

Abstract
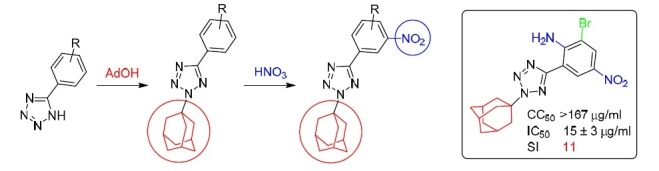
The reaction of 5-aryl-NH-tetrazoles with adamantan-1-ol in concentrated sulfuric acid proceeds regioselectively with the formation of the corresponding 2-adamantyl-5-aryl-2H-tetrazoles. Nitration of these compounds leads to 2-(adamantan-1-yl)-5-(3-nitroaryl)-2Htetrazoles. The structures and composition of the obtained novel 2-adamantyl-5-aryltetrazoles were proven by IR spectroscopy, 1H and 13C NMR spectroscopy, high-resolution mass spectrometry, and also by X-ray structural analysis. According to the simultaneous thermal analysis data, the obtained compounds are thermally stable up to a temperature of about 150°C. In vitro studies have shown that some of the 2-adamantyl-5-aryltetrazoles exhibit moderate inhibitory activity against influenza A (H1N1) virus. The antiviral selectivity index (SI) of 2-[2-(adamantan-1-yl)-2H-tetrazol-5-yl]-6-bromo-4-nitroaniline is significantly higher (SI 11) than that of the reference drug rimantadine (SI 5).
Supplementary Information
The online version contains supplementary material available at 10.1007/s10593-021-02931-5.
Keywords: 2-(adamantan-1-yl)-5-aryl-2H-tetrazoles, adamantylation, anti-influenza activity, nitration, X-ray structural analysis
It is known that a number of adamantane derivatives and other cage hydrocarbons exhibit high antiviral activity.1 Some of these compounds are known drugs against influenza virus (amantadine, rimantadine).4 (Adamantan-2-yl)-1-(2-oxoindolin-3-ylidene)thiosemicarbazide A (Fig. 1) reduces the reproducibility of vaccinia virus, and also exhibits activity in vitro against some Gram-positive and Gramnegative bacteria and fungi.2, 3 Substituted aminoacetyladamantylamines B exhibit inhibitory activity against vaccinia virus (Fig. 1).3 1-Adamantanecarboxylic acid amide containing the (–)-10-amino-2-pinene fragment (structure C)4 and N-(1-adamantyl)-2,2-diphenylacetamide D proved to be effective inhibitors of smallpox virus.5 In addition, compounds containing the adamantyl fragment such as memantine E (Fig. 1) are also capable of inhibiting the N-methyl-D-aspartate receptor, which allows them to be considered as potential agents for the treatment of moderate and severe Alzheimer's disease.6 Adamantane derivatives have shown in vitro activity against various coronaviruses.7, 8 For instance, amantadine analogs are moderately active against human coronavirus 229E.7 Moderate antiviral activity has also been found for amantadine, rimantadine, memantine in bovine models.8
Figure 1.
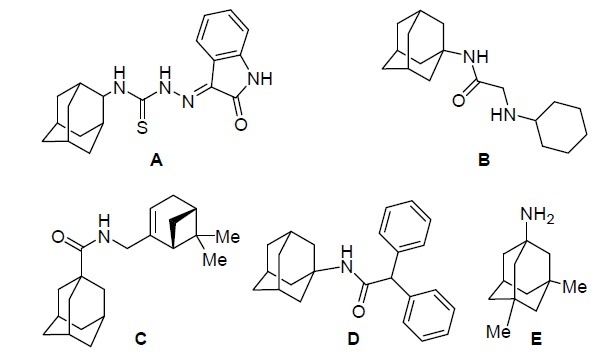
Adamantane-based biologically active compounds.
One of the promising design options for new highly effective anti-influenza drugs is the replacement of the amino group with polynitrogen heterocyclic fragments.9–11 This fragment can be the tetrazole ring, a well-known pharmacophore9–11 due to its high stability in metabolic processes and low toxicity. Tetrazole derivatives containing the adamantyl moiety showed significant activity against influenza A virus, including rimantadine-resistant influenza virus strain A/Puerto Rico/8/34 (Fig. 2).12, 13
Figure 2.
