

Summary
Glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) regulate glucose and energy homeostasis. Targeting both pathways with GIP receptor (GIPR) antagonist antibody (GIPR-Ab) and GLP-1 receptor (GLP-1R) agonist, by generating GIPR-Ab/GLP-1 bispecific molecules, is an approach for treating obesity and its comorbidities. In mice and monkeys, these molecules reduce body weight (BW) and improve many metabolic parameters. BW loss is greater with GIPR-Ab/GLP-1 than with GIPR-Ab or a control antibody conjugate, suggesting synergistic effects. GIPR-Ab/GLP-1 also reduces the respiratory exchange ratio in DIO mice. Simultaneous receptor binding and rapid receptor internalization by GIPR-Ab/GLP-1 amplify endosomal cAMP production in recombinant cells expressing both receptors. This may explain the efficacy of the bispecific molecules. Overall, our GIPR-Ab/GLP-1 molecules promote BW loss, and they may be used for treating obesity.
Keywords: diet-induced obese mice, glucose-dependent insulinotropic polypeptide, glucagon-like peptide-1, monkeys, obesity, weight loss, antibody, cAMP, Cyclic adenosine monophosphate
Graphical abstract
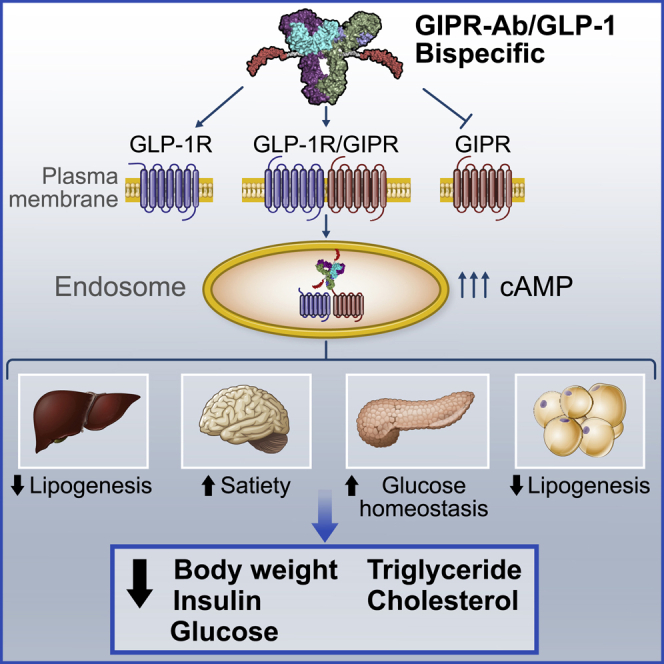
Highlights
GIPR-Ab/GLP-1 is a bispecific molecule used for the treatment of obesity
GIPR-Ab/GLP-1 antagonizes GIPR and agonizes GLP-1R in vitro
GIPR-Ab/GLP-1 synergistically reduces body weight and metabolic values in animals
Greater receptor internalization with GIPR-Ab/GLP-1 amplifies endosomal cAMP levels
Lu et al. show that tackling obesity with bispecific molecules that antagonize/agonize GIPR/GLP-1R pathways decreases body weight and metabolic parameters in obese mice and monkeys. Mechanistic studies suggest that such molecules bind to GIPR and GLP-1R simultaneously and trigger receptor internalization, amplifying endosomal cAMP signaling in cells expressing both receptors.
Introduction
Obesity and its comorbidities have significant impacts on the global economic state.1,2 Current pharmacotherapies show modest weight-lowering capacity, need frequent dosing, and are often accompanied with significant adverse events.3 Thus, there is an unmet medical need for developing safe and effective anti-obesity agents.
Glucagon-like peptide-1 (GLP-1) is an incretin secreted by intestinal L cells within minutes following ingestion of a meal. In addition to its incretin function, GLP-1 has been reported to delay gastric emptying and promote satiety and has thus become an attractive approach to treat patients with type 2 diabetes (T2D) and obesity.4 Several GLP-1 receptor agonists (GLP-1Ras) are marketed for T2D and obesity, many of which contain modifications that allow extended circulating half-life. In patients with T2D and obesity, GLP-1Ras lowers hemoglobin A1c and weight, with low risk of hypoglycemia.3
Another incretin, glucose-dependent insulinotropic polypeptide (GIP) is secreted from the enteroendocrine K cells in the small intestine following food ingestion.5, 6, 7 Studies have shown that GIP analogs reduce body weight (BW),8 and a GIP/GLP-1 co-agonist augments incretin response and BW reduction compared with GLP-1Ras in rodents9,10 and in clinical trials.10,11 However, GIP promotes adipogenesis in vivo and ex vivo,12,13 and circulating GIP levels are elevated in obese mice and humans,14, 15, 16 indicating a pro-obesogenic state associated with GIP. Further, genetic ablation of GIP receptor (GIPR) led to a decrease in BW in diet-induced obese (DIO) mice.16, 17, 18, 19 Moreover, analysis of genome-wide association studies (GWASs) have identified variants with reduced activity at the human GIPR (hGIPR) locus that are associated with reduced body mass index.20 Hence, antagonizing GIP/GIPR may also be a suitable strategy for generating anti-obesity therapies. In line with this, pharmacological inhibition using GIPR-neutralizing monoclonal antibodies (mAbs) protected against BW gain in DIO mice and obese monkeys.21 Similarly, central inhibition of GIPR reduced BW and adiposity through a leptin-dependent mechanism.22 In addition, BW loss was significantly enhanced in DIO mice and monkeys when anti-GIPR antibodies (Abs) were co-administered with GLP-1Ras.21 These results support the development of single molecules that target multiple pathways for improving efficacy in treating obesity.23 In this study, we combined GIPR antagonism with GLP-1R agonism by generating GIPR-Ab/GLP-1 bispecific molecules (hereinafter referred to as GIPR-Ab/GLP-1) with GLP-1 peptides containing amino-acid modifications to extend half-life while optimizing potency. GIPR-Ab/GLP-1 exhibited long-acting pharmacokinetic (PK) properties and synergistically promoted BW reduction in DIO mice and monkeys. Mechanistically, GIPR-Ab/GLP-1 induced internalization of both GLP-1R and GIPR accompanied by amplified cyclic adenosine monophosphate (cAMP) production. Inhibition of receptor internalization reduced cAMP production and the internalized bispecific molecule was co-localized with GLP-1R and GIPR in early and recycling endosomes. This sustained cAMP response suggested that our GIPR-Ab/GLP-1 induced cAMP production in the endosomes. Collectively, our data show that monomeric anti-GIPR-Ab/GLP-1 enhances BW loss in two different preclinical models of obesity: DIO mice and obese monkeys.
Results
Generation of GIPR-Ab/GLP-1
We previously reported the generation of a fully human anti-human GIPR-Ab (hGIPR-Ab).21 During the same GIPR-Ab campaign, we identified an hGIPR-Ab that cross-reacts with mouse GIPR (mGIPR-Ab). To design GIPR-Ab/GLP-1 molecules, GLP-1 peptides (see P1 or P2, Figure 1A) containing a (GGGGS)3 linker were tethered by chemical conjugation to site-specific engineered cysteines (E384C) to either hGIPR-Ab (hGIPR-Ab/P1 and hGIPR-Ab/P2) or mGIPR-Ab (mGIPR-Ab/P1 and mGIPR-Ab/P2; Figure 1A). The site E384C was selected to conjugate GLP-1 peptides as it allowed desirable alkylation efficiency, and, after conjugation, the molecules showed a more favorable PK profile than when conjugation occurred at some other sites.24 A non-GIPR binding control Ab was also engineered to allow conjugation of P1 (control-Ab/P1), and this molecule was used as a long-acting GLP-1 control molecule. GLP-1 peptide P1 was designed to improve metabolic stability by incorporating 2-aminoisobutyric acid (Aib) into positions 8 and 22 of the peptide.25 Arginine-36 was substituted with glycine to improve in vivo stability. In addition to the modifications in the P1 peptide, we designed peptide P2 to attenuate GLP-1 potency by adding a D15E mutation (Figure 1A). We aimed to generate molecules with different GLP-1 potency to mitigate the potential gastrointestinal side effects.26
Figure 1.

GIPR-Ab/GLP-1 bispecific molecules exhibit GIPR antagonist and GLP-1R agonist activities in vitro
(A) Structure and nomenclature of GIPR-Ab/GLP-1.
(B–D) Representative dose-response curves of cAMP assays with GIP (agonist mode) or bispecific molecules + 50 pM GIP (antagonist mode) in cells expressing human (B), monkey (C), or mouse (D) GIPR.
(E–G) Representative dose-response curves of cAMP assays with GLP-1 or bispecific molecules in cells expressing human (E), monkey (F), or mouse (G) GLP-1R.
Data represent mean ± SEM of n = 2 replicates per treatment.
GIPR-Ab/GLP-1 exhibits GIPR antagonist and GLP-1R agonist activities in vitro
As reported earlier,21 hGIPR-Ab antagonized hGIPR activity by inhibiting GIP-induced cAMP production in cells expressing hGIPR (half maximal inhibitory concentration [IC50] = 136.1 nM; Figure 1B). Antagonist activities toward GIPR of hGIPR-Ab/P1 and hGIPR-Ab/P2 are comparable with that of hGIPR-Ab (IC50 = 103.6 and 121.8 nM, respectively), indicating that the conjugation of P1 or P2 did not impact the antagonist hGIPR-Ab activity (Figure 1B). Antagonist activities of hGIPR-Ab, hGIPR-Ab/P1, and hGIPR-Ab/P2 are also comparable in cells expressing monkey GIPR (IC50 = 24.6, 17.1, and 16.1 nM, respectively) (Figure 1C). Similarly, mGIPR-Ab/P1 and mGIPR-Ab/P2 retained the antagonist activity of mGIPR-Ab (IC50, mGIPR-Ab = 2.7 nM; mGIPR-Ab/P1 = 2.8 nM, and mGIPR-Ab/P2 = 6.4 nM) (Figure 1D). As expected, the control-Ab/P1 did not show any antagonist activity in cells expressing mGIPR (Figure 1D). For GLP-1R agonist activity, all GIPR-Ab/GLP-1 molecules and control conjugate showed 20- to 40-fold less potency to stimulate cAMP production than native GLP-1(7-37) peptide in cells expressing human, monkey, or mouse GLP-1R (Figures 1E–1G). Although P2 was designed to demonstrate attenuated potency than P1, no significant differences in potency were seen between hGIPR-Ab/P1 and hGIPR-Ab/P2 or between mGIPR-Ab/P1 and mGIPR-Ab/P2 in these assays (Figures 1E–1G). We hypothesize that the high expression levels of the GLP-1R in our cell system did not allow to detect differences for GLP-1 potency.
GIPR-Ab/GLP-1 showed extended PK profiles
To determine the PK properties of GIPR-Ab/GLP-1, exposure levels of mGIPR-Ab/P1 and hGIPR-Ab/P1 were assessed by measuring the concentration of intact GIPR-Ab/GLP-1 in mouse and monkey plasma after intravenous (i.v.) or subcutaneous (s.c.) administration. The specificity of the PK assay enabled the differentiation of intact GIPR-Ab/GLP-1 from entities that may have been altered or degraded in vivo. Mean plasma concentration-time profiles of mGIPR-Ab/P1 and hGIPR-Ab/P1 are presented in Figures 2A–2D. PK parameters obtained from noncompartmental analysis are summarized in Figure 2E. After a single i.v. injection of mGIPR-Ab/P1 (5 mg/kg) in mice, the mean terminal half-life (t1/2,z) was 5.3 days and mean systemic clearance (CL) was 15.0 mL/day/kg (Figures 2A and 2E). For hGIPR-Ab/P1 at 5 mg/kg, the mean t1/2,z was 6.2 and 5.3 days, and mean CL or apparent clearance (CL/F) was 14.1 and 17.2 mL/day/kg after i.v. and s.c. single administration, respectively (Figures 2B and 2E). Bioavailability of hGIPR-Ab/P1 after s.c. administration was 82%. When hGIPR-Ab/P1 was administered as a single s.c. dose of 3 mg/kg in monkeys, it showed a mean t1/2,z of 8.7 days and a mean CL/F of 7.0 mL/day/kg (Figures 2C and 2E). After a single s.c. injection of hGIPR-Ab/P1 at 1 mg/kg in obese monkeys, the mean t1/2,z was 9.1 days and mean CL/F was 11.3 mL/day/kg (Figures 2D and 2E). PK profiles and parameters indicated that test articles were remarkably stable in preclinical species, consistent with the slow clearance and long half-life typically observed for therapeutic mAbs.27,28 Notably, GIPR-Ab/GLP-1 showed superior PK properties in preclinical species than that published for marketed GLP-1Ras liraglutide and dulaglutide.29,30
Figure 2.

GIPR-Ab/GLP-1 bispecific molecules showed extended pharmacokinetic profiles and biodistribution of GIPR-Ab/GLP-1
(A) mGIPR-Ab/P1 PK in mice. Data represent mean ± SEM of plasma concentration-time profile after single i.v. administration at 5 mg/kg (n = 2).
(B–D) hGIPR-Ab/P1 PK in mice (B), monkeys (C), or obese monkeys (D). Data represent mean ± SEM of plasma concentration-time profile after single i.e. (B) or s.c. (B–D) administration at 5 (B), 3 (C), or 1 (D) mg//kg (n = 3–5).
(E) Summary of PK characteristics of mGIPR-Ab/P1 and hGIPR-Ab/P1.
(F) Tissue to blood AUC ratio of mGIPR-Ab and mGIPR-Ab/P1. Data represent mean ± SEM, n = 3 mice per time point and 6 time points in total.
(G) Tissue to blood AUC ratio side-by-side comparison in the pancreas, liver, WAT, BAT, brain, muscle, bone marrow, and lung. ∗∗p < 0.01, ∗∗∗p < 0.001 for mGIPR-Ab/P1 versus mGIPR-Ab. Data represent mean ± SEM, n = 3 mice per time point and 6 time points in total.
See also Figure S1.
Biodistribution of GIPR-Ab/GLP-1
The biodistribution study aimed to characterize tissue PK of mGIPR-Ab/P1 and mGIPR-Ab, identify their sites of catabolism, and compare the influence of target expression on tissue distribution in organs of interest. 111In-DOTA-labeled mGIPR-Ab/P1 and mGIPR-Ab were used for this purpose. Serum concentration-time profiles of labeled mGIPR-Ab and mGIPR-Ab/P1 were found to be similar (Figure S1A). The difference between serum area under the concentration-time curve (AUC0–120h) for mGIPR-Ab and mGIPR-Ab/P1 was not significant (2.21 × 103 versus 2.24 × 103 injected dose [ID] h/mL, p > 0.05). More than 89% of the protein was trichloroacetic acid (TCA) precipitable, suggesting stability of 111In-protein conjugate (Figure S1B). Key organs of interest to evaluate (based on known expression of GIPR and GLP-1R) were pancreas, brain, white adipose tissue (WAT), and brown adipose tissue (BAT). Rank ordering of the tissue to blood AUC ratios in these tissues for mGIPR-Ab was BAT>pancreas>WAT>brain, while for mGIPR-Ab/P1, it was pancreas>BAT>WAT>brain (Figure 2F). The tissue to blood AUC ratio for the pancreas was 61.2% higher for mGIPR-Ab/P1 than that for mGIPR-Ab (p < 0.001, Figure 2G). The tissue to blood AUC ratio for WAT, BAT, muscle, and brain between the two groups was not significant (p > 0.05, Figure 2G). The liver to blood AUC ratio was ∼61.7% higher for mGIPR-Ab/P1 than that for mGIPR-Ab (p < 0.001, Figure 2G). The lung to blood AUC ratio was ∼60% higher for mGIPR-Ab/P1 than that for mGIPR-Ab (p < 0.001, Figure 2G). Similarly, bone marrow to blood AUC ratio was ∼48.7% higher for mGIPR-Ab/P1 than that for mGIPR-Ab (p < 0.01, Figure 2G).
