

Abstract
Background
P-glycoprotein (P-gp) is an Adenosine triphosphate (ATP) dependent drug-efflux pump which is located abundantly in the stomach and protects the gut mucosa from xenobiotic.
Objective
The purpose of this study was to investigate the influence of P-gp modulation on the efficacy of treatment regimen.
Method
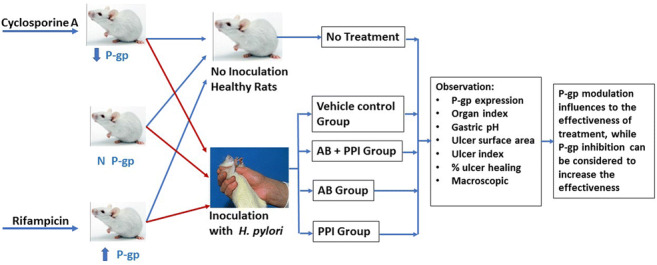
P-gp modulation in rats was performed by using P-gp inducer (150 mg/kg rifampicin) and P-gp inhibitor (10 mg/kg cyclosporine A) for 14 days prior to be infected with Helicobacter pylori (H. pylori). The rats were further divided into groups, which were normal control, vehicle control, antibiotics and omeprazole, antibiotics only and omeprazole only for another 2 weeks of treatment. The ulcer formation and P-gp expression were determined by using macroscopic evaluation and western blot analysis, respectively.
Results
The highest P-gp expression was shown in the induced P-gp rats (2.00 ± 0.68) while the lowest P-gp expression was shown in the inhibited P-gp rats (0.45 ± 0.36) compared to the normal P-gp rats. In all groups, the rats which were infected with H. pylori, had a significant increase (p < 0.05) in P-gp expression level and a more severe ulcer formation compared to the healthy rats. The ulcer developed at different levels in the rats with inhibited, induced, or normal P-gp expression. After receiving the standard therapy for H. pylori, it was observed that the healing rate for ulcer was increased to 91% (rats with inhibited P-gp expression), 82% (rats with induced P-gp expression) and 75% in rats with normal P-gp. The use of rifampicin to induce P-gp level was also shown to be effective in eradicating the H. pylori infection.
Conclusion
The synergism in the standard therapy by using two antibiotics (clarithromycin and amoxicillin) and proton pump inhibitor (omeprazole) have shown to effectively eradicate the H. pylori infection. Thus, P-gp expression influenced the effectiveness of the treatment.
Graphical abstract
Keywords: P-glycoprotein, Helicobacter pylori, Rifampicin, Cyclosporine a, Amoxicillin, Clarithromycin, Omeprazole
Introduction
P-glycoprotein (P-gp) is an Adenosine triphosphate-binding (ATP-binding) protein and acts as a drug transporter, encoded by the multidrug resistance 1 (MDR1) gene [1]. Recently, more attention has been focused on the potential impact of P-gp expression on drug absorption and bioavailability of various drugs [2]. Higher P-gp may lead to poor bioavailability of substrate drug and lower P-gp may lead to toxic effect on tissues. Broad range substrate specificity may lead to drug interaction when co-administered together with the drug P-gp inducer or inhibitor [3].
In normal human tissues, P-gp is located at the liver, stomach, intestine, kidney and blood brain barrier [4]. It limits cellular uptake of drugs from gastrointestinal tract to enterocyte [5]. In the presence of P-gp, the elimination of certain drugs into the hepatocytes, renal tubules, and intestinal epithelial cell adjacent to the luminal space would be enhanced [5, 6]. The P-gp level was gradually reported to be increased from the stomach to the duodenum [7].
Helicobacter pylori (H. pylori) is a gram-negative bacterium which is colonized in the upper part of the stomach (fundus area). Cag A, Vac A, Bab A and Sab A genes in H. pylori are virulent factors that cause gastric cancer [8]. H. pylori caused gastric cancer in 5.5% out of 25% of H. pylori infection cases [9]. The eradication of H. pylori infection was done using a standard regimen therapy. According to Chey et al. (2017), the gold standard of the therapy could be accomplished by combining the use of antibiotics such as amoxicillin and clarithromycin and proton pump inhibitor (PPI) [10]. However, there are reports of ineffectiveness of the treatment regimen, where the unsuccessful eradication of H. pylori could be attributed by the antibiotic resistance, variation of H. pylori strains, host condition, or low compliance for the high number of pills to be taken daily [11]. As for the second line therapy, rifabutin has indicated a good eradication rate when administered in the combination with amoxicillin and PPI.
Rifampin and rifabutin belong to the rifamycins which was noted as the moderate to potent inducers of drugs undergoing metabolism by the cytochrome P450 enzyme system (notably CYP3A4). Rifampicin has shown good efficacy against H. pylori in various in-vitro studies [12]. It has been reported that rifampicin has the ability to induce the P-gp expression in colon cancer-derived adenocarcinoma cell line such as Caco-2 cells [13]. It prevented the H. pylori attachment on the cell lines and enhanced the levels of P-gp in the Caco-2 cells. While rifampicin induces P-gp expression, the P-gp inhibitor such as PSC-833 will inhibit the P-gp activity on the cell line, and thus allows bacteria attachment. PSC-833 is a cyclosporine A analogue with lower immunosuppressant effect compared to cyclosporine A [14]. However, cyclosporine A was widely used to inhibit P-gp level in rats due to its immediate decreasing effects on P-gp [15, 16]. Therefore, the modulation of P-gp at the gut mucosa is important.
The purpose of this study was to investigate the influence of P-gp expression on the efficacy of standard therapy regimen of H. pylori infection in Sprague Dawley rat model and to evaluate whether proton pump inhibitors (PPIs) were required in the standard therapy.
