a promising candidate to treat obesity23, 24, 25, 26. However, the underlying mechanism by which celastrol reduces MetS remains unclear.
In this study, we have investigated the effect of celastrol on high fat diet-induced MetS. Our studies show that celastrol protects mice from high fat diet-induced obesity, insulin resistance, hepatic steatosis, and inflammation. Mechanistically, we have discovered that celastrol provides these MetS-protective effects by binding to CAP1 and inhibiting CAP1‒resistin interaction. Our work identifies that celastrol can directly target CAP1 and inhibit resistin-induced inflammation and offer a therapy for MetS.
2. Materials and methods
2.1. Mice
C57BL/6J mice were purchased from GemPharmatech Co., Ltd. (Nanjing, China). Age- and sex-matched mice 6–10 weeks of age were used. All mice except the normal control group were fed 60% fat feed for 16 weeks, providing that obesity, high blood sugar, and at least one lipid abnormality were considered to meet the MetS standard. Mice with successful molding were randomly divided into high-fat diet (HFD) feed group, celastrol high-dose group (celastrol, 3 mg/kg/day), medium dose group (celastrol, 1.5 mg/kg/day), low-dose group (celastrol, 0.75 mg/kg/day), and C57BL/6J mice fed normally for a normal control group of five groups. The mice in the administration group were given the indicated dose of the subject drug daily, and the normal group and the HFD group filled the stomach with volume solvents daily, for 25 consecutive days. The weight of the mice was measured every 5 days from the time of the drug was given and the food intake of each cage of mice was recorded to determine whether obesity in the mice had improved after the drug was given. All mice were housed in individually ventilated cages and maintained on a 12 h light–dark cycle. Animal welfare and experimental procedures were approved by the Institutional Animal Care and Use Committee at Nanjing University (Nanjing, China). All efforts were made to reduce the number of animals used and to minimize animal suffering.
2.2. Cell lines and cell culture
Human myeloid leukemia mononuclear cells (THP-1) and HEK293T cells were purchased from Shanghai Institute of Cell Biology (Shanghai, China). THP-1 cells were cultured in RPMI 1640 (Biological Industries, Kibbutz Beit Haemek, Israel) culture media containing 10% fetal bovine serum (Biological Industries, Kibbutz Beit Haemek, Israel) at 37 °C in 5% CO2. HEK293T cells were maintained in Dulbecco's Modified Eagle's Medium (Biological Industries, Kibbutz Beit Haemek, Israel) culture media supplemented with 10% fetal bovine serum (Biological Industries, Kibbutz Beit Haemek, Israel), 100 U/mL streptomycin-penicillin (Beyotime, Shanghai, China) at 37 °C in 5% CO2.
2.3. Glucose tolerance test
Mice were fasted for 12 h (free drinking water), and then intraperitoneally administered dextrose (2 g/kg body weight). Blood glucose levels were measured with a glucometer (Johnson, USA) using the blood obtained from the tail before dextrose administration, and 30, 60, 90, and 120 min after administration. Plotted with the blood sugar time curve, and calculated area under curve (AUC) using GraphPad Prism 8.0.
2.4. Insulin tolerance test
Mice were fasted for 6 h. Recombinant human insulin (1 IU/kg body weight) was intraperitoneally administrated. Blood glucose levels were measured with a glucometer using the blood obtained from the tail before insulin administration, and 30, 60, 90, and 120 min after administration. Plotted the blood sugar changes over time curve, and calculate the area under the curve AUC.
2.5. Serum triglycerides (TG) and total cholesterol (T-CHO) measurements
The mice took blood from the eyeballs, placed the blood in a refrigerator at 4 °C for 3 h, centrifuged at 1000×g for 15 min, and isolated the mouse serum as a sample. Detections were all performed according to the manufacturer's instructions of TG kits (Nanjing Jiancheng Bioengineering Institute, China) and T-CHO kits (Nanjing Jiancheng Bioengineering Institute, China).
2.6. Enzyme linked immunosorbent assay (ELISA)
ELISA assays were used to detect for the presence of various cytokines in mouse serum isolated from imiquimod (IMQ)-induced murine model animals. ELISA kits for mouse cytokines tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin 1 beta (IL-1β), and resistin were purchased from Dakewe Biotech, China. ELISA detections were all performed according to the manufacturer's instructions.
2.7. Histological analysis
Harvested mouse livers and epididymal visceral adipose tissue were flushed with phosphate buffer saline (PBS), fixed with 4% formaldehyde overnight and embedded in paraffin. Sections (5 μm thick) were stained with hematoxylin and eosin (H&E) according to standard procedures.
For immunohistochemistry, the mouse liver and epididymis paraffin sections were deparaffinized, rehydrated, and antibody retrieved with sodium citrate, blocked, and then stained with anti-CD11c (Proteintech, China) and anti-resistin (ABclonal, USA) antibodies at 1:100 overnight at 4 °C. After 3 rinses of 1 × PBS, the slides were detected using Real Envision Detection kit (GeneTech, China) according to the manufacturer's instructions.
For immunofluorescence, paraffin-embedded human and mouse skin sections were deparaffinized and rehydrated, antigen retrieved with sodium citrate, blocked with 5% goat serum, and incubated with primary Abs overnight at 4 °C. Anti-caveolin-1 (Santa Cruz Biotechnology, USA), anti-CAP1 (Abcam, USA), anti-p-p65 (Santa Cruz Biotechnology, USA) were used in 1:100. After 3 rinses of 1 × PBST, sections were treated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (H+L) cross-adsorbed secondary Ab (1:500; Invitrogen, USA), Alexa Fluor 488-conjugated goat anti-mouse IgG (H+L) cross-adsorbed secondary Ab (1:500; Invitrogen, USA), Alexa Fluor 546-conjugated goat anti-mouse IgG (H+L) cross-adsorbed secondary Ab (1:500; Invitrogen, USA), and Alexa Fluor 594-conjugated goat anti-rabbit IgG (H+L) cross-adsorbed secondary Ab (1:500; Invitrogen, USA) at room temperature for 2 h in the dark, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Beyotime). All the cells were imaged by an inverted confocal microscope (Carl Zeiss, Germany).
