

Abstract
Summary: A 5-year-old girl with no preexisting systemic or CNS neoplasm presented with a large right temporal mass lesion, the histopathology of which proved to be a poorly differentiated adenosquamous carcinoma, a highly unusual primary intracranial tumor. The tumor recurred despite radical resection, chemotherapy, and radiation therapy.
Malignant transformation of benign intracranial dysembryogenetic tumors is known to occur infrequently. A primary intracranial squamous cell carcinoma that develops in the absence of a preexisting tumor is extremely rare, previously reported in only a few adult patients (1). We describe the imaging findings associated with this very rare primary intracranial tumor arising de novo in a child.
Case Report
A 5-year-old left-handed girl presented with a 2-month history of intermittent headache that was associated with nausea and vomiting during the week before diagnosis. The patient had no history of fever, otorrhea, seizures, visual complaints, or other neurologic symptoms. Her neurodevelopmental history was normal. Physical examination revealed that the vital signs were normal and that the patient had no skin lesions suggestive of any neurocutaneous syndrome. A neurologic examination revealed that her mental status, cranial nerve function, funduscopic findings, sensory function, and gait all were normal. The results of a motor examination were normal except for the presence of a subtle left pronator drift.
Contrast-enhanced CT of the head showed a large, ill-defined, heterogeneously enhancing mass containing calcification and cystic areas, located in the posterior right temporal lobe and extending to the petrous apex. Dedicated temporal bone CT showed minimal bone remodeling along the petrous apex (Fig 1A) and bilateral mastoid air cell opacification but no bone destruction.
Fig 1.

Images from the case of a 5-year-old left-handed girl who presented with a 2-month history of intermittent headache that was associated with nausea and vomiting during the week before diagnosis.
A, Coronal CT bone window scan shows a large, ill-defined, intra-axial mass located in the right temporal lobe (arrowheads) with remodeling of the tegmen tympani (arrows) but no direct invasion of the temporal bone.
B, Axial T2-weighted MR image shows a heterogeneous mass in the right temporal lobe.
C, Coronal contrast-enhanced T1-weighted MR image shows a heterogeneously enhancing mass with mild dural enhancement (arrowheads) but no extension into the underlying petrous temporal bone.
Contrast-enhanced MR imaging of the brain confirmed the large heterogeneously enhancing intra-axial mass that contained solid and cystic components and was associated with surrounding edema (Fig 1B and C). It occupied most of the posterior right temporal lobe extending anteriorly almost to the temporal pole. The mass extended medially to the basal ganglia. Dural enhancement was noted along the petrous apex, but no temporal bone invasion could be seen. 2D chemical shift imaging multivoxel spectroscopy performed by using a 1.5-T magnet showed the lack of N-acetylaspartate peak, increased lactate, and slightly elevated choline:creatine ratio within the mass, suggestive of a neoplasm. The differential diagnoses based on the imaging characteristics were considered to include primitive neuroectodermal tumor, ependymoma, and high-grade glioma.
The patient underwent right temporal craniotomy for radical resection of the tumor, which was grossly observed to include solid regions of dense fibrous tissue and cystic pockets containing clear brown fluid. The lesion involved the underlying dura but did not invade the temporal bone. Histologically, the neoplasm consisted of cohesive nests and sheets of poorly differentiated malignant epithelial cells with minimal glandular differentiation. Cytologic features of the malignant cells included nuclear pleomorphism and hyperchromasia, a few prominent nucleoli, and clear to eosinophilic cytoplasm. Tumor cells expressed abundant cytokeratin 7, CAM 5.2, focal keratin 903, and carcinoembryonic antigen (Fig 2). The tumor contained scattered mitotic figures and a very high Mib-1 proliferation index (>90% in some microscopic fields). It had regions of necrosis, desmoplasia, calcification, cholesterol clefts, and hemosiderin deposition from previous hemorrhage. The margin of tumor in brain was distinct and well circumscribed. The results of a metastatic workup, including CT of the chest, were negative; thus, the final pathologic finding was consistent with a primary intracranial malignant neoplasm with a prominent squamous cell component, similar to a primary squamous cell carcinoma or a malignant craniopharyngioma. Considering its intra-axial location in the temporal lobe, the lesion was thought to be most consistent with a primary intracranial squamous cell carcinoma.
Fig 2.