Methodology
Animal
The study was approved by Universiti Kebangsaan Malaysia (UKM) Animal Ethics Committee (UKMAEC) under the approval number, FF/2015/ENDANG/29-SEPT./699-SEPT.-2015-SEPT, and in accordance with the international guidelines for animal studies [17]. A total of 90 male Sprague Dawley rats with an average weight of 150–250 g were used in this study. They were divided randomly into groups of three rats per cage for the adaptation period. The rats were placed in wired tapered cage that was exposed to light for 12 h and a further 12 h in dark condition. They were fed with commercial rodent diet (Altormin International, Germany) and water ad libitum. They were acclimatized for 7 days prior to the experiment.
Experimental design
The rats were divided into three large groups (n = 30), in which the rats in the first group were given cyclosporine A with a dose of 10 mg/kg as the P-gp inhibitor [18], the second group was the normal rats which were given sterile distilled water and the third group was given rifampicin with a dose of 150 mg/kg as P-gp inducer [19], once a day for 14 days. Each of the groups was then further divided into five groups (n = 6): Group 1, no bacterial inoculation without any treatment; Group 2, vehicle control, bacterial inoculation and 0.25% carboxymethylcellulose; Group 3, bacterial inoculation with the treatment of amoxicillin (90 mg/kg), clarithromycin (135 mg/kg) and omeprazole 3.6 mg/kg (standard treatment); Group 4, bacterial inoculation and with the treatment of amoxicillin (90 mg/kg) and clarithromycin (135 mg/kg); Group 5, bacterial inoculation and with the treatment of omeprazole (3.6 mg/kg).
H. pylori Oral inoculation
H. pylori was kindly supplied by the Department of Medical Microbiology and Immunology, UKM Medical Centre, Cheras, Malaysia. The bacteria were cultured on Columbia blood agar (Merck) supplemented with 10% lake horse blood (Thermo fisher) and selective medium (Oxoid) at 37 °C (atmosphere: 5% CO2 or 95% air) for five to 7 days. The bacteria were identified as H. pylori strain by gram staining, oxidase, catalase, and urease positive test. A few H. pylori colonies were scrapped into 15 mL Muller Hinton broth (Merck) supplemented with 10% fetal calf serum (Sigma Aldrich) and 0.1% yeast extract. The broth was further incubated in a CO2 incubator at 37 °C with 5% CO2 and 95% air for 24–48 h. Prior to the usage, the bacteria were adjusted to an optical density at 600 nm of 1.0, which corresponded to 1 × 108 CFU/mL. The bacteria were inoculated into the rats with the volume of 1 mL using oral gavage twice daily at an interval of 4 h for 7 days. On the first day before H. pylori inoculation, the rats were fasted for 16 h. For the subsequent dose, the rats were fasted for 3 h prior to H. pylori inoculation.
Macroscopic evaluation
The rats were sacrificed by spinal dislocation after anesthesia by using ketamine-xylazine cocktail after 14 days of treatment. The stomach was dissected along the greater curvature and observed under macroscopic observation. The gastric juice was collected, and pH of the sample was measured by using a pH meter. Gastric mucosa that had been cleaned from food contaminant and blood clot was rinsed by using 0.9% sodium chloride. After the macroscopic observation, the ulcer parts were chopped by using a knife into one (1) mm2 in diameter and kept at -80 °C for western blot analysis. Previously, every part of the ulcer was confirmed to be induced by H. pylori. The sterile tip on the ulcer part was carefully scrapped without damaging the surface area of the gastric mucosa. Thereafter, it was put into urease broth and incubated for 24 h. The gastric mucosa was observed under a 10 X magnifier lens to determine ulcer formation prior to ulcer image captured by using a SONY digital camera with 20 Mega pixels. The ulcer formation was observed and analyzed for ulcer score which was calculated according to Geometric formula as described by Mahmood et al. [20]:
(a) Scoring ulcer was observed on the stomach for score 0.0 = Normal color stomach; 0.5 = Red coloration/petechiae; 1.0 = Spot ulcer; 1.5 = Hemorrhagic streak; 2.0 = Deep ulcer; 3.0 = Perforation.
(b) Mean ulcer score for each animal will be expressed as ulcer index, UI = (UN + US +UP) × 10−1; UI = Ulcer index; UN = Average number of ulcer per animal; US = Average number of severity score; UP = Percentage of animal with ulcer.
(c) Percentage inhibition of ulceration was calculated as below:
Usc = Ulcer surface area control.
Ust = Ulcer surface area in treated animals.
(d) Organ Index = weight of organs/weight of rats before sacrificed.
Sample lysis
Ulcer samples that were kept at −80 °C were rapidly thawed prior to the addition of cold lysis buffer radioimmunoprecipitation assay (RIPA), phenylmethylsulfonyl fluoride (PMSF) and protease inhibitor. A weight of 10 mg ulcer sample was mixed with 100 μl cold buffer then sonicated for 2 min on ice. The amplitude of sonicator was set at 35% with the frequency sonication of 20 kHz. The lysate was incubated on ice for 5 min. Next, it was centrifuged at 11,000 rpm for 15 min at 4 °C. Then, the supernatant was collected and frozen at -80 °C until it was used for Bradford assay and western blot analysis [21].