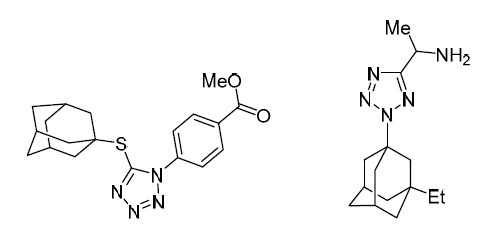
Adamantyltetrazoles showing high inhibitory activity against influenza virus.
In this work, we obtained a series of NH-unsubstituted 5-aryltetrazoles 2a–h by azidation of aromatic nitriles 1a–h via the 1,3-dipolar cycloaddition reaction (Scheme 1). Azidation was carried out in DMF according to a known method at 115°C.11–14 The reaction of 5-aryl-NH-tetrazoles 2a–h with adamantan-1-ol in concentrated H2SO4 gave a series of novel 2-adamantyl-5-aryl-2Н-tetrazoles 3a–h. The adamantylation of NH-tetrazoles 2a–h in H2SO4 proceeds exclusively at the N-2 nitrogen atom of the tetrazole ring with the formation of 2-(adamantan-1-yl)-5-aryl-2Н-tetrazoles 3a–h in high (81–92%) yields. Compounds 3c,d were nitrated with a sulfuric-nitric acid mixture at 8–10°С according to the previously described method15 to give 2-(adamantan-1-yl)-5-(nitroaryl)-2Н-tetrazoles 3j,k. Compound 3i was obtained by reduction of nitro compound 3g with metallic zinc.
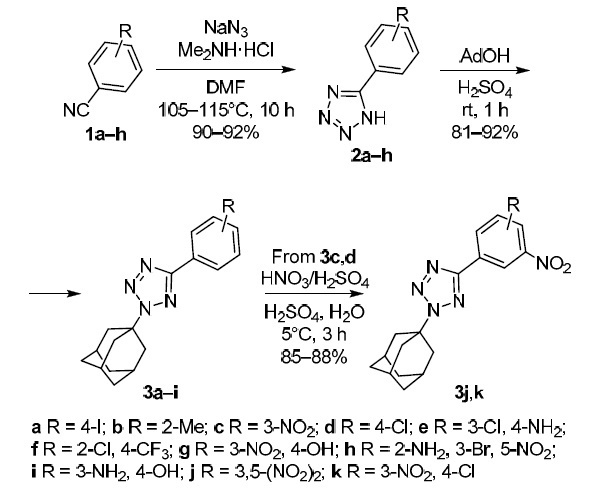
Scheme 1
The structure, composition, and thermal stability of the obtained compounds were studied by IR spectroscopy, 1H, 13C NMR spectroscopy, high-resolution mass spectrometry, X-ray structural analysis, and simultaneous thermal analysis. The 1H and 13C NMR spectra confirm the structures of all the obtained compounds. In the 1Н NMR spectra of 2-(adamantan-1-yl)-5-aryl-2Н-tetrazoles 3a–k, the adamantyl moiety appears as broadened singlets at 1.76–1.87 (compounds 3a–g,i–k), 2.16 (compound 3h), and 2.22–2.36 ppm and in the form of broadened singlets or multiplets in the 2.29–2.43 ppm range. In the 13C NMR spectra of compounds 3a–k, the signals of the endocyclic carbon atom of the tetrazole fragment were observed at 161.1–164.9 ppm which reliably confirms the preparation of 2H-regioisomers.14
The IR spectra of compounds 3a,b,e,h contain the characteristic absorption bands of the cage hydrocarbon group in the range of 3346–2850 cm–1 which is consistent with previously published data.16
The data of X-ray structural analysis of compounds 3b,e–g also unambiguously confirm the structure of the obtained compounds (Fig. 3). According to these data, the 2H-tetrazole ring is a planar highly aromatic system. The dihedral angle between the planes of the benzene and tetrazole rings in compounds 3b,e–g is small, which may indicate π–π conjugation between unsaturated fragments. In all of the studied compounds, the tetrazole ring appears to be partially sterically blocked by a cage hydrocarbon fragment.
Figure 3.
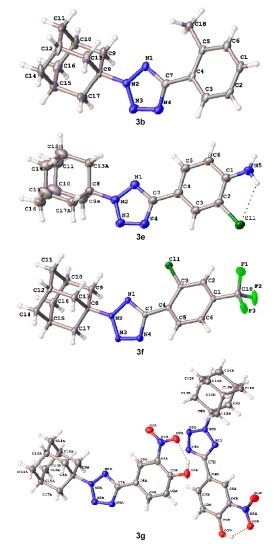
Molecular structure of compounds 3b,e,f,g with atoms represented as thermal vibration ellipsoids of 50% probability.