GIPR-Ab/GLP-1 reduced BW in lean and DIO mice and decreased BW more than dulaglutide in DIO mice
To further characterize and understand the efficacy of mGIPR-Ab/P1, BW and metabolic parameters were measured throughout an 18-day study in lean and DIO mice (Figure 3A). mGIPR-Ab, mGIPR-Ab/P1, or control-Ab/P1 was administered in equimolar doses, and these doses were adjusted to match exposure for each treatment. Monotherapy with mGIPR-Ab showed no effect on BW in DIO mice (−1.2%). Additionally, control-Ab/P1 monotherapy reduced BW by 5.1% in lean mice and 15.3% in DIO mice. The mGIPR-Ab/P1 molecule showed a synergistic reduction in BW compared with either monotherapy in DIO mice. A suboptimal dose (0.5 mg/kg) of the molecule was able to reduce BW to the maximum achieved by GLP-1R agonist monotherapy (12.2% versus 15.3%). At 2.5 mg/kg (the exposure-matched dose), the bispecific molecule nearly doubled the weight loss in the DIO mice to 29.3%. The weight loss was coupled with improved metabolic parameters including circulating insulin and cholesterol levels in DIO mice (Figure 3A).
Figure 3.

mGIPR-Ab/P1 dose-dependently reduced BW and showed greater effects on BW loss than mGIPR-Ab or control-Ab/P1 administered alone in lean and DIO mice, and the effects are independent of pancreatic β cells
(A) BW percentage change was measured over time and terminal plasma insulin, triglycerides, and total cholesterol were measured in lean or DIO mice dosed with vehicle, mGIPR-Ab (2.5 mg/kg), control-Ab/P1 (2 mg/kg), and mGIPR-Ab/P1 (0.5 mg/kg and 2.5 mg/kg). n = 7 mice/group for lean and 7–8 mice/group for DIO, not all lean mice produced enough plasma for analysis (n = 4–6 mice/group for insulin, and n = 4–7 mice/group for triglycerides and cholesterol). Two-way repeated-measures ANOVA with Dunnett’s multiple comparisons for BW analysis and one-way ANOVA with Sidak’s test for multiple comparisons were done for glucose, insulin, triglycerides, and total cholesterol; #p < 0.05, ####p < 0.0001 vehicle versus mGIPR-Ab (2.5 mg/kg); ++++p < 0.0001 vehicle versus control-Ab/P1; ˆˆp < 0.001, ˆˆˆp < 0.001, ˆˆˆˆp < 0.0001 vehicle versus mGIPR-Ab/P1 (0.5 mg/kg); or ∗∗p < 0.01, ∗∗∗∗p < 0.0001 vehicle versus mGIPR-Ab/P1 (2.5 mg/kg); ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 treatment versus vehicle or between different groups as indicated in the graph with a bracket.
(B) Giprfl/fl and GiprβCell−/− male littermates were fed HFD for 12 weeks and then IP dosed with the vehicle or mGIPR-Ab/P1 (0.5 mg/kg or 2.5 mg/kg) every 6 days for 18 days. BW percentage change was measured over time, and terminal blood glucose and plasma insulin, triglycerides, and total cholesterol were determined. n = 8 mice/group for all measurements and for triglycerides, n = 6–8 mice/group. For BW analysis, two-way repeated-measures ANOVA with Tukey’s HSD for multiple comparisons, ˆp < 0.05, ˆˆˆˆp < 0.0001 Giprfl/fl mGIPR-Ab/P1 (0.5 mg/kg) versus Giprfl/fl Vehicle; +++p < 0.001, ++++p < 0.0001 GiprβCell−/− mGIPR-Ab/P1 (0.5 mg/kg) versus GiprβCell−/− vehicle; #p < 0.05 Giprfl/fl mGIPR-Ab/P1 (0.5 mg/kg) versus GiprβCell−/− mGIPR-Ab/P1 (0.5 mg/kg); ∗∗∗∗p < 0.0001 Giprfl/fl mGIPR-Ab/P1 (2.5 mg/kg) versus Giprfl/fl vehicle; &&&&p < 0.0001 GiprβCell−/− mGIPR-Ab/P1 (2.5 mg/kg) versus GiprβCell−/− vehicle. For glucose, insulin, triglycerides, and total cholesterol, one-way ANOVA with Sidak’s test for multiple comparisons; ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001 for treatment versus vehicle.
See also Figure S2.
In a separate study, dulaglutide was injected twice weekly in one group of mice and once weekly in another group of mice to follow the dose regimen of Ab-treated mice. After 28 days, mice treated with mGIPR-Ab and control-Ab/P1 lost 0.6% and 7% of BW, respectively (Figure S2). Mice treated with dulaglutide administered twice weekly or once weekly lost 10% or 0.9% of BW, respectively (Figure S2). Mice treated with mGIPR-Ab/P1 lost 16.4% of BW, a 2-fold greater BW loss than the sum of both monotherapies and significantly greater than the loss in dulaglutide-treated mice (Figure S2). Plasma total cholesterol was significantly reduced in the mGIPR-Ab/P1 and dulaglutide twice-weekly treated mice, while plasma triglycerides were not significantly altered (Figure S2). Plasma insulin levels were reduced in all treated groups; however, statistical significance was not reached (Figure S2). Overall, mGIPR-Ab/P1-treated mice showed the greatest BW loss and metabolic parameters than all other treatment groups.
GIPR activity has been shown to be fundamental for pancreatic β cell function.31,32 We used β cell-specific Gipr knockout mice to determine whether GIPR activity in pancreatic β cells caused the potent effects of mGIPR-Ab/P1 on BW reduction and improved metabolic parameters. A study similar to the 18-day study described above (Figure 3A) was conducted in DIO mice with Gipr knockout in pancreatic β cells (GiprβCell−/−) and their wild-type littermates (Giprfl/fl) (mice described earlier21). After 18 days of treatment, both vehicle-treated Giprfl/fl and GiprβCell−/− mice gained 0.6% and 0.5% of their starting BWs, respectively, while Giprfl/fl mice treated with mGIPR-Ab/P1 lost 8.4% and 19.8% of BW with 0.5 mg/kg and 2.5 mg/kg treatments, respectively (Figure 3B). Notably, GiprβCell−/− mice also lost 10.5% and 23.6% of BW with 0.5 mg/kg and 2.5 mg/kg, respectively, indicating that GIPR activity in pancreatic β cells is not necessary for the activity of mGIPR-Ab/P1. As reported earlier,21 GiprβCell−/− mice had significantly lower fasting blood glucose than Giprfl/fl mice as seen in the vehicle-treated mice, and mGIPR-Ab/P1 was able to reduce blood glucose in Giprfl/fl mice to the same level as GiprβCell−/− mice at 0.5 mg/kg and further lower blood glucose in both lines of mice at 2.5 mg/kg compared with vehicle. Consistent with studies in wild-type DIO mice, mGIPR-Ab/P1 significantly reduced fasting plasma insulin and cholesterol in both lines of mice. Overall, these data indicated that the effect of mGIPR-Ab/P1 on BW reduction and improved metabolic parameters is independent of GIPR activity in pancreatic β cells.
Chronic administration of GIPR-Ab/GLP-1 reduced BW in DIO mice and obese monkeys
As both of our bispecific molecules performed equally well in our in vitro cAMP assay for GLP-1R response (Figures 1E–1G), we tested their efficacy in two preclinical models of obesity: DIO mice (Figure 4A) and obese monkeys (Figure 4B). A dose-dependent BW loss was observed with both molecules. Although mGIPR-Ab/P1 and mGIPR-Ab/P2 showed similar potency in vitro (Figures 1D and 1G), mGIPR-Ab/P1 was more potent than mGIPR-Ab/P2 in vivo. On average, obese mice lost ∼18.0% of their BW by day 18 with the high dose of mGIPR-Ab/P2 (2.5 mg/kg) and ∼16.2% of BW with the low dose of mGIPR-Ab/P1 (0.5 mg/kg, Figure 4A). Both the low-dose mGIPR-Ab/P1 and high-dose mGIPR-Ab/P2 significantly reduced BW from day 3 until the end of the study. The high dose of mGIPR-Ab/P1 significantly reduced BW from day 3 and low dose of mGIPR-Ab/P2 from day 10 onward (Figure 4A). A similar dose-dependent profile was observed for the food intake reduction. All treatment groups significantly reduced food intake during each of the 3-day measurements that were collected with the greatest reduction observed after the first dose on days 0–3. Food intake gradually increased thereafter at subsequent measurements (Figure 4A). Mice treated with the high dose of mGIPR-Ab/P1 showed the greatest food intake inhibition and mice treated with the low dose of mGIPR-Ab/P2 showed the least food intake inhibition compared with the group of mice treated with vehicle. Mice treated with low dose of mGIPR-Ab/P1 and high dose of mGIPR-Ab/P2 showed similar food intake inhibition. A similar reduction in plasma insulin and lipid levels was also seen (Figure 4A).
Figure 4.

Chronic administration of GIPR-Ab/GLP-1 bispecific molecules reduced BW in obese monkeys
(A) Dose response of mGIPR-Ab/P1 and mGIPR-Ab/P2 on BW, food intake, insulin, triglycerides, and total cholesterol in DIO mice. Statistical analysis was performed using GraphPad Prism V7.04. Two-way ANOVA with Tukey’s HSD for multiple comparisons was performed for BW (repeated-measures) and food intake data. One-way ANOVA with Sidak’s test for multiple comparisons was performed for all other parameters. For Figure 4A BW data, statistical significance is denoted as ˆˆp < 0.01, ˆˆˆp < 0.001, and ˆˆˆˆp < 0.0001 vehicle versus mGIPR-Ab/P1 (0.5 mg/kg); ∗∗∗∗p < 0.0001 vehicle versus mGIPR-Ab/P1 (2.5 mg/kg); #p < 0.05, and ##p < 0.01 vehicle versus mGIPR-Ab/P2 (0.5 mg/kg); 2+p < 0.01, +++p < 0.001, and ++++p < 0.0001 vehicle versus mGIPR-Ab/P2 (2.5 mg/kg). For food intake and metabolic parameters, statistical significance is denoted as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 versus vehicle.
(B) Compounds exposure and the effects of hGIPR-Ab/P1 and hGIPR-Ab/P2 on BW, food intake, insulin, triglycerides, and total cholesterol in obese cynomolgus monkeys. All data are represented as group mean ± SEM. Two-way repeated-measures ANOVA with Dunnett’s multiple comparisons was performed using GraphPad Prism V7.04 and statistical significance is denoted as ˆp < 0.05, ˆˆp < 0.01, ˆˆˆp < 0.001, and ˆˆˆˆp < 0.0001 versus vehicle for hGIPR-Ab/P1 and ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 versus vehicle for hGIPR-Ab/P2.
To assess whether the efficacy observed in mice translated into monkeys, hGIPR-Ab/P1 and hGIPR-Ab/P2 were tested in obese monkeys. Weekly dosing was sufficient to achieve steady-state levels of both compounds with similar exposure (Figure 4B). The treatment was well tolerated, and, as expected, total energy intake and water intake (data not shown) decreased once treatment started; however, all animals continued to eat and drink throughout the study. Weekly administration of hGIPR-Ab/P1 and hGIPR-Ab/P2 for 6 weeks led to a reduction in BW (Figure 4B), food intake (Figure 4B), fasting insulin and triglycerides, and total cholesterol (Figure 4B). Compared with vehicle, at the end of the treatment phase, hGIPR-Ab/P2 decreased BW by 8.4% and hGIPR-Ab/P1 decreased BW by 14.4%. The difference between the tested molecules was obvious for the BW reduction; however, it was observed to a lesser extent with total energy intake and fasting triglycerides but not with fasting insulin or total cholesterol (Figure 4B). All measured parameters began rebounding toward baseline at varying rates once treatment ended, but baseline BW was not regained by the end of the 4-week washout period (Figure 4B).
GIPR-Ab/GLP-1 reduced the respiratory exchange ratio and changed gene expression and protein levels of metabolic pathways in DIO mice
To investigate the physiological changes related to treatment with GIPR-Ab/GLP-1, we performed indirect calorimetry measurement for 6 days in DIO mice treated with a single injection of vehicle, mGIPR-Ab, control-Ab/P1, or mGIPR-Ab/P1. Mice treated with control-Ab/P1 and mGIPR-Ab/P1 showed significant reduction in oxygen consumption, carbon dioxide production, and respiratory exchange ratio (RER) compared with mice treated with vehicle or mGIPR-Ab (Figures 5A–5E). RER values during the day (Figure 5D) and the night (Figure 5E) were significantly reduced in the mGIPR-Ab/P1 and control-Ab/P1 treatment groups than in the vehicle-treated group. No changes were observed in the group of animals treated with mGIPR-Ab compared with the group of animals treated with vehicle (Figures 5D and 5E). Interestingly at day 6, the BW and food intake of mice treated with mGIPR-Ab/P1 were significantly further decreased than that of mice treated with control-Ab/P1 (Figures 5F and 5G). This decrease translated into a significantly lower RER for animals treated with mGIPR-Ab/P1 than for animals treated with control-Ab/P1 (Figure 5D). The latter group showed rebounding RER values starting on day 5, suggesting that mice treated with mGIPR-Ab/P1 presented a greater lipid oxidation during the resting phase, and this effect was directly associated with reduced food intake and enhanced BW reduction.
Figure 5.

mGIPR-Ab/P1 prolonged reduction of light-cycle RER associated with enhanced BW loss and food intake reduction
(A–C) DIO mice (23 weeks old) were dosed with vehicle, mGIPR-Ab (2.5 mg/kg), control-Ab/P1 (2 mg/kg), or mGIPR-Ab/P1 (2.5 mg/kg), and indirect calorimetry was conducted continuously for 6 days. (A) Oxygen consumption, (B) carbon dioxide production, and (C) RER measurements were taken continuously every 11 min. Each data point represents a rolling average of six time points, and dark cycles (6:00 p.m. to 6:00 a.m.) are shown by a shaded gray box.
(D–G) RER (D) light-cycle and (E) dark-cycle measurement were averaged and displayed as mean ± SEM for each day or night over time, respectively. On day 6 (F), BW change and (G) food intake were measured.
n = 5–6 mice/group; two-way repeated-measures ANOVA with Tukey’s HSD for multiple comparisons were performed. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 vehicle versus treatment or mGIPR-Ab/P1 versus control-Ab/P1 indicated with bracket.
To further assess the metabolic changes induced by the different compounds, the expression levels in liver and adipose tissues of genes involved in glucose, lipid, and cholesterol metabolism were measured in DIO mice after 3 weeks of treatment (Figure S3). Blood changes of metabolic hormones levels were also determined (Figure S4). The expression levels of glucose 6-phosphatase (G6Pase) in liver trended to a reduction in the control-Ab/P1 and mGIPR-Ab/P1 treatment groups than in the vehicle and mGIPR-Ab treatment groups (Figure S3A). Interestingly, mGIPR-Ab and mGIPR-Ab/P1 significantly increased cytochrome c oxidase subunit 8B (Cox8b, Figure S3B), a marker of mitochondrial biogenesis. mGIPR-Ab/P1 independently increased peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Ppargc1a). Control-Ab/P1 and mGIPR-Ab/P1 significantly reduced the expression of hepatic genes involved in lipogenesis, such as fatty acid synthase (Fasn), stearoyl-CoA desaturase 1 (Scd1), acetyl-CoA carboxylase 1 (Acaca, Acc1), elongation long-chain fatty acids family member 6 (Elovl6), 3-hydroxy-3-methylglutaryl-coenzyme A reductase (Hmgcr), and low-density lipoprotein receptor (Ldlr) (Figure S3C). mGIPR-Ab alone reduced the expression of Fasn, Acc1, and Hmgcr in the liver. In adipose tissues, the expression of Fasn, Acc1, and leptin (Lep) were reduced only in the control-Ab/P1 and mGIPR-Ab/P1 treatment groups, while no significant changes were observed in adipogenic genes in any group (Figures S3D and S3E). The metabolic hormonal changes are represented in Figure S4. Peptide YY, pancreatic polypeptide, ghrelin, glucagon, and amylin were not changed by mGIPR-Ab, control-Ab/P1, or GIPR-Ab/P1 treatments (Figure S4). GIP concentrations were not altered by mGIPR-Ab but were significantly reduced in the control-Ab/P1 and GIPR-Ab/P1-treated groups (Figure S4). GLP-1 levels were increased only in the control-Ab/P1 or GIPR-Ab/P1 treatment groups, possibly a result of assay cross-reactivity with the conjugated GLP-1 peptide portion of the molecules (Figure S4). Consistent with the observed insulin changes, C-peptide levels were reduced in control-Ab/P1 or GIPR-Ab/P1 treatment groups (Figure S4). Finally, resistin, insulin, and leptin levels were significantly decreased in the control-Ab/P1 and GIPR-Ab/P1 treatment groups (Figure S4).