Western blot analysis
The total protein was quantified by using Bradford assay and P-gp expression was detected via western blot analysis. The protein concentration of 3 mg/ml was loaded into 10% Tris gel. Protein separation was done at 150 V for 67 min. The protein on the gel was transferred on polyvinylidene fluoride (PVDF) membrane by using Mini Chamber Transblot Bio Rad (California, USA) at 32 V for 105 min. The membrane was blocked with antibody solution from Western Breeze® Invitrogen, Victoria, containing of P-gp Sc13131 Mdr [G-1] mouse monoclonal IgG2b and mouse anti-B-actin (Sigma-Aldrich, St Louis MO). After an overnight incubation, the membrane was washed with tris-buffered saline Tween-20 (TBST) and thereafter incubated with the secondary antibody (Goat anti mouse IgG AP Pre-diluted, Western Breeze® Invitrogen, Victoria). The PVDF membrane was washed with TBST before being incubated with 200 μl ECL substrate for 5 min. The protein band on the membrane was detected by using the Fusion fx1 gel documentation system (Vilber Lourmat, Marne-La-Vallée, France) [21].
Statistical analysis
Data was presented as mean ± standard error mean (n = 6) and analyzed by using one-way analysis of variance (ANOVA), post hoc Dunnet test by GraphPad Prism 5 Software (CA, USA). The differences between the experimental groups were considered significant if p < 0.05 and indicated as follows: ***(p < 0.001), **(p < 0.01) and *(p < 0.05) compared to the normal controlled rats, while ###p < 0.001, ##p < 0.01 and #p < 0.05 were statistically significant compared to negative/vehicle control and or induced P-gp rats.
Results
Ulcer induction by H. pylori inoculation in P-gp modulated rats
Stomach ulcer formation of vehicle control was compared with non-infected rat’s stomach in P-gp modulated groups or non-modulated groups (normal rats). The results showed that there was no ulcer formation on the gastric mucosa of normal P-gp groups (Fig. 1a) and all P-gp modulated groups, either P-gp induced with rifampicin (Fig. 2a) or P-gp inhibited with cyclosporine A (Fig. 3a) of non-infected rats. On the contrary, the ulcer formation on the rat’s stomach due to H. pylori infection in P-gp normal group, the ulcer index was 9.14 ± 0.41 (Fig. 1b and Table 1), P-gp modulated groups with ulcer index for P-gp induced was 3.50 ± 0.18 (Fig. 2b and Table 2), and for P-gp inhibited was 11.23 ± 0.39 (Fig. 3b and Table 3). All results were significant with p < 0.001 for normal and modulated P-gp rats compared to their own non-infected group with ulcer index was 0. These results showed that the lowest P-gp expression caused severe H. pylori infection with the highest value of ulcer index.
Fig. 1.

Macroscopic appearance of the H. pylori ulcer induction on the gastric mucosa in normal P-gp rats. (a) Healthy rats showed no ulcer formation on the gastric mucosa. (b) Ulcers were shown on the gastric mucosa of vehicle control rats (0.25% Carboxymethylcellulose as vehicle). (c) Rats were treated with a combination of antibiotic and omeprazole (standard regimen). (d) Rats were treated with antibiotic only. (e) Rats were treated with omeprazole only
Fig. 2.

Macroscopic appearance of the H. pylori ulcer induction on the gastric mucosa in induced P-gp rats. (a) Healthy rats showed no ulcer formation on the gastric mucosa. (b) Ulcers were shown on the gastric mucosa of vehicle control rats (0.25% Carboxymethylcellulose as vehicle). (c) Rats were treated with a combination of antibiotic and omeprazole (standard regimen). (d) Rats were treated with antibiotic only. (e) Rats were treated with omeprazole only
Fig. 3.

Macroscopic appearance of the H. pylori ulcer induction on the gastric mucosa in inhibited P-gp rats. (a) Healthy rats showed no ulcer formation on the gastric mucosa. (b) Ulcers were shown on the gastric mucosa of vehicle control rats (0.25% Carboxymethylcellulose as vehicle). (c) Rats were treated with a combination of antibiotic and omeprazole (standard regimen). (d) Rats were treated with antibiotic only. (e) Rats were treated with omeprazole only
Table 1.
The effects of combination therapy amoxicillin, clarithromycin and/or omeprazole on ulcer formation in normal P-gp rats
| No | Treatment/ Group | Parameters | ||||
|---|---|---|---|---|---|---|
| Organ index | Gastric pH | Ulcer surface area (cm2) | Ulcer index | % ulcer healing | ||
| 1. | Normal control (Healthy, no treatment) | 0.007 ± 0.001 | 3.35 ± 1.1 | 0 | 0 | 0 |
| 2. | Vehicle control group | 0.009 ± 0.002 | 6.14 ± 0.7*** | 0.1133 ± 0.09 | 9.14 ± 0.41 | 0 |
| 3. | Positive control group | 0.007 ± 0.0005 | 5.22 ± 0.3** | 0.028 ± 0.05 | 5.22 ± 0.21### | 75% |
| 4. | AB group | 0.008 ± 0.002 | 4.62 ± 0.4* | 0.0457 ± 0.06 | 6.87 ± 0.12## | 60% |
| 5. | PPI group | 0.008 ± 0.0012 | 5.63 ± 0.4** | 0.056 ± 0.06 | 8.65 ± 0.17# | 50% |
Positive control (standard regimen) group treated with 90 mg/kg amoxicillin, 135 mg/kg clarithromycin, 3.6 mg/kg omeprazole. AB group treated by amoxicillin + clarithromycin; PPI group treated by omeprazole; Vehicle used is 0.25% carboxymethylcellulose. Value are mean ± standard error mean of six animals in each group. All comparisons were performed using one-way ANOVA (dunnet’s test). *** p < 0.001, ** p < 0.01 and *p < 0.05 vs normal control rats. ### p < 0.001, ## p < 0.01 and # p < 0.05 vs vehicle control rats
Table 2.