2.8. Immunocytochemistry
Phorbol ester (PMA, Sigma–Aldrich, USA)-differentiated THP-1 cells were incubated in 24-well culture dishes at a density of 1 × 105 cells per well. Cells were washed by PBS and then fixed by 4% paraformaldehyde for 10 min at room temperature. Following 3 rinses in 1 × PBS, cells were permeabilized using 0.5% Triton X-100 (Beyotime) for 30 min at 4 °C. After blocking cells with 5% BSA for 1 h, cells were cultured with primary Abs overnight at 4 °C. Anti-CAP1 and anti-p-p65 Abs were used at 1:100. After 3 rinses of 1 × PBST, coverslips were treated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (H+L) cross-adsorbed secondary Ab (1:500), Alexa Fluor 488-conjugated goat anti-mouse IgG (H+L) cross-adsorbed secondary Ab (1:500), Alexa Fluor 546-conjugated goat anti-mouse IgG (H+L) cross-adsorbed secondary Ab (1:500), and Alexa Fluor 594-conjugated goat anti-rabbit IgG (H+L) cross-adsorbed secondary Ab (1:500) at room temperature for 2 h in the dark, and the nuclei were stained with DAPI. All the cells were imaged by an inverted confocal microscope (Carl Zeiss, Germany).
2.9. Quantitative real-time polymerase chain reaction PCR (qRT-PCR)
Total RNA was extracted from THP-1 cells and HEK293T cells using TRIzol (TaKaRa, Japan) as described by the manufacturer. Single-stranded cDNA was synthesized from 1 μg of total RNA by reverse transcription. Real-time PCR was performed with SYBR Green Realtime PCR Master Mix (Vazyme Biotech Co., Ltd., China) on a CFX 100 (Bio-Rad, Hercules, CA, USA) cycler. The amplification program was as follows: 95 °C for 2.5 min, and 44 cycles at 95 °C for 15 s, 60 °C for 30 s. Dissociation curves were analyzed at the end of the amplification. The level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or actin RNA expression was used to normalize the data.
2.10. Target discovery via a target-responsive accessibility profiling approach
The screening of celastrol binding proteins was performed as described previously27. Briefly, Target-Responsive Accessibility Profiling (TRAP) approach was employed to discover the binding proteins for celastrol in cell milieu by monitoring ligand engagement-induced lysine accessibility changes on a proteome-level. Briefly, two dishes of cells were treated with 30 nmol/L celastrol and dimethyl sulfoxide (DMSO), respectively. After 1 h incubation, cells were permeabilized by M-PER buffer (Thermo Scientific) and the resultant lysates were covalently labeled by the addition of formaldehyde and borane pyridine complex that together specifically label proteinaceous lysine residues at room temperature for accessibility profiling. Then, the lysates were precipitated by organic solvent, and the collected pellets were redissolved in 8 mol/L urea, reduced by dithiothreitol (DTT) at 56 °C for 30 min followed by alkylation using iodoacetamide (IAA) in dark for 30 min. Appropriate amount of DTT solution was added again to react with excess IAA. Subsequently, the proteome was diluted with ammonium bicarbonate solution until the final concentration of urea reaches 1 mol/L. The collected protein digests were desalted on C18 HLB columns (Waters, Milford, MA, USA), and the enriched peptides were dried and reconstituted in 0.1% formic acid (FA) aqueous solution. A nanoLC-SYNAPT G2 Si Q-TOF system (Waters) was employed to analyze the samples for quantitative profiling the lysine accessibility changes in response to celastrol binding for target discovery. Data dependence acquisition (DDA) in the positive mode was employed for data acquisition. Data analysis was performed using PEAKS Studio 8.5 (BSI solutions, Waterloo, Canada). Specifically, cys alkylation was selected as fixed modification, and methionine oxidation and lysine dimethylation, achieved by TRAP labeling, were set as variable modifications. Briefly, peptides that contain TRAP-induced dimethylation and exhibited significant abundance changes with and without celastrol incubation were assigned as target responsive peptides. The ratio of the abundance of each TRAP-labeled peptide indicates the extent of accessibility change, and is intimately associated with ligand-binding affinity. Student's t-test was carried out to assess whether the detected accessibility changes of labeled peptides are statistically significant. An inter-group P value (P < 0.001) and R value (TRAP ratio>2 or <0.5) was set as the cutoff to screen the target responsive peptides belonging to the celastrol-binding proteins from the whole quantified proteome.
2.11. Cellular thermal shift assay (CETSA)
CETSA was performed as described previously reported14. PMA-differentiated THP-1 cells were divided into 2 groups, one group hatched with 30 nmol/L celastrol, and the control group added DMSO of the same volume, and two groups of cells were collected after 2 h. Two groups of cells were washed twice and re-suspended with 600 μL of PBS at the last time. Both groups are divided into 10 servings, each of which is 50 μL, and loaded into a PCR tube. Set the PCR meter to 10 temperature gradients (40, 42, 44, 46, 48, 50, 52, 54, 56, and 58 °C) to each sample in the control group corresponds to a temperature, which is heated: after the PCR instrument temperature rises to the specified temperature, a PCR tube is placed in each of the corresponding experimental group and the dosing group, heated 3 min, and next placed at room temperature for 3 min, and then placed in an ice bath. After all the samples have been processed, the samples are repeatedly frozen and melted: −80 °C refrigerator placed overnight, taken out and placed at room temperature, dissolved and then put in −80 °C frozen for 2 h, repeat the operation twice. Transfer the processed sample to an imported eppendorf tube, centrifuge at 20,000×g and 4 °C for 20 min, add 6×loading buffer, heat the mixed sample for 5 min, and then perform a sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel electrophoresis analysis.