It is known that some 2H-tetrazoles, especially those containing electron-withdrawing substituents, can relatively easily undergo thermal decomposition with the dissociation of the N–N bonds and release of molecular nitrogen.11, 14 The thermal stability of compound 3g was studied by the method of simultaneous thermal analysis (Fig. S54, Supplementary information file). This compound contains an electron-withdrawing nitro group in the aryl moiety and is a good model for assessing the stability of compounds of this series. According to the loss of weight, in the case of compound 3g molecular nitrogen is released at 156.2°C. This process is accompanied by a strong exothermic effect. The complete decomposition of adamantyltetrazole 3g occurs starting with 250°C with a loss of 33.6% of its mass.
In this work, we studied the antiviral activity of adamantyltetrazole derivatives 3а,b,d,e,h,i against influenza A/Puerto Rico/8/34 (H1N1) virus (Table 1). Analysis of the obtained data shows that compounds 3а,d,e containing a halogen atom as a substituent mainly showed moderate inhibitory activity against the virus (IC50 31–2 μg/ml) and rather high toxicity against cells (CC50 46–4 μg/ml), which is expressed by the low values of the selectivity index (SI). At the same time, tetrazole derivative 3h containing an amino group and a bromine atom turned out to be the least toxic to cells (CC50 167 μg/ml) in combination with moderate activity against the virus (IC50 15 μg/ml), which is expressed by the relatively high value of the selectivity index of SI 11 exceeding the SI value of the reference drug rimantadine.
Table 1.
Cytotoxic (CC50), antiviral (IC50) activity, selectivity index (SI) of adamantyltetrazole derivatives 3а,b,d,e,h,i against the H1N1 influenza A virus strain
| Compound | CС50, μg/ml | IC50, μg/ml | SI |
|---|---|---|---|
| 3a | 46 ± 3 | 31 ± 4 | 1 |
| 3b | >300 | >300 | 1 |
| 3d | 4 ± 0.5 | 2 ± 0.3 | 2 |
| 3e | <3 | <3 | – |
| 3h | >167 | 15 ± 3 | 11 |
| 3i | 36 ± 2 | 15 ± 2 | 2 |
| Rimantadine | 60 ± 4 | 12 ± 2 | 5 |
The influenza virus used in the study shows resistance to rimantadine due to the S31N mutation in the viral protein M2. The activity of compound 3h demonstrated against this virus may indicate both that it is a specific inhibitor of such a mutant ion channel and that the target of its action is another viral protein and the mechanism of action of compounds of this class is not related to blocking the M2 proton channel.
To conclude, employing the regioselective alkylation of 5-aryl-NH-tetrazoles with adamantan-1-ol in sulfuric acid, a series of new 2-adamantyl-5-aryl-2H-tetrazoles were synthesized. The structure of the obtained compounds was proved by spectral methods and by the X-ray structural analysis technique. Some of the 2-adamantyl-5-aryltetrazoles exhibit moderate inhibitory activity against influenza A (H1N1) virus and good selectivity. 2-Adamantyl-5-aryl-2H-tetrazoles are promising compounds which can display high anti-influenza activity.
Experimental
IR spectra were registered on an IRPrestige-21 Fourier transform spectrometer in KBr pellets. 1H and 13C NMR spectra were acquired on a Bruker Avance III 400 spectrometer (400 and 100 MHz, respectively) at 25°C. High-resolution mass spectrometry was performed on a Bruker MicroTOF spectrometer (electrospray ionization). Melting points were determined on a PTP apparatus using 1°C/min heating rate in the melting range. Simultaneous thermal analysis was performed on a Netzsch STA 449F1 apparatus under a flow of argon (100 ml/min–1) with a heating rate of 10°С/min in aluminum crucibles. TLC was performed on Merck KGaA 60 F254 plates, eluent CHCl3–MeOH, 19:1.