The effects of combination therapy amoxicillin, clarithromycin and/or omeprazole on ulcer formation in Induced P-gp rats
| No | Treatment/ Group | Parameters | ||||
|---|---|---|---|---|---|---|
| Organ index | Gastric pH | Ulcer surface area (cm2) | Ulcer index | % ulcer healing | ||
| 1 | Normal control (Healthy, no treatment) | 0.007 ± 0.0008 | 5.33 ± 1.6 | 0 | 0 | 0 |
| 2 | Vehicle control group | 0.007 ± 0.0008 | 5.32 ± 1.0 | 0.0187 ± 0.03 | 3.50 ± 0.18 | 0 |
| 3 | Positive control group | 0.007 ± 0.001 | 4.68 ± 1.0 | 0.0033 ± 0.008 | 1.77 ± 0.13### | 82% |
| 4 | AB group | 0.007 ± 0.0004 | 5.07 ± 0.47 | 0.0050 ± 0.01 | 1.79 ± 0.19### | 73% |
| 5 | PPI group | 0.007 ± 0.0005 | 5.26 ± 1.33 | 0.0067 ± 0.012 | 3.48 ± 0.21 | 64% |
Positive control (standard regimen) group treated with 90 mg/kg amoxicillin, 135 mg/kg clarithromycin, 3.6 mg/kg omeprazole. AB group treated by amoxicillin + clarithromycin; PPI group treated by omeprazole; Vehicle used is 0.25% carboxymethylcellulose. Value are mean ± standard error mean of six animals in each group. All comparisons were performed using one-way ANOVA (dunnet’s test). *** p < 0.001, ** p < 0.01 and *p < 0.05 vs normal control rats. ### p < 0.001, ## p < 0.01 and # p < 0.05 vs vehicle control rats
Table 3.
The effects of combination therapy amoxicillin, clarithromycin and/or omeprazole on ulcer formation in inhibited P-gp rats
| No | Treatment/ Group | Parameters | ||||
|---|---|---|---|---|---|---|
| Organ index | Gastric pH | Ulcer surface area (cm2) | Ulcer index | % ulcer healing | ||
| 1 | Normal control (Healthy, no treatment)) | 0.007 ± 0.001 | 5.14 ± 0.6 | 0 | 0 | 0 |
| 2 | Negative control group | 0.009 ± 0.0006** | 7.66 ± 0.9*** | 0.3118 ± 0.13 | 11.23 ± 0.39 | 0 |
| 3 | Positive control group | 0.008 ± 0.0005 | 5.17 ± 0.6 | 0.0273 ± 0.03### | 6.96 ± 0.25### | 91% |
| 4 | AB group | 0.009 ± 0.0005* | 4.06 ± 0.73* | 0.0344 ± 0.04## | 7.09 ± 0.32### | 89% |
| 5 | PPI group | 0.009 ± 0.002* | 7.51 ± 0.45* | 0.0896 ± 0.10# | 8.79 ± 0.3### | 71% |
Positive control (standard regimen) group treated with 90 mg/kg amoxicillin, 135 mg/kg clarithromycin, 3.6 mg/kg omeprazole. AB group treated by amoxicillin + clarithromycin; PPI group treated by omeprazole; Vehicle used is 0.25% carboxymethylcellulose. Value are mean ± standard error mean of six animals in each group. All comparisons were performed using one-way ANOVA (dunnet’s test). *** p < 0.001, ** p < 0.01 and *p < 0.05 vs normal control rats. ### p < 0.001, ## p < 0.01 and # p < 0.05 vs vehicle control rats
Efficacy of standard treatment in P-gp modulated rats
The efficacy of standard treatment was indicated by decreasing or the disappearance of ulcer formation through ulcer index values. This study compared ulcer index of Group 3 treated with amoxicillin (90 mg/kg) and clarithromycin (135 mg/kg) and omeprazole (3.6 mg/kg), standard regimen, Group 4 treated with amoxicillin (90 mg/kg) and clarithromycin (135 mg/kg) and Group 5 treated with omeprazole (3.6 mg/kg) only. The experimental Group 2 was defined as vehicle control that was for normal P-gp, P-gp induced and P-gp inhibited rats. Figure 1 and Table 1 show the data of normal P-gp rats and there was a significant decrease in ulcer index in Group 3 (treated with standard regimen) 5.22 ± 0.21, p < 0.001, Group 4 (treated with antibiotic only) 6.87 ± 0.12, p < 0.01 and Group 5 (treated with omeprazole only) 8.65 ± 0.17, p < 0.05, compared to the vehicle control with ulcer index was 9.14 ± 0.41. The percentages of ulcer healing showed in Group 3 (standard regimen), Group 4 (antibiotic only) and Group 5 (omeprazole only) were 75%, 60% and 50%, respectively (Table 1). These results showed that the standard regimen (Group 3) produced the best outcome of recovery after 14 days of treatment for normal P-gp rats.
The outcomes of the treatment in P-gp induced rats are shown in Fig. 2 and Table 2. There was a significant decrease in ulcer index in Group 3 (standard regimen) 1.77 ± 0.13, p < 0.001, Group 4 (antibiotic only) 1.79 ± 0.19, p < 0.001 compared to vehicle control with ulcer index was 3.50 ± 0.18. Meanwhile, no significant difference in the ulcer index between Group 5 (omeprazole only), 3.48 ± 0.21, compared to vehicle control. The percentages of ulcer healing showed in Group 3 (standard regimen), Group 4 (antibiotic only) and Group 5 (omeprazole only) were 82%, 73% and 64%, respectively (Table 2). The standard regimen produced the best outcome of recovery after 14 days of treatment in P-gp induced rats with the highest percentage of ulcer healing. Furthermore, all treatment groups in P-gp induced rats showed the higher percentage of ulcer healing compared to the normal P-gp treatment groups.