2.12. Microscale thermophoresis (MST)
HEK293T cells were transfected with plenti-mCherry-CAP1, after 48 h, cells were lysed and centrifuged at 13,000×g and 4 °C for 10 min. For the measurement, each ligand dilution was mixed with one volume of cell lysis, which led to a final fluorescence of 6000 and final ligand concentrations in the nmol/L to mmol/L range. After 10 min incubation followed by centrifugation at 10,000×g and 4 °C for 10 min, the samples were loaded into Monolith NT.LabelFree zero-background capillaries (NanoTemper Technologies). The MST experiment was performed with a Monolith NT.LabelFree instrument (NanoTemper Technologies) at 25 °C. For each ligand-binding curve, data from at least three independently pipetted measurements were included in the analysis with the MO.Affinity Analysis v2.3.
2.13. Isothermal titration calorimetry (ITC)
Protein samples were dialyzed using buffer (20 mmol/L Tris-HCl pH 8.0, 100 mmol/L NaCl, 0.1 mmol/L Tris-(carboxyethyl) phosphine hydrochloride (TCEP), and 0.5% DMSO), and celastrol powder was dissolved in the same buffer. ITC experiments were carried out in a Nano ITC instrument (TA Instruments) at 25 °C. The titrations were performed by injecting 1.0 μL aliquots of SHP099 into the calorimeter cell containing a 300 μL solution of CAP1 (30 μmol/L CAP1 and 600 μmol/L celastrol) with a constant stirring speed at 200×g. The data were analyzed with the NanoAnalyze using the independent fit model. All the uncertainties were estimated by the native Statistics module with 1000 synthetic trials and 95% confidence level.
2.14. Immunoprecipitation
THP-1 cells were infected with CAP1 lentiviruses. PMA-differentiated CAP1-overexpressed THP-1 cells were treated with or without celastrol (30 nmol/L) for 6 h. Then we extracted protein using lysis buffer (Beyotime) supplemented with protease and phosphatase inhibitors (MCE, China). Each sample was incubated with anti-CAP1 antibodies overnight at 4 °C, then incubated with magnetic beads (Millipore) at room temperature for 1 h. Proteins not immobilized on beads were removed by 5 times washes with cold lysis buffer. Next, the precipitates were boiled for 10 min in SDS-loading buffer. The presence or absence of the target protein was evaluated by Western blot using the indicated Abs.
2.15. Western blot
Cells were lysed in lysis buffer supplemented with protease and phosphatase inhibitor (MCE). Proteins were quantified by the Bradford assay (HyClone-Pierce). The proteins were then separated by SDS-PAGE and electrophoretically transferred onto polyvinylidene difluoride membranes. The membranes were probed with Abs overnight at 4 °C, and then incubated with a horseradish peroxidase-coupled secondary Ab. Detection was performed using a LumiGLO chemiluminescent substrate system. Anti-p-IGF-I receptor β (IRβ) Cell Signal Technology, USA anti-p-protein kinase B (AKT; 1:1000, Proteintech, China), anti-CAP1 (Abcam, USA), anti-resistin (1:1000, Abcam, USA), anti-p-vasodilator stimulated phosphoprotein (VASP; 1:1000; Cell Signal Technology, USA), anti-p-p65 (1:1000; Cell Signal Technology, USA), anti-Na+/K+-ATPase (1:1000; Cell Signal Technology, USA), anti-β-actin (1:2000, Abmart, USA), anti-GAPDH (1:2000, Abmart, USA) were used.
2.16. Statistics
Statistical analysis was performed using GraphPad Prism 8.0. Data are presented as the mean ± standard error of mean (SEM). We assessed data for normal distribution and similar variance between groups. Statistical significance (∗P < 0.05, ∗∗P < 0.01, ns, not significant) was assessed using two-tailed unpaired Student's t-test for comparisons between 2 groups. No animals were excluded from statistical analysis. Quantifications of data in representative Western blot figures were shown in Supporting Information Fig. S1.
3. Results
3.1. Celastrol reduces body weight of HFD-induced MetS mice
The structure of celastrol is shown in Fig. 1A. To investigate the effects of celastrol on MetS, we first established a MetS murine model by feeding HFD for 16 weeks and then intraperitoneally treated with 0.75, 1.5, and 3 mg/kg celastrol for 25 days continuously. Celastrol drastically reduced body weight in the group with a dose of 3 mg/kg (Fig. 1B and C), indicating that celastrol can significantly improve obesity of the MetS mice. At the same time, the daily food intake of mice in each group was counted. The results showed that celastrol could not affect the food intake of mice (Fig. 1D). These data suggest that celastrol can ameliorate obesity in HFD-fed MetS mice.
Figure 1.

Celastrol (Cel) reduces body weight of high fat diet (HFD)-induced metabolic syndrome (MetS) mice. (A) The chemical structure of celastrol. (B)–(D) Analysis of mice on HFD, either without (HFD, n = 7) or treated with celastrol at doses of 0.75 mg/kg/day (HFD+Cel 0.75, n = 7), 1 mg/kg/day (HFD+Cel 1.5, n = 7), or 3 mg/kg/day (HFD+Cel 3, n = 7), including: (B) phenotypic presentation; (C) body weight; (D) food intake (n = 4). Data represent as mean ± standard error of mean (SEM) in (C) (n = 7) and (D) (n = 4). P values are determined by two-tailed Student's t test. ∗P < 0.05, ∗∗P < 0.01.
3.2. Celastrol protects mice from HFD-induced insulin resistance
To evaluate the glucose homeostasis, we measured the levels of glucose tolerance (GTT) and insulin tolerance (ITT). Celastrol effectively improved the insulin resistance of mice at the doses of 1.5 and 3 mg/kg (Fig. 2A–D). Consistent with this reduction, celastrol inhibited the phosphorylation of IR and AKT, and restrained glycogen synthesis (Fig. 2E). We also observed that celastrol treatment could significantly reduce the fasting blood glucose level compared with the model group (Fig. 2F).
Figure 2.

