

Abstract
Myeloproliferative neoplasm (MPN) is a category in the World Health Organization classification of myeloid tumors. BCR-ABL1–negative MPN is a subcategory that includes primary myelofibrosis (MF), post–essential thrombocythemia MF, and post–polycythemia vera MF. These disorders are characterized by stem cell–derived clonal myeloproliferation. Clinically, these diseases present with anemia and splenomegaly and significant constitutional symptoms such as severe fatigue, symptoms associated with an enlarged spleen and liver, pruritus, fevers, night sweats, and bone pain. Multiple treatment options may provide symptom relief and improved survival; however, allogeneic stem cell transplantation (HCT) remains the only potentially curative option. The decision for a transplant is based on patient prognosis, age, comorbidities, and functional status. This review describes the recent data on various peritransplantation factors and their effect on outcomes of patients with MF and new therapeutic areas, such as the use and timing of Janus kinase inhibitors with HCT and gives overall conclusions from the available data in the published literature.
Keywords: Allogeneic stem cell, transplantation, Myelofibrosis
INTRODUCTION
Myelofibrosis (MF) is a clonal myeloproliferative neoplasm (MPN) that can arise de novo or result from previous polycythemia vera or essential thrombocythemia (post-ET MF). MF is characterized by a clonal stem cell process, resulting in ineffective erythropoiesis, reactive fibrosis in bone marrow, and extramedullary hematopoiesis in the spleen or in multiple organs [1]. The disease process causes debilitating symptoms as a consequence of anemia and splenomegaly, leading to fatigue, abdominal discomfort, early satiety, cachexia, constitutional symptoms, and eventually death. Reported causes of death include transformation to acute leukemia, progression of primary disease, thrombosis and cardiovascular complications, and infection or bleeding [2]. Herein, we discuss prognostic factors; current therapeutic options, including nonallogeneic stem cell transplant and allogeneic stem cell transplant (HCT); pre-HCT factors; and post-HCT factors. Future directions and some ongoing studies are also discussed. A PubMed search was conducted, using the keywords “myelofibrosis,” “allogeneic stem cell transplantation” and “Janus kinase (JAK) inhibitors.” Published abstracts related to the search were also reviewed.
Symptom Burden
Patients with MF can have a significant symptom burden, which can include severe fatigue, symptoms associated with an enlarged spleen and liver, pruritus, fevers, night sweats, and bone pain. To quantify the symptoms associated with MF, the Myelofibrosis Symptom Assessment Form (SAF) was created in 2009 [3]. This survey asks a series of 20 questions, and patients rate their symptom score on a scale of 0 to 10, with 0 being “not a problem” and 10 being “the worst imaginable symptom.” In 2011, this survey was expanded to become the 27-question Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF), created to encompass MF as well as polycythemia vera and ET [4]. This form included assessment of microvascular symptoms, such as insomnia, difficulty concentrating, sexual dysfunction, vertigo, headaches, and numbness/tingling. The MPN-SAF was subsequently consolidated into a 10-question survey known as the MPN-SAF-Total Symptoms Scale (MPN-SAF-TSS), which captured relevant data in a shorter format [5]. A clinical response is indicated by a 50% reduction in the total score. The current guidelines recommend collecting the MPN-SAF for the initial evaluation and MPN-SAF-TSS for subsequent follow-up assessments [6].
Prognostic Factors
Clinical prognostic scoring systems
The median survival of patients with MF varies from 1.5 years to more than a decade, depending on the severity of disease. Multiple scoring systems have been used over the years to guide patients and providers in treatment choices. The International Prognostic Scoring System (IPSS) uses age over 65 years, presence of constitutional symptoms, hemoglobin less than 10 g/dL, WBC count greater than 25 × 10.9/L, and circulating blasts over 1% as risk factors for determining survival [2]. Using this system, median survival in patients with low risk (0 risk factors) was 135 months; intermediate-1 risk (1 risk factor), 95 months; intermediate-2 risk (2 risk factors), 48 months; and high risk (≥3 risk factors), 27 months.
In 2010 the Dynamic IPSS (DIPSS) was developed. This prognostic model could be used to guide decision-making at any time during the clinical course of primary MF [7]. Median survival of patients with low risk (score of 0) was not reached; for intermediate-1 risk (score of 1 or 2), median survival was 14.2 years; for intermediate-2 risk (score of 3 or 4), 4 years; and for high risk (score of 5 or 6), 1.5 years. In addition to the DIPSS variables as predictors of overall survival in primary MF, DIPSS plus included patients with an unfavorable karyotype, a platelet count less than 100 × 109/L, and a need for RBC transfusion [8]. An additional point was assigned to each of these additional variables. Unfavorable karyotype included a complex karyotype or 1 or 2 abnormalities, including +8, −7/7q-, i(17q), −5/5q-, 12p-, inv(3), or 11q23 rearrangement.
Driver mutations
Driver mutations, such as those in JAK2, myeloproliferative leukemia receptor (MPL) and calreticulin receptor (CAL-R), have been shown to be associated with poor survival and leukemic transformation. These mutations are crucial for decision-making because they substantially affect disease biology and outcomes. Primary MF with triple-negative mutation status (ie, negative for JAK2, MPL, or CAL-R mutations) has a poorer prognosis and higher risk of leukemic transformation. Data from patients at the Mayo Clinic Rochester were used to estimate overall survival from the date of diagnosis or first referral and showed a median overall survival of 4.3 years for patients with JAK2 mutant, 4.1 years for patients with MPL mutant, 8.2 years for patients with CAL-R mutant, and 2.5 years for patients with a triple-negative mutation [9]. In an Italian study the estimated overall median survival from time of diagnosis was 9.2 years for patients with JAK2 mutant, 9.1 years for patients with MPL mutant, 17.7 years for patients with CAL-R mutant, and 3.2 years for patients with a triple-negative mutation [10]. The CAL-R mutation was shown to have a relatively indolent course compared with the JAK2 mutation in ET and primary MF.