Figure 3 and Table 3 show the data of P-gp inhibited rats and there was a significant decrease with p < 0.001 for all treatment groups in the ulcer index of Group 3 (standard regimen) 6.96 ± 0.25, Group 4 (antibiotic only) 7.09 ± 0.32, and Group 5 (omeprazole only) 8.79 ± 0.3, compared to vehicle control with ulcer index value of 11.23 ± 0.39 (p < 0.001). The percentages of ulcer healing in Group 3 (standard regimen), Group 4 (antibiotic only) and Group 5 (omeprazole only) were 91%, 89% and 71%, respectively (Table 3). These results indicated that the standard regimen produced the best outcome of recovery after 14 days of treatment in P-gp inhibited rats with the highest percentage of ulcer healing. Despite P-gp inhibited groups showed severe infection of H. pylori (ulcer index 11.23 ± 0.39), they produced the best outcome of treatment with standard regimen (91% ulcer healing), compared to the induced and normal P-gp rat groups.
Effect of P-gp modulation on the efficacy of treatment in H. pylori infection
Table 1, Table 2 and Table 3 show the ulcer index of non-infected normal P-gp and P-gp modulated rats were 0. Figure 1a, 2a and 3a, indicated the modulation of P-gp was not affected on the gastric mucosa of non-infected rats as there was no ulcer formation observed on the mucosa of the normal P-gp and P-gp modulated rats.
Figure 1b, Fig. 2b, and Fig. 3b show the vehicle control of P-gp inhibited rats contributed the worst ulcer formation than the normal P-gp and P-gp induced rats. These results corresponded with the ulcer index values of 11.23 ± 0.39 for P-gp inhibited rats, 9.14 ± 0.41 for normal P-gp rats and 3.5 ± 0.18 for P-gp induced rats (Table 1, Table 2 and Table 3). Different from the standard regimen, the P-gp inhibited rats showed the highest percentage of ulcer healing (91%, 89% and 71%, respectively), compared to P-gp induced rats (82%, 73% and 64%, respectively) and normal P-gp rats (75%, 60% and 50%, respectively), when only antibiotic and omeprazole were administered into the rats. From these findings, it was suggested that although the lower P-gp expression caused more severe infection, it provided a better outcome of the treatment.
Figures 1 to 3 and Tables 1 to 3 show the tested different three regimens have the same pattern of outcomes where the standard regimen gave the best efficacy compared to only antibiotic and omeprazole in normal P-gp rats, P-gp induced rats and P-gp inhibited rats.
The P-gp expression in P-gp modulated rats with H. pylori infection treated with standard regimen
Figure 4a shows after 14 days of modulation P-gp in the rats, the induced P-gp rats showed an increase of P-gp expression (2.00 ± 0.68) than normal P-gp rats (1.00 ± 0.00), while the inhibited P-gp rats showed lower P-gp expression (0.45 ± 0.36) than the normal P-gp rats. Higher P-gp expression was shown in all modulated P-gp rats that had been infected with H. pylori (Fig. 4b). The lowest P-gp expression was shown in modulated P-gp rats that had been treated with amoxicillin, clarithromycin and omeprazole (Fig. 4c), followed by amoxicillin and clarithromycin (Fig. 4d) and omeprazole only (Fig. 4e).
Fig. 4.

The expression of modulated P-gp rats with H. pylori induced ulcer formation treated with triple therapy. (a) P-gp expression on normal rats and modulated P-gp rats treated with rifampicin (P-gp inducer) and cyclosporine A (P-gp inhibitor). P-gp expression on normal and modulated P-gp rats with H. pylori induced ulcer formation in rats treated with (b) 0.25% carboxymethylcellulose (negative control), (c) amoxicillin, clarithromycin and omeprazole (standard regimen), (d) antibiotic (AB), and (e) omeprazole (PPI). *p < 0.05, **p < 0.01 vs normal P-gp rats; #p < 0.05, ##p < 0.01 vs induced P-gp rats
Discussion
Modulating the P-gp level in rats is important to eliminate the causal of multidrug resistance by increasing the bioavailability of drugs and enhancing their effectiveness for recovery. In this study, normal P-gp rats showed higher P-gp expression compared to the inhibited P-gp rats, suggesting normal P-gp rats probably refused the intake of xenobiotics or drugs. Besides, the normal tissue also contained pregnane-x-receptor (PXR) that might prevent entrance of xenobiotics into the gastric mucosal cells [22]. According to Kuster et al., cancer drug would modify P-gp expression through stress response, rather than cell destruction or apoptosis [23]. Since P-gp is well known as the first responder that receives xenobiotic as well as chemicals on cancer cells, and thus increases the P-gp level in normal tissue which removes the unwanted drug from entering to the cells, impairing the efficacy of the treatment regimen. To enhance the efficacy of drug treatment, cyclosporine A was given as inhibitor P-gp to increase the bioavailability and pharmacological effects of drugs [18]. Cyclosporine A is a substrate of P-gp, but not to CYP3A [24]. Therefore, when the P-gp level was inhibited, it would reduce the drug metabolism in the gut by reducing the drug from being exposed to CYP3A, thereby increasing the bioavailability of the drug. While high P-gp expression could be treated with P-gp inhibitor, low P-gp expression could be treated with P-gp inducer such as rifampicin [25]. Rifampicin is P-gp inducer that could activate PXR. PXR played an important role in the metabolism and secretion of xenobiotic by inducing CYP enzyme and protein transporter [26]. Both the CYP enzyme and P-gp transporter have been regulated by nuclear receptor such as PXR [27]. The activation of nuclear receptor by P-gp inducer might increase mRNA P-gp expression [28].