GIPR-Ab/GLP-1 induced receptor internalization and amplified cAMP response in recombinant cells expressing GLP-1R and GIPR and in INS1 832/3 cells
To study potential mechanisms by which GIPR-Ab/GLP-1 promoted synergistic BW reduction compared with GIPR-Ab or GLP-1 peptide alone, we first examined whether GIPR-Ab/GLP-1 showed differential activity on stimulating cAMP production. Control-Ab/P1 and hGIPR-Ab/P1 showed similar activity on cAMP production (half maximal effective concentration [EC50] = 49.6 and 40.1 pM, respectively) in Chinese hamster ovary K1 (CHOK1) cells expressing human GLP-1R (hGLP-1R). In contrast, hGIPR-Ab/P1 was ∼100-fold more potent (EC50 = 0.9 pM) than control-Ab/P1 (EC50 = 85.7 pM) in CHOK1 cells recombinantly co-expressing both hGLP-1R and hGIPR (hGLP-1R/hGIPR) (Figure 6A). To investigate whether a similar potency shift would occur in cells expressing GLP-1R and GIPR endogenously, we studied cAMP production in the presence of mGIPR-Ab/P1 and control-Ab/P1 in INS1 832/3 cells. mGIPR-Ab/P1 showed a more potent cAMP production than control-Ab/P1 (EC50 = 0.73 and 35.1 nM, respectively). Notably, mGIPR-Ab/P1 was also more potent than control-Ab/P1 in stimulating insulin secretion from INS1 832/3 (Figure 6A, EC50 = 0.19 and 5.3 nM, respectively), suggesting that the superior cAMP response induced by mGIPR-Ab/P1 translated into increased glucose-induced insulin secretion in INS1 832/3 cells (Figure 6A).
Figure 6.

GIPR-Ab/GLP-1 molecules induced receptor internalization and amplified cAMP response in recombinant cells expressing GLP-1R and GIPR and in INS1 832/3 cells
(A) Representative dose-response curves of cAMP assays and insulin secretion assay with GLP-1, control-Ab/P1, and hGIPR-Ab/P1 in cells expressing hGLP-1R or hGLP-1R/hGIPR or with GLP-1, control-Ab/P1, and mGIPR-Ab/P1 in INS1 832/3 cells. Data shown are representative of n ≥ 3 experiments.
(B) FACS analysis of control human Fc antibody (hFc-Ab), hGIPR-Ab, control-Ab/P1, and hGIPR-Ab/P1 in cells expressing hGIPR, hGLP-1R, and hGLP-1R/hGIPR. The schematic figures represent the proposed receptor binding model for hGIPR-Ab/P1. Data represent mean ± SEM of n = 2 replicates per treatment.
(C) Comparison of GLP-1, hGIPR-Ab/P1, and control-Ab/P1 induced hGLP-1R (orange-left) and hGIPR (red-right) receptor internalization in a CHOK1 cell line stably expressing both receptors. Cells were fixed, permeabilized, and stained at indicated time points after ligand (5 nM) stimulation. Image data shown are representative of n ≥ 3 experiments.
(D) Pretreatment of CHOK1 cells expressing both hGLP-1R and hGIPR with 0.4 M sucrose for 15 min prevented hGIPR-Ab/P1 (5 nM) induced hGLP-1R (orange) and hGIPR (red) receptor and ligand (green) internalization (30 min time point shown) (left panel) and reduced cAMP production by >90% upon treatment at all concentrations of hGIPR-Ab/P1 tested (3, 10, 30 pM) at 15 min (right panel, ± SEM). Data shown are representative of n ≥ 3 experiments.
See also Figure S7.
We used a fluorescence-activated cell sorting (FACS)-based binding assay to show that the hGIPR-Ab/P1 molecule presents equivalent binding to controls when the cells express only one receptor; however, a distinct binding pattern was seen in cells expressing both GLP-1R and GIPR (Figure 6B). In cells expressing hGIPR alone, the shift in intensity for hGIPR-Ab/P1 was equivalent to hGIPR-Ab, indicating that both molecules bind to hGIPR similarly. In cells expressing hGLP-1R alone, control-Ab/P1, and hGIPR-Ab/P1 showed a similar binding pattern (Figure 6B). Although the shift in binding for hGIPR-Ab/P1 overlapped partially with control-Ab/P1 or hGIPR-Ab, some populations of cells showed distinct binding patterns in cells expressing hGLP-1R/hGIPR when assessed by flow cytometry. We hypothesized that some hGIPR-Ab/P1 molecules may bind to GLP-1R or GIPR independently or to both receptors simultaneously in cells expressing both receptors (Figure 6B). To further explain our FACS binding data, the interaction between GLP-1R and GIPR was studied after co-immunoprecipitation/immunoblotting western blot analysis of U2OS cells that stably expressed SNAP-tagged GLP-1R and GIPR (Figure S5). For both immunoblots probed with either anti-GIPR or anti-SNAP, GIPR and SNAP-GLP-1R bands were seen only in clones 16-1 and 16-9. The intensity of the SNAP-GLP-1R band was darker for clone 16-9 than for clone 16-1 (Figure S5A). This finding correlated with the level of receptor expression measured in U2OS cells (Figure S5B). In addition, GLP-1R or GIPR β-arrestin recruitment was studied in a PathHunter system (DiscoveRx, Fremont, CA) (Figure S6). In this system, cells expressed enzyme acceptor (EA)-tagged β-arrestin and ProLink-tagged GLP-1R (GLP-1R-ProLink) or GIPR (GIPR-ProLink). To study β-arrestin recruitment in cells co-expressing both receptors, we generated cell lines that expressed GIPR in GLP-1R-ProLink cells (GLP-1R-ProLink/GIPR) and GLP-1R in GIPR-ProLink cells (GIPR-ProLink/GLP-1R) (Figure S6). GLP-1 showed similar β-arrestin recruitment in GLP-1R-ProLink and GLP-1R-ProLink/GIPR cells (Figure S6A and S6B). Similarly, GIP showed β-arrestin recruitment in GIPR-ProLink and GIPR-ProLink/GLP-1R cells (Figures S6C and S6D). Control-Ab/P1 and hGIPR-Ab/P1 showed similar β-arrestin recruitment in cells expressing GLP-1R-ProLink and GLP-1R-ProLink/GIPR; however, none of them generated any signal in cells expressing GIPR-ProLink (Figures S6A–S6C), and, as expected, β-arrestin recruitment are ligand dependent. Intriguingly, hGIPR-Ab/P1, but not control-Ab/P1, showed β-arrestin recruitment in cells expressing GIPR-ProLink/GLP-1R (Figure S6D), which indicated that hGIPR-Ab/P1 bound to two receptors simultaneously and triggered signals through GIPR-ProLink. Together, the co-immunoprecipitation and the β-arrestin recruitment data suggest that GIPR and GLP-1R may form dimers, and that hGIPR-Ab/P1 binds to two receptors simultaneously, triggering β-arrestin recruitment.
Ligand-induced GLP-1R and GIPR receptor internalization was determined using high-content imaging with validated GLP-1R and GIPR antibodies (Figure S7A) in CHOK1 cells stably expressing hGLP-1R alone (Figure S7B) and both receptors (Figure 6C and Figure S7C). Consistent with our efforts to reduce the potency of GLP-1R agonism, native GLP-1 induced a faster and greater degree of GLP-1R internalization in cells expressing hGLP-1R only than with hGIPR-Ab/P1 or control-Ab/P1 (Figure S7B). However, in CHOK1 cells expressing both GLP-1R and GIPR, hGIPR-Ab/P1 induced greater GLP-1R internalization than GLP-1 or control-Ab/P1 (Figures 6C, left, and S7C for a complete time course). In cells expressing both receptors, the maximal amount of GLP-1R internalization achieved by the GLP-1-treated cells was less than half of the GLP-1R internalization reached by hGIPR-Ab/P1-treated cells (Figure S7C). Limited GLP-1R internalization was observed with control-Ab/P1 (Figures 6C, left, and S7B). Similar findings were also observed with ligand-induced GIPR internalization (Figure 6C, right). hGIPR-Ab/P1 induced a faster and greater degree of GIPR internalization in CHOK1 cells stably expressing hGLP-1R/hGIPR than GIP or control-Ab/P1 (Figure 6C).
As the degree of hGIPR-Ab/P1-induced receptor internalization correlated with cAMP production, we tested whether inhibition of receptor internalization reduced cAMP production upon GLP-1R activation. Pretreatment of CHOK1 cells stably expressing hGLP-1R/hGIPR with a hypertonic solution of sucrose inhibited ligand-induced internalization as illustrated by the disappearance of intracellular staining of receptor puncta (Figure 6D) comparing the left panel with the right panel. A dose response of hGIPR-Ab/P1 showed that inhibiting receptor internalization decreased cAMP production by >90% (Figure 6D). Together, these data highlight the importance of receptor internalization for cellular function.
Co-localization of hGIPR-Ab/P1 with hGLP-1R and hGIPR receptors and with early and recycling endosome markers
To understand whether the bispecific molecule hGIPR-Ab/P1 co-localized with its receptors GLP-1R and GIPR, U2OS cells stably expressing SNAP-tagged GLP-1R and GIPR were used to enable simultaneous detection of GLP-1R or GIPR. We used U2OS cells as their cytoplasm is larger than that of CHOK1 cells; this enabled better visualization of the subcellular localization of the internalized receptors and conjugates. Simultaneous detection of SNAP-Alexa Fluor 546-labeled GLP-1R, GIPR (anti-GIPR Ab), and hGIPR-Ab/P1 (anti-human immunoglobulin G [IgG] Ab) showed that hGIPR-Ab/P1, GLP-1R, and GIPR are co-localized upon internalization (Figure 7A). To further expand on the internalization of the hGIPR-Ab/P1 molecule and its receptor GLP-1R, cells were co-stained with early endosome auto-antigen 1, an early endosome marker, and Rab11, a recycling endosome marker (Figures 7B and 7C). Both markers showed co-localization with GLP-1R and hGIPR-Ab/P1, supporting the internalization of a receptor-ligand complex within an endosomal pathway (Figures 7B and 7C).
Figure 7.

Co-localization of hGIPR-Ab/P1 with hGLP-1R and hGIPR receptors and with early and recycling endosome markers
U2OS cells stably expressing SNAP-tagged hGLP-1R and hGIPR were fixed and permeabilized at indicated time points after hGIPR-Ab/P1 stimulation.
(A) hGIPR-Ab/P1 (green) co-localized with hGLP-1R (orange) and hGIPR (red) in dual receptor-expressing cells (30 min after stimulation).
(B) hGIPR-Ab/P1 (green) co-localized with hGLP-1R (orange) and EEA1 (red), an early endosome marker.
(C) hGIPR-Ab/P1 (green) co-localized with hGLP-1R (orange) and Rab11 (red), a perinuclear recycling endosome marker. Data shown are representative of n ≥ 3 experiments.
Discussion
The demand for developing safe and effective anti-obesity agents is increasing, and new compounds are being tested through clinical trials.23 An approach to generate such agents targets GIPR and GLP-1R pathways as the GIP/GLP-1 dual agonist has shown BW loss in rodents and humans.10,11 Conversely, we and others reported that GIPR antagonism, rather than agonism, attenuated weight gain or stimulated BW loss in preclinical obesity models.21,33 Co-administration of anti-GIPR antagonist Abs and GLP-1Ras showed enhanced weight loss in DIO mice and monkeys compared with either of these compounds administered alone.21 Given the BW loss in these models,21 we engineered monomeric GIPR/GLP-1R by conjugating GLP-1 peptides to anti-GIPR Abs. These molecules target both pathways to increase BW loss and improve obesity-related metabolic comorbidities. GIPR-Ab/GLP-1 showed potent GIPR antagonist and GLP-1R agonist activities in vitro, which translated to effective BW reduction in two preclinical models of obesity. The bispecific molecule was more effective than additive effects of monotherapies targeting either GLP-1 or GIPR.
An advantage of Ab therapeutics is their long half-life. We used this property to engineer GLP-1 peptides with extended half-life compared with the native GLP-1 peptide to improve the duration of the GLP-1 activity closer to the duration of the anti-GIPR Ab activity (Figure 2). Modifications applied to the GLP-1 peptide included Aib substitution at positions 8 and 22 and glycine substitution at position 36. When these modified peptides were conjugated to the anti-GIPR Ab, they were more stable than when unconjugated. The PK data suggest that our bispecific molecules showed comparable PK profiles to typical Abs (Figure 2), indicating that these molecules may be suitable for weekly to monthly administration. Further studies are needed to find out which regimen will be the most suitable to treat patients.
Tissue biodistribution of the mGIPR-Ab and mGIPR-Ab/P1 was evaluated using gamma-emitting radiohalogen Indium-111 (111In, half-life of 3 days). 111In is known to be a residualizing probe; once the mAb is taken up by receptor-mediated endocytosis, the radioactive catabolites of the mAb labeled with 111In get trapped inside cells and accumulate.34 Hence, tissue PK observed with 111In probes does not represent real-time PK but represents cumulative tissue uptake of mAbs. This approach may help identify tissues that contribute most toward mAb uptake and catabolism.35 The mAb distribution rate is determined by convective transport, extravasation, receptor-mediated endocytosis, extent of binding to tissues, and eventual elimination.27 Both GLP-1R and GIPR are expressed in the pancreas, stomach, small intestine, lung, heart, and many regions of the brain.36, 37, 38, 39 In line with these findings, the biodistribution study showed that 111In-DOTA-labeled molecules accumulated in the tissues listed above in a time-dependent manner. Notably, the mGIPR-Ab/P1 tissue to blood AUC ratio was higher in the pancreas, followed by liver, bone marrow, and lung compared with the ratio for mGIPR-Ab. The difference in the ratio may reflect the dual receptor-binding ability of the mGIPR-Ab/P1 molecule. Notably, the tissue to blood AUC ratio for the pancreas was 61.2% higher for mGIPR-Ab/P1 than for mGIPR-Ab. As GIPR and GLP-1R are co-expressed in pancreatic islets, we hypothesized that other tissues co-expressing these receptors may accumulate higher concentrations of mGIPR-Ab/P1 than mGIPR-Ab. However, we did not measure mGIPR-Ab/P1 in the other tissues as they were either not collected (the signal was diluted as it was measured in the entire organ, e.g., brain instead of localized area of the brain) or the gamma-emitting radiohalogen 111In technique may not have been sensitive enough to detect the signal above background.