Celastrol protects mice from HFD-induced insulin resistance. (A)‒(D) Glucose tolerance test and insulin tolerance test were performed on HFD-fed mice treated with celastrol or vehicle. (E) Western blot analysis of insulin signaling in mice (Normal, HFD, HFD+Cel 3 mg/kg) liver. (F) Plasma glucose was determined in HFD-fed mice and celastrol treated mice. Data represent as mean ± SEM in (A) and (B) (n = 7), and (C)–(F) (n = 4). P values are determined by two-tailed Student's t test. ∗P < 0.05, ∗∗P < 0.01. GTT, glucose tolerance; ITT, insulin tolerance; AUC, calculated area under curve.
3.3. Celastrol protects against HFD-induced hepatic steatosis
Compared with normal group, the liver lobule structures of HFD-fed mice had varying degrees of looseness or clear-like changes, a large number of vacuoles were generally present in the cytoplasm, and the infiltration of inflammatory cells was increased, suggesting that fat and inflammatory lesions occurred in the livers of the HFD-fed mice. In celastrol administration groups, the livers had different degrees of recovery, the tissue structures were relatively complete, lipid droplet vacuoles and infiltrated inflammatory cells were reduced, indicating that celastrol can effectively alleviate fat and inflammatory lesions in the liver tissue of mice with MetS (Fig. 3A). The celastrol-treated mice also showed a dramatic downregulation in the expression of caveolin-1, a lipid raft associated with lipid dynamics that has emerged as a key player in obesity and insulin resistance, indicating attenuated hepatic steatosis (Fig. 3B). In addition, lipid metabolism disorders also are features of MetS, with abnormally elevated levels of T-CHO and TG. We measured the levels of T-CHO and TG in the serum of each group of mice by ELISA (Fig. 3C and D), and the results show that the levels of T-CHO and TG were significantly decreased under 1.5 and 3 mg/kg celastrol treatments. Significantly, administration with 3 mg/kg celastrol, T-CHO and TG levels were reduced by about 50%, indicating that 3 mg/kg celastrol has a better effect on improving dyslipidemia in mice with MetS.
Figure 3.

Celastrol protects against HFD-induced hepatic steatosis. (A) and (B) Representative hematoxylin and eosin (H&E) staining and caveolin-1 staining of liver from HFD-fed mice treated with celastrol or vehicle. (C) and (D) total cholesterol (T-CHO) and triglycerides (TG) in serum from mice were measured. Data represent as mean ± SEM in (C) and (D) (n = 4). P values are determined by two-tailed Student's t test. ∗P < 0.05, ∗∗P < 0.01, ns, not significant.
3.4. Celastrol attenuates HFD-induced inflammation
Chronic inflammation in white adipose tissue plays an important role in the occurrence and development of obesity. To investigate the effect of celastrol on the degree of inflammation in white adipose tissue, we first isolated the epididymal adipose tissue of mice and found that celastrol can significantly reduce the hypertrophy of epididymal adipose tissue in mice with MetS (Fig. 4A). Next, we performed H&E staining on the epididymal adipose tissue of mice. After celastrol treatment, the number of infiltrated inflammatory cells on adipocyte interstitium is reduced, indicating that celastrol can effectively improve the level of inflammatory cell infiltration in the epididymal adipose tissue of mice with MetS (Fig. 4B). Macrophages (adipose tissue macrophages, ATMs) infiltrating adipose tissue are the main source of inflammatory factors in obese and insulin resistant patients. CD11c is a characteristic marker of M1 type macrophages. The number of M1 macrophages in the epididymal adipose tissues of mice was significantly reduced in celastrol treated mice (Fig. 4C). We detected the levels of pro-inflammatory factors IL-1β, IL-6, and TNF-α in the serum of mice. At the dose of 3 mg/kg, the levels of these pro-inflammatory factors were significantly reduced (Fig. 4D), indicating that celastrol can inhibit the secretion of anti-inflammatory factors to improve the overall inflammatory state of mice with MetS.
Figure 4.

Celastrol attenuates HFD-induced inflammation. (A)‒(C) Phenotypic presentation, H&E staining, CD11c staining of epididymal adipose tissue from HFD-fed mice treated with celastrol or vehicle. (D) ELISA quantification of protein levels of cytokines in mouse serum. Data represent as mean ± SEM in D (n = 4). P values are determined by two-tailed Student's t test. ∗P < 0.05, ∗∗P < 0.01, ns, not significant.
3.5. Celastrol binds CAP1 protein
To identify the molecular mechanism by which celastrol prevents HFD-induced MetS, we screened for potential celastrol binding proteins. The result of 3-(4,5-dimethyl-2-thiazyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay showed that low dose of celastrol did not affect THP-1 and HEK293T cells proliferation (Supporting Information Fig. S2). Then, we used TRAP approach to discover the celastrol-binding proteins by probing the lysine accessibility changes on a proteome-level (Fig. 5A). Among the proteins that exhibit accessibility changes in response to celastrol administration via TRAP analysis, the CAP1 protein showed a pronounced reduction in intensity. This result indicated that the accessibility of the probed region displayed the marked decrease due to celastrol binding-induced steric repulsion, and hence assigned CAP1 as the binding protein (Fig. 5B). TRAP analysis showed that celastrol bound to the src homology 3 (SH3) domain of CAP1 (Supporting Information Fig. S3). We then confirmed the engagement between celastrol and CAP1 using CETSA. Data exhibited that celastrol significantly enhanced the thermal stability of CAP1 even at higher temperatures, compared to control group (DMSO treatment) (Fig. 5C). Celastrol treatment preserved CAP1 stability in a dose-dependently manner (Fig. 5D). Next, we conducted biotin labeled celastrol (Cel-biotin, Fig. 5E), and its functional activity was affected to a certain extent as can be seen by its inhibition of IL-1β in THP-1 cells. When the concentration of Cel-biotin reached 1 μmol/L, the inhibitory effect on IL-1β was equivalent to that of celastrol at 30 nmol/L (Supporting Information Fig. S4). Biotinylated–protein interaction pull-down assays showed that Cel-biotin could bind to CAP1 in lysates from THP-1 cells (Fig. 5F). The fluorescence result was consistent with pull-down assay result (Fig. 5G). Results of microscale thermophoresis (MST) assay showed a higher binding affinity for celastrol and CAP1, the KD value was 16.5 nmol/L (Fig. 5H). Next, we expressed and purified CAP1 protein (Supporting Information Fig. S5). ITC results show that the KD value between celastrol and CAP1 was 110 nmol/L (Fig. 5I). For MST, we used CAP1-overexpressed cell lysates, and for ITC, we used CAP1 purified protein. In addition, the sensitivities of these two assays are different, therefore, the KD values of MST and ITC are not consistent. Collectively, these studies show that celastrol directly binds to CAP1 at both cellular and molecular levels.