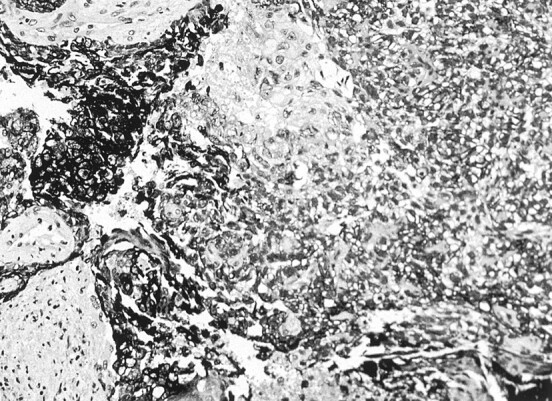
Carcinoma cells express CAM 5.2 low molecular weight cytokeratin as dark cytoplasmic staining. Negative zones of desmoplasia flank the darkest carcinoma cells.
The child was treated with four cycles of regular dose multi-agent chemotherapy with cisplatin, VP-16, and ifosfamide and two cycles of dose-intensified chemotherapy with Cytoxan and carboplatin supported by autologous stem cells. Radiation therapy was then administered to a wide involved field. Three months after therapy and 10 months after diagnosis, the tumor progressed locally, with the appearance of enhancing nodules at the edge of the residual resection cavity, for which the patient again underwent tumor debulking and stereotactic radiosurgery. Further local tumor progression was confirmed by follow-up MR imaging. At the time of this writing, 15 months after initial diagnosis, the patient was undergoing salvage adjuvant chemotherapy.
Discussion
Most the squamous cell carcinomas that occur in the CNS are metastases from other organs or result from direct invasion from a primary head and neck malignancy. Primary intracranial squamous cell carcinomas are very rare, and it seems that most arise from preexisting benign intracranial dysembryogenetic tumors, such as epidermoid or dermoid cysts, and craniopharyngiomas. They may also rarely arise as areas of metaplastic transformation in glioblastomas. The present case of an intracranial squamous cell carcinoma that seems to have arisen de novo in the absence of a preexisting tumor is, to our knowledge, only the fifth such reported case and is the first such pediatric case to be reported (1).
Dysembryogenetic tumors can be classified into three main categories: 1) tumors derived from embryonal cell rests in situ, such as craniopharyngiomas, Rathke’s cleft cysts, or chordomas; 2) tumors derived from embryonal cells straying into the tissue (“verirrte Keime”), such as so-called germ cell tumors, dermoid or epidermoid cysts, lipomas, or hamartomas; and 3) included twin (fetus-in-fetu) tumors. The pathogenesis hypothesized for the second group is that the embryonic cells at various stages of embryogenesis stray into the bilaminar embryonic disk at the time of primitive streak formation during the third week of gestation, join the stream of lateral mesoderm that gets incorporated into the neural plate, and thus become incorrectly enfolded into the brain at the time of neural tube formation (2). These misplaced cells, if not killed by the body’s normal immune mechanisms, may later develop into various types of tumors. The third group of included twin tumors is hypothesized to arise from the sequestration of blastocyst cells, before differential blocking of the genome has occurred (2).
Malignant transformation of some benign intracranial dysembryogenetic tumors, such as epidermoid or dermoid cysts and craniopharyngiomas, is a rare but known phenomenon with no proved explanation. It has been postulated that a prolonged or reparative process, particularly with the introduction of foreign material, leads to development of cellular atypia and eventual neoplasia in epidermoid or dermoid cysts (3, 4), whereas irradiation has been proposed as a carcinogenic factor in craniopharyngiomas (5). Squamous cell carcinomas are reported to arise in intracranial epidermoid cysts undergoing malignant transformation. These seem to result from malignant change in the squamous epithelial lining of the normally thin-walled cysts, which become thickened and nodular with the neoplastic squamous cell division, forming a solid and infiltrative tumor (3, 6). Malignant change in extracranial dermoid cysts has been described to occur in its constituent structures, such as squamous epithelium, sebaceous glands, or hair follicles. Most (approximately 75%) are squamous cell carcinomas, and 15% are adenocarcinomas (7, 8). Intracranial dermoids have been reported to give rise to squamous cell carcinomas in adult patients (3, 4).
Histopathology in the present case shows squamous cell predominance with some glandular differentiation, most suggestive of a poorly differentiated squamous cell carcinoma with an adenomatous component. This is consistent with the possibility that the tumor developed in a preexisting epidermoid or dermoid cyst. The histologically and radiologically confirmed calcifications in the tumor would favor the latter because of the greater likelihood of calcifications occurring in a dermoid. However, the absence of a known preexisting tumor makes it impossible to confirm the hypothesis that the tumor arose from either type of the cyst. An alternative hypothesis for pathogenesis of the tumor is that it represents a malignant teratoma, a tumor that has been proposed to arise de novo, like benign teratomas and dermoid or epidermoid cysts, from stray embryonal cells mistakenly enfolded into the brain during embryogenesis, as described above. However, malignant teratomas contain representative tissue from all three embryonic germ cell layers. The tumor in our case had only two—a squamous cell component from ectoderm and an adenocarcinoma from mesoderm—but no tissue representing endoderm. It is possible that a sampling error or an unusual variant that simply failed to develop completely could account for this histologic appearance.
The following criteria have been proposed to define primary intracranial squamous cell carcinomas: 1) the tumor must be restricted to the intracranial, intradural compartment without invasion of, or extension beyond, the dura or cranial bones; 2) there must be no extension or invasion through intracranial orifices; 3) there must be no communication or connection with the middle ear, air sinuses, or sella turcica; and 4) there must be no evidence of a nasopharyngeal tumor (9). In the present case, although there was dural invasion, as shown on images and at surgery, we think that the neoplasm was primarily intracranial, because the center of the mass lesion was in the temporal lobe with no invasion of the underlying petrous apex or the mastoid air cells. Partial opacification of the bilateral middle ear cavities seen on CT scans and MR images is probably incidental mucosal thickening and is not related to the primary lesion. Moreover, a thorough examination did not reveal an extracranial primary site.
Conclusion
Most intracranial squamous cell carcinomas are either metastases or direct invasions of head and neck carcinomas, and most primary intracranial squamous cell carcinomas arise from preexisting neoplasms, such as dermoids, epidermoids, and craniopharyngiomas. We think that the present case of a de novo intracranial squamous cell carcinoma and the cases previously reported in the literature are most likely to have developed in preexisting tumors that remained asymptomatic and undiagnosed until they underwent malignant transformation.
References
- 1.Murase S, Yamakawa H, Ohkuma A, et al. Primary intracranial squamous cell carcinoma. Neurol Med Chir (Tokyo) 1999;39:49–54 [DOI] [PubMed] [Google Scholar]
- 2.Sano K. Intracranial dysembryogenetic tumors: pathogenesis and their order of malignancy. Neurosurg Rev 2001;24:162–167 [DOI] [PubMed] [Google Scholar]
- 3.Nishio S, Takeshita I, Morioka T, Fukui M. Primary intracranial squamous cell carcinomas: report of two cases. Neurosurgery 1995;37:329–332 [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi T, et al. A squamous cell carcinoma originated from intracranial dermoid cyst. Neurochirurgia 1993;36:26–29 [DOI] [PubMed] [Google Scholar]
- 5.Kristopaitis T, Thomas C, Petruzzelli GJ, Lee JM. Malignant craniopharyngioma. Arch Pathol Lab Med 2000;124:1356–1360 [DOI] [PubMed] [Google Scholar]
- 6.Giangaspero F, Manetto V, Ferracini R, Piazza G. Squamous cell carcinoma of the brain with sarcoma-like stroma. Virchows Arch A Pathol Anat Histopathol 1984;402:459–464 [DOI] [PubMed] [Google Scholar]
- 7.Klionsky BL, Nickens O, Amortegui AJ. Squamous cell carcinoma in situ arising in adult cystic teratoma of the ovary. Arch Pathol 1972;93:161–163 [PubMed] [Google Scholar]
- 8.Peterson WF. Malignant degeneration of benign cystic teratomas of the ovary: a collective review of the literature. Obstet Gynecol Surv 1957;12:793–830 [DOI] [PubMed] [Google Scholar]
- 9.Garcia CA, McGarry PA, Rodriguez F. Primary intracranial squamous cell carcinoma of the right cerebellopontine angle. J Neurosurg 1981;54:824–828 [DOI] [PubMed] [Google Scholar]