Other somatic mutations
Somatic mutations other than JAK2, MPL, and CAL-R are frequently observed by next-generation sequencing in patients with MF. Mutations such as ASXL1, SRSF2, and EZH2 mutations independently and negatively affect survival, whereas IDH1/2, SRSF2, and ASXL1 mutations were associated with leukemic transformation [11]. Also, patients with CAL-R unmutated and ASXL1 mutant (CAL-R-/ASXL+) primary MF had a particularly poor survival (median, 2.3 years) [9]. Next-generation sequencing in 189 patients identified variants in SCRIB, MIR662, BARD1, TCF12, FAT4, DAP3, POLG, and NRAS, which were recurrent and occurred in more than 3% of patients with MPN who were tested [12]. In addition, 8 patients (4.7%) in this study with primary MF who harbored a heterozygous NRAS mutation in codon 12 had a poorer prognosis and were associated with a higher risk category (intermediate-2 and higher).
Additional somatic mutations are being identified. These mutations may add to the current scoring systems and may identify patients in the intermediate-risk or low-risk categories who have a greater chance of disease progression. For these patients an HCT would be considered earlier in their treatment course. The Mutation-Enhanced IPSS (MIPSS) has more recently been described and includes mutations such as JAK2, MPL, CAL-R, EZH2, ASXL1, IDH1/2, and SRSF2 [13]. In this scoring system age over 60 years, constitutional symptoms, hemoglobin level less than 10 g/dL, platelet count less than 200 × 109/L, triple-negative mutation status, JAK2 or MPL mutation, and ASXL1 or SRSF2 mutations were found to be significant risk factors for poor survival.
CURRENT THERAPEUTIC OPTIONS
Current treatment recommendations are based on the risk stratification from the DIPSS or DIPSS plus score and on the patient’s symptom burden (Figure 1) [6]. The available pharmacologic therapies for MF are aimed at improving symptoms, quality of life, and overall survival. HCT remains the only potentially curative treatment modality for patients with MF [14].
Figure 1.
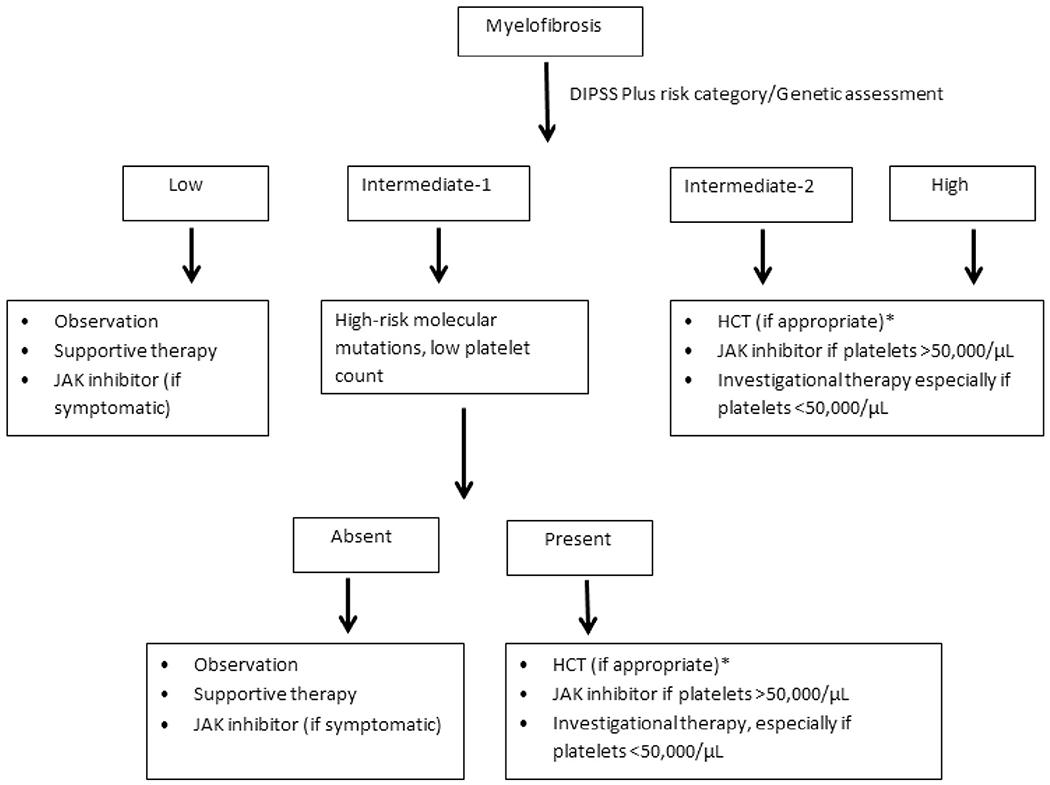
Treatment algorithm for MF. *Directly to HCT or after bridging therapy to decrease marrow blasts.
Nontransplantation Options
Patients in low-risk and intermediate-1 risk categories can be observed for disease progression or given erythropoietin or hydroxyurea. For patients with an erythropoietin level less than 500 mU/mL, erythropoietin-stimulating agents may ameliorate the anemia [15]. IFN-α, pegylated IFN-alfa-2a, and pegylated iIFN-alfa-2b have also been evaluated in several series of patients with MF and have been shown to improve cellularity, splenomegaly, and bone marrow morphology [16–18].
Role of JAK inhibitors
Ruxolitinib is a selective JAK1/JAK2 inhibitor approved by the US Food and Drug Administration in 2011 for intermediate- and high-risk MF patients, including those with primary MF, post-polycythemia vera MF, and post-ET MF. The US Food and Drug Administration approval was based on improvement in spleen size and quality of life in patients taking the drug compared with those given placebo and was the best available therapy in 2 phase III studies: COMFORT-I and COMFORT-II (Controlled Myelofibrosis Study With Oral JAK Inhibitor) [19,20]. The COMFORT studies included patients with intermediate-2 and high-risk disease. In the 3-year follow-up to COMFORT-II, there was a survival advantage with the use of ruxolitinib [21]. The most commonly reported hematologic adverse effects in the COMFORT studies were anemia and thrombocytopenia, which improved after prolonged therapy beyond 8 to 12 weeks. Retrospective data also show a benefit for ruxolitinib in symptomatic patients with low-risk disease [22]. The ROBUST trial included patients with intermediate-1, intermediate-2, and high-risk MF, with improvement noted in the symptom score in approximately 80% of patients with intermediate-1 risk disease [23]. The role and efficacy in patients with low-risk MF is yet to be evaluated in a prospective trial. Recently published National Comprehensive Cancer Network guidelines recommend its use in intermediate-and high-risk disease and recommend consideration of use in patients who are symptomatic with low-risk disease [6].