Figure 5.

Celastrol binds adenylyl cyclase-associated protein 1 (CAP1) protein. (A) Schematic representation of the TRAP experiment. (B) Celastrol binding proteins using target-responsive accessibility profiling (TRAP) experiment. (C) and (D) Cells were incubated with celastrol or DMSO for 2 h, and cellular thermal shift assays (CETSA) analyzed the thermal stabilization of CAP1 protein at different temperatures and concentrations. (E) Chemical structure of the biotin labeled celastrol (Cel-biotin). (F) Cel-biotin was added to streptavidin-agarose beads and incubated. Biotin alone was used as a control. Lysates prepared from THP-1 cells were added to the streptavidin-agarose beads with Cel-biotin. Eluent was then loaded on a polyacrylamide gel for Western blot analysis. Total lysates were used as an input control. (G) Immunofluorescence staining of phorbol ester (PMA)-differentiated THP-1 cells were treated with celastrol for 2 h, and then stimulated with biotin or Cel-biotin for 4 h. (H) The interaction of celastrol with CAP1 or vector was measured by microscale thermophoresis (MST). The KD value of celastrol and CAP1 interaction was determined with MO.Affinity Analysis Software. (I) Isothermal titration calorimetry (ITC) enthalpogram of the interaction between celastrol and CAP1 at 25 °C. The titration curve is depicted as a function of the molar ratio between CAP1 and the calculated concentration of celastrol in the assay. Data represent as mean ± SEM. P values are determined by two-tailed Student's t test (n = 3). ∗∗P < 0.01.
3.6. Celastrol inhibits resistin-induced cytokines levels
Lee et al.19 found that resistin can bind to the CAP1 receptor in human peripheral blood mononuclear macrophages. Resistin activated macrophages and produced IL-1β, IL-6, and TNF-α through the cAMP‒PKA‒NF-κB signaling pathway (Supporting Information Fig. S6). Previous studies28 have shown that the level of resistin is closely related to the blood glucose concentration. Normalizing plasma resistin through certain means can relieve insulin resistance. First, we detected the content of resistin in the serum, epididymal adipose tissues, and liver tissues of mice. Data showed that HFD could increase the expression of resistin in these tissues, and after celastrol administration, the levels of resistin decreased significantly (Fig. 6A–C). We then tested the activation of resistin on the CAP1‒cAMP‒PKA‒NF-κB signaling pathway in THP-1 cells, using forskolin as a positive control. Forskolin is a specific agonist of adenylate cyclase, which directly activates adenylate cyclase through its catalytic subunit. Forskolin increased the level of cAMP in the cell, and activated downstream signaling pathways (Supporting Information Fig. S7). Celastrol could significantly reduce the cAMP content in the cell supernatant (Fig. 6D) and inhibit the activation of the downstream PKA‒NF-κB pathway (Fig. 6E and F). Both 10 and 30 nmol/L celastrol could effectively reduce the content of IL-1β, and 30 nmol/L celastrol could effectively reduce the content of IL-6 and TNF-α (Fig. 6G).
Figure 6.

Celastrol inhibits resistin-induced cytokines levels. (A) ELISA quantification of resistin level in mouse serum. (B) Resistin staining in epididymal adipose tissue and liver tissue of mice. (C) Resistin expression in mice liver was determined by Western blot. (D) ELISA quantification of cAMP level in cell supernatant. (E) PKA‒NF-κB signaling in PMA-differentiated THP-1 cells were determined by Western blot analysis after celastrol treating. (F) Immunofluorescence staining of PMA-differentiated THP-1 cells were treated with celastrol for 2 h, and then stimulated with resistin for 15 min. (G) ELISA quantification of protein levels of cytokines in cell supernatant. Data represent as mean ± SEM. P values are determined by two-tailed Student's t test (n = 3). ∗P < 0.05, ∗∗P < 0.01, ns, not significant.
3.7. Celastrol inhibits CAP1‒resistin interaction
CAP1 mainly localized near the plasma membrane in THP-1 cells (Fig. 7A). We isolated membrane and cytoplasm from THP-1 cells. Previous studies19 reported that the interaction of resistin and CAP1 activates downstream cAMP‒PKA‒NF-κB pathway and exacerbates inflammation. Therefore, we first measured whether celastrol affects the interaction of resistin and CAP1. Our data show that celastrol inhibited the interaction between CAP1 and resistin (Fig. 7B). Then we conducted CAP1 knocked-down and overexpressed THP-1 cells (Fig. 7C and D). Knockdown of CAP1 in macrophages abrogated the resistin-mediated inflammatory activity (Fig. 7E). In contrast, overexpression of CAP1 in macrophages aggravated inflammation (Fig. 7F). These results indicate that celastrol ameliorates HFD-induced MetS through suppressing the interaction of CAP1 and resistin.
Figure 7.