H. pylori invaded the gastric mucosa to initiate its pathogenesis [23]. Upon H. pylori colonization, the normal and inhibited P-gp rats showed higher ulcer index in the gastric mucosa. Higher colonization of H. pylori in the gastric mucosa of inhibited P-gp rats was due to the low protection of P-gp against xenobiotic (H. pylori) compared to normal P-gp rats. Basically, P-gp was reduced following H. pylori infection as P-gp was localized at the epithelial cell membrane of the gastric mucosa that controlled the entrance of xenobiotic [29]. However, in this study it indicated higher P-gp expression in the inhibited and normal P-gp rats that had been infected with H. pylori. This finding is in accordance with the findings reported by several researchers, in which H. pylori were able to stimulate P-gp expression [6, 13, 30]. H. pylori stimulated P-gp expression through COX-2 expression since both are involved in tissue inflammation [31]. The COX-2 expression was shown in H. pylori positive patient with gastritis and the expression was enhanced significantly in gastric cancer [32]. Another study by Patel et al. (2002), reported that increased mRNA level in MDR1 might increase COX-2 expression [33]. In this study, high P-gp expression was not only detected in the normal and inhibited P-gp rats, but also in the induced P-gp rats infected by H. pylori. Nevertheless, the rats with P-gp induced by rifampicin showed a mild H. pylori infection on the gastric mucosa. Besides its role as P-gp inducer, rifampicin also has bactericidal effects by disturbing nucleic acid synthesis in H. pylori, subsequently limits the H. pylori colonization into the gastric mucosal cells [34].
There were cases of treatment failure reported for the H. pylori eradication therapy despite the use of the standard regimen [35]. Therefore, modulating the P-gp expression prior by giving a treatment is one of the options for H. pylori eradication therapy. In this current study, the inhibited P-gp expression rats that were being infected with H. pylori and treated with antibiotic and omeprazole (standard regimen) showed the highest recovery (91%). The absorption of drugs into the gastric mucosal cell was based on the pKa principle, in which basic drugs were ionized at acidic pH while acid drugs were ionized at basic pH [36]. During H. pylori infection, the gastric environment was at alkaline pH. Therefore, acid drug (amoxicillin) would be ionized, while basic drug (chlaritromycin) would not be ionized in the stomach [37, 38]. Since drugs easily pass through the gastric mucosal cells at low ionic environment with highest membrane permeability, therefore clarithromycin was readily absorbed into the cells compared to amoxicillin. Besides, P-gp has membrane non-ionic transporter, allowing only drug that has the highest lipid permeability to pass through the cell, and thus increases the amount of clarithromycin in the cell. Despite omeprazole’s role in blocking the H+/K + - ATPase and P-gp expression, it also has the ability to prevent or hold a drug that already entered the cells [13, 39]. Therefore, omeprazole indirectly retains clarithromycin and amoxicillin were removed from the cell. Clarithromycin, which was also an inhibitor to P-gp, gave a double inhibition of P-gp expression when combined with omeprazole [40]. This subsequently increased the absorption of amoxicillin into the gastric mucosal cell. Since amoxicillin has bactericidal effect, it will eradicate H. pylori effectively [41].
Even though all modulated P-gp rats showed an increase in eradication of H. pylori when treated with antibiotic and omeprazole, there was variation in P-gp expression within those groups. Reducing the P-gp expression facilitated the retainment of drugs in the gastric mucosal cells, which subsequenly eradicated the H. pylori more effectively. Our findings suggested that rifampicin has the ability to eradicate H. pylori in the group of rats with induced P-gp level. Therefore, P-gp expression played an important role in the treatment of H. pylori infection, particularly in the formation of ulcer. The induction of P-gp expression in the mucosa gut of the rat has been shown to lower down the rate of ulcer in the presence of H. pylori. Nevertheless, as the gene polymorphism such as MDR1 C3435T was not measured in this study, hence our findings should be interpreted with caution as the presence of the polymorphism could directly affect the P-gp expression and pharmacokinetics of drugs.
Conclusion
The synergism between amoxicillin, clarithromycin and omeprazole as a standard therapy still provides the best outcome in the eradication of H. pylori infection. This study suggested that P-gp expression affected the effect of medication regimen used in the treatment of H. pylori infection, where in the inhibition P-gp expression caused severe infection of H. pylori with high value of ulcer index, although it produced the best outcome of treatment. In contrast, high expression of P-gp results in the least infection of H. pylori with low value of ulcer index, although no antibiotic treatment was given, but still showed a lower outcome of treatment compared to inhibited P-gp condition. In this study, the use of rifampicin has also indicated the eradication of H. pylori infection effectively by inducing the P-gp expression.
Acknowledgments
This study was funded by Universiti Kebangsaan Malaysia with grant no. GGPM 2013-094. Special thanks to Dr. Alfizah Hanafiah (PPUKM) for providing the bacterial sample.
Availability of data and materials
Data generated or analyzed during this study was included in this article.
Authors’ contributions
EK and MSO planned the experimental phase and NSD and AHM performed the experiments and carried out the statistical analysis. EK and MSO conceptualized the study design and AFAR coordinated the research activities. The manuscript was written by NSD, MSO and reviewed by EK. All authors have read and approved the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declared that they have no conflict of interest.
Ethical approval
The study was approved by Universiti Kebangsaan Malaysia Animal Ethics Committee, (UKMAEC) under the approval number FF/2015/ENDANG/29-SEPT./699-SEPT.-2015-SEPT in accordance with the international guidelines for animal studies (WHO Chronicle, 1985) [17].