Other JAK2 inhibitors being developed that have shown promising early results are pacritinib and momelotinib. Pacritinib is a JAK2/FLT-3 inhibitor that is better tolerated in patients with cytopenias and has shown spleen and symptom response in patients with MF in early-phase studies [24]. Phase III trials (PERSIST-1 and PERSIST-2) of pacritinib currently underway are including patients with platelet counts less than 50,000/μL. Likewise, momelotinib has improved anemia, splenomegaly, and constitutional symptoms in patients and is currently in phase III trials (NCT01969838, NCT02101268). Additional therapies, such as imetelstat (a telomerase inhibitor, NCT02426086) and PRM-151 (an antifibrotic agent, NCT01981850), are also in early phases of development.
Management of MPNs in Blast Phase
As a clonal myeloid neoplasm, MF (and, rarely, ET and polycythemia vera) has a risk of progression to acute leukemia, with more than 20% blasts in peripheral blood or bone marrow, a phase referred to as MPNs in blast phase [25]. MPNs in blast phase progresses rapidly and has a poor prognosis, with median survival for patients of less than 3 months [26]. There is currently an unmet need for therapeutic options for this phase of disease. HCT has been recognized as a potential salvage option, with improved results after a response from induction therapy [27]. Nontransplant options, such as hypomethylating agents and JAK2 inhibitors, have been somewhat successful [28–30]. A better understanding of the pathogenesis of the blast phase is needed to help develop novel treatment strategies for this group of patients.
Role of HCT
HCT has an established role in long-term remission and potential cure of patients with MF. Current recommendations from the European Blood and Marrow Transplantation Group and the European Leukemia Net international working groups include considering HCT for patients less than 70 years old and with intermediate-2 or high-risk disease [31]. In addition, patients who have intermediate-1 risk disease and are less than 65 years old are considered for HCT if they have refractory, transfusion-dependent anemia or a blast percentage greater than 2% in peripheral blood or unfavorable cytogenetic characteristics [31]. The benefit of an HCT is derived from the cytotoxic effect of the pretransplantation conditioning regimen and, subsequently, to the alloimmune graft-versus-leukemia effect. Despite the benefit of long-term, relapse-free survival with HCT, it can be associated with substantial treatment-related morbidity and mortality. Nonrelapse mortality associated with HCT is about 20% [32,33] and is related to a variety of host-, disease-, and transplant-related factors. Therefore, it is imperative to select the “right patients” at the “right time” in their disease course to gain the most benefit from this valuable therapeutic option.
MAKING THE BEST OF HCT: THE CHALLENGE AND THE SUCCESS
Patient and Disease Factors
Patient age and comorbidities
The median age of patients diagnosed with MF is more than 65 years [34]. Given the older age and coexisting medical conditions, most patients are not considered for HCT because of concerns regarding treatment-related toxicities. Patients older than 55 years have been shown to have poorer survival in some previous studies [35,36]. However, once a patient is older than 60, there appears to be little difference in survival. A study by the Center for International Bone Marrow Transplant Research (CIBMTR) of over 1200 patients with myelodysplastic syndrome showed no difference in survival in patients 55 to 64 versus 65 years or older [37]. A retrospective analysis of 30 patients aged 60 to 78 years who underwent HCT for MF showed that 27 patients had donor engraftment and hematopoietic recovery [38]. Forty-five percent of patients were alive at a median of 22 months, with a progression-free survival of 40% at 3 years. More importantly, survival was not significantly different in patients between ages 60 and 65 years and those between 66 and 78 years. Despite a suspected selection bias in including healthier older patients for HCT and in the choice of conditioning regimen, the results are encouraging for considering lower-intensity conditioning regimens for pursuing HCT in older patients with MF who have minimal comorbidities. In our practice age 65 to 70 years is commonly used as a threshold for considering HCT. There is currently an ongoing CIBMTR study to assess the benefit of HCT in patients with MF, comparing the outcomes of a prospectively enrolled cohort of HCT recipients with an age-matched cohort who did not undergo HCT (NCT02934477).
A patient’s chronologic age is not always a good guide for making a decision for HCT. Biologic age is equally important as disease status in assessing suitability for HCT. In addition, the severity of comorbidities increases the risk of treatment-related toxicity and, hence, decreases a patient’s chance of survival. The hematopoietic cell transplantation-specific comorbidity index is a tool used to assess the risk of nonrelapse mortality and survival by accounting for medical conditions before HCT [39]. Common comorbidities in older patients, such as pulmonary dysfunction (diffusing capacity of the lungs for carbon monoxide and/or forced expiratory volume in 1 second ≤ 65%, dyspnea at rest, or dyspnea requiring oxygen), valvular heart disease (except mitral valve prolapse), and hepatic dysfunction (cirrhosis, bilirubin > 1.5 times the upper limit of normal, or aspartate transaminase/alanine transaminase ratio > 2.5 times the upper limit of normal) were among the highest-weighted factors for influencing outcomes. A modified hematopoietic cell transplantation–specific comorbidity index model that included risk stratification can also serve as a predictor for nonrelapse mortality [40].
In addition to comorbidities, a comprehensive geriatric assessment is a potential way to select older patients for HCT. In a prospective study of approximately 200 patients, poorer survival after HCT, especially in patients older than 60 years, was associated with instrumental activities of daily living, slow walking speed, a high comorbidity index, a poor mental health score on the Short Form-36 Mental Component Summary, and elevated C-reactive protein level [41].
Disease status
Various studies have looked at correlations of outcomes and pretransplant DIPSS score (Table 1). Most of these studies have shown better outcomes for patients with a lower DIPSS score. Patients with low-risk disease are generally not considered for transplant because they have better survival rates with pharmacologic and supportive therapy alone. High treatment-related complication and mortality rates are associated with HCT, which is concerning because of the curative potential of HCT in this group of patients. A retrospective study compared outcomes of patients younger than 65 years with primary MF on the basis of pretreatment DIPSS and HCT versus conventional therapies [45]. Despite an inherent selection bias and inclusion of only primary MF patients, the study showed a benefit of HCT for patients with intermediate-2 and high-risk disease. This study included 438 patients; 190 underwent HCT and 248 received conventional therapies. Relative risk of death after HCT versus conventional therapies for patients with low risk per the DIPSS model was 5.6 (95% confidence interval [CI], 1.7 to 19; P = .005); for intermediate-1 risk, 1.6 (95% CI, .79 to 3.2; P = .19); for intermediate-2 risk, .55 (95% CI, .36 to .83; P = .005); and for high risk, .37 (95% CI, .21 to .66; P = .0007), thus suggesting the benefit of HCT is limited to patients with a higher risk based on the DIPSS system.