Celastrol inhibits CAP1‒resistin interaction. (A) Confocal microscopy imaging of CAP1 in PMA-differentiated THP-1 cells. (B) PMA-differentiated THP-1 cells were untreated or treated with celastrol. Cell lysates underwent immunoprecipitation with CAP1 antibodies and the immunoblotting with indicated antibodies. (C) and (D) CAP1 protein and mRNA levels in PMA-differentiated THP-1 cells infected with shRNA-Control, shRNA-CAP1, vector, or CAP1 lentiviruses. (E) and (F) PKA‒NF-κB signaling in PMA-differentiated THP-1 cells infected with shRNA-Control, shRNA-CAP1, vector, or CAP1 lentiviruses were determined by Western blot analysis after celastrol treating. Data represent as mean ± SEM. P values are determined by two-tailed Student's t test (n = 3). ∗∗P < 0.01.
In obesity, adipocytes become hypertrophy owing to the increased triglyceride storage compare to normal. As obesity develops, the macrophages’ accumulation and pro-inflammatory cytokines’ secretion in adipose tissue lead to macrophage infiltration and aggravate the development of chronic low-grade inflammation. Specifically, in states of obesity, adipose tissue generates resistin, which binds to CAP1 receptors on macrophages and activates the cAMP‒PKA‒NF-κB signaling pathway and promotes the ensuing expression of proinflammatory cytokines. These pro-inflammatory factors further promote inflammation and insulin resistance of adipose tissue on the one hand, and also aggravate liver insulin resistance and hepatic steatosis on the other hand. Celastrol binds to CAP1, and inhibits the interaction between resistin and CAP1. Thus, celastrol effectively prevents the subsequent inflammatory response and ameliorates MetS (Fig. 8).
Figure 8.

The graphic illustration of the mechanism of celastrol ameliorating metabolic syndrome. In obesity, adipose tissue generates resistin, which binds to CAP1 on macrophages and activates the PKA‒NF-κB pathway and promotes the expression of pro-inflammatory cytokines. These pro-inflammatory factors further promote inflammation and insulin resistance of adipose tissue on the one hand, and also aggravate liver insulin resistance and hepatic steatosis on the other hand. Celastrol effectively prevents inflammatory response through binding to CAP1 and inhibiting the interaction between resistin and CAP1. Created in BioRender.com.
4. Discussion
In this study, we identified that celastrol, a Tripterygium wilfordii-derived constituent of traditional Chinese medicine21, protects mice from HFD-induced MetS, including obesity, insulin resistance, hepatic steatosis, and inflammation. Mechanistically, we found celastrol directly binds to CAP1 protein and reduces interaction between resistin and CAP1 and the ensuing inflammation response. Importantly, we knocked down CAP1 in macrophages, and the resistin-mediated inflammatory activity was abrogated. In contrast, overexpression of CAP1 in macrophages aggravated inflammation. Our results identify celastrol targets CAP1 to ameliorate HFD-induced MetS in mice.
MetS is closely related to the chronic low-grade inflammation12,29,30. Recruitment and infiltration of ATMs lead to adipocyte inflammation31. There are 5% macrophages in lean adipose tissue, whereas, during obesity, the ratio rises dramatically up to 50%32. In obesity, fat accumulation causes adipocytes to secrete the pro-inflammatory cytokines like as IL-6, TNF-α, IL-1β, and monocyte chemoattractant protein 1 (MCP-1), which results in the accumulation of macrophages in adipose tissue and contributes to the development of insulin resistance33. An interesting study found that macrophages’ deficient in fatty acid synthase protects HFD-induced mice from insulin resistance, macrophage recruitment in adipose tissue, and chronic inflammation34. Therefore, inhibition of obesity-related inflammation might be a promising strategy to improve MetS.
Celastrol has many pharmacological activities including anti-inflammatory and anti-cancer effects35. Celastrol‒albumin nanoparticles are potent in treating glomerulonephritis in rat anti-Thy1.1 nephritis models36. Celastrol suppresses the progression of rheumatoid arthritis (RA) by inhibiting the secretion of pro-inflammatory cytokines through restraining the repolarization of macrophages toward M1 phenotype37. Moreover, celastrol and mitoxantrone together successfully inhibit tumor metastasis to major organs and prolongs progression-free survival in desmoplastic melanoma38. Celastrol-loaded neutrophil membrane-coated nanoparticles effectively reduce pancreatic carcinoma39. In addition, celastrol was identified as a potent anti-obesity agent23. Celastrol increases the sensitivity of leptin via interleukin 1 receptor type 1 (IL1R1) to protect against obesity24. Celastrol ameliorates obesity through activating heat shock factor 1 (HSF1) to increase mitochondrial function in adipose tissue and muscle25. Celastrol significantly improves HFD-induced nonalcoholic fatty liver disease (NAFLD) by activating nicotinamide adenine dinucleotide (NAD)-dependent protein deacetylase sirtuin 1 (SIRT1)40. In addition, celastrol also inhibits the differentiation of adipocytes and extenuates obesity41. Therefore, celastrol may be a promising therapeutic agent to improve the pathological states that result from obesity-related metabolic disorders, such as MetS. However, despite these promising results, the underlying mechanisms have not been identified.
Small molecules usually have the characteristics of multiple targets, and celastrol is no exception. Commonly, small molecules’ targets are not the same in different diseases. Zhang's group42 proved that celastrol binds to and induces Nur77 translocation and interaction with tumor necrosis factor receptor-associated factor 2 (TRAF2), inhibiting the chronic inflammation in obese animals and the acute liver inflammation. Liang and colleagues43 found that celastrol directly binds to and inhibits the phosphorylation and nuclear translocation of signal transducer and activator of transcription-3 (STAT3), improving the hypertrophic fibrosis caused by STAT3, and ultimately protected heart function in angiotensin II-induced cardiac dysfunction. They also demonstrated that celastrol directly binds to peroxiredoxin-2, which then increased cellular ROS levels and led to ROS-mediated apoptosis in gastric cancer cells44. In our study, we identified that celastrol ameliorates HFD-induced MetS by inhibiting macrophages-mediated inflammation. Specifically, celastrol targets CAP1 and suppresses the interaction with resistin, which inhibits the cAMP‒PKA‒NF-κB signaling pathway and the ensuing expression of pro-inflammatory cytokines.