Synthesis of 5-aryl- NH -tetrazoles 2a–h (General method). The corresponding nitrile 1a–h (10 mmol) was added to a solution of NaN3 (11.4 mmol) and Me2NH·HCl (11.2 mmol) in DMF (40 ml). The resulting suspension was heated at 105–115°C for 10 h. Then, the reaction mixture was cooled, the precipitate was filtered off and additionally washed with DMF (2×5 ml). Then cold Н2О (50 ml) was added to the filtrate and acidified with a solution of 10% HCl to pH 2–3. The resulting suspension was filtered, and the precipitate was washed with cold H2O. The product was air-dried. The physical and spectral characteristics of the obtained and previously described compounds 2a–d,h correspond to the literature.13, 15, 17
2-Chloro-4-(1 H -tetrazol-5-yl)aniline (2e). Yield 1.76 g (90%), white crystals, mp 169–170°C. 1H NMR spectrum (DMSO-d6), δ, ppm (J , Hz): 6.05 (2H, s, NH2); 6.94 (1H, d, J = 8.5, H Ar); 7.71 (1H, dd, J = 8.5, J = 2.0, Н Ar); 7.88 (1H, d, J = 2.0, Н Ar). 13C NMR spectrum (DMSO-d6), δ, ppm: 112.2, 115.8, 117.4, 127.3, 128.3, 147.9 (С Ar); 156.1 (C tetrazole). Found, m/z: 196.0379 [M+Н]+. C7H7ClN5. Calculated, m/z: 196.0385.
5-[2-Chloro-4-(trifluoromethyl)phenyl]-1 H -tetrazole (2f). Yield 2.29 g (92%), beige crystals, mp 179–181°C. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.72 (1H, dd, J = 8.2, J = 1.8, H Ar); 7.86 (1H, s, H Ar); 8.07 (1H, d, J = 8.2, H Ar). 13C NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 122.6 (d, J = 270.4, CF3); 124.0, 125.4, 127.4, 132.1 (2C), 136.4 (C Ar); 158.8 (C tetrazole). Found, m/z: 270.9969 [M+Na]+. C8H4ClF3N4Na. Calculated, m/z: 270.9969.
2-Nitro-4-(1 H -tetrazol-5-yl)phenol (2g). Yield 1.88 g (91%), yellow crystals, mp 179–181°C. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.34 (1H, d, J = 8.7, H Ar); 8.18 (1H, dd, J = 8.7, J = 2.3, H Ar); 8.55 (1H, d, J = 2.2, H Ar); 11.84 (1H, br. s, OH). 13C NMR spectrum (DMSO-d6), δ, ppm: 120.8, 124.6, 133.7, 137.6, 138.3, 154.6 (C Ar); 155.8 (C tetrazole). Found, m/z: 206.0318 [M–Н]–. C7H4N5O3. Calculated, m/z: 206.0309.
Synthesis of 2-(adamantan-1-yl)-5-aryl-2 H -tetrazoles 3a–h (General method). Adamantan-1-ol (1.37 g, 9 mmol) was added to a solution of 5-arylterazole 2a–h (9 mmol) in 94% H2SO4 (25 ml). The reaction mixture was kept at room temperature for 60 min and poured into H2O/ice (60 ml). The product was extracted with CHCl3, the organic phase was washed with 5% aqueous NaHCO3 (2×25 ml) followed by H2O. It was then dried over anhydrous MgSO4 and evaporated under reduced pressure. The physical and spectral characteristics of the obtained and previously described compounds 3b–d correspond to the literature.13, 15
2-(Adamantan-1-yl)-5-(4-iodophenyl)-2 H -tetrazole (3a). Yield 3.22 g (88%), light-orange crystals, mp 118–120°C. IR spectrum, ν, cm–1: 3295, 2917, 2850, 1697, 1449, 1353, 1302, 1116, 1088, 927. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 1.85 (6H, br. s, H Ad); 2.33 (3H, br. s, H Ad); 2.41 (6H, br. s, H Ad); 7.47 (2H, d, J = 8.4, H Ar); 8.12 (2H, d, J = 8.8, H Ar). 13C NMR spectrum (CDCl3), δ, ppm: 29.7, 36.1, 42.3, 64.9 (C Ad); 127.5, 128.4, 137.6, 138.0 (C Ar); 163.5 (C tetrazole). Found, m/z: 429.0545 [M+Na]+. C17H19IN4Na. Calculated, m/z: 429.0547.
2-(Adamantan-1-yl)-5-( о -tolyl)-2 H -tetrazole (3b). Yield 2.44 g (92%), light-beige crystals, mp 85–87°C (mp 84–86°C13). IR spectrum, ν, cm–1: 3292, 2924, 2911, 2856, 1476, 1453, 1356, 1316, 1021, 746. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 1.77 (6H, br. s, H Ad); 2.24 (3H, br. s, H Ad); 2.33 (6H, d, J = 2.8, H Ad); 2.55 (3H, s, CH3); 7.33–7.43 (3H, m, Н Ar); 7.90 (1H, d, J = 7.6, Н Ar). 13C NMR spectrum (DMSO-d6), δ, ppm: 21.5 (СH3); 29.3, 35.6, 42.1, 64.0 (С Ad); 126.6, 126.9, 129.4, 130.4, 131.9, 137.1 (С Ar); 164.2 (C tetrazole). Found, m/z: 295.1923 [M+Н]+. C18H23N4. Calculated, m/z: 295.1917.