Table 1.
Outcomes of Allogeneic Stem Cell Transplant Based on Pretransplant DIPSS Score
| Study | No. of Patients, Type of Study | Type of MF | Donor Type, % | DIPSS Score Pretransplant, % | OS outcomes |
|---|---|---|---|---|---|
| Gupta et al [42] | 233, Retrospective | Primary MF | MRD, 34 MUD, 45 MMUD, 21 |
Low, 12 Intermediate-1, 49 Intermediate-2, 37 High, 1 NA, 1 |
Relative risk: Low/intermediate-1 1 Intermediate-2/high, 1.37 (P = .10) Survival at 5 yr (95% CI): Low/intermediate-1, 51% (42–59%) Intermediate-2/high, 41% (30–52%) |
| Alchalby et al [36] | 150, Retrospective | Primary MF Post-ET MF Post-PV MF |
Sibling, 28 Unrelated, 72 Matched, 80 Mismatched, 20 |
Low, 2 Intermediate-1, 29 Intermediate-2, 58 High, 11 |
Relative risk: Low/intermediate-1 1 Intermediate-2, 1.52 High, 2.86 5-yr OS (95% CI): Low/intermediate-1, 70% (57–83%) Intermediate-2, 60% (50–70%) High, 47% (29–65%) |
| Ditschkowski et al [43] | 76, Retrospective | Primary MF Post-ET MF Post-PV MF |
Sibling, 39 Unrelated, 61 |
Low, 10 Intermediate-1, 28 Intermediate-2, 49 High, 13 |
5-yr OS rate: Low, 76% Intermediate-1, 48% Intermediate-2/high, 38% Median OS: Intermediate-1, 30 mo Intermediate-2/high, 35 mo |
| Scott et al [44] | 170, Retrospective | Primary MF Post-ET MF Post-PV MF |
MRD, 47 MMRD, 2 MUD, 40 MMUD, 11 |
Low, 12 Intermediate-1, 28 Intermediate-2, 30 High, 30 |
Relative risk: Low, 1 Intermediate-1, 1.97 (P = .22) Intermediate-2, 3.15 (P = .04) High, 4.01 (P = .01) Median OS: Low, NR at 5.2 yr Intermediate-1, NR at 6.3 yr Intermediate-2, 7 yr High, 2.5 yr |
OS indicates overall survival; MRD, matched-related donor; MUD, matched unrelated donor; MMUD, mismatched-unrelated donor; NA, not available; post-PV MF, post-polycythemia vera myelofibrosis; MMRD, mismatched-related donor; NR, not reached.
The DIPSS does not take into consideration somatic mutations that can impact outcomes. In younger patients, who have high-risk molecular mutations, HCT is often considered earlier in the disease course. Preliminary data suggest that HCT improves outcomes in patients with SRSF2, EZH2, and IDH1 mutations; however, it is less clear whether it improves outcomes in patients with ASXL1, U2AF1, IDH2, or DNMT3A mutations [46].
JAK2 therapy before HCT
The impact of treatment with JAK2 inhibitors like ruxolitinib before HCT was addressed in 2 retrospective studies and 1 prospective study [47–49]. One of the retrospective studies included 22 patients who underwent HCT and who had previously been treated with JAK2 inhibitors [47]. In 12 of these patients, spleen size responded to ruxolitinib, and they had superior overall survival at 1 year compared with the 10 patients who did not respond to ruxolitinib therapy (100% versus 60%, P=.02). In another larger retrospective study that included 100 patients, 23 patients who improved clinically with JAK1/JAK2 inhibitors had an overall survival rate of 91% at 2 years compared with 32% for the 13 patients who had leukemic transformation while undergoing therapy [48]. Clinical improvement was defined as at least 50% improvement in spleen size for a spleen of at least 10 cm or complete resolution of splenomegaly for a spleen less than 10 cm. Although these numbers are small, the favorable outcome with HCT for patients who have a clinical response to JAK1/JAK2 inhibitor therapy is encouraging and should be further validated. These 2 studies also indicated that ruxolitinib should be continued until close to the time conditioning regimens are initiated. Symptoms from discontinuing the drug were more common in patients with a longer duration between discontinuation of therapy and the start of conditioning chemotherapy.
The first prospective study to assess the impact of ruxolitinib in MF included 23 patients who were candidates for HCT [49]. Of the 16 patients assessed for response, 8 (50%) had a decrease in spleen size of 25% and improved constitutional symptoms and another 8 patients (50%) had stable disease. Unexpected adverse effects reported after ruxolitinib withdrawal included febrile cardiogenic shock and tumor lysis syndrome with acute renal failure during conditioning. Unfortunately, despite amending the protocol by including tumor lysis prophylaxis, ruxolitinib tapering with steroids, and changing the conditioning regimen, 2 additional patients developed tumor lysis and 1 developed cardiogenic shock. As a result, study enrollment was discontinued.
Transplant-Related Factors
Donor selection
Donor source is an important question before transplant because HCT with an HLA-mismatched unrelated to related donor has been reported to be an independent risk factor for both disease-free survival and overall survival in MF (Table 2) [35,42,50]. These reports did not include cord blood or haploidentical donors. In a study from the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation, nonrelapse mortality at 1 year after HCT was similar for the HLA-identical sibling and matched unrelated donor groups (10% versus 13%, P = .05); however, it was significantly higher for patients with mismatched versus matched donor HCT (38% versus 12%, P = .003) [35]. Similarly, disease-free survival and overall survival at 5 years were improved in HLA-matched versus mismatched transplants: disease-free survival, 59% versus 40% (P = .01); and overall survival, 74% versus 38% (P = .003). Data from the Myeloproliferative Disease Research Consortium also showed that overall survival at a median follow-up of 25 months was higher in sibling donor transplants (75%, median not reached) versus unrelated donor transplants (32%; median, 6 months; P < .0001) [50]. The donor source affected survival in this study, but no difference was determined between HLA-matched and -mismatched unrelated donor transplants. Retrospective data from CIBMTR reported that adjusted probability of survival at 5 years was 56% for a matched-related donor, 48% for a matched-unrelated donor, and 34% for partially matched/mismatched unrelated donor (P = .002) [42].