In lean and DIO mice, mGIPR-Ab/P1 promoted greater BW loss than mGIPR-Ab alone, control-Ab/P1 alone, or the sum of the weight loss obtained with both molecules (Figure 3A), indicating a synergistic effect of mGIPR-Ab/P1 on weight loss. Also, in the separate study performed in DIO mice with dulaglutide as a comparator, mGIPR-Ab/P1 showed greater BW loss than dulaglutide, a commercialized GLP-1 analog (Figure S2), confirming that these findings were not unique to the control-Ab/GLP-1 molecule. Interestingly, we showed that the cAMP response induced by hGIPR-Ab/P1 was comparable with that induced by control-Ab/P1 but weaker than that induced by native GLP-1 in recombinant cells expressing GLP-1R only. As bispecific molecules target both the GLP-1R and the GIPR, we determined the cAMP activity in recombinant cells that we engineered to co-express both receptors. In these co-expressing cells, hGIPR-Ab/P1 showed a 100-fold increase in cAMP activity compared with the control-Ab/P1. Consistent with findings of previous studies,40 we showed that GIPR and GLP-1R may form dimers (Figure S5). Further, the results from FACS analysis and β-arrestin recruitment indicated that hGIPR-Ab/P1 may bind to GIPR and GLP-1R simultaneously. Furthermore, in INS1 832/3, a rat insulinoma cell line known to endogenously express both GLP-1R and GIPR,41 mGIPR-Ab/P1 showed ∼30-fold greater activity on cAMP production than control-Ab/P1. Notably, mGIPR-Ab/P1 was also more potent in stimulating insulin secretion in INS1 832/3 than in control-Ab/P1. These data suggest that the effect of GIPR/GLP-1R on BW efficacy may be steered by tissues co-expressing both receptors. Furthermore, our biodistribution study indicated that pancreas contained the highest levels of mGIPR-Ab/P1, leading us to determine the effects of bispecific molecule in GiprβCell−/− mice. Surprisingly, mGIPR-Ab/P1 showed similar effects on BW reduction in GiprβCell−/− mice compared with that in Giprfl/fl mice, suggesting that tissues other than pancreatic β cells were responsible for the observed synergistic BW loss. Importantly, the Cre recombinase driven by the rat insulin promoter (RIP-Cre) has been reported to display Cre activity in the brain,42 and the RIP-Giprβcell−/− may not be β cell specific. However, we previously reported21 that Gipr deletion using RIP-Cre does not impact BW regulation. Similarly, in the model presented by Campbell et al.31 utilizing Cre recombinase driven by the mouse insulin promoter (MIP-Cre) to induce deletion of Gipr, the MIP-Gipr βcell−/− did not display differences in BW during high-fat diet (HFD) feeding over time compared with MIP-Cre controls. Given the reproducibility of the phenotype from the two previously described mouse models of GIPR β cell knockout mice when fed HFD, we believe that GIPR activity in pancreatic β cells does not regulate BW or fat mass in DIO mice.
Studies have shown that GIPR and GLP-1R are co-expressed in a subset of cells from human and mouse hypothalamus,43 and as mGIPR-Ab/P1 decreased food intake to a greater extent than the other molecules, the brain may be the organ that drives the synergistic effect on BW and food intake. In addition, antagonizing GIP signaling in the central nervous system (CNS) enhanced the effects of leptin on weight loss in DIO mice.22 These authors reported that food intake decreases with GIPR antagonism22 or agonism,43 but it was not additive to concomitant GLP-1R activation. However, these data are not in line with our previously reported data, as we have already shown an additive effect on food intake reduction when combining GIPR antagonism with GLP-1R activation21 and are confirming this effect in this report with mGIPR-Ab/P1. The data discrepancy between the other studies22,43 and our independent studies may be explained by the fact that Kaneko et al.22 used a low dose of a GIPR-neutralizing Ab, and Adriaenssens et al.43 determined food intake acutely. We anticipate neither the GIPR-Ab nor the mGIPR-Ab/P1 molecule has direct access to the CNS as both molecules have large molecular weights. However, we cannot rule out that the synergistic effects we observed may have arisen from signals through other tissues. Further studies are needed to determine whether mGIPR-Ab/P1 acts through peripheral tissues or other CNS areas accessible by large molecules, such as the area postrema, to regulate food intake.
GIPR-Ab/GLP-1 appears to promote BW reduction in DIO mice compared to that achieved by the GIP/GLP-1 dual agonist LY3298176. LY3298176 lowered BW by 25%–28% in DIO mice after day 15 of weekly injection at the 10 nmol/kg or 30 nmol/kg doses,10 which is similar to our observation with mGIPR-Ab/P1 at the 2.5 mg/kg dose (∼16 nmol/kg). In vitro, LY3298176 has shown similar potency to native GIP and about 13-fold weaker potency than native GLP-1.10 Similarly, GIPR-Ab/GLP-1 showed about 20- to 40-fold weaker cAMP potency compared with native GLP-1 peptide in cells expressing GLP-1R. We could not compare GIP potency between GIPR-Ab/GLP-1 and co-agonist as our molecule antagonizes the GIPR, whereas LY3298176 agonizes it. It has now been shown44 that chronic GIPR agonism and antagonism efficacy prevent similar weight gain alone or weight loss when combined with GLP-1Ra. The authors hypothesized that agonist induced desensitization. The desensitization of the GIPR may explain the decreased response to an agonist upon repeated stimulation44 and why it generated similar weight loss to a GIPR antagonist. Mice treated with either control-Ab/P1 or mGIPR-Ab/P1 showed RER values reflective of increased lipid oxidation in accordance with RER values measured in DIO mice treated with semaglutide.45 Though mGIPR-Ab showed no difference on RER values compared with the vehicle, RER values of mice treated with control-Ab/P1 significantly increased at day 5 than that of mice treated with mGIPR-Ab/P1. This increase correlated with a rebound effect on BW and food intake, a phenomenon observed earlier with long-acting GLP-1Ra.46 Hepatic and adipose lipogenesis are inhibited with GLP-1.47 In this study, mGIPR-Ab, control-Ab/P1, and mGIPR-Ab/P1 showed significant reduction in the expression of genes involved in hepatic lipogenesis. However, no synergistic effects similar to the synergistic effect that we observed on BW with mGIPR-Ab/P1 was seen on genes involved in hepatic lipogenesis molecules. Interestingly, the expression of mitochondrial biogenesis markers was enhanced in the liver of mGIPR-Ab or mGIPR-Ab/P1 treatment group than in vehicle and control Ab/P1 groups. Further studies are needed to understand the mechanism of action of GIPR antagonism on mitochondrial biogenesis.
Upon ligand binding, G-protein-coupled receptors (GPCRs) undergo agonist-mediated internalization.48 Studies have shown that many GPCRs mediate G-protein signaling at both the plasma membrane and the endosomal membrane, a step that seems important to prolong G-protein signaling.49 These authors also showed that sustained cAMP signaling is physiologically relevant as it resulted in prolonged responses in vivo.49 GLP-1R is internalized when activated by its agonist50 and it mediates endosomal cAMP generation.51 Once internalized, GIPR co-localizes with adenylate cyclase in early endosomes, and it continues to trigger cAMP production, resulting in a sustained cAMP response.52 As GIPR-Ab/GLP-1 binds to both GLP-1R and GIPR and promotes a sustained cAMP response, we examined subsequent signaling events in cells that co-express both receptors. We found that hGIPR-Ab/P1 triggers earlier and greater GLP-1R and GIPR internalization than native GLP-1, GIP, or control-Ab/P1, and this effect lasted up to 60 min. Inhibition of receptor internalization with sucrose significantly reduced cAMP production, suggesting that hGIPR-Ab/P1 induced receptor internalization, a step critical for pronounced cAMP response. In addition, the internalized bispecific molecule was co-localized with GLP-1R and GIPR in early and recycling endosomes. Taken together, GIPR-Ab/GLP-1 acts by promoting receptor internalization and sustained cAMP response, and these responses may explain their efficacy in reducing BW.
Here, we report two separate bispecific molecules, mGIPR-Ab/P1 and mGIPR-Ab/P2, that cross-react with mouse GLP-1R and GIPR, and hGIPR-Ab/P1 and hGIPR-Ab/P2 that cross-react with monkey hGIPR and hGLP-1R. Importantly, GLP-1 peptide potency cannot be indefinitely increased, as GLP-1 analogs are known to induce nausea and vomiting in humans and may become intolerable.53 To mitigate the potential for nausea, we intended to reduce potency by introducing D15E substitution in P2. In our in vitro assays, we could not differentiate GLP-1 agonist potency between P1 and P2 as the expression levels of GLP-1R in the recombinant cells were higher than in cells that expressed endogenous GLP-1R. However, in DIO mice or in obese monkey models, mGIPR-Ab/P1 and hGIPR-Ab/P1 showed greater BW reduction than mGIPR-Ab/P2 and hGIPR-Ab/P2. This greater effect was also observed on food intake but not on plasma metabolic parameters. These findings show that the synergic effects were observed only on BW and food intake. Both the DIO mice and obese monkeys models allowed us to rank the efficacy of the two bispecific molecules on BW. Notably, our bispecific molecules were well tolerated in obese monkeys.
In summary, our monomeric GIPR-Ab/GLP-1 molecules (antagonist/agonist) greatly decreased BW in both mouse and monkey preclinical models of obesity. GIPR-Ab/GLP-1 reduced the RER and decreased the levels of many metabolic hormones in DIO mice. FACS analysis and β-arrestin recruitment indicated that hGIPR-Ab/P1 may bind to GIPR and GLP-1R simultaneously. We provide data suggesting that the GIPR-Ab/GLP-1 molecule binds to GIPR and GLP-1R simultaneously and triggers receptor internalization to amplify endosomal cAMP signaling in cells expressing both receptors. This may possibly explain the efficacy of the bispecific molecule. However, further studies are needed to substantiate this and further elucidate the mechanism for the apparent advantage that the combination of GIPR antagonism and GLP-1R agonism has compared with GLP-1R agonism alone.
Limitations of study
Limitations of this study include using recombinant cells and INS1 832/3 cells instead of primary islet cells for mechanistic studies. We and others54 have found high batch-to-batch and donor-to-donor variability and small assay windows when using mouse or human primary islets. Allowing the islets to recover from damages caused during the isolation period is important; however, increasing recovery period is a proportional time-dependent process involving deprivation of nutrients and oxygen to β cells, which makes them less responsive than cell lines. A perifusion system where the islets are exposed to a continuously flowing equilibrated medium would be an ideal system to mimic the time points and conditions used with the recombinant and INS1 832/3 cells.
Another limitation is how we validated the commercial GIPR and GLP-1R antibodies. We used parental cells and cells recombinantly expressing GIPR or GLP-1R to show that these antibodies detect each receptor. The use of knockout tissues and/or tissues that do not express GIPR or GLP-1R to show complete lack of signal detection may have further provided evidence for each antibody specificity.
Our study was not designed to demonstrate how the bispecific molecules reduce BW or why they appear to do so to a greater degree compared with GLP-1R agonism alone. These questions need further study.
STAR★Methods
Key resources table
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human GLP-1R Mouse Monoclonal IgG2B clone#197920 | R&D Systems | Cat#MAB2814; RRID:AB_2109906 |
| Human GIPR Mouse Monoclonal IgG1 clone#591853 | R&D Systems | Cat#MAB8210 |
| Goat anti-Human IgG Antibody, Alexa Fluor® 488 | Thermo Fisher Scientific | Cat#A11013; RRID:AB_141360 |
| Goat anti-Mouse IgG Alexa Fluor® Plus 488 | Thermo Fisher Scientific | Cat#A32723; RRID:AB_2633275 |
| Goat anti-Mouse IgG Alexa Fluor® Plus 555 | Thermo Fisher Scientific | Cat#A232727; RRID:AB_2633276 |
| Goat anti-Mouse IgG Alexa Fluor® 647 | Thermo Fisher Scientific | Cat#A21235; RRID:AB_2535804 |
| Goat anti-Rabbit IgG Alexa Fluor® 647 | Thermo Fisher Scientific | Cat#A21245; RRID:AB_2535813 |
| Rabbit EEA1 Polyclonal Antibody | Thermo Fisher Scientific | Cat#PA1-063A; RRID:AB_2096819 |
| Rabbit Rab11 Monoclonal Antibody (D4F5) | Cell Signaling Technology | Cat#5589; RRID:AB_10693925 |
| SNAP-Surface® Alexa Fluor® 546 | New England Biolabs | Cat#S9132S |
| SNAP-Surface® Alexa Fluor® 647 | New England Biolabs | Cat#S9136S |
| Mouse monoclonal anti-human IgG Fc | Amgen | Clone 1.35 |
| Mouse monoclonal anti-human GLP-1 | Thermo Fisher Scientific | Cat#ABS 033-04-02; Clone 4, RRID:AB_876534 |
| hGIPR-Ab | Killion et al.21 (Amgen) | https://stm.sciencemag.org/content/10/472/eaat3392.short |
| mGIPR-Ab | This paper | N/A |
| Anti-GIPR antibody | Abcam | Cat#ab136266 |
| Anti-SNAP-tag antibody | New England Biolabs | Cat#P310S |
| Biological samples | ||
| Cynomolgus monkey plasma (K2EDTA) | BioIVT | N/A |
| CD-1 mouse plasma (K2EDTA) | BioIVT | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Human GIP | Phoenix Pharmaceuticals | Cat#027-02 |
| GLP-1 | Phoenix Pharmaceuticals | Cat#028-13 |
| GLP-1 analog | This paper | N/A |
| hGIPR-Ab/P1 | This paper | N/A |
| hGIPR-Ab/P2 | This paper | N/A |
| mGIPR-Ab/P1 | This paper | N/A |
| mGIPR-Ab/P2 | This paper | N/A |
| Control-Ab/P1 | This paper | N/A |
| Sucrose | Sigma | Cat#S0389 |
| Hoechst 33342, trihydrochloride trihydrate | Thermo Fisher Scientific | Cat#H3570; RRID:AB_2651135 |
| p-SCN-Bn-DOTA (chemical name: S-2-(4-isothiocyanatobenzyl)-1,4,7,10-tetraazacyclododecane tetraacetic acid | Macrocyclics | Cat#B-205 |
| Indium chloride (111In) | IPG-In-111, Nordion (Canada) Inc. | https://www.nordion.com/wp-content/uploads/2014/10/MI_Indium-111_Canada.pdf |
| Streptavidin-HRP | R&D Systems | Cat#DY998 |
| Rink-Amide-MBHA Resin (100-200 mesh) 1% DVB | Peptides International | RFR-1063-PI |
| 20% 4-Methylpiperidine in DMF | Sigma-Aldrich | 792152 |
| N,N-Diisopropylcarbodiimide | Sigma-Aldrich | CAS#693-13-0 |
