

Supplemental Digital Content is available in the text.
Keywords: adult-onset Still’s disease, autoimmune diseases, case report, critical illness, macrophage-activation syndrome, pregnancy trimester, third
Abstract
BACKGROUND:
We report a case of a young woman with adult-onset Still’s disease presenting as macrophage-activation syndrome complicated by shock and respiratory failure during the third trimester of pregnancy.
CASE SUMMARY:
A previously healthy 26-year-old woman at 35 weeks of gestation presented with 1 week of constitutional symptoms and was found to be febrile, tachycardic, and hypotensive. She delivered a healthy neonate by cesarean section. Following delivery, she had worsening hypotension and fevers despite fluid resuscitation and antibiotics, and developed progressive hypoxemia requiring up to 60% Fio2, with bilateral upper-lobe predominant opacities on chest CT. She also had laboratory derangements including anemia, thrombocytopenia, low fibrinogen, elevated ferritin, and abnormal liver chemistries. After extensive testing to exclude infections, hemolysis, and other pertinent disorders, the development of polyarthralgias and a characteristic rash fulfilled criteria for adult-onset Still’s disease complicated by macrophage-activation syndrome. Her condition improved with immunosuppressive therapy.
CONCLUSION:
To our knowledge, this is the first report of new-onset adult-onset Still’s disease during the third trimester of pregnancy, which presented as macrophage-activation syndrome. In the context of late pregnancy, macrophage-activation syndrome can mimic or raise concern for hemolysis, elevated liver enzymes, and low platelet syndrome and other peripartum disorders. Furthermore, the characteristic articular symptoms of adult-onset Still’s disease may be mild and/or delayed, and pulmonary involvement with severe hypoxemia can occur. Clinicians should consider this diagnosis when evaluating a pregnant patient with unexplained fever and multiorgan dysfunction.
CASE REPORT
A previously healthy 26-year-old woman (gravida 2, para 1, prior pregnancy uncomplicated) at 35 weeks of gestation presented to a local hospital with 1 week of malaise, myalgias, and vague abdominal pain. She was febrile, tachycardic, and hypotensive, and received fluid resuscitation and empiric antibiotics. The next day, she delivered a healthy neonate by cesarean section. Pathology of the placenta, cord, and fetal membranes was unremarkable.
After delivery, daily fever spikes continued to a peak of 40.6°C. She also developed an intermittent pale-pink macular rash, hypoxemia requiring up to 60% Fio2 by high-flow nasal cannula, and distributive shock requiring low-dose norepinephrine. She was transferred to the ICU at our tertiary center.
Pertinent laboratory findings included hemoglobin 8.2 g/dL, platelets 132,000/μL and rapidly declining to 55,000, and normal leukocyte count with 82.5% neutrophils. Ferritin was 14,462 ng/mL; erythrocyte sedimentation rate and C-reactive protein were 7 mm/hr and 27.5 mg/dL, respectively. Lactate dehydrogenase was elevated to 1,023 U/L and haptoglobin undetectably low. International normalized ratio (INR) was 1.1 and partial thromboplastin time was 29 s, with d-dimer units elevated to 9,163 ng/mL and fibrinogen decreased to 135 mg/dL. ADAMTS13 (disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) activity was 63%. A peripheral smear showed no schistocytes or fragments; findings included normocytic anemia with mild anisopoikilocytosis, occasional ovalocytes, burr cells, and teardrop forms; neutrophils with toxic changes; and thrombocytopenia with unremarkable platelet morphology.
Alkaline phosphatase was elevated to 168 U/L and aspartate aminotransferase to 165, with normal alanine aminotransferase and bilirubin. Albumin was decreased to 2.3 g/dL. Triglycerides were 209 mg/dL. Antinuclear antibody was positive at 1:160 titer with negative full autoimmune panel, complement levels, rheumatoid factor, and cyclic citrullinated peptide. Urinalysis showed 30+ proteins, otherwise bland. Noninvasive infectious studies were unrevealing (Table 1).
Table 1.