Table 2.
Donor Source and Conditioning Regimens
| Study | No. of Patients/Disease at Baseline | Conditioning Regimen | Donor Distribution | Nonrelapse Mortality | OS | Comments |
|---|---|---|---|---|---|---|
| Kroger et al [35] | 103/PMF, post-ET MF, post-PV MF |
Bu-Flu-ATG | HLA-matched, 82 |
Overall NRM at 1 y, 16% HLA MRD, 10% 10/10 MUD, 13% MMUD, 38% Completely matched, 12% |
OS at 5 yr, 67% HLA-matched, 74% HLA-mismatched, 38% (P = .03) |
OS was significantly better in patients <55 yr vs. >55 yr and HLA-matched vs. mismatched donor |
| Rondelli et al [50] | 66/PMF, post-ET MF, post-PV MF | Flu-Mel ATG in unrelated donor HCT | HLA-matched sibling, 30 HLA-mismatched sibling, 2 HLA-matched unrelated, 25 HLA-mismatched unrelated, 9 |
Median follow-up, 25 mo Sibling donor, 22% Unrelated donor, 59% |
Median follow-up, 25 mo Sibling donor, 75% Unrelated donor, 32% |
Median for OS not reached in the sibling cohort vs. 6 mo in the unrelated cohort (P < .0001) |
| Gupta et al [42] | 233/PMF | Flu-TBI, 22% Flu-Mel, 28% Bu-Flu, 38% Other RIC, 12% |
MRD, 79 MUD, 104 MMUD, 50 |
Cumulative incidence of NRM at 1 y, 18%; 5 yr, 24% RR for NRM in MUD, 3.92; MMUD, 9.37 (P < .0001) |
Adjusted probabilities for survival at 5 yr: MRD, 56% MUD, 48% MMUD, 34% (P = .002) |
Donor type was an independent risk factor for NRM and OS |
PMF indicates primary myelofibrosis; Bu-Flu, busulfan + fludarabine; ATG, antithymocyte globulin; Flu-Mel, fludarabine + melphalan; Flu-TBI, fludarabine + total body irradiation; NRM, nonrelapse mortality; RR, relative risk.
Other possibilities for donor source include haploidentical-related donors and umbilical cord blood. Haploidentical-related donors have become an alternate option for donor source, especially after effective control of graft-versus-host (GVHD) disease with post-transplantation cyclophosphamide [51]. However, there are no studies yet that exclusively describe results of haploidentical donor transplantation for MF patients. In a study from an Italian group, 20 of 37 patients who underwent HCT for MF between 2011 and 2014 were from haploidentical family donors compared with 3 of 58 patients between 2000 and 2010 [52]. This study did not evaluate the outcomes specific to haploidentical transplantation; however, overall deaths within 1 year of HCT in alternate donors in recent years (after 2011) were about half of those in alternate donor HCTs in the years 2000 to 2010 (31% versus 56%, P = .06). This difference existed despite a higher proportion of haploidentical HCTs in the more recent years, suggesting haploidentical donor HCTs as a potential option for MF as is for other hematologic malignancies. Umbilical cord blood transplantation was studied in 35 patients with MF, including patients with transformation to acute leukemia [53]. Platelet and neutrophil counts recovered in all patients who received total body irradiation (2 to 12 Gy), fludarabine, and a cyclophosphamide-conditioning regimen. Two-year overall survival and event-free survival rates were 44% and 30%, respectively, despite a concern for graft failure.
Conditioning regimens
Patient comorbidity and functional status must be carefully considered in choosing a conditioning regimen for HCT in patients with MF. A myeloablative conditioning (MAC) regimen has been shown to have curative potential, with 5-year survival rates of between 47% and 61% [54,55]. This regimen, however, is associated with a nonrelapse mortality ranging from 20% to 48% at 1 year. Reduced-intensity conditioning (RIC) regimens have been described in various studies and have been shown to be beneficial for older patients who cannot tolerate myeloablative regimens (Table 2) [35,42,50]. The RIC regimens described have been busulfan-fludarabine or fludarabine-melphalan-based. The mechanism of action with a MAC regimen is to eliminate tumor cells before HCT; with RIC regimen, it is to use the graft-versus-leukemia effect after HCT. No prospective studies have compared MAC versus RIC regimens for HCT in patients with MF, especially to understand if RIC regimens would be a better choice than MAC for younger patients. That said, retrospective reports from centers in Italy, Canada/Europe, and the United Kingdom have not suggested that intensity of a conditioning regimen affected outcomes after transplantation [56–58]. It is likely that in these studies RIC regimens were used in patients who were older with a poorer functional status at baseline. These older patients receiving RIC still managed to achieve a similar survival as fitter patients who received MAC. This supports the use of RIC regimens in patients undergoing HCT for MF.
The RIC regimen that would have the most favorable outcomes for patients with MF is not yet clearly known. No randomized prospective trials have been done. In a retrospective study, fludarabine-busulfan was compared with fludarabine-melphalan [59]. The 7-year progression-free survival of the fludarabine-busulfan versus fludarabine-melphalan groups was 33% versus 52%; 7-year overall survival, 59% versus 52%; and nonrelapse mortality, 31% versus 43% (P = .068), respectively.
Post-Transplantation Care: The Challenges
Graft failure
Graft failure is a detrimental consequence of HCT in MF and is reported at rates varying from 2% to as high as 28% in various settings. The risk factors for graft failure are not yet well known. Some studies have suggested donor source and pretransplantation splenectomy affect leucocyte engraftment [35,50]. Rates of primary graft failure were as low as 2% in the study from the European Group for Blood and Marrow Transplantation [35]. Of note, however, 11% of patients in this study received a stem cell boost after HCT because of poor engraftment or decreasing donor chimerism. Patients in this study received antithymocyte globulin along with a conditioning regimen for T cell depletion, suggesting a potential benefit in preventing graft failure. Data from the Myeloproliferative Disease Research Consortium 101 showed rates of primary graft failure of 3% in sibling donor HCT and 24% in unrelated donor HCT [50]. Secondary graft failure occurred in 3% of sibling donor HCTs and in 12% of unrelated donor HCTs. This higher rate of graft failure occurred despite the use of antithymocyte globulin for patients undergoing unrelated donor HCT. Data from CIBMTR showed a probability of neutrophil engraftment of 84% at 28 days and 97% at 100 days; the probability of platelet engraftment was 47% at 28 days and 77% at 100 days [42]. This study did not show a difference in engraftment due to donor type. In a cohort of patients from a study in the Netherlands (53 patients across 3 centers), the rate of primary graft failure at 60 days was 28% [60]. Patients receiving a nonmyeloablative conditioning regimen had a lower rate of neutrophil engraftment than patients who received RIC (56% versus 84%, P = .003).