2-(Adamantan-1-yl)-5-(3-nitrophenyl)-2 H -tetrazole (3c). Yield 2.52 g (86%), light-yellow crystals, mp 192–193°C (mp 192–193°C15). 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 1.87 (6H, br. s, H Ad); 2.35 (3H, br. s, H Ad); 2.43 (6H, br. s, H Ad); 7.70 (1H, t, J = 8.0, Н Ar); 8.31–8.35 (1Н, m, Н Ar); 8.37 (1Н, s, Н Ar); 8.52–8.55 (1H, m, Н Ar). 13C NMR spectrum (CDCl3), δ, ppm: 29.4; 35.8; 42.3; 64.5 (С Ad); 121.8, 124.1, 124.5, 127.6, 129.9, 132.4 (С Ar); 162.4 (C tetrazole). Found, m/z: 348.0814 [M+Na]+. C17H19N5NaO2. Calculated, m/z: 348.1431.
2-(Adamantan-1-yl)-5-(4-chlorophenyl)-2 H -tetrazole (3d). Yield 2.38 g (84%), white crystals, mp 134–136°C (mp 131–133°C13). 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 1.85 (6H, t, J = 3.2, H Ad); 2.33 (3H, br. s, H Ad); 2.41 (6H, d, J = 2.8, H Ad); 7.47 (2H, d, J = 8.8, Н Ar); 8.12 (2H, d, J = 8.8, Н Ar). 13C NMR spectrum (CDCl3), δ, ppm: 29.4, 35.9, 42.3, 64.1 (С Ad); 126.5, 128.1 (2C), 129.0, 129.1, 135.9 (С Ar); 163.3 (C tetrazole). Found, m/z: 315.1375 [M+Н]+. C17H20ClN4. Calculated, m/z: 315.1371.
4-[2-(Adamantan-1-yl)-2 H -tetrazol-5-yl]-2-chloroaniline (3e). Yield 2.47 g (83%), light-yellow crystals, mp 190–192°C. IR spectrum, ν, cm–1: 3202, 2941, 2916, 1635, 1606, 1466, 1420. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 1.77 (6H, br. s, H Ad); 2.24 (3H, br. s, H Ad); 2.29 (6H, br. s, H Ad); 5.87 (2H, br. s, NH2), 6.91 (1H, d, J = 8.4, Н Ar); 7.70 (1H, d, J = 8.4, Н Ar); 7.82 (1H, d, J = 1.6, Н Ar). 13C NMR spectrum (DMSO-d6), δ, ppm: 29.3, 35.7, 42.1, 63.8 (С Ad); 115.9, 117.4, 126.5, 127.4, 131.0, 147.1 (С Ar); 163.5 (C tetrazole). Found, m/z: 330.1496 [M+Н]+. C17H21ClN5. Calculated, m/z: 330.1480.
2-(Adamantan-1-yl)-5-[2-chloro-4-(trifluoromethyl)-phenyl]-2 H -tetrazole (3f). Yield 2.79 g (81%), white crystals, mp 126–128°C. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 1.76 (6H, br. s, H Ad); 2.24 (3H, br. s, H Ad); 2.33 (6H, d, J = 2.8, H Ad); 7.89–7.92 (1H, m, Н Ar); 8.08 (1H, s, Н Ar); 8.15 (1H, d, J = 8.0, H Ar). 13C NMR spectrum (DMSO-d6), δ, ppm: 29.3, 35.5, 42.1, 64.8 (С Ad); 125.0 (в, J = 270.4, CF3); 125.8, 128.1, 130.6, 132.8 (2С), 133.2 (С Ar); 161.1 (C tetrazole). Found, m/z: 383.1252 [M+Н]+. C18H19ClF3N4. Calculated, m/z: 383.1245.