Consent for publication
Not applicable.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Silva R, Vilas-Boas V, Carmo H, Dinis-Oliveria RJ, Carvalho F, De Lourdes BM, et al. Modulation of P-glycoprotein efflux pump: induction and activation as a therapeutic strategy. Pharmacol Ther. 2015;149:1–123. doi: 10.1016/j.pharmthera.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 2.Elmeliegy M, Vourhavis M, Guo C, Wang DD. Effect of P-glycoprotein (P-gp) inducers on exposure of p-gp substrates. Review of clinical drug-drug interaction studies. Clin Pharmacokinet. 2020;59(2726):699–714. doi: 10.1007/s40262-020-00867-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palatini P, Martin SR. Pharmacokinetic drug interactions in liver disease: an update. World J Gastroenterol. 2016;22(3):1260–1278. doi: 10.3748/wjg.v22.i3.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu D. Where is it and how does it get there – intracellular localization and traffic p-glycoprotein. Front Oncol. 2013;3(321):1–5. doi: 10.3389/fonc.2013.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin JH. Drug-drug interaction mediated by inhibition and induction of p-glycoprotein. Adv Drug Deliv Rev. 2003;55(1):53–81. doi: 10.1016/s0169-409x(02)00171-0. [DOI] [PubMed] [Google Scholar]
- 6.Babic Z, Svoboda-Beusan L, Kuicisec-Tepes N, Dekaris D, Troskot R. Increased activity of P-glycoprotein multidrug transporter in patient with Helicobacter pylori infection. World J Gastroenterol. 2005;11:2720–2725. doi: 10.3748/wjg.v11.i18.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mouly S, Paine MF. P-glycoprotein increases from proximal to distal regions of human small intestine. Pharm Res. 2003;20(10):1595–1599. doi: 10.1023/A:1026183200740. [DOI] [PubMed] [Google Scholar]
- 8.Wessler S, Krisch LM, Elmer DP, Aberger F. From inflammation to gastric cancer – the importance of hedgehog/GLI signaling in Helicobacter pylori-induced chronic inflammatory and neoplastic diseases. Cell Commun Signal. 2017;15:15. doi: 10.1186/s12964-017-0171-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mbulaiteye SM, Hisada M, El-Omar EM. Helicobacter Pylori associated global gastric cancer burden. Front Biosci. 2009;14:1490–1504. doi: 10.2741/3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chey WD, Leontiadis GI, Howden CW, Steven F, Moss SF. ACG clinical guideline: treatment of Helicobacter pylori infection. Am J Gastroenterol. 2017;112:212–238. doi: 10.1038/ajg.2016.563. [DOI] [PubMed] [Google Scholar]
- 11.Zhang M. High antibiotic resistance rate: a difficult issue for Helicobacter pylori eradication treatment. World J Gastroenterol. 2015;21(48):13432–13437. doi: 10.3748/wjg.v21.i48.13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cianci R, Montalto M, Pandolfi F, Gasbarrini GB, Cammarota G. Third-line rescue therapy for helicobacter pylori infection. World J Gastroenterol. 2006;12(15):2313–2319. doi: 10.3748/wjg.v12.i15.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Omar MS, Damanhuri NS, Kumolosasi E. Influences of proton pump inhibitor on Helicobacter pylori adherence to the gastrointestinal cell lines. Turk J Gastroenterol. 2017;28(1):55–61. doi: 10.5152/tjg.2016.0409. [DOI] [PubMed] [Google Scholar]
- 14.Twentyman PR, Bleehen NM. Resistance modification by PSC-833, a novel non-immunosupressive cyclosporine a. Eur J Cancer. 1991;27(12):1639–1642. doi: 10.1016/0277-5379(91)90435-G. [DOI] [PubMed] [Google Scholar]
- 15.Colabufo NA, Francesco B, Maria GP, Elena C, Mariangela C, Carmela I, et al. Substrates, inhibitors and activators of P-glycoprotein: candidates for radiolabeling and imaging perspectives. Curr Top Med Chem. 2010;10:1703–1714. doi: 10.2174/156802610792928022. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Wang H, Zhifa L, Wang Y, Wang B, Xie Y, et al. Rescue therapy with a proton pump inhibitor plus amoxicillin and rifabutin for Helicobacter pylori infection: a systematic review and meta-analysis. Gastroenterol Res Pract. 2015:1–11. 10.1155/2015/415648. [DOI] [PMC free article] [PubMed]
- 17.Howard-Jones N. A CIOMS Ethical Code for Animal Experimentation. WHO Chronicle. 1985;39(2):51–56. [PubMed] [Google Scholar]
- 18.Luo X, Yang T, Yang C, Zhou J, Liu Y, Huang Y, et al. Efffect of multiple oral dosing of cyclosporie on the pharmacokinetics of quercetin in rats. Int J Clin Exp Med. 2016;9(6):5880–5890. [Google Scholar]
- 19.Sandström R, Lennernäs H. Repeated oral rifampicin decreases the jejunal permeability of R/S- verapamil in rats. Drug Metab Dispos. 1999;27(8):951–955. [PubMed] [Google Scholar]
- 20.Mahmood AA, Mariod AA, Al-Bayaty F, Abdel-Wahab SI. Antiulcerogenic activity of Gynura procumbens leaf extract against experimentally-induced gastric lesions in rats. J Med Plant Res. 2010;4(8):685–691. [Google Scholar]