Relapse
Relapse after HCT for MF can be clinical or molecular, the latter indicated by an increased JAK2 V617F mutation. Cumulative relapse rates have been reported from 22% to 29% at 3 to 5 years [35,61]. Donor source and HLA-matched status did not appear to affect relapse rates [35,42]. In 2 retrospective studies, the intensity of the conditioning regimen was not significantly associated with relapse [56,57].
Data from a study by Stewart et al [57] showed a nonsignificant tendency toward a higher relapse rate at 3 years in a RIC group than in a MAC group (46% versus 15%, P = .06). However, the median time to transplant from diagnosis in this study was 54 months in the RIC group and 9.2 months in the MAC group. A longer time to transplant was described as an unfavorable factor for relapse-free survival in the Italian study [56]. Hence, the higher relapse in RIC group could, at least in part, be a result of this difference in the 2 groups.
Donor lymphocyte infusion
For patients who relapse or undergo graft rejection, an option for donor lymphocyte infusion exists. In a 2-step approach described by a German group, patients received a dose-escalated donor lymphocyte infusion at a median 1.2 × 106 T cells/kg [62]. Of 26 patients in their study, 10 (39%) who received a median of 3 doses had a complete response to donor lymphocyte infusion. An expected complication of acute GVHD was seen in 12% of patients at grades II to IV and chronic GVHD occurred in 36% of patients. Patients who did not respond to donor lymphocyte infusion had a second HCT. The complete response rate with a second HCT was 60%, with an overall response rate of 80%. Two-year overall survival and progression-free survival for the entire cohort of 30 patients were 70% and 67%, respectively.
Graft-versus-host disease
GVHD is the most common complication of HCT, requiring treatment with immunosuppressive agents and resulting in complications, including life-threatening infections. Whether patients with MF who undergo HCT are at a higher risk for GVHD is not yet clearly understood. However, theoretically, the inflammatory cytokines that mediate acute GVHD also correlate with the symptom complex in MF [63]. In studies using RIC regimens, the rates of acute GVHD were 27% to 41% for grades II to IV and 11% to 21% for grades III to IV, whereas rates of chronic GVHD were 36% to 51% [35,42,50]. The CIBMTR and European study data also suggested that patients who develop acute GVHD have a lower rate of relapse [42] and higher 3-year survival [35], suggesting the possibility of a concurrent graft-versus-leukemia (MF) effect.
JAK2 V617F status post-transplantation/minimal residual disease
Prognostic indicators after HCT are not well understood in MF, and the role of follow-up for JAK2 V617F mutation status post-transplantation is currently being debated. In 30 patients measurement of JAK2 V617F was followed post-transplantation, and an allele burden of more than 1% was shown to be associated with a higher risk of relapse and poorer overall survival [64]. From the data of 63 patients who had a JAK2 V617F mutation positive at diagnosis, 45 became negative at a median of 96 days (range, 20 to 427), 7 became negative after donor lymphocyte infusion, and 11 never became JAK2 V617F negative [65].JAK2 V617F status was reviewed at 3 and 6 months. Patients with a JAK2 V617F negative status at 3 months after HCT had a decreased incidence of relapse versus patients who were JAK2 positive at 3 months (5% versus 31%, P = .06). Relapses were also higher in patients who stayed JAK2 V617F positive at 6 months versus those who were JAK2 V617F negative at that time (35% versus 5%, P = .03). Assessment of minimal residual disease with repeat cytogenetic testing, JAK2 V617/CAL-R/MPL mutation status, and assessment of donor chimerism is generally recommended.
CONCLUSION
With the advent of novel therapies in the past decade, more treatment options exist for patients with MF. At the same time, overall outcomes from HCT have also improved because of improved conditioning regimens, more advanced antimicrobial therapies, and better GVHD prophylactic regimens. As the only curative option, HCT remains the best hope for long-term remission and relapse-free survival for a select group of patients with MF. At the same time, new therapies with existing and forthcoming JAK inhibitors have shown promise in symptom control, with improved survival shown with ruxolitinib. Long-term outcomes with the JAK inhibitors in development remain to be seen. Patient selection and treatment within the best timeframe for transplant remain critical for best outcomes.
FUTURE DIRECTIONS
Improved understanding of the molecular basis of the disease will help with risk stratification and identifying the appropriate timing for transplant. This will also identify patients who are at high risk of relapse and may benefit from post-transplant treatment to reduce that risk. Another question moving forward will be how to incorporate transplant in the era of evolving non-HCT treatment strategies. These questions require a concerted effort by the transplant community to move forward with prospective, multicenter trials that collect comprehensive data on patients.
ACKNOWLEDGMENTS
Financial disclosure:
T.J.: nothing to disclose. R.A.M.: (Consultant) Novartis, Ariad, Galena; (Research Support) Incyte, Gilead, CTI, Promedior, Celgene. J.M.P.: nothing to disclose.
Footnotes
Conflict of interest statement: There are no conflicts of interest to report.