4-[(2-Adamantan-1-yl)-2 H -tetrazol-5-yl]-2-nitrophenol (3g). Yield 2.64 g (86%), light-orange crystals, mp 256–258°C. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 1.79 (6H, br. s, H Ad); 2.26 (3H, br. s, H Ad); 2.33 (6H, d, J = 2.4, H Ad); 7.33 (1H, d, J = 8.4, Н Ar); 8.19 (1H, dd, J = 8.8, J = 2.0, Н Ar); 8.48 (1H, d, J = 2.0, Н Ar); 11.63 (1H, s, OH). 13C NMR spectrum (DMSO-d6), δ, ppm: 29.3, 35.6, 42.1, 64.4 (С Ad); 105.0, 120.3, 123.6, 133.1, 139.6, 154.0 (С Ar); 164.9 (C tetrazole). Found, m/z: 340.1400 [M–Н]–. C17H18N5O3. Calculated, m/z: 340.1415.
2-[2-(Adamantan-1-yl)-2 H -tetrazol-5-yl]-6-bromo-4-nitroaniline (3h). Yield 3.13 g (83%), light-yellow crystals, mp 172–174°C. IR spectrum, ν, cm–1: 3346, 2908, 1611, 1594, 1495, 1323, 1303, 1273, 1142, 1020. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 2.16 (6H, br. s, H Ad); 2.36 (3H, br. s, H Ad); 2.43 (6H, br. s, H Ad); 8.47 (1H, d, J = 2.4, Н Ar); 9.07 (1H, d, J = 2.8, H Ar). 13C NMR spectrum (CDCl3), δ, ppm: 29.4, 36.1, 42.3, 64.9 (С Ad); 109.0, 109.9, 124.3, 124.5, 129.4, 148.2 (С Ar); 162.1 (C tetrazole). Found, m/z: 417.0677 [M–Н]–. C17H18BrN6O2. Calculated, m/z: 417.0680.
4-[2-(Adamantan-1-yl)-2 H -tetrazol-5-yl]-2-aminophenol (3i). HCl and metallic Zn powder in small portions were successively added to a suspension of 4-[(2-adamantan-1-yl)-2H-tetrazol-5-yl]-2-nitrophenol (3g) (2 mmol) in a mixture of i-PrOH (30 ml) and H2O (50 ml). The reaction mixture was heated to 50°C and maintained until the disappearance of the starting compound (TLC control, eluent CHCl3–MeOH, 19:1). The reaction mixture was then evaporated under reduced pressure. The residue was neutralized with an aqueous NaOAc to pH 4–5. The formed precipitate was filtered off, air-dried, and recrystallized from MeOH. Yield 2.30 g (82%), light-brown crystals, mp 157–159°C. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 1.78 (6H, br. s, H Ad); 2.25 (3H, br. s, H Ad); 2.30–2.34 (6H, m, H Ad); 4.77 (2H, br. s, NH2); 6.76 (1H, d, J = 8.4, Н Ar); 7.15 (1H, dd, J = 6.0, J = 2.0, Н Ar); 7.34 (1H, d, J = 2.0, H Ar). 13C NMR spectrum (DMSO-d6), δ, ppm: 29.3, 35.6, 42.1, 64.4 (С Ad); 107.5, 123.7, 128.1, 133.1, 137.5, 150.8 (С Ar); 162.5 (C tetrazole). Found, m/z: 312.1814 [M+Н]+. C17H22N5O. Calculated, m/z: 312.1819.
Nitration of 5-aryltetrazole adamantyl derivatives (General method). A mixture of concentrated HNO3 (7 ml) and concentrated H2SO4 (5 ml) was added to a cooled (5°C) solution of adamantyl derivative of tetrazole 3с,d (6 mmol) in 75% H2SO4 (7 ml). The reaction mixture was stirred at 8–10°С for 3 h, then the pH of the reaction mixture was adjusted to 7 by adding an aqueous NaOH. The precipitated product was filtered off and dried.
2-(Adamantan-1-yl)-5-(3,5-dinitrophenyl)-2 H -tetrazole (3j). Yield 1.84 g (85%), white crystals, mp 276–277°C (mp 276–277°C15). 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 1.75–1.85 (6H, m, H Ad); 2.29 (3H, br. s, H Ad); 2.39 (6H, br. s, H Ad); 8.96 (1H, t, J = 2.1, Н Ar); 9.07 (2H, d, J = 2.1, Н Ar). 13C NMR spectrum (CDCl3), δ, ppm: 29.3, 35.5, 45.7, 65.2 (С Ad); 120.3, 126.5, 130.3, 149.3 (С Ar); 161.1 (C tetrazole). Found, m/z: 393.1250 [M+Na]+. C17H17N6NaO4. Calculated, m/z: 393.1282.