- 21.Mahmood T, Yang PC. Western blot: technique, theory, and trouble shooting. N Am J Med Sci. 2012;4(9):429–434. doi: 10.4103/1947-2714.100998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhuo W, Hu L, Lv J, Wang H, Zhou H, Fan L. Role of pregnane-x-receptor in chemotherapeutic treatment. Cancer Chemoth Pharm. 2014;74(2):217–227. doi: 10.1007/s00280-014-2494-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuster JG, Van Vliet HMA, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19(3):449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christians U, Schmitz V, Haschke M. Functional interactions between P-glycoprotein and CYP3A in drug metabolism. Expert Opin Drug Metab Toxicol. 2005;1(4):641–654. doi: 10.1517/17425255.1.4.641. [DOI] [PubMed] [Google Scholar]
- 25.Oesch F, Arand M, Benedetti MS, Castelli MG, Dostert P. Inducing properties of rifampicin and Rifabutin for selected enzyme activities of the cytochrome P-450 and UDP-glucuronosyltransferase Superfamilies in female rat liver. J Antimicrob Chemother. 1996;37:1111–1119. doi: 10.1093/jac/37.6.1111. [DOI] [PubMed] [Google Scholar]
- 26.Luo G, Cunningham M, Kim S, Burn T, Lin J, Sinz M, Hamilton G, Rizzo C, Jolley S, Gilbert D, Downey A, Mudra D, Graham R, Carroll K, Xie J, Madan A, Parkinson A, Christ D, Selling B, LeCluyse E, Gan LS. CYP3A4 induction by drugs: correlation between a pregnane-x-receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug Metab Dispos. 2002;30:795–804. doi: 10.1124/dmd.30.7.795. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Sykes DB, Miller DS. Constitutive androstane receptor-mediated up-regulation of ATP-deriven xenobiotic efflux transporters at the blood brain barrier. Mol Pharmacol. 2010;78:376–383. doi: 10.1124/mol.110.063685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowest KS, Haslam IS, Fihn B, Hilgendorf C, Karlsson JE, Simmons NL, et al. The effect of pregnanolone 16α-carbonitrile dosing on digoxin pharmacokinetics and intestinal absortion in the rat. Pharmaceutics. 2010;2:61–77. doi: 10.3390/pharmaceutics2010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferreira RJ, Dos Santos DJ, Ferreira MJ. P-glicoprotein and membrane roles in multidrug resistance. Future Med Chem. 2015;7(7):929–946. doi: 10.4155/fmc.15.36. [DOI] [PubMed] [Google Scholar]
- 30.Omar M, Crowe A, Richard P, Hooi EE, Tay CY, Hughes J. P-glycoprotein expression in Helicobacter pylori – positive patients: the influence of MDR1 C345TT polymorphism. J Dig Dis. 2012;13:414–420. doi: 10.1111/j.1751-2980.2012.00606.x. [DOI] [PubMed] [Google Scholar]
- 31.Nardone G, Rocco A, Vaira D, Staibano S, Budillon A, Tatangelo F, Sciulli MG, Perna F, Salvatore G, di Benedetto M, de Rosa G, Patrignani P. Expression of COX-2, mPGE-synthase1, MDR-1 (P-gp), and Bcl-x L : a molecular pathway of Helicobacter pylori related gastric carcinogenesis. J Pathol. 2004;202(3):305–312. doi: 10.1002/path.1512. [DOI] [PubMed] [Google Scholar]
- 32.Shao Y, Sun K, Xu W, Li X, Shen H, Sun W. Helicobacter pylori infection, gastrin and Cyclooxygenase-2 in gastric carcinogenesis. World J Gastroenterol. 2014;20(36):12860–12873. doi: 10.3748/wjg.v20.i36.12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel VA, Dunn MJ, Sorokin A. Regulation of MDR1 (P-glycoprotein) by Cyclooxygenase-2. J Biol Chem. 2002;277(41):38915–38920. doi: 10.1074/jbc.M206855200. [DOI] [PubMed] [Google Scholar]
- 34.Wu W, Yang Y, Sun G. Recent insights into antibiotic resistance in Helicobacter pylori eradication. Gastroenterol Res Pract. 2012;8:1–8. doi: 10.1155/2012/723183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tay CY, Windsor HM, Thirriot F, Lu W, Conway C, Perkins TT, Marshall BJ. Helicobacter pylori eradication in Western Australia using novel quadruple therapy combination. Aliment Pharmacol Ther. 2012;36:1076–1083. doi: 10.1111/apt.12089. [DOI] [PubMed] [Google Scholar]
- 36.Helmenstine AM. pKa Definition in Chemistry. ThoughtCo, thoughtco.com/what-is-pka-in-chemistry-605521. [Feb. 11, 2020].
- 37.Babic S, Horvat AJM, Pavlovic DM, Macan MK. Determination of Pka value of active pharmaceutical ingredients. Trac- Trend Anal Chem. 2007;26(11):1043–1061. doi: 10.1016/j.trac.2007.09.004. [DOI] [Google Scholar]
- 38.Hancu G, Naecsu A, Papp LA, Ciurba A. Simultaneous determination of amoxicillin and Calvulanic acid in pharmaceutical preparation by capillary zone electrophoresis. Braz J Pharm Sci. 2016;52(2):281–286. doi: 10.1590/S1984-82502016000200006. [DOI] [Google Scholar]
- 39.Paulli-Magnus C, Rekersbrink S, Klotz U, Fromm MF. Interaction of omeprazole, Lansoprazole, and pantoprazole with P-glycoprotein. Naunyn Schmiedeberg’s Arch Pharmacol. 2001;364:551–557. doi: 10.1007/s00210-001-0489-7. [DOI] [PubMed] [Google Scholar]
- 40.Lam A, Hoang JD, Singleton A, Han X, Bleier BS. Itraconazole and Chlaritromycin inhibit P-glycoprotein activity in primary human Sinonasal epithelial cell. Int Forum Allergy Rhinol. 2015;5(6):477–480. doi: 10.1002/alr.21454. [DOI] [PubMed] [Google Scholar]
- 41.Marcus EA, Sachs G, Scott DR. Eradication of Helicobacter pylori infection. Curr Gastroenterol Rep. 2016;18(7):33. doi: 10.1007/s11894-016-0509-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data generated or analyzed during this study was included in this article.