Noninvasive Infectious Studies Performed
| Negative Studies | ||
|---|---|---|
| Blood cultures | Urine legionella antigen | Herpes simplex virus serologies and PCR |
| Urine cultures | Urine histoplasma antigen | Hepatitis B virus serologies |
| Stool cultures | Urine blastomyces antigen | Hepatitis C virus serologies |
| Respiratory viral panel | Quantiferon gold | Varicella zoster virus serologies |
| Rectovaginal group B Streptococcus culture | Syphilis chemiluminescence immunoassay | Parvovirus B19 serologies |
| Nasal methicillin-resistant Staphylococcus aureus PCR | HIV antibody/antigen screen | Beta-D-glucan |
| Cervical gonorrhea and chlamydia nucleic acid amplification test | Cytomegalovirus serologies and PCR | Serum galactomannan |
| Urine strep pneumoniae antigen | Epstein-Barr virus serologies and PCR |
PCR = polymerase chain reaction.
CT scan of the chest, abdomen, and pelvis revealed bilateral upper-lobe predominant patchy ground glass opacities, bilateral pleural effusions, and splenomegaly to 14 cm (Fig. 1). Both CT angiography of the chest and duplex ultrasound of the legs showed no clots. Transthoracic echocardiography revealed a moderately reduced left ventricular ejection fraction of 49% and was otherwise unremarkable.
Figure 1.
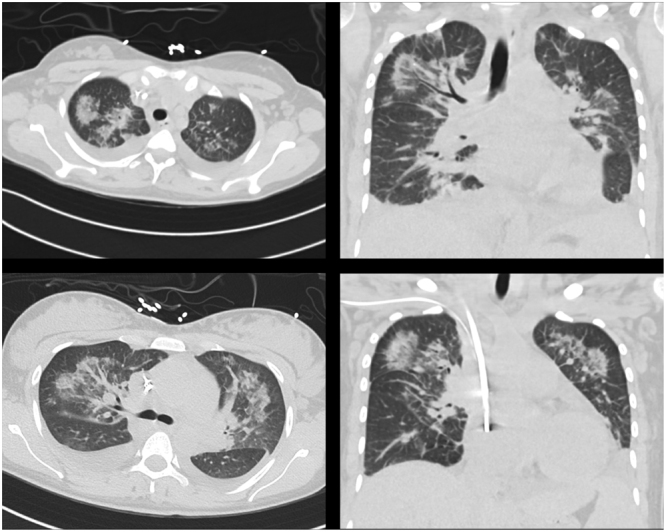
Pulmonary findings on CT scan include bilateral upper-lobe predominant nodular and ground-glass opacities, and bilateral pleural effusions with compressive atelectasis.
Bronchoscopy with right upper lobe lavage was performed. Cell counts showed 182 WBCs (16% neutrophil, 62% macrophage) and 673 RBCs. One fungal culture grew rare Candida albicans and was felt to be a contaminant; cultures were negative for aerobic, anaerobic, acid-fast bacteria, and fungi; Aspergillus galactomannan and Pneumocystis direct fluorescent antigen were also negative. Cytology showed alveolar macrophages, benign bronchial cells, and few benign squamous cells.
With supportive care, the patient became hemodynamically stable with steadily improving oxygen requirement. On hospital day 10, she was saturating well on room air, and empiric antibiotics were stopped. Around the same time, she developed new right shoulder pain and was started on low-dose prednisone (20 mg daily) and colchicine. She was discharged on hospital day 14 after several days afebrile.
Eight days later, the patient returned to the emergency department with relapse of daily fevers and rash (Supplemental Fig. 1, http://links.lww.com/CCX/A646), and new migratory polyarthralgias in the bilateral knees, wrists, and elbows. Studies on readmission revealed worsened transaminases and alkaline phosphatase compared with levels at discharge. Biopsy of the rash revealed neutrophil-rich urticaria.