| 6-Chloro-1-hydroxybenzotriazole | Chem Impex International | CAS#26198-19-6 |
| Fmoc-Lys(ivDde)-OH | ChemPep Inc | CAS#204777-78-6 |
| Cysteamine hydrochloride | Sigma-Aldrich | CAS#156-57-0 |
| Cystamine dihydrochloride | Sigma-Aldrich | CAS#56-17-7 |
| Tris(2-carboxyethyl)phosphine hydrochloride | Sigma-Aldrich | CAS#51805-45-9 |
| Dehydroascorbic acid | Biosynth International | CAS#490-83-5; MD16669 |
| Complete, ETDA-free protease inhibitor cocktail tablets | Roche | Cat#04-693-132-001 |
| Recombinant protein G agarose | Invitrogen | Cat#15920010 |
| Protein A agarose | Invitrogen | Cat#15918014 |
| 2X Laemmli sample buffer | Bio-Rad | Cat#1610737 |
| 2-Mercaptoethanol | Bio-Rad | Cat#1610710 |
| DC protein assay kit II | Bio-Rad | Cat#5000112 |
| Critical commercial assays | ||
| Cyclic AMP Dynamic 2 Kit | Cisbio | Cat#62AM4PEJ |
| pGloSensor-22F cAMP Plasmid | Promega | Cat#E2301 |
| GloSensor cAMP Reagent | Promega | Cat#E1290 |
| Insulin High Range kit | Cisbio | Cat#62IN1PEG |
| Mouse Insulin ELISA | ALPCO | Cat#80-INSMS-E01 |
| Triglyceride Quantification Colorimetric Kit | BioVision | Cat#K622 |
| Total Cholesterol | Wako | Cat#999-02601 |
| PathHunter® Detection Kit | DiscoverX | Cat#93-0001 |
| Milliplex Mouse Metabolic Magnetic Bead Panel (amylin (Active), C-Peptide, ghrelin (Active), GIP (Total), GLP-1 (Active), glucagon, insulin, leptin, PP, PYY and resistin) | EMD Millipore | Cat#MMHMAG-44K |
| Milliplex Mouse adiponectin Single Plex Magnetic Bead Kit | EMD Millipore | Cat#MADPNMAG-70K |
| TaqMan™ RNA-to-CT™1-Step Kit | Thermo Fisher Scientific | Cat# 4392938 |
| RNeasy 96 Universal Tissue Kit | QIAGEN | Cat#74881 |
| Gene Expression Assay- G6pc (G6Pase) | Thermo Fisher Scientific | Mm00839363_m1 |
| Gene Expression Assay- Pck1 (PEPCK) | Thermo Fisher Scientific | Mm01247058_m1 |
| Gene Expression Assay- Cox8b | Thermo Fisher Scientific | Mm00432648_m1 |
| Gene Expression Assay- Ppargc1a (Pgc1α) | Integrated DNA Technologies | Mm.PT.5828716430 |
| Gene Expression Assay- Fasn | Integrated DNA Technologies | Mm.PT.58.14276063 |
| Gene Expression Assay- Scd1 | Integrated DNA Technologies | Mm.PT.58.8351960 |
| Gene Expression Assay- Acc1 | Integrated DNA Technologies | Mm.PT.58.12492865 |
| Gene Expression Assay- Elovl6 | Thermo Fisher Scientific | Mm00851223_s1 |
| Gene Expression Assay- Hmgcr | Integrated DNA Technologies | Mm.PT.58.31538611 |
| Gene Expression Assay- Ldlr | Integrated DNA Technologies | Mm.PT.58.23359070 |
| Gene Expression Assay- Lpl | Thermo Fisher Scientific | Mm00434764_m1 |
| Gene Expression Assay- Lipe (HSL) | Thermo Fisher Scientific | Mm00495359_m1 |
| Gene Expression Assay- Pnpla2 (Atgl) | Thermo Fisher Scientific | Mm00503040_m1 |
| Gene Expression Assay- Lep (Leptin) | Thermo Fisher Scientific | Mm00434759_m1 |
| Gene Expression Assay- AdipoQ | Thermo Fisher Scientific | Mm00456425_m1 |
| Gene Expression Assay- Cebpa | Integrated DNA Technologies | Mm.PT.58.30061639.g |
| Gene Expression Assay- Pparg | Integrated DNA Technologies | Mm.PT.58.31161924 |
| Gene Expression Assay- Fabp4 (aP2) | Thermo Fisher Scientific | Mm00445878_m1 |
| Gene Expression Assay- Ucp1 | Thermo Fisher Scientific | Mm01244861_m1 |
| Gene Expression Assay- Ppia | Integrated DNA Technologies | Mm.PT.39a.2.gs |
| Experimental models: cell lines | ||
| CHOK1 cells, stably expressing human GLP-1R, clone H20 | Amgen | N/A |
| CHOK1 cells, stably expressing SNAP-tagged hGLP-1R, clone 10 | Amgen | N/A |
| HEK293T cells, stably expressing human GIPR, clone 10 | Amgen | N/A |
| CHOK1 cells, stably expressing human GLP-1R and GIPR, clone M1 | Amgen | N/A |
| CHOK1 cells, stably expressing mouse GLP-1R, clone 8 | Amgen | N/A |
| CHO AM1D cells, stably expressing monkey GLP-1R, clone 6 | Amgen | N/A |
| CHO AM1D cells, stably expressing mouse GIPR, clone 3 | Amgen | N/A |
| HEK293T cells, stably expressing monkey GIPR, clone 3 | Amgen | N/A |
| INS1 832/3 | EMD Millipore | Cat#SCC208 |
| U2OS | ATCC | Cat#HTB-96 |
| U2OS SNAP-tagged hGLP-1R, clone 15 | Amgen | N/A |
| U2OS SNAP-tagged hGLP-1R + hGIPR, clone 16-1 | Amgen | N/A |
| U2OS SNAP-tagged hGLP-1R + hGIPR, clone 16-9 | Amgen | N/A |
| PathHunter® CHOK1 human GIPR β-Arrestin Cell Line (GIPR-PK) | DiscoverX | Cat#93-1095C2 |
| PathHunter® CHOK1 human GLP1R β-Arrestin Cell Line (GLP-1R-PK) | DiscoverX | Cat# 93-0300C2 |
| CHOK1 cells stably expressing human GLP-1R-PK and GIPR, clone 1 (GLP-1R-PK/GIPR) | Amgen | N/A |
| CHOK1 cells stably expressing human GIPR-PK and GLP-1R, clone 12 (GIPR-PK/GLP-1R) | Amgen | N/A |
| Experimental models: organisms/strains | ||
| Giprfl/fl: B6.Cg-Tg(Ins2-cre)25Mgn/J | Killion et al.,21 (Amgen) | https://stm.sciencemag.org/content/10/472/eaat3392.short |
| Naive C57BL/6 mice used in biodistribution study | University of Massachusetts Medical School | NA |
| Male CD-1 IGS mice (Crl:CD1(ICR)) | Charles River | Strain code: 022 |
| C57BL/6NHsd (naive male C57BL/6 mice) | Envigo | RRID#565547 |
| C57BL/6NHsd (naive male C57Bl6 fed high fat diet) | Envigo | RRID#565547 |
| Naive female cynomolgus monkeys (Macaca fascicularis) | MPI Research stock colony | NA |
| Naive male obese cynomolgus monkeys (Macaca fascicularis) | Kunming Biomed International stock colony | NA |
| Software and algorithms | ||
| GraphPad Prism v7.02 | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID:SCR_002798 |
| Harmony High Content Imaging and Analysis Software | PerkinElmer | HH17000001 |
| Watson LIMS v7.4 | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/INF-21000#/INF-21000 |
| Phoenix WinNonlin v6.4 | Certara USA, Inc | https://www.certara.com/phoenix-winnonlin-primary/?ap=PMX |
| Phoenix WinNonlin v8.1 | Certara USA, Inc | https://www.certara.com/phoenix-winnonlin-primary/?ap=PMX |
| Oxymax for Windows v5.53 | Columbus Instruments | 0233-128M |
| Other | ||
| Operetta | PerkinElmer | HH12000001 |
| Operetta CLS | PerkinElmer | HH16000000 |
| Roche C311/C501 clinical chemistry analyzers | Roche | Model C311/C501 |
| Clinical Chemistry Analyzer | Siemens Medical Solutions) | Model 1800 |
| Comprehensive Lab Animal Monitoring System (CLAMS / Oxymax Model 2018) | Columbus Instruments | 0233-004M, Serial No 190395 |
| QuantStudio 7 Flex Real-Time PCR System | Thermo Fisher Scientific | 4485701 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Murielle M. Véniant (mveniant@amgen.com).
Materials availability
Upon execution of a material transfer agreement, Amgen is willing to distribute materials and protocols to qualified researchers. Requests from an academic or nonprofit institution should be submitted at https://wwwext.amgen.com:443/partners/academic-collaborations/new-requests/. Requests from a for-profit entity should be submitted to BDopportunities@amgen.com.
Data and code availability
This study did not generate any unique datasets or code.
Experimental model and subject details
Cell lines
CHOK1 stably expressing hGLP-1R cells or mouse GLP-1R cells were cultured in Ham’s F12 media (Thermo Fisher, Waltham, MA) supplemented with 1% penicillin/streptomycin/L-glutamine (PSG; Thermo Fisher), 10% fetal bovine serum (FBS; Thermo Fisher), 250 μg/mL zeocin (Thermo Fisher). CHO AM1D cells stably expressing monkey GLP-1R cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher) supplemented with 1% PSG, 10% dialyzed FBS, 1% nonessential amino acids (NEAA, Thermo Fisher), 1 mM sodium pyruvate, 1% sodium hypoxanthine and thymidine supplement (HT supplement, Thermo Fisher), 400 μg/mL hygromycin (Thermo Fisher). CHOK1 hGLP-1R/hGIPR cell line was cultured in Ham’s F12 media supplemented with 1% PSG, 10% FBS, 5 μg/mL puromycin (Thermo Fisher), 250 μg/mL zeocin. HEK293T hGIPR cells were cultured in DMEM, 1% PSG, 10% FBS, and 5 μg/mL puromycin. CHO AMID mouse GIPR cells were cultured in DMEM supplemented with 1% PSG, 10% dialyzed FBS, 1% NEAA, 1 mM sodium pyruvate, 1% HT supplement, and 400 μg/mL hygromycin. 293T monkey GIPR cells were cultured in DMEM supplemented with 1% PSG, 10% FBS, 2 μg/mL puromycin. U2OS cells were cultured in McCoy’s 5A medium (Thermo Fisher) supplemented with 1% PSG and 10% FBS. U2OS cells stably expressing SNAP-tagged hGLP-1R were cultured in McCoy’s 5A medium supplemented with 1% PSG, 10% FBS, and 500 μg/mL Geneticin (Thermo Fisher). U2OS cells stably expressing SNAP-tagged hGLP-1R and hGIPR were cultured in McCoy’s 5A medium supplemented with 1% PSG, 10% FBS, 500 μg/mL Geneticin, 5 μg/mL puromycin. INS1 832/3 cells were cultured in RPMI-1640 medium (Thermo Fisher) supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 10 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 0.05 mM β-mercaptoethanol. CHOK1 cells expressing GLP-1R-PK (DiscoverX, 93-0300C2) or GIPR-PK cells (DiscoverX, 93-1095C2) were cultured in Ham’s F12 media supplemented with 1% PSG, 10% FBS, 1% NEAA, 300 μg/mL hygromycin, 800 μg/mL Geneticin. GLP-1R-PK/GIPR cells were cultured in Ham’s F12 media supplemented with 1% PSG, 10% FBS, 1% NEAA, 300 μg/mL hygromycin, 800 μg/mL Geneticin, and 5 μg/mL puromycin. GIPR-PK/GLP-1R cells were cultured in Ham’s F12 media supplemented with 1% PSG, 10% FBS, 1% NEAA, 300 μg/mL hygromycin, 800 μg/mL Geneticin, and 330 μg/mL Zeocin. CHOK1 cells were cultured in Ham’s F12 media supplemented with 1% PSG, 10% FBS; CHOK1 cells expressing SNAP-tagged hGIPR were cultured in the same media with the addition of 500 μg/mL Geneticin. All cells were cultured in humidified incubators maintained at 37°C and 5% CO2. The sex of CHOK1, CHO AM1D, HEK293T and U2OS cell lines was female and the INS1 832/3 cell line was male. Further authentication of these cell lines was not performed.
Normal mice
All mouse studies using non-DIO mice for BW measurements and PK study were conducted at Amgen Inc. (Thousand Oaks, CA), an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International accredited facility. Animals were cared for in accordance with the Guide for the Care and Use of Laboratory Animals, 8th Edition. All research protocols were reviewed and approved by the Amgen Institutional Animal Care and Use Committee. Male C57BL/6 (Envigo, Indianapolis, IN) and CD-1 mice (Charles River, Hollister, CA) were delivered at approximately 4 weeks of age (26 days of age to be exact) and housed 2–3 mice per cage with littermates in static caging on an irradiated corncob bedding (Envigo Teklad 7097). Lighting in animal holding rooms was maintained on 12:12 hour light:dark cycle, and the ambient temperature and humidity were maintained at 68°F–79°F and 30%–70%, respectively. Animals had ad libitum access to irradiated pelleted feed (Envigo Teklad Global Soy Protein-free Extruded Rodent Diet 2020X) and) and reverse osmosis (RO)-chlorinated (0.3–0.5 ppm) water via an automatic watering system. Cages were changed weekly. After 9 weeks (1-week) acclimation plus 8 weeks of feeding phase for DIO mice described below), mice were single housed, and cages were changed once weekly.
Mice were acclimated to handling and BW measurements. The day before study initiation (day −1; study initiation was day 0), all mice were weighed and sorted into treatment groups based on BW so that all groups had identical starting BW before treatment (n = 7/group). Mice were intraperitoneally (IP) injected with vehicle, mGIPR-Ab (2.5 mg/kg), control-Ab/P1 (2 mg/kg), or mGIPR-Ab/P1 (0.5 or 2.5 mg/kg) every 6 days starting on day 0. BW was measured every 2 days. Terminal trunk blood was collected in ethylenediaminetetraacetic acid (EDTA) filled microtainer tubes (365974, Becton Dickinson, Franklin Lakes, NJ). Plasma insulin and lipids were measured according to the manufacturer’s guidelines. Plasma insulin was measured by enzyme-linked immunosorbent assay (ELISA; ALPCO, Salem, NH, 80-INSMS-E01), and plasma lipids were measured using a colorimetric assay (triglycerides – BioVision, Milpitas, CA, K622; total cholesterol – Fujifilm Wako, Osaka, Japan, 999-02601).
Mouse PK study was conducted in male CD-1 mice (8–12 weeks old) following a 1-week acclimation. The mGIPR-Ab/P1 and hGIPR-Ab/P1 test articles were formulated in 10 mM sodium acetate with 9% sucrose, pH 5.2. Mice received a single 5 mg/kg injection with the appropriate test article and route of administration as follows: mGIPR-Ab/P1 (IV), hGIPR-Ab/P1 (IV), and hGIPR-Ab/P1 (SC). Vascular delivery was achieved via the lateral tail, and the extravascular dose was administered in the mid-scapular region. Blood samples were collected at predetermined time points up to 7 days and 14 days after the dose for mGIPR-Ab/P1 and hGIPR-Ab/P1, respectively, by submandibular venipuncture. Whole blood was collected, placed into Microvette® 500 μl, K3 EDTA plasma separator tubes (20.1341.102, Sarstedt, Newton, NC), gently mixed by 8–10 manual inversions, and centrifuged at 11,500 x g at 4°C for 5 minutes. The resulting plasma was stored at −70°C (±10°C) until analysis.
DIO mice
All mouse studies using DIO mice were conducted at Amgen Inc., an AAALAC International accredited facility. Animals were cared for in accordance with the Guide for the Care and Use of Laboratory Animals, 8th Edition. All research protocols were reviewed and approved by the Amgen Institutional Animal Care and Use Committee.
DIO mice prepared at Amgen Inc. Mice (male C57BL/6, Envigo) were delivered at approximately 4 weeks of age (26 days of age to be exact) and housed 2–3 mice per cage with littermates in static caging on an irradiated corncob bedding (Envigo Teklad 7097). Lighting in animal holding rooms was maintained on 12:12 hour light:dark cycle, and the ambient temperature and humidity were maintained at 68°–79°F and 30%–70%, respectively. Animals had ad libitum access to irradiated pelleted feed (Envigo Teklad Global Soy Protein-free Extruded Rodent Diet 2020X) and RO-chlorinated (0.3–0.5 ppm) water via an automatic watering system. Cages were changed twice weekly. Following 1 week of acclimation, mice were introduced to 60% kcal HFD (Research Diets D12492) ad libitum. Mice were single housed after 8 weeks of HFD feeding and continued on HFD feeding for the duration of the study. Cages were changed once, then changed once weekly. Mice were randomized to experimental groups based on BW so that all groups had identical starting BW before treatment.