2-(Adamantan-1-yl)-5-(4-chloro-3-nitrophenyl)-2 H tetrazole (3k). Yield 1.95 g (88%), white crystals, mp 149–151°C. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 1.52–1.80 (6H, m, H Ad); 2.18–2.45 (3H, m, H Ad); 2.33 (6H, br. s, H Ad); 7.97 (1H, d, J = 8.4, Н Ar); 8.33 (1H, dd, J = 8.4, J = 2.0, Н Ar); 8.63 (1H, d, J = 2.0, Н Ar). 13C NMR spectrum (DMSO-d6), δ, ppm: 29.3, 35.6, 42.1, 64.9 (С Ad); 105.0, 114.8, 123.6, 131.7, 133.4, 148.5 (С Ar); 162.9 (C tetrazole). Found, m/z: 360.1225 [M+Н]+. C17H19ClN5O2. Calculated, m/z: 360.1222.
The study of the biological activity of compounds 3а,b,d,e,h,i. The cytotoxic properties of compounds were assessed using the methyl tetrazolium (MTT) assay. A series of threefold dilutions (300–3.7 μg/ml) were prepared from the studied compounds, after which they were introduced into the wells of plates with a monolayer of MDCK cells. The plates were incubated at 36°C for 72 h. Thereafter, MTT solution was added to the wells which by the action of mitochondrial enzymes transformed into an insoluble violet formazan derivative.18 Plates with cells were kept for 2 h. The sediment in each well was dissolved in DMSO (0.1 ml). The optical density in the wells was measured using a Thermo Multiskan FC plate photometer (Thermo Fisher Scientific, USA) with a wavelength of 540 nm. Based on the obtained data, the CC50 value was calculated as the concentration of the compound resulting in a decrease in optical density by half compared to the wells without addition of compounds.
The study of the antiviral activity of compounds 3а,b,d,e,h,i was carried by determining the reduction of the degree of cytopathic effect. The experiments used the influenza A/Puerto Rico/8/34 (H1N1) virus. The studied compounds in various concentrations were added to the cells in the wells of the plate, incubated for 1 h, then the cells were infected with the virus at a dose of 0.01 TCID50 per cell. The cells were incubated for 72 h, after which the cell viability was determined by the MTT assay as described above. Based on the obtained data for each compound, 50% inhibitory concentration (IC50) was calculated as the concentration of the compound that reduces the degree of viral cell destruction by 50%, and the selectivity index (SI) as the ratio of CC50 to IC50.
X-ray structural analysis of compounds 3b,e–g. Crystals suitable for X-ray structural analysis were obtained by slow evaporation of solutions of compounds 3b,e–g in CHCl3–MeOH, 1: 2 mixture at low temperature.
X-ray structural analysis of compounds 3b,e–g was done on a Rigaku Oxford Diffraction XtaLAB SuperNova diffractometer with a HyPix3000 CCD detector (compounds 3b,e,f) and on an Agilent Technologies Xcalibur diffractometer with an Atlas CCD detector (compound 3g) at 100K using monochromatic focused CuKα radiation. The obtained data were integrated in the CrysAlis software package.19 Absorption was corrected semiempirically. The signs of disordering of the adamantane group are removed from Figure 3 (compound 3e) for the sake of clarity. The unit cell parameters were specified by the least-squares method. The structures were solved and refined using the SHELX software package20 included in the OLEX221 interface. Crystallographic data of compounds 3b,e–g were deposited at the Cambridge Crystallographic Data Center (deposits CCDC 1984233 (compound 3b), CCDC 1984234 (compound 3e), CCDC 1984236 (compound 3f), CCDC 1984237 (compound 3g)).
Supplementary information file containing the IR, 1H and 13С NMR, and mass spectra of all the synthesized compounds, simultaneous thermal analysis data for compound 3g, as well as X-ray structural analysis data for compounds 3b,e–g is available at the journal website at http://link.springer.com/journal/10593.
This work was financially supported by the Russian Foundation for Basic Research (project 20-53-00039-Bel_a).
The work was carried out using the equipment of the resource centers of Saint Petersburg State University “Magnetic Resonance Research Center”, “Chemical Analysis and Materials Research Center”, “Chemistry Educational Center”.