At this point, a final diagnosis was made of adult-onset Still’s disease (AOSD) presenting as reactive hemophagocytic lymphohistiocytosis (HLH), also known as macrophage-activation syndrome (MAS). Prednisone was increased to 60 mg daily, with both improvement of symptoms and resolution of abnormal liver chemistries. At outpatient follow-up, prednisone dose was successfully tapered. Therapy was transitioned to methotrexate, then later to etanercept, and subsequently tocilizumab.
The patient gave written informed consent for publication of this article and associated images.
DISCUSSION
AOSD is a rare inflammatory disorder classically characterized by high fever, arthritis, and an evanescent salmon-colored rash. HLH is a systemic immune hyperactivation syndrome that can be a primary process in genetically predisposed individuals or can be triggered by another process, often a viral infection or malignancy. Secondary HLH is termed “MAS” when the trigger is a rheumatologic disorder. Although the full pathways remain unknown, decreased natural killer (NK) cell activity, elevated soluble interleukin-2 receptor (sIL-2R), and broadly excessive cytokine production appear to be elements of both AOSD and MAS pathogenesis (1, 2).
We did not perform bone marrow biopsy, NK cell activity, or sIL-2R assays. However, available results were sufficient to diagnose HLH/MAS by both HLH-2004 criteria (Table 2) and the HScore validated for adult patients, indicating a 98–99% probability of reactive HLH (4, 5). With recent evidence that hemophagocytosis is a relatively nonspecific finding (6), we find it reasonable to defer bone marrow biopsy when noninvasive evidence already points strongly to this diagnosis.
Table 2.
Pertinent Diagnostic Criteria, Categorized by Patient’s Results
| Features | HLH-2004 Criteria for HLH | Criteria for Adult-Onset Still’s Disease | Criteria for Overt DIC | Society for Maternal-Fetal Medicine Criteria for Amniotic Fluid Embolism | ||
|---|---|---|---|---|---|---|
| Yamaguchi | Fautrel | ISTH Score | Modified ISTH for Pregnancy (3) | |||
| Requirement for diagnosis | At least five criteria | At least five criteria, with at least 2 major | Either four major, or three major and two minor | At least 5 points | At least 3 points | All four criteria |
| Diagnosis satisfied? | Yes: 5 | Yes: 5, 3 major | Yes: 4 major, 1 minor | No: 4 | No: 2 | No: 0 |
| Criteria satisfied | Fever ≥38.5°CSplenomegalyBicytopenia (at least 2 of: hemoglobin <9 g/dL, Plt <100,000/μL, neutrophils <1,000/μL) Triglycerides >265 mg/dL and/or fibrinogen <150 mg/dL Ferritin >500 ng/mL |
Fever ≥39°C at least 1 wk Arthralgias at least 2 wk Typical rash during feverAbnormal liver function tests Hepatomegaly and/or splenomegaly |
Fever ≥ 39 C Arthralgias Transient erythema Neutrophils ≥ 80% Maculopapular rash |
Plt < 100,000/μL (1 point), <50,000/μL (2 points) d-dimer “moderate increase” (2 points) or “severe increase” (3 point) |
Plt < 100,000/μL (1 point), <50,000/μL (2 points) Fibrinogen < 200 mg/dL (1 point) |
|
| Criteria not satisfied | [Hemophagocytosis in biopsy of bone marrow, spleen, lymph node, or liver] [Low natural killer cell activity] [Elevated soluble interleukin-2 receptor] |
Leukocytes ≥ 10,000/μL with ≥ 80% neutrophils Pharyngitis Lymphadenopathy Both antinuclear antibody and rheumatoid factor negative |
Leukocytes ≥ 10,000/μL Pharyngitis[Glycosylated ferritin < 20%] |
PT prolonged by 3-6 s (1 point), ≥ 6 (2 points)Fibrinogen < 100 mg/dL (1 point) | PT prolonged by 25-50% above normal (1 point), >50% (2 points) | Sudden, not gradual, onset of hypotension and respiratory failure Onset during labor or within 30 min of delivery Overt DIC No fever during labor |
DIC = disseminated intravascular coagulation, HLH = hemophagocytic lymphohistiocytosis, ISTH = International Society on Thrombosis and Haemostasis, Plt = platelets, PT = prothrombin time
Italics indicate minor criteria; normal text indicates major criteria. Square brackets indicate tests not performed.