For the study in Figure 3A, a subset of age-matched mice (described above) remained on 2020X standard diet for the duration of feeding and study period (mice on HFD for 15 weeks at study initiation). Mice were acclimated to handling and BW measurements. The day before study initiation (day −1; study initiation was day 0), all mice were weighed and sorted into treatment groups with n = 8/group. Mice were injected (IP) with vehicle, mGIPR-Ab (2.5 mg/kg), control-Ab/P1 (2 mg/kg), or mGIPR-Ab/P1 (0.5 or 2.5 mg/kg) every 6 days starting on day 0. BW was measured every 2 days. Terminal trunk blood was collected in EDTA-filled microtainer tubes (365974, Becton Dickinson). Plasma insulin and lipids were measured according to the manufacturer’s guidelines. Plasma insulin was measured by ELISA (ALPCO, 80-INSMS-E01) and plasma lipids were measured using a colorimetric assay (triglycerides – BioVision, K622; total cholesterol – Fujifilm Wako, 999-02601). One DIO mouse from the hGIPR-Ab/P1 0.5 mg/kg group was excluded from study/data analysis because of loss of exposure (antidrug antibody [ADA]–mediated clearance).
For the study in Figure 4A, 20-week-old DIO mice were injected (IP) with vehicle, mGIPR-Ab/P1 (0.5 or 2.5 mg/kg), or mGIPR-Ab/P2 (0.5 or 2.5 mg/kg) every 6 days. BW was measured every 1–2 days. Cumulative food intake was measured between days 0 and 3, between days 12 and 15, and between days 15 and 18, and represented as average daily food intake. Terminal trunk blood was collected in EDTA-filled microtainer tubes (365974, Becton Dickinson) with protease inhibitor (05056489001, Roche Diagnostics, Santa Clara, CA) added prior to collection. Plasma insulin and lipids were measured according to the manufacturer’s guidelines. Plasma insulin was measured by ELISA (ALPCO, 80-INSMS-E01), and plasma lipids were measured using a clinical chemistry analyzer (ADVIA 1800, Siemens Medical Solutions, Malvern, PA).
For the study in Figure S2, 18-week-old DIO mice were injected (IP) with vehicle (twice-weekly), mGIPR-Ab, control-Ab/P1, or mGIPR-Ab/P1 at 0.75 mg/kg (once-weekly), or an equimolar dose of dulaglutide (once-weekly or twice-weekly at 0.3 mg/kg). To ensure that all mice were consistently handled and underwent the same number of injections, weekly treated groups were administered vehicle (IP) in between their scheduled weekly doses to align with the twice-weekly treated groups. Terminal trunk blood was collected in EDTA-filled microtainer tubes (365974, Becton Dickinson) with protease inhibitor (05056489001, Roche Diagnostics) added prior to collection. Plasma insulin and lipids were measured according to the manufacturer’s guidelines. Plasma insulin was measured by ELISA (ALPCO, 80-INSMS-E01), and plasma lipids were measured using a clinical chemistry analyzer (ADVIA 1800, Siemens Medical Solutions).
DIO mice indirect calorimetry
DIO mice as prepared above were acclimated to water bottles in their home cages for 5 days and were then transferred to the Comprehensive Lab Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH) and acclimated to CLAMS cages for an additional 7 days. Mice that remained weight neutral after acclimation (n = 22 out of 24 total) were randomized based on BW on day 0 (23 weeks old) to treatment groups, CLAMS cages changed, BW measured, and IP injected with vehicle, mGIPR-Ab (2.5 mg/kg), control-Ab/P1 (2 mg/kg), or mGIPR-Ab/P1 (2.5 mg/kg). Oxygen consumption, CO2 production, and RER measurements were collected continuously every 11 minutes using the Oxymax Software (Columbus Instruments). Mice had free access to food (60% kcal HFD, Research Diets D12492) and RO-chlorinated (0.3–0.5 ppm) water via water bottle throughout the study. Mice were removed from the CLAMS 144 hours after injection, and BW change and food intake were measured. Data were exported directly from the Oxymax software to Microsoft Excel and a rolling average of six time points was calculated for each measurement.
Gipr knockout mice
Mice with Gipr knockout in pancreatic β-cells (GiprβCell−/−) and their wild-type littermates (Giprfl/fl) have been previously described.21 Mice were bred at Charles River (San Diego, CA) and male mice were shipped to Amgen (Thousand Oaks, CA) at 7 weeks old and immediately started with feeding of HFD (Research Diets D12492) for 12 weeks. Mice were randomized to experimental groups based on BW so that all groups had identical starting BW before treatment. Mice were injected IP with vehicle or mGIPR-Ab/P1 (0.5 or 2.5 mg/kg) every 6 days for 18 days total. BW was measured every 3 days for the first 9 days of the study, then measured every day for the final 9 days of the study. On day 18, mice were fasted in the morning for 4 hours and then terminal trunk blood was collected in EDTA-filled microtainer tubes (365974, Becton Dickinson). Blood glucose from terminal trunk blood was measured by glucometer at the time of sacrifice. Plasma insulin and lipids were measured according to the manufacturer’s guidelines. Plasma insulin was measured by ELISA (ALPCO, 80-INSMS-E01), and plasma lipids were measured using a colorimetric assay (triglycerides – BioVision, K622; total cholesterol – Fujifilm Wako, 999-02601).
Normal cynomolgus monkeys
The PK study in normal monkeys was performed at MPI Research (Mattawan, MI). Animal care was in accordance with the Guide for the Care and Use of Laboratory Animals, 8th Edition, and the study was conducted per protocols approved by the Institutional Animal Care and Use Committee at MPI Research. Animals used on study were females weighing from 2 to 3 kg (young adults) from the MPI Research stock colony of naive cynomolgus monkeys (Macaca fascicularis). Prior to assignment to study, monkeys were quarantined and acclimated per MPI Research procedures. Monkeys were housed individually in stainless steel cages and were provided environmental enrichment during the study. Lighting was provided via automatic timer for approximately 12 hours per day. Food was offered twice daily (Lab Diet® Certified Primate Diet #5048, PMI Nutrition International) and water was available ad libitum. Temperature and humidity were maintained in the range of 64°F–79°F and 30%–70%, respectively. Following an 8-hour fasting period prior to dosing, monkeys (n = 3) received a single SC bolus dose of hGIPR-Ab/P1 at 3 mg/kg in the scapular region on the back of each animal. Blood samples (∼1 mL) were collected from the femoral vein/artery at predetermined time points up to 35 days after the dose. Blood samples were processed to K2 EDTA plasma and stored at −70°C (±10°C) until analysis.
Obese cynomolgus monkeys
Studies using obese cynomolgus monkeys were performed at Kunming Biomed International (KBI) in China. Monkeys were housed in an AAALAC International accredited facility. All research protocols were reviewed and approved by KBI’s Institutional Animal Care and Use Committee.
Naive spontaneously obese male cynomolgus monkeys (Macaca fascicularis, male, > 7 kg, body mass index > 41 kg/m2, 10–12 years of age) were obtained from KBI’s stock colony. Lighting in animal holding rooms was maintained on 12:12 hour light:dark cycle, and the ambient temperature and humidity range were set at 64°F–84°F and 30%–70%, respectively. Monkeys were fed standard chow twice daily and received apple snack once daily and had continuous access to clean water via water bottles (filled four times per day or as needed). The monkeys were provided with cage toys for environmental enrichment to ensure adequate welfare and psychological well-being. Food treats (2–3 peanuts) were given as a reward after each procedure or activity, such as blood draws, injections, and BW measurements.
Monkeys were sorted into treatment groups of n = 10 monkeys/group based on data (BW and blood chemistries) collected during a 4-week acclimation/training phase prior to treatment initiation. Monkeys were then subcutaneously injected once weekly for 6 weeks (on days 1, 8, 15, 22, 29, and 36) with vehicle, hGIPR-Ab/P1 0.75 mg/kg, or hGIPR-Ab/P2 0.75 mg/kg followed by a 3-week washout period. BW, blood chemistries, and exposure were monitored weekly; total energy and water intake were monitored daily (water intake data not shown). Blood was collected at trough period (6 days after previous injection) after a 16-hour fast and processed for serum (chemistries) and plasma (exposure). One monkey from the hGIPR-Ab/P2 0.75 mg/kg group was excluded from study/data analysis because of loss of exposure (ADA-mediated clearance). Blood chemistries were measured at KBI using a clinical chemistry analyzer (Roche C311/C501).
Method details
Peptide synthesis
Peptides with linkers were prepared by standard fluorenylmethoxycarbonyl (Fmoc)–based solid peptide synthesis using 4-(2′,4′-dimethoxyphenyl-Fmoc-aminmethyl)-phenoxyacetamido-methylbenzhydryl amine resin (Rink-amide-MBHA resin, Peptides International) on an Intavis MultiPep RSi synthesizer (Koln, Germany). The synthesizer utilized 20% 4-methyl piperidine in N, N-dimethylformamide (DMF) for Fmoc removal and 1,3-diisopropylcarbodiimide (DIC)/6-chloro-1-hydroxybenzotriazole (6-Cl−HOBt) for amino acid coupling. Each residue was coupled with an excess of coupling solution (5.0 eq), and each coupling reaction was performed twice at each position. The lysine residue was protected with (4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)-3-methylbutyl (ivDde), and the ivDde group was selectively removed with 2% hydrazine in DMF. Bromoacetyl group was introduced with DIC (10 eq)/bromoacetic acid (20 eq) in a mixture of methylene chloride and DMF.
GIPR-Ab/GLP-1 bispecific molecule synthesis
GIPR-Ab with specific cysteine mutation (cys-mAb) was incubated with a solution of 2.5 mM cystamine and 2.5 mM cysteamine in 40 mM HEPES buffer (pH 8.2) at 2.5 mg/mL concentration for 15–20 hours. The reaction mixture was filtered through a 0.22 μm polyethersulfone (PES) filter and diluted with 100 mM sodium acetate buffer pH 5.0. The reaction mixture was purified by cation exchange chromatography (GE custom packed 240 mL SP/HP, A: 100 mM NaOAc, pH 5.0, B: A + 1.2 M NaCl, 0%–20% over 10 CV, 20 mL/min). The main peak containing bis-cysteamine-capped GIPR-Ab cys-mAb was collected and buffer exchanged to 10 mM sodium acetate with 9% sucrose pH 5.2 using tangential flow filtration (Millipore Pellicon® 3, Ultracel® 30 kDa Membrane, 88 cm2).
Bis-cysteamine-capped GIPR-Ab cys-mAb (6 mg/mL in 10 mM sodium acetate with 9% sucrose) was partially reduced using four equivalents of tris(2-carboxyethyl) phosphine (TCEP) at room temperature (RT) for 60–90 minutes. Reaction completion was determined by analytical cation exchange chromatography (YMC BioPro SP-F, 30 × 4.6 mm, A: 20 mM NaOAc, pH 5.0, B: A + 1.0 M NaCl, 1.5 mL/min, Gradient: 10%–30% B over 4 minutes). TCEP was removed by using a centrifugal filter with molecular weight cutoff at 30 kDa (Millipore Amicon Ultra-15) or tangential flow filtration (Millipore Pellicon® 3, Ultracel® 30 kDa Membrane, 88 cm2), clean 10 mM NaOAc, 9% sucrose, pH 5.2. The reduced GIPR-Ab cys-mAb was diluted with 50 mM sodium phosphate buffer containing 2 mM EDTA pH 7.5 and treated with eight equivalents of dehydroascorbic acid (DHAA, BioSynth International) at RT until only trace amount of partially reduced cys-mAb species was observed (30–120 minutes, monitored by RP-HPLC, Agilent PLRPS 4000A 5 μm 50 × 2.1 mm, A: 0.1% trifluoroacetic acid (TFA) in H2O, B: 0.1% TFA in CH3CN, gradient: 2%–50% B over 3 min). Without removing DHAA, 3–8 equivalents of bromoacetyl-GLP-1 peptides were added to the reaction mixture and incubated at RT for 15–20 hours. All the reagents were removed, and the anti-GIPR/GLP-1 peptide conjugate product was buffer exchanged to final formulation using centrifugal filter or tangential flow filtration.
In vitro cAMP assay
For GLP-1R agonist activity, CHO cells stably expressing human, mouse, and monkey GLP-1R, CHOK1 cells stably expressing both hGLP-1R and hGIPR and INS1 832/3 cells were used to measure peptide or bispecific molecule–induced cAMP production in a homogeneous time-resolved fluorescence (HTRF) assay (Cisbio, Bedford, MA, cat# 62AM4PEJ). Serial diluted peptides or bispecific molecules were incubated with 40,000 cells in assay buffer (0.1% bovine serum albumin [BSA], 500 μM 3-Isobutyl-1-methylxanthine [IBMX] in F12 media) for 15 minutes for CHOK1 cells or 5 minutes for INS1 832/3 cells at 37°C. Cells were then lysed with lysis buffer containing cAMP-d2 and cAMP cryptate (Cisbio) and incubated for 1 hour at RT before measurement in the EnVision plate reader (PerkinElmer, Waltham, MA). The cAMP levels are expressed as a fluorescence ratio of 665/620 nm.
For GIPR antagonist activity, HEK293T cells stably expressing human or monkey GIPR and CHO AM1D stably expressing mouse GIPR were used to measure peptide or bispecific molecule–induced cAMP production in HTRF assay (Cisbio, cat# 62AM4PEJ). Serial diluted conjugates or GIPR-Ab were incubated with 30,000 cells in assay buffer (0.1% BSA, 500 μM IBMX in F12 media) for 30 minutes at 37°C before treatment with GIP at final concentration of 0.05 nM. Cells were incubated for 30 minutes at 37°C and then lysed in lysis buffer containing cAMP-d2 and cAMP cryptate (Cisbio) for 1 hour at RT. The fluorescence was measured in an EnVision plate reader (PerkinElmer), and the cAMP levels are expressed as a ratio of 665/620 nm.
GloSensor cAMP assay
The GloSensor cAMP assay was conducted per the manufacturer’s protocol (Promega, Madison, WI). In brief, CHOK1 cells stably expressing both hGLP-1R and hGIPR were seeded at 15,000 cells per well in a 96-well plate and incubated for 16 hours in a humidified 37°C, 5% CO2 incubator. Cells were transiently transfected with 100 ng pGloSensor-22F cAMP plasmid (Promega) using FuGENE HD for 24 hours. Cells were then equilibrated with 0.1% BSA, 2% GloSensor cAMP reagent in CO2 independent media (Invitrogen, Waltham, MA) for 2 hours at RT. Cells were preincubated for 15 minutes with or without 0.43 M sucrose (Sigma, St. Louis, MO). Basal luminescence measurements were taken for 10 minutes prior to addition of hGIPR-Ab/P1 in conditions with or without sucrose. Immediately after addition of hGIPR-Ab/P1, kinetic luminescence was measured using an integration time of 0.1–1 s every 60 s in the TECAN Infinite M1000 instrument (San Jose, CA). Data are presented as an endpoint measurement at 15 minutes.
FACS analysis
CHOK1 stably expressing hGLP-1R, 293T stably expressing hGIPR, and CHOK1 stably expressing both hGLP-1R and hGIPR cells were suspended at a density of 1 × 106 cells in 100 μL of F12 assay media containing 1% FBS, 0.05% sodium azide and incubated with 10 μg/mL of indicated Abs or GIPR-Ab/GLP-1 bispecific molecules for 1 hour at 4°C. Cells were washed one time with assay buffer and then incubated with 10 μg/mL of Alexa Fluor 647-goat anti–human Fc (Jackson, West Grove, PA) for 30 minutes at 4°C. The fluorescence was measured using BD LSR II Flow Cytometer (BD Biosciences, San Jose, CA).
Insulin secretion assay
INS1 832/3 cells were seeded at 250,000 cells per well in a 48-well poly-d-lysine–coated plate and incubated in a humidified, 37°C, 5% CO2 incubator for 48 hours. Prior to the assay, cells were washed with 1 mL assay buffer (Krebs Ringer buffer: 98.5 mM NaCl, 4 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 20 mM HEPES, 25.9 mM NaHCO3, 2.6 mM CaCl2, 0.2% BSA, pH 7.4) and incubated for 1.5 hours at 37°C in assay buffer containing 0.5 mM glucose. Cells were washed again with 1 mL of assay buffer to remove any insulin secreted during this incubation period. Cells were then incubated with serial diluted peptides or bispecific molecules in assay buffer containing 8.3 mM glucose for 1 hour at 37°C. Insulin secretion was measured by high range insulin HTRF assay (Cisbio, cat# 62IN1PEG) and expressed as normalized ratio relative to the amount of insulin secretion at 8.3 mM glucose.