5. Conclusions
In summary, we unraveled a novel target of celastrol in HFD-induced MetS. We found that celastrol protects against MetS by binding to CAP1 and restraining macrophages-mediated inflammation in adipose tissues. Celastrol might be a promising drug for treatment of inflammatory metabolic diseases.
Acknowledgments
This work was supported by National Natural Science Foundation of China (Nos. 81673436, 91853109, 81872877, and 81872838), Natural Science Foundation of Jiangsu Province (BK20180079, China), and Mountain-Climbing Talents Project of Nanjing University (China).
Author contributions
Yang Sun conceived this project and designed the study. Yuyu Zhu, Ning Wan, Xinni Shan, and Guoliang Deng performed the experiments and analyzed the data. Hui Ye and Qiang Xu gave methodological support and conceptual advice. Yang Sun and Yuyu Zhu wrote the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.12.008.
Contributor Information
Hui Ye, Email: cpuyehui@cpu.edu.cn.
Yang Sun, Email: yangsun@nju.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Samson S.L., Garber A.J. Metabolic syndrome. Endocrinol Metab Clin N Am. 2014;43:1–23. doi: 10.1016/j.ecl.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 2.Rochlani Y., Pothineni N.V., Kovelamudi S., Mehta J.L. Metabolic syndrome: Pathophysiology, management, and modulation by natural compounds. Ther Adv Cardiovas Dis. 2017;11:215–225. doi: 10.1177/1753944717711379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCracken E., Monaghan M., Sreenivasan S. Pathophysiology of the metabolic syndrome. Clin Dermatol. 2018;36:14–20. doi: 10.1016/j.clindermatol.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Eckel R.H., Grundy S.M., Zimmet P.Z. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 5.Sherling D.H., Perumareddi P., Hennekens C.H. Metabolic syndrome. J Cardiovasc Pharmacol Therapeut. 2017;22:365–367. doi: 10.1177/1074248416686187. [DOI] [PubMed] [Google Scholar]
- 6.Saklayen M.G. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. 2018;20:12. doi: 10.1007/s11906-018-0812-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tune J.D., Goodwill A.G., Sassoon D.J., Mather K.J. Cardiovascular consequences of metabolic syndrome. Transl Res. 2017;183:57–70. doi: 10.1016/j.trsl.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pucci G., Alcidi R., Tap L., Battista F., Mattace-Raso F., Schillaci G. Sex- and gender-related prevalence, cardiovascular risk and therapeutic approach in metabolic syndrome: A review of the literature. Pharmacol Res. 2017;120:34–42. doi: 10.1016/j.phrs.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 9.Sun L., Pang Y., Wang X., Wu Q., Liu H., Liu B. Ablation of gut microbiota alleviates obesity-induced hepatic steatosis and glucose intolerance by modulating bile acid metabolism in hamsters. Acta Pharm Sin B. 2019;9:702–710. doi: 10.1016/j.apsb.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Depommier C., Everard A., Druart C., Plovier H., Van Hul M., Vieira-Silva S. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat Med. 2019;25:1096–1103. doi: 10.1038/s41591-019-0495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hendrickx J.O., van Gastel J., Leysen H., Martin B., Maudsley S. High-dimensionality data analysis of pharmacological systems associated with complex diseases. Pharmacol Rev. 2020;72:191–217. doi: 10.1124/pr.119.017921. [DOI] [PubMed] [Google Scholar]
- 12.Mogilenko D.A., Haas J.T., L’homme L., Fleury S., Quemener S., Levavasseur M. Metabolic and innate immune cues merge into a specific inflammatory response via the UPR. Cell. 2019;177:1201–1216. doi: 10.1016/j.cell.2019.03.018. [DOI] [PubMed] [Google Scholar]
- 13.Gluvic Z., Zaric B., Resanovic I., Obradovic M., Mitrovic A., Radak D. Link between metabolic syndrome and insulin resistance. Curr Vasc Pharmacol. 2017;15:30–39. doi: 10.2174/1570161114666161007164510. [DOI] [PubMed] [Google Scholar]
- 14.Benomar Y., Amine H., Crépin D., Al Rifai S., Riffault L., Gertler A. Central resistin/TLR4 impairs adiponectin signaling, contributing to insulin and FGF21 resistance. Diabetes. 2016;65:913–926. doi: 10.2337/db15-1029. [DOI] [PubMed] [Google Scholar]
- 15.Rodríguez M., Pintado C., Moltó E., Gallardo N., Fernández‒Martos C.M., López V. Central s-resistin deficiency ameliorates hypothalamic inflammation and increases whole body insulin sensitivity. Sci Rep. 2018;8:3921. doi: 10.1038/s41598-018-22255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo J., Huang L., Wang A., Liu Y., Cai R., Li W. Resistin-induced endoplasmic reticulum stress contributes to the impairment of insulin signaling in endothelium. Front Pharmacol. 2018;9:1226. doi: 10.3389/fphar.2018.01226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hastuti P., Tasmini T., Utami R.F., Riwa M.R.K., Steven S., Sadewa A.H. Variation of resistin gene is correlated with insulin resistance in obese people of Indonesia. Open Access Maced J Med Sci. 2019;7:1891–1895. doi: 10.3889/oamjms.2019.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park H.K., Kwak M.K., Kim H.J., Ahima R.S. Linking resistin, inflammation, and cardiometabolic diseases. Korean J Intern Med. 2017;32:239–247. doi: 10.3904/kjim.2016.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S., Lee H.C., Kwon Y.W., Lee S.E., Cho Y., Kim J. Adenylyl cyclase-associated protein 1 is a receptor for human resistin and mediates inflammatory actions of human monocytes. Cell Metabol. 2014;19:484–497. doi: 10.1016/j.cmet.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdin A.A., Hasby E.A. Modulatory effect of celastrol on Th1/Th2 cytokines profile, TLR2 and CD3+ T-lymphocyte expression in a relapsing-remitting model of multiple sclerosis in rats. Eur J Pharmacol. 2014;742:102–112. doi: 10.1016/j.ejphar.