REFERENCES
- 1.Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med. 2000;342:1255–1265. [DOI] [PubMed] [Google Scholar]
- 2.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113:2895–2901. [DOI] [PubMed] [Google Scholar]
- 3.Mesa RA, Schwager S, Radia D, et al. The Myelofibrosis Symptom Assessment Form (MFSAF): an evidence-based brief inventory to measure quality of life and symptomatic response to treatment in myelofibrosis. Leuk Res. 2009;33:1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood. 2011;118:401–408. [DOI] [PubMed] [Google Scholar]
- 5.Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol. 2012;30:4098–4103. [Erratum in: J Clin Oncol. 2012 Dec 20;30(36):4590.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mesa R, Jamieson C, Bhatia R, et al. Myeloproliferative neoplasms, version 2.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2016;14:1572–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115:1703–1708. [DOI] [PubMed] [Google Scholar]
- 8.Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29:392–397. [DOI] [PubMed] [Google Scholar]
- 9.Tefferi A, Lasho TL, Finke CM, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014;28:1472–1477. [DOI] [PubMed] [Google Scholar]
- 10.Rumi E, Pietra D, Pascutto C, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood. 2014;124:1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27:1861–1869. [DOI] [PubMed] [Google Scholar]
- 12.Tenedini E, Bernardis I, Artusi V, et al. Targeted cancer exome sequencing reveals recurrent mutations in myeloproliferative neoplasms. Leukemia. 2014;28:1052–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vannucchi AM, Guglielmelli P, Rotunno G, et al. Mutation-enhanced International Prognostic Scoring System (MIPSS) for primary myelofibrosis: an AGIMM and IWG-MRT project [abstract]. Blood. 2014;124:405. [Google Scholar]
- 14.Deeg HJ, Gooley TA, Flowers ME, et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood. 2003;102:3912–3918. [DOI] [PubMed] [Google Scholar]
- 15.Cervantes F, Alvarez-Larran A, Hernandez-Boluda JC, et al. Darbepoetin-alpha for the anaemia of myelofibrosis with myeloid metaplasia. Br J Haematol. 2006;134:184–186. [DOI] [PubMed] [Google Scholar]
- 16.Silver RT, Lascu E, Feldman EJ, et al. Recombinant interferon alpha (rIFN) may retard progression of early myelofibrosis by reducing splenomegaly and by decreasing marrow fibrosis. Blood. 2013;122:4053. [Google Scholar]
- 17.Jabbour E, Kantarjian H, Cortes J, et al. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer. 2007;110:2012–2018. [DOI] [PubMed] [Google Scholar]
- 18.Ianotto JC, Boyer-Perrard F, Gyan E, et al. Efficacy and safety of pegylated-interferon α-2a in myelofibrosis: a study by the FIM and GEM French cooperative groups. Br J Haematol. 2013;162:783–791. [DOI] [PubMed] [Google Scholar]
- 19.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–798. [DOI] [PubMed] [Google Scholar]
- 21.Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122:4047–4053. [DOI] [PubMed] [Google Scholar]
- 22.Davis KL, Kaye JA, Cote I, et al. Real-world assessment of clinical outcomes in lower-risk myelofibrosis patients receiving treatment with ruxolitinib [abstract]. Blood. 2014;124:1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mead AJ, Milojkovic D, Knapper S, et al. Response to ruxolitinib in patients with intermediate-1-, intermediate-2-, and high-risk myelofibrosis: results of the UK ROBUST Trial. Br J Haematol. 2015; 170:29–39. [DOI] [PubMed] [Google Scholar]
- 24.Jain T, Mesa R. The development, safety and efficacy of pacritinib for the treatment of myelofibrosis. Expert Rev Anticancer Ther. 2016;16:1101–1108. [DOI] [PubMed] [Google Scholar]
- 25.Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): consensus on terminology by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT). Leuk Res. 2007;31:737–740. [DOI] [PubMed] [Google Scholar]
- 26.Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105:973– 977. [DOI] [PubMed] [Google Scholar]
- 27.Cherington C, Slack JL, Leis J, et al. Allogeneic stem cell transplantation for myeloproliferative neoplasm in blast phase. Leuk Res. 2012;36:1147–1151. [DOI] [PubMed] [Google Scholar]
- 28.Mascarenhas J, Navada S, Malone A, Rodriguez A, Najfeld V, Hoffman R. Therapeutic options for patients with myelofibrosis in blast phase. Leuk Res. 2010;34:1246–1249. [DOI] [PubMed] [Google Scholar]
- 29.Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood. 2010;116:3735–3742. [DOI] [PubMed] [Google Scholar]
- 30.Eghtedar A, Verstovsek S, Estrov Z, et al. Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood. 2012;119:4614–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroger NM, Deeg JH, Olavarria E, et al. Indication and management of allogeneic stem cell transplantation in primary myelofibrosis: a consensus process by an EBMT/ELN international working group. Leukemia. 2015;29:2126–2133. [DOI] [PubMed] [Google Scholar]
- 32.Gupta V, Hari P, Hoffman R. Allogeneic hematopoietic cell transplantation for myelofibrosis in the era of JAK inhibitors. Blood. 2012;120:1367–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ballen KK, Shrestha S, Sobocinski KA, et al. Outcome of transplantation for myelofibrosis. Biol Blood Marrow Transplant. 2010;16:358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tefferi A, Lasho TL,Jimma T, et al. One thousand patients with primary myelofibrosis: the Mayo Clinic experience. Mayo Clin Proc. 2012;87:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kroger N, Holler E, Kobbe G, et al. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2009;114:5264–5270. [DOI] [PubMed] [Google Scholar]
- 36.Alchalby H, Yunus DR, Zabelina T, et al. Risk models predicting survival after reduced-intensity transplantation for myelofibrosis. Br J Haematol. 2012;157:75–85. [DOI] [PubMed] [Google Scholar]
- 37.Atallah E, Horowitz MM, Logan B, et al. Outcome of patients 65 years and older with myelodysplastic syndrome (MDS) receiving allogeneic hematopoietic stem cell transplantation compared to patients 55-64 years of age. Blood. 2015;126:193. [Google Scholar]
- 38.Samuelson S, Sandmaier BM, Heslop HE, et al. Allogeneic haematopoietic cell transplantation for myelofibrosis in 30 patients 60-78 years of age. Br J Haematol. 2011;153:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood. 2005;106:2912–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barba P, Pinana JL, Martino R, et al. Comparison of two pretransplant predictive models and a flexible HCT-CI using different cut off points to determine low-, intermediate-, and high-risk groups: the flexible HCT-CI is the best predictor of NRM and OS in a population of patients undergoing allo-RIC. Biol Blood Marrow Transplant. 2010;16:413–420. [DOI] [PubMed] [Google Scholar]