Expression determination and co-immunoprecipitation of GLP-1R with GIPR
Parental U2OS cells, U2OS cells stably expressing SNAP-tagged hGLP-1R (clone 15), and U2OS cells stably expressing both SNAP-tagged hGLP-1R and hGIPR (clone 16-1 and 16-9) were grown in 96-well plates for expression determination and 6-well plates for immunoprecipitation experiments. For expression determination, cells were fixed with 4% paraformaldehyde for 20 minutes and permeabilized with 0.2% Triton X-100 in PBS for 10 minutes. Cells were incubated with mouse anti–GLP-1R (MAB2814, R&D Systems, Minneapolis, MN) or mouse anti-GIPR antibody (MAB2810, R&D Systems) at 4°C overnight. Cells were incubated with Alexa Fluor 555 or Alexa Fluor 647 conjugated anti-mouse secondary antibodies (Thermo Fisher) for GLP-1R or GIPR detection, respectively. Hoechst 33342 (Thermo Fisher) was used for nuclei detection. Images were captured using Operetta CLS high content imaging system (PerkinElmer, Waltham, MA) and analyzed by using the Harmony High-Content Imaging and Analysis Software (PerkinElmer) to quantify the total cellular GLP-1R or GIPR expression represented as fluorescence intensities.
For co-immunoprecipitation experiment, cell monolayers were lysed with lysis buffer containing 30 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5 mM EDTA, 10% glycerol, 1% Igepal 630, 0.5 mM PMSF, 1 mM activated sodium orthovanadate, 40 mM sodium fluoride (Sigma), and protease inhibitor cocktail tablets (Roche). Cell lysates were harvested, centrifuged for 5 minutes at 10,000 rpm and normalized to 1 mg/mL using the DC protein assay kit (Bio-Rad, Hercules, CA). Each clarified cell lysate was then incubated with 1 mg/mL hGIPR antibody overnight with gentle rocking at 4°C. Protein A agarose and recombinant protein G agarose (Invitrogen) were mixed and washed twice with lysis buffer. Agarose beads were then added to each cell lysate for an additional 4-hour incubation. Immunoprecipitate complexes were washed five times with lysis buffer and after the final wash, Laemmli sample buffer and 2-mercaptoethanol (Bio-Rad) were added to the dried complexes. For western blot, samples were heated at 95°C for 5 minutes and separated using gel electrophoresis. Gels were transferred to a nitrocellulose membrane and probed for anti-SNAP-tag antibody (New England Biolabs, Ipswich, MA) or anti-GIPR antibody (Abcam, Cambridge, MA).
β-arrestin recruitment assay
β-arrestin-2 recruitment was determined by enzyme fragment complementation assay using the PathHunter system (DiscoverX, 93-0001). CHOK1 cells expressing GLP-1R-ProLink, GIPR-ProLink, GLP-1R-ProLink/GIPR, and GIPR-ProLink/GLP-1R were plated at a density of 20,000 cells/well in assay buffer (0.1% BSA in F12 media) in 96-well plates and cultured overnight at 37°C, 5% CO2. Cells were incubated with serial diluted peptides or bispecific molecules in assay buffer for 90 minutes before adding working detection solution. Chemiluminescent signal was developed for 60 minutes at room temperature and quantified using an EnVision plate reader (PerkinElmer).
Receptor internalization
Antibodies used for the detection of GLP-1R and GIPR internalization were validated in U2OS cells expressing SNAP-tagged GLP-1R and CHOK1 cells expressing SNAP-tagged GIPR, respectively. Parental U2OS and CHOK1 cells were used as negative controls. Briefly, cells were incubated with SNAP Surface-Alexa Fluor 647 substrate (New England Biolabs) for 30 minutes and washed to remove excess label. Cell were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. For antibody detection, cells were incubated with mouse anti-GLP-1R (MAB2814, R&D Systems) or GIPR (MAB8210, R&D Systems), followed by detection with secondary anti-mouse Alexa Fluor plus 488 conjugated antibodies (Thermo Fisher). Hoechst 33342 (Thermo Fisher) was used for nuclei detection. Images were captured using Operetta or Operetta CLS high content imaging system (PerkinElmer) and analyzed using Harmony high-content imaging and analysis software (PerkinElmer). Validation of the receptor detection by anti-GLP-1R (MAB2814, R&D Systems) or anti-GIPR (MAB8210, R&D Systems) antibodies was done by confirming the absence of antibody staining in parental cells with staining detected only in GLP-1R- or GIPR-expressing cells. Furthermore, similar staining profiles were observed when comparing the antibody staining profile with the Alexa Fluor 647 staining profile of SNAP-tagged receptors.
GLP-1R and GIPR internalization was assessed in CHOK1 cells expressing both hGLP-1R and hGIPR or U2OS cells expressing GIPR and SNAP-tagged GLP-1R. Cells were plated at a density of 25,000 cells/well in 96-well plates and cultured overnight at 37°C, 5% CO2. Cells were serum starved in F12 media (CHOK1) or McCoy’s 5A media (U2OS) with 0.1% BSA for 4 hours. Prior to treatment, SNAP-tagged GLP-1R on the surface of U2OS cells was labeled with Alexa Fluor 564 by incubating with SNAP-Surface Alexa Fluor 546 substrate for 30 minutes. Cells were then washed to remove excess label. In the endocytosis inhibition experiments, cells were preincubated with 0.4 M sucrose for 15 minutes prior to treatment. Cells were treated with 5 nM of GLP-1, GIP, or bispecific conjugates hGIPR-Ab/P1 or control-Ab/P1 for specified time. Cells were washed and fixed with 4% paraformaldehyde for 20 minutes and permeabilized with 0.2% Triton X-100 in PBS for 10 minutes. To detect GIPR or GLP-1R, cells were first blocked with Odyssey blocking buffer (LI COR, Lincoln, NE) for 1 hour at RT and incubated with the indicated Abs:GLP-1R (CHOK1 only, R&D Systems), GIPR (R&D Systems), EEA1 (Thermo Fisher), or Rab11 (Cell Signaling) at 4°C overnight followed by 1-hour incubation with Alexa Fluor 555 or Alexa Fluor 647 conjugated anti-mouse or rabbit secondary Abs for detection. Control-Ab/P1 and hGIPR-Ab/P1 were detected using Alexa Fluor 488 labeled anti-human IgG Ab (Thermo Fisher), and Hoechst 33342 (Thermo Fisher) was used for nuclei detection. Images were captured using Operetta or Operetta CLS high content imaging system (PerkinElmer) and analyzed by using the Harmony High-Content Imaging Analysis and Software (PerkinElmer) to quantify the intracellular GLP-1R or GIPR content as fluorescent spot areas in pixel units as the readout parameter for the degree of internalization.
Bioanalytical methods and pharmacokinetics
Concentrations of mGIPR-Ab/P1, hGIPR-Ab/P1, and hGIPR-Ab/P2 in mouse and cynomolgus monkey plasma specimens were determined by ELISA specific for intact full-length test article. Microtiter plates were passively coated with a mouse mAb directed against human IgG Fc (clone no. 1.35, Amgen Inc.) in phosphate-buffered saline overnight at 4°C. Coated plates were blocked with blocking buffer overnight at 4°C. Calibration standards were prepared in a range from 30 to 2,000 ng/mL in mouse or monkey plasma (BioIVT, Estbury, NY). After dilution in blocking buffer, standards, controls, and unknown samples were added and incubated for ∼2 hours at RT. After washing, a biotin-conjugated mouse mAb directed against free N terminus of GLP-1 (clone no. 4, Thermo Fisher) was added and incubated for ∼1 hour at RT. Following an additional wash step, a streptavidin-horseradish peroxidase conjugate (R&D Systems, Inc., Minneapolis, MN) was added and incubated for ∼30 minutes at RT. After a final wash step, a tetramethylbenzidine peroxide substrate solution (SeraCare, Milford, MA) was added and incubated for ∼10 minutes at RT. The chromogenic reaction was stopped by addition of H2SO4, and absorbance values were determined at 450 nm with reference to 650 nm using a SpectraMax microtiter plate reader (Molecular Devices, San Jose, CA). Sample concentrations were interpolated from a standard curve fit to a four-parameter logistic model using Watson LIMS (v7.4; Thermo Fisher). PK parameters were estimated from individual plasma concentration-nominal time data by noncompartmental analysis using Phoenix® WinNonlin® (v6.4; Certara, Princeton, NJ). Plasma concentration-time profiles presented in Figures 2 and 4 were generated using GraphPad Prism (v7.02; GraphPad Software, Inc., San Diego, CA).
Gene expression analysis
Total RNA was isolated from frozen liver and epididymal WAT using RNeasy® 96 Universal tissue kit with DNase I treatment (QIAGEN, Valencia, CA). Gene expression was determined by quantitative real-time polymerase chain reaction (qRT-PCR) with 50–100 ng of total RNA in 20 μL volume in 384-well plates using TaqMan RNA-to-CT 1-step kit (Thermo Fisher) and analyzed using QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher). Relative gene expression (to vehicle treated group was determined using the comparative CT method with Cyclophilin A (Ppia) as the reference gene.
Systemic biomarkers of metabolism
Plasma concentration of amylin (active), C-peptide, ghrelin (active), GIP (total), GLP-1 (active), glucagon, insulin, leptin, PP, PYY, and resistin were evaluated using the Milliplex Mouse Metabolic Magnetic Bead Panel and systemic level of adiponectin by the Milliplex Mouse Adiponectin Single Plex Magnetic Bead Kit (both kits were provided by Millipore [EMD Millipore, Billerica, MA]). All measurements were performed according to manufacturer’s protocols. Plasma biomarker datasets were plotted and analyzed in GraphPad Prism (v8.04 GraphPad Software, La Jolla, CA).
Conjugation of mGIPR-Ab and mGIPR-Ab/P1
A stock solution of p-SCN-Bn-DOTA [chemical name: S-2-(4-isothiocyanatobenzyl)-1, 4, 7, 10-tetraazacyclododecane tetraacetic acid, purchased from Macrocyclics, cat no. B-205, Dallas, TX] was made in 0.1 M sodium bicarbonate solution. To facilitate the conjugation, mGIPR-Ab (stock solution: 11.1 mg/mL, 10 mM sodium acetate, pH 5.2, 0.9% sucrose) and mGIPR-Ab/P1 (stock solution: 22.1 mg/mL, 10 mM sodium acetate, pH 5.2, 0.9% sucrose) were also adjusted to pH 7.5–8.0 with 0.1 M sodium bicarbonate. The target molar ratio of DOTA to Ab was kept at 1:1 to preserve the biological activity. To achieve this final ratio, the conjugation reaction of SCN-DOTA to Ab was set to 5:1. The SCN-DOTA stock solution was added to the Ab solution with repeated pipetting and incubated at 4°C for 4 hours with occasional shaking. The final pH was set to be between 7.0 and 8.0. The conjugation efficiency was determined by size exclusion high-performance liquid chromatography (HPLC).
Radiolabeling with Indium-111
mGIPR-Ab and mGIPR-Ab/P1 were radiolabeled with Indium-111 (111In) following the same procedure. The DOTA conjugated Ab/Ab-P1 was mixed with HEPES (pH 5) followed by addition of 111In chloride. The sample mixture was set in a 40°C water bath for 2 hours. The radiochemical purity was determined by HPLC and was found to be greater than 98.5% for both proteins. Trichloroacetic acid precipitation of the protein was carried out through the course of the study (0.083–120 hours) to remove the free 111In and to determine the stability of the protein-111In. The samples were stored at 4°C.
In vivo biodistribution design and analysis
Mice (naive C57BL/6 mice) were divided into two arms of the study receiving 111In-labeled mGIPR-Ab and mGIPR-Ab/P1 (n = 18/group). Mice in each arm received a single IV injection of 111In-labeled protein at 0.5 mg/kg. For every time point, blood and tissue samples for PK analysis were collected at 0.083, 4, 24, 48, 72, and 120 hours. Tissue samples collected were lymph nodes, spleen, liver, pancreas, stomach, thymus, femur (bone marrow), cecum, duodenum, jejunum, ileum, brain, kidney, heart, lung, muscle, skin (between shoulder blades), BAT (interscapular), and WAT (flank). All the samples were counted for radioactivity using a gamma counter with correction for background and decay. Animals were cared for in accordance with the Guide for the Care and Use of Laboratory Animals, 8th Edition. All research protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Massachusetts Medicine School. Concentration of the protein in serum and tissues was reported in terms of % ID/mL. Densities of all tissues were assumed to be 1 g/mL. The blood and tissue PK data were analyzed using noncompartmental analysis using Phoenix WinNonlin v6.1 (Pharsight, Mountain View, CA). The area under the concentration-time curve (AUC0–120h) was calculated using the linear trapezoidal rule. Standard error on the AUC0–120h was calculated using the modified Bailer’s method. The Bailer’s method was applied to PK data using the in-built sparse sample module in Phoenix WinNonlin v6.1. Tissue to blood AUC ratios were also calculated and the corresponding standard error was calculated using the method of error propagation of ratios. Student’s t test was performed to test the statistical significance of differences between the two arms of the study receiving 111In-labeled mGIPR-Ab and mGIPR-Ab/P1. A threshold of p < 0.05 was set for Student’s t test.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism or Phoenix WinNonlin software. Data are presented as mean ± SEM, and statistical significance are reported in figures and figure legends. Data are presented to be statistically significant when p < 0.05 by Student’s t test, one-way or two-way ANOVA, where appropriate, and detailed methods and p values for the statistical significance are described in the figure legends for each figure.
Acknowledgments
The authors thank Mary Rusckowski, PhD, and Yuzhen Wang, PhD, from the University of Massachusetts Medical School for their technical assistance with the biodistribution study. The authors thank Briana Griego for her help with collecting tissues from in vivo studies. The authors also thank Cathryn M. Carter, MS, Stephen Lee, PhD for editorial support; they received compensation as an Amgen employee. This work was supported by Amgen Inc.
Author contributions
S.-C.L., M.C., E.A.K., L.A., M.S., D.D., R.K., and S.S. designed the study, carried out the research, analyzed and interpreted the results, and wrote the manuscript. Y.C., C.N., and J.F. designed and engineered constructs and provided reagents. T.H. and V.A.T. designed the study, analyzed and interpreted the results, and wrote the manuscript. D.J.L., C.H., and J.M.H. supervised the study and edited the manuscript. S.-C.L. and M.M.V. conceived and supervised the study and wrote and edited the manuscript.
Declaration of interests
The authors declare no competing interests. All authors are employees and shareholders of Amgen. A patent related to this work has been filed: International Application No. PCT/US18/13918.
Published: April 30, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100263.