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Astry B., Venkatesha S.H., Laurence A., Christensen-Quick A., Garzino-Demo A., Frieman M.B. Celastrol, a Chinese herbal compound, controls autoimmune inflammation by altering the balance of pathogenic and regulatory T cells in the target organ. Clin Immunol. 2015;157:228–238. doi: 10.1016/j.clim.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venkatesha S.H., Dudics S., Astry B., Moudgil K.D. Control of autoimmune inflammation by celastrol, a natural triterpenoid. Pathog Dis. 2016;74:ftw059. doi: 10.1093/femspd/ftw059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J., Lee J., Salazar Hernandez M.A., Mazitschek R., Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161:999–1011. doi: 10.1016/j.cell.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng X., Guan D., Auen T., Choi J.W., Salazar Hernández M.A., Lee J. IL1R1 is required for celastrol's leptin-sensitization and antiobesity effects. Nat Med. 2019;25:575–582. doi: 10.1038/s41591-019-0358-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma X., Xu L., Alberobello A.T., Gavrilova O., Bagattin A., Skarulis M. Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1‒PGC1α transcriptional axis. Cell Metabol. 2015;22:695–708. doi: 10.1016/j.cmet.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Saito K., Davis K.C., Morgan D.A., Toth B.A., Jiang J., Singh U. Celastrol reduces obesity in MC4R deficiency and stimulates sympathetic nerve activity affecting metabolic and cardiovascular functions. Diabetes. 2019;68:1210–1220. doi: 10.2337/db18-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian Y., Wan N., Ding M., Shao C., Wang N., Bao Q. Chemoproteomics maps glycolytic targetome in cancer cells. bioRxiv. 2020 doi: 10.1101/2020.11.18.387670. Available from: [DOI] [PubMed] [Google Scholar]
- 28.Steppan C.M., Bailey S.T., Bhat S., Brown E.J., Banerjee R.R., Wright C.M. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 29.Iyengar N.M., Gucalp A., Dannenberg A.J., Hudis C.A. Obesity and cancer mechanisms: Tumor microenvironment and inflammation. J Clin Oncol. 2016;34:4270–4276. doi: 10.1200/JCO.2016.67.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saltiel A.R., Olefsky J.M. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. 2017;127:1–4. doi: 10.1172/JCI92035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Permana P.A., Menge C., Reaven P.D. Macrophage-secreted factors induce adipocyte inflammation and insulin resistance. Biochem Biophys Res Commun. 2006;341:507–514. doi: 10.1016/j.bbrc.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 32.Weisberg S.P., McCann D., Desai M., Rosenbaum M., Leibel R.L., Ferrante A.W. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chylikova J., Dvorackova J., Tauber Z., Kamarad V. M1/M2 macrophage polarization in human obese adipose tissue. Biomed Pap Med Fac Univ Palacky Olomouc Czech. 2018;162:79–82. doi: 10.5507/bp.2018.015. [DOI] [PubMed] [Google Scholar]
- 34.Wei X., Song H., Yin L., Rizzo M.G., Sidhu R., Covey D.F. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. 2016;539:294–298. doi: 10.1038/nature20117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kannaiyan R., Shanmugam M.K., Sethi G. Molecular targets of celastrol derived from Thunder of God Vine: Potential role in the treatment of inflammatory disorders and cancer. Canc Lett. 2011;303:9–20. doi: 10.1016/j.canlet.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 36.Guo L., Luo S., Du Z., Zhou M., Li P., Fu Y. Targeted delivery of celastrol to mesangial cells is effective against mesangioproliferative glomerulonephritis. Nat Commun. 2017;8:878. doi: 10.1038/s41467-017-00834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.An L., Li Z., Shi L., Wang L., Wang Y., Jin L. Inflammation-targeted celastrol nanodrug attenuates collagen-induced arthritis through NF-κB and Notch1 pathways. Nano Lett. 2020;20:7728–7736. doi: 10.1021/acs.nanolett.0c03279. [DOI] [PubMed] [Google Scholar]
- 38.Liu Q., Chen F., Hou L., Shen L., Zhang X., Wang D. Nanocarrier-mediated chemo-immunotherapy arrested cancer progression and induced tumor dormancy in desmoplastic melanoma. ACS Nano. 2018;12:7812–7825. doi: 10.1021/acsnano.8b01890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao X., Hu Y., Luo S., Wang Y., Gong T., Sun X. Neutrophil-mimicking therapeutic nanoparticles for targeted chemotherapy of pancreatic carcinoma. Acta Pharm Sin B. 2019;9:575–589. doi: 10.1016/j.apsb.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y., Geng C., Liu X., Li M., Gao M., Liu X. Celastrol ameliorates liver metabolic damage caused by a high-fat diet through Sirt1. Mol Metab. 2017;6:138–147. doi: 10.1016/j.molmet.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi S.K., Park S., Jang S., Cho H.H., Lee S., You S. Cascade regulation of PPARγ2 and C/EBPα signaling pathways by celastrol impairs adipocyte differentiation and stimulates lipolysis in 3T3-L1 adipocytes. Metabolism. 2016;65:646–654. doi: 10.1016/j.metabol.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 42.Hu M., Luo Q., Alitongbieke G., Chong S., Xu C., Xie L. Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol Cell. 2017;66 doi: 10.1016/j.molcel.2017.03.008. 141‒53.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye S., Luo W., Khan Z.A., Wu G., Xuan L., Shan P. Celastrol attenuates angiotensin II-induced cardiac remodeling by targeting STAT3. Circ Res. 2020;126:1007–1023. doi: 10.1161/CIRCRESAHA.119.315861. [DOI] [PubMed] [Google Scholar]
- 44.Chen X., Zhao Y., Luo W., Chen S., Lin F., Zhang X. Celastrol induces ROS-mediated apoptosis via directly targeting peroxiredoxin-2 in gastric cancer cells. Theranostics. 2020;10:10290–10308. doi: 10.7150/thno.46728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.