By its nature, HLH/MAS is often difficult to distinguish from sepsis and other conditions that cause systemic inflammation, but the setting of late pregnancy introduced an even broader differential diagnosis for our patient’s constellation of problems. In particular, her initial laboratory studies could be consistent with hemolysis, elevated liver enzymes, and low platelet (HELLP) syndrome; however, the peripheral smear showed no microangiopathic hemolytic anemia, and HELLP should improve after delivery. Her undetectable haptoglobin presumably reflected not hemolysis, but liver dysfunction from MAS involvement.
Another catastrophic diagnosis under early consideration, given peripartum shock and respiratory failure, was amniotic fluid embolism (AFE). This condition has been historically overdiagnosed, with retrospective reviews suggesting that 30–50% of patients labeled with AFE had a more likely alternative diagnosis (3). In response, the Society of Maternal-Fetal Medicine (SMFM) has proposed diagnostic criteria arguing that true AFE must have “sudden” onset of hypotension and hypoxemia without fever, during or just after labor, and provoke overt disseminated intravascular coagulation (DIC) (3). As our case satisfies none of the mandatory features, this definition strongly rules out AFE (Table 2).
In turn, identifying DIC during the third trimester can be difficult, because both fibrin degradation products and fibrinogen are physiologically elevated during this period (making our patient’s low fibrinogen especially ominous). However, the strikingly normal INR keeps our patient short of overt DIC by both the original International Society on Thrombosis and Haemostasis criteria (7) and a proposed modified version for term pregnancy by the SMFM/AFE authors (3) (Table 2). The question of whether she had a “low-grade DIC” lingers, but especially in the absence of apparent bleeding or thrombosis, does not affect management.
Other serious differential diagnoses included thrombotic thrombocytopenic purpura, pulmonary embolism, endometritis, and other causes of sepsis. Once these were ruled out, a final challenge was the late onset of joint symptoms, which delayed the diagnosis of AOSD. At the time of the first hospital discharge, our working diagnosis had been HLH secondary to pregnancy, as systemic inflammation appeared to be spontaneously resolving and the isolated shoulder pain was not clearly related to the broader syndrome. Only after her return with fevers, polyarthralgias, and a typical rash were Yamaguchi and Fautrel classification criteria for AOSD satisfied (Table 2) (8, 9). In fact, almost all documented AOSD in pregnancy has the so-called “systemic” subphenotype, characterized by marked systemic inflammation and mild or nonexistent joint symptoms. This contrasts with the “articular” phenotype, in which arthritis is prominent and systemic manifestations limited (1, 10).
To our knowledge, this is the first report of new-onset AOSD during the third trimester of pregnancy. Previous case series have documented flares in the first and second trimesters and postpartum period, with greatest incidence in the second trimester (10, 11). HLH/MAS of other causes has occasionally been reported in the third trimester but also seems disproportionately to favor the second trimester and postpartum period (12, 13).
Although pulmonary involvement is not characteristic of either AOSD or MAS, it can occur in both disorders (14, 15). In one series, about 5% of patients with AOSD developed parenchymal lung involvement (15). Of those, 40% had acute respiratory distress syndrome (ARDS); among the remaining 60%, the most common CT finding was bilateral lower lobe predominant interstitial pneumonia. Notably, these patients were disproportionately likely to present with MAS and to have the systemic subphenotype of AOSD, perhaps hinting that lung injury in this disorder could occur by an ARDS-like mechanism (in the context of a systemic inflammatory state) even in those not meeting the Berlin criteria.