Supplementary Information
(PDF 2818 kb)
Footnotes
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2021, 57(4), 442–447
Contributor Information
Olga V. Mikolaichuk, Email: st801293@spbu.ru
Vladimir V. Zarubaev, Email: zarubaev@gmail.com
Ilya V. Kornyakov, Email: i.kornyakov@spbu.ru
Rostislav Е. Trifonov, Email: rost_trifonov@mail.ru
References
- 1.Stockdale TP, Williams CM. Chem. Soc. Rev. 2015;44:7737. doi: 10.1039/C4CS00477A. [DOI] [PubMed] [Google Scholar]
- 2.Suslov EV, Mozhaytsev ES, Korchagina DV, Bormotov NI, Yarovaya OI, Volcho KP, Serova OA, Agafonov AP, Maksyutov RA, Shishkina LN, Salakhutdinov NF. RSC Med. Chem. 2020;11:1185. doi: 10.1039/D0MD00108B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pham VH, Phan TPD, Phan DC, Vu BD. Molecules. 2020;25:324. doi: 10.3390/molecules25020324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kreutzberger A, Schröders H-Н, Stratmann J. Arch. Pharm. 1984;317:767. doi: 10.1002/ardp.19843170907. [DOI] [PubMed] [Google Scholar]
- 5.Kreutzberger A, Schröders H-Н. Arch. Pharm. 1974;307:766. doi: 10.1002/ardp.19743071007. [DOI] [PubMed] [Google Scholar]
- 6.Parsons CG, Gruner R, Rozental J, Millar J, Lodge D. Neuropharmacology. 1993;32:1337. doi: 10.1016/0028-3908(93)90029-3. [DOI] [PubMed] [Google Scholar]
- 7.Rejdak K, Grieb P. Mult. Scler. Relat. Disord. 2020;42:102163. doi: 10.1016/j.msard.2020.102163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cimolai N. J. Med. Virol. 2020;92:531. doi: 10.1002/jmv.25752. [DOI] [PubMed] [Google Scholar]
- 9.Ostrovskii, V. A.; Trifonov, R. E.; Popova, E. A. Russ. Chem. Bull., Int. Ed.2012, 61, 768. [Izv. Akad. Sci., Ser. Khim.2012, 765.]
- 10.Popova, E. A.; Trifonov, R. E.; Ostrovskii, V. A. Russ. Chem. Rev. 2019, 88, 644. [Usp. Khim.2019, 88, 644.]
- 11.Ostrovskii VA, Popova EA, Trifonov RE. Adv. Heterocycl. Chem. 2017;123:1. doi: 10.1016/bs.aihch.2016.12.003. [DOI] [Google Scholar]
- 12.Zarubaev VV, Golod EL, Anfimov PM, Shtro AA, Saraev VV, Gavrilov AS, Logvinov AV, Kiselev OI. Bioorg. Med. Chem. 2010;18:839. doi: 10.1016/j.bmc.2009.11.047. [DOI] [PubMed] [Google Scholar]
- 13.Seliverstova, D. V.; Suslonov, V. V.; Zarubaev V. V.; Trifonov R. E. Russ. J. Org. Chem. 2018, 54, 633. [Zh. Org. Khim.2018, 54, 630.]
- 14.Ostrovskii, V. A.; Koldobskii, G. I.; Trifonov, R. E. In Сomprehensive Heterocyclic Chemistry III; Zhdankin, V. V., Ed.; Elsevier: Amsterdam, 2008, Vol. 6, p. 257.
- 15.Mikolaichuk, O. V.; Spasibenko, D. V.; Trifonov, R. E. Chem. Heterocycl. Compd.2020, 56, 961. [Khim. Geterotsikl. Soedin. 56, 961.]
- 16.D'yachenko, V. S.; Danilov, D. V.; Shkineva, T. K.; Vatsadze, I. A.; Burmistrov, V. V.; Butov, G. M. Chem. Heterocycl. Compd. 2019, 55, 129. [Khim. Geterotsikl. Soedin.2019, 55, 129.]
- 17.Mikolaichuk, O. V.; Batyrenko A. A.; Kornyakov I. V.; Trifonov, R. E. J. Heterocycl. Chem.2021, DOI: doi/10.1002/jhet.4232. [DOI] [PMC free article] [PubMed]
- 18.Mosmann T. J. Immunol. Methods. 1983;65:55. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 19.CrysAlisPro Software system, version 1.171.39.50a; Rigaku Oxford Diffraction: Oxford, 2019.
- 20.(a) Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv.2015, A71, 3. (b) Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem.2015, C71, 3.
- 21.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. J. Appl. Crystallogr. 2009;42:339. doi: 10.1107/S0021889808042726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 2818 kb)