- 41.Muffly LS, Kocherginsky M, Stock W, et al. Geriatric assessment to predict survival in older allogeneic hematopoietic cell transplantation recipients. Haematologica. 2014;99:1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta V, Malone AK, Hari PN, et al. Reduced-intensity hematopoietic cell transplantation for patients with primary myelofibrosis: a cohort analysis from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant. 2014;20:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ditschkowski M, Elmaagacli AH, Trenschel R, et al. Dynamic International Prognostic Scoring System scores, pre-transplant therapy and chronic graft-versus-host disease determine outcome after allogeneic hematopoietic stem cell transplantation for myelofibrosis. Haematologica. 2012;97:1574–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scott BL, Gooley TA, Sorror ML, et al. The Dynamic International Prognostic Scoring System for myelofibrosis predicts outcomes after hematopoietic cell transplantation. Blood. 2012;119:2657–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kroger N, Giorgino T, Scott BL, et al. Impact of allogeneic stem cell transplantation on survival of patients less than 65 years of age with primary myelofibrosis. Blood. 2015;125:3347–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kroeger N, Panagiota V, Zabelina T, et al. Impact of molecular genetics on disease-free survival in myelofibrosis patients following allogeneic stem cell transplantation. Blood. 2015;126:352. [Google Scholar]
- 47.Stubig T, Alchalby H, Ditschkowski M, et al. JAK inhibition with ruxolitinib as pretreatment for allogeneic stem cell transplantation in primary or post-ET/PV myelofibrosis.Leukemia.2014;28:1736–1738. [DOI] [PubMed] [Google Scholar]
- 48.Shanavas M, Popat U, Michaelis LC, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with myelofibrosis with prior exposure to Janus Kinase 1/2 Inhibitors. Biol Blood Marrow Transplant. 2016;22:432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robin M, Francois S, Huynh A, et al. Ruxolitinib before allogeneic hematopoietic stem cell transplantation (HCST) in patients with myelofibrosis: a preliminary descriptive report of the JAK ALLO study, a phase II trial sponsored by Goelams-FIM in collaboration with the SFGMTC [abstract]. Blood. 2013;122:306.23869074 [Google Scholar]
- 50.Rondelli D, Goldberg JD, Isola L, et al. MPD-RC 101 prospective study of reduced-intensity allogeneic hematopoietic stem cell transplantation in patients with myelofibrosis. Blood. 2014;124:1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luznik L, O’Donnell PV, Symons HJ, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2008;14:641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bregante S, Dominietto A, Ghiso A, et al. Improved outcome of alternative donor transplantations in patients with myelofibrosis: from unrelated to haploidentical family donors. Biol Blood Marrow Transplant. 2016; 22:324–329. [DOI] [PubMed] [Google Scholar]
- 53.Robin M, Giannotti F, Deconinck E, et al. Unrelated cord blood transplantation for patients with primary or secondary myelofibrosis. Biol Blood Marrow Transplant. 2014;20:1841–1846. [DOI] [PubMed] [Google Scholar]
- 54.Guardiola P, Anderson JE, Bandini G, et al. Allogeneic stem cell transplantation for agnogenic myeloid metaplasia: a European Group for Blood and Marrow Transplantation, Societe Francaise de Greffe de Moelle, Gruppo Italiano per il Trapianto del Midollo Osseo, and Fred Hutchinson Cancer Research Center Collaborative Study. Blood. 1999; 93:2831–2838. [PubMed] [Google Scholar]
- 55.Kerbauy DM, Gooley TA, Sale GE, et al. Hematopoietic cell transplantation as curative therapy for idiopathic myelofibrosis, advanced polycythemia vera, and essential thrombocythemia. Biol Blood Marrow Transplant. 2007;13:355–365. [DOI] [PubMed] [Google Scholar]
- 56.Patriarca F, Bacigalupo A, Sperotto A, et al. Allogeneic hematopoietic stem cell transplantation in myelofibrosis: the 20-year experience of the Gruppo Italiano Trapianto di Midollo Osseo (GITMO). Haematologica. 2008;93:1514–1522. [DOI] [PubMed] [Google Scholar]
- 57.Stewart WA, Pearce R, Kirkland KE, et al. The role of allogeneic SCT in primary myelofibrosis: a British Society for Blood and Marrow Transplantation study. Bone Marrow Transplant. 2010;45:1587–1593. [DOI] [PubMed] [Google Scholar]
- 58.Gupta V, Kroger N, Aschan J, et al. A retrospective comparison of conventional intensity conditioning and reduced-intensity conditioning for allogeneic hematopoietic cell transplantation in myelofibrosis. Bone Marrow Transplant. 2009;44:317–320. [DOI] [PubMed] [Google Scholar]
- 59.Robin M, Porcher R, Wolschke C, et al. Outcome after transplantation according to reduced-intensity conditioning regimen in patients undergoing transplantation for myelofibrosis. Biol Blood Marrow Transplant. 2016;22:1206–1211. [DOI] [PubMed] [Google Scholar]
- 60.Slot S, Smits K, van de Donk NW, et al. Effect of conditioning regimens on graft failure in myelofibrosis: a retrospective analysis. Bone Marrow Transplant. 2015;50:1424–1431. [DOI] [PubMed] [Google Scholar]
- 61.Robin M, Tabrizi R, Mohty M, et al. Allogeneic haematopoietic stem cell transplantation for myelofibrosis: a report of the Societe Francaise de Greffe de Moelle et de Therapie Cellulaire (SFGM-TC). Br J Haematol. 2011;152:331–339. [DOI] [PubMed] [Google Scholar]
- 62.Klyuchnikov E, Holler E, Bornhauser M, et al. Donor lymphocyte infusions and second transplantation as salvage treatment for relapsed myelofibrosis after reduced-intensity allografting. Br J Haematol. 2012;159:172–181. [DOI] [PubMed] [Google Scholar]
- 63.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29:1356–1363. [DOI] [PubMed] [Google Scholar]
- 64.Lange T, Edelmann A, Siebolts U, et al. JAK2 p.V617F allele burden in myeloproliferative neoplasms one month after allogeneic stem cell transplantation significantly predicts outcome and risk of relapse. Haematologica. 2013;98:722–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alchalby H, Badbaran A, Zabelina T, et al. Impact of JAK2V617F mutation status, allele burden, and clearance after allogeneic stem cell transplantation for myelofibrosis. Blood. 2010;116:3572–3581. [DOI] [PubMed] [Google Scholar]