To comment briefly on management, hindsight suggests our patient could have benefited from earlier and more aggressive immunosuppressive therapy. However, during the initial severe phase of her course, concern for infectious causes was high enough that the risk of high-dose glucocorticoids seemed to outweigh benefit. By the time a satisfactory infectious evaluation could be concluded with bronchoalveolar lavage results, her overall condition had greatly improved, so we trialed only low-dose prednisone to target the early articular symptoms. The timing and selection of empiric immunosuppression are frequent challenges in the management of suspected HLH/MAS, given the near-ubiquitous concern for infection in such cases, and remain highly dependent on individual patient factors and clinician judgment.
In summary, we illustrate that AOSD can present as MAS during the third trimester, and can mimic or raise concern for peripartum disorders such as HELLP syndrome. The characteristic articular symptoms of AOSD may be absent or delayed, and pulmonary involvement with severe hypoxemia can occur. Clinicians should consider this rare diagnosis when evaluating a patient with unexplained fever and multiorgan dysfunction, even in the context of pregnancy.
Supplementary Material
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website (http://journals.lww.com/ccejournal).
The authors have disclosed that they do not have any potential conflicts of interest.
REFERENCES
- 1.Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still’s disease. J Autoimmun. 2018; 93:24–36 [DOI] [PubMed] [Google Scholar]
- 2.Gerfaud-Valentin M, Jamilloux Y, Iwaz J, et al. Adult-onset Still’s disease. Autoimmun Rev. 2014; 13:708–722 [DOI] [PubMed] [Google Scholar]
- 3.Clark SL, Romero R, Dildy GA, et al. Proposed diagnostic criteria for the case definition of amniotic fluid embolism in research studies. Am J Obstet Gynecol. 2016; 215:408–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: Long-term results of the cooperative HLH-2004 study. Blood. 2017; 130:2728–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014; 66:2613–2620 [DOI] [PubMed] [Google Scholar]
- 6.Goel S, Polski JM, Imran H. Sensitivity and specificity of bone marrow hemophagocytosis in hemophagocytic lymphohistiocytosis. Ann Clin Lab Sci. 2012; 42:21–25 [PubMed] [Google Scholar]
- 7.Toh CH, Hoots WK; SSC on Disseminated Intravascular Coagulation of the ISTH. The scoring system of the scientific and standardisation committee on disseminated intravascular coagulation of the International Society on Thrombosis and Haemostasis: A 5-year overview. J Thromb Haemost. 2007; 5:604–606 [DOI] [PubMed] [Google Scholar]
- 8.Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992; 19:424–430 [PubMed] [Google Scholar]
- 9.Lebrun D, Mestrallet S, Dehoux M, et al. Validation of the Fautrel classification criteria for adult-onset Still’s disease. Semin Arthritis Rheum. 2018; 47:578–585 [DOI] [PubMed] [Google Scholar]
- 10.Gerfaud-Valentin M, Hot A, Huissoud C, et al. Adult-onset Still’s disease and pregnancy: About ten cases and review of the literature. Rheumatol Int. 2014; 34:867–871 [DOI] [PubMed] [Google Scholar]
- 11.Plaçais L, Mekinian A, Bornes M, et al. Adult onset Still’s disease occurring during pregnancy: Case-report and literature review. Semin Arthritis Rheum. 2018; 47:575–577 [DOI] [PubMed] [Google Scholar]
- 12.Song Y, Wang Z, Hao Z, et al. Requirement for etoposide in the treatment of pregnancy related hemophagocytic lymphohistiocytosis: A multicenter retrospective study. Orphanet J Rare Dis. 2019; 14:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parrott J, Shilling A, Male HJ, et al. Hemophagocytic lymphohistiocytosis in pregnancy: A case series and review of the current literature. Case Rep Obstet Gynecol. 2019; 2019:9695367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seguin A, Galicier L, Boutboul D, et al. Pulmonary involvement in patients with hemophagocytic lymphohistiocytosis. Chest. 2016; 149:1294–1301 [DOI] [PubMed] [Google Scholar]
- 15.Gerfaud-Valentin M, Cottin V, Jamilloux Y, et al. Parenchymal lung involvement in adult-onset Still disease: A STROBE-compliant case series and literature review. Medicine (Baltimore). 2016; 95:e4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.