Supplemental information
References
- 1.Finkelstein E.A., Trogdon J.G., Cohen J.W., Dietz W. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Aff. (Millwood) 2009;28:w822–w831. doi: 10.1377/hlthaff.28.5.w822. [DOI] [PubMed] [Google Scholar]
- 2.Valsamakis G., Konstantakou P., Mastorakos G. New targets for drug treatment of obesity. Annu. Rev. Pharmacol. Toxicol. 2017;57:585–605. doi: 10.1146/annurev-pharmtox-010716-104735. [DOI] [PubMed] [Google Scholar]
- 3.Prasad-Reddy L., Isaacs D. A clinical review of GLP-1 receptor agonists: efficacy and safety in diabetes and beyond. Drugs Context. 2015;4:212283. doi: 10.7573/dic.212283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drucker D.J. The biology of incretin hormones. Cell Metab. 2006;3:153–165. doi: 10.1016/j.cmet.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Inagaki N., Seino Y., Takeda J., Yano H., Yamada Y., Bell G.I., Eddy R.L., Fukushima Y., Byers M.G., Shows T.B. Gastric inhibitory polypeptide: structure and chromosomal localization of the human gene. Mol. Endocrinol. 1989;3:1014–1021. doi: 10.1210/mend-3-6-1014. [DOI] [PubMed] [Google Scholar]
- 6.Takeda J., Seino Y., Tanaka K., Fukumoto H., Kayano T., Takahashi H., Mitani T., Kurono M., Suzuki T., Tobe T. Sequence of an intestinal cDNA encoding human gastric inhibitory polypeptide precursor. Proc. Natl. Acad. Sci. USA. 1987;84:7005–7008. doi: 10.1073/pnas.84.20.7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lardinois C.K., Starich G.H., Mazzaferri E.L. The postprandial response of gastric inhibitory polypeptide to various dietary fats in man. J. Am. Coll. Nutr. 1988;7:241–247. doi: 10.1080/07315724.1988.10720241. [DOI] [PubMed] [Google Scholar]
- 8.Mroz P.A., Finan B., Gelfanov V., Yang B., Tschöp M.H., DiMarchi R.D., Perez-Tilve D. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol. Metab. 2019;20:51–62. doi: 10.1016/j.molmet.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finan B., Ma T., Ottaway N., Müller T.D., Habegger K.M., Heppner K.M., Kirchner H., Holland J., Hembree J., Raver C. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci. Transl. Med. 2013;5:209ra151. doi: 10.1126/scitranslmed.3007218. [DOI] [PubMed] [Google Scholar]
- 10.Coskun T., Sloop K.W., Loghin C., Alsina-Fernandez J., Urva S., Bokvist K.B., Cui X., Briere D.A., Cabrera O., Roell W.C. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol. Metab. 2018;18:3–14. doi: 10.1016/j.molmet.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frias J.P., Nauck M.A., Van J., Kutner M.E., Cui X., Benson C., Urva S., Gimeno R.E., Milicevic Z., Robins D., Haupt A. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. 2018;392:2180–2193. doi: 10.1016/S0140-6736(18)32260-8. [DOI] [PubMed] [Google Scholar]
- 12.Beck B., Max J.P. Direct metabolic effects of gastric inhibitory polypeptide (GIP): dissociation at physiological levels of effects on insulin-stimulated fatty acid and glucose incorporation in rat adipose tissue. Diabetologia. 1986;29:68. doi: 10.1007/BF02427284. [DOI] [PubMed] [Google Scholar]
- 13.Murphy M.C., Isherwood S.G., Sethi S., Gould B.J., Wright J.W., Knapper J.A., Williams C.M. Postprandial lipid and hormone responses to meals of varying fat contents: modulatory role of lipoprotein lipase? Eur. J. Clin. Nutr. 1995;49:578–588. [PubMed] [Google Scholar]
- 14.Bailey C.J., Flatt P.R., Kwasowski P., Powell C.J., Marks V. Immunoreactive gastric inhibitory polypeptide and K cell hyperplasia in obese hyperglycaemic (ob/ob) mice fed high fat and high carbohydrate cafeteria diets. Acta Endocrinol. (Copenh.) 1986;112:224–229. doi: 10.1530/acta.0.1120224. [DOI] [PubMed] [Google Scholar]
- 15.Salera M., Giacomoni P., Pironi L., Cornia G., Capelli M., Marini A., Benfenati F., Miglioli M., Barbara L. Gastric inhibitory polypeptide release after oral glucose: relationship to glucose intolerance, diabetes mellitus, and obesity. J. Clin. Endocrinol. Metab. 1982;55:329–336. doi: 10.1210/jcem-55-2-329. [DOI] [PubMed] [Google Scholar]
- 16.Miyawaki K., Yamada Y., Ban N., Ihara Y., Tsukiyama K., Zhou H., Fujimoto S., Oku A., Tsuda K., Toyokuni S. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat. Med. 2002;8:738–742. doi: 10.1038/nm727. [DOI] [PubMed] [Google Scholar]
- 17.Miyawaki K., Yamada Y., Yano H., Niwa H., Ban N., Ihara Y., Kubota A., Fujimoto S., Kajikawa M., Kuroe A. Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc. Natl. Acad. Sci. USA. 1999;96:14843–14847. doi: 10.1073/pnas.96.26.14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naitoh R., Miyawaki K., Harada N., Mizunoya W., Toyoda K., Fushiki T., Yamada Y., Seino Y., Inagaki N. Inhibition of GIP signaling modulates adiponectin levels under high-fat diet in mice. Biochem. Biophys. Res. Commun. 2008;376:21–25. doi: 10.1016/j.bbrc.2008.08.052. [DOI] [PubMed] [Google Scholar]
- 19.Pamir N., Lynn F.C., Buchan A.M., Ehses J., Hinke S.A., Pospisilik J.A., Miyawaki K., Yamada Y., Seino Y., McIntosh C.H., Pederson R.A. Glucose-dependent insulinotropic polypeptide receptor null mice exhibit compensatory changes in the enteroinsular axis. Am. J. Physiol. Endocrinol. Metab. 2003;284:E931–E939. doi: 10.1152/ajpendo.00270.2002. [DOI] [PubMed] [Google Scholar]
- 20.Killion E.A., Lu S.C., Fort M., Yamada Y., Véniant M.M., Lloyd D.J. Glucose-dependent insulinotropic polypeptide receptor therapies for the treatment of obesity, do agonists = antagonists? Endocr. Rev. 2020;41:bnz002. doi: 10.1210/endrev/bnz002. [DOI] [PubMed] [Google Scholar]
- 21.Killion E.A., Wang J., Yie J., Shi S.D., Bates D., Min X., Komorowski R., Hager T., Deng L., Atangan L. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci. Transl. Med. 2018;10:eaat3392. doi: 10.1126/scitranslmed.aat3392. [DOI] [PubMed] [Google Scholar]
- 22.Kaneko K., Fu Y., Lin H.Y., Cordonier E.L., Mo Q., Gao Y., Yao T., Naylor J., Howard V., Saito K. Gut-derived GIP activates central Rap1 to impair neural leptin sensitivity during overnutrition. J. Clin. Invest. 2019;129:3786–3791. doi: 10.1172/JCI126107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tschöp M.H., Finan B., Clemmensen C., Gelfanov V., Perez-Tilve D., Müller T.D., DiMarchi R.D. Unimolecular polypharmacy for treatment of diabetes and obesity. Cell Metab. 2016;24:51–62. doi: 10.1016/j.cmet.2016.06.021. [DOI] [PubMed] [Google Scholar]
- 24.Biswas K., Nixey T.E., Murray J.K., Falsey J.R., Yin L., Liu H., Gingras J., Hall B.E., Herberich B., Holder J.R. Engineering antibody reactivity for efficient derivatization to generate NaV1.7 inhibitory GpTx-1 peptide-antibody conjugates. ACS Chem. Biol. 2017;12:2427–2435. doi: 10.1021/acschembio.7b00542. [DOI] [PubMed] [Google Scholar]
- 25.Miranda L.P., Winters K.A., Gegg C.V., Patel A., Aral J., Long J., Zhang J., Diamond S., Guido M., Stanislaus S. Design and synthesis of conformationally constrained glucagon-like peptide-1 derivatives with increased plasma stability and prolonged in vivo activity. J. Med. Chem. 2008;51:2758–2765. doi: 10.1021/jm701522b. [DOI] [PubMed] [Google Scholar]
- 26.Sun F., Chai S., Yu K., Quan X., Yang Z., Wu S., Zhang Y., Ji L., Wang J., Shi L. Gastrointestinal adverse events of glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: a systematic review and network meta-analysis. Diabetes Technol. Ther. 2015;17:35–42. doi: 10.1089/dia.2014.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang W., Wang E.Q., Balthasar J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008;84:548–558. doi: 10.1038/clpt.2008.170. [DOI] [PubMed] [Google Scholar]
- 28.Deng R., Iyer S., Theil F.P., Mortensen D.L., Fielder P.J., Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs. 2011;3:61–66. doi: 10.4161/mabs.3.1.13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King-Ahmad A.J., Kalgutkar A.S., Niosi M., Eng H., Holliman C. A sensitive method for the quantitation of the peptide-based glucagon-like peptide-1 receptor agonist liraglutide in plasma using microfluidics chromatography tandem MS. Bioanalysis. 2018;10:357–368. doi: 10.4155/bio-2017-0239. [DOI] [PubMed] [Google Scholar]
- 30.Glaesner W., Vick A.M., Millican R., Ellis B., Tschang S.H., Tian Y., Bokvist K., Brenner M., Koester A., Porksen N. Engineering and characterization of the long-acting glucagon-like peptide-1 analogue LY2189265, an Fc fusion protein. Diabetes Metab. Res. Rev. 2010;26:287–296. doi: 10.1002/dmrr.1080. [DOI] [PubMed] [Google Scholar]
- 31.Campbell J.E., Ussher J.R., Mulvihill E.E., Kolic J., Baggio L.L., Cao X., Liu Y., Lamont B.J., Morii T., Streutker C.J. TCF1 links GIPR signaling to the control of beta cell function and survival. Nat. Med. 2016;22:84–90. doi: 10.1038/nm.3997. [DOI] [PubMed] [Google Scholar]
- 32.Gasbjerg L.S., Christensen M.B., Hartmann B., Lanng A.R., Sparre-Ulrich A.H., Gabe M.B.N., Dela F., Vilsbøll T., Holst J.J., Rosenkilde M.M., Knop F.K. GIP(3-30)NH2 is an efficacious GIP receptor antagonist in humans: a randomised, double-blinded, placebo-controlled, crossover study. Diabetologia. 2018;61:413–423. doi: 10.1007/s00125-017-4447-4. [DOI] [PubMed] [Google Scholar]
- 33.Svendsen B., Capozzi M.E., Nui J., Hannou S.A., Finan B., Naylor J., Ravn P., D’Alessio D.A., Campbell J.E. Pharmacological antagonism of the incretin system protects against diet-induced obesity. Mol. Metab. 2020;32:44–55. doi: 10.1016/j.molmet.2019.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yip V., Palma E., Tesar D.B., Mundo E.E., Bumbaca D., Torres E.K., Reyes N.A., Shen B.Q., Fielder P.J., Prabhu S. Quantitative cumulative biodistribution of antibodies in mice: effect of modulating binding affinity to the neonatal Fc receptor. MAbs. 2014;6:689–696. doi: 10.4161/mabs.28254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boswell C.A., Bumbaca D., Fielder P.J., Khawli L.A. Compartmental tissue distribution of antibody therapeutics: experimental approaches and interpretations. AAPS J. 2012;14:612–618. doi: 10.1208/s12248-012-9374-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Usdin T.B., Mezey E., Button D.C., Brownstein M.J., Bonner T.I. Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology. 1993;133:2861–2870. doi: 10.1210/endo.133.6.8243312. [DOI] [PubMed] [Google Scholar]
- 37.Yip R.G., Boylan M.O., Kieffer T.J., Wolfe M.M. Functional GIP receptors are present on adipocytes. Endocrinology. 1998;139:4004–4007. doi: 10.1210/endo.139.9.6288. [DOI] [PubMed] [Google Scholar]
- 38.Campos R.V., Lee Y.C., Drucker D.J. Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology. 1994;134:2156–2164. doi: 10.1210/endo.134.5.8156917. [DOI] [PubMed] [Google Scholar]
- 39.Göke R., Larsen P.J., Mikkelsen J.D., Sheikh S.P. Distribution of GLP-1 binding sites in the rat brain: evidence that exendin-4 is a ligand of brain GLP-1 binding sites. Eur. J. Neurosci. 1995;7:2294–2300. doi: 10.1111/j.1460-9568.1995.tb00650.x. [DOI] [PubMed] [Google Scholar]
- 40.Whitaker G.M., Lynn F.C., McIntosh C.H., Accili E.A. Regulation of GIP and GLP1 receptor cell surface expression by N-glycosylation and receptor heteromerization. PLoS ONE. 2012;7:e32675. doi: 10.1371/journal.pone.0032675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maida A., Lamont B.J., Cao X., Drucker D.J. Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-α in mice. Diabetologia. 2011;54:339–349. doi: 10.1007/s00125-010-1937-z. [DOI] [PubMed] [Google Scholar]
- 42.Johnson J.D. A practical guide to genetic engineering of pancreatic β-cells in vivo: getting a grip on RIP and MIP. Islets. 2014;6:e944439. doi: 10.4161/19382014.2014.944439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adriaenssens A.E., Biggs E.K., Darwish T., Tadross J., Sukthankar T., Girish M., Polex-Wolf J., Lam B.Y., Zvetkova I., Pan W. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab. 2019;30:987–996. doi: 10.1016/j.cmet.2019.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Killion E.A., Chen M., Falsey J.R., Sivits G., Hager T., Atangan L., Helmering J., Lee J., Li H., Wu B. Chronic glucose-dependent insulinotropic polypeptide receptor (GIPR) agonism desensitizes adipocyte GIPR activity mimicking functional GIPR antagonism. Nat. Commun. 2020;11:4981. doi: 10.1038/s41467-020-18751-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gabery S., Salinas C.G., Paulsen S.J., Ahnfelt-Rønne J., Alanentalo T., Baquero A.F., Buckley S.T., Farkas E., Fekete C., Frederiksen K.S. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. 2020;5:e133429. doi: 10.1172/jci.insight.133429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams J.M., Pei H., Sandoval D.A., Seeley R.J., Chang R.B., Liberles S.D., Olson D.P. Liraglutide modulates appetite and body weight through glucagon-like peptide 1 receptor-expressing glutamatergic neurons. Diabetes. 2018;67:1538–1548. doi: 10.2337/db17-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Svegliati-Baroni G., Patrício B., Lioci G., Macedo M.P., Gastaldelli A. Gut-pancreas-liver axis as a target for treatment of NAFLD/NASH. Int. J. Mol. Sci. 2020;21:5820. doi: 10.3390/ijms21165820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krupnick J.G., Benovic J.L. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- 49.Vilardaga J.P., Jean-Alphonse F.G., Gardella T.J. Endosomal generation of cAMP in GPCR signaling. Nat. Chem. Biol. 2014;10:700–706. doi: 10.1038/nchembio.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Widmann C., Dolci W., Thorens B. Agonist-induced internalization and recycling of the glucagon-like peptide-1 receptor in transfected fibroblasts and in insulinomas. Biochem. J. 1995;310:203–214. doi: 10.1042/bj3100203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuna R.S., Girada S.B., Asalla S., Vallentyne J., Maddika S., Patterson J.T., Smiley D.L., DiMarchi R.D., Mitra P. Glucagon-like peptide-1 receptor-mediated endosomal cAMP generation promotes glucose-stimulated insulin secretion in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 2013;305:E161–E170. doi: 10.1152/ajpendo.00551.2012. [DOI] [PubMed] [Google Scholar]
- 52.Ismail S., Gherardi M.J., Froese A., Zanoun M., Gigoux V., Clerc P., Gaits-Iacovoni F., Steyaert J., Nikolaev V.O., Fourmy D. Internalized Receptor for Glucose-dependent Insulinotropic Peptide stimulates adenylyl cyclase on early endosomes. Biochem. Pharmacol. 2016;120:33–45. doi: 10.1016/j.bcp.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 53.Ratner R., Nauck M., Kapitza C., Asnaghi V., Boldrin M., Balena R. Safety and tolerability of high doses of taspoglutide, a once-weekly human GLP-1 analogue, in diabetic patients treated with metformin: a randomized double-blind placebo-controlled study. Diabet. Med. 2010;27:556–562. doi: 10.1111/j.1464-5491.2010.02990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pagliuca F.W., Millman J.R., Gürtler M., Segel M., Van Dervort A., Ryu J.H., Peterson Q.P., Greiner D., Melton D.A. Generation of functional human pancreatic β cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.