

Abstract
Glasdegib is a potent and selective oral inhibitor of the Hedgehog pathway. We report data from the single-arm, lead-in cohort of an open-label phase lb/2 trial of glasdegib in patients with primary/secondary myelofibrosis (MF) previously treated with at least one Janus kinase inhibitor (JAKi). Patients received glasdegib 100 mg orally once daily until there was no further clinical benefit. Primary endpoints included adverse events (AEs). Secondary endpoints included patients with spleen volume reduction (SVR) ≥35% at week 24, patients with ≥50% total symptom score (TSS) reduction, and pharmacokinetics. All 21 treated patients had one or more AE and five (23.8%) had serious AEs. Most common (> 30%) AEs were dysgeusia (61.9%), muscle spasms (57.1%), alopecia (38.1%), fatigue (33.3%), and decreased appetite (33.3%). Although no patient had ≥35% SVR at week 24, one patient previously treated with ruxolitinib had an SVR of 32.9%. At week 12, two (9.5%) patients had ≥50% reduction in TSS from baseline and ~40% had ≥20% reduction. One patient had an anaemia response. Following administration of glasdegib 100 mg once daily, the median time to peak plasma concentrations at steady-state generally occurred at 1 h post-dose. The safety profile of glasdegib monotherapy was manageable in patients with primary/secondary MF. Further study of glasdegib in combination with JAKi in a MF population may be warranted.
Keywords: Glasdegib, Hedgehog inhibitor, Smoothened inhibitor, Myelofibrosis
1. Introduction
Primary and secondary (i.e., post-polycythemia vera and post-essential thrombocythemia) myelofibrosis (MF) is characterised by a clonal proliferation of myeloid hematopoietic cells leading to bone marrow fibrosis [1,2]. Clinical features include abnormal blood counts, hepatosplenomegaly, bleeding, thrombosis, and debilitating constitutional symptoms (e.g., fatigue, weight loss, night sweats, fever, pruritus, bone pain, abdominal pain, and early satiety) [2].
Ruxolitinib, a selective inhibitor of Janus kinase (JAK) 1 and JAK2, is approved for first-line treatment of patients with intermediate- or high-risk MF. Ruxolitinib can reduce spleen volume and ameliorate disease-related symptoms [3,4]; however, significant clinical benefit is not seen in all patients. In the COMFORT trials [4]. The proportion of patients with spleen volume reduction (SVR) ranged between 28–42%, and improvement ≥50% in total symptom score (TSS) occurred in 46% of patients. Pooled data from these studies suggest ruxolitinib prolongs survival in some patients, but lacks curative potential [5–7]. Regulatory-approved treatment options following ruxolitinib treatment failure are lacking; hence, second-line therapy for MF is a significant unmet need.
Translational studies have shown a 20- to 100-fold increase in expression of target genes of the Hedgehog (Hh) signalling pathway, including glioma-associated oncogene (GLI1) and protein-patched homolog 1 (PTCH1), in granulocytes isolated from patients with MF [8]. In a preclinical mouse model of MF, inhibition of the Hh pathway combined with ruxolitinib demonstrated greater reduction of mutant allele burden, reduced bone marrow fibrosis, and lowered white blood cells and platelet count compared with ruxolitinib alone [8].
Glasdegib is a potent and selective oral inhibitor of Hh pathway activity that acts by binding to Smoothened (SMO), a G-protein-coupled receptor, and blocking signal transduction. The United States Food and Drug Administration recently approved glasdegib in combination with low-dose cytarabine for the treatment of newly diagnosed acute myeloid leukaemia (AML) in adult patients who are ≥75 years old or who have comorbidities that preclude the use of intensive induction chemotherapy. This study evaluated safety and tolerability of glasdegib in an open-label, lead-in cohort of patients with MF previously treated with at least one JAK inhibitor (JAKi), followed by a phase 2, randomized portion. Herein, we report data from the lead-in portion of this phase 1b/2 trial.
2. Material and methods
2.1. Study design
This open-label, lead-in study in patients with primary or secondary MF previously treated with one or more JAKi was conducted at ten centres in Japan and the United States. The following were collected at screening: date of diagnosis; Dynamic International Prognosis Scoring System [9] risk category at enrolment; complete history, best response, and reason for discontinuation of prior MF therapies; and events leading to intolerance of prior MF therapies, including severity and outcome of event(s).
The primary objective of the lead-in cohort was to assess safety and tolerability of glasdegib in patients with primary or secondary MF previously treated with one of more JAKi. Secondary objectives included SVR assessed by magnetic resonance imaging (MRI) or computed tomography (CT) following 24 weeks of treatment, proportion of patients with ≥50% reduction in TSS at week 24 measured by the Myeloproliferative Neoplasm Symptom Assessment Diary (MPN-SAD), haematologic improvement (i.e., peripheral blood), and pharmacokinetics (PK) of glasdegib. Exploratory objectives included patient-reported, health-related quality of life (HRQoL) and overall health status, reduction of JAK2 V617F mutant allele burden (in patients with JAK2 V617F-mutant MF), and bone marrow and blood-based molecular markers of response and resistance to glasdegib.
This study was approved by the Institutional Review Board and/or Independent Ethics Committee at each investigational centre and was conducted in compliance with the Declaration of Helsinki and all International Council for Harmonization Good Clinical Practice Guidelines. All patients provided informed consent. The study is registered at ClinicalTrials.gov (NCT02226172).
2.2. Patients
Eligible patients were aged ≥18 years with a diagnosis of primary or secondary MF defined by World Health Organization 2008 criteria [10]. Patients were previously treated with one or more JAKi (licensed or experimental) for ≥4 weeks and had failed to achieve or sustain adequate symptom control and/or adequate reduction of splenomegaly based on investigator discretion, or had JAKi therapy discontinuation for unacceptable toxicity irrespective of therapy duration. Also, patients had to have spleen volume ≥5 cm below the inferior left costal margin, measured by manual palpation, and severe MF symptoms (per investigator judgment). Patients were excluded if they had prior treatment with a licensed or experimental SMO inhibitor or splenic irradiation ≤3 months prior to enrolment.
2.3. Treatment
Patients received open-label oral glasdegib 100 mg once daily for up to 24 weeks. Patients could remain on treatment for as long as they tolerated it and derived clinical benefit. To manage safety and tolerability, dose reduction to 75 mg or 50 mg once daily or alternative dosing schedules were explored. Patients were followed-up for at least 12 weeks after their last glasdegib dose.
2.4. Assessments
2.4.1. Safety
Safety assessments included recording of adverse events (AEs), their type, incidence, severity (graded by the National Cancer Institute Common Terminology Criteria for Adverse Events v4.03), timing, seriousness, and relationship to study drug. Other assessments included red blood cells and platelet transfusions (number of units) during treatment.
2.4.2. Efficacy
To measure spleen volume, abdominal MRI (or CT, if MRI was contraindicated) was completed at baseline, every three cycles, and at confirmation of progression. Imaging studies were evaluated for change from baseline in spleen and liver volume by a blinded independent central review.
To assess symptom burden, patients completed the MPN-SAD daily via an electronic handheld device (eDiary) beginning at least 7 days before cycle 1 day 1 and continuing through cycle 7 day 1 (a total of 25 weeks). Thereafter, patients recorded symptoms weekly until treatment discontinuation and then monthly until study discontinuation. MPN-SAD is a ten-item instrument that assesses MF symptoms including early satiety, abdominal discomfort, inactivity, problems with concentration, night sweats, pruritus, bone pain, fever (> 37.8 °C), unintentional weight loss, and fatigue.
The key secondary efficacy endpoint was symptom improvement, defined as achieving ≥50% reduction from baseline in TSS at week 24, measured by the MPN-SAD. TSS at week 24 was defined as the average of daily total scores from the 28 days of symptom scores immediately prior to week 24 (cycle 7 day 1). Disease response, measured from baseline through end of treatment, was evaluated using Revised International Working Group for Myelofibrosis Research and Treatment Response Criteria.
An ad hoc analysis was conducted on a subset of patients with severe symptom burden at baseline, defined as MPN-SAD severity score ≥5 (on 0–10 scale) on at least one item, or a severity score ≥3 on at least two of the following: fatigue, early satiety, abdominal discomfort, inactivity, night sweats, pruritus, and bone pain.
2.4.3. Patient-reported outcomes
Other patient-reported outcomes included HRQoL assessed by the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLQ-C30) [11], Patient Global Impression of Change (PGIC), and overall health status assessed by the EuroQol-5 Dimension Questionnaire, 5-level version (EQ-5D-5L) [12].
2.4.4. Pharmacokinetics
Blood samples (1.5 ml whole blood, sufficient to provide a minimum of 0.6 ml of plasma) were collected for PK analysis of glasdegib at 0.25, 1, 2, and 4 h postdose on cycle 1 day 1; at predose, 0.25, 0.5,1, 2, 4, and 6 h postdose on cycle 1 day 15; at 24 h postdose on cycle 1 day 16; and at predose and 1 and 4h postdose on day 1 of cycles 2 and 3. The predose PK sample was collected within 30 min before administration of glasdegib to capture trough levels accurately. Plasma samples were analysed for glasdegib concentrations using a validated, sensitive, and specific high-performance liquid chromatography-tandem mass spectrometric method.
Glasdegib PK parameters were calculated for each patient using non-compartmental analysis of steady state plasma concentration–time data and included maximum observed plasma concentration (Cmax); area under the concentration–time curve (AUC) from time zero to time τ; the dosing interval (AUCtau), where τ = 24 h for once-daily dosing; lowest concentration observed during the dosing interval τ; and, if measured at end of dosing interval, equivalent to trough plasma concentration and average concentration at steady-state (calculated as AUCtau/τ).
3. Results
3.1. Patients
A total of 21 patients were screened and treated with glasdegib. Patient demographic and baseline characteristics are presented in Table 1. Fifteen (71.4%) patients received one prior JAKi therapy; three (14.3%) received two prior JAKi therapies, and three (14.3%) received three or more prior JAKi therapies. The most common reason for discontinuing prior JAKi therapy was inadequate response/primary resistance (n = 11 [52.4%]), followed by toxicity and “other” (n = 8 [38.1%], each).
Table 1.
Patient demographic and baseline characteristics.
| Parameter, n (%) | Glasdegib lead-in N = 21 |
|---|---|
| Body mass index,* kg/m2 | |
| Mean (SD) | 26.6 (4.1) |
| Range | 20.8–38.2 |
| Number of prior JAKi therapies | |
| 1 | 15 (71.4) |
| 2 | 3 (14.3) |
| ≥3 | 3 (14.3) |
| Ruxolitinib only | 14 (66.7) |
| DIPSS risk category | |
| Intermediate 1 | 2 (9.5) |
| Intermediate 2 | 15 (71.4) |
| High risk | 4 (19.0) |
| Reason for stopping prior JAKi therapy† | |
| Inadequate response / primary resistance | 11 (52.4) |
| Disease progression / acquired resistance | 3 (14.3) |
| Toxicity | 8 (38.1) |
| Other | 8 (38.1) |
| Not reported | 4 (19.0) |
| Duration of treatment of prior JAKi, days | |
| Mean (SD) | 709.9 (647.2) |
| Range | 77–2600 |
| Response to last prior JAKi treatment | |
| Complete remission | 0 |
| Partial remission | 1 (4.8) |
| Clinical improvement | 10 (47.6) |
| Stable disease | 3 (14.3) |
| Not reported | 7 (33.3) |
| Best response to prior JAKi treatment‡ | |
| Complete remission | 0 |
| Partial remission | 2 (9.5) |
| Clinical improvement | 13 (61.9) |
| Stable disease | 2 (9.5) |
| Not reported | 4 (19.0) |
| Transfusion independent | |
| Yes | 17 (81.0) |
| No | 4 (19.0) |
DIPSS = Dynamic International Prognostic Scoring System; JAKi = Janus kinase inhibitor; SD = standard deviation.
Body mass index is defined as weight / (height × 0.01)2.
Patients may have more than one reason for stopping prior JAKi therapies.
Best response to prior therapy includes all responses prior to start of study therapy.
As of the data cut-off, December 2016, eight patients completed the study, ten patients discontinued from the study, and three patients were still receiving study treatment. Eighteen (85.7%) patients discontinued treatment due to treatment-related AEs (n = 10), insufficient clinical response (n = 3), progressive disease (n = 2), global deterioration of health status (n = 1), patient refused further follow-up (n = 1), and an AE unrelated to study drug (n = 1).
3.2. Safety
Median duration of glasdegib treatment was 85.0 days (range, 22–512 days), median daily dose was 96.0 mg (range, 61–101 mg), and median relative dose intensity was 97.0% (range, 60–100%). Six (28.6%) patients had a dose reduction and five (23.8%) patients experienced at least one treatment delay (i.e., delay of the start of a cycle); median duration of delay was 8.0 days (range, 6–11 days). Ten (47.6%) patients had at least one dose interruption (i.e., missing a total of ≥6 doses of glasdegib within a cycle); median duration of interruption was 15.0 days (range, 8–29 days). Four patients received more than six cycles of glasdegib, three of whom were ongoing patients at the data cutoff date. The fourth patient received seven cycles of glasdegib. All 21 patients had one of more treatment-emergent AE (TEAE); 190 TEAEs were reported. Fourteen (66.7%) patients had grade 3/4 TEAEs and one (4.8%) patient had a grade 5 TEAE of respiratory failure. All-causality TEAEs experienced by > 10% of patients are presented in Table 2. The most common treatment-related TEAEs were dysgeusia (61.9%), muscle spasms (52.4%), alopecia (38.1%), decreased appetite (33.3%), and fatigue (23.8%).
Table 2.
Treatment-emergent all-causalities adverse events reported by > 10% of patients.
| Adverse event, n (%) | Glasdegib lead-in (N = 21) |
||||
|---|---|---|---|---|---|
| All Grades* | Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
| Dysgeusia | 13 (61.9) | 9 (42.9) | 4 (19.0) | 0 | 0 |
| Muscle spasms | 12 (57.1) | 5 (23.8) | 5 (23.8) | 2 (9.5) | 0 |
| Alopecia | 8 (38.1) | 5 (23.8) | 3 (14.3) | 0 | 0 |
| Decreased appetite | 7 (33.3) | 6 (28.6) | 1 (4.8) | 0 | 0 |
| Fatigue | 7 (33.3) | 1 (4.8) | 5 (23.8) | 1 (4.8) | 0 |
| Lipase increased | 5 (23.8) | 0 | 0 | 4 (19.0) | 1 (4.8) |
| Weight decreased | 5 (23.8) | 2 (9.5) | 3 (14.3) | 0 | 0 |
| Anaemia | 4 (19.0) | 0 | 1 (4.8) | 2 (9.5) | 1 (4.8) |
| Hyperuricaemia | 4 (19.0) | 2 (9.5) | 0 | 0 | 2 (9.5) |
| Nausea | 4 (19.0) | 4 (19.0) | 0 | 0 | 0 |
| Pyrexia | 4 (19.0) | 2 (9.5) | 2 (9.5) | 0 | 0 |
| Asthenia | 3 (14.3) | 1 (4.8) | 1 (4.8) | 1 (4.8) | 0 |
| Constipation | 3 (14.3) | 1 (4.8) | 1 (4.8) | 1 (4.8) | 0 |
| cough | 3 (14.3) | 3 (14.3) | 0 | 0 | 0 |
| Dehydration | 3 (14.3) | 2 (9.5) | 1 (4.8) | 0 | 0 |
| Electrocardiogram QT prolonged | 3 (14.3) | 1 (4.8) | 2 (9.5) | 0 | 0 |
| Lymphocyte count decreased | 3 (14.3) | 0 | 0 | 2 (9.5) | 1 (4.8) |
| Myalgia | 3 (14.3) | 3 (14.3) | 0 | 0 | 0 |
| Pain in extremity | 3 (14.3) | 2 (9.5) | 1 (4.8) | 0 | 0 |
| Thrombocymiddleenia | 3 (14.3) | 0 | 3 (14.3) | 0 | 0 |
| Upper respiratory tract infection | 3 (14.3) | 1 (4.8) | 1 (4.8) | 1 (4.8) | 0 |
Grade 5 adverse event of respiratory failure occurred in one patient throughout the study.
Twelve (57.1%) patients permanently discontinued study treatment due to TEAEs, mostly due to muscle spasms (n = 6: one grade 3; four grade 2; one grade 1) and dysgeusia (n = 3). One of these twelve patients discontinued treatment due to grade 1 pyrexia; however, relatedness was not entered into the case report form, and the site recorded the reason for discontinuation as progressive disease. A notable number of discontinuations due to muscle spasms occurred at a single site (n = 5 of 6). No concurrent creatine kinase increases to suggest rhab-domyolysis were reported during the study. Per protocol, patients experiencing muscle spasms or myalgia were advised to consume oral rehydration solutions or salts containing electrolytes.
One (4.8%) patient had a dose reduction due to an AE, four (19.0%) patients had a temporary discontinuation due to AEs, and four (19.0%) patients had both.
No patients died within 28 days of the last dose of glasdegib. During the follow-up period, one patient died due to disease progression, and one patient died due to unknown cause. Five (23.8%) patients experienced serious AEs (SAEs), including one patient each: memory impairment and fatigue; gastric varices haemorrhage, portal hypertension, respiratory failure, and oesophageal varices; mental status change; failure to thrive; and anaemia. No SAE was reported in more than one patient. Most SAEs were grade 2 or 3; one patient each had a grade 4 gastric variceal haemorrhage and a grade 5 SAE of respiratory failure, both judged to be unrelated to study treatment. Two patients had treatment-related SAEs (fatigue, memory impairment, and mental status change).
Grade 3 haematological toxicities included: anaemia (n = 1), lymphopenia (n = 3), neutropenia (n = 1), and leukopenia (n = 1). One patient each had grade 4 lymphopenia and neutropenia. Grade 3 and 4 hyperlipasemia was seen in five and one patients, respectively.
3.3. Efficacy
On physical examination, patients with ≥50% decrease in spleen size were reported at cycle 4 day 1 (n = 1) and at end of treatment (n = 2). Median change from baseline in spleen size was −28.6% (range, −100% to 0%; Fig. 1A). On MRI/CT, mean spleen volume at week 24 was increased from baseline for the six patients evaluated. Mean (standard deviation [SD]) absolute change from baseline was 116.7 cm3 (148.7), and mean (SD) percent change from baseline was 8.8% (9.0%). The best post-baseline spleen measurements over time by MRI/CT are presented in Fig. 1B. Based on MRI/CT assessment, no patient had an SVR ≥35% at any time during study.
Fig. 1.
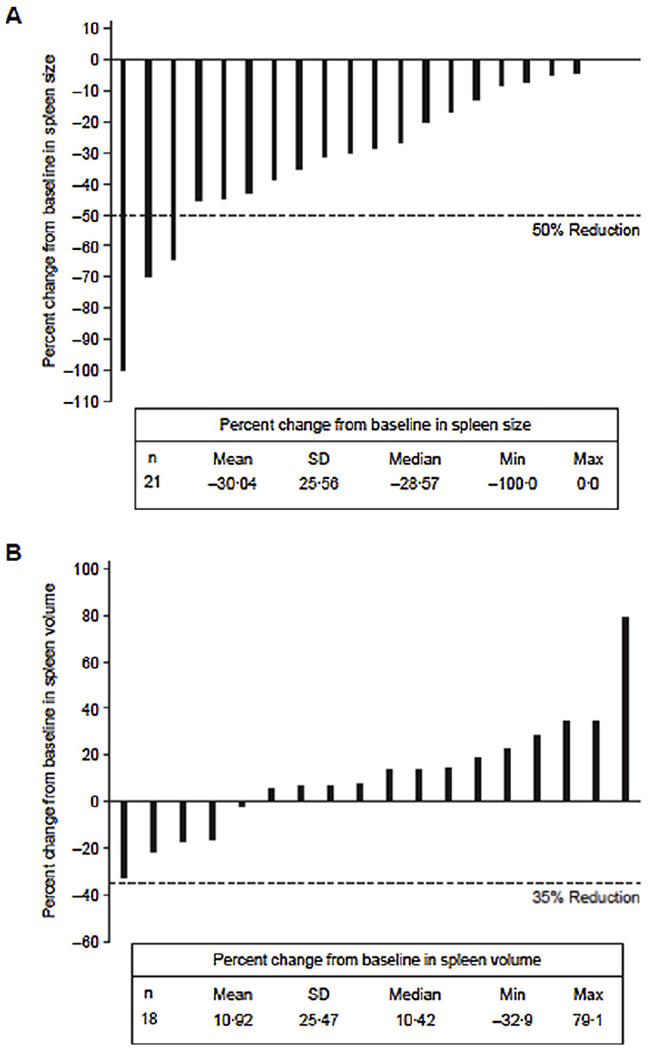
Percentage change from baseline in spleen volume. Best post-baseline spleen measurements by (A) physical examination and (B) MRI/CT. max = maximum; min = minimum; MRI/CT = magnetic resonance imaging/computed tomography; SD = standard deviation.
A 10.6% and 7.6% median decrease from baseline in spleen size based on MRI/CT assessment was reported at weeks 36 and 48, respectively, for the three patients still receiving glasdegib at these time points. These patients had a relatively short duration of prior therapy. Notably, one patient had an SVR of 32.9%; this patient was previously treated with five doses of ruxolitinib. A second patient had splenomegaly and spleen volume stabilised; this patient was previously treated with two doses of ruxolitinib. In a third patient, spleen volume stabilised; this patient was previously treated with four doses of an experimental medication INC424.
One (5.9%) patient had anaemia response, defined as ≥20 g/l increase in haemoglobin level wherein baseline haemoglobin level was < 100 g/l, or transfusion-dependent patients becoming transfusion-independent. This patient also had splenomegaly and thrombocytopenia; prior treatments included ruxolitinib and fedratinib (SAR 302503). On day 1, baseline haemoglobin was 97 g/l. On day 92, haemoglobin was 117 g/l (reference range, 117–155 g/l). On day 99, haemoglobin was 105 g/l and no longer constituted an anaemia response. The patient permanently discontinued treatment due to muscle spasms and dizziness in cycles 3 through 6; the patient’s last dose was on day 169.
3.4. Symptom score and patient-reported outcomes
Baseline symptom scores on MPN-SAD were low and most averaged < 4 (i.e., mild) with minimal severity (Table 3). Fatigue was the only symptom with an average score > 4 at baseline. Baseline symptom scores in the severe subset (n = 14) were higher than those in the entire lead-in cohort: fatigue remained the worst symptom, followed by inactivity and spleen-related symptoms of early satiety and abdominal discomfort (Table 3).
Table 3.
Baseline MPN-SAD symptom scores*.
| Leadin (N = 21) |
Severe Subset† (n = 14) |
|||
|---|---|---|---|---|
| Mean | Median | Mean | Median | |
| Bone pain | 1.87 | 1.00 | 2.71 | 2.36 |
| Problems with concentration | 2.52 | 1.86 | 3.36 | 3.29 |
| Abdominal discomfort | 3.65 | 2.86 | 4.73 | 5.00 |
| Fatigue | 4.34 | 4.86 | 5.84 | 5.64 |
| Fever (> 37.8 °C) | 0.37 | 0.00 | 0.56 | 0.00 |
| Early satiety | 3.69 | 3.86 | 4.74 | 4.93 |
| Inactivity | 3.45 | 3.29 | 4.76 | 5.07 |
| Pruritus | 1.51 | 0.86 | 1.83 | 1.14 |
| Night sweats | 1.95 | 1.14 | 2.79 | 3.36 |
| Unintentional weight loss | 0.66 | 0.14 | 0.83 | 0.36 |
| Spleen-related symptom score‡ | 7.33 | 6.86 | 9.48 | 9.86 |
| Constitutional symptom score§ | 9.67 | 7.71 | 13.16 | 14.07 |
| Total symptom score | 24.00 | 24.86 | 32.14 | 30.36 |
| nTssǁ | 17.00 | 17.43 | 22.64 | 22.00 |
MPN-SAD = Myeloproliferative Neoplasm Symptom Assessment Diary; nTSS = new total symptom score.
Large values correspond to worse symptoms.
Severe Subset included patients with a baseline (7-day average) MPN-SAD severity score ≥ 5 (on 0–10 scale) on at least one, or a severity score ≥3 on at least two of the following items: fatigue, early satiety, abdominal discomfort, inactivity, night sweats, pruritus, and/or bone pain.
Spleen-related symptom score is the sum of early satiety and abdominal discomfort.
Constitutional symptom score is the sum of fatigue, night sweats, pruritus, and bone pain.
nTSS is the sum of spleen-related symptoms and constitutional symptoms.
Using the prespecified criteria of ≥50% improvement from baseline, the monthly mean symptom response rate at weeks 12 and 24, respectively, was approximately 10% and 5% in the lead-in cohort and 14% and 7% in the severe subset (Table 4). Two (9.5%) patients, who were also included in the severe subset, had a reduction ≥50% in MPN-SAD (mean change from baseline, −67.5%).
Table 4.
Symptom improvement: MPN-SAD and PGIC.
| Time point, % improvement, cumulative* | Lead-in cohort (N = 21) | |||||
|---|---|---|---|---|---|---|
| MPN-SAD†n (%) |
PGIC improvement **n (%) |
|||||
| TSS | Spleen‡ | Constitutional§ | nTSSǁ | |||
| Monthly mean at week 12 | Cycle 4 day 1 | |||||
| ≥50% | 2 (9.52) | 6 (28.57) | 3 (14.29) | 4 (19.05) | Very much | 1 (4.76) |
| ≥30% | 5 (23.81) | 7 (33.33) | 4 (19.05) | 6 (28.57) | Much | 0 |
| ≥20% | 8 (38.10) | 8 (38.10) | 6 (28.57) | 7 (33.33) | Minimally | 4 (19.05) |
| Monthly mean at week 24 | Cycle 7 day 1 | |||||
| ≥50% | 1 (4.76) | 2(9.52) | 2(9.52) | 1 (4.76) | Very much | 0 |
| ≥30% | 3 (14.29) | 4 (19.05) | 3 (14.29) | 3 (14.20) | Much | 1 (4.76) |
| ≥20% | 4 (19.05) | 5 (23.81) | 3 (14.29) | 4 (19.05) | Minimally | 2 (9.52) |
| Severe Subset†† (n = 14) | ||||||
| Monthly mean at week 12 | Cycle 4 day 1 | |||||
| ≥50% | 2 (14.29) | 4 (28.57) | 2 (14.29) | 2 (14.29) | Very much | 1 (7.14) |
| ≥30% | 4 (28.57) | 5 (35.71) | 3 (21.43) | 4 (28.57) | Much | 0 |
| ≥20% | 6 (42.86) | 6 (42.86) | 5 (35.71) | 5 (35.71) | Minimally | 2 (14.29) |
| Monthly mean at week 24 | Cycle 7 day 1 | |||||
| ≥50% | 1 (7.14) | 2 (14.29) | 0 | 1 (7.14) | Very much | 0 |
| ≥30% | 1 (7.14) | 3 (21.43) | 1 (7.14) | 1 (7.14) | Much | 1 (7.14) |
| ≥20% | 2 (14.29) | 3 (21.43) | 1 (7.14) | 2 (14.29) | Minimally | 1 (7.14) |
MPN-SAD = Myeloproliferative Neoplasm Symptom Assessment Diary; nTSS = new total symptom score; PGIC = Patient Global Impression of Change.
The cumulative frequency of patients is the total of each frequency added to its predecessor. The cumulative percentage of patients divides the cumulative frequency by the total number of observations.
Higher scores are associated with better health for functional scales and worse health for symptom scales.
Spleen-Related Symptom Score is the sum of early satiety and abdominal discomfort.
Constitutional Symptom Score is the sum of fatigue, night sweats, pruritus, and bone pain.
nTSS is the sum of spleen-related symptoms and constitutional symptoms.
Since starting the study medication, my myelofibrosis symptoms are: 1 = Very much improved; 2 = Much improved; 3 = Minimally improved; 4 = No change; 5 = Minimally worse; 6 = Much worse; 7 = Very much worse.
Severe Subset are patients with a baseline (7-day average) MPN-SAD severity score ≥5 (on 0–10 scale) on at least one, or a severity score ≥3 on at least two of the following items: fatigue, early satiety, abdominal discomfort, inactivity, night sweats, pruritus, and/or bone pain.
Using alternative responder definitions of ≥30% or ≥20% improvement from baseline, or focusing on the spleen-related subscale, monthly mean response rates were higher. The highest response rate, which was observed for ≥20% improvement in the TSS and also the spleen-related subscale at week 12, was 38.1% in the lead-in cohort and 42.9% in the severe subset (Table 4).
In the lead-in cohort, mean TSS score and all subscale scores showed a general trend of improvement over time. Individual scores of spleen-related symptoms and inactivity showed improvement at weeks 12 and 24. Of the constitutional symptoms, fatigue showed improvement at weeks 12 and 24, whereas bone pain and pruritus worsened (data not shown). In the severe subset, a clearer trend of symptom improvement and TSS reduction emerged. All individual symptoms except bone pain and pruritus showed greater improvement compared with the lead-in cohort, particularly spleen-related symptoms, fatigue, and inactivity (data not shown).
Results of the EORTC QLQ-30 questionnaire showed an overall favourable trend over time in QoL and functional improvement in the entire lead-in cohort. Patients in the severe subset demonstrated a mostly favourable trend in QoL and functional improvement over time. The mean (SD) change from baseline in EQ-5D-5 L rating up to cycle 10 day 1 was 0.78 (0.10) in the entire lead-in cohort. The greatest reported change from baseline was −0.18 (0.49) at cycle 19 day 1, when only three patients remained in the study. Results were similar for the severe subset. The EQ visual analogue scale yielded inconsistent results. Symptom improvement in PGIC at weeks 12 and 24 is presented in Table 4.
3.5. Pharmacokinetics
Nineteen patients provided plasma PK data considered evaluable (at steady-state for glasdegib) for purposes of glasdegib PK analysis on cycle 1 day 15. Representative individual plasma glasdegib concentration time profiles are shown in Fig. 2. Following administration of 100 mg once-daily oral dose of glasdegib for 15 days, Cmax occurred at 1.02 h postdose (range, 0.48–4.00). Geometric mean (%coefficient of variation [CV] of geometric means) values for AUCtau and Cmax were 13,150 ng.h/ml (50%) and 996.8 ng/ml (45%), respectively. Pre-dose concentrations (Ctrough) following daily dosing were consistent across multiple cycles, with geometric mean (%CV) Ctrough values of 204.1 ng/ml (70%), 176.1 ng/ml (113%) and 189.5 ng/ml (88%) on cycle 1 day 15, cycle 2 day 1, and cycle 3 day 1, respectively.
Fig. 2.
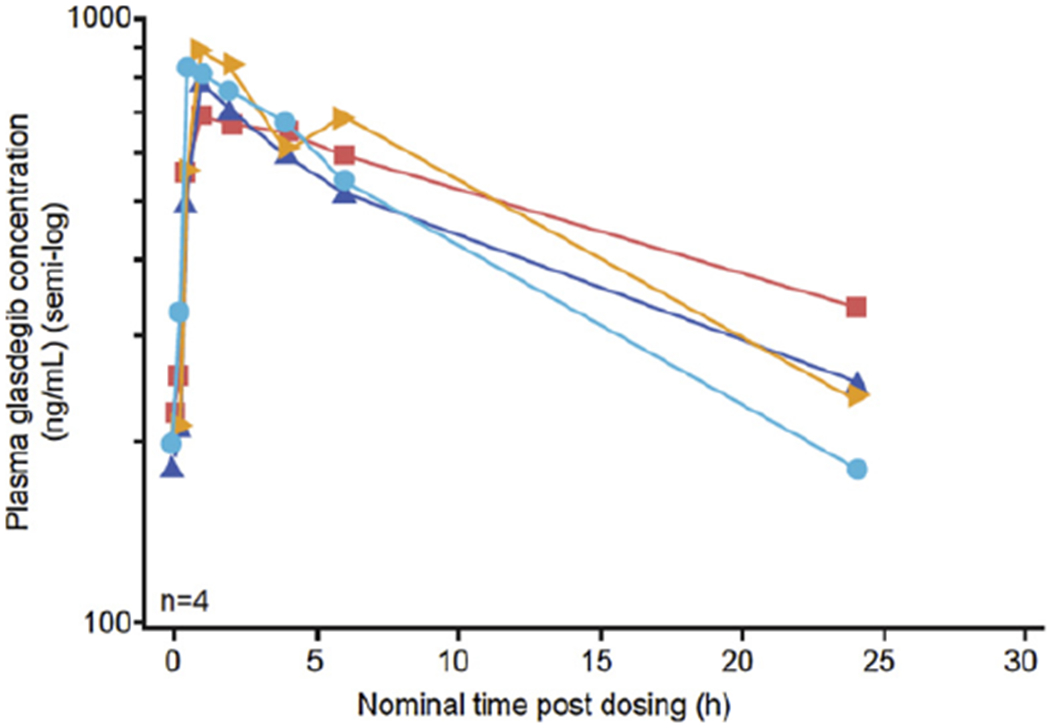
Representative individual plasma glasdegib concentration–time profiles following 15 days of glasdegib 100 mg once-daily oral dosing.
4. Discussion
This open-label, lead-in cohort was designed to evaluate the safety and tolerability of glasdegib in patients with primary/secondary MF previously treated with JAKi. Although glasdegib was considered safe and tolerable, prespecified secondary TSS reduction and SVR endpoints were not met. Therefore, this study did not proceed to the randomised phase.
For pre-treated patients with relapsed and refractory MF, glasdegib demonstrated an acceptable toxicity profile. The most commonly reported TEAEs were dysgeusia, muscle spasms, alopecia, decreased appetite, and fatigue, which are consistent with prior studies of glasdegib and other SMO inhibitors [13–17]. The frequencies of “on target” TEAEs in patients with MF were higher than those reported for glasdegib in patients with other myeloid malignancies (i.e., AML or myelodysplastic syndrome), respectively: dysgeusia (62% vs 28%), muscle spasms (57% vs 9%), alopecia (38% vs 15%), decreased appetite (33% vs 19%), and fatigue (33% vs 11%) [16]. Most of these AEs were grade 1 or 2, with the exception of two cases of grade 3 muscle spasms [16]. The small number of patients, different nature of disease under study, and prior exposure to ruxolitinib may have contributed to differences in the frequency and severity of AEs. Over the course of the study, the investigators became more attuned to recognizing, and better able to address, emerging AEs (i.e., electrolyte solution for muscle spasms). No deaths occurred within 28 days of the last dose of glasdegib, and the number of SAEs reported was low and with no discernible pattern.
Although results from physical examination indicated there may be decreases in spleen size from baseline, they were generally not sustainable as indicated by subsequent MRI/CT assessment. The discrepancy in measurements is most likely due to the fact that physical examination is subjective whereas MRI/CT gives precise measurements. The results imply that MRI/CT is the more accurate modality to make these types of assessments. Mean spleen volume measured by MRI/CT at week 24 suggested glasdegib as monotherapy may not sustainably decrease spleen volume in patients with primary/secondary MF, although two patients imaged at later time points showed evidence of SVR. These results are consistent with a phase 2 study in which patients with MF treated with SMO inhibitor IPI-926 demonstrated reductions in spleen size < 50% from baseline [18]. Anaemia response was observed in 1 patient.
Approximately 40% of patients achieved > 20% to 30% reduction in symptoms, suggesting glasdegib may have a true treatment benefit in improving MF-related symptoms. This is supported by overall consistency of the results from other patient-reported outcome measurements (EORTC QLQ-30, EQ-5D-5 L, and PGIC questionnaires).
Following oral administration of glasdegib, 100 mg once daily to steady-state (cycle 1 day 15), Cmax generally occurred around 1 h postdose. The geometric mean AUCtau and Cmax values were 13,150 ngh/ml and 996.8 ng/ml, respectively, consistent with the observed PK of glasdegib administered as monotherapy [16,17].
Due to secondary efficacy endpoints not being met in the lead-in cohort, the study was terminated early, and the primary efficacy endpoint of SVR with glasdegib vs placebo was not analysed. Patient-reported outcomes were variable.
The population in this study was heavily pre-treated with multiple JAKi relative to the patient population in the COMFORT trial [4]. The majority (67.1%) of patients in the COMFORT study received only previous hydroxyurea, whereas in the current study, 71.4% of patients received 1 prior JAKi therapy and 14.3% each received two and three or more prior JAKi therapies. Therefore, traditional endpoints (i.e., 35% reduction in spleen volume, 50% improvement in symptoms) used to measure efficacy in treatment-naïve patients may not be appropriate to determine efficacy in patients refractory to previous treatments.
4.1. Conclusions
The safety profile of glasdegib is manageable as monotherapy in patients with primary/secondary MF, and no new safety concerns have arisen during this study. However, TEAEs limited treatment duration in some patients. One potential strategy to increase tolerability of glasdegib in this and similar populations may be to consider alternative dosing schedules. Although no patients sustained an SVR ≥ 35%, three patients had stabilisation or reduction in spleen size. Moreover, patient-reported outcome measures and long duration of therapy in select patients, indicative of continued clinical response, suggest glasdegib may improve MF symptoms in this population of pretreated patients. Preclinical studies combining treatment with an Hh inhibitor and JAKi demonstrated increased efficacy in MF [8], suggesting that further study of glasdegib in combination with JAKi in MF populations may be warranted. Furthermore, traditional efficacy endpoints used in treatment-naïve patients may not capture the entire spectrum of clinical benefit, and a new characterisation of efficacy, such as molecular biomarkers of response, may need to be developed for heavily pre-treated patients.
Acknowledgements
Medical writing support was provided by Vardit Dror, PhD, and Emily Balevich, PhD, of Engage Scientific Solutions and was funded by Pfizer.
Funding
This study was sponsored by Pfizer. The sponsor participated in study design and collection, analysis, and interpretation of data. All authors had full access to study data, and the corresponding author had final responsibility for the decision to submit for publication.
Abbreviations:
- AE
adverse event
- AML
acute myeloid leukaemia
- CT
computed tomography
- CV
coefficient of variation
- EQ-5D-5L
EuroQol-5 Dimension Questionnaire, 5-level version
- EORTC QLQ-C30
European Organization for Research and Treatment of Cancer Quality of Life Questionnaire
- Hh
Hedgehog
- HRQoL
health-related quality of life
- JAK
Janus kinase
- JAKi
JAK inhibitor
- MF
myelofibrosis
- MPN-SAD
Myeloproliferative Neoplasm Symptom Assessment Diary
- MRI
magnetic resonance imaging
- PGIC
Patient Global Impression of Change
- PK
pharmacokinetics
- SAE
serious adverse event
- SD
standard deviation
- SMO
Smoothened
- SVR
spleen volume reduction
- TEAE
treatment-emergent adverse event
- TSS
total symptom score
Footnotes
Data sharing statement
Upon request, and subject to certain criteria, conditions and exceptions see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Declaration of interest
ATG declares receiving consulting fees and research funding from Celgene, Incyte, and CTI Biopharma, consulting fees from Apexx Oncology, and research funding from Gilead, Genentech, Roche, Imago Biosciences, Pfizer, and Samus Therapeutics. TT declares receiving research funding from Pfizer. ER declares receiving consulting fees and research funding from Novartis, honoraria from Incyte and Pfizer, and speaker’s bureau from Celgene. MD has no conflict of interests to disclose. CJ declares having equity ownership from Impact Biomedicines and Wintherix, receiving research funding from Celgene and Johnson & Johnson, and patents & royalties from Forty Seven Inc. RM declares receiving consulting fees from Novartis, Ariad, and Galena, and research funding from Incyte, Gilead, CTI, Promedior, and Celgene. MH declares receiving consulting fees from Novartis. NK has no conflict of interests to disclose. HM declares receiving research funding from Pfizer. YS was an employee of Pfizer at the time of manuscript development and owns stock in Pfizer. NS, XZ, CD, MZ, and GC are employees of and own stock in Pfizer. MT declares receiving research funding from Pfizer.
References
- [1].Tefferi A, Myelofibrosis with myeloid metaplasia, N. Engl. J. Med. 342 (17) (2000) 1255–1265, 10.1056/NEJM200004273421706. [DOI] [PubMed] [Google Scholar]
- [2].Savona MR, Are we altering the natural history of primary myelofibrosis? Leuk. Res. 38 (9) (2014) 1004–1012, 10.1016/j.leukres.2014.04.012. [DOI] [PubMed] [Google Scholar]
- [3].Harrison C, Kiladjian JJ, Al-Ali HK, et al. , JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis, N. Engl. J. Med. 366 (9) (2012) 787–798, 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- [4].Verstovsek S, Mesa RA, Gotlib J, et al. , A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis, N. Engl. J. Med. 366 (9) (2012) 799–807, 10.1056/NEJMoal110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Barosi G, Zhang MJ, Peter Gale R, Does ruxolitinib improve survival of persons with MPN-associated myelofibrosis? should it? Leukemia 28 (11) (2014) 2267–2270, 10.1038/leu.2014.220. [DOI] [PubMed] [Google Scholar]
- [6].Cervantes F, Pereira A, Does ruxolitinib prolong the survival of patients with myelofibrosis? Blood 129 (7) (2017) 832–837, 10.1182/blood-2016-11-731604. [DOI] [PubMed] [Google Scholar]
- [7].Verstovsek S, Gotlib J, Mesa RA, et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses, J. Hematol. Oncol. 10 (1) (2017) 156, 10.1186/sl3045-017-0527-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bhagwat N, Keller MD, Rampal RK, et al. , Improved efficacy of combination of JAK2 and Hedgehog inhibitors in myelofibrosis, Blood 122 (21) (2013) 666.23794067 [Google Scholar]
- [9].Passamonti F, Cervantes F, Vannucchi AM, et al. , A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment), Blood. 115 (9) (2010) 1703–1708, 10.1182/blood-2009-09-245837. [DOI] [PubMed] [Google Scholar]
- [10].Vardiman JW, Thiele J, Arber DA, et al. , The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes, Blood 114 (5) (2009) 937–951, 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- [11].Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology, J. Natl. Cancer Inst. 85 (5) (1993) 365–376. [DOI] [PubMed] [Google Scholar]
- [12].Rabin R, de Charro F, EQ-5D: a measure of health status from the EuroQol Group, Ann. Med. 33 (5) (2001) 337–343. [DOI] [PubMed] [Google Scholar]
- [13].Axelson M, Liu K, Jiang X, et al. , U.S. Food and Drug Administration approval: vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma, Clin. Cancer Res. 19 (9) (2013) 2289–2293, 10.1158/1078-0432.CCR-. [DOI] [PubMed] [Google Scholar]
- [14].Lacouture ME, Dreno B, Ascierto PA, et al. , Characterization and management of Hedgehog pathway inhibitor-related adverse events in patients with advanced basal cell carcinoma, Oncologist 21 (10) (2016) 1218–1229, 10.1634/theoncologist.2016-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lin TL, Matsui W, Hedgehog pathway as a drug target: smoothened inhibitors in development, Onco Targets Ther. 5 (2012) 47–58, 10.2147/OTT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Martinelli G, Oehler VG, Papayannidis C, et al. , Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: a phase 1 safety and pharmacokinetics study, Lancet Haematol. 2 (8) (2015) 00096–00094. [DOI] [PubMed] [Google Scholar]
- [17].Wagner AJ, Messersmith WA, Shaik MN, et al. , A phase I study of PF-04449913, an oral hedgehog inhibitor, in patients with advanced solid tumors, Clin. Cancer Res. 21 (5) (2015) 1044–1051, 10.1158/1078-0432.CCR-14-1116. [DOI] [PubMed] [Google Scholar]
- [18].Sasaki K, Gotlib JR, Mesa RA, et al. , Phase II evaluation of IPI-926, an oral Hedgehog inhibitor, in patients with myelofibrosis, Leuk. Lymphoma 56 (7) (2015) 2092–2097, 10.3109/10428194.2014.984703. [DOI] [PMC free article] [PubMed] [